
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExpanding the chemical space for antiviral discovery: the potential of twistenediones†
Aida
Jaafar‡
a,
Daniel
Guerra-González‡
a,
Ana
Pascual
a,
Ana M.
Ortuño
b,
Carlos M.
Cruz
 b,
Juan M.
Cuerva
b,
Paula
Bueno
c,
Victoria
Castro
c,
Urtzi
Garaigorta
c,
Pablo
Gastaminza
*c,
Javier
Adrio
*ad and
María
Ribagorda
*ad
b,
Juan M.
Cuerva
b,
Paula
Bueno
c,
Victoria
Castro
c,
Urtzi
Garaigorta
c,
Pablo
Gastaminza
*c,
Javier
Adrio
*ad and
María
Ribagorda
*ad
aDepartamento de Química Orgánica, Facultad de Ciencias, Universidad Autónoma de Madrid, C/ Francisco Tomás y Valiente 7, 28049-Madrid, Spain. E-mail: javier.adrio@uam.es; maria.ribagorda@uam.es
bDepartamento de Química Orgánica, Facultad de Ciencias, Unidad de Excelencia de Química (UEQ), Universidad de Granada, Avda. Fuente Nueva s/n, 18071 Granada, Spain
cDepartamento de Biología Molecular y Celular, Centro Nacional de Biotecnología (CNB), Consejo Superior de Investigaciones Científicas (CSIC), C/ Darwin 3, 28049 Madrid, Spain. E-mail: pgastaminza@cnb.csic.es
dInstitute for Advanced Research in Chemical Sciences (IAdChem), Universidad Autónoma de Madrid, Spain
First published on 22nd January 2025
Abstract
Despite significant progress in drug discovery, there remains an urgent need to identify new structures capable of targeting drug-resistant diseases, as well as novel pathogens, to address the growing challenges in global health. This work highlights the underexplored potential of twistane-like structures as promising candidates for drug development, particularly as antiviral agents. We provide the first comprehensive study of their antiviral activity, in particular against SARS-CoV-2. We report the synthesis of a family of chiral indolyl-twistenediones, with the separation and characterization of both enantiomers via chiral semipreparative HPLC. The absolute configurations were determined using experimental and theoretical ECD techniques, supported by DFT calculations. A detailed biological study of their antiviral activity against various pathogenic RNA viruses demonstrates selective efficacy against members of the Coronaviridae family, specifically targeting a post-entry step in the viral replication cycle. Further investigation revealed a remarkable chiral distinction in the antiviral activity between the two enantiomers, opening new avenues for research in the 3D space of chiral cage compounds.
Introduction
The discovery of novel chemical entities is a dynamic field that continually seeks to identify innovative molecules with therapeutic potential. This effort aims to address various medical challenges and improve the effectiveness of treatments. A great deal of interest has been placed on incorporating a three-dimensional (3D) character into screening libraries, as emphasized by the concept of “Escape from Flatland”.1 This approach highlights the importance of increasing the fraction of sp3-hybridized carbons in drug discovery, leading to the generation of new saturated molecules with improved medicinal properties.2 Furthermore, the role of chirality and stereochemistry is vital in drug design, as the 3D arrangement of chiral molecules critically influences their interactions with biological targets. Polycyclic cage-shaped compounds represent a burgeoning area of research and development in medicinal chemistry, materials science, and other interdisciplinary fields. These cage-shaped structures, characterized by their unique geometry and properties, serve as versatile building blocks for creating innovative molecules with diverse applications. Despite the historical focus on certain cage-shaped compounds like adamantane,3 more recent efforts have expanded the scope to include other derivatives such as bicyclo[1.1.1]pentanes, cubanes or carboranes which have opened the door to unexplored chemical space. Twistane (tricyclo[4.4.0.03,8]decane) is a cage twist-boat isomer of adamantane that exists in two enantiomeric forms due to its inherent chirality (D2-symmetry) (Fig. 1).4 In contrast to the extensive attention received by adamantane analogs, twistane derivatives have been the subject of limited research, primarily due to the challenges associated with their synthesis.5 Twistane was first synthetized by Whitlock et al.6 in 1962 using an intramolecular enolate alkylation as a key step to form the twistane scaffold in a total synthesis of eleven steps. Since then, only a limited number of methodologies,7–10 including one developed by our group,11 have described the preparation of these tricyclic cage compounds. Therefore, the development of cage-shape derivatives including chiral derivatives will boost the discover of new biological relevant molecules.12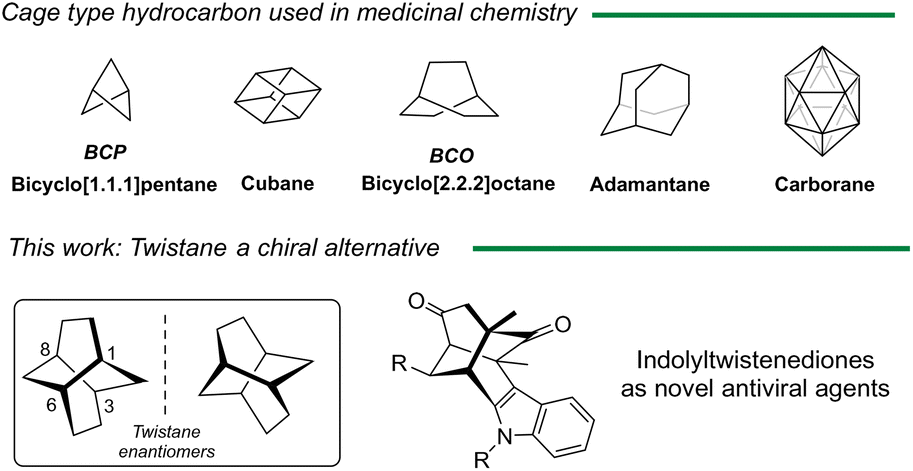 | ||
| Fig. 1 Main cage hydrocarbon structures as potential drugs and novel indole twistene like structures. | ||
In 1968, Deslongchamps patented the preparation of tricyclo[4.4.0.0]decan-1-amine and reported its potential activity against certain strains of influenza, in particular influenza A and A′.13 Despite these notable features, no further biological studies have been reported of such derivatives, nor explore the chiral impact on the biological activity. Inspired by these results, and taking into account the pharmacological versatility of indole, we reasoned that indole twistenedienones (Fig. 1) could behave as novel antiviral agents. Herein, we report the preparation of different functionalized twistenedienones structures and their effectiveness against SARS-CoV-2 virus. Moreover, we described the first enantiopure derivative and report the chiral distinction in the viral activity against major human viral pathogens including SARS-CoV-2 and MERS-CoV (middle-east respiratory syndrome) coronavirus, and non-related RNA viruses like West Nile virus (WNV) or a recombinant vesicular stomatitis virus (VSV).
Results and discussion
Our group reported the direct synthesis of indolyl substituted twistenediones 1 by a domino process involving 2-quinonyl boronic acid 2 and (E)-2-arylalkenylindoles 3.11 This 6/6/6 cage skeleton are formed via domino sequence that involves three different transformations: the regioselective 1,4-addition of the indole to the C-3 quinone position, followed by an intramolecular regioselective Diels–Alder cycloaddition (IMDA) and protodeboronation step (Scheme 1). The boronic acid on the benzoquinone proved essential for controlling regioselectivity and enhancing reactivity, while also acting as a traceless directing agent.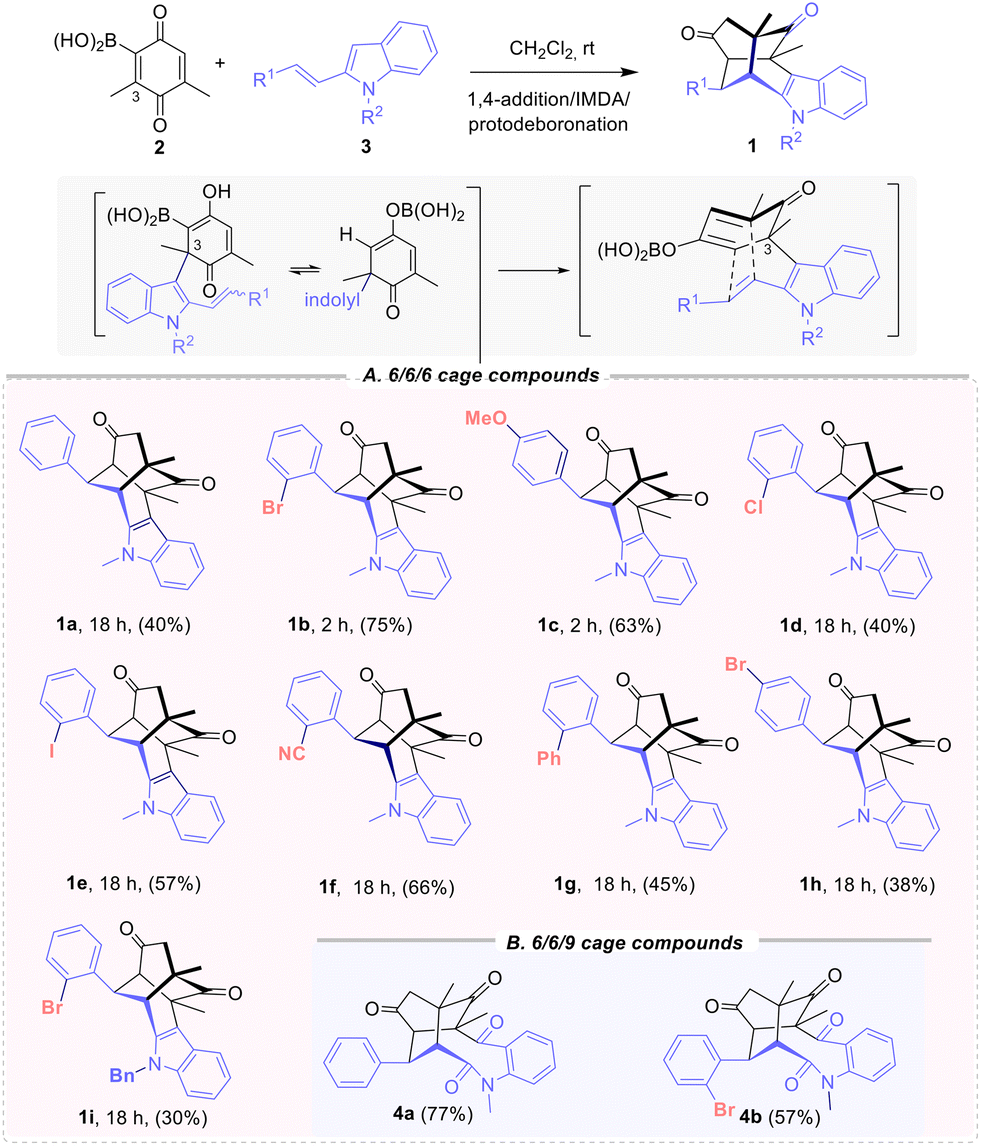 | ||
| Scheme 1 Synthesis of twistendienones 1 and structures for antiviral evaluation (A) 6/6/6 cage 1a–i and (B) 6/6/9 cage compound 4a,b. | ||
The initial evaluation of the antiviral activity was conducted with twistenediones 1a–c (R1 = Ph, ortho-bromophenyl and para-methoxyphenyl) (Scheme 1), which were prepared from the corresponding N-methyl 2-alkenyl-indole 3, using DCM as solvent at room temperature.14
The antiviral activity was initially evaluated using A549-ACE2 cell monolayers. Viral propagation was monitored by quantitation of the viral antigen in cultures treated with the vehicle (100%) or cultures that were not inoculated with the virus (0%). To assess the antiviral activity of 1a–c, serial dilutions of the compounds were mixed with infectious SARS-CoV2 before inoculation to the cell monolayers. Table 1 shows potency indexes (EC50, EC90 effective concentrations capable of reducing viral propagation by 50% or 90%) of the compounds and their interference with cell viability, as determined by an MTT-based assay (CC50 cytotoxic concentration reducing cell viability by 50%). Gratifyingly, compounds 1a–c interfered with viral propagation at concentrations <22 μM for EC50 and <35 μM for EC90 (Table 1, entries 1–3). Moreover, the measurement of the CC50/EC50 ratio, which reflects the degree of toxicity versus the level of protection against the virus, displayed some degree of cytotoxicity, resulting in narrow therapeutic windows. Compounds showed substantial antiviral activity at concentrations that reduced viability by more than 20% (CC20/EC90 < 1), a cut-off commonly established for the viability assays.
| Entry | 1 | EC50 | EC90 | CC50 | CC20 | CC50/EC50 | CC20/EC90 | VEROb |
|---|---|---|---|---|---|---|---|---|
| a Antiviral efficacy (EC50 and EC90; μM) and cytotoxicity (CC50 and CC20; μM) indexes were determined in A549-ACE2 cells and are shown as mean and the SD of a minimum of three biological replicates, see ESI† for details. b Cell monolayer integrity experiments were carried out in Vero-E6 cell and are expressed qualitatively. c Range of concentration studied 0.8–50 μM, protective at doses ranging from 12.5 to 50 μM. | ||||||||
| 1 | 1a | 13.4 | 34.8 | 25.7 | 12.7 | 1.9 | 0.4 | No |
| 2 | 1b | 15.5 | 35.0 | 70.4 | 32.2 | 4.5 | 0.9 | Yes |
| 3 | 1c | 21.0 | 30.3 | 17.7 | 5.1 | 0.8 | 0.2 | No |
| 4 | 1d | 13.0 | 35.2 | 62.3 | 20.7 | 4.8 | 0.6 | No |
| 5 | 1e | 10.6 | 23.4 | 61.7 | 10.6 | 5.8 | 0.5 | No |
| 6 | 1f | 18.8 | 36.0 | 100 | 61.7 | 5.3 | 1.7 | No |
| 7 | 1g | 22.0 | 100 | 76.7 | 26.7 | 3.5 | 0.3 | No |
| 8 | 1h | 8.7 | 48.3 | 77.6 | 56.6 | 8.9 | 1.2 | No |
| 9 | 1i | 19.0 | 66.5 | 70.7 | 17.1 | 3.7 | 0.3 | No |
| 10 | 4a | 43.7 | 89.0 | 24.8 | 89.0 | 2.3 | 1.0 | No |
| 11 | 4b | >100 | >100 | 100 | 3.7 | — | — | No |
In view of these limitations, compounds were subsequently tested for their ability to protect Vero-E6 cell monolayers from SARS-CoV-2-induced cell death. This is a semi-quantitative phenotypic assay that assists in the identification of candidates with robust antiviral activity.15 A positive protection result requires that the compound does not substantially interfere with cell viability for a period of 72 hours and that it interferes sufficiently with viral propagation so that the monolayer is intact after the same period of time. Among the tested compounds, only 1b was capable of completely protecting the VeroE6 monolayer in a relatively wide range of concentrations, from 50 to 6.25 μM. These preliminary results indicated that 1b exhibits antiviral activity against SARS-CoV-2 both in human and African green monkey cells.
A comparison of derivatives 1a–c revealed that ortho substitution has a positive influence on antiviral activity. Subsequently, various ortho-phenyl substituted 2-(aryl)vinylindoles were synthetized to prepare the corresponding twistenediones. Thus, derivatives with pedant ortho-chloro (1d), iodo (1e), cyano (1f) or phenyl (1g) substituents were prepared (Scheme 1). Moreover, to assess the impact of the bromo substituent at the para position, derivative (1h) was also synthetized. Additionally, the N-benzyl ortho-bromophenyl derivative 1i was also prepared. The new 6/6/6 cage-derivatives 1d–i and expanded 6/6/9 tricyclic derivative 4a–b were tested for their ability to interfere with viral propagation of SARS-CoV-2 in A549-ACE2 cells (Table 1, entries 4–11). All twistenedienones 1d–i interfered at concentrations <22 μM (Table 1, EC50, EC90). The o-cyano 1f and the p-bromo 1h derivatives presented the best values (Table 1, entries 6 and 8 respectively). Most compounds showed antiviral activity at concentrations that reduced viability by more than 20% (CC20/EC90 < 1). To assess the impact of the 6/6/6 cage skeleton, expanded 6/6/9 tricyclic derivative 4a and ortho-bromo 4b, lacking the twistene framework, were also evaluated. The ketolactams derivatives 4a–b14 were prepared through the oxidative cleavage of the C-2–C-3 indole bond (Witkop type oxidation)16 promoted by MCPBA and catalytic p-TsOH. Both 6/6/9 tricyclic cage derivatives 4a and 4b showed a complete lack of reactivity, pointing out the importance of the twistene framework. Further evaluation of the Vero-E6 cell monolayers protection from SARS-CoV-2-induced cell death, revealed that exclusively the ortho-bromo derivative 1b provided protection with a sufficient therapeutic window (Table 1, entry 2). Therefore, compound 1b was selected for further biological characterization.
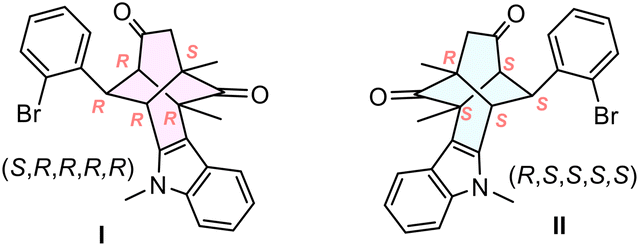
Next, we investigated the relationship between the configuration of the twistenedione 1b and the antiviral activity. Both enantiomers of 1b (namely I and II) were separated from the racemic mixture by semipreparative scale chiral stationary phase HPLC (CSP-HPLC) (Fig. 2a). The absolute configuration was established by comparison of the experimental ECD spectra of pure enantiomers with the simulated ECD spectra. The experimental ECD spectra exhibits two main absorption bands centered at 275 and 320 nm with opposite sign for each enantiomer (Fig. 2c, black and blue solid lines). The geometry of the enantiomer (S,R,R,R,R) was optimized by DFT calculations (Fig. 2b), and the corresponding UV-vis and ECD spectra were simulated with the aid of TD-DFT (Fig. 2c, red dash line), matching the experimental ECD of I enantiomer, thereby proving the (S,R,R,R,R) configuration to I and (R,S,S,S,S) to II.
 | ||
Fig. 2 a) CSP-HPLC separation chromatogram of chiral racemic 1b using a Chiralpak-IA column, hexane/EtOAc (7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3), 3.4 mL min−1, retention time I = 8.90 min and II = 9.85 min and assignment of absolute configuration; b) optimized structure of (R,R,R,R,R)-1b (CAM-B3LYP/6-31G++(d,p)); c) simulated ECD spectrum of (S,R,R,R,R) (CAM-B3LYP/6-31G++(d,p)/PCM, dashed red line) and experimental ECD spectra of both enantiomers of I and II in DCM at ca. 10−5 M (black and blue lines). :3), 3.4 mL min−1, retention time I = 8.90 min and II = 9.85 min and assignment of absolute configuration; b) optimized structure of (R,R,R,R,R)-1b (CAM-B3LYP/6-31G++(d,p)); c) simulated ECD spectrum of (S,R,R,R,R) (CAM-B3LYP/6-31G++(d,p)/PCM, dashed red line) and experimental ECD spectra of both enantiomers of I and II in DCM at ca. 10−5 M (black and blue lines). | ||
Enantiomers I and II were then subjected to the same biological characterization using A549-ACE2 cell monolayers, and the results indicate that antiviral activity segregates with I, improving all EC50 and EC90 values, as well as the therapeutic window (CC20/EC90) compared to II (Table 2, entries 2–3).
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | 1 | EC50 | EC90 | CC50 | EC20 | CC50/EC50 | CC20/EC90 | VEROb |
| a Antiviral efficacy (EC50 and EC90; μM) and cytotoxicity (CC50 and CC20; μM) indexes were determined in A549-ACE2 cells and are shown as mean; SD of a minimum of three biological replicates, see ESI† for details. b Cell monolayer integrity experiments were carried out in Vero-E6 cell and are expressed qualitatively. Range of concentration studied. c Racemic mixture. d 50–12.5 μM. e 25–6.25 μM. | ||||||||
| 1 | 1b | 15.5 | 35.0 | 70.4 | 32.2 | 4.5 | 0.9 | Yesd |
| 2 | I | 5.8 | 8.6 | 100 | 36 | 17.2 | 4.2 | Yes |
| 3 | II | 28.9 | 47.0 | 100 | 71.3 | 3.5 | 1.5 | No |
Moreover, only the enantiomer I provided protection of the Vero-E6 cell monolayers from SARS-CoV-2-induced cell death, at reasonable lower concentrations of the racemic sample, demonstrating a clear selectivity of the virus for this enantiomer (Table 2, VERO column). This phenomenon was verified in three different cell lines (Fig. 3) (Vero-E6, A549-ACE2 and human lung epithelial cell line Calu 3). Both compounds also show some degree of associated cytotoxicity with a measurable impact on cell viability (CC20 = 36 ± 10 μM for I and 71 ± 8 μM for II). All subsequent characterization experiments were performed below 30 μM to ensure a minimal impact on cell viability of either compound.
 | ||
| Fig. 3 Stereoselective evaluation of enantiopure I and II in different cell lines. Target cells were inoculated with SARS-CoV-2 in multiple cycle infection experiments in the presence of I (blue) or II (grey) at 10 μM, using solvent (DMSO, white) as a control. Relative infection efficiency was determined as described in the Experimental section (ESI†). Data is shown as mean and SD of three biological replicates. Statistical significance was determined by one-way ANOVA and a Dunnett's post hoc analysis (*p < 0.05). | ||
To determine if the antiviral activity of both enantiomers 1b was restricted to SARS-CoV-2, cells were infected with a pathogenic middle-east respiratory syndrome coronavirus (MERS-CoV) in the presence of increasing doses of I and II (Fig. 4). Moreover, both enantiomers were also tested against non-related RNA viruses like West Nile virus (WNV) or a recombinant vesicular stomatitis virus (VSV), bearing a reporter green fluorescent protein (GFP) (Fig. 4). Viral propagation efficiency was determined by measuring the relative viral RNA load at 48 hours post-inoculation in cells infected with SARS-CoV-2, MERS-CoV or WNV (MOI – multiplicity of infection experiment – 0.01) by real-time quantitative PCR. VSV–GFP propagation efficiency was determined by the relative GFP expression at 16 hours post-inoculation (MOI 0.01) using automated fluorescence microscopy. The results indicated that I caused more than 3log viral load reduction in SARS-CoV-2 and MERS-infected cells, while the same concentration only marginally interfered with WNV or VSV replication (Fig. 4). On the contrary, II did not significantly interfere with infection by any of these viruses, confirming that antiviral activity segregates with I. These results together with the mock control experiments and solvent (DMSO) assays and experiments suggest a selective antiviral mechanism and strongly reduces the concern that antiviral activity is solely due to compound cytotoxicity.
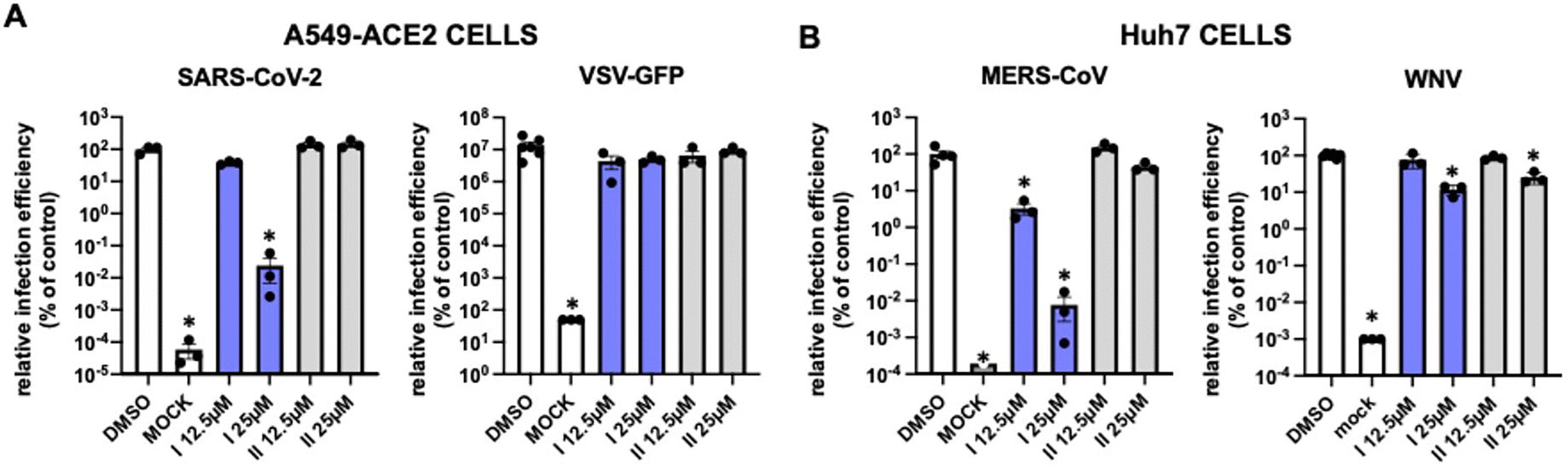 | ||
| Fig. 4 Evaluation of the selectivity of chiral enantiomers I and II for coronaviruses. Target cells were inoculated with different viruses in multiple cycle infection experiments in the presence of the candidate compounds (white, controls; blue I; grey, II). Relative infection efficiency was determined as described in the Experimental section (ESI†). A) SARS-CoV-2 (MOI 0.001) or VSV-GFP (MOI 0.01) were used to inoculate A549-ACE2 cells. B) MERS-CoV (MOI 0.01) or WNV (MOI 0.01) were used to inoculate Huh7 cells. Data is shown as mean and SD of three biological replicates. Statistical significance was determined by one-way ANOVA and a Dunnett's post hoc analysis (*p < 0.05). | ||
Finally, we set out to determine if both enantiomers interfered with viral entry or with a post-entry event of coronaviruses. To this aim, we performed a time-of-addition relative to human coronavirus hCoV-229E inoculation and determine the relative efficiency of infection in the presence of antiviral doses of I or II. For these experiments, Huh7 cells (human hepatoma cells expressing ACE2) were used, as they are susceptible to a recombinant hCoV-229E expressing GFP as reporter gene.17 Three different evaluations were run: i) I or II present at all times (Fig. 5, ALL), ii) I or II added only during virus inoculation (Fig. 5, ENTRY), and iii) I or II added only after the virions had effectively penetrated the cells in single-cycle infection experiments (Fig. 5, POST-ENTRY). For the ENTRY and ALL experiments, compounds were mixed with sufficient virions to achieve MOI 2 and produce a single cycle infection experiment. After 1 h at 37 °C, cells were washed and replenished with media devoid of antivirals (Fig. 5, ENTRY) or left with the media until the end of the experiment (Fig. 5, ALL). To determine if the compounds interfered with infection POST-ENTRY, target cells were inoculated with the virions (MOI 2) for 1 h at 37 °C in the absence of compounds. Then, the inoculum was removed, cells were washed, and the inoculated cultures were subsequently treated with the compounds I and II.
 | ||
| Fig. 5 Time-of-addition experiments: schematic diagram of the times where enantiomers I or II were present in the assay (yellow fill arrows) Huh7 cells were inoculated at MOI 2 with a recombinant human alphacoronavirus expressing GFP (hCoV-229E-GFP) in the presence or absence of the compounds. After 1 hour at 37 °C, media were either left until the end of the experiment (ALL) or removed and replenished with normal media (ENTRY). A set of untreated samples was treated after the inoculation period (POST-ENTRY) until the end of the experiment. 2mAde (25 μM) (Boc Sciences) was used as a positive post-entry replication inhibitor in the experiment. Samples were fixed 36 hours post-inoculation and infection efficiency was determined by quantitative fluorescence microscopy. Data is shown as mean and SD of three biological replicates. Statistical significance was determined by one-way ANOVA and a Dunnett's post hoc analysis (*p < 0.05). | ||
The relative infection efficiency was measured 36 hours post-inoculation using automated fluorescence microscopy. 2mAde (2′-c-methyladenosine) (Boc Sciences) was used as a positive post-entry replication inhibitor in the experiment and DMSO as a negative inhibition control. Enantiomers I or II did not interfered with infection when added during viral ENTRY experiment (Fig. 5, ENTRY). Interestingly, enantiomer I demonstrated full antiviral activity at a POST-ENTRY step (Fig. 5, POST-ENTRY), with antiviral activity comparable to that observed when the compound was kept for the entire duration of the experiment (Fig. 5, ALL). These experiments validate the antiviral activity of (S,R,R,R,R)-1b (I) while demonstrating the absence of activity for II (Fig. 5, ALL), further extending the breadth of the anti-coronavirus spectrum.
Conclusions
This study has brought to light the potential of indolyl twistenedione cage compounds, both in their racemic form and as enantiopure chiral entities, as novel drug candidates, particularly as antiviral agents. In particular, a series of indolyl twistenediones has been prepared and their activity against SARS-CoV-2 has been evaluated for the first time. The results highlighted the potential of the ortho-bromo phenyl derivative 1b providing the best results as the antiviral candidate. Both enantiomers of twistenedione 1b were separated from the racemic mixture by semipreparative chiral HPLC (CSP-HPLC) and the absolute configuration was established by comparison of the experimental ECD spectra of pure enantiomers with the simulated ECD spectra. The antiviral activity of a chiral enantiopure 6/6/6 cage derivative has been evaluated for the first time, showing an outstanding chiral differentiation using the (S,R,R,R,R)-enantiomer (I), while demonstrating the absence of activity for (R,S,S,S,S)-II enantiomer. The efficiency of both enantiomers was tested in three different cell lines (A549-ACE2, Vero-E6 and Calu3), and with three different coronavirus (SARS-CoV-2, MERS-CoV or hCoV-229-GFP) and two non-related RNA viruses (West Nile virus or a recombinant vesicular stomatitis virus). Our results indicate that enantiomer (S,R,R,R,R)-(I) showed a remarkable antiviral activity in all the cell lines evaluated. In particular, enantiomer I caused more than 3log viral load reduction in SARS-CoV-2 and MERS-infected cells, while the same concentration slightly interfered with WNV or VSV replication. Regarding the interaction with the virus replication cycle, the time-of-addition experiments showed a selective post-entry event of enantiomer I with the virus. Our results highlight the potential use of chiral enantiopure twistene-like compounds, with unique geometries, for antiviral therapy, demonstrating its selectivity against coronaviruses. One of the main limitations of the study is the current therapeutic window (CC50/EC90), of the lead compound. The differential efficacy of the compounds against different viruses is probably due to their differential replication strategies and host cell requirements. We ruled out that the host cell line contributes to the differential efficacy by testing VSV and SARS-CoV2 in A549-ACE2 cells and MERS and WNV in Huh7 cells, further supporting the notion for a selective antiviral activity and a family-specific mode-of-action. Thus, further endeavours should aim at preparing derivatives with larger therapeutic window, which will constitute valuable tools to determine the antiviral spectrum quantitatively. Additionally, such compounds could also be used for genetic resistance profiling as well as other mode of action and target deconvolution approaches. This study paves the way for further applications of enantiopure cage-shape compounds in antiviral research and drug development.
Data availability
Cell culture, viruses, evaluation of the antiviral activity, cytotoxicity, viral quantitation, general synthetic procedures and analytical data including copies of 1H and 13C-NMR spectra are provided in the ESI.†Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. Conceptualization was done by U. G., P. G., J. A. and M. R.; methodology and characterization was performed by A. J., D. G.-G. and A. P.; separation of enantiomers, photophysical characterization and theoretical computation was performed by A. M. O., C. M. C., J. M. C., biological evaluations was done by P. B. V. C. U. G. P. G. Manuscript preparation was done by J. A., P. G. and M. R.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank MICINN (Grant PID2023-146801NB-C32, PID2023-146801NB-C31, PID2020-113059GB-C22, PID2021-1248553NB-100), the Comunidad de Madrid (S2022/BMD-7403 RENIM-CM) co-financed by the European Structural and Investment Fund for financial support. This study was supported by Spanish National Research Council (PIE-RD-COVID-19 ref. E202020E079 and PIE_CNB_SC Ref. 202320E187 to the CNB-CSIC). This research work was also funded by the European Commission – NextGenerationEU (Regulation EU 2020/2094), through CSIC's Global Health Platform (PTI Salud Global). We acknowledge R. Molenkamp (Erasmus University Medical Center, Rotterdam, The Netherlands; participant of the EU-funded EVA-GLOBAL project, grant agreement 871029) for the SARS-CoV-2 strain NL/2020 virus; Dr. Pei-Yong Shi (UTMB; Galveston, TX,USA) for the WNV (NY99) plasmid construct, Dr. Rodriguez (CNB-CSIC, Madrid, SPAIN) for the VSV-GFP virus, Dr. Volker Thiel (U. of Lausanne, CH) for the hCoV-229E-GFP virus and Dr. Enjuanes (CNB-CSIC, Madrid, SPAIN) for the MERS-CoV-EMC12 virus. Paula Bueno is supported by PIPF2022/SAL-GL-25488 fellowship from Comunidad de Madrid.References
- (a) F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752–6756 CrossRef CAS PubMed; (b) F. Lovering, Med. Chem. Commun., 2013, 4, 515–519 RSC.
- (a) B. A. Wright and R. Sarpong, Nat. Rev. Chem., 2024, 8, 776–792 CrossRef CAS PubMed; (b) D.-H. Liu, P. M. Pflüger, A. Outlaw, L. Lückemeier, F. Zhang, C. Regan, H. R. Nodeh, T. Cernak, J. Ma and F. Glorius, J. Am. Chem. Soc., 2024, 146(17), 11866–11875 CrossRef CAS PubMed; (c) A. Krzyzanowski, A. Pahl, M. Grigalunas and H. Waldmann, J. Med. Chem., 2023, 66, 12739–12750 CrossRef CAS PubMed; (d) M. Ishikawa and Y. Hashimoto, J. Med. Chem., 2011, 54, 1539–1554 CrossRef CAS PubMed.
- L. Wanka, K. Iqbal and P. Schreiner, Chem. Rev., 2013, 113, 3516–3604 CrossRef CAS PubMed.
- (a) D. M. McCann, P. J. Stephens and J. R. Cheeseman, J. Org. Chem., 2004, 69, 8709–8717 CrossRef CAS PubMed; (b) R. Ch. Geivandov, I. V. Goncharova and V. V. Titov, Mol. Cryst. Liq. Cryst., 1989, 166, 101–103 CrossRef CAS; (c) M. Nakazaki, H. Chikamatsu and M. Taniguchi, Chem. Lett., 1982, 11, 1761–1764 CrossRef.
- D. P. G. Hamon and R. N. Young, Aust. J. Chem., 1976, 29, 145–161 CAS.
- H. W. Whitlock Jr, J. Am. Chem. Soc., 1962, 84, 3412–3413 CrossRef.
- (a) J. Gauthier and P. Deslongchamps, Can. J. Chem., 1967, 45, 297–300 CrossRef CAS; (b) A. Bélanger, J. Poupart and P. Deslongchamps, Tetrahedron Lett., 1968, 9, 2127–2128 CrossRef; (c) A. Bélanger, Y. Lambert and P. Deslongchamps, Can. J. Chem., 1969, 47, 795–802 CrossRef; (d) H. Greuter and H. Schmid, Helv. Chim. Acta, 1972, 55, 2382–2400 CrossRef CAS; (e) E. Osawa, P. V. R. Schleyer, L. W. K. Chang and V. V. Kane, Tetrahedron Lett., 1974, 15, 4189–4192 CrossRef; (f) A. S. Secco and J. Trotter, Acta Crystallogr., Sect. C:Cryst. Struct. Commun., 1983, 39, 362–365 CrossRef; (g) H.-G. Capraro and C. Ganter, Helv. Chim. Acta, 1980, 63, 1347–1351 CrossRef CAS.
- For asymmetric synthesis, see: (a) M. Tichý and J. Sicher, Tetrahedron Lett., 1969, 10, 4609–4613 CrossRef; (b) K. Adachi, K. Naemura and M. Nakazaki, Tetrahedron Lett., 1968, 9, 5467–5470 CrossRef.
- For synthesis of ditwistanes, see: (a) T. Berkenbusch, A. C. Laungani, R. Brückner and M. Keller, Tetrahedron Lett., 2004, 45, 9517–9520 CrossRef CAS; (b) E. LeGoff and S. Oka, J. Am. Chem. Soc., 1969, 91, 5665–5667 CrossRef CAS.
- For a topological and computational analysis of polytwistane, see: S. R. Barua, H. Quanz, M. Olbrich, P. R. Schreiner, D. Trauner and W. D. Allen, Chem. – Eur. J., 2014, 20, 1638–1645 CrossRef CAS PubMed.
- J. Rojas-Martín, M. Veguillas, M. Ribagorda and M. C. Carreño, Org. Lett., 2013, 15, 5686–5689 CrossRef PubMed.
- I. Proietti Silvestri and P. J. J. Colbon, ACS Med. Chem. Lett., 2021, 12(8), 1220–1229 CrossRef PubMed.
- P. Deslongchamps, (to Ayerst, McKenna, and Harrison Ltd.), US Pat., 3579567, 1971 Search PubMed.
- See ESI† for details.
- G. H. Jimenez-Aleman, V. Castro, A. Londaitsbehere, M. Gutierrez-Rodríguez, U. Garaigorta, R. Solano and P. Gastaminza, Pharmaceuticals, 2021, 14, 1048 CrossRef CAS PubMed.
- (a) B. Witkop, J. B. Patrick and M. Rosenblum, J. Am. Chem. Soc., 1951, 73, 2641–2647 CrossRef CAS; (b) M. Mentel and R. Breinbauer, Curr. Org. Chem., 2007, 11, 159–176 CrossRef CAS.
- L. Cervantes-Barragan, R. Züst, R. Maier, S. Sierro, J. Janda, F. Levy, D. Speiser, P. Romero, P.-S. Rohrlich, B. Ludewig and V. Thiel, MBio, 2010, 1, e00171-e210 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4md00891j |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |