
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
New acridone derivatives to target telomerase and oncogenes – an anticancer approach†
Tiago J. S.
Marques
 a,
Diana
Salvador
bc,
Helena
Oliveira
b,
Vanda V.
Serra
d,
Nicholas
Paradis
e,
Chun
Wu
e,
Vera L. M.
Silva
*a and
Catarina I. V.
Ramos
*a
a,
Diana
Salvador
bc,
Helena
Oliveira
b,
Vanda V.
Serra
d,
Nicholas
Paradis
e,
Chun
Wu
e,
Vera L. M.
Silva
*a and
Catarina I. V.
Ramos
*a
aLAQV-REQUIMTE, Department of Chemistry, University of Aveiro, 3810-193 Aveiro, Portugal. E-mail: c.ramos@ua.pt
bCESAM-Centre for Environmental and Marine Studies, Department of Biology, University of Aveiro, 3810-193 Aveiro, Portugal
cCICECO, Aveiro Institute of Materials, Department of Chemistry, University of Aveiro, 3810-193 Aveiro, Portugal
dCentro de Química Estrutural, Institute of Molecular Sciences, Instituto Superior Técnico, Universidade de Lisboa, Av. Rovisco Pais 1, 1049-001, Lisboa, Portugal
eDepartment of Chemistry and Biochemistry, Rowan University, Glassboro, New Jersey, USA
First published on 17th April 2025
Abstract
In this work, two new acridone derivatives, AcridPy and AcridPyMe, were synthesized, for the first time, aiming to evaluate their potential as quadruplex stabilizers and anticancer agents. AcridPy was synthesized through a very straightforward one-pot sequential chemical reaction involving the Heck cross-coupling reaction of (E)-3-iodo-2-(4-methoxystyryl)-1-methylquinolin-4(1H)-one with a vinyl pyridine followed by in situ electrocyclization and oxidation, while the synthesis of AcridPyMe involved an additional N-methylation of the pyridine ring. Their ability to stabilize G-quadruplex DNA structures, which are associated with the regulation of oncogenes, was assessed using biophysical methods. Both compounds demonstrated significant quadruplex stabilization properties, showing selectivity to G-quadruplexes over duplex DNA. Molecular dynamics simulation experiments supported the preferential binding of AcridPyMe to MYC. The cytotoxicity of these derivatives was further evaluated in vitro in two distinct pancreatic tumor cell lines, PanC-1 and MIA PaCa-2, the lung tumor A549 cell line, the melanoma A375 cell line, and the immortalized human keratinocyte HaCaT cell line, through the evaluation of cell viability. For PanC-1 and MIA PaCa-2, the cell cycle dynamics and apoptotic cell death along with colocalization were also evaluated. The results revealed that AcridPyMe exhibited anticancer activity, correlated with its quadruplex stabilization ability and, although not exclusive, nuclear co-localization was observed. These findings suggest that the newly synthesized cationic acridone is a promising candidate for the development of novel anticancer therapies targeting G-quadruplex structures.
1. Introduction
Despite huge efforts in the last decades aiming at cancer eradication, this complex and challenging disease is still growing in a large group of countries, especially in the developed ones, due to the prevalence of unhealthy lifestyles that increase the risk of cancer onset and development.1,2 Furthermore, tumor formation can also occur due to the modification of certain molecular mechanisms that can promote the development of this disease.One of the most challenging cancers is pancreatic cancer, which can include pancreatic ductal adenocarcinoma, a disease responsible for the death of almost 470![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 people in 2022, being ranked as the sixth most deadly cancer in the world, according to the World Health Organization (WHO).3
000 people in 2022, being ranked as the sixth most deadly cancer in the world, according to the World Health Organization (WHO).3
The overexpression of the enzyme telomerase, which is responsible for the elongation of telomeres and is present in about 90% of all human cancers, is one of the molecular mechanisms responsible for the onset of cancer.4 Telomerase is constituted by two major subunits – a telomerase reverse transcriptase (TERT) and a telomerase RNA component (TERC) – both associated with important structural proteins (NHP2, NOP10, GAR and dyskerin). Together, this enzymatic complex is responsible for the addition of telomeric repeats TTAGGG in 3′ ends of the telomere.5 The expression of this enzyme in most cancers allows the “end-replication problem” to be overcome, leading to a massive growth and uninterrupted cell division scenario of cancer cells.6,7
In addition, oncogenes, such as MYC or KRAS, are also closely associated with the initiation of cancer due to their active role in cell division and proliferation. Before undergoing mutation, these genes are present in the human genome in the form of proto-oncogenes, which can participate in some important cellular processes, such as growth and nuclear transcription factors.8
The presence, in both oncogene and telomeric regions, of repetitive genomic sequences containing high abundance of guanine (G), able to form secondary DNA structures, known as G-quadruplexes (G4s), makes oncogenes and telomeres potential antitumor targets.9,10 G4s are assembled by the stacking of G-quartets (Fig. 1a), formed by four guanines, and supported by Hoogsteen bonds between these nitrogen bases, enabling the establishment of unconventional hydrogen bonds with other purines.11 These genomic elements are highly associated with the regulation of some important cellular processes, such as translation, processing of mRNA, and replication initialization, among others.12,13
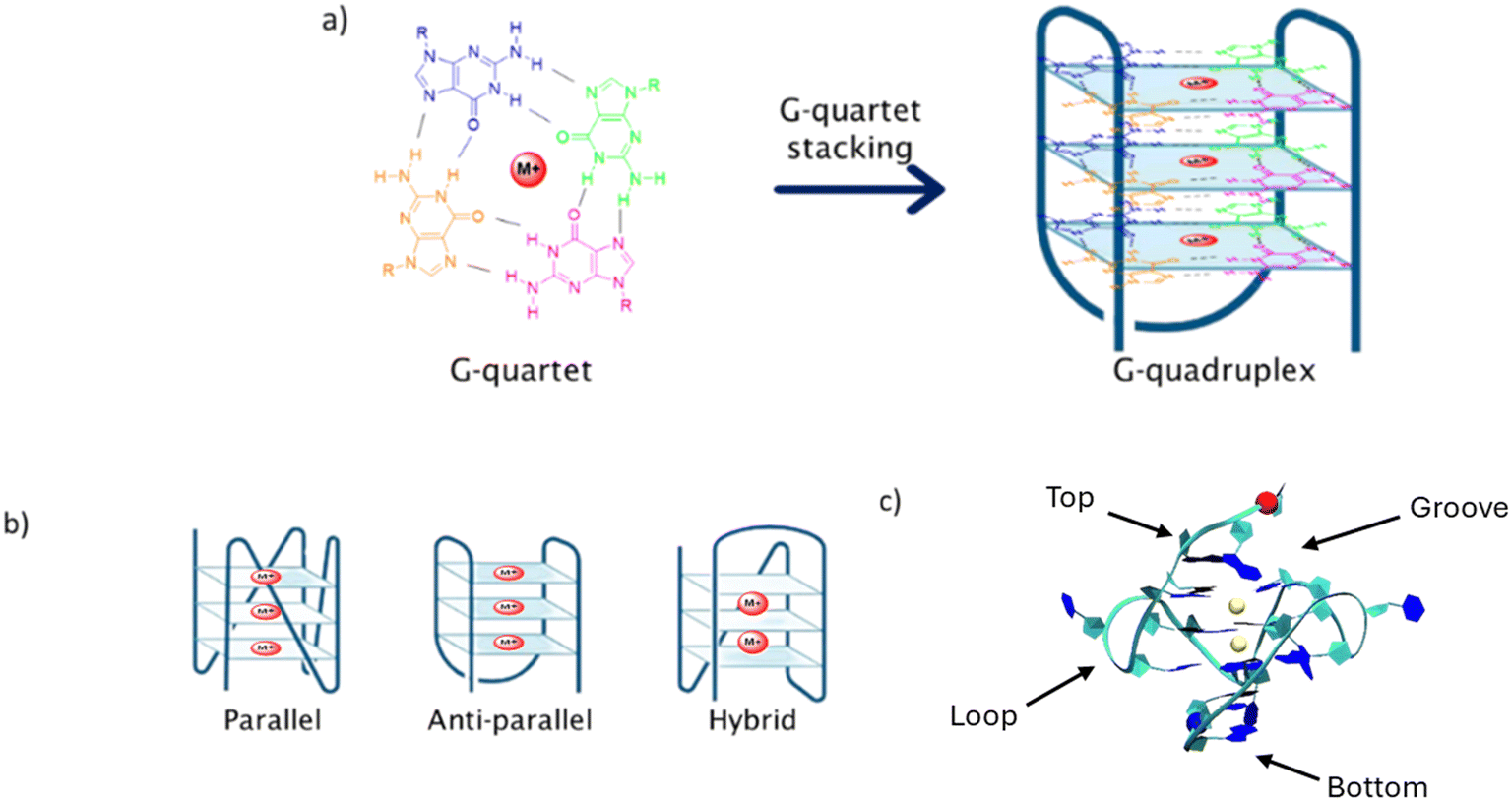 | ||
| Fig. 1 a) Formation of the G-quadruplex structure by stacking of G-quartet units and stabilization by cations (such as Na+ and K+), represented by the red circle M+. Each guanine is represented by a different color. b) Three main structural conformations adopted by G4 structures. c) Four different binding sites of a ligand in the G4 structure. | ||
G4s are highly polymorphic and can adopt different structural conformations depending on the DNA strand orientation, the presence of different ionic neighborhood and internal or external elements, such as temperature. Thus, G4s can be found in three main conformational arrangements: parallel, anti-parallel or hybrid (Fig. 1b).14 Highly abundant cations, such as Na+ and K+, which play important roles in metabolic processes, are usually involved in G4 stabilization. It is interesting to note that the presence of a different cation will give rise to different G4 structural conformation. For example, for the telomeric G4 DNA d[AG3(T2AG3)3], the presence of K+ results in the formation of a hybrid topology, while Na+ gives rise to an antiparallel conformation (Fig. 1b).15
Due to the importance of these secondary genomic structures in cell regulation, the stabilization or unfolding of G4s may help in the enhancement or control of certain processes. This relatively new approach to cancer growth control is focused on the stabilization of G4 structures, therefore preventing the enzymatic activity of telomerase in telomeres or of DNA polymerase II in oncogenes.16,17
In the past years, several ligands have been synthesized and evaluated as potential G4 stabilizing agents, with the aim of interrupting telomerase/polymerase activity.18–22 These ligands can interact with the G4 structure through different modes: in the upper or in the lower end of the G4 tetrad core, known as end-stacking, the interaction occurring with the G4 backbone, so-called groove or loop binding, and between the tetrads, recognized as intercalation (Fig. 1c). The process of intercalation between tetrads is considered a difficult process, since the structural conformation of the G4 is stable and presents some rigidity. Therefore, this specific type of binding should involve a high amount of energy, thus end-stacking, loop or groove binding modes are considered energetically more favorable.23
A large number of organic compounds have been evaluated as G4 stabilizers and as potential anticancer drugs, with the 2,6-diaminoanthraquinone derivative (Fig. 2) identified as the first G4-stabilizing organic ligand.24Fig. 2 shows examples of other promising ligands studied since then, along with their given/commercial name. Some of these molecules, such as CX-5461, are already in clinical phase trials and present favorable interactions with the G4 DNA and direct correlation with the tumor's growth control.20,25,26 Other well-known G4 ligands include large heterocyclic compounds like phthalocyanines and porphyrins, such as P2-PPh3. These two families of ligands are widely studied as G4 stabilizers due to their particular planar core, similar to the G-quartet design.22,27–30
 | ||
| Fig. 2 Some examples of G4 stabilizing ligands. | ||
Among G4 stabilizers, special attention is being focused on acridones. These heterocyclic derivatives are well-known for their diverse biological activities, including antimicrobial, antiviral, and anticancer properties.31–33 The planar structure of acridones allows for effective interaction with DNA, making them particularly attractive as candidates for G4 stabilization. Previous studies have shown that certain acridone derivatives can interact with nucleic acids,34–37 but there is a limited understanding of their specific interactions with G4 structures and their potential as anticancer agents. By comparing the structures presented in Fig. 2, it is possible to observe a similarity between the acridine core (in red) of BRACO-19 and the central core of the acridones AcridPy and AcridPyMe (vide infraScheme 1) described herein. In addition, it is interesting to note that several studies indicate that ligands having charged substituents exhibit higher affinity towards G4 than its neutral analogues.22,38 This characteristic was also considered in the design of the ligands described in this study.
 | ||
| Scheme 1 Synthesis steps of both AcridPy and AcridPyMe. | ||
The present work aims to evaluate the ability of the two novel acridone derivatives, AcridPy and AcridPyMe, to stabilize G4 DNA structures (G4 Tel, MYC and KRAS) and to identify their potential to be used in anticancer therapies. The interaction between the neutral and cationic acridone derivatives with synthetic DNA structures was assessed through several biophysical and biochemical techniques, including fluorescence spectroscopy, fluorescence intercalator displacement (FID) and circular dichroism (CD).39–42 Molecular Dynamics (MD) simulations were also conducted to assess the preferential binding of the ligands to the selected G4 sequences in comparison to a double-stranded sequence (ds26). Cytotoxicity was evaluated in two distinct pancreatic tumor cell lines, PanC-1 and MIA PaCa-2, the lung tumor A549 cell line, the melanoma A375 cell line, and the immortalized human keratinocyte HaCaT cell line, through the evaluation of cell viability. For PanC-1 and MIA PaCa-2, cell cycle analysis, apoptosis, and colocalization by FLIM microscopy were also evaluated.
All together the results demonstrated the potential of the novel cationic acridone AcridPyMe to target G4 structures and to be used as a new therapeutic agent against pancreatic cancer cells.
2. Results and discussion
2.1. Synthesis and characterization of AcridPy and AcridPyMe
AcridPy was obtained starting from the (E)-3-iodo-2-(4-methoxystyryl)-1-methylquinolin-4(1H)-one precursor via the Heck reaction with 4-vinylpyridine using a similar procedure to that reported for the synthesis of (E,E)-2,3-distyrylquinolin-4(1H)-one derivatives (Scheme 1).43 However, after the coupling reaction, an in situ electrocyclization and oxidation took place affording the acridone derivative as the main reaction product. The obtained compound was then methylated with iodomethane, resulting in the formation of AcridPyMe. The structures of these compounds (Scheme 1) were confirmed by 1H and 13C NMR spectroscopy as well as by mass spectrometry (ESI-MS and ESI-HRMS) (Fig. S1–S6†).The 1H NMR spectrum of AcridPy showed two singlets in the aliphatic region, corresponding to the resonance of the protons of the methoxy (–OCH3) group, at δ = 3.84 ppm, and of the methyl group linked to the nitrogen (–NCH3) in the acridone core, at δ = 4.04 ppm. In the aromatic region the singlets appearing at δ = 8.56 ppm and δ = 7.82 ppm were ascribed to the resonance of protons 1 and 4, respectively, from the new fused aromatic ring (please see numbering of both compounds in Fig. S1 and S3†). The other aromatic protons were identified based on their chemical environment and showed the expected splitting pattern. The analysis of the 13C NMR spectrum, with the aid of HSQC and HMBC spectra, showed the presence of two characteristic peaks in the aliphatic region, corresponding to the methoxy group (–OCH3), at δ = 33.2 ppm, and the methyl group (–NCH3), δ = 54.4 ppm.
The main difference between the 1H NMR spectra of AcridPy and AcridPyMe is the presence of an additional singlet at δ = 4.36 ppm due to the resonance of the protons from the extra N-methyl group in the pyridinium unit. In both spectra, the remaining peaks show a similar pattern, apart from small deviations caused by the presence of the new positive charge (Fig. S1 and S3†). Also, in the 13C NMR spectrum of AcridPyMe it was possible to find an extra peak, at δ = 46.6 ppm, corresponding to the new N-methyl group in the pyridine moiety.
The mass spectrum of AcridPy showed a peak at m/z 393.2, corresponding to its protonated molecular ion [M + H]+, while for AcridPyMe the peak at m/z 407.2, corresponding to M+, confirmed the presence of the additional methyl group (Fig. S5 and S6†).
Regarding the photophysical properties of both ligands, their absorption spectra, recorded in DMSO and PBS, showed an intense band at ca. 320 nm, which is characteristic of typical transitions (π → π* or n → π*) in aromatic and conjugated molecules. Less intense bands between 380 and 420 nm reflect the presence of heteroatoms in the ligand structures. The emission spectrum of AcridPy shows a band with a shoulder in the 400 to 500 nm range, while the cationic counterpart shows a broadband between 430 and 630 nm. In both the absorption and emission spectra, the cationic ligand AcridPyMe shows less intense peaks (Fig. 3). It is worth noting that a higher concentration of AcridPyMe (100 μM) was required since the absorbance at 20 μM is around 0.1 thus being in the error range of the equipment and compromising the band definition.
 | ||
| Fig. 3 Absorption spectra a) and emission spectra b) of AcridPy and AcridPyMe. | ||
Considering the absorption and emission spectra of both molecules in DMSO (Fig. 3), a Stokes shift of approximately 120 and 220 nm can be noticed in AcridPy and AcridPyMe, respectively. This large difference between the maximum absorption wavelength and the emission wavelength presents an advantage for the use of these molecules in biological applications, such as cell imaging and tracing.44,45 The fluorescence of both ligands under UV light (∼365 nm) in different solvents is shown in Fig. 4. In PBS, although a considerable decrease of fluorescence was observed, both ligands remained emissive. The fluorescence quantum yield (ΦS) values in DMSO, determined according to the procedure detailed in the Experimental Section, were 0.210 for AcridPy and 0.675 for AcridPyMe (see Fig. S10†).
 | ||
| Fig. 4 Photographs of AcridPyMe (indicated by the red plus sign) and AcridPy solutions in dichloromethane (DCM), methanol (MeOH), dimethyl sulfoxide (DMSO) and phosphate-buffered saline (PBS), taken under UV light (∼365 nm). | ||
2.2. Fluorescence spectroscopy
Usually, during fluorometric titrations, the occurrence of DNA–ligand interactions can be easily detected by an increase (hyperchromism) or a decrease (hypochromism) in fluorescence intensity. The formation of the ligand–DNA complex can lead to structural compactness in both the drug and/or the DNA, which may result in hypochromism. Furthermore, red (bathochromic effect) or blue shifts (hypsochromic effect) can also occur simultaneously, as a result of changes in the electronic environment. The red shift is indicative of a coupling between the π orbital of the DNA and the π* orbital of the ligand, reducing the π–π* transition energy.46 On the other hand, when a ligand binds closely to DNA, the π-electrons in the DNA bases can modify the ligand's electronic transitions, increasing the energy gap between the ligand's ground and excited states which results in a hypsochromic shift (blue shift) in the fluorescence spectrum.42,47,48
These titrations also allow the determination of the apparent dissociation constant (KD), which is indicative of the tendency of the DNA–ligand adduct to dissociate, with lower values of KD corresponding to stronger interactions.49 In addition, the Hill coefficient (n), indicative of the binding cooperativity between the ligand and the G4 DNA, was determined using the Hill saturation model.
In this work, the fluorescence titrations were performed in the presence of G4-Tel, MYC, KRAS and also of the double stranded DNA conformation (ds26) (see oligonucleotide sequences in Table 1) in order to evaluate the selectivity of the new acridones AcridPy and AcridPyMe to G4 structures. Since a large percentage of DNA in the cellular environment exists in the double-stranded form, ligands must exhibit higher affinity and selectivity for G4 DNA structures. Otherwise, their availability to interact with G4 structures will be limited by their interactions with dsDNA.
| Abbreviation | Oligonucleotide sequence | Incidence |
|---|---|---|
| ds26 | 5′-CAA TCG GAT CGA ATT CGA TCC GAT TG-3′ | Duplex DNA |
| MYC | 5′-TGA GGG TGG GTA GGG TGG GTA A-3′ | Oncogene promoter |
| KRAS | 5′-AGG GCG GTG TGG GAA GAG GGA AGA GGG GGA GG-3′ | Oncogene promoter |
| G4 Tel | 5′-AGG GTT AGG GTTAGG GTT AGGG-3′ | Telomeric regions |
For the sake of simplicity, the hybrid conformation of telomeric G4 obtained in the presence of K+ ions will be represented as G4 Tel and the antiparallel conformation obtained when Na+ ions are present will be represented as G4 Tel in Na+.
The spectra obtained from the titrations of AcridPy and AcridPyMe with G4 Tel and MYC are shown in Fig. 5. The spectra obtained for the telomeric G4 Tel in Na+, KRAS, and ds26 are presented, as ESI,† in Fig. S11 and S12, respectively.
 | ||
| Fig. 5 Fluorescence titration spectra of AcridPy with a) G4 Tel, b) MYC, and AcridPyMe with c) G4 Tel and d) MYC. | ||
All titrations resulted in a quenching of the fluorescence intensity, though this quenching had different values for the various oligonucleotides. AcridPy showed a more pronounced quenching when interacting with G4 Tel (∼94%), with a slight quenching being observed for MYC (∼45%). In the case of AcridPyMe although a considerable quenching was observed with G4 Tel (∼73%), a higher quenching was observed towards MYC (∼88%). As can be seen in Fig. 5c and d, S12b (KRAS) and S12c (ds26), along with the above-mentioned quenching effect, pronounced blue shifts, from 13 to 19 nm, were also observed in the titrations of the cationic derivative AcridPyMe with the sequences able to form G4 structures. The occurrence of a blue shift can be correlated with the ligand–DNA binding strength, where larger shifts suggest stronger binding, whereas smaller shifts may indicate weaker or more distant binding.
From the data obtained, it is clear that, when compared to AcridPy, the cationic acridone, AcridPyMe, presents considerable high affinity and selectivity for the G4 structures, the higher blue shifts observed with the sequences able to form G4 structures, pointing to the occurrence of strong interactions. The obtained quenching percentages, spectroscopic shifts and dissociation constants are summarized in Table 2.
| G4 Tel | G4 Tel Na+ | MYC | KRAS | ds26 | ||
|---|---|---|---|---|---|---|
| Note: The second value of quenching corresponds to the observed quenching subtracted from the quenching of titrations made with PBS/Tris-NaCl with both ligands. | ||||||
| AcridPy | Quenching (%) | 94/91 | 87/43 | 45/42 | 86/83 | 93/90 |
| Blueshift (nm) | 1 | 1 | 0 | 2 | 1 | |
| K D (μM) ± error | 2.58 | 3.45 | 10.85 | 5.51 | 3.73 | |
| 0.03 | 0.05 | 0.30 | 0.079 | 0.02 | ||
| Hill coefficient (n) | 7.83 | 4.66 | 4.68 | 8.07 | 15.27 | |
| AcridPyMe | Quenching (%) | 73/47 | 72/41 | 88/62 | 69/43 | 43/17 |
| Blueshift (nm) | 13 | 17 | 19 | 3 | 10 | |
| K D (μM) ± error | 3.30 | 2.08 | 1.86 | 9.62 | 14.95 | |
| 0.11 | 0.07 | 0.04 | 0.70 | 1.00 | ||
| Hill coefficient (n) | 0.98 | 0.80 | 1.33 | 0.97 | 0.98 | |
The AcridPy derivative showed almost no selectivity towards G4 structures, since it also showed a great binding affinity towards duplex DNA (ds26), with a quenching of ∼93%, a slight blueshift of 1 nm and a dissociation constant (KD) of 3.73 μM (see Fig. S11c†). In contrast, AcridPyMe is clearly more selective towards G4, especially for the MYC oncogene, with a KD of 1.86 μM, eight-fold lower than the KD of ds26 (KD = 14.95 μM), therefore revealing a promising and important feature of selectivity of this ligand. Regarding the Hill coefficient (n), AcridPy showed all coefficients higher than 1, indicating a positive cooperativity between the neutral ligand and the selected DNA structures. On the other hand, AcridPyMe presented only a value of n higher than 1 when interacting with MYC structures, showing values lower than 1 when interacting with the remaining DNA sequences, which is indicative of a negative cooperativity.50
Titrations with PBS and Tris-NaCl buffer solutions (blank experiments) were also performed with both ligands to determine the contribution of buffer to the fluorescence quenching observed in each titration (please see Fig. S13†). The observed quenching values in the titrations with PBS solution were 3% and 26% for AcridPy and AcridPyMe, respectively. In the case of Tris-NaCl, values of 44% for AcridPy and 31% for AcridPyMe were observed, showing a higher contribution of this buffer to fluorescence quenching.
To better understand the data obtained and to evaluate the influence of time on the ligand–G4 DNA interaction, the fluorescence intensity of the G4–ligand adducts formed by addition of 2 equiv. of different oligonucleotides to both compounds was monitored for 12 h (see Fig. 6 for G4 Tel and Fig. S14† for the other oligonucleotides). It is interesting to note that AcridPyMe had no or little variation in fluorescence intensity after 12 h of interaction, revealing that all interactions occur almost immediately upon addition of the ligand. On the other hand, the results obtained for AcridPy point to a slower interaction, with a significant fluorescence intensity decrease after a period of 12 h of interaction with the oligonucleotides.
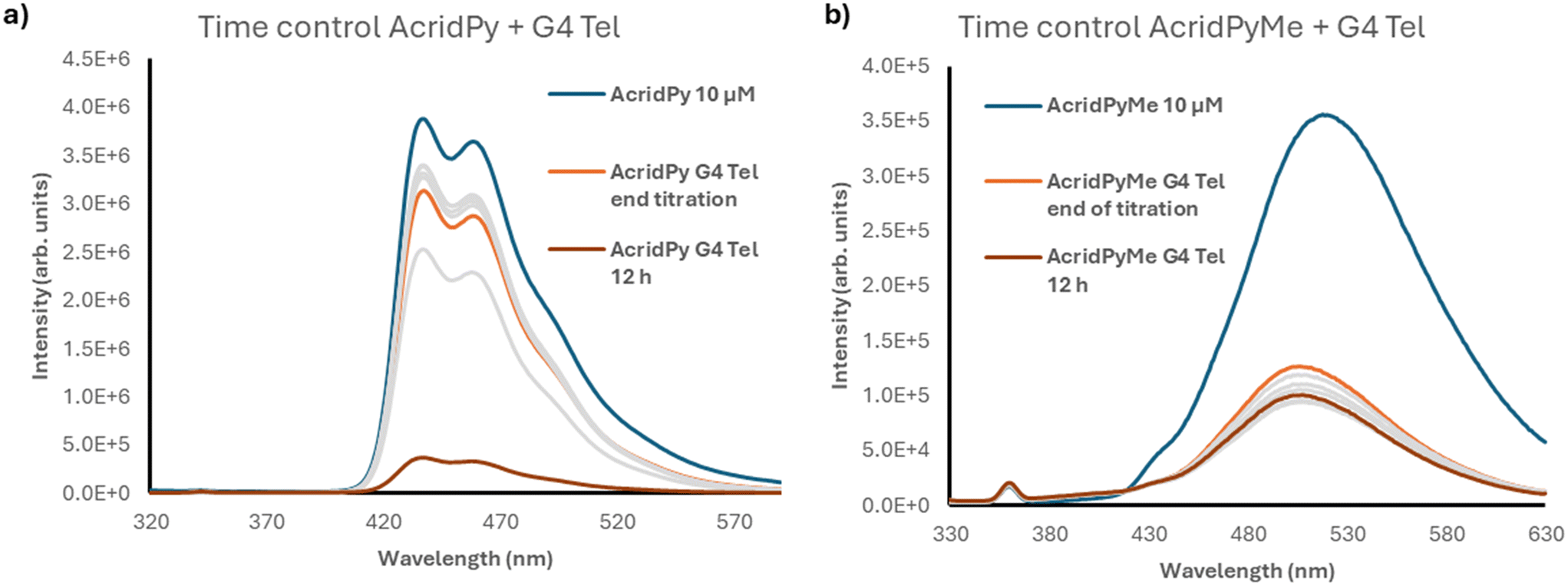 | ||
| Fig. 6 Time control spectra of G4 Tel with 10 μM of a) AcridPy and b) AcridPyMe, respectively. | ||
As depicted in Fig. 6, when comparing the fluorescence intensity of AcridPyMe in the end of the titration and after 12 h of interaction with G4 Tel, a quenching of 20% in the fluorescence occurred. For AcridPy, after 12 h of interaction, a decrease of about 4 times in the fluorescence intensity, corresponding to a quenching percentage of 88.3%, was observed.
These interesting differences observed in time-controlled measurements may be explained by the presence of the positive charge in AcridPyMe, favoring electrostatic interactions with the negative net charge of the oligonucleotide backbone. This correlation between the efficiency of the binding and the presence of positive charges was previously described in porphyrins, where multicharged porphyrins have shown a higher stabilization of the DNA adduct, when compared with the neutral ligands.22
2.2.2.1. Thiazole Orange. Thiazole orange (TO) is a well-known end-stacking binding probe that only emits fluorescence when bound to DNA structures.51 G4-FID assay was performed to assess the ability of both ligands to displace the TO probe from the G4 DNA structures and to obtain the DC50 value, which corresponds to the concentration of the ligand necessary to displace TO by 50% from the preformed TO–DNA adduct. The displacement can be followed by adding increasing amounts of the ligand to the preformed TO–DNA solution. The obtained DC50 reflects the affinity of ligands to the different DNA structures, with a lower concentration of the ligand being indicative of higher affinity of the ligand to DNA structures.
Surprisingly, no significant alteration in fluorescence intensity occurred when AcridPy or AcridPyMe was added to the adduct solutions (Fig. S15†), indicating that TO has not been displaced from the preformed TO–DNA adducts. This might indicate that both ligands preferentially bind DNA through a process different from end-stacking.
Considering the results obtained from the fluorescence titrations, which indicated the occurrence of an interaction between the ligands and G4, we postulate that an adduct comprising TO–DNA–Ligand is probably being formed. This interaction may occur through ligand binding at alternative sites on the G4 structures, possibly through groove or loop binding. It is interesting to note that a decrease in fluorescence intensity is expected when electrostatic, hydrogen bonding or hydrophobic interaction with the sugar-phosphate backbone occurs, as in the case of groove.42
To evaluate the occurrence of a process of groove binding, FID was also performed using the fluorescent probe Hoechst 33258 in substitution of TO, due to its proven groove binding ability.52
2.2.2.2. Hoechst 33258. In this experiment it was possible to observe the displacement of Hoechst 33258 from the Hoechst–G4 adduct, by AcridPyMe, which was shown by the decrease in the fluorescence of the adduct, with a quenching value of nearly 35% (Fig. S16†). Interestingly, a red shift is seen as the titration is performed, indicating that the AcridPyMe–MYC adduct presents a higher maximum wavelength than the Hoechst–MYC adduct. However, after a few more ligand additions, the displacement could not be tracked due to the appearance of a new species in the spectra that presented a larger red shift, probably corresponding to an excess of free ligand that interferes with the fluorescence of the DNA adduct. This displacement may indicate a possible competition between AcridPyMe and Hoechst for the groove binding of the G4 adduct.
2.3. Circular dichroism
The G4 formed in oncogene MYC presents a parallel conformation, characterized by a particular CD spectrum, with a positive and a negative band at about 260 and 240 nm, respectively. On the other hand, the antiparallel conformation, in the case of G4 Tel Na+, displays a positive band at 295 nm and a negative band at 260 nm, whilst hybrid structures present two positive bands at 295 and 260 nm and a negative one at 245 nm.53
CD titrations were employed to evaluate the ability of ligands to bind and/or induce conformational changes in the selected G4 structures (G4 Tel, G4 Tel Na+, and MYC). The spectra were recorded in the absence of ligands and in the presence of increasing concentrations of both ligands, as can be seen in Fig. S17† (G4 Tel and MYC) and in Fig. S18† (G4 Tel Na+). The titrations were performed within a range of 0 to 24 μM concentration, corresponding to 0 to 6 molar equivalents of the ligand when compared to the 4 μM of DNA solutions.
The results show that the CD spectra of all the G4 oligonucleotides did not show significant changes in the presence of either ligand, revealing that both can interact with each DNA sequence without damaging the established conformation. The fact that none of the ligands present the ability to change the established conformation of the chosen G4 DNA in the range of concentrations studied also points that the characteristic conformation of the quadruplex is not compromised by the presence of the ligands.
The ellipticity of these measurements was registered at different wavelengths, according to the topology of G4: 290 nm for parallel topology (MYC) and 295 nm for hybrid structures (G4 Tel). In Fig. 7, the obtained melting spectra of MYC and G4 Tel in the absence or presence of AcridPyMe are presented.
 | ||
| Fig. 7 CD melting spectra of a) MYC and b) G4 Tel in the absence and presence of increased concentrations of AcridPyMe. | ||
The melting temperature (Tm) obtained for MYC and for G4 Tel in the absence of ligand was 76.1 °C and 61.4 °C, respectively. An excess of ligand (8 equiv.) was added to ensure that all possible interactions with the G4 structures took place. After incubation with 8 equiv. of the ligands, the Tm of the MYC–AcridPyMe adduct increased by 10.7 °C to 86.8 °C, while G4 Tel–AcridPyMe showed a less expressive increment of 0.9 °C, giving a Tm of 62.3 °C. These results are in line with the earlier data obtained using fluorescence spectroscopy, confirming the binding ability and higher affinity of AcridPyMe to MYC structures.
On the other hand, the presence of AcridPy in the MYC structure with formation of the MYC–AcridPy adduct resulted in an increase in the Tm of 6.2 °C and a slight increase of 1.7 °C in the case of the adduct G4 Tel–AcridPy (Fig. S19†). These results point to the lower affinity of AcridPy for G4 structures and are again in line with previous data from fluorescence titrations, described in section 3.2.1. These obtained data are summarized in Table 3.
| MYC | G4 Tel | |||||
|---|---|---|---|---|---|---|
| T mi | T mf | ΔTm | T mi | T mf | ΔTm | |
| Note: Tmi – initial melting temperature (G4 alone); Tmf – final melting temperature (G4 + Ligand); ΔTm – melting temperature difference (Tmf − Tmi). All values are expressed in °C. | ||||||
| AcridPy | 76.3 | 82.5 | 6.2 | 61.4 | 63.1 | 1.7 |
| AcridPyMe | 76.1 | 86.8 | 10.7 | 61.4 | 62.3 | 0.9 |
Given the proven stabilization of G4 structures in the presence of the ligands, we decided to complement our studies by performing molecular dynamics to identify the preferential binding modes between both partners, as well as to conduct in vitro assays with two different pancreatic tumor cell lines (see sections 2.4 and 2.5).
2.4. Molecular dynamics of AcridPy and AcridPyMe
Additionally, the Ligand-RMSD (L-RMSD) of AcridPy and AcridPyMe was calculated to check for ligand binding convergence, stability and length of duration for particular binding poses. Similarly, L-RMSD plots were generated (Fig. S20†) and their average values for the last 50 ns of trajectory data were tabulated (Table 4).
| R-RMSD | ||||
|---|---|---|---|---|
| MYC | G4 Tel | KRAS | ds26 | |
| AcridPy | 2.7 | 2.2 | 2.4 | 3.5 |
| AcridPyMe | 2.7 | 2.3 | 3.2 | 3.6 |
| L-RMSD | ||||
|---|---|---|---|---|
| MYC | G4 Tel | KRAS | ds26 | |
| AcridPy | 40.7 | 34.9 | 40.3 | 80.8 |
| AcridPyMe | 50.5 | 37.7 | 51.2 | 53.8 |
| Atom contacts | ||||
|---|---|---|---|---|
| MYC | G4 Tel | KRAS | ds26 | |
| AcridPy | 16.5 | 29.6 | 12.8 | 12.8 |
| AcridPyMe | 41.1 | 15.5 | 20.6 | 13.0 |
For AcridPy, the L-RMSD convergence was achieved at different time points for MYC (>220 ns), G4 Tel (<50 to ∼300 ns, and >420 ns), KRAS (>260 ns) and ds26 (>180 ns). For AcridPyMe, the L-RMSD convergence was similar for MYC (>220 ns), G4 Tel (<50 to ∼300 ns, and >420 ns). KRAS (>260 ns) and ds26 (>50 ns). Within the last 100 ns of simulation time, the L-RMSD values for AcridPy and AcridPyMe were generally consistent for G4 Tel (34.9 Å and 37.7 Å), but not for MYC (40.7 Å and 50.5 Å), KRAS (40.3 Å and 51.2 Å) or ds26 (80.8 Å and 53.8 Å). The much higher AcridPy and AcridPyMe L-RMSD values for ds26 systems than the G4 systems are apparent here. Please note that the high L-RMSD values relative to the R-RMSD values are due to the 10 Å ligand displacement distance from the nucleic acid structures at the beginning of the simulation to allow for global ligand sampling. Some interruptions in the L-RMSD plot for certain systems indicate switching of ligand binding poses. For instance, AcridPy–MYC exhibits two periods of L-RMSD change (0 to ∼60 ns and ∼130 to ∼220 ns); AcridPy–MYC exhibits one period of L-RMSD change (∼210 ns); AcridPyMe–G4 Tel demonstrates a large L-RMSD disturbance late into the simulation before relaxing (∼300 ns to ∼410 ns). Additionally, the global ligand sampling for AcridPy and AcridPyMe for the KRAS system converges much later relative to the MYC, G4 Tel and ds26 systems (>200 ns), suggesting that these ligands have a longer time to binding to KRAS. The binding modes for AcridPy and AcridPyMe to each nucleic acid structure will be examined.
The distribution of AcridPy and AcridPyMe binding to each nucleic structure is shown in Fig. 8.
 | ||
| Fig. 8 Ligand clustering analysis of AcridPy (orange beads) and AcridPyMe (ice blue beads) for MYC, G4 Tel, KRAS and ds26 nucleic acid structures. Each bead represents 1 ns snapshot of simulation time of the C1 ligand atom, within 5 Å of the nucleic acid structure, to simplify the clustering viewpoint. | ||
Generally, different binding modes were observed between AcridPy and AcridPyMe for each nucleic acid system. In MYC, AcridPy showed two binding cluster nodes: a dense cluster at the bottom G-tetrad layer and a much looser cluster at both the top G4 layer and 3rd groove pocket closer towards the 3′ terminus; in contrast, AcridPyMe exhibited three distinct binding clusters at the top G4 layer, the first loop and the 3rd groove pocket. In G4 Tel, AcridPy exhibited a single loose cluster at the top G4 layer, whereas AcridPyMe exhibited two denser clusters at the top and bottom G4 layers. In KRAS, AcridPy and AcridPyMe both exhibited two binding clusters, one located near the first groove region closest to the 5′ termini; however, the second cluster was located at the 3rd groove region (AcridPy) or the bottom G4 layer (AcridPyMe). Lastly in ds26, AcridPy exhibits two binding clusters at both termini of ds26, whereas AcridPyMe exhibits two binding clusters at one of the termini and another cluster at a groove region.
Atom contact analysis was also performed to provide additional insight into the binding preferences of the compounds to each nucleic acid structure. The atom contact plots and average values of the total contacts between AcridPy/AcridPyMe and nucleic acid structure over the last 100 ns of simulation time were both generated (Fig. S21† and Table 4). For MYC, AcridPyMe exhibited greater contacts (41) compared to AcridPy (17). For G4 Tel, the reverse is true for AcridPy (30) compared to AcridPyMe (16). For KRAS, AcridPyMe demonstrated greater contacts (21) than AcridPy (13). Lastly, for ds26, AcridPy and AcridPyMe exhibited the same contact number (13). Conversely, AcridPyMe made most contacts with KRAS (69), followed by ds26 (43), MYC (38) and G4 Tel (22). Hence, AcridPy and AcridPyMe generally engage in more binding with G4s rather than ds26. Combined with the ligand clustering analysis, the additional methyl moiety on AcridPyMe seems to confirm the difference in the ligand binding modes compared to AcridPy for MYC, G4 Tel, KRAS and ds26 systems.
2.5. Biological assays
 | ||
| Fig. 9 MTT assay to assess the AcridPyMe effect on the cell viability of a) PanC-1, b) MIA PaCa-2, c) A549, d) A375, and e) HaCaT cell lines. Data is represented by mean values ± standard deviation of three independent experiments with three technical replicates each. “*” is indicative of statistical significance in comparison to control (p < 0.05). | ||
As can be seen in Fig. 9 and Tables S4 and S5,† the cell viability of both pancreatic cancer cell lines seems to be inversely proportional to the concentration of the ligand, with the 72 h of exposure showing the most promising results. In contrast, AcridPyMe had none to little effect on the cell viability of the A549 (Fig. 9c, Table S6†), A375 (Fig. 9d, Table S7†) and HaCaT (Fig. 9e, Table S8†) cells, with viability values above 84%. In the PanC-1 cell line (Fig. 9a), cell viability decreased with statistical significance when exposed to AcridPyMe concentrations equal or superior to 25 μM for 24 h. After 72 h of exposure, a subtle cytotoxic effect can be seen at a concentration higher than 5 μM.
Regarding the MIA PaCa-2 cells (Fig. 9b), after 24 h of exposure, only the highest concentration of 100 μM showed reduced cell viability with statistical significance. After 72 h of exposure, only concentrations equal or above 10 μM affected MIA PaCa-2 cell viability. Comparing all cell lines, AcridPyMe showed a greater cytotoxic effect on the MIA PaCa-2 cell line above 50 μM, with a calculated IC50 of 72.69 μM after 72 h exposure. This value was not calculated for the other four cell lines since for the range of concentrations tested the decrease in viability of these cell lines did not reach 50%.
AcridPy showed a higher extent of cellular damage, when compared to AcridPyMe, decreasing cell viability of A549, HaCaT and A375 by about 50%, 60% and 30%, respectively, and by about 70% in the case of PanC-1 and MIA PaCa-2, after 72 h exposure (see Fig. S23 and Tables S9–S13†). However, these results were not considered for further in vitro experiments due to the evidence of formation of crystal-like aggregates in cellular medium (Fig. S23f†) in concentrations higher than 10 μM. To better understand their structure and formation process, the observed aggregates will be further studied.
In order to get time-resolved information about AcridPyMe spatial distribution in the cellular microenvironment, fluorescence lifetime images of MIA PaCa-2 cells incubated with AcridPyMe were acquired (Fig. 10b) and compared to fluorescence lifetime images obtained for single cells (Fig. 10a) and to cells incubated with propidium iodide (Fig. 10c), a well-known red fluorescent nuclear and chromosome counterstain.55 Fluorescence lifetime images obtained under similar experimental conditions (fluence rate and λexc), as well as the histograms of average fluorescence lifetimes of each image are shown in Fig. 10a–c and e, respectively.
 | ||
| Fig. 10 Fluorescence lifetime imaging microscopy of a) single cells; b) cells incubated with AcridPyMe; c) cells incubated with PI; d) AcridPyMe in incubation media; e) fluorescence lifetime histograms obtained for 1-D images. | ||
FLIM images of single cells show only residual fluorescence (τm = 3.7 ns) due to intrinsic fluorescence of endogenous fluorophores.56 After AcridPyMe incubation a significant increase in the fluorescence intensity of the cell occurs, as expected for the interaction with the fluorescent AcridPyMe. Simultaneously, an increase in the average fluorescence lifetime is observed (τm = 4.2 ns). The average fluorescence lifetimes of AcridPyMe in cells are slightly lower than the ones obtained in the ensemble measurements of an aqueous medium solution (τm = 5.8 ns), which agrees well with changes in the fluorophore's microenvironment after incubation. Although the spatial distribution of AcridPyMeτm correlates with distinct cell regions (cytoplasm and nucleus), it seems to be preferentially located in the area that overlaps the nucleus region (green spots, Fig. 10b) as compared with the cell region marked with PI (Fig. 10c, red spots, τm = 8 ns). Interestingly, a dividing cell can be observed in Fig. 10b with the two nuclear regions marked with green spots (τm = 4.2 ns, area marked with a white dashed line).
 | ||
| Fig. 11 Cytotoxic effects of AcridPyMe on cell cycle distribution of MIA PaCa-2 cells. a) Cell cycle distribution (%); b) representative histograms of cell cycle analysis (arbitrary units). Data is represented by mean values ± standard deviation of two independent experiments with three technical replicates each and each replicate with at least 5000 events. “*” is indicative of statistical significance in comparison to control (p < 0.05). | ||
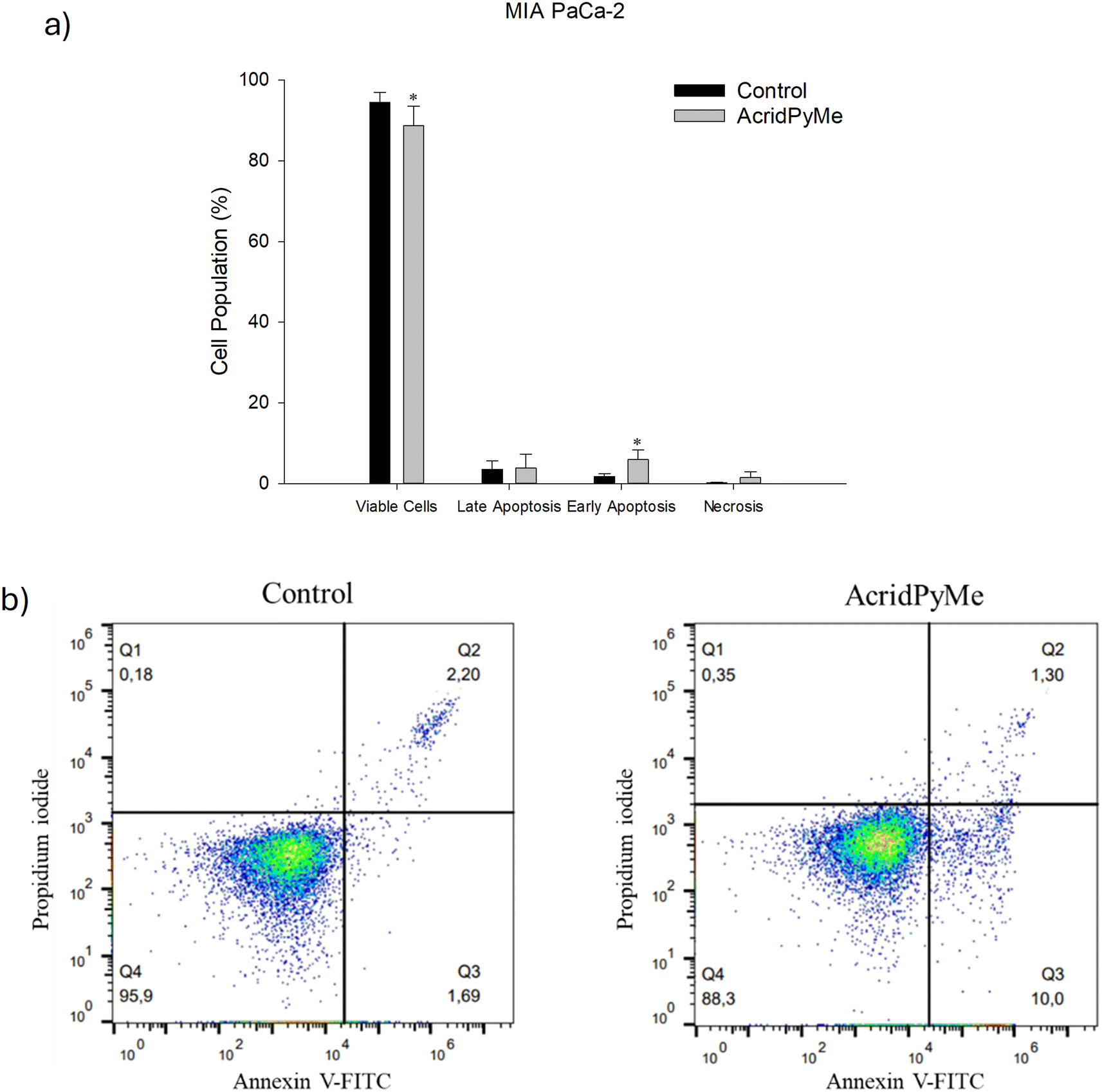 | ||
| Fig. 12 Cytotoxic effects of AcridPyMe on apoptosis in MIA PaCa-2 cells. a) Apoptotic cells (%) after treatment in groups analogous to viable and non-apoptotic; early and late apoptotic cells; b) representative histograms of Annexin V-FITC/propidium iodide. Data is represented by mean values ± standard deviation of two independent experiments with three technical replicates each and each replicate with at least 5000 events. “*” is indicative of statistical significance in comparison to control (p < 0.05). | ||
3. Materials and methods
3.1. Chemicals
All commercial chemicals, solvents and reagents were acquired and used without any further purification. The DNA oligonucleotides were purchased from Eurogentec (Belgium). The DNA stock solutions (1 mM) were prepared in MilliQ waster, using the provided values for the molar extinction coefficient, and working solutions with desired concentrations were prepared with adequate buffer solutions. Stock solutions of AcridPy and AcridPyMe were prepared in dimethyl sulfoxide (DMSO) and stored at room temperature.Thin layer chromatography (TLC) was carried out on precoated sheets with silica gel (Macherey-Nagel UV254). Column chromatography was carried out using silica gel (Merck, 35–70 mesh). 1H and 13C NMR spectra were recorded on a Bruker Avance 500 [500.16 MHz (1H), 125.77 MHz (13C)]. Deuterated methanol (CD3OD) was used as a solvent, and the internal reference was tetramethylsilane (TMS). The chemical shifts of the NMR spectra are expressed in δ (ppm) and the coupling constants (J) in Hz.
ESI-MS spectra were obtained using a linear ion trap mass spectrometer LXQ (ThermoFinnigan, San Jose, CA). Data were acquired, in positive mode, using a source voltage of 5 kV, capillary temperature of 275 °C and a sheath gas flow of 25 U. The Xcalibur data system (version 2.0, ThermoFinnigan, San Jose, CA) was used for data analysis.
High-resolution mass spectra (HRMS) were obtained, in positive mode, in a Q-Exactive® hybrid quadrupole Orbitrap® mass spectrometer (Thermo Fisher Scientific, Bremen, Germany). Data were acquired using 3.0 kV as spray voltage and interfaced with a HESI II ion source, capillary temperature of 320 °C and S-lens rf level 50. ESI analysis was performed at a flow rate of 10 μL min−1, nitrogen was used as sheath and auxiliary gas at flow rates of 5 and 1 (arb. units), respectively. Spectra were analyzed using the Xcalibur version 4.0 (Thermo Scientific, San Jose, CA, USA).
The melting point was obtained using a Büchi Melting Point B-540 within a range of 100 °C to 295 °C, with a gradient of 5 °C min−1. All the compounds are >95% pure by HPLC.
3.2. Preparation of DNA structures
A phosphate buffered saline (PBS) solution containing 20 mM phosphate buffer (10 mL of KH2PO4 1 M and 200 μL of K2HPO4 1 M) and 100 mM KCl was prepared with an adjusted pH of 6.8. Similarly, tris(hydroxymethyl)aminomethane (Tris)-NaCl buffer was prepared from a solution of 10 mL of Tris-HCl 100 mM and 10 mL of NaCl 1 M or LiCl 1 M.The oligonucleotide solutions were heated at 90 °C for 10 minutes and left in ice for cooling for 15 minutes. All solutions were stored at −20 °C. The studied oligonucleotide sequences and corresponding abbreviations are presented in Table 1.
3.3. Synthesis of AcridPy and AcridPyMe
:0.5) as eluent. The compound AcridPy was obtained with a yield of 52%. A molar extinction coefficient (ε306) of 22637 M−1 cm−1, in PBS, was found (Fig. S7†). The purity of AcridPy was determined through HPLC/UV-vis analysis, obtaining a purity higher than 99% (Fig. S8†).
3-(4-Methoxyphenyl)-10-methyl-2-(pyridin-4-yl)acridin-9(10H)-one (AcridPy), brownish solid (yield 52%, 24.3 mg), mp: >235.0 °C (decomposition); 1H NMR (500.16 MHz, CD3OD), δH = 8.56 (s, 1H, H-1), 8.54 (d, J = 5.8 Hz, 2H, H-3′,5′), 8.50 (dd, J = 7.6, 1.6 Hz, 1H, H-8), 7.92–7.86 (m, 2H, H-6, H-5), 7.82 (br s, 1H, H-4), 7.55 (d, J = 5.8 Hz, 2H, H-2′,6′), 7.43 (t, J = 7.6 Hz, 1H, H-7), 7.26 (d, J = 8.6 Hz, 2H, H-2′′,6′′), 6.95 (d, J = 8.6 Hz, 2H, H-3′′,5′′), 4.04 (s, 3H, –NCH3), 3.84 (s, 3H, 4′-OCH3) ppm. 13C NMR (CD3OD, 125.77 MHz), δC = 178.0 (C-9), 160.0 (C-4′′), 154.0 (C-2), 146.5 (C-3), 145.0 (C-1), 143.1 (C-4b), 142.9 (C-4a), 134.6 (C-6), 131.6 (C-1′′), 131.0 (C-2′′,6′′), 129.8 (C-1′), 129.3 (C-2′,6′), 126.6 (C-8), 126.1 (C-3′,5′), 122.1 (C-8a), 121.9 (C-7), 120.5 (C-9a), 117.9 (C-4), 115.7 (C-5), 113.7 (C-3′′,5′′), 54.4 (NCH3), 33.2 (4′′′-OCH3) ppm. MS (ESI) m/z: 393.3 [M + H]+. HRMS-ESI(+): m/z calcd. for C26H20N2O2 393.1598 [M + H]+, found 393.1595.
4-[3-(4-Methoxyphenyl)-10-methyl-9-oxo-9,10-dihydroacridin-2-yl]-1-methylpyridin-1-ium, yellow solid (yield 67%, 17.41 mg), mp: >258.0 °C (decomposition); 1H NMR (500.16 MHz, CD3OD), δH = 8.73 (s, 1H, H-1), 8.72 (d, J = 6.8 Hz, 2H, H-3′,5′), 8.52 (dt, J = 8.0, 1.2 Hz, 1H, H-8), 7.95–7.93 (m, 2H, H-5, H-6), 7.91 (br s, 1H, H-4), 7.88 (d, J = 6.8 Hz, 2H, H-2′,6′), 7.47 (ddd, J = 8.0, 4.7, 3.2 Hz, 1H, H-7), 7.32 (d, J = 8.7 Hz, 2H, H-2′′,6′′), 7.00 (d, J = 8.7 Hz, 2H, H-3′′,5′′), 4.36 (s, 3H, NCH3-Py), 4.12 (s, 3H, NCH3), 3.85 (s, 3H, 4′′-OCH3) ppm. 13C NMR (CD3OD, 125.77 MHz), δC = 178.0 (C-9), 160.1 (C-4′′), 157.7 (C-2), 146.4 (C-3), 144.4 (C-1), 143.9 (C-1′), 143.9 (C-4a), 143.0 (C-4b), 134.8 (C-6), 131.0 (C-2′′,6′′), 130.8 (C-1′′), 130.2 (C-3′,5′), 127.7 (C-2′,6′), 126.7 (C-8), 122.3 (C-9a), 122.3 (C-7), 120.7 (C-8a), 118.5 (C-4), 116.0 (C-5), 114.1 (C-3′′,5′′), 54.6 (4′′-OCH3), 46.65 (NCH3-Py), 33.7 (NCH3) ppm. MS (ESI) m/z: 407.3 M+. HRMS-ESI(+): m/z calcd. for C27H23N2O2 407.1760 M+, found 407.1747.
3.4. UV–vis spectroscopy
The UV–vis absorption spectra were recorded on a Shimadzu UV-2501-PC spectrophotometer. All measurements were restricted to a range from 250 to 550 nm, using a quartz cuvette of 1 cm length, at room temperature. Successive additions of AcridPy or AcridPyMe were made in PBS to determine the extinction coefficients (ε) of these molecules by a calibration curve, using the Beer–Lambert Law| A = ε × b × c | (1) |
3.5. Fluorescence studies
 | (2) |
Rearranging the previous equation, we can determine the Φ of our sample by measuring the absorbance and fluorescence intensity of several standard solutions (in DMSO) with known concentration and by plotting the integrated fluorescence intensity as a function of absorbance (Fig. S10†), resulting in
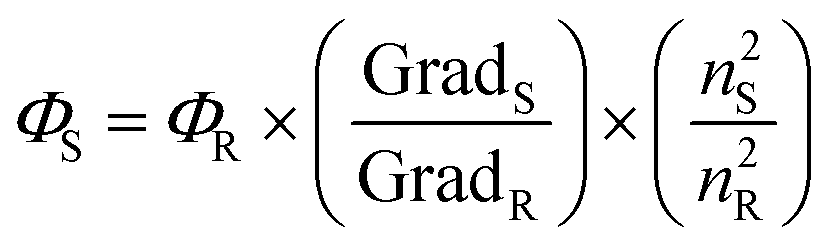 | (3) |
Titrations of ligand solutions at 10 μM with the appropriate oligonucleotide at concentrations ranging from 0 to 16.7 μM were performed in the same equipment. The apparent dissociation constants (KD) were obtained from the changes observed in fluorescence spectra of each oligonucleotide–ligand interaction. The obtained data was converted into the fraction of ligand that is bound to the DNA (α), and plotted using the expression:
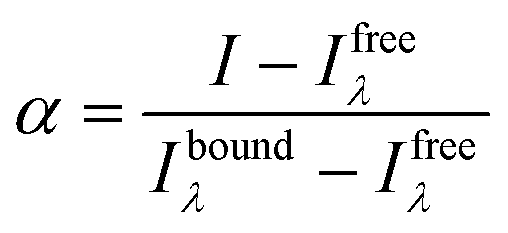 | (4) |
Each KD was then determined following the saturation binding model, using OriginPro 2016, described by the following equation:
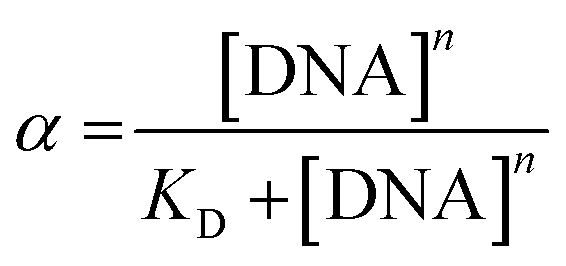 | (5) |
:1 proportion and shaken at 500 rpm for 10 min. The fluorescence was then measured in a Horiba FluoroMax-4 spectrofluorometer, with an excitation wavelength of 485 nm in the emission range of 510–750 nm and slits at 10 nm.
Similarly, FID was also performed using Hoechst 33258 (3.5 μM) and maintaining the other conditions mentioned above. In this case, the spectra were recorded at an excitation wavelength of 340 nm and an emission range between 350 and 600 nm with both excitation and emission slits set at 3 nm.
3.6. Circular dichroism spectroscopy
For CD titration, solutions of oligonucleotides (4 μM) were prepared in 20 mM phosphate buffer (10 mM KH2PO4, 10 mM K2HPO4, pH 7.1) containing 100 mM KCl for c-MYC and G4 Tel, and 100 mM NaCl also for G4 Tel Na+. The ligands were added to the quartz cell solution within a range of 0 to 24 μM.
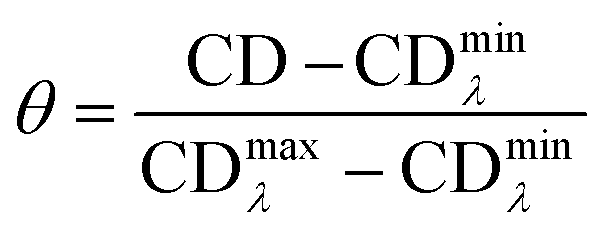 | (6) |
3.7. In silico studies
3.8. Biological assays
 | (7) |
Prior to analysis, samples were centrifuged at 700 g for 5 min, and ethanol was removed. Cells were resuspended with PBS, filtered with nylon filter membranes, and incubated with 50 μg mL−1 of RNase (Merck KGaA, Darmstadt, Germany) for 10 min. Next, 50 μg mL−1 of propidium iodide (PI, ≥94%, Merck KGaA, Darmstadt, Germany) was added and samples were incubated for at least 20 min in the dark. Cell cycle distributions were analyzed on an Attune® Acoustic Focusing Cytometer (Applied Biosystems, Thermo Fischer Scientific, Waltham, MA, USA) flow cytometer. Two independent assays with three replicates each were completed, and at least 5000 events were obtained. The percentage of cells in each cell cycle phase was determined using the FlowJo software (FlowJo LLC, Ashland, OR, USA).
4. Conclusions and future perspectives
The results presented in this work show that these novel acridone derivatives are promising G4 binding ligands, with the ability to stabilize these secondary DNA structures. Fluorescence experiments demonstrated their ability to bind and stabilize G4 DNA, with a particular affinity of AcridPy for telomeric G4 DNA and of AcridPyMe for MYC oncogene sequences. The cationic derivative AcridPyMe showed high selectivity towards G4 structures, rather than duplex DNA. CD melting assays have also shown an enhancement in the melting temperature of the ligand–G4 adduct, especially in the case of the AcridPyMe–MYC adduct, with a difference greater than 10 °C confirming the stabilization of MYC induced by the presence of the ligand. The results observed from in silico studies point to the occurrence of binding on top and/or groove binding modes of both ligands, which was supported by the fluorescent displacement assays performed earlier.The biological in vitro assays also demonstrated a good cytotoxic effect of AcridPyMe in the MIA PaCa-2 cell line, which was sustained by cell viability, cell cycle and apoptosis assays, that showed cell cycle arrest at the G0/G1 phase, a decrease in viable cells and an increase in cells in early apoptosis when treated with AcridPyMe. Internalization showed that AcridPyMe appears to be distributed in the cellular cytoplasm and nucleus, apparently being preferentially located in the area that overlaps the nucleus region.
Overall, the results of this study led to the proposal of the new AcridPyMe ligand as a promising candidate to target the MYC oncogene, effectively stabilizing the signature G4 structures present in this type of DNA sequence and being able to compromise cell proliferation. These findings may contribute, in the future, to the development of G4-targeted cancer therapeutics.
Funding
This work received financial support from PT national funds (FCT/MCTES, Fundação para a Ciência e Tecnologia and Ministério da Ciência, Tecnologia e Ensino Superior) through the project unit UID/50006/2020 (Laboratório Associado para a Química Verde - Tecnologias e Processos Limpos ) through national funds and UID Centro de Estudos do Ambiente e Mar (CESAM) + LA/P/0094/2020. CIV Ramos thanks FCT for funding through program DL 57/ 2016 (CDLCTTRI-047-88-ARH/2018). D. Salvador thanks FCT for the PhD grant (2022.11049.BD). V. V. Serra thanks FCT for the institutional funds provided to Centro de Química Estrutural (UIDB/00100/2020 and UIDP/00100/2020), to the Institute of Molecular Sciences (LA/P/0056/2020), and her research contract (IST/ID/113/2018) funded by national funds (OE), through FCT.Data availability
Data will be made available on request.Conflicts of interest
There is no conflict of interest to declare.Acknowledgements
The authors would like to acknowledge Professor M. Graça Neves for her invaluable contributions for the discussion of this work, which greatly contribute to its success and to Doctor M. Q. Alves for confocal images. The authors acknowledge the support and help from FCT/MCTES (LA/P/0008/2020 https://doi.org/10.54499/LA/P/0008/2020, UID/50006), from UID Centro de Estudos do Ambiente e Mar (CESAM) + LA/P/0094/2020 through national funds, and the Portuguese NMR Network. CIV Ramos thanks FCT for funding through program DL 57/2016 (CDL-CTTRI-047-88-ARH/2018), H. Oliveira thanks FCT for the research contract under the Scientific Employment Stimulus (CEECIND/04050/2017; https://doi.org/10.54499/CEECIND/04050/2017/CP1459/CT0023) and D. Salvador thanks FCT for the PhD grant (2022.11049.BD). Vera L. M. Silva also thanks funding through FCT under the Scientific Employment Stimulus – Institutional Call – reference CEECINST/00026/2018 (https://doi.org/10.54499/CEECINST/00026/2018/CP1521/CT0013). V. V. Serra thanks FCT for the institutional funds provided to Centro de Química Estrutural (UIDB/00100/2020 and UIDP/00100/2020), to the Institute of Molecular Sciences (LA/P/0056/2020), and her research contract (IST/ID/113/2018) funded by national funds (OE), through FCT.References
- L. A. Torre, R. L. Siegel, E. M. Ward and A. Jemal, Global Cancer Incidence and Mortality Rates and Trends—An Update, Cancer Epidemiol., Biomarkers Prev., 2016, 25(1), 16–27, DOI:10.1158/1055–9965.EPI-15–0578
.
- E. J. Gallagher and D. LeRoith, Obesity and Diabetes: The Increased Risk of Cancer and Cancer-Related Mortality, Physiol. Rev., 2015, 95(3), 727–748, DOI:10.1152/physrev.00030.2014
- Cancer Today, https://gco.iarc.fr/today/fact-sheets-cancers (accessed 2024-07-04).
- T. Liu, X. Yuan and D. Xu, Cancer-Specific Telomerase Reverse Transcriptase (TERT) Promoter Mutations: Biological and Clinical Implications, Genes, 2016, 7(7), 38, DOI:10.3390/genes7070038
- I. Udroiu, J. Marinaccio and A. Sgura, Many Functions of Telomerase Components: Certainties, Doubts, and Inconsistencies, Int. J. Mol. Sci., 2022, 23(23), 15189, DOI:10.3390/ijms232315189
- H. Banu Lateef, P. Mitta Suresh, P. Bharathi, S. Pathak and A. Banerjee, A Brief Overview of Telomeres and Telomerase in Aging and Cancer, Curr. Appl. Sci. Technol., 2022, 23(4), 1–24, DOI:10.55003/cast.2023.04.23.015
- M. Gao, G. Zheng, Y. Li, Y. Zhang, P. Hu and Y. Pan, Telomere Length in Multiple Cancer: Insight into Recurrence Risk from a Meta-analysis, J. Gastroenterol. Hepatol., 2023, 38(6), 844–853, DOI:10.1111/jgh.16186
- E. N. Kontomanolis, A. Koutras, A. Syllaios, D. Schizas, A. Mastoraki, N. Garmpis, M. Diakosavvas, K. Angelou, G. Tsatsaris, A. Pagkalos, T. Ntounis and Z. Fasoulakis, Role of Oncogenes and Tumor-Suppressor Genes in Carcinogenesis: A Review, Anticancer Res., 2020, 40(11), 6009–6015, DOI:10.21873/anticanres.14622
- K. D. Makova and M. H. Weissensteiner, Noncanonical DNA Structures Are Drivers of Genome Evolution, Trends Genet., 2023, 39(2), 109–124, DOI:10.1016/j.tig.2022.11.005
- D. J. Patel, A. T. Phan and V. Kuryavyi, Human Telomere, Oncogenic Promoter and 5′-UTR G-Quadruplexes: Diverse Higher Order DNA and RNA Targets for Cancer Therapeutics, Nucleic Acids Res., 2007, 35(22), 7429–7455, DOI:10.1093/nar/gkm711
- E. N. Nikolova, E. Kim, A. A. Wise, P. J. O'Brien, I. Andricioaei and H. M. Al-Hashimi, Transient Hoogsteen Base Pairs in Canonical Duplex DNA, Nature, 2011, 470(7335), 498–502, DOI:10.1038/nature09775
- R. Hänsel-Hertsch, D. Beraldi, S. V. Lensing, G. Marsico, K. Zyner, A. Parry, M. Di Antonio, J. Pike, H. Kimura, M. Narita, D. Tannahill and S. Balasubramanian, G-Quadruplex Structures Mark Human Regulatory Chromatin, Nat. Genet., 2016, 48(10), 1267–1272, DOI:10.1038/ng.3662
- T. M. Bryan, G-Quadruplexes at Telomeres: Friend or Foe?, Molecules, 2020, 25(16), 3686, DOI:10.3390/molecules25163686
- Y. Luo, A. Granzhan, J. Marquevielle, A. Cucchiarini, L. Lacroix, S. Amrane, D. Verga and J.-L. Mergny, Guidelines for G-Quadruplexes: I. In Vitro Characterization, Biochimie, 2023, 214, 5–23, DOI:10.1016/j.biochi.2022.12.019
-
E. Largy, J.-L. Mergny and V. Gabelica, Role of Alkali Metal Ions in G-Quadruplex Nucleic Acid Structure and Stability, in Met Ions Life Sci, Walter de Gruyter GmbH, 2016, vol. 16, pp. 203–258, DOI:10.1007/978-3-319-21756-7_7
- T. Tian, Y.-Q. Chen, S.-R. Wang and X. Zhou, G-Quadruplex: A Regulator of Gene Expression and Its Chemical Targeting, Chem, 2018, 4(6), 1314–1344, DOI:10.1016/j.chempr.2018.02.014
- A. L. Moye, K. C. Porter, S. B. Cohen, T. Phan, K. G. Zyner, N. Sasaki, G. O. Lovrecz, J. L. Beck and T. M. Bryan, Telomeric G-Quadruplexes Are a Substrate and Site of Localization for Human Telomerase, Nat. Commun., 2015, 6(1), 7643, DOI:10.1038/ncomms8643
- C. I. V. Ramos, V. A. S. Almodôvar, N. R. Candeias, T. Santos, C. Cruz, M. G. P. M. S. Neves and A. C. Tomé, Diketopyrrolo[3,4-c]Pyrrole Derivative as a Promising Ligand for the Stabilization of G-Quadruplex DNA Structures, Bioorg. Chem., 2022, 122, 105703, DOI:10.1016/j.bioorg.2022.105703
- G. Zhou, X. Liu, Y. Li, S. Xu, C. Ma, X. Wu, Y. Cheng, Z. Yu, G. Zhao and Y. Chen, Telomere Targeting with a Novel G-Quadruplex-Interactive Ligand BRACO-19 Induces T-Loop Disassembly and Telomerase Displacement in Human Glioblastoma Cells, Onco Targets Ther, 2016, 7(12), 14925–14939, DOI:10.18632/oncotarget.7483
- A. M. Burger, F. Dai, C. M. Schultes, A. P. Reszka, M. J. Moore, J. A. Double and S. Neidle, The G-Quadruplex-Interactive Molecule BRACO-19 Inhibits Tumor Growth, Consistent with Telomere Targeting and Interference with Telomerase Function, Cancer Res., 2005, 65(4), 1489–1496, DOI:10.1158/0008-5472.CAN-04-2910
- C. I. V. Ramos, S. P. Almeida, L. M. O. Lourenço, P. M. R. Pereira, R. Fernandes, M. A. F. Faustino, J. P. C. Tomé, J. Carvalho, C. Cruz and M. G. P. M. S. Neves, Multicharged Phthalocyanines as Selective Ligands for G-Quadruplex DNA Structures, Molecules, 2019, 24(4), 733, DOI:10.3390/molecules24040733
- C. I. V. Ramos, J. P. C. Tomé and M. G. Santana-Marques, Charge and Substituent Effects on the Stability of Porphyrin/G-quadruplex Adducts, J. Mass Spectrom., 2012, 47(2), 173–179, DOI:10.1002/jms.2048
- T. Ou, Y. Lu, J. Tan, Z. Huang, K. Wong and L. Gu, G-Quadruplexes: Targets in Anticancer Drug Design, ChemMedChem, 2008, 3(5), 690–713, DOI:10.1002/cmdc.200700300
- D. Sun, B. Thompson, B. E. Cathers, M. Salazar, S. M. Kerwin, J. O. Trent, T. C. Jenkins, S. Neidle and L. H. Hurley, Inhibition of Human Telomerase by a G-Quadruplex-Interactive Compound, J. Med. Chem., 1997, 40(14), 2113–2116, DOI:10.1021/jm970199z
- H. Xu, M. Di Antonio, S. McKinney, V. Mathew, B. Ho, N. J. O'Neil, N. Dos Santos, J. Silvester, V. Wei, J. Garcia, F. Kabeer, D. Lai, P. Soriano, J. Banáth, D. S. Chiu, D. Yap, D. D. Le, F. B. Ye, A. Zhang, K. Thu, J. Soong, S. Lin, A. H. C. Tsai, T. Osako, T. Algara, D. N. Saunders, J. Wong, J. Xian, M. B. Bally, J. D. Brenton, G. W. Brown, S. P. Shah, D. Cescon, T. W. Mak, C. Caldas, P. C. Stirling, P. Hieter, S. Balasubramanian and S. Aparicio, CX-5461 Is a DNA G-Quadruplex Stabilizer with Selective Lethality in BRCA1/2 Deficient Tumours, Nat. Commun., 2017, 8(1), 14432, DOI:10.1038/ncomms14432
- M.-Z. Li, T. Meng, S.-S. Song, X.-B. Bao, L.-P. Ma, N. Zhang, T. Yu, Y.-L. Zhang, B. Xiong, J.-K. Shen, Z.-H. Miao and J.-X. He, Discovery of MTR-106 as a Highly Potent G-Quadruplex Stabilizer for Treating BRCA-Deficient Cancers, Invest. New Drugs, 2021, 39(5), 1213–1221, DOI:10.1007/s10637-021-01096-4
- J. Lopes-Nunes, J. Carvalho, J. Figueiredo, C. I. V. Ramos, L. M. O. Lourenço, J. P. C. Tomé, M. G. P. M. S. Neves, J.-L. Mergny, J. A. Queiroz, G. F. Salgado and C. Cruz, Phthalocyanines for G-Quadruplex Aptamers Binding, Bioorg. Chem., 2020, 100, 103920, DOI:10.1016/j.bioorg.2020.103920
- C. Wei, G. Han, G. Jia, J. Zhou and C. Li, Study on the Interaction of Porphyrin with G-Quadruplex DNAs, Biophys. Chem., 2008, 137(1), 19–23, DOI:10.1016/j.bpc.2008.06.006
- P. Sengupta, S. Chattopadhyay and S. Chatterjee, G-Quadruplex Surveillance in BCL-2 Gene: A Promising Therapeutic Intervention in Cancer Treatment, Drug Discovery Today, 2017, 22(8), 1165–1186, DOI:10.1016/j.drudis.2017.05.001
- N. M. M. Moura, S. Guedes, D. Salvador, H. Oliveira, M. Q. Alves, N. Paradis, C. Wu, M. G. P. M. S. Neves and C. I. V. Ramos, Oncogenic and Telomeric G-Quadruplexes: Targets for Porphyrin-Triphenylphosphonium Conjugates, Int. J. Biol. Macromol., 2024, 277, 134126, DOI:10.1016/j.ijbiomac.2024.134126
- S. Sulthanudeen, P. M. Imran, M. Selvakumaran and A. Kubaib, Novel Acridone Derivatives Probed Using DFT, Including Design, Synthesis, Characterization with Anti-Oxidant and Anti-Mitotic Screening, Results Chem., 2023, 5, 100753, DOI:10.1016/j.rechem.2022.100753
- A. Begum, Synthesis of Acridone Base Phenolic Compounds for Antibacterial Activity, Organic and Medicinal Chemistry International Journal, 2019, 9(02), 32, DOI:10.19080/OMCIJ.2019.09.555756
- S. Padigela, B. R. RM and R. P. VVS, Synthesis, Characterization, and Anticancer Activity of Some Novel Acridine Derivatives, Asian J. Pharm. Clin. Res., 2020, 13(6), 166–169, DOI:10.22159/ajpcr.2020.v13i6.35794
- K. Thimmaiah, A. G. Ugarkar, E. F. Martis, M. S. Shaikh, E. C. Coutinho and M. C. Yergeri, Drug–DNA Interaction Studies of Acridone-Based Derivatives, Nucleosides, Nucleotides Nucleic Acids, 2015, 34(5), 309–331, DOI:10.1080/15257770.2014.992531
- A. Askari, A. A. Entezari, M. Pordel, S. Beigoli, Z. Nezafat Yazdi, A. Jahani Moghaddam and J. Chamani, Design, Synthesis and Investigation of the Interaction Behavior between Two Acridone Derivatives, 8-Chloro Acridone and Nitrile Cyanide Acridone with Calf Thymus DNA, by Different Spectroscopic Techniques, J. Iran. Chem. Soc., 2020, 17(1), 135–149, DOI:10.1007/s13738-019-01757-5
- A. Stankiewicz-Drogon, L. G. Palchykovska, V. G. Kostina, I. V. Alexeeva, A. D. Shved and A. M. Boguszewska-Chachulska, New Acridone-4-Carboxylic Acid Derivatives as Potential Inhibitors of Hepatitis C Virus Infection, Bioorg. Med. Chem., 2008, 16(19), 8846–8852, DOI:10.1016/j.bmc.2008.08.074
- T. T. Yadav, M. Murahari, G. J. Peters and M. YC, A Comprehensive Review on Acridone Based Derivatives as Future Anti-Cancer Agents and Their Structure Activity Relationships, Eur. J. Med. Chem., 2022, 239, 114527, DOI:10.1016/j.ejmech.2022.114527
- S. Alsaedi, B. A. Babgi, M. H. Abdellatif, A.-H. Emwas, M. Jaremko, M. G. Humphrey and M. A. Hussien, Effect of Net Charge on DNA-Binding, Protein-Binding and Anticancer Properties of Copper(I) Phosphine-Diimine Complexes, J. Inorg. Organomet. Polym. Mater., 2021, 31(10), 3943–3952, DOI:10.1007/s10904-021-02063-5
- A. I. Karsisiotis, N. M. Hessari, E. Novellino, G. P. Spada, A. Randazzo and M. Webba da Silva, Topological Characterization of Nucleic Acid G-Quadruplexes by UV Absorption and Circular Dichroism, Angew. Chem., Int. Ed., 2011, 50(45), 10645–10648, DOI:10.1002/anie.201105193
- T. Santos, G. F. Salgado, E. J. Cabrita and C. Cruz, G-Quadruplexes and Their Ligands: Biophysical Methods to Unravel G-Quadruplex/Ligand Interactions, Pharmaceuticals, 2021, 14(8), 769, DOI:10.3390/ph14080769
- D. Monchaud, C. Allain and M.-P. Teulade-Fichou, Development of a Fluorescent Intercalator Displacement Assay (G4-FID) for Establishing Quadruplex-DNA Affinity and Selectivity of Putative Ligands, Bioorg. Med. Chem. Lett., 2006, 16(18), 4842–4845, DOI:10.1016/j.bmcl.2006.06.067
- M. Sirajuddin, S. Ali and A. Badshah, Drug–DNA Interactions and Their Study by UV–Visible, Fluorescence Spectroscopies and Cyclic Voltametry, J. Photochem. Photobiol., B, 2013, 124, 1–19, DOI:10.1016/j.jphotobiol.2013.03.013
- V. L. M. Silva and A. M. S. Silva, New Synthetic Methods for 2,3-Diarylacridin-9(10H)-One and (E)-2-Aryl-4-Styrylfuro[3,2-c]Quinoline Derivatives, Tetrahedron, 2014, 70(34), 5310–5320, DOI:10.1016/j.tet.2014.05.040
- Z. Gao, Y. Hao, M. Zheng and Y. Chen, A Fluorescent Dye with Large Stokes Shift and High Stability: Synthesis and Application to Live Cell Imaging, RSC Adv., 2017, 7(13), 7604–7609, 10.1039/C6RA27547H
- K. Li, W. Qin, D. Ding, N. Tomczak, J. Geng, R. Liu, J. Liu, X. Zhang, H. Liu, B. Liu and B. Z. Tang, Photostable Fluorescent Organic Dots with Aggregation-Induced Emission (AIE Dots) for Noninvasive Long-Term Cell Tracing, Sci. Rep., 2013, 3(1), 1150, DOI:10.1038/srep01150
- C. I. V. Ramos, A. R. Monteiro, N. M. M. Moura, M. A. F. Faustino, T. Trindade and M. G. P. M. S. Neves, The Interactions of H2TMPyP, Analogues and Its Metal Complexes with DNA G-Quadruplexes—An Overview, Biomolecules, 2021, 11(10), 1404, DOI:10.3390/biom11101404
- A. A. Almaqwashi, T. Paramanathan, I. Rouzina and M. C. Williams, Mechanisms of Small Molecule–DNA Interactions Probed by Single-Molecule Force Spectroscopy, Nucleic Acids Res., 2016, 44(9), 3971–3988, DOI:10.1093/nar/gkw237
-
J. R. Lakowicz, in Principles of Fluorescence Spectroscopy, ed. J. R. Lakowicz, Springer US, Boston, MA, 2006, DOI:10.1007/978-0-387-46312-4
- E. C. Hulme and M. A. Trevethick, Ligand Binding Assays at Equilibrium: Validation and Interpretation, Br. J. Pharmacol., 2010, 161(6), 1219–1237, DOI:10.1111/j.1476–5381.2009.00604.x
- M. I. Stefan and N. Le Novère, Cooperative Binding, PLoS Comput. Biol., 2013, 9(6), e1003106, DOI:10.1371/journal.pcbi.1003106
- D. Monchaud, C. Allain and M. P. Teulade-Fichou, Thiazole Orange: A Useful Probe for Fluorescence Sensing of G-Quadruplex-Ligand Interactions, Nucleosides, Nucleotides Nucleic Acids, 2007, 26(10–12), 1585–1588, DOI:10.1080/15257770701548212
- L. H. Fornander, L. Wu, M. Billeter, P. Lincoln and B. Nordén, Minor-Groove Binding Drugs: Where Is the Second Hoechst 33258 Molecule?, J. Phys. Chem. B, 2013, 117(19), 5820–5830, DOI:10.1021/jp400418w
- J. Carvalho, J. A. Queiroz and C. Cruz, Circular Dichroism of G-Quadruplex: A Laboratory Experiment for the Study of Topology and Ligand Binding, J. Chem. Educ., 2017, 94(10), 1547–1551, DOI:10.1021/acs.jchemed.7b00160
- N. Hatami, Fluorescence Lifetime Imaging Microscopy for Brain Tumor Image-Guided Surgery, J. Biomed. Opt., 2010, 15(5), 056022, DOI:10.1117/1.3486612
- L. C. Crowley, A. P. Scott, B. J. Marfell, J. A. Boughaba, G. Chojnowski and N. J. Waterhouse, Measuring Cell Death by Propidium Iodide Uptake and Flow Cytometry, Cold Spring Harb. Protoc., 2016, 2016(7), pdb.prot087163, DOI:10.1101/pdb.prot087163
- M. W. Conklin, P. P. Provenzano, K. W. Eliceiri, R. Sullivan and P. J. Keely, Fluorescence Lifetime Imaging of Endogenous Fluorophores in Histopathology Sections Reveals Differences Between Normal and Tumor Epithelium in Carcinoma In Situ of the Breast, Cell Biochem. Biophys., 2009, 53(3), 145–157, DOI:10.1007/s12013-009-9046-7
- S. Pavey, L. Spoerri, N. K. Haass and B. Gabrielli, DNA Repair and Cell Cycle Checkpoint Defects as Drivers and Therapeutic Targets in Melanoma, Pigm. Cell Melanoma Res., 2013, 26(6), 805–816, DOI:10.1111/pcmr.12136
- M. van Engeland, L. J. W. Nieland, F. C. S. Ramaekers, B. Schutte and C. P. M. Reutelingsperger, Annexin V-Affinity Assay: A Review on an Apoptosis Detection System Based on Phosphatidylserine Exposure, Cytometry, 1998, 31(1), 1–9, DOI:10.1002/(SICI)1097-0320(19980101)31:1<1::AID-CYTO1>3.0.CO;2-R
- K. Kupcho, J. Shultz, R. Hurst, J. Hartnett, W. Zhou, T. Machleidt, J. Grailer, T. Worzella, T. Riss, D. Lazar, J. J. Cali and A. Niles, A Real-Time, Bioluminescent Annexin V Assay for the Assessment of Apoptosis, Apoptosis, 2019, 24(1–2), 184–197, DOI:10.1007/s10495-018-1502-7
- R. E. Kellogg and R. G. Bennett, Radiationless Intermolecular Energy Transfer. III. Determination of Phosphorescence Efficiencies, J. Chem. Phys., 1964, 41(10), 3042–3045, DOI:10.1063/1.1725672
- W. H. Melhuish, Quantum Efficiencies of Fluorescence of Organic Substances: Effect of Solvent and Concentration of the Fluorescenct Solute, J. Phys. Chem., 1961, 65(2), 229–235, DOI:10.1021/j100820a009
- Y. Wang and D. J. Patel, Solution Structure of the Human Telomeric Repeat d[AG3(T2AG3)3] G-Tetraplex, Structure, 1993, 1(4), 263–282, DOI:10.1016/0969-2126(93)90015-9
- B. Chen, G. Fountain, H.-J. Sullivan, N. Paradis and C. Wu, To Probe the Binding Pathway of a Selective Compound (D089-0563) to c-MYC Pu24 G-Quadruplex Using Free Ligand Binding Simulations and Markov State Model Analysis, Phys. Chem. Chem. Phys., 2020, 22(39), 22567–22583, 10.1039/D0CP03863F
- A. Ambrus, D. Chen, J. Dai, R. A. Jones and D. Yang, Solution Structure of the Biologically Relevant G-Quadruplex Element in the Human c-MYC Promoter. Implications for G-Quadruplex Stabilization, Biochemistry, 2005, 44(6), 2048–2058, DOI:10.1021/bi048242p
- A. Kerkour, J. Marquevielle, S. Ivashchenko, L. A. Yatsunyk, J.-L. Mergny and G. F. Salgado, High-Resolution Three-Dimensional NMR Structure of the KRAS Proto-Oncogene Promoter Reveals Key Features of a G-Quadruplex Involved in Transcriptional Regulation, J. Biol. Chem., 2017, 292(19), 8082–8091, DOI:10.1074/jbc.M117.781906
- M. Zgarbová, F. J. Luque, J. Šponer, T. E. Cheatham, M. Otyepka and P. Jurečka, Toward Improved Description of DNA Backbone: Revisiting Epsilon and Zeta Torsion Force Field Parameters, J. Chem. Theory Comput., 2013, 9(5), 2339–2354, DOI:10.1021/ct400154j
- M. Zgarbová, J. Šponer, M. Otyepka, T. E. Cheatham, R. Galindo-Murillo and P. Jurečka, Refinement of the Sugar–Phosphate Backbone Torsion Beta for AMBER Force Fields Improves the Description of Z- and B-DNA, J. Chem. Theory Comput., 2015, 11(12), 5723–5736, DOI:10.1021/acs.jctc.5b00716
- M. Krepl, M. Zgarbová, P. Stadlbauer, M. Otyepka, P. Banáš, J. Koča, T. E. Cheatham, P. Jurečka and J. Šponer, Reference Simulations of Noncanonical Nucleic Acids with Different χ Variants of the AMBER Force Field: Quadruplex DNA, Quadruplex RNA, and Z-DNA, J. Chem. Theory Comput., 2012, 8(7), 2506–2520, DOI:10.1021/ct300275s
- A. Pérez, I. Marchán, D. Svozil, J. Sponer, T. E. Cheatham, C. A. Laughton and M. Orozco, Refinement of the AMBER Force Field for Nucleic Acids: Improving the Description of α/γ Conformers, Biophys. J., 2007, 92(11), 3817–3829, DOI:10.1529/biophysj.106.097782
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, Comparison of Simple Potential Functions for Simulating Liquid Water, J. Chem. Phys., 1983, 79(2), 926–935, DOI:10.1063/1.445869
- I. S. Joung and T. E. Cheatham, Determination of Alkali and Halide Monovalent Ion Parameters for Use in Explicitly Solvated Biomolecular Simulations, J. Phys. Chem. B, 2008, 112(30), 9020–9041, DOI:10.1021/jp8001614
- N. J. Paradis, A. Clark, H. Gogoj, P. M. Lakernick, T. D. Vaden and C. Wu, To Probe the Binding of TMPyP4 to C-MYC G-Quadruplex with in Water and in Imidazolium-Based Ionic Liquids Using Spectroscopy Coupled with Molecular Dynamics Simulations, J. Mol. Liq., 2022, 365, 120097, DOI:10.1016/j.molliq.2022.120097
- K. Mulholland and C. Wu, Binding of Telomestatin to a Telomeric G-Quadruplex DNA Probed by All-Atom Molecular Dynamics Simulations with Explicit Solvent, J. Chem. Inf. Model., 2016, 56(10), 2093–2102, DOI:10.1021/acs.jcim.6b00473
- B. Machireddy, H.-J. Sullivan and C. Wu, Binding of BRACO19 to a Telomeric G-Quadruplex DNA Probed by All-Atom Molecular Dynamics Simulations with Explicit Solvent, Molecules, 2019, 24(6), 1010, DOI:10.3390/molecules24061010
- J.-P. Ryckaert, G. Ciccotti and H. J. C. Berendsen, Numerical Integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of n-Alkanes, J. Comput. Phys., 1977, 23(3), 327–341, DOI:10.1016/0021-9991(77)90098-5
- U. Essmann, L. Perera, M. L. Berkowitz, T. Darden, H. Lee and L. G. Pedersen, A Smooth Particle Mesh Ewald Method, J. Chem. Phys., 1995, 103(19), 8577–8593, DOI:10.1063/1.470117
- P. Procacci and B. J. Berne, Multiple Time Scale Methods for Constant Pressure Molecular Dynamics Simulations of Molecular Systems, Mol. Phys., 1994, 83(2), 255–272, DOI:10.1080/00268979400101241
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4md00959b |
| This journal is © The Royal Society of Chemistry 2025 |