
In vitro and in vivo ADME of heterobifunctional degraders: a tailored approach to optimize DMPK properties of PROTACs©†
Christine Katharina
Maurer
a,
Zhizhou
Fang
a,
Christina
Schindler
 b,
Gianna
Pohl
b,
Fouzia
Machrouhi-Porcher
c,
Marc
Lecomte
a,
Carl
Petersson
a and
Heide Marika
Duevel
*a
b,
Gianna
Pohl
b,
Fouzia
Machrouhi-Porcher
c,
Marc
Lecomte
a,
Carl
Petersson
a and
Heide Marika
Duevel
*a
aNCE DMPK, Merck Healthcare KGaA, Frankfurter Straße 250, 64293 Darmstadt, Germany. E-mail: Heide.Duevel@merckgroup.com
bMedicinal Chemistry & Drug Design, Merck Healthcare KGaA, Frankfurter Straße 250, 64293 Darmstadt, Germany
cSite Management Analytics, Merck KGaA, Frankfurter Straße 250, 64293 Darmstadt, Germany
First published on 21st February 2025
Abstract
Proteolysis-targeting chimeras (PROTACs©) have recently emerged as a promising new drug modality. Residing beyond the rule-of-five space, they pose challenges in terms of physicochemical properties. With this study, we contribute to enhancing the understanding of their early ADME characterization. For permeability assessment, transwell assays such as Caco-2 remain challenging. Although the addition of serum may reduce unspecific binding and improve recovery, the assay was not found predictive for absorption. As a surrogate, we propose to focus optimization on molecular descriptors and support a preferred space for oral PROTACs© with ≤3 H-bond donors (HBDs), molecular weight (MW) ≤950 Da and number of rotatable bonds ≤12. We have developed a predictive score serving as initial guidance for design and prioritization according to this property space. In addition, the reduction of exposed polar surface area, e.g. through shielding of HBDs, is a powerful approach to optimize permeability. Using standard small molecule-based methods for in vitro–in vivo extrapolation (IVIVE) of intrinsic clearance (CLint) with experimentally determined hepatocyte CLint and fraction unbound in plasma, and predicted fraction unbound in the incubation (fu,inc), a systematic under-prediction from mouse hepatocytes was observed for PROTACs©. In line with our observation that the Kilford equation was not suitable for PROTAC© fu,inc prediction, this bias could be overcome by using experimentally determined fu,inc. Taken together, this study suggests a tailored in vitro DMPK discovery assay cascade and frontloading in vivo studies. It also underlines the need for inclusion of surrogate permeability descriptors and experimentally determined values for IVIVE of CLint.
Introduction
Over the past few years, the development of small molecule therapeutics has undergone a paradigm shift through the rise of heterobifunctional degraders, also referred to as Proteolysis Targeting Chimeras (PROTACs©, trademarked by Arvinas, is hereafter used as PROTACs© to describe the modality). These molecules harness the cellular ubiquitin–proteasome system to selectively degrade target proteins, presenting a novel strategy for drug discovery in various disease areas, predominantly in cancer1 but also emerging in immunology.2 As for all non-topical therapeutics, a comprehensive understanding of their absorption, distribution, metabolism, and excretion (ADME) properties enables the clinical translation of pharmacokinetic (PK) behavior, efficacy, and safety profile. However, due to the physicochemical properties of PROTACs©, not all methodologies established for classical small molecules can be applied to this modality. Heterobifunctional degraders consist of two binding moieties, one for the target protein of interest (POI), the other for the E3 ligase, which are connected through a linker. Hence, PROTACs© reside within the beyond-rule-of-5 (bRo5) space and, due to high lipophilicity, size and hydrogen bond donor (HBD) count, pose challenges in terms of solubility and permeability resulting in a low probability for oral absorption.3–5 Also, low solubility and/or unspecific binding to in vitro assay systems can confound or even prevent reliable experimental results and need to be accounted for.6,7 So far, little has been reported on systematic metabolic stability assessment, in vitro–in vivo extrapolation (IVIVE) of intrinsic clearance (CLint), and excretion pathways for PROTACs©.8,9 For permeability assessment, several approaches for adaptation of the Caco-2 transwell assay6,7,10 as well as surrogate methods such as exposed polar surface area (ePSA) determination11 are being described. Despite these efforts, rational design strategies to optimize the drug-like properties of PROTACs© based on in vitro assay results remain scarce. Hence, the field is still relying largely on in vivo studies as optimization strategy to identify orally absorbed PROTACs©. Recently, several reports established upper limits of physicochemical properties that seem to be suitable guidelines for design of oral PROTACs©. Hornberger et al. defined 2 unsatisfied HBDs, 15 hydrogen bond acceptors (HBA), 14 rotatable bonds, a molecular weight (MW) of 950 Da and a topological polar surface area (TPSA) of 200 Å2 as well as a calculated log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P/D (clogP/D) of 7 as upper boundaries, derived from a large dataset of 1806 PROTACs© with intravenous (iv) and oral PK data in rats.12 In another report, most emphasis is put on the number of exposed HBDs, with a number of ≤2 being recommended.13 For the other parameters, Schade et al. widen the boundaries of MW, chromatographic logD (ChromlogD), ePSA, experimental rotational bond count (eRotB) and solvent-exposed HBA (eHBA) to 1000 Da, 7, 170 Å2, 13 and 16, respectively, when the upper limit of solvent-exposed HBD (eHBD) to ≤2 is not exceeded.
P/D (clogP/D) of 7 as upper boundaries, derived from a large dataset of 1806 PROTACs© with intravenous (iv) and oral PK data in rats.12 In another report, most emphasis is put on the number of exposed HBDs, with a number of ≤2 being recommended.13 For the other parameters, Schade et al. widen the boundaries of MW, chromatographic logD (ChromlogD), ePSA, experimental rotational bond count (eRotB) and solvent-exposed HBA (eHBA) to 1000 Da, 7, 170 Å2, 13 and 16, respectively, when the upper limit of solvent-exposed HBD (eHBD) to ≤2 is not exceeded.
However, even though solutions have been published to solve issues in specific cases or for certain chemical matter, they are not universally applicable. In this article, we present our efforts to adapt our established small molecule in vitro ADME assays to the chemical space PROTACs© reside in and assess feasibility of methods published by others to our internal PROTAC© dataset. We evaluate various methods for permeability assessment such as Caco-2 transwell assay and surrogate determination of ePSA. In addition, we have developed a predictive model that serves as an early prioritization tool for design of oral PROTACs©. This model is based on molecular descriptors including MW and HBD count. Lastly, we describe our key findings on IVIVE of CLint and suggest a tailored discovery assay cascade for early ADME characterization of PROTACs©. As a perspective, we describe alternative routes of administration as a surrogate to overcome the issue of low oral bioavailability.
Experimental procedures
Details on materials used in this paper and additional experimental procedures (ePSA, ChromlogD, and kinetic solubility assays, PK in mice, UHPLC-MS/MS method for CLint assay) are described in the ESI.†
Caco-2 assay
The Caco-2 assays were performed in a transwell assay setup using Caco-2 cells (TC7 clone). The Caco-2 cells were seeded into the apical wells (125000 cells per well) in DMEM (Dulbecco's Modified Eagle's Medium) with 20% FBS (fetal bovine serum) into Corning 24-well transwell plates, and cultured for 14–21 days. Prior to the experiment, the plates were washed with HBSS (Hanks' balanced salt solution). Apparent permeability Papp was determined from apical-to-basolateral direction (Papp,AB) by adding 1 μL of the test compound in HBSS into the apical compartment, and HBSS into the basolateral compartment, and vice versa for Papp,BA from basolateral-to-apical. Apical volumes were 250 μL, basolateral volumes were 750 μL. DMSO content was <1% (v/v) in all wells. Monolayer tightness was controlled using melagatran as tightness marker.
Samples were taken from both compartments at timepoint t0 and after 2 hours incubation at 37 °C in 5% CO2 and 100% humidity, and analyzed via UHPLC-MS/MS. The apparent permeability Papp was calculated as follows:

Mass balance was checked after the experiment by determining the recovery after the experiment:

The geometric mean of Papp,AB and Papp,BA was used as measure of passive permeability Papp,pass.

For some experiments, following modifications were made: In the FCS (fetal calf serum) experiment, the HBSS buffer contained 10% FCS on both sides. In the pre-incubation experiment, 1 μM of the respective compounds was added as DMSO stock on day 13, before the actual experiment. The buffer during the experiment contained 0.25% BSA. In the pH 6.5 experiment, 10 mM HEPES was added to the HBSS buffer, and the apical compartments were adjusted to pH 6.5 instead of pH 7.4. In the FaSSIF experiment, the apical buffer was FaSSIF instead of HBSS. The basolateral wells were supplemented with 0.5% BSA. In the mucin experiment, only direction A-to-B was measured. After washing of the cells, 50 mg mL−1 of mucin was applied on top of the Caco-2 cells in the apical wells. The apical buffer was FaSSIF instead of HBSS. The basolateral wells were supplemented with 1% BSA.
Determination of CLint in mouse hepatocytes
The CLint values were determined using cryopreserved female CD-1 mouse hepatocytes in suspension on a liquid handling platform (Hamilton Microlab VANTAGE, Bonaduz, Switzerland). Viability was determined in a Neubauer chamber by trypan blue staining and always above 70%. Compounds (1 μM final) were incubated with hepatocytes at a cell density of 0.2 × 106 cells per mL in Krebs–Henseleit buffer (pH 7.4). The final DMSO concentration did not exceed 1% (v/v). Incubations were carried out in duplicate at 37 °C under an atmosphere of 5% CO2 and 95% humidity. Aliquots were taken at time points 0, 10, 20, 40, 60, and 90 min and quenched with the two-fold volume of an ACN solution containing 1.5 μM of internal standard. The supernatants were further diluted 1:3 (v/v) with supernatants from other compound incubations for bioanalytical cocktailing designed to avoid analytical interference. The samples were analyzed by a standard reversed phase liquid chromatography assay coupled to tandem mass spectrometry (UHPLC-MS/MS) (for details see supplementary information).
For quantitation, MS peak area ratios of compounds and internal standard were calculated and transformed to nanomolar concentrations determined based on a four-point calibration comprising 1.2% to 150% of the incubation concentration. Calibration standards were prepared using heat-inactivated (60 °C for at least 15 min) hepatocytes as biomatrix and processed in duplicate as described for the test samples.
For data analysis, concentration data was plotted against incubation time, the first order elimination rate constant (kel) determined by non-linear regression using the software GraphPad Prism version 10.2.1, and CLint values (expressed in μL min−1 per 106 cells) obtained by normalizing kel with the hepatocytes concentration.14
IVIVE of mouse CLint from hepatocytes
For IVIVE of CLint, we used the ‘regression offset approach’ described essentially by Sohlenius-Sternbeck et al.15 using experimentally determined hepatocyte CLint and fraction unbound in plasma (fu,p) data, fraction unbound in the hepatocyte incubation (fu,inc) data either predicted by the Kilford equation16 with calculated logD at pH 7.4 (clogD7.4) or determined experimentally, blood-to plasma ratio Rb assumed to be 1 for bases and neutrals, regression correction based on a small molecule regression line8,14,15 (slope: 0.85, intercept: 0.25), and back-calculation of CLint by means of the well-stirred model.
Prediction of fu,inc
The free fraction in the hepatocyte incubation (fu,inc) was predicted using equation 5 published by Kilford et al.16 using Vr of 0.001 (given the cell density of 0.2 × 106 cells per mL) and clogD7.4 for neutral and basic ion classes. The clogD7.4 was calculated using Percepta (ACD Labs)17 models for pKa and lipophilicity, retrained with in-house experimental data (GALAS method18).
Determination of fraction unbound in plasma (fu,p) and in the hepatocyte incubation (fu,inc)
Free fraction in mouse or human plasma was determined using rapid equilibrium dialysis (RED) in teflon-coated 96-well RED devices using serum as surrogate, as in-house data showed that fu values do not differ between serum and plasma. Mouse or human serum is dialyzed overnight to exchange the carbonate with a phosphate buffer, and then used as donor matrix in the experiment. Test compounds are added as DMSO stock to the donor matrix (50% serum in phosphate buffer pH 7.4) at 0.5 μM (final DMSO concentration 0.5% and dialyzed against phosphate buffer pH 7.4 in the receiver compartment for 4 hours at 37 °C.f u,inc is determined using RED in teflon-coated 96-well RED devices. Mouse hepatocytes were inactivated by incubating over night at RT, and then used as donor matrix in the experiment. Test compounds are added as DMSO stock to the donor matrix (0.2 million cells per mL in phosphate buffer pH 7.4) at 0.5 μM (final DMSO concentration 0.5%), and dialyzed against phosphate buffer pH 7.4 in the receiver for four hours at 37 °C.
Samples were taken from both compartments after 4 hours incubation, and analyzed via UHPLC-MS/MS. The fraction unbound fu was calculated as follows:
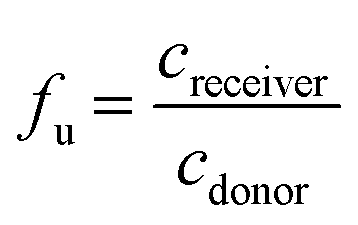
where fu2 at matrix concentration M2 can be calculated from the fu1 value at matrix concentration M1.

Ethical statement
All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Merck KGaA/EMD Serono and approved by the internal Animal Ethics Committee of Merck/EMD Serono. All used laboratories and CROs have been audited by the Merck/EMD Serono animal welfare office, and the animal work has been approved by the local ethics committees responsible for the respective CROs/laboratories.Results & discussion
To evaluate the applicability of our early in vitro ADME assay cascade, we selected representative heterobifunctional degraders from internal drug discovery projects as well as external molecules known to be in pre-clinical or clinical development. We have compiled several datasets, one for Caco-2 measurements comprising 57 heterobifunctional degraders, for which Papp,AB and Papp,BA were determined (37% rigid linker, 65% CRBN, 33% VHL). For CLint IVIVE, the dataset contained 25 PROTACs© (20 rigid linkers, 5 flexible PEG or alkyl linkers) mostly relying on CRBN as the E3 ligase (80% of PROTACs©). 8 PROTACs©, including KT-474 and ARV-110, are published examples, and 17 are from 3 internal programs. A table in the supplemental information summarizes the physicochemical properties of the PROTACs© used in this study (Tables S6 and S7†).Caco-2 adaptions and results
Caco-2 cells are widely employed in a transwell setup to determine bi-directional permeation rates in apical-to-basolateral direction and vice versa. The apparent permeability (Papp) values can be used to quantify passive permeability (Papp,pass) and transporter activity (efflux ratio ER). Papp,pass is typically used for assessing oral absorption properties of small molecules as it correlates with fraction absorbed (fa). Due to the lack of human fa data for PROTACs©, mouse fafg derived from oral bioavailability in in-house PK studies was used as surrogate to assess predictiveness of Caco-2 for oral absorption of PROTACs©. We have evaluated both passive permeability as a measure of absorption rate when active transport in the GI tract is saturated, as well as Papp,AB for assuming transport is not saturated.Under standard conditions that we employ for other small molecules, no clear correlation of mouse fafg with neither Papp,pass (Fig. 1A) nor Papp,AB (Fig. 1B) could be established for PROTACs©.
 | ||
| Fig. 1 Correlation of fafg from mouse PK studies vs. Papp,pass in presence of CsA (A) or Papp,AB without inhibitor (B) reveal a lack of predictiveness of oral absorption for PROTACs©. Colour-coding indicates ER under respective conditions: ≤3 (green) via yellow to ≥20 (red). Triangles represent qualified data, i.e. the Papp,pass/Papp,AB values are actually lower than where they are depicted. | ||
PROTACs© often exhibit high lipophilicity, high MW, low permeability, and low solubility. Therefore, they pose various challenges to in vitro models. In transwell setups such as the Caco-2 assays, these challenging properties can result in loss of free compound from incubation by non-specific binding (NSB) to plasticware or precipitation, or in slow equilibration rates. Several modifications to the standard protocol have been described in the literature to overcome these limitations. Cantrill et al.3 have reported a method utilizing the addition of 10% FCS to mitigate NSB and precipitation. Cui et al.19 and Muschong et al.6 have addressed a potentially slow equilibration of intra- and extracellular compartments of bRo5 compounds (such as PROTACs©) by pre-incubating cells with the respective compound of interest prior to running a bidirectional transwell assay. For the PROTACs© in our study, we found that these modifications provided only limited benefits.
While the addition of 10% FCS into the Caco-2 assay increased recovery values (Fig. 2A), this increase did not result in elevated Papp values in general. While some PROTACs© exhibited increases in both recovery and Papp, there were equally as many PROTACs© in which recovery improvement did not affect the Papp value, and vice versa (Fig. 2B). We therefore conclude that Papp values in the absence of FCS had not been under-estimated due to loss of compound in the receiver compartment after permeating through the cell layer. In fact, the compounds that were most affected by the FCS addition and exhibited the biggest changes in Papp were low solubility PROTACs© (Fig. 2C). This suggests that the increase in Papp is linked to a higher free concentration of the PROTAC© in the donor compartment, aided by the solubilizing effect of FCS. Without FCS, the actual donor concentration of PROTAC© was presumably below the targeted 1 μM, leading to low compound flux rates.
 | ||
| Fig. 2 (A) Average recovery in the Caco-2 assay with and without 10% FCS in the incubation. The addition of FCS increased recovery values to 80–100%. Colour-coding indicates clogD: from ≤1 (green) via yellow to ≥6 (red). (B) Correlation of fold-changes in Pappvs. changes in recovery. The black lines represent the respective 95% confidence intervals for the 2 parameters. Colour-coding indicates average recovery under standard conditions from colour-coding indicates clogD: from ≤20% (red) via yellow to ≥80% (green). (C) Fold-changes in Pappvs. kinetic solubility. While those exhibiting a kinetic solubility of >4 μM largely did not show differences in Papp, most PROTACs© with a kinetic solubility <4 μM showed a significantly different Papp value. Colour-coding indicates clogD: from ≤1 (green) via yellow to ≥6 (red). | ||
Nevertheless, despite increasing Papp values for some PROTACs©, no correlation between mouse fafg and Papp,pass (Fig. 3A) nor Papp,AB (Fig. 3B) was observed. Similarly, when we pre-incubated Caco-2 cells with PROTACs© for 24 hours prior to running the assay in order to ensure equilibrium between the intracellular and extracellular compartments, we observed higher Papp values in general for all compounds; however, neither the Papp,pass (Fig. 3C) nor the Papp,AB values (Fig. 3D) were predictive of mouse fafg under these pre-equilibrated conditions. The accuracy of correctly predicting well-absorbed PROTACs© was similar to what was reported by Muschong et al.:6 56% probability of a PROTAC© exhibiting an fafg >0.5 when Papp,pass is >5 × 10−6 cm s−1, and a 43% probability of a PROTAC© exhibiting an fafg >0.3 when Papp,AB is >3 × 10−6 cm s−1.
 | ||
| Fig. 3 Correlation of fafg from mouse PK studies vs. Papp,pass in presence of CsA (A and C) and Papp,AB without inhibitor (B and D), when co-incubated with 10% FCS in the buffer (A and B) or when pre-incubated with the respective compound of interest for 24 h in the culture medium (C and D). Colour-coding indicates ER under respective conditions: ≤3 (green) via yellow to ≥20 (red). Triangles represent qualified data, i.e. the Papp,pass/Papp,AB values are actually lower than where they are depicted. | ||
Due to their large size and elongated shape, PROTACs© are known to adopt different conformations depending on their environment, forming secondary structures via e.g. intramolecular HBDs (chameleonicity), which could influence their permeability.20 It is hence possible, that the conformation that PROTACs© adopt in in vitro systems such as in the Caco-2 assay is not representative of the one exhibited in the gut environment. Therefore, additional modifications to the Caco-2 assays have been explored, aiming to provide incubation conditions in vitro that have a better resemblance of the in vivo conditions in the GI tract. However, neither the introduction of a pH gradient, nor the utilization of FaSSIF as apical incubation buffer, nor the addition of mucin10 yielded a predictive Caco-2 model for our selection of PROTACs© (data shown in ESI†).
In conclusion, the regular Caco-2 assay protocol has limited applicability to PROTACs©. Various modifications to the assay have been described in the literature to address these limitations encountered in the laboratories of the authors. However, none of those modifications was effective in enhancing the Caco-2 assay's ability to predict the in vivo behaviour of PROTACs©. The observed discrepancies may stem from differences in the test substances employed. This underlines the chemical diversity among PROTACs©, each presenting unique challenges, e.g. instability or low solubility, binding to the assay system, or inconclusive permeability. This observation is not exclusive to the Caco-2 assay, but was also found in other in vitro assays. Consequently, published adapted methods may not be universally applicable to all PROTACs©. A fit-for-purpose assay setup is often required to effectively address the distinct chemical space of PROTACs©, and alternative systems need to be considered.
Experimental polar surface area as surrogate for intestinal permeability
As outlined before, PROTACs© can form intramolecular contacts and adopt a more compact structure, reducing PSA. Astra Zeneca recently described an NMR-based method to determine the number of exposed HBDs13 and showed that all four clinical PROTACs© studied expose fewer HBDs in solution than present in the chemical structure reducing their polar surface area. In line with this finding, we established a method to determine the ePSA (see ESI† for experimental procedure). This is a chromatographic technique employing a column mimicking the passage through the cellular lipid bilayer. A polar column is eluted with a non-polar solvent which enables a shorter residence of molecules harboring the ability to reduce their polar surface area by e.g. intramolecular HBD formation.21 Since the cell membrane is lipophilic, a less polar surface in this surrounding should allow for a better passive permeability. Based on results from two very similar series within one internal project, we found that ePSA was inversely correlated with observed mouse fafg (low to moderate correlation confirmed by linear regression, r = −0.38). Hence a lower ePSA value resulted in a higher fraction absorbed (Fig. 4). However, as for many in vitro techniques, the correlation needs to be investigated for its suitability to the respective chemical series and cutoffs can often not be transferred across projects. Often, every chemical series has its own “sweet spot” of physicochemical properties that needs to be identified during optimization. While we think ePSA is a very useful parameter and should be included in PROTAC© optimization strategies in general, its importance and relevant numeric values need to be assessed for each chemical series individually. Strategies to lower ePSA could be shielding of HBDs, reducing polar groups or introducing other intramolecular interactions that reduce the ePSA.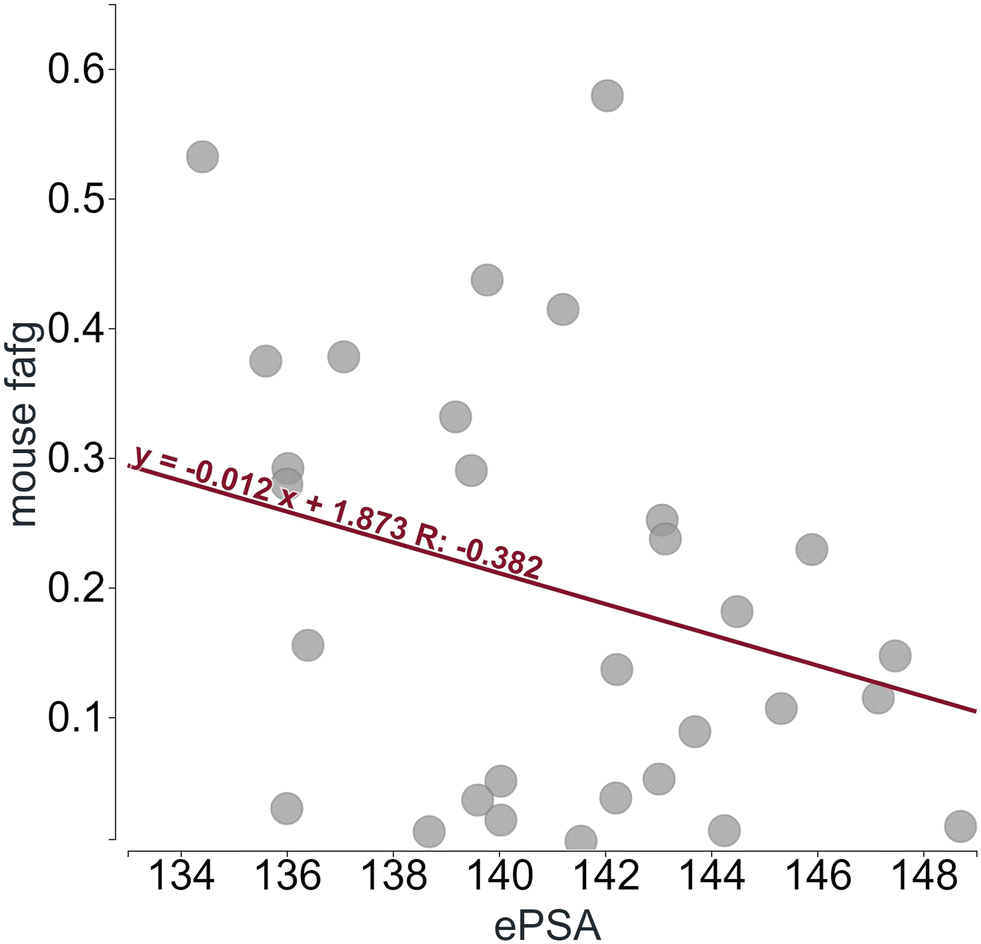 | ||
| Fig. 4 Experimental polar surface area (ePSA) as optimization parameter. Data from one in-house PROTAC© project is shown where smaller ePSA enhances the likelihood of observing increased fafg. PK studies were performed in mice at an oral dose of 10 mg kg−1. | ||
Development of a predictive score for mouse oral bioavailability based on molecular descriptors
We sought for physicochemical or structural characteristics elevating the likelihood of a PROTAC© being orally bioavailable in response to the challenges associated with in vitro permeability. Several publications showed the benefit of reducing and/or shielding HBDs to keep the number of exposed HBDs as low as possible. For other physicochemical parameters such as MW and PSA a beneficial property space was established.12,13 Our mouse in vivo dataset comprised 11 reference PROTACs© (e.g. ARV-110, ARV-471, KT-474) and 22 PROTACs© from two internal projects, with rigid linkers and a total CL of less than 33 mL min−1 kg−1. The exclusion of flexible linkers and moderate to high clearance (CL) compounds was undertaken to be able to directly relate oral bioavailability (F%) to molecular descriptors since we assumed in this case oral F was mostly determined by fa. We related oral bioavailability to several structural parameters such as number of HBDs, MW and number of rotatable bonds (Fig. 5). Importantly, all these parameters are derived from the chemical formula and no intramolecular connections or shielding of HBDs was considered. As a result of the analysis, we identified thresholds of several physicochemical parameters below which the likelihood for oral bioavailability was increased in mice. We selected to take 10% oral F as an arbitrary threshold. This led us to an upper border of 3 HBD, a MW of 950 Da and 12 rotatable bonds which is in line with the property budget and boundaries published by Hornberger et al.12 | ||
| Fig. 5 Predictive physicochemical parameters for oral bioavailability identified. 33 reference and internal PROTACs© were profiled in mouse PK (iv/po) and molecular determinants as number of rotatable bonds (A), MW (B) and HBD (C) were plotted against oral bioavailability. To reduce impact of fh on the correlation, only PROTACs© with low to moderate CL (<33 mL min−1 kg−1) were chosen. PROTACs© were administered at 0.2–1 mg kg−1 intravenously and at 10 mg kg−1 orally. | ||
When examining individual plots, we realized that an orally bioavailable PROTAC© with a relatively high MW would most likely have a low HBD count or lower number of rotatable bonds suggesting that properties could compensate for each other. Therefore, the probability of a PROTAC© being orally available is a composite of individual parameters. Hence, we tested if a composite score could be developed to predict oral bioavailability based on calculated molecular descriptors similar to previously proposed scores for the bRo5 chemical space (e.g. AB-MPS22). We decided to concentrate on calculated values for ChromlogD and standard molecular descriptors (MW, TPSA, HBA, HBD, N_Aro, N_rot) to be able to already apply such a score in silico before synthesizing a molecule. A number of regression models and the transformation into a sigmoidal model for different combinations of the above-described parameters were tested and the final empirical model was chosen for high performance and minimal number of descriptors. Here we found that the inclusion of MW, HBA and HBD to the model in contrast to the AB-MPS score that only considers clogD, N_Aro and N_rot, greatly enhanced the predictive power. Despite the small dataset, we achieved a high confidence (R2 = 0.74) and the model became an integral part of our early PROTAC© design and optimization strategy (Fig. 6). We obtained the following equation:

 | ||
| Fig. 6 A weighted, composite score for prediction of oral bioavailability (oral F score). Using a subset of the mouse PK data, a predictive score was developed based on molecular descriptors. The score helps to prioritize design ideas and facilitate compound selection for animal studies. 95% confidence interval of the regression line is depicted in light blue. | ||
Systematic under-prediction of mouse CLint from hepatocytes using standard small molecule IVIVE approach
Residing beyond the rule-of-five chemical space, PROTACs© may exhibit physicochemical properties outside of the property space of classical small molecules. This warrants investigation of the applicability of small molecule-based standard methods for IVIVE of metabolic CLint to PROTACs©. While IVIVE of CLint for PROTACs© is subject to on-going discussions in the scientific community,23,24 little has been published about it. Cantrill et al. reported a three-fold under-prediction of CLint using scaling from rat hepatocytes for one internal compound.3 Using standard IVIVE method15 Pike et al. reported the scaling of CLint from mouse hepatocytes for 27 internal compounds with 16 compounds (59%) predicted within 3-fold of unity, concluding that the approach had performed as expected for typical small molecules.8 However, in vivo CLint values of the selected data set were >316 mL min−1 kg−1 (i.e. logCLint > 2.5) and thus not applicable to our desired ADME space.
Therefore, we investigated the applicability of classical in silico and in vitro methods required for IVIVE of PROTACs©. To this end, we selected 25 compounds, among them 17 molecules from internal projects representing the Merck chemical space and 8 published reference PROTACs©. In vivo CLint data was collected from female CD-1 mice, typically used for in vivo pharmacology testing. We chose mouse hepatocytes as in vitro system, as they covered the full spectrum of drug-metabolizing liver enzymes and have been used for metabolic stability assessment of PROTACs©.3,8,24,25 For IVIVE, we applied the approach suggested by Sohlenius-Sternbeck et al.,15 using experimentally determined hepatocyte CLint and fu,p and fu,inc predicted by the Kilford equation.16 Plotting in vivo against in vitro CLint data (Fig. 10) demonstrated a systematic under-prediction of CLint for both, internal and external compounds with only 4 of 25 compounds (16%) predicted within 2-fold and 6 (24%) within 3-fold of unity (Fig. 7).
 | ||
| Fig. 7 In vitro–in vivo extrapolation of intrinsic clearance from mouse hepatocytes using predicted fraction unbound in the incubation (standard approach). CLint, intrinsic clearance; obs., observed in mice; pred., predicted from mouse hepatocytes; black line, unity; red solid line, two-fold of unity; red dotted line, three-fold of unity; filled dots, internal compounds; open squares, external compounds. | ||
Experimental fu,inc is required for IVIVE of CLint
As discussed by Pike et al., fu,inc is an important factor to be considered for scaling of CLint for PROTACs© in particular, given their potential for high binding to lipid membranes.8 Accordingly, we suspected that the under-prediction of CLint was due to an over-prediction of fu,inc, given that the underlying Kilford equation was built on small molecules only and inaccurate for compounds with logD ≥3,16 a lipophilicity range often observed for PROTACs©26 (Table S7†).
To confirm our hypothesis, we determined the fu,inc experimentally using RED. When comparing the experimental data with the predicted ones, no correlation was observed. While experimental values ranged from very low (0.05%) to high (81%) free fractions, predicted values considering hepatocyte incubation conditions (i.e. cell density of 0.2 × 106 cells per mL) limited themselves to free fractions from 44 to 98%, demonstrating an over-prediction of PROTAC© fu,inc by the Kilford equation (Fig. 8).
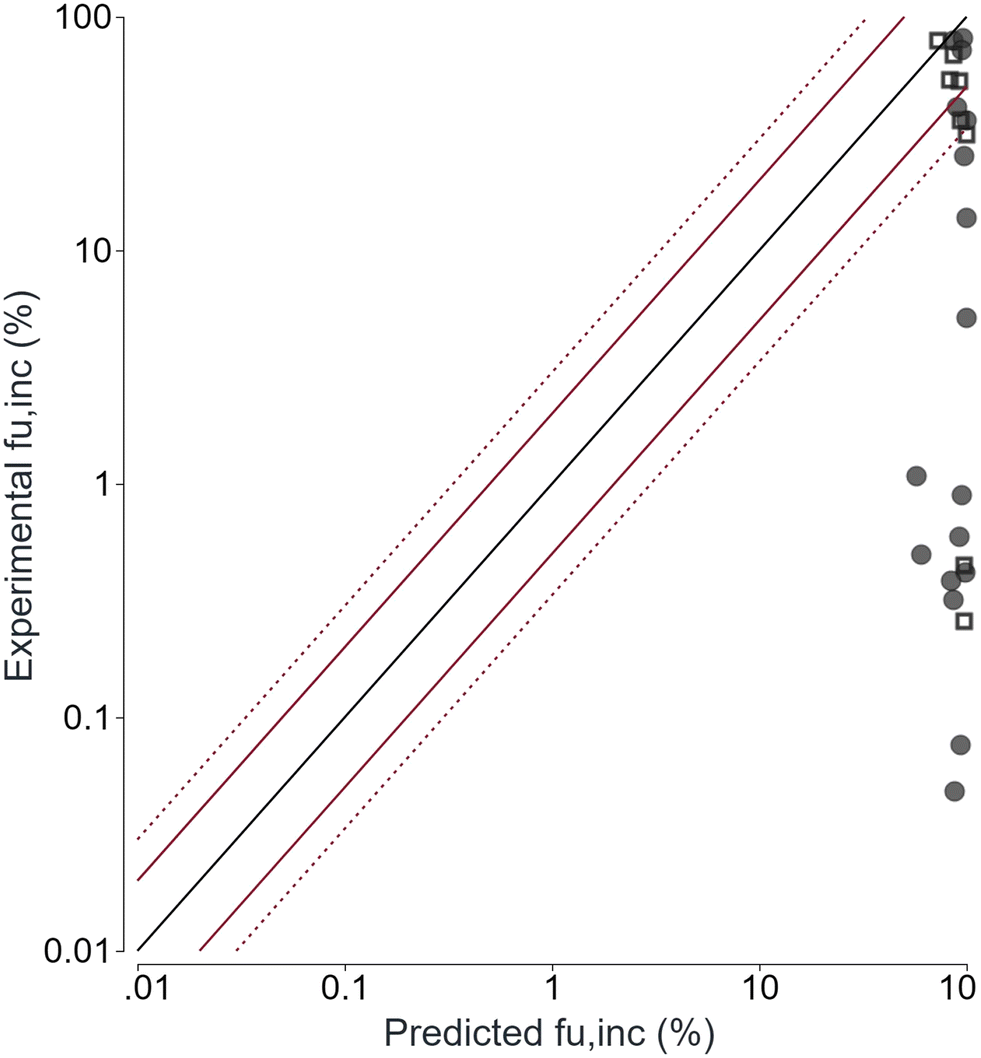 | ||
| Fig. 8 Correlation between experimentally determined and predicted fraction unbound in the incubation. fu,inc, fraction unbound in the hepatocyte incubation; black line, unity; red solid line, two-fold of unity; red dotted line, three-fold of unity; filled dots, internal compounds; open squares, external compounds. | ||
Consequently, we updated the IVIVE by exchanging predicted with experimental fu,inc data. As expected, this improved the under-prediction with 8 compounds (32%) falling within 2-fold and 10 compounds (40%) within 3-fold of unity and led to a normal distribution around the line of unity (Fig. 9). Therefore, we suggested the use of experimentally determined fu,inc data as best practice for IVIVE of CLint for PROTACs©.
 | ||
| Fig. 9 In vitro–in vivo extrapolation of intrinsic clearance from mouse hepatocytes using experimentally determined fraction unbound in the incubation. CLint, intrinsic clearance; obs., observed in mice; pred., predicted from mouse hepatocytes; black line, unity; red solid line, two-fold of unity; red dotted line, three-fold of unity; filled dots, internal compounds; open squares, external compounds. | ||
Despite the improvement of IVIVE by using experimental fu,inc values, only 40% (10 of 25) of the compounds were predicted within 3-fold of unity. Observed over-prediction of CLint might be due to an over-determination of fu,p values (ranging from 0.02 to 0.43%), the assessment of which is discussed to be challenging for PROTACs©.8,27 An alternative ‘membrane-free’ assay suggested by He et al. might help to accurately determine very low fu,p down to 0.00001%.27 Under-prediction of CLint may be related to further in vitro or in vivo challenges. Non-specific binding to the incubation plate,8 which is not covered with the fu,inc determination, may present an additional source of error in the extrapolation from in vitro to in vivo. Binding to red blood cells, i.e. when Rb does not equal unity (as assumed for neutrals and bases), might additionally confound clearance prediction, especially for high clearance drugs.28 Extrahepatic metabolic or transport-limited elimination may contribute to an under-estimation of clearance. Of the 12 under-predicted PROTACs©, 8 contain a CRBN-based E3 ligase binder. PROTACs© with such imid structures have been reported as prone to degradation by hydrolysis occurring either chemically or by hydrolytic enzymes also outside of the liver.8,29 Transporter-mediated elimination (i.e. active biliary or renal excretion) has been discussed for PROTACs© given their high MW (Table S7†) and low permeability.3,8,9 However, reported data sets are limited and the extent of non-metabolic elimination has been reported as minor.
Generally, for an improved understanding of PROTAC© IVIVE of CLint, we focus our current activities on enlarging the compound set to a broader chemical space, putting the PROTAC© data into perspective with small molecule data, and exploring ways to achieve fu,inc values accurately reflecting the binding in the in vitro CLint assay. Furthermore, we are developing an improved model for in silico prediction of PROTAC© fu,inc. In addition, for selected lead compounds, we are deciphering the predominant elimination routes to support CLint IVIVE with the incorporation of all relevant elimination mechanisms.
A tailored discovery ADME assay cascade is required for PROTACs©
Based on the assessment of our standard small molecule in vitro assay cascade for its suitability to guide and direct the optimization of PROTACs© described in this report, we have adapted our strategy to support optimization campaigns (Fig. 10). When selecting suitable POI ligand (target protein) starting points for PROTACs©, we focus on chemical matter with a low HBD count or on identifying a path forward to reducing or shielding HBD to have a good basis for acceptable permeability. During PROTAC© design, we rely on estimating probability of oral absorption by our in-house developed oral F score and predicted ePSA. However, the oral F score applicability is limited due solely taking computed molecular descriptors and predicted ChromlogD into account. Subtle changes in the molecule e.g. electron density or impact of the addition heteroatoms will not be reflected in detail. Hence, supportive physicochemical characterization and cellular models are needed. In this regard, cellular degradation potency evaluation and optimization stays the first priority during the drug discovery process. Due to their high lipophilicity, PROTACs© are prone to binding to assay systems and e.g. serum proteins present in pharmacological assays. This can significantly influence the amount of free, pharmacologically active PROTAC© and needs to be considered when interpreting these results. Potency determination is followed by an assessment of lipophilicity (ChromlogD), solubility in FaSSIF as well as ePSA measurements. Thermodynamic solubility in FaSSIF is preferred over kinetic solubility in PBS since it represents better the physiological situation in the gastro-intestinal system. Metabolic stability is monitored subsequently. We do not only rely on IVIVE for selecting compounds for in vivo PK studies in early discovery phases but also select diverse chemical matter for PK since CLint IVIVE is still challenging. We then seek to identify chemical patterns that are beneficial for low in vivo CLint and run exploratory oral PKs. In general, we are relying on more and earlier in vivo PK studies by taking advantage of iv cassette studies to reduce the number of animals. Subsequently, we concentrate preferably on those series where in vivo CLint is in the well-predicted range. Caco-2 transwell assays are not run routinely for PROTACs© for absorption prediction and it remains to be seen if it can support rank-ordering of compounds. Overall, it is critical for each project to identify the optimization-relevant physicochemical and ADME parameters. These can differ depending on the chemical space employed in the respective programs.
 | ||
| Fig. 10 Tailored in vitro and in vivo ADME assay cascade applied in early PROTAC© discovery projects. | ||
Alternative delivery routes overcoming susceptibility for low oral bioavailability
While we could establish design guidelines for oral PROTACs©, alternative routes of administration (RoA) might become relevant if oral bioavailability cannot be achieved, or pharmacological considerations favor delivery via parenteral routes. In our hands, subcutaneous administration has resulted in favorable exposures in mice (Fig. 11) for several VHL- or CRBN-based PROTACs©. Here, for a VHL-based PROTAC©, the oral AUC0−∞was 28 h* ng mL−1 at 10 mg kg−1 with a bioavailability (based on AUC 0−∞) of 0.3% whereas the AUC0−∞ was 10, 406 h* ng mL−1 at 10 mg kg−1 for subcutaneous RoA (bioavailability (based on AUC0−∞) of 126%). The AUC0-∞ after intravenous dosing (0.5 mg kg−1) was 414 h* ng mL−1. Total clearance was determined 1.21 L h−1 kg−1 and Vss 0.27 L kg−1. Due to a plateau in the exposure profile (depot and zero order skin absorption kinetic30,31) observed after subcutaneous administration of several PROTACs©, this RoA could allow for longer action of the heterobifunctional degraders if required pharmacologically. Also, coupling (covalently or non-covalently) with an antibody can overcome pharmacokinetic hurdles and allow for selective delivery to the target tissue.32,33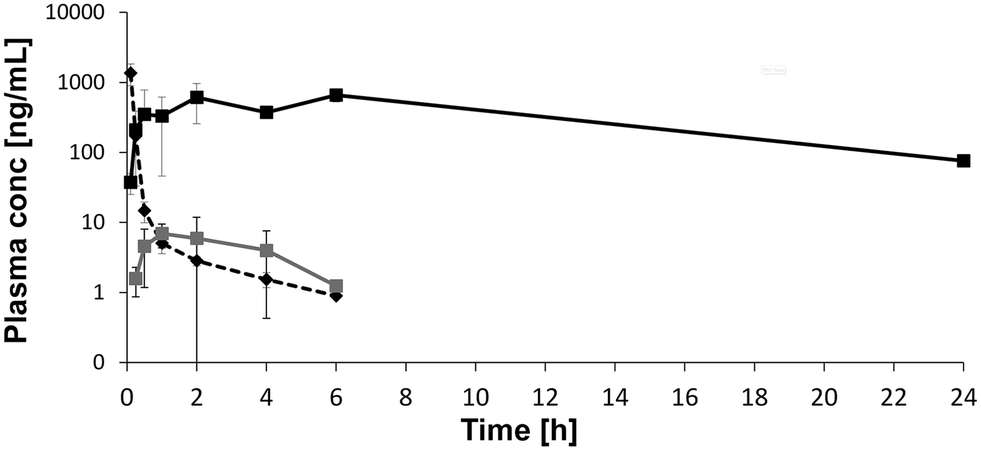 | ||
| Fig. 11 Subcutaneous dosing of a VHL-based PROTAC© results in higher exposure than oral administration in mice. A VHL-based degrader was administered in a single dose pharmacokinetic study to mice (iv 0.5 mg kg−1 - dashed black; po 10 mg kg−1 - grey; sc 10 mg kg−1 - black. The vehicle was 5% DMSO and 20% Kolliphor HS15 in water for all RoAs. Total plasma concentrations are plotted as a function of time. | ||
Conclusion
Heterobifunctional degraders are a promising modality to enrich the molecular toolbox to treat diseases and broaden the druggable space. With this paper, we contribute to enhancing the understanding of in vitro and early in vivo characterization and optimization of these molecules. For permeability assessment, transwell assays such as Caco-2 still remain challenging, although the addition of serum may reduce unspecific binding and improve recovery. Nevertheless, we did not find the predictive power for absorption as for standard small molecules. As a surrogate, we propose to focus optimization on molecular parameters and support a preferred space for oral PROTACs© with ≤3 HBD, molecular weight ≤950 and number of rotatable bonds ≤12. We have developed a composite, predictive score that serves as first guidance for design and prioritization of suitable chemical space according to this property space. In addition, the reduction of exposed polar surface area, e.g. through shielding of HBDs, is a powerful approach to improve permeability during compound optimization. Using classical IVIVE methods for CLint, as generally applied to small molecules, a systematic under-prediction from mouse hepatocytes was observed for PROTACs©. This bias could be overcome by using experimentally determined fu,inc values, while we demonstrated that the Kilford equation was not suitable for PROTAC© fu,inc prediction. Therefore, we advise project teams to use experimental fu,inc data as input parameters for IVIVE. For the future, we are working on the development of an alternative model for fu,inc prediction of PROTACs©. Furthermore, towards a refined PROTAC© IVIVE, we are aiming at expanding the PROTAC© data set, putting PROTAC© data into perspective with small molecules, improving the assessment of fu,inc accurately reflecting the binding in the in vitro CLint assay, and elucidating possible further routes of elimination. The tailored in vitro and early in vivo ADME assay cascade reflects the findings of this manuscript. In addition, alternative parenteral and targeted delivery options are actively explored.Data availability
The physicochemical data supporting this article have been included as part of the ESI.† Structural information is included for molecules publicly available.Author contributions
C. K. M. planned and performed CLint IVIVE analysis and interpretation and wrote the manuscript, Z. F. was responsible for planning and designing Caco-2 assays and result interpretation and wrote the manuscript, H. M. D. conducted PK studies, analyzed the influence of physicochemical properties and wrote the manuscript, G. P. and C. S. modelled the score for oral bioavailability, F. M.-P. established and performed ePSA and ChromlogD measurements. M. L. conducted PK studies and contributed to the design principles and alternative delivery routes for PROTACs©, C. P. contributed to the in vitro data interpretation and supported the PROTAC© CLint IVIVE strategy. H. M. D. steered and supervised the study.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Marlene Hagel, Dominik Jacob, Christian Stelz, Ulrich Seibl, and Sabrina Kaiser for generating the in vitro ADME data, Heike Ruetzel and Jan Selonke for performing chromatographic logD assays, Dieter Spuck for performing ePSA assays, and Kai Koedderitzsch, Xiaohua Zhu and Mary-Jo Miller for supervising the generation of mouse in vivo clearance data. Floriane Lignet for data sharing and discussion. Ulrike Gradhand, Stephanie Harlfinger, Dominique Perrin and Holger Scheible for critical review of the manuscript.References
- M. H. Brodermann, E. K. Henderson and R. S. Sellar, The emerging role of targeted protein degradation to treat and study cancer, J. Pathol., 2024, 263, 403–417 CrossRef PubMed.
- X. Zheng, N. Ji, V. Campbell, A. Slavin, X. Zhu, D. Chen, H. Rong, B. Enerson, M. Mayo, K. Sharma, C. M. Browne, C. R. Klaus, H. Li, G. Massa, A. A. McDonald, Y. Shi, M. Sintchak, S. Skouras, D. M. Walther, K. Yuan, Y. Zhang, J. Kelleher, G. Liu, X. Luo, N. Mainolfi and M. M. Weiss, Discovery of KT-474–a Potent, Selective, and Orally Bioavailable IRAK4 Degrader for the Treatment of Autoimmune Diseases, J. Med. Chem., 2024, 67, 18022−18037 Search PubMed.
- C. Cantrill, P. Chaturvedi, C. Rynn, J. Petrig Schaffland, I. Walter and M. B. Wittwer, Fundamental aspects of DMPK optimization of targeted protein degraders, Drug Discovery Today, 2020, 25, 969–982 CrossRef CAS PubMed.
- G. Apprato, G. Ermondi and G. Caron, The Quest for Oral PROTAC drugs: Evaluating the Weaknesses of the Screening Pipeline, ACS Med. Chem. Lett., 2023, 14, 879–883 CrossRef CAS PubMed.
- D. García Jiménez, M. Rossi Sebastiano, M. Vallaro, V. Mileo, D. Pizzirani, E. Moretti, G. Ermondi and G. Caron, Designing Soluble PROTACs: Strategies and Preliminary Guidelines, J. Med. Chem., 2022, 65, 12639–12649 CrossRef.
- P. Muschong, K. Awwad, E. Price, M. Mezler and M. Weinheimer, Conquering the beyond Rule of Five Space with an Optimized High-Throughput Caco-2 Assay to Close Gaps in Absorption Prediction, Pharmaceutics, 2024, 16, 846 CrossRef CAS.
- C. Kofink, N. Trainor, B. Mair, S. Wöhrle, M. Wurm, N. Mischerikow, M. J. Roy, G. Bader, P. Greb, G. Garavel, E. Diers, R. McLennan, C. Whitworth, V. Vetma, K. Rumpel, M. Scharnweber, J. E. Fuchs, T. Gerstberger, Y. Cui, G. Gremel, P. Chetta, S. Hopf, N. Budano, J. Rinnenthal, G. Gmaschitz, M. Mayer, M. Koegl, A. Ciulli, H. Weinstabl and W. Farnaby, A selective and orally bioavailable VHL-recruiting PROTAC achieves SMARCA2 degradation in vivo, Nat. Commun., 2022, 13, 5969 CrossRef.
- A. Pike, B. Williamson, S. Harlfinger, S. Martin and D. F. McGinnity, Optimising proteolysis-targeting chimeras (PROTACs) for oral drug delivery: a drug metabolism and pharmacokinetics perspective, Drug Discovery Today, 2020, 25, 1793–1800 Search PubMed.
- D. Zhang, B. Ma, P. S. Dragovich, L. Ma, S. Chen, E. C. Chen, X. Ye, J. Liu, J. Pizzano, E. Bortolon, E. Chan, X. Zhang, Y.-C. Chen, E. S. Levy, R. L. Yauch, S. C. Khojasteh and C. E. C. A. Hop, Tissue distribution and retention drives efficacy of rapidly clearing VHL-based PROTACs, Commun. Med., 2024, 4, 87 CrossRef CAS.
- D. Ye, Á. López Mármol, V. Lenz, P. Muschong, A. Wilhelm-Alkubaisi, M. Weinheimer, M. Koziolek, K. A. Sauer, L. Laplanche and M. Mezler, Mucin-Protected Caco-2 Assay to Study Drug Permeation in the Presence of Complex Biorelevant Media, Pharmaceutics, 2022, 14, 699 CrossRef CAS PubMed.
- Y.-T. Wang, E. Price, M. Feng, J. Hulen, S. Doktor, D. M. Stresser, E. M. Maes, Q. C. Ji and G. J. Jenkins, High-Throughput SFC-MS/MS Method to Measure EPSA and Predict Human Permeability, J. Med. Chem., 2024, 67, 13765–13777 CrossRef CAS PubMed.
- K. R. Hornberger and E. M. V. Araujo, Physicochemical Property Determinants of Oral Absorption for PROTAC Protein Degraders, J. Med. Chem., 2023, 66, 8281–8287 CrossRef CAS.
- M. Schade, J. S. Scott, T. G. Hayhow, A. Pike, I. Terstiege, M. Ahlqvist, J. R. Johansson, C. R. Diene, C. Fallan, A. Y. S. Balazs, E. Chiarparin and D. Wilson, Structural and Physicochemical Features of Oral PROTACs, J. Med. Chem., 2024, 67, 13106–13116 CrossRef CAS.
- T. Yamagata, U. Zanelli, D. Gallemann, D. Perrin, H. Dolgos and C. Petersson, Comparison of methods for the prediction of human clearance from hepatocyte intrinsic clearance for a set of reference compounds and an external evaluation set, Xenobiotica, 2017, 47, 741–751 CrossRef CAS.
- A.-K. Sohlenius-Sternbeck, C. Jones, D. Ferguson, B. J. Middleton, D. Projean, E. Floby, J. Bylund and L. Afzelius, Practical use of the regression offset approach for the prediction of in vivo intrinsic clearance from hepatocytes, Xenobiotica, 2012, 42, 841–853 CrossRef CAS PubMed.
- P. J. Kilford, M. Gertz, J. B. Houston and A. Galetin, Hepatocellular binding of drugs: correction for unbound fraction in hepatocyte incubations using microsomal binding or drug lipophilicity data, Drug Metab. Dispos., 2008, 36, 1194–1197 CrossRef CAS PubMed.
- Percepta v2022, Advanced Chemistry Development, Inc. (ACD/Labs), Toronto, Canada, 2022.
- GALAS, Global Adjusted Locally According to Similarity.
- Y. Cui, C. Desevaux, I. Truebenbach, P. Sieger, K. Klinder, A. Long and A. Sauer, A Bidirectional Permeability Assay for beyond Rule of 5 Compounds, Pharmaceutics, 2021, 13, 1146 CrossRef CAS PubMed.
- Y. Atilaw, V. Poongavanam, C. Svensson Nilsson, D. Nguyen, A. Giese, D. Meibom, M. Erdelyi and J. Kihlberg, Solution Conformations Shed Light on PROTAC Cell Permeability, ACS Med. Chem. Lett., 2021, 12, 107–114 Search PubMed.
- G. H. Goetz, W. Farrell, M. Shalaeva, S. Sciabola, D. Anderson, J. Yan, L. Philippe and M. J. Shapiro, High throughput method for the indirect detection of intramolecular hydrogen bonding, J. Med. Chem., 2014, 57, 2920–2929 CrossRef CAS.
- D. A. DeGoey, H.-J. Chen, P. B. Cox and M. D. Wendt, Beyond the Rule of 5: Lessons Learned from AbbVie's Drugs and Compound Collection, J. Med. Chem., 2018, 61, 2636–2651 CrossRef CAS PubMed.
- C. Rynn and H. M. Duevel, Meeting report: DMPK optimisation strategies and quantitative translational PKPD frameworks to predict human PK and efficacious dose of targeted protein degraders, Xenobiotica, 2024, 1–5 Search PubMed.
- L. P. Volak, H. M. Duevel, S. Humphreys, D. Nettleton, C. Phipps, A. Pike, C. Rynn, P. Scott-Stevens, D. Zhang and M. Zientek, Industry Perspective on the Pharmacokinetic and Absorption, Distribution, Metabolism, and Excretion Characterization of Heterobifunctional Protein Degraders, Drug Metab. Dispos., 2023, 51, 792–803 CrossRef PubMed.
- T. G. Hayhow, B. Williamson, M. Lawson, N. Cureton, E. L. Braybrooke, A. Campbell, R. J. Carbajo, A. Cheraghchi-Bashi, E. Chiarparin, C. R. Diène, C. Fallan, D. I. Fisher, F. W. Goldberg, L. Hopcroft, P. Hopcroft, A. Jackson, J. G. Kettle, T. Klinowska, U. Künzel, G. Lamont, H. J. Lewis, G. Maglennon, S. Martin, P. M. Gutierrez, C. J. Morrow, M. Nikolaou, J. W. M. Nissink, P. O'Shea, R. Polanski, M. Schade, J. S. Scott, A. Smith, J. Weber, J. Wilson, B. Yang and C. Crafter, Metabolism-driven in vitro/in vivo disconnect of an oral ERα VHL-PROTAC, Commun. Biol., 2024, 7, 563 Search PubMed.
- S. D. Edmondson, B. Yang and C. Fallan, Proteolysis targeting chimeras (PROTACs) in 'beyond rule-of-five' chemical space: Recent progress and future challenges, Bioorg. Med. Chem. Lett., 2019, 29, 1555–1564 CrossRef PubMed.
- Z. He, J. Tucker and A. Smith, Novel plasma protein binding ‘membrane-free’ assay delivers Fraction unbound values for beyond Rule of 5’s compounds, 6th RSC-BMCS / DMDG New Perspectives in DMPK meeting, Liverpool, UK, 2024.
- J. Yang, M. Jamei, K. R. Yeo, A. Rostami-Hodjegan and G. T. Tucker, Misuse of the well-stirred model of hepatic drug clearance, Drug Metab. Dispos., 2007, 35, 501–502 Search PubMed.
- S. E. Fabro, H. Schumacher, R. L. Smith and R. T. Williams, Metabolismo della talidomide. I. L'idrolisi spontanea della talidomide, Boll. - Soc. Ital. Biol. Sper., 1963, 39, 1921–1925 Search PubMed.
- S. Yamamoto, M. Karashima, Y. Arai, K. Tohyama and N. Amano, Prediction of Human Pharmacokinetic Profile After Transdermal Drug Application Using Excised Human Skin, J. Pharm. Sci., 2017, 106, 2787–2794 Search PubMed.
- W. Cawello, M. Braun and J.-O. Andreas, Drug Delivery and Transport into the Central Circulation: An Example of Zero-Order In vivo Absorption of Rotigotine from a Transdermal Patch Formulation, Eur. J. Drug Metab. Pharmacokinet., 2018, 43, 475–481 CrossRef PubMed.
- H. Schneider, S. Jäger, D. Könning, N. Rasche, C. Schröter, D. Elter, A. Evers, M. Lecomte, F. R. Sirtori, D. Schwarz, A. Wegener, I. Hartung and M. Rieker, PROxAb Shuttle: A non-covalent plug-and-play platform for the rapid generation of tumor-targeting antibody-PROTAC conjugates, bioRxiv, 2023, preprint, DOI:10.1101/2023.09.29.558399.
- P. S. Dragovich, Degrader-antibody conjugates, Chem. Soc. Rev., 2022, 51, 3886–3897 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4md00854e |
| This journal is © The Royal Society of Chemistry 2025 |