
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMembrane lipid composition directs the cellular selectivity of antimicrobial metallohelices†
Nicola J.
Rogers
 *ab,
Miles L.
Postings
b,
Ann M.
Dixon
b,
John
Moat
c,
Georgia
Shreeve
b,
Louise
Stuart
b,
Nicholas R.
Waterfield
d and
Peter
Scott
b
*ab,
Miles L.
Postings
b,
Ann M.
Dixon
b,
John
Moat
c,
Georgia
Shreeve
b,
Louise
Stuart
b,
Nicholas R.
Waterfield
d and
Peter
Scott
b
aDepartment of Chemistry, Hong Kong Baptist University, Kowloon Tong, Hong Kong. E-mail: nicolarogers@hkbu.edu.hk
bDepartment of Chemistry, University of Warwick, Coventry CV4 7AL, UK
cSchool of Life Sciences, University of Warwick, Gibbet Hill Campus, Coventry, UK
dWarwick Medical School, University of Warwick, Coventry, UK
First published on 5th March 2025
Abstract
Two enantiomeric pairs of iron(II) metallohelices, available as water-soluble, stable, and optically pure bimetallic complexes, differ principally in the length of the central hydrophobic region between two cationic domains, and have distinct activity and cell selectivity profiles against Gram-positive and Gram-negative microbes. The effects of dose concentration and temperature on levels of intracellular accumulation in E. coli and S. aureus, studied via isotopic labelling, indicate that the metallohelices enter the microbial cells via passive diffusion, whereupon (as previously determined) they act on intracellular targets. Whilst the metallohelices with the shorter central hydrophobic regions accumulate less readily than those with the longer hydrophobic bridge in both E. coli and S. aureus cells when incubated at the same concentration, an order of magnitude less is actually required per cell to inhibit growth in E. coli, hence they are more active. Furthermore, these more Gram-negative active compounds (with the shorter central hydrophobic region) are less toxic towards human APRE-19 mammalian cells and equine red blood cells. We hypothesise that these cell selectivities originate from the membrane composition. Dynamic light scattering and zeta potential measurements demonstrate that the more lipophilic metallohelices interact more strongly with the membrane-mimetic vesicles, notably in the charge-neutral mammalian model; thus the selectivity is not simply a result of electrostatic effects. For the less lipophilic metallohelices we observe that the binding affinity with the E. coli model vesicles is greater than with S. aureus vesicles, despite the lower negative surface charge, and this corresponds with the cellular accumulation data and the measured MICs. Specifically, the presence of membrane phosphatidylethanolamine (POPE) significantly increases the binding affinity of these metallohelices, and we postulate that a high proportion of such conical, non-lamellar phospholipids is important for metallohelix transport across the membrane. The metallohelices with the shorter hydrophobic bridge studied have a balance of charge and lipophilicity which allows selective cell entry in E. coli over mammalian cells, while the more lipophilic metallohelices are membrane promiscuous and unselective.
Introduction
Natural antimicrobial peptides (AMPs) are produced by most organisms as a first line of defence against infection and are known to exhibit broad-spectrum activity principally via destabilisation of the membrane, but also by action on intracellular targets and modulation of the host immune system.1–4 Although the many reported AMPs5–7 include various sequences, lengths, and secondary structures, they share two key properties; a positive net charge (often in the +4 to +6 range) leading to selective accumulation at anionic bacterial cell surfaces over neutral mammalian cells, and amphiphilicity via spatially separated hydrophobic sections within their active region8 assisting the interaction with the lipid bilayer whilst maintaining good solubility in water.9–11The clinical development of AMPs (e.g. LL-37 (ref. 12) and LTX-109 (ref. 13)) has been largely limited to use as topical medicines due to toxicity and unfavourable pharmacokinetics when administered systemically. The lack of selectivity is primarily due to their mode of action, targeting ubiquitous structural components of cell membranes, such as lipids, rather than specific proteins or receptors. This does not readily furnish selectivity between microbial and host mammalian cells. AMPs are nevertheless recognised as templates for prospective antimicrobial agents, and the development of ‘synthetic mimics’ has greatly expanded over the last three decades, leading to improvements in potency and reduced size to minimise the cost of production.14 Peptidomimetics range from modified natural peptides,15de novo peptide systems,16 non-peptidic scaffolds that emulate the AMP pharmacocores, including peptide-like oligomers,17 and synthetic polymers and foldamers.16 Whilst these approaches generate molecules that vary significantly in their chemical constitution, the crux of the work is in tuning the amphiphilic balance for all these systems.18
Among the helicate and similar architectures, comprising chiral arrays of organic ligands about multiple metal centres,19 our metallohelices, accessed using highly efficient one-pot self-assembly reactions of pre-programmed fragments at Fe(II) metal centres,20–24 provide uniquely stable, diverse and optically-pure structures (e.g.Scheme 1) reminiscent of the α-helical active units of many AMPs. They constitute an attractive synthetic platform for the generation of water-soluble molecules with net positive charge and readily tuneable patchy hydrophobicity. Being non-peptidic they are expected to resist protease degradation, and being permanently ‘folded’, like stapled peptides,25 the entropic penalties for binding experienced by flexible AMPs are not present. Various architectures of this type have been studied in cancer26–32 and DNA motif binding,33–43 Alzheimer's disease,44–46 diabetes,47 gene delivery,38 and inhibition of ice recrystallisation.48
 | ||
| Scheme 1 Self-assembly iron(II) metallohelix enantiomers 1 and 2 and the 57Fe isotopologues 1′ and 2′via optically pure diamines. Rings colorised pink and green indicate relatively hydrophobic and hydrophilic regions respectively. Insets show space-fill plots of the molecular structures of Zn(II) analogues from published X-ray crystallography data.49 | ||
Thus, while our metallohelices may be considered to emulate19 the properties of AMPs and cell-penetrating peptide domains,20–24 we were intrigued as to why so few examples have thus far shown significant antimicrobial activity.49 We report here on the activities, selectivities, cellular accumulation and model membrane affinities in Gram-negative and Gram-positive bacteria and mammalian cells, of two pairs of iron metallohelix enantiomers (Scheme 1) formed from meta-xylene (1) and dibenzofuran-bridged (2) strands. This work indicates that as with AMPs, the amphipathic architecture of the metallohelix has a profound effect on the ability to enter microbial cells, as well as selectivity versus host cells.
Results and discussion
The four optically pure compounds, Λ-1, Δ-1, Λ-2, and Δ-2 are synthesised directly by self-assembly49 without the need for resolution (Scheme 1); the configuration (Λ or Δ) at each metal centre is determined by the absolute configuration of the diamine. Molecular structures of the Zn(II) metallohelix analogues49 – isostructural with the Fe(II) systems – show that the longer dibenzofuran bridge in 2 results in an intermetallic distance of 14.4 vs. 12.4 Å in 1 and thus greater separation of the centres of charge. Compounds 2 correspondingly have larger hydrophobic regions (dibenzofuran and benzene have log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P values of 4.12 and 2.13 respectively)50 as evidenced by the lack of anion or other close polar contacts in the crystal structures. It is also evident that the torsion angle between the Zn–N(imine) bonds for the same ligand (along the intermetallic axis) are greater for 2 than in 1 (ca. 160° vs. 101°), which results in there being a greater helical ‘twist’ for 2, such that the relative orientations of the tris(pyridyl) ends are not the same in the two structures. Metallohelix 2 is also slightly wider in diameter than 1, due to the dibenzofuran bridge.
P values of 4.12 and 2.13 respectively)50 as evidenced by the lack of anion or other close polar contacts in the crystal structures. It is also evident that the torsion angle between the Zn–N(imine) bonds for the same ligand (along the intermetallic axis) are greater for 2 than in 1 (ca. 160° vs. 101°), which results in there being a greater helical ‘twist’ for 2, such that the relative orientations of the tris(pyridyl) ends are not the same in the two structures. Metallohelix 2 is also slightly wider in diameter than 1, due to the dibenzofuran bridge.
The metallohelices studied have exceptional stability towards hydrolysis (t1/2 of >10 d even at pH 1.5 (ref. 49)). We also tested the stability under antimicrobial experimental conditions (vida infra) and observed the same antimicrobial activity against E. coli ATCC 25922 with compounds that were pre-incubated for a week in Mueller Hinton broth at 37 °C prior to the experiment as with fresh samples.
Compounds 1 show promising activity and selectivity
Using standard microbial assays, minimum inhibitory concentrations (MICs) for the four compounds were determined (Table 1) against the highly virulent antimicrobial susceptibility control strains S. aureus ATCC 29213 and E. coli ATCC 25922, as well as the further ESKAPE51 pathogens E. faecium ATCC 700221 and K. pneumoniae K6. Further, MICs are reported against the E. coli TOP10 and S. aureus USA300 strains that are used in later analytical studies.| MICa in mg mL−1 (μM) | IC50/μM | ||||||
|---|---|---|---|---|---|---|---|
| Gram− bacteria | Gram+ bacteria | ||||||
| Compound | E. coli ATCC 25922 | E. coli TOP10 | K. pneumoniae K6 | S. aureus ATCC 29213 | S. aureus USA300 | E. faecium ATCC 70022 | ARPE-19b |
| a MICs determined via a standard broth microdilution method, following 20 h incubation with compound. b Measured using MTT assay following 96 h incubation of cells with compound. c Positive control standard drugs. | |||||||
| Λ-1 | 4 (2.0) | 2 (1.0) | 32 (15.7) | 16 (7.8) | 16 (7.8) | 4 (2.0) | 6.4 ± 2.4 |
| Δ-1 | 8 (3.9) | 4 (2.0) | 16 (7.8) | 32 (15.7) | 16 (7.8) | 8 (3.9) | 22.4 ± 2.6 |
| Λ-2 | 32 (13.2) | 16 (6.6) | >256 (>106) | 2 (0.8) | 2 (0.8) | 2 (0.8) | 2.0 ± 0.5 |
| Δ-2 | 32 (13.2) | 16 (6.6) | >256 (>106) | 4 (1.7) | 2 (0.8) | 2 (0.8) | 1.9 ± 0.6 |
| Cefoxitinc | 2 | 32 | 32 | 2 | 32 | — | |
| Ampicillinc | — | — | — | — | — | 64 | |
Consistent with previous observations,49 enantiomers 1 were the more active in the Gram-negative bacteria, whilst 2 were the more active in Gram-positive. Minimum bactericidal concentrations (MBCs) were also measured in E. coli and S. aureus (see ESI,† Table S1) and all compounds have MBC/MIC ratios ≤4, indicating that they are bactericidal. There are observable enantiomeric differences between compounds 1 in almost all cell lines, whereas the activities of Λ-2 and Δ-2 perform very similarly.
In preliminary work on assessment of toxicity of the compounds, we measured the concentrations of compounds required to cause 10% haemolysis of equine erythrocytes. This gave promisingly high values for Λ-1 and Δ-1 of 512 μg mL−1, and while those for the enantiomers of 2 were lower at 256 μg mL−1 both are an order of magnitude higher than similar measurements for antimicrobial peptides.52
The toxicity against human epithelial retinal pigment ARPE-19 cells was also measured (Table 1), revealing the significantly lower toxicity of compounds 1, and in particular the Δ-enantiomer, compared with compounds 2. For the purposes of comparison, and noting that the activities are determined in different assay types, we define a dimensionless selectivity index SI = (IC50 in ARPE19)/(MIC in cell line) as depicted in Fig. 1 for each compound and relevant microbial cell line (Gram- positive in black/grey and Gram-negative in red/pink). By this measure, both enantiomers of 1 are selective towards Gram-negative E. coli (SI of Λ-1 = 3.2, thus the MIC concentration is 3.2 times lower than the concentration that inhibits 50% ARPE-19 cells), while only Δ-1 is selective towards K. pneumoniae K6. In contrast, enantiomers 2 show no selectivity towards these Gram-negative strains (SI < 1). Interestingly, while only Λ-2 has significant selectivity for Gram-positive S. aureus, all compounds have good selectivity towards the E. faecium strain.
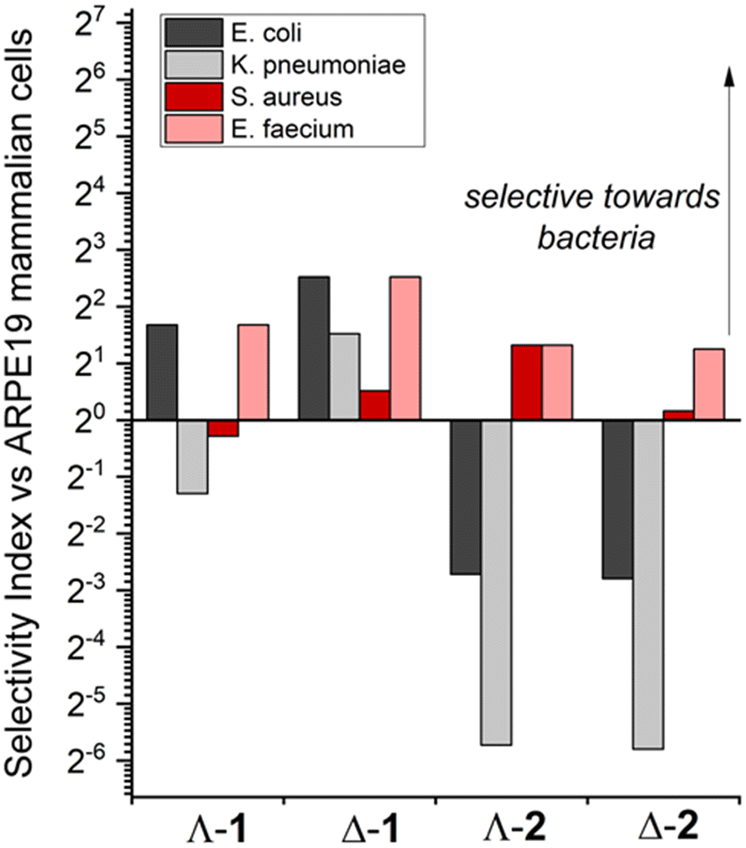 | ||
| Fig. 1 Selectivity of metallohelices for bacterial vs. mammalian cells. The selectivity indices, defined as [mean 96 h IC50 (APRE-19)]/[mean MIC (bacteria)] towards Gram-negative E. coli ATCC 25922 (black) and K. pneumonia K6 (grey), and Gram-positive S. aureus ATCC 29213 (red), and E. faecium SKB (pink). | ||
Passive diffusion leading to selective cellular accumulation
We previously demonstrated via ‘click’ fluorescence microscopy that an alkyne-appended analogue of Λ-1 crossed the intact double membrane of E. coli bacteria, and results from genomic analyses suggest that this compound acts upon intracellular targets (vide infra).49 In order to facilitate a quantitative study of intracellular accumulation of the pairs of enantiomers 1 and 2, 57Fe isotopologues 1′ and 2′ were prepared as shown in Scheme 1 using 57Fe labelled iron(II) chloride as the metal ion source. The compounds were thoroughly characterised (see Materials and methods, and Fig. S1–S12†). Bacterial inhibition assays of these 57Fe complexes were also performed and the MIC values were found to be the same as the corresponding 56Fe compounds (see Tables S2 and S3†).Cultures of non-pathogenic E. coli TOP10 and S. aureus USA300 bacteria were treated with the 57Fe enantiomers at various concentrations and incubation temperatures for 30 min only before inactivation with 4% formaldehyde, pelleting by centrifugation and removal of supernatant. The cell pellets were subject to two further resuspension/centrifugation cycles in buffer to remove any extracellular compound, before digestion in nitric acid and 57Fe quantification (Fig. 2) by ICP-MS.
 | ||
| Fig. 2 Cellular accumulation (ng 57Fe per 108 cells) in E. coli TOP10 bacteria (grey bars) and S. aureus USA300 (red bars) of 57Fe metallohelix isotopologues Λ-1′, Δ-1′, Λ-2′, and Δ-2′ when dosed at (a) equimolar (8 μg mL−1) concentrations, (b) equitoxic (MIC and 0.5 MIC) concentrations, and (c) equitoxic (MIC) concentration, at both 37 °C (solid fill) and 4 °C (dashed fill). Incubation time = 30 min. | ||
As can be seen in Fig. 2(a) compounds 1′ accumulate significantly more in the E. coli (black bars) than in S. aureus (red bars) per cell at the same incubation concentration (8 μg mL−1), whereas for compounds 2′ the accumulations achieved are similar. It is also evident that the more lipophilic compounds 2′ accumulate in the S. aureus cells to a far greater extent (∼20-fold higher) than do compounds 1′ under these conditions. There is also a notable difference in the accumulation of enantiomers of 1′ in E. coli, with Λ-1 achieving ca. double the accumulation of Δ-1, whereas for enantiomers 2′ there is no significant difference in either bacterium.
The cells were also incubated at the corresponding MICs for each compound (Table 1) to establish the concentration required to kill cells [black and red bars, Fig. 2(b)]. In both E. coli and S. aureus the compounds 2′ are far less efficient in acting on the target than 1′; in E. coli the accumulation at the MIC is ca. one order of magnitude higher for compounds 2 than compounds 1, while in S. aureus the difference is a factor of ca. 2. It thus appears that the better activity of compounds 2 against S. aureus is due to their ability to accumulate in the cells more readily, rather than being more potent actors on the target than 1. On the other hand, the better activity of compounds 1 in E. coli is due to these metallohelices being far more potent inhibitors of intracellular targets than compounds 2 in that bacterium. Interestingly, the enantiomeric differences in accumulation of 1′ seen at 8 μg mL−1 are no longer observed at equipotent dose; the two enantiomers are required at the same internalised concentration to inhibit the cell growth, and regardless of the molecular mechanism of action, it is thus this difference in accumulation that underpins the difference in MIC between Δ- and Λ-1 (vide infra).
It is generally considered that a drug molecule either diffuses directly across the lipid membrane or is transported via pores and proteins embedded within the lipid bilayer, although there is some debate on this matter.53–56 In Fig. 2(b), we see that in all cases the accumulation reduces by ∼50% when bacteria are dosed at 0.5× MIC (grey and pink bars). Further, as shown in Fig. 2(c), there is very little effect on accumulation when the incubation temperature is reduced to 4 °C, where ATP-dependent transport mechanisms would be inactive. Hence the transport of these metallohelices across the membrane bilayer appears to be “passive” in nature. Notably the uptake mechanisms for some non-lytic AMPs (e.g. indolicidin and buforin II) have also been shown to be transporter-independent and ascribed to passive translocation directly across membranes.57,58
Considering the selectivities observed above, and the energy-independent accumulation mechanisms implied, we set out to investigate interactions between each compound and membrane bilayers in vitro using vesicle models and circular dichroism (CD) spectroscopy, dynamic light scattering (DLS) and zeta potential titrations.
Membrane- and compound-dependent solvatochromism
While the plasma membrane of most eukaryotic cells contains predominantly zwitterionic phospholipids at the outer leaflet, bacterial cells carry negative surface charge arising variously from anionic phospholipids, plus lipopolysaccharides in Gram-negative bacteria and teichoic acids and lipoteichoic acids in Gram-positive bacteria. Small unilamellar vesicles were thus prepared using outer-leaflet membrane lipid (Scheme 2) compositions59–62 as cytosolic membrane models as follows: (a) E. coli – POPE: 13.40 mg (67.0 wt%), POPG: 4.64 mg (23.2 wt%), CL: 1.96 mg (9.8 wt%); (b) S. aureus – POPG: 11.60 mg (58.0 wt%), CL: 8.40 mg (42.0 wt%); and (c) a generic ‘healthy’ mammalian cell – POPC: 14.22 mg (71.1 wt%), POPE: 1.78 mg (8.9 wt%), cholesterol: 4.00 mg (20.0 wt%). While these models cannot realistically address the complexities of cell surfaces containing microdomains, minor lipid species, embedded proteins and glycolipids, differences in fluidity, diverse fatty acid chain composition, transmembrane potential and pH gradients,59 they do allow us to evaluate the role of the phospholipids within the lipid bilayer of the cytosolic membrane outer leaflet. | ||
| Scheme 2 Phospholipid structures used in model unilamellar vesicles. Ratios shown refer to numbers of atoms and double bonds in the carbon chains. | ||
The Fe(II) metallohelices exhibit strong metal-to-ligand charge-transfer (MLCT) absorption bands in the visible region (450–700 nm), and since these are centred in the most hydrophilic regions of the structures we considered that they may be subject to changes in wavelength (solvatochromism) on association with hydrophobic membrane regions that could be monitored using UV-vis spectroscopy. However, in preliminary experiments we found that this was hampered by scattering from the vesicle particles, and thus moved to CD spectroscopy where only the optically pure helix is detectable in the visible region. Using this technique, we observed significant bathochromic shifts (Fig. 3) in the MLCT peaks at ca. 596 nm for Λ-1 and Λ-2 in the presence of both model S. aureus (6.0 ± 0.5 nm and 9.5 ± 0.5 nm) and E. coli vesicles (4.0 ± 0.5 nm and 6.0 ± 0.5 nm). Both compounds showed a larger shift upon exposure to the most negatively charged vesicles (S. aureus), and in general the most significant changes to the CD spectrum upon addition of vesicles were observed for the more lipophilic compound Λ-2. In contrast, negligible shifts were observed in the presence of model mammalian vesicles.
 | ||
| Fig. 3 CD spectra of (a) Λ-1 and (b) Λ-2 in the presence of no vesicles (green dashes), E. coli vesicles (black solid line), S. aureus vesicles (red solid line), and mammalian vesicles (blue solid line). Metallohelix concentration = 50 μM, lipid concentration = 4 mM, measured in 25 mM sodium phosphate buffer, pH 7.4. Raw data smoothed using Savitzky–Golay method. | ||
Membrane affinity correlates with microbial activity and selectivity
Membrane–metallohelix interactions were further investigated using DLS measurements of vesicle size upon titration with compounds Λ-1 and Λ-2 (for the enantiomers, see Fig. S13–S24†). In Fig. 4(a) and (b) we see that the hydrodynamic diameter of the model E. coli (black lines) and model S. aureus vesicles (red lines) increased with Λ-1 and Λ-2 concentration, indicative of interactions between the cationic metallohelices and the anionic vesicle surfaces causing colloidal instability and vesicle aggregation/fusion. In contrast the neutral (zwitterionic) mammalian vesicles (blue lines) are almost unaffected. In the E. coli vesicles, addition of Λ-1 above a concentration of 75 μM [Fig. 4(a)] leads to a sharp increase in diameter, while for Λ-2 [Fig. 4(b)] an increase in vesicle size is seen at a lower concentration (ca. 40 μM) likely reflecting the more lipophilic nature of this compound. A similar trend is observed in the S. aureus vesicles but at higher compound concentrations, with an increase in vesicle size occurring at concentrations above 150 μM for Λ-1 and ca. 90 μM for Λ-2. | ||
| Fig. 4 DLS Z-average particle diameters of 0.3 mg mL−1 (∼0.4 mM lipid) E. coli (black squares), S. aureus (red circles), and mammalian (blue triangles) membrane-mimetic unilamellar vesicles) in sodium phosphate buffer (25 mM, pH 7.4), upon addition of 0–440 μM Λ-1 (a) and 0–192 μM Λ-2 (b). Zeta potential measurements of 0.5 mg mL−1 (∼0.6 mM lipid) E. coli (black squares), S. aureus (red circles), and mammalian (blue triangles) membrane-mimetic unilamellar vesicles) in sodium phosphate buffer (25 mM, pH 7.4), upon addition of 0–192 μM Λ-1 (solid symbols) and Λ-2 (open symbols). Samples measured at 25 °C, and presented as a mean of 10 (for sizing) or 5 (for zeta potential) independent measurements ±1 standard deviation. | ||
These observations are corroborated by the changes in the zeta potential measurements as shown in Fig. 4(c); the initial potentials for each vesicle system reflect the order of increasing negative surface charge, from mammalian (essentially neutral) via E. coli to S. aureus (most negative). The titrations reveal a striking difference between the interaction of compounds Λ-1 (filled shapes) and Λ-2 (hollow shapes) with the zwitterionic mammalian vesicles, such that while Λ-1 had little effect, addition of Λ-2, which has the same molecular charge, immediately increased the zeta potential (the enantiomers behave similarly – see Fig. S25†). An apparent binding constant (Kapp = 7.2 ± 1.0 × 104 M−1) was estimated from these data (see Fig. S26–28†) as described in Materials and methods, assuming that the change in surface charge is proportional to the number of metallohelix–vesicle interactions.‡ This greater affinity of the more lipophilic 2 than 1 with the mammalian model membranes corresponds with the greater toxicity to ARPE-19 cells (Table 1) and the higher haemolytic activity.
For the microbial membrane models, both compounds increase the zeta potentials in a similar fashion at low concentration. This is as expected since at this point the interaction will be driven by electrostatics, which are similar for all the metallohelices. At higher concentrations, when the membrane negative charge (if any) has been titrated out by adsorption of the metallohelix, so that hydrophobic interactions between the metallohelices and the lipid tails feature more strongly, the effect of Λ-2 (hollow points) is greater in all cases.
Correspondingly, with E. coli vesicles (black lines), the apparent binding constant Kapp estimated for Λ-1 (3.1 ± 0.4 × 104 M−1) is of the same order of magnitude as that estimated for Λ-2 (1.2 ± 0.5 × 104 M−1), i.e. electrostatic effects are dominant throughout. In the S. aureus model membrane (red lines), Kapp for Λ-1 (5.1 ± 1.2 × 103 M−1) is an order of magnitude lower, and in the case of Λ-2 we were not able to fit the binding data as a result of the dominance of the second (presumably hydrophobic) binding event. The greater hydrophobic influence of 2 than 1 is readily observable in these data.
Conclusions
Compounds 1 are more potent than 2 against Gram-negative E. coli and K. pneumoniae, while compounds 2 are the more potent in Gram-positive S. aureus and E. faecium. At the same time, 2 have higher toxicity, as indicated by tests against human ARPE19 and equine red blood cells.Concentration- and temperature-dependence observations of cellular accumulation using 57Fe labelled compounds 1′ and 2′ in E. coli and S. aureus clearly point towards passive translocation of metallohelices rather than energy-dependent (“active”) mechanisms via transporter proteins.56 Fascinatingly, while enantiomers of 2′ accumulate with relative ease in either microbe, the amounts of these compounds required to effect inhibition of E. coli are an order of magnitude higher than those of 1′ (and double in the case of S. aureus) i.e. the antimicrobial effects of 2 are far less efficient than 1 once they enter the cell. We also see that although the enantiomer Λ-1′ achieves higher accumulation than Δ-1′ in E. coli cells at the same dose, the internal concentration at the MIC is the same for both enantiomers, i.e. the higher potency of enantiomer Λ-1 is principally a result of better accumulation efficiency rather than the dynamics at the intracellular target(s).
In solution studies, significant solvatochromic effects are observed in the CD spectra of metallohelices 1 and 2 upon addition of model membrane vesicles, with the greatest shifts observed with the most negatively charged system (S. aureus) and the most lipophilic compounds 2. Measurement of zeta potential changes in the same system also showed that enantiomers 2 interact more strongly than 1 with all the membrane models, particularly in the charge-neutral mammalian mimetic vesicles where electrostatic interactions between the membrane and metallohelix are unimportant. This membrane promiscuity of compounds 2 is at the core of their lack of selectivity.
Zeta potential measurements further indicate that binding constants for 1 in the E. coli vesicles are an order of magnitude higher than they are in S. aureus. This mirrors their far higher accumulation in E. coli cells [Fig. 2(a)] and lower MICs. Since the S. aureus model vesicles carry more surface charge than the E. coli vesicles, this selectivity is not a result of electrostatics. The E. coli inner membrane contains a significant amount of ‘non-lamellar’ phospholipids i.e. POPE and CL cone-shaped lipids with small head-groups that induce negative curvature, which can lower the interfacial lateral pressure when constrained within a bilayer. POPE has been reported to mediate membrane insertion and binding of proteins e.g. the catalytic domain of leader peptidase63 and prePhoE protein in E. coli,64 and to enhance permeation of the drug doxorubicin.65
We propose that a high proportion of POPE/CL increases the permeability of the E. coli membrane for the less lipophilic compounds 1, whilst the stronger hydrophobic interaction between the lipid bilayer and compounds 2 drives their permeation of the S. aureus and mammalian systems that contain more cylindrical ‘lamellar’ lipids (i.e. 58 wt% POPG in the S. aureus model, 71 wt% POPC in the mammalian model). Consistent with this, Λ-1 and Δ-1 also have activity against the POPE-rich K. pneumoniae K6 bacteria (Table 1), with membranes containing ca. 60% POPE (35% POPG).66 Notably, an alkyne derivative of 1 was shown to accumulate preferentially in dividing E. coli cells,49 where the membrane undergoes changes in composition and fluidity, and for example accumulates the dimeric, dianionic lipid CL at the external negative curvature of the septum. Preferential uptake of the AMPs including Cecropin A has also been observed in exponentially-growing bacteria cf. stationary phase bacteria.67
Overall, the metallohelices 1 have far closer to an optimal balance of charge and lipophilicity to furnish selective cell entry in E. coli over mammalian cells, due to their differences in lipid composition, while 2 more freely enters all cell types tested.
The difference in accumulation of enantiomers 1′ (Λ > Δ) in E. coli mentioned above must arise from interaction with some chiral species, and there are various candidates. Compound Λ-1 has an apparent binding constant Kapp of ca. 108 M−1 at calf thymus DNA, with a particular affinity for G-quadruplex structures68 also competing favourably with DNA-binding DAPI stain in E. coli cells and inhibiting both relaxation and supercoiling of pBR322 plasmid DNA by E. coli topoisomerase I and DNA gyrase respectively.49 Given that genetic material constitutes ca. 1/4 of the dry mass of E. coli cells, differential binding could at least contribute to the observed enantioselective accumulation. We also note Paegel and co-workers' recently demonstrated that enantiomers of neutral mono- and di-peptides permeate at rates that differ by up to an order of magnitude.69 It is thus highly feasible that such phenomena will affect transport of the cationic and permanently folded enantiomers 1.
The cell-uptake mechanism of an antimicrobial agent is important for long-term clinical viability; resistance-development for antimicrobial drugs with transporter-mediated uptake mechanisms is relatively straight forward (i.e. down-regulation of the relevant transporter/receptor), whilst resistance against direct lipoidal diffusion is non-trivial and likely to afford high fitness cost. The proposal that our metallohelices enter bacteria via direct membrane crossing is consistent with the rapid response to sub-lethal levels of Λ-1 observed via transcriptomics analysis in the E. coli strain EHEC O157:H7 Sakai;49 the cells modified gene expression so as to reduce the net surface negative charge and upregulate lipopolysaccharide (LPS) modification and unsaturated fatty acid biosynthesis, whilst down-regulating production of OmpW porins, i.e. pore structures in the outer membrane. Further to this the E. coli did not evolve fully resistant mutants to Λ-1, and slightly tolerant sub-type isolates acquired enzyme-related mutations that would alter the biophysical properties of the LPS outer membrane.
Finally, we note that the potency and selectivity of 1 in E. coli and the potential revealed here for modulation of hydrophobicity suggests that we should explore the chemical space of this very accessible structural platform and undertake further mechanistic studies.
Materials and methods
Metallohelices 1 and 2 were synthesised as described previously49 and 57Fe isotopologues were synthesised on a smaller scale using isotopically enriched 57FeCl2 purchased from CK Isotopes. Phospholipids, cardiolipin, and cholesterol were purchased from Sigma-Aldrich or Avanti Polar Lipids, Inc.Synthesis of 57Fe-metallohelices
In brief, diamine (3.0 eq.) and corresponding pyridinecarboxaldehyde (6.0 eq.) were dissolved in methanol (25 mL) and stirred for 24 h at ambient temperature. Anhydrous 57FeCl2 (2.0 eq.) was added and an instantaneous colour change to deep purple was observed. The solution was then heated at reflux (80 °C) for 48 hours (open to air) then concentrated under reduced pressure. The crude was dissolved in minimum methanol (∼2 mL) then pipetted into ethyl acetate (75 mL). The precipitate was filtered by fine filter paper and washed with ethyl acetate (3 × 25 mL) to give the desired product as a dark purple solid. The solid was dried overnight at 50 °C under vacuum. 57Fe complex NMR spectra very similar to the previously published 56Fe analogues.49![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH), 8.37 (s, 3H, OCH2CCHC), 7.77 (t, 3JHH = 7.5 Hz, 6H, NCHCH), 7.68 (d, 3JHH = 7.0 Hz, 6H, NCHCH), 7.55–7.15 (m, 21H, OCH2CCHCH, OCH2CHCCH, NCHCHCH), 7.04 (t, 3JHH = 7.0 Hz, 6H, OCH2CHCCH), 6.88 (d, 3JHH = 7.5 Hz, 12H, OCH2CHCCHCH), 6.79 (d, 3JHH = 7.5 Hz, 6H, NCCH), 6.56 (s, 6H, OCH2CHCCHCHCH), 5.84 (d, 3JHH = 11.0 Hz, 6H, OCH2CH), 5.05 (d, 2JHH = 10.5 Hz, 6H, OCH2C), 4.40 (t, 2JHH/3JHH = 11.0 Hz, 6H, OCH2CH), 3.09 (d, 2JHH = 10.5 Hz, 6H, OCH2CH). 13C{1H} NMR (126 MHz, 298 K, MeOD): δC 172.9 (NCH), 160.3 (NCHC), 154.5 (NCHCCH, 139.9 (NCHCH), 139.4 (OCH2C), 135.8 (OCH2CHC), 130.6 (OCH2CHCCH, OCH2CCHCH), 130.3 (NCHCH), 130.2 (OCH2CHCCHCH, NCHCHCH), 129.3 (OCH2CHCCHCHCH), 128.8 (CCHC), 76.0 (OCH2C), 74.4 (OCH2CH), 72.4 (OCH2CH). HRMS: calculated for [Fe2L3]4+m/z 444.1683, found m/z 444.1647. FTIR: ν cm−1 3353 w (br), 3028 w, 2860 w, 1610 w, 1593 w, 1494 w, 1470 m, 1458 m, 1385 w, 1357 m, 1300 m, 1239 w, 1102 m, 1070 s, 1001 m, 930 w, 887 w, 831 w, 751 s. Elemental analysis found (calculated for C108H102Cl457Fe2N12O6·6H2O): % C 63.68 (63.97), H 5.82 (5.67), N 8.25 (8.29).
CH), 8.39 (s, 3H, OCH2CCHC), 7.76 (t, 3JHH = 7.5 Hz, 6H, NCHCH), 7.68 (s, 6H, NCHCH), 7.57–7.17 (m, 21H, OCH2CCHCH, OCH2CHCCH, NCHCHCH), 7.04 (t, 3JHH = 7.5 Hz, 6H, OCH2CHCCH), 6.84 (m, 18H, OCH2CHCCHCH, NCCH), 6.55 (s, 6H, OCH2CHCCHCHCH), 5.88 (d, 3JHH = 10.5 Hz, 6H, OCH2CH), 5.06 (d, 2JHH = 10.5 Hz, 6H, OCH2C), 4.40 (t, 2JHH/3JHH = 11.0 Hz, 6H, OCH2CH), 3.08 (d, 2JHH = 10.5 Hz, 6H, OCH2CH). 13C{1H} NMR (126 MHz, 298 K, MeOD): δC 172.9 (NCH), 160.3 (NCHC), 154.5 (NCHCCH, 139.9 (NCHCH), 139.4 (OCH2C), 135.8 (OCH2CHC), 130.5 (OCH2CHCCH, OCH2CCHCH), 130.3 (NCHCH), 130.2 (OCH2CHCCHCH, NCHCHCH), 129.3 (OCH2CHCCHCHCH), 128.8 (CCHC), 75.9 (OCH2C), 74.4 (OCH2CH), 72.4 (OCH2CH). HRMS: calculated for [Fe2L3]4+m/z 444.1683, found m/z 444.1673. FTIR: ν cm−1 3350 w (br), 3026 w, 2861 w, 1610 w, 1592 w, 1498 w, 1469 m, 1458 m, 1385 w, 1355 m, 1298 m, 1237 w, 1100 m, 1069 s, 1004 m, 930 w, 887 w, 831 w, 752 s. Elemental analysis found (calculated for C108H102Cl457Fe2N12O6·6H2O): % C 63.59 (63.97), H 5.87 (5.67), N 8.18 (8.29).
CH), 9.02 (s, 6H, OCH2CCHC), 7.77 (d, 3JHH = 8.0 Hz, 6H, OCH2CCH), 7.71 (t, 3JHH = 6.5 Hz, 6H, NCCH), 7.62 (d, 3JHH = 8.0 Hz, 6H, OCH2CCHCH), 7.47 (s, 6H, Py), 7.18–7.14 (m, 6H, Py), 7.07 (t, 3JHH = 7.0 Hz, 6H, Ph), 6.97 (t, 3JHH = 7.0 Hz, 12H, Ph), 6.67 (d, 3JHH = 5.0 Hz, 6H, NCH), 6.52 (s, 6H, Ph), 5.57 (d, 3JHH = 9.0, 6H, CH2CH), 4.52 (d, 2JHH = 9.0 Hz, 6H, OCH2–DBF), 4.04 (t, 3JHH = 10.5 Hz, 6H, OCH2CH), 3.78 (d, 2JHH = 8.5 Hz, 6H, OCH2–DBF), 2.37 (d, 2JHH = 8.0 Hz, 6H, OCH2CH). 13C{1H} NMR (126 MHz, 298 K, MeOD): δC 171.3 (NCH), 159.0 (NCHC), 156.8 (DBF–C), 153.0 (Py–CH), 138.3 (Py–CH), 134.4 (Py–C), 132.2 (DBF–C), 130.3 (DBF–CH), 128.9/128.7/128.0/127.2 (Ar–CH), 123.6 (DBF–C), 121.8 (DBF–CH), 111.7 (DBF–CH), 73.5 (CH2–DBF), 72.5 (CH2CH), 70.9 (CH2CH). HRMS: calculated for [Fe2L3]4+m/z 511.6776, found m/z 511.6760. FTIR: ν cm−1 3369 w (br), 3031 w, 2857 m, 1608 w, 1589 w, 1484 w, 1474 m, 1450 m, 1389 m, 1363 w, 1240 w, 1208 m, 1183 m, 1102 m, 1073 s, 1028 m, 998 m, 800 w, 757 s, 699 s. Elemental analysis found (calculated for C126H108Cl457Fe2N12O9·11H2O): % C 63.77 (63.37), H 5.76 (5.49), N 6.82 (7.04).
CH), 9.02 (s, 6H, OCH2CCHC), 7.77 (d, 3JHH = 8.0 Hz, 6H, OCH2CCH), 7.71 (t, 3JHH = 6.0 Hz, 6H, NCCH), 7.61 (d, 3JHH = 8.0 Hz, 6H, OCH2CCHCH), 7.47 (d, 3JHH = 5.5 Hz, 6H, Py), 7.19–7.14 (m, 6H, Py), 7.07 (t, 3JHH = 7.0 Hz, 6H, Ph), 6.97 (t, 3JHH = 7.0 Hz, 12H, Ph), 6.67 (d, 3JHH = 5.0 Hz, 6H, NCH), 6.52 (s, 6H, Ph), 5.57 (d, 3JHH = 9.0, 6H, CH2CH), 4.52 (d, 2JHH = 8.5 Hz, 6H, OCH2–DBF), 4.04 (t, 3JHH = 8.5 Hz, 6H, OCH2CH), 3.78 (d, 2JHH = 8.5 Hz, 6H, OCH2–DBF), 2.37 (d, 2JHH = 8.5 Hz, 6H, OCH2CH). 13C{1H} NMR (126 MHz, 298 K, MeOD): δC 171.3 (NCH), 159.0 (NCHC), 156.8 (DBF–C), 152.9 (Py–CH), 138.3 (Py–CH), 134.4 (Py–C), 132.2 (DBF–C), 130.3 (DBF–CH), 128.9/128.7/128.0/127.2/127.0 (Ar–CH), 123.6 (DBF–C), 121.8 (DBF–CH), 111.7 (DBF–CH), 73.5 (CH2–DBF), 72.5 (CH2CH), 70.9 (CH2CH). HRMS: calculated for [Fe2L3]4+m/z 511.6776, found m/z 511.6760. FTIR: ν cm−1 3362 w (br), 3029 w, 2854 m, 1608 w, 1589 m, 1486 w, 1472 m, 1452 m, 1385 m, 1361 w, 1241 w, 1210 m, 1183 m, 1100 m, 1071 s, 1029 m, 999 m, 801 w, 757 s, 698 s. Elemental analysis found (calculated for C126H108Cl457Fe2N12O9·11H2O): % C 63.32 (63.37), H 5.85 (5.49), N 6.86 (7.04).
CH), 8.37 (s, 3H, OCH2CCHC), 7.77 (t, 3JHH = 7.5 Hz, 6H, NCHCH), 7.68 (d, 3JHH = 7.0 Hz, 6H, NCHCH), 7.55–7.15 (m, 21H, OCH2CCHCH, OCH2CHCCH, NCHCHCH), 7.04 (t, 3JHH = 7.0 Hz, 6H, OCH2CHCCH), 6.88 (d, 3JHH = 7.5 Hz, 12H, OCH2CHCCHCH), 6.79 (d, 3JHH = 7.5 Hz, 6H, NCCH), 6.56 (s, 6H, OCH2CHCCHCHCH), 5.84 (d, 3JHH = 11.0 Hz, 6H, OCH2CH), 5.05 (d, 2JHH = 10.5 Hz, 6H, OCH2C), 4.40 (t, 2JHH/3JHH = 11.0 Hz, 6H, OCH2CH), 3.09 (d, 2JHH = 10.5 Hz, 6H, OCH2CH). 13C{1H} NMR (126 MHz, 298 K, MeOD): δC 172.9 (NCH), 160.3 (NCHC), 154.5 (NCHCCH, 139.9 (NCHCH), 139.4 (OCH2C), 135.8 (OCH2CHC), 130.6 (OCH2CHCCH, OCH2CCHCH), 130.3 (NCHCH), 130.2 (OCH2CHCCHCH, NCHCHCH), 129.3 (OCH2CHCCHCHCH), 128.8 (CCHC), 76.0 (OCH2C), 74.4 (OCH2CH), 72.4 (OCH2CH). HRMS: calculated for [Fe2L3]4+m/z 444.1683, found m/z 444.1647. FTIR: ν cm−1 3353 w (br), 3028 w, 2860 w, 1610 w, 1593 w, 1494 w, 1470 m, 1458 m, 1385 w, 1357 m, 1300 m, 1239 w, 1102 m, 1070 s, 1001 m, 930 w, 887 w, 831 w, 751 s. Elemental analysis found (calculated for C108H102Cl457Fe2N12O6·6H2O): % C 63.68 (63.97), H 5.82 (5.67), N 8.25 (8.29).
CH), 8.39 (s, 3H, OCH2CCHC), 7.76 (t, 3JHH = 7.5 Hz, 6H, NCHCH), 7.68 (s, 6H, NCHCH), 7.57–7.17 (m, 21H, OCH2CCHCH, OCH2CHCCH, NCHCHCH), 7.04 (t, 3JHH = 7.5 Hz, 6H, OCH2CHCCH), 6.84 (m, 18H, OCH2CHCCHCH, NCCH), 6.55 (s, 6H, OCH2CHCCHCHCH), 5.88 (d, 3JHH = 10.5 Hz, 6H, OCH2CH), 5.06 (d, 2JHH = 10.5 Hz, 6H, OCH2C), 4.40 (t, 2JHH/3JHH = 11.0 Hz, 6H, OCH2CH), 3.08 (d, 2JHH = 10.5 Hz, 6H, OCH2CH). 13C{1H} NMR (126 MHz, 298 K, MeOD): δC 172.9 (NCH), 160.3 (NCHC), 154.5 (NCHCCH, 139.9 (NCHCH), 139.4 (OCH2C), 135.8 (OCH2CHC), 130.5 (OCH2CHCCH, OCH2CCHCH), 130.3 (NCHCH), 130.2 (OCH2CHCCHCH, NCHCHCH), 129.3 (OCH2CHCCHCHCH), 128.8 (CCHC), 75.9 (OCH2C), 74.4 (OCH2CH), 72.4 (OCH2CH). HRMS: calculated for [Fe2L3]4+m/z 444.1683, found m/z 444.1673. FTIR: ν cm−1 3350 w (br), 3026 w, 2861 w, 1610 w, 1592 w, 1498 w, 1469 m, 1458 m, 1385 w, 1355 m, 1298 m, 1237 w, 1100 m, 1069 s, 1004 m, 930 w, 887 w, 831 w, 752 s. Elemental analysis found (calculated for C108H102Cl457Fe2N12O6·6H2O): % C 63.59 (63.97), H 5.87 (5.67), N 8.18 (8.29).
CH), 9.02 (s, 6H, OCH2CCHC), 7.77 (d, 3JHH = 8.0 Hz, 6H, OCH2CCH), 7.71 (t, 3JHH = 6.5 Hz, 6H, NCCH), 7.62 (d, 3JHH = 8.0 Hz, 6H, OCH2CCHCH), 7.47 (s, 6H, Py), 7.18–7.14 (m, 6H, Py), 7.07 (t, 3JHH = 7.0 Hz, 6H, Ph), 6.97 (t, 3JHH = 7.0 Hz, 12H, Ph), 6.67 (d, 3JHH = 5.0 Hz, 6H, NCH), 6.52 (s, 6H, Ph), 5.57 (d, 3JHH = 9.0, 6H, CH2CH), 4.52 (d, 2JHH = 9.0 Hz, 6H, OCH2–DBF), 4.04 (t, 3JHH = 10.5 Hz, 6H, OCH2CH), 3.78 (d, 2JHH = 8.5 Hz, 6H, OCH2–DBF), 2.37 (d, 2JHH = 8.0 Hz, 6H, OCH2CH). 13C{1H} NMR (126 MHz, 298 K, MeOD): δC 171.3 (NCH), 159.0 (NCHC), 156.8 (DBF–C), 153.0 (Py–CH), 138.3 (Py–CH), 134.4 (Py–C), 132.2 (DBF–C), 130.3 (DBF–CH), 128.9/128.7/128.0/127.2 (Ar–CH), 123.6 (DBF–C), 121.8 (DBF–CH), 111.7 (DBF–CH), 73.5 (CH2–DBF), 72.5 (CH2CH), 70.9 (CH2CH). HRMS: calculated for [Fe2L3]4+m/z 511.6776, found m/z 511.6760. FTIR: ν cm−1 3369 w (br), 3031 w, 2857 m, 1608 w, 1589 w, 1484 w, 1474 m, 1450 m, 1389 m, 1363 w, 1240 w, 1208 m, 1183 m, 1102 m, 1073 s, 1028 m, 998 m, 800 w, 757 s, 699 s. Elemental analysis found (calculated for C126H108Cl457Fe2N12O9·11H2O): % C 63.77 (63.37), H 5.76 (5.49), N 6.82 (7.04).
CH), 9.02 (s, 6H, OCH2CCHC), 7.77 (d, 3JHH = 8.0 Hz, 6H, OCH2CCH), 7.71 (t, 3JHH = 6.0 Hz, 6H, NCCH), 7.61 (d, 3JHH = 8.0 Hz, 6H, OCH2CCHCH), 7.47 (d, 3JHH = 5.5 Hz, 6H, Py), 7.19–7.14 (m, 6H, Py), 7.07 (t, 3JHH = 7.0 Hz, 6H, Ph), 6.97 (t, 3JHH = 7.0 Hz, 12H, Ph), 6.67 (d, 3JHH = 5.0 Hz, 6H, NCH), 6.52 (s, 6H, Ph), 5.57 (d, 3JHH = 9.0, 6H, CH2CH), 4.52 (d, 2JHH = 8.5 Hz, 6H, OCH2–DBF), 4.04 (t, 3JHH = 8.5 Hz, 6H, OCH2CH), 3.78 (d, 2JHH = 8.5 Hz, 6H, OCH2–DBF), 2.37 (d, 2JHH = 8.5 Hz, 6H, OCH2CH). 13C{1H} NMR (126 MHz, 298 K, MeOD): δC 171.3 (NCH), 159.0 (NCHC), 156.8 (DBF–C), 152.9 (Py–CH), 138.3 (Py–CH), 134.4 (Py–C), 132.2 (DBF–C), 130.3 (DBF–CH), 128.9/128.7/128.0/127.2/127.0 (Ar–CH), 123.6 (DBF–C), 121.8 (DBF–CH), 111.7 (DBF–CH), 73.5 (CH2–DBF), 72.5 (CH2CH), 70.9 (CH2CH). HRMS: calculated for [Fe2L3]4+m/z 511.6776, found m/z 511.6760. FTIR: ν cm−1 3362 w (br), 3029 w, 2854 m, 1608 w, 1589 m, 1486 w, 1472 m, 1452 m, 1385 m, 1361 w, 1241 w, 1210 m, 1183 m, 1100 m, 1071 s, 1029 m, 999 m, 801 w, 757 s, 698 s. Elemental analysis found (calculated for C126H108Cl457Fe2N12O9·11H2O): % C 63.32 (63.37), H 5.85 (5.49), N 6.86 (7.04).
Preparation of model vesicles
Lipid films were formulated by dissolving the chosen lipids (20 mg total, see below) in chloroform:methanol (2:1, v:v, 1.5 mL), and removing the solvent using a rotary evaporator (bath at 20 °C, 1 h) to deposit a thin film on the wall of a round-bottomed flask. The films were hydrated with sodium phosphate buffer (6.7 mL, 25 mM, pH 7.4), to give stock solutions (3 mg mL−1 lipid). To form unilamellar vesicles, the aqueous lipid suspensions were subjected to four freeze/thaw/sonication cycles: sonication of the sample (4 × 30 s); freezing the sample at −20 °C; thawing the sample at room temperature. The following phospholipids which were used for each membrane–mimetic system: E. coli – POPE: 13.40 mg (67.0 wt%), POPG: 4.64 mg (23.2 wt%), CL: 1.96 mg (9.8 wt%); S. aureus – POPG: 11.60 mg (58.0 wt%), CL: 8.40 mg (42.0 wt%); mammalian – POPC: 14.22 mg (71.1 wt%), POPE: 1.78 mg (8.9 wt%), cholesterol: 4.00 mg (20.0 wt%).
Microbiology and cell culture
Bacterial strains E. coli ATCC 25922 (Castellani and Chalmers), E. faecium ATCC 700221 (Orla-Jensen/Schleifer and Kilpper-Balz), K. pneumoniae ATCC 700603 (K6) and S. aureus ATCC 29213 (Rosenbach), and antibiotic stock solutions were provided by John Moat (Antimicrobial Screening Facility, University of Warwick). Bacterial strains E. coli ATCC PTA-10989 (TOP10) and S. aureus ATCC BAA-1717 (USA300) and were provided by Professor Nick Waterfield (Warwick Medical School, University of Warwick).ARPE-19 human retinal epithelial cells were obtained from ATCC and cultured in DMEM/F12 culture medium containing L-glutamine (2.5 mM), sodium pyruvate (0.5 mM), HEPES buffer (15 mM) and foetal calf serum (10% v/v). Fresh equine blood was purchased from Fisher Scientific.
Antimicrobial activity
A standard broth microdilution method was employed, in agreement with the Clinical and Laboratory Standards Institute (CLSI) guidelines.A 3.2 mg mL−1 stock solution of each compound was prepared in water, corresponding to a 12.5-fold concentration of the highest concentration tested, 256 μg mL−1. In a sterile 96-well plate, 32 μL of each 3.2 mg mL−1 stock was added to 168 μl of antibiotic-free CAMHB to return 512 μg mL−1 solutions. These solutions were subject to twofold serial dilution in CAMHB. Overnight cultures of each bacterial strain in CAMHB were diluted in the same medium to a cell concentration of 1 × 106 CFU ml−1, before 100 μL addition of this culture to each compound well, allowing a 0.008–256 μg mL−1 compound concentration range to be tested. Plates were incubated at 37 °C for 20 h without shaking, and the lowest concentration deemed to inhibit >99% of bacterial growth for each compound was judged to be the MIC. Positive (culture only, no antimicrobial) and negative controls (CAMHB only) were used to ensure suitable bacterial growth and no contamination of media, respectively. Appropriate clinical antimicrobials (ampicillin, tetracycline, ticarcillin, etc.) were used as quality controls and compared to literature values to validate MIC values. Results were repeated in triplicate.
Antiproliferative activity in mammalian cells
ARPE-19 (human retinal pigment epithelial) cells were incubated in 96-well plates at a cell concentration of 0.5 × 104 cells per mL. The cells were used when between 50 and 80% confluent in the stock flasks. Complete cell media containing DMEM, supplemented with 10% foetal calf serum and L-glutamine (2 mM), was used to prepare the desired cell concentration and reference wells. Plates containing cells were incubated for 24 h at 37 °C in 5% CO2 atmosphere, prior to drug exposure. Cell media (200 μL) was added to the reference cells and differing concentrations of drug solution (200 μL) were added to the remaining wells. The plates were incubated for a further 96 h at 37 °C in 5% CO2 atmosphere. 3-(4,5-Dimethylthiazol-1-yl)-2,5-diphenyltetrazolium bromide (MTT) solution (0.5 mg mL−1, 20 μL per well) was added to each well and incubated for 4 h at 37 °C in 5% CO2 atmosphere. Upon completion all solutions were removed from the wells and DMSO (150 μL) was added to each well to dissolve the purple formazan crystals. A ThermoScientific Multiskan EX microplate photometer was used to measure the absorbance at 540 nm.Lanes containing 100% cell media and untreated cells were used as a blank and 100% cell survival respectively. Cell survival was determined as the absorbance of treated cells minus the blank cell media, divided by the absorbance of the untreated control; this value was expressed as a percentage. The IC50 values were determined from a plot of percentage cell survival against drug concentration (μM). Assays were conducted in triplicate and the mean IC50 ± standard deviation was determined.
Haemolysis assays
Fresh equine blood was centrifuged (1000 × g, 10 min) and the supernatant was removed. Harvested erythrocytes were washed three times with PBS and then resuspended to a 5% erythrocyte concentration in PBS. Metallohelix compounds were dissolved in PBS to form 3.2 mg mL−1 stock solutions. These stock solutions were used to prepare 1–1024 μg mL−1 serial dilution ranges in 96-well round bottom plates using PBS. The suspended erythrocytes (100 μL) were added to the compound solution wells (100 μL) and incubated without agitation (310 K, 1 h). The haemolytic concentration for each compound was determined for each compound by visual lysis inspection of wells was performed after the incubation period, in which the lowest concentration deemed to cause >10% cell lysis for each compound was judged to be the haemolytic concentration. Controls included a PBS and 1% Triton X-100 as 0 and 100% haemolysis, respectively. Each measurement was performed in triplicate.57Fe-isotopologue cellular accumulation experiments
Overnight cultures of S. aureus USA300 and E. coli TOP10 were grown in CAMHB (310 K) to exponential phase (OD600 ≈ 0.5). Once at exponential phase, each compound was added to culture (1 mL) to afford the desired compound concentration (8 μg mL−1, MIC or 0.5× MIC) and samples were incubated with shaking (277 K or 310 K, 30 min). Samples were then pelleted by centrifugation (8000 × g, 5 min), supernatant removed, and a twofold repeat of resuspension and pelleting was performed. Following resuspension of all pellets in PBS, samples were subject to a final centrifugation, removal of supernatant, and the resultant pellets were frozen (253 K) until digestion.68% v/v nitric acid (300 μL) was used to digest bacteria pellets before the suspensions were heated overnight at 348 K to ensure complete digestion. Each sample was diluted 20-fold using 18.2 MΩ cm Milli-Q water to attain concentrations of 3.4% v/v nitric acid (total dissolved solids <0.2% w/v). 57Fe calibration solutions were prepared in the range 0.1–1000 ppb using 10 ppm (10 μg mL−1) 57Fe plasma standard solution (Thermo Fisher Scientific) using 3.4% v/v nitric acid. 57Fe accumulation was recorded using an Agilent 7900 ICP-MS spectrometer running in He gas mode.
Dynamic light scattering (DLS) experiments
DLS spectra were measured on a Malvern ZetaSizer Nano-Series DLS instrument. Measurements were performed using disposable micro polystyrene cuvettes using the following standard parameters: dispersant refractivity index = 1.331, viscosity = 0.9074 cP, material RI = 1.35, material absorption = 0.000, temperature = 25 °C, equilibrium time = 300 s, with 12 measurements taken per sample.Electrophoretic mobility (zeta potential) measurements
Zeta potential measurements were collected on an Anton-Paar particle size analyser instrument using an Omega cuvette and the following standard parameters: dispersant refractive index = 1.331, viscosity = 0.9041 cP, relative permittivity = 76.64, temperature = 25 °C, equilibration time = 30 s, with 5 measurements taken per sample. Data presented as the mean of 5 measurements ±1 standard deviation.Apparent binding constants were estimated by fitting the change in zeta potential as a function of metallohelix concentration using eqn (1), assuming that the change in the surface charge is proportional to the number of metallohelix–vesicle interaction:
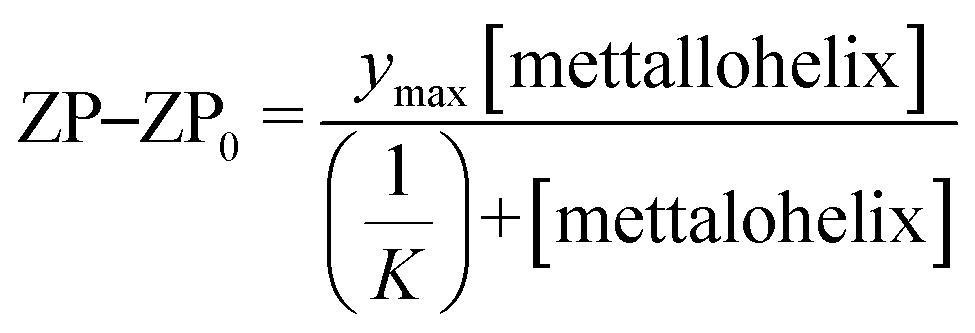 | (1) |
Circular dichroism
CD spectra were measured on Jasco J-1500 spectrometer. Measurements were collected in a 0.1 cm path-length quartz cuvette using the following standard parameters: photometric mode = CD, measurement range = 800–190 nm, data pitch = 0.2 nm, D.I.T. = 1 s, bandwidth = 2.00 nm, scanning speed = 100 nm min−1, with 9 accumulations taken per sample to give an average spectrum with reduced noise.Metallohelix stock solutions (0.5 mM in 10% CH3OH, water) were diluted to 4.91 × 10−5 M using either sodium phosphate buffer (25 mM, pH 6.8) or membrane-mimetic vesicle stock (2.725 mg mL−1; ∼3.64 × 10−3 M). Sodium phosphate baselines were collected using the same cuvette and parameters and subtracted from raw spectra to reduce background noise. Solutions containing vesicles were rescanned after one hour and after two hours to ensure that changes were not occurring over this time frame.
Data availability
All data are contained in the manuscript and ESI.†Author contributions
NJR, PS, and AMD designed the research, NJR, MLP, JM, GS, and LS performed experiments, NJR, MLP, AMD, PS, and NW analysed data, NJR, PS, and AMD wrote the paper. NJR supervised researchers and coordinated the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
NJR thanks Hong Kong Baptist University and The University of Warwick for support. The authors would like to acknowledge the University of Warwick Antimicrobial Screening Facility, Dr Hannah Bridgewater for cell cytotoxicity studies, and the Polymer Research Technology Platform at Warwick for assistance in the research described in this publication. We dedicate this publication to John Moat – a kind and diligent scientist with a wealth of knowledge and experience in microbiology.Notes and references
- R. E. W. Hancock and A. Rozek, FEMS Microbiol. Lett., 2002, 206, 143–149 CrossRef CAS PubMed.
- K. A. Brogden, Nat. Rev. Microbiol., 2005, 3, 238–250 CrossRef CAS PubMed.
- M. R. Yeaman and N. Y. Yount, Pharmacol. Rev., 2003, 55, 27–55 CrossRef CAS PubMed.
- J. F. Marcos and M. Gandía, Expert Opin. Drug Discovery, 2009, 4, 659–671 CrossRef CAS PubMed.
- G. Wang, X. Li and Z. Wang, Nucleic Acids Res., 2016, 44, D1087–D1093 CrossRef CAS PubMed.
- https://aps.unmc.edu/ .
- M. Zasloff, Nature, 2002, 415, 389–395 CrossRef CAS PubMed.
- R. E. W. Hancock, Lancet, 1997, 349, 418–422 CrossRef CAS PubMed.
- Z. Oren and Y. Shai, Peptide Sci., 1998, 47, 451–463 CrossRef CAS.
- J.-P. S. Powers and R. E. W. Hancock, Peptides, 2003, 24, 1681–1691 CrossRef CAS PubMed.
- L. Zhang, A. Rozek and R. E. W. Hancock, J. Biol. Chem., 2001, 276, 35714–35722 CrossRef CAS PubMed.
- A. Grönberg, M. Mahlapuu, M. Ståhle, C. Whately-Smith and O. Rollman, Wound Repair Regen., 2014, 22, 613–621 CrossRef PubMed.
- A. C. Nilsson, H. Janson, H. Wold, A. Fugelli, K. Andersson, C. Håkangård, P. Olsson and W. M. Olsen, Antimicrob. Agents Chemother., 2015, 59, 145–151 CrossRef PubMed.
- F. Azmi, M. Skwarczynski and I. Toth, Curr. Med. Chem., 2016, 23, 4610–4624 CrossRef CAS PubMed.
- A. Tossi, in Prokaryotic Antimicrobial Peptides: From Genes to Applications, ed. D. Drider and S. Rebuffat, Springer New York, New York, NY, 2011, pp. 81–98 Search PubMed.
- G. N. Tew, R. W. Scott, M. L. Klein and W. F. DeGrado, Acc. Chem. Res., 2010, 43, 30–39 CrossRef CAS PubMed.
- J. A. Patch and A. E. Barron, Curr. Opin. Chem. Biol., 2002, 6, 872–877 CrossRef CAS PubMed.
- E. S. Jayawant, J. Hutchinson, D. Gašparíková, C. Lockey, L. Pruñonosa Lara, C. Guy, R. L. Brooks and A. M. Dixon, ChemBioChem, 2021, 22, 2430–2439 CrossRef CAS PubMed.
- H. Song, M. Postings, P. Scott and N. J. Rogers, Chem. Sci., 2021, 12, 1620–1631 RSC.
- P. G. Dougherty, A. Sahni and D. Pei, Chem. Rev., 2019, 119, 10241–10287 CrossRef CAS PubMed.
- C. Foerg and H. P. Merkle, J. Pharm. Sci., 2008, 97, 144–162 CrossRef CAS PubMed.
- J. P. Richard, K. Melikov, E. Vives, C. Ramos, B. Verbeure, M. J. Gait, L. V. Chernomordik and B. Lebleu, J. Biol. Chem., 2003, 278, 585–590 CrossRef CAS PubMed.
- K. Splith and I. Neundorf, Eur. Biophys. J., 2011, 40, 387–397 CrossRef CAS PubMed.
- R. Tréhin and H. P. Merkle, Eur. J. Pharm. Biopharm., 2004, 58, 209–223 CrossRef PubMed.
- L. D. Walensky and G. H. Bird, J. Med. Chem., 2014, 57, 6275–6288 CrossRef CAS PubMed.
- V. Brabec, S. E. Howson, R. A. Kaner, R. M. Lord, J. Malina, R. M. Phillips, Q. M. A. Abdallah, P. C. McGowan, A. Rodger and P. Scott, Chem. Sci., 2013, 4, 4407–4416 RSC.
- R. A. Kaner, S. J. Allison, A. D. Faulkner, R. M. Phillips, D. I. Roper, S. L. Shepherd, D. H. Simpson, N. R. Waterfield and P. Scott, Chem. Sci., 2016, 7, 951–958 RSC.
- A. D. Faulkner, R. A. Kaner, Q. M. A. Abdallah, G. Clarkson, D. J. Fox, P. Gurnani, S. E. Howson, R. M. Phillips, D. I. Roper, D. H. Simpson and P. Scott, Nat. Chem., 2014, 6, 797–803 CrossRef CAS PubMed.
- H. Song, N. J. Rogers, S. J. Allison, V. Brabec, H. Bridgewater, H. Kostrhunova, L. Markova, R. M. Phillips, E. C. Pinder, S. L. Shepherd, L. S. Young, J. Zajac and P. Scott, Chem. Sci., 2019, 10, 8547–8557 RSC.
- H. Song, S. J. Allison, V. Brabec, H. E. Bridgewater, J. Kasparkova, H. Kostrhunova, V. Novohradsky, R. M. Phillips, J. Pracharova, N. J. Rogers, S. L. Shepherd and P. Scott, Angew. Chem., Int. Ed., 2020, 59, 14677–14685 CrossRef CAS PubMed.
- J. P. C. Coverdale, H. Kostrhunova, L. Markova, H. Song, M. Postings, H. E. Bridgewater, V. Brabec, N. J. Rogers and P. Scott, Dalton Trans., 2023, 52, 6656–6667 RSC.
- H. Song, H. Kostrhunova, J. Cervinka, J. Macpherson, J. Malina, T. Rajan, R. Phillips, M. Postings, S. Shepherd, X. Zhang, V. Brabec, N. J. Rogers and P. Scott, Chem. Sci., 2024, 15, 11029–11037 RSC.
- J. Malina, P. Scott and V. Brabec, Nucleic Acids Res., 2015, 43, 5297–5306 CrossRef CAS PubMed.
- A. D. Zhao, S. E. Howson, C. Q. Zhao, J. S. Ren, P. Scott, C. Y. Wang and X. G. Qu, Nucleic Acids Res., 2017, 45, 5026–5035 CrossRef CAS PubMed.
- C. Zhao, H. Song, P. Scott, A. Zhao, H. Tateishi-Karimata, N. Sugimoto, J. Ren and X. Qu, Angew. Chem., Int. Ed., 2018, 57, 15723–15727 CrossRef CAS PubMed.
- J. Malina, H. Kostrhunova, H. Song, P. Scott and V. Brabec, J. Enzyme Inhib. Med. Chem., 2023, 38, 2198678 CrossRef PubMed.
- J. Malina, H. Kostrhunova, P. Scott and V. Brabec, Nucleic Acids Res., 2023, 51, 7174–7183 CrossRef CAS PubMed.
- J. Malina, H. Kostrhunova, V. Novohradsky, P. Scott and V. Brabec, Nucleic Acids Res., 2022, 50, 674–683 CrossRef CAS PubMed.
- O. Hrabina, J. Malina, P. Scott and V. Brabec, Chem. – Eur. J., 2020, 26, 16554–16562 CrossRef CAS PubMed.
- J. Malina, P. Scott and V. Brabec, Sci. Rep., 2020, 10, 14543 CrossRef CAS PubMed.
- J. Malina, P. Scott and V. Brabec, Chem. – Eur. J., 2020, 26, 8435–8442 CrossRef CAS PubMed.
- O. Hrabina, J. Malina, H. Kostrhunova, V. Novohradsky, J. Pracharova, N. Rogers, D. H. Simpson, P. Scott and V. Brabec, Inorg. Chem., 2020, 59, 3304–3311 CrossRef CAS PubMed.
- J. Malina, P. Scott and V. Brabec, Dalton Trans., 2015, 44, 14656–14665 RSC.
- M. Li, S. E. Howson, K. Dong, N. Gao, J. Ren, P. Scott and X. Qu, J. Am. Chem. Soc., 2014, 136, 11655–11663 CrossRef CAS PubMed.
- Y. Guan, Z. Du, N. Gao, Y. Cao, X. Wang, P. Scott, H. Song, J. Ren and X. Qu, Sci. Adv., 2018, 4, eaao6718 CrossRef PubMed.
- Z. Du, C. Liu, Z. Liu, H. Song, P. Scott, X. Du, J. Ren and X. Qu, Chem. Sci., 2023, 14, 506–513 RSC.
- Z. Liu, D. Yu, H. Song, M. L. Postings, P. Scott, Z. Wang, J. Ren and X. Qu, ACS Nano, 2023, 17, 8141–8152 CrossRef CAS PubMed.
- D. E. Mitchell, G. Clarkson, D. J. Fox, R. A. Vipond, P. Scott and M. I. Gibson, J. Am. Chem. Soc., 2017, 139, 9835–9838 CrossRef CAS PubMed.
- D. H. Simpson, A. Hapeshi, N. J. Rogers, V. Brabec, G. J. Clarkson, D. J. Fox, O. Hrabina, G. L. Kay, A. K. King, J. Malina, A. D. Millard, J. Moat, D. I. Roper, H. Song, N. R. Waterfield and P. Scott, Chem. Sci., 2019, 10, 9708–9720 RSC.
- C. Hansch, A. Leo and D. Hoekman, Exploring QSAR - Hydrophobic, Electronic, and Steric Constants, American Chemical Society, USA, 1995 Search PubMed.
- L. B. Rice, J. Infect. Dis., 2008, 197, 1079–1081 CrossRef PubMed.
- J. Lei, L. Sun, S. Huang, C. Zhu, P. Li, J. He, V. Mackey, D. H. Coy and Q. He, Am. J. Transl. Res., 2019, 11, 3919–3931 CAS.
- D. B. Kell, P. D. Dobson and S. G. Oliver, Drug Discovery Today, 2011, 16, 704–714 CrossRef CAS PubMed.
- P. D. Dobson and D. B. Kell, Nat. Rev. Drug Discovery, 2008, 7, 205–220 CrossRef CAS PubMed.
- L. Di, P. Artursson, A. Avdeef, G. F. Ecker, B. Faller, H. Fischer, J. B. Houston, M. Kansy, E. H. Kerns, S. D. Krämer, H. Lennernäs and K. Sugano, Drug Discovery Today, 2012, 17, 905–912 CrossRef CAS PubMed.
- K. Sugano, M. Kansy, P. Artursson, A. Avdeef, S. Bendels, L. Di, G. F. Ecker, B. Faller, H. Fischer, G. Gerebtzoff, H. Lennernaes and F. Senner, Nat. Rev. Drug Discovery, 2010, 9, 597–614 CrossRef CAS PubMed.
- J. Frimodt-Møller, C. Campion, P. E. Nielsen and A. Løbner-Olesen, Curr. Genet., 2022, 68, 83–90 CrossRef PubMed.
- S. Kobayashi, A. Chikushi, S. Tougu, Y. Imura, M. Nishida, Y. Yano and K. Matsuzaki, Biochemistry, 2004, 43, 15610–15616 CrossRef CAS PubMed.
- R. M. Epand and R. F. Epand, J. Pept. Sci., 2011, 17, 298–305 CrossRef CAS PubMed.
- D. Ciumac, H. Gong, X. Hu and J. R. Lu, J. Colloid Interface Sci., 2019, 537, 163–185 CrossRef CAS PubMed.
- P. L. Yeagle, The Structure of Biological Membranes, CRC Press, Boca Raton, 3rd edn, 2011 Search PubMed.
- S. Morein, A.-S. Andersson, L. Rilfors and G. Lindblom, J. Biol. Chem., 1996, 271, 6801–6809 CrossRef CAS PubMed.
- W. van Klompenburg, M. Paetzel, J. M. de Jong, R. E. Dalbey, R. A. Demel, G. von Heijne and B. de Kruijff, FEBS Lett., 1998, 431, 75–79 CrossRef CAS PubMed.
- A. L. J. Van Raalte, R. A. Demel, G. Verberkmoes, E. Breukink, R. C. A. Keller and B. De Kruijff, Eur. J. Biochem., 1996, 235, 207–214 CrossRef CAS PubMed.
- G. Speelmans, R. W. H. M. Staffhorst and B. de Kruijff, Biochemistry, 1997, 36, 8657–8662 CrossRef PubMed.
- H. S. Aluyi, V. Boote, D. B. Drucker, J. M. Wilson and Y. H. Ling, J. Appl. Bacteriol., 1992, 73, 426–432 CrossRef CAS PubMed.
- A. Agrawal, N. Rangarajan and J. C. Weisshaar, Biochim. Biophys. Acta, 2019, 1861, 182990 CrossRef CAS PubMed.
- J. Malina, H. Kostrhunova, P. Scott and V. Brabec, Chem. – Eur. J., 2021, 27, 11682–11692 CrossRef CAS PubMed.
- J. Hu, W. G. Cochrane, A. X. Jones, D. G. Blackmond and B. M. Paegel, Nat. Chem., 2021, 13, 786–791 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Including compound characterisation data, antimicrobial data, DLS and zeta potential data. See DOI: https://doi.org/10.1039/d4md00973h |
| ‡ While acknowledging the limitations of this approach i.e. that it does not discriminate between surface adhesion and lipid insertion, and that relatively few data points are available for fitting, the Kapp values (where 1/Kapp indicates the concentration at which the metallohelices occupy half the ‘binding sites’ of the vesicles under the experimental conditions) are useful for comparison. |
| This journal is © The Royal Society of Chemistry 2025 |