DOI:
10.1039/D4MD00849A
(Research Article)
RSC Med. Chem., 2025,
16, 1397-1409
Development of small-molecule fluorescent probes targeting neutrophils via N-formyl peptide receptors†
Received
31st October 2024
, Accepted 19th December 2024
First published on 14th January 2025
Abstract
N-Formyl peptide receptors (FPRs) are membrane receptors that are abundantly expressed in innate immune cells, including neutrophils and platelets, demonstrating potential new targets for immune system regulation and the treatment of inflammatory conditions. We report here the development and bio-physical validation of new FPR imaging agents as effective tools to track FPR distribution, localisation and functions, ultimately helping to establish FPR exact roles and functions in pathological and physiological conditions. The new series of probes feature a small molecule-based FPR address system conjugated to suitable fluorophores, resulting in highly specific FPR agents, including a partial agonist endowed with high affinity (i.e. low/sub-nanomolar potency) on FPR-transfected cells and human neutrophils. Preliminary imaging studies via multiphoton microscopy demonstrate that the probes enable the visualisation of FPRs in live cells, thus representing valid bio-imaging tools for the analysis of FPR-mediated signalling, such as the activation of neutrophils in inflammatory events.
Introduction
Human N-formyl peptide receptors (FPR1, FPR2, and FPR3) are a small class of remarkably versatile G-protein coupled receptors (GPCRs) involved in a wide range of cellular processes at the basis of immunity and host defence.1 Firstly identified in phagocytic leukocytes (i.e. neutrophils and monocytes),2 FPRs have been also found to regulate the activation of several other classes of immune system cells,3 including white blood cells (e.g. macrophages and dendritic cells) and platelets.4,5 By actively participating in complex cell signalling pathways, often initiated by pro- and anti-inflammatory stimuli, FPRs modulate both the innate immune responses and the infiltration of immune cells following tissue damage.6–8 Moreover, several studies also demonstrate the over-expression of FPRs in various tissues and nonhematopoietic cells ranging from osteoblasts to lung epithelial cells and hepatocytes,9–11 suggesting possible wider roles beyond immunomodulation and anti-inflammation.12 In this context, FPRs have been extensively investigated as potential new targets to interfere with the inflammatory states of a variety of diseases, including respiratory disorders,6,9 stroke and ischemia–reperfusion injury,13,14 as well as many metastatic cancers.15–17
Although several classes of FPR ligands have been identified to date,18–20 further evidence is required to establish structural and functional features of this GPCR sub-family,21 in order to accurately clarify their contribution to the biological signalling in both physiological and pathological contexts linked to inflammation and immunity. To this end, targeted imaging probes that allow direct real-time visualisation of FPRs through high-throughput and advanced imaging methodologies represent valid tools to fully determine cellular specificity and distribution,22 as well as functional roles, including receptor biosynthesis, ligand binding, internalisation and degradation, or recycling,23–26 which in turn can influence cellular states and behaviours. In this regard, a number of high-throughput strategies and techniques have been used lately to optically visualise FPRs, such as automated flow cytometry and microfluidic platforms.9,27–32 In parallel, several peptide-based FPR imaging conjugates have also been developed for fluorescence and near-infrared (NIR) spectroscopy,23–26 positron emission tomography (PET),33 single-photon emission computed tomography (SPECT),34,35 and magnetic resonance imaging (MRI).35 Nonetheless, these peptide-based agents have some limitations, such as the large size, low specificity in cell membrane targeting, and low stability (i.e., degradation by proteases). In contrast, small molecule-based probes are typically endowed with higher stability in bio-matrices, along with other benefits, such us a non-immunogenic character and the overall ‘ease of use’ in biological assays. We expect that the development of effective small-molecule fluorescent ligands targeted to FPRs can lead to the identification of enhanced molecular tools useful to univocally clarify the biology of the receptors, for instance in inflammation, allowing selective monitoring of signalling responses, as well as other related events, at the cellular and subcellular levels. However, the development of small molecule-based fluorescent probes remains limited to date. To our knowledge, only one fluorescent probe has been reported, namely a Quin-C1 derivative selective for FPR2,36 while small-molecule imaging agents targeted to FPR1 are currently lacking.
In previous research, we have established that pyridazin-3(2H)-one-based small-molecules are effective FPR agonists (see e.g. in Fig. 1A),37–40 demonstrating high affinity (e.g., low nanomolar potency) and in some cases a good level of selectivity through FPR isoforms.41,42
 |
| | Fig. 1 A) Examples of established pyridazin-3(2H)-one-based FPR ligands and agonist activity (EC50) in FPR1/FPR2 transfected cells and human neutrophils (hPMN).37,43 B) Overview of the molecular design to develop fluorescently labelled analogues; X = linker. | |
Here we report the development of four FPR small-molecule imaging agents obtained through further elaboration of previous series of ligands in order to introduce selected fluorophores (i.e., anthranyl, dansyl, EDANS and BODIPY; Fig. 1B) in selected positions of the lead chemotype adopted here as the FPR ‘address system’ (Fig. 1), thereby enabling to simultaneously achieve FPR binding (via the address system) and direct fluorescence visualisation (via the fluorescent tag). The biological activity was initially measured through Ca2+ mobilization functional assays in FPR-transfected cells and supported by molecular modelling simulations, establishing that the new probes effectively interact with FPRs, and two of them resulted in pure antagonist effects. Moreover, to confirm the suitability to target neutrophils, we tested the new probes in these primary human isolated cells and recorded high values of activity, with one probe exhibiting picomolar values of potency (i.e. via Ca2+ mobilization) as an antagonist. Lastly, in two-photon microscopy experiments, we confirmed that the probes bind to FPR-transfected live cells and maintain their fluorescent signal to enable effective visualisation (and tracking) upon interaction with the receptors in the cells. Overall, our results demonstrate the suitability of these probes to selectively target FPR-expressing cells, such as neutrophils. The versatile FPR-targeted agents reported herein open possibilities of generating advanced imaging probes based on the same molecular architecture and bearing fluorophores with enhanced optical efficiency (e.g., suitable for single-molecule and super-resolution imaging), to support the development of microscopy methods useful for tracking and visualising neutrophils in real-time within drug discovery research programmes in the field of inflammation.
Results and discussion
Rational design and synthesis of FPR-targeted optical probes
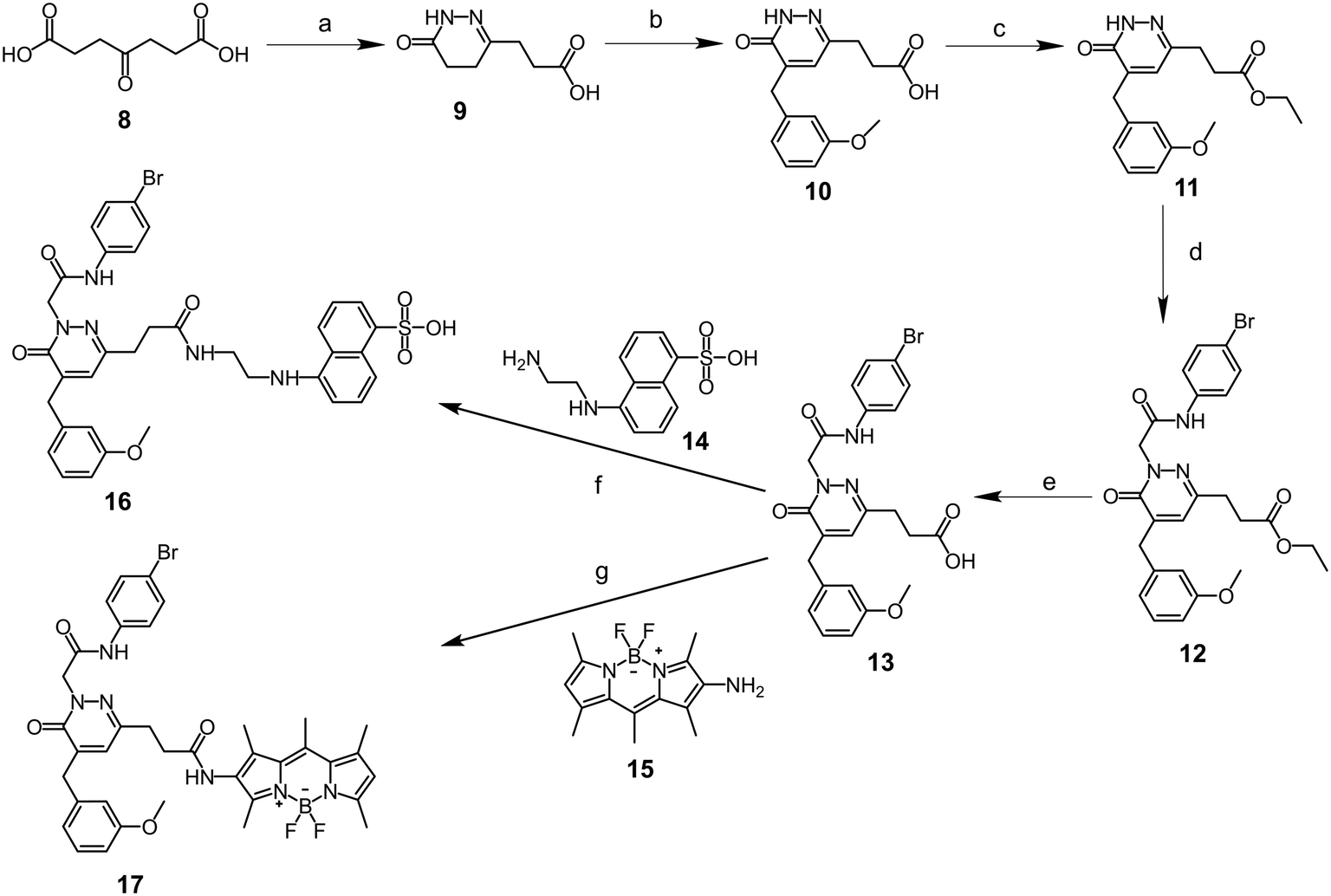
The synthetic approach adopted to obtain the new FPR fluorescent ligands is shown in Schemes 1 and 2, and the structures were confirmed on the basis of analytical and spectral data. The optical probes (4, 7, 16 and 17) are structurally based on the known FPR targeting module (i.e., pyridazinone-based address system; Fig. 1). These probes contain the groups and/or functions known as crucial for FPR binding (i.e., an aryl residue linked to an acetamide or sulphonamide in position N-2, an aromatic or polyaromatic group in position C-4 of the heterocycle) and suitable fluorescent tags (in positions N-2, C-4 or C-6) having sizes compatible with small-molecule ligands. Due to their electronic and structural similarities to the residues present in the reference compounds (Fig. 1), the anthranyl and dansyl fluorophores were introduced in C-4 (probe 4, where anthracene fulfils the requirement of having an aromatic group in this position) and N-2 (probe 7, where the sulphonamide function substitutes the acetamide in the previous FPR ligand series).
 |
| | Scheme 1 Reagents and conditions: a) 9-anthracenecarboxaldehyde or 3-methoxybenzaldehyde, KOH/EtOH (5% w/v), reflux, 2.5 h; b) N-(4-bromophenyl)-2-chloroacetamide 3, K2CO3, CH3CN, reflux, 3.5 h; c) dansyl chloride 6, Et3N, CH3CN, rt, 16 h. | |
 |
| | Scheme 2 Reagents and conditions: a) N2H4·H2O, EtOH, 60 °C, 1.5 h; b) 3-methoxybenzaldehyde, KOH/EtOH (5% w/v), reflux, 5 h; c) H2SO4, EtOH, reflux, 4 h; d) N-(4-bromophenyl)-2-chloroacetamide 3, K2CO3, anhydrous CH3CN, reflux 5–7 h; e) tetrahydrofuran/2 M NaOH (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v), rt, overnight; f) HOBt, Et3N, EDC, 14, anhydrous DMF, N2, rt, 48 h; g) Et3N, anhydrous tetrahydrofuran, −5 °C, 1 h → ethyl chloroformate, 0 °C, 1 h → 15, rt, 16 h. :1, v/v), rt, overnight; f) HOBt, Et3N, EDC, 14, anhydrous DMF, N2, rt, 48 h; g) Et3N, anhydrous tetrahydrofuran, −5 °C, 1 h → ethyl chloroformate, 0 °C, 1 h → 15, rt, 16 h. | |
The synthetic pathway leading to the final compounds 4 and 7 (Scheme 1), bearing anthranyl and dansyl fluorophores in positions C-4 and N-2 of the pyridazinone ring, respectively, involved firstly a Knoevenagel condensation (followed by isomerization) on the 6-methyl-4,5-dihydropyridazin-3(2H)-one 137,39 using the appropriate aromatic aldehyde in the presence of KOH. Subsequently, compound 2 was alkylated under standard conditions with N-(4-bromophenyl)-2-chloroacetamide 342 to obtain the anthranyl optical probe 4, while intermediate 537 was reacted with dansyl chloride in the presence of triethylamine to obtain imaging probe 7.
The procedure adopted to synthesise optical probe 16 is shown in Scheme 2. The commercially available 4-oxoheptanedioic acid 8 was reacted with hydrazine hydrate to obtain dihydropyridazinone 9,44 bearing a carboxylic acid function in position C-6. After Knoevenagel condensation with 3-methoxybenzaldehyde, intermediate 10 was converted into the ethyl ester derivative 11 to protect the carboxylic group. Subsequently, compound 11 was reacted via standard alkylation with N-(4-bromophenyl)-2-chloroacetamide 343 to obtain compound 12. After removing the ethyl group in 12 by NaOH-mediated hydrolysis, the resulting acid intermediate 13 was coupled to the 5-[(2-aminoethyl)-amino]naphthalene-1-sulfonyl (EDANS) 14 through an amidation reaction using 1-hydroxybenzotriazole (HOBt) and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) as coupling reagents, leading to the formation of the optical probe 16. Lastly, 5,5-difluoro-1,3,7,9,10-pentamethyl-5H-4λ4,5λ4-dipyrrolo[1,2-c:2′,1′-f][1,3,2]diazaborinin-2-amine (“amino-BODIPY”) 1545 was reacted with intermediate 13 adopting the mixed anhydride method, with ethyl chloroformate in the presence of triethylamine, to obtain optical probe 17.
Biological activity in FPR-transfected cells
Optical probes 4, 7, 16, and 17 were primarily evaluated for their ability to produce changes in intracellular Ca2+ levels in human HL-60 cells transfected with FPR1 or FPR2.37,43 The reference peptide agonists fMLF and WKYMVm (W-peptide) were used as controls, and the results are reported as EC50 (and relative efficacy) and IC50 values in Table 1 (and Fig. S3.2–3.5 in ESI,† for representative dose–response curves). For the optical probe 17, to overcome issues of fluorescence interference between test probe (17) and Ca2+ indicator, we used a spectroscopy procedure based on a different Ca2+ chelator for read out,46i.e. Fura-2AM, since the two methods are both established approaches for Ca2+ measurement and typically give comparable outcomes. In agreement with the activity recorded for previously developed ligands,37,38,43,47 all the four probes demonstrated binding towards FPR1/FPR2 (as indicated by the EC50/IC50 values; Table 1), although with very different activity profiles. In parallel, no activity was observed in non-transfected (wild type) cells, clearly confirming the apparent specificity of the optical probes for FPRs. Probe 7 is a selective pure antagonist for FPR1 over FPR2 with IC50 at micromolar range. In similar fashion, probe 17 is also a pure antagonist, but for both FPR1 and FPR2, with comparable potency (in the low micromolar range; Table 1) on the two isoforms. Differently, probes 4 and 16 are two partial agonists for FPR1 and FPR2, one of which (probe 16) has high apparent affinity on both FPR-transfected cells [i.e. low nanomolar potency for FPR1 and 100-fold (as agonist) or 1000-fold (as antagonist) selectivity vs. FPR2] and isolated human neutrophils (i.e. sub-nanomolar potency; see next section ‘Biological activity in primary human neutrophils’).
Table 1 Biological activity of probes 4, 7, 16 and 17 in FPR1/2-transfected HL-60 cells
| Cmpd |
FPR1 |
FPR2 |
| EC50, μM (efficacy, %) |
IC50, μM |
EC50, μM (efficacy, %) |
IC50, μM |
| Values are expressed as EC50 (μM) and efficacy (% in brackets) as determined in Ca2+ flux assay. Efficacy (in brackets) is expressed as % of the response induced by 5 nM fMLF in human polymorphonuclear neutrophils (hPMN) and FPR1-HL60 cells or by 5 nM WKYMVm in FPR2-HL60 cells. No activity was found in non-transfected HL-60 cells. N.A. = no activity (no response was observed during first 2 min after addition of probes under investigation) considering the limits of efficacy <20% and EC50 <50 μM. |
|
4
|
4.0 ± 0.7 (115) |
0.5 ± 0.2 |
6.7 ± 2.2 (120) |
17.5 ± 3.5 |
|
7
|
N.A. |
17.7 ± 3.7 |
N.A. |
N.A. |
|
16
|
0.01 ± 0.005 (140) |
0.002 ± 0.001 |
1.4 ± 0.7 (145) |
1.3 ± 0.6 |
|
17
|
N.A. |
2.6 ± 2.1 |
N.A. |
1.6 ± 1.4 |
The overall high potency of 16, as well as the higher inhibitory effect of 17 (compared to 7), can be explained via SAR analysis, taking into consideration the activity recorded for previously developed ligands.37,38,43,47 In particular, the optical probes 16 and 17 exhibit no structural variation in the FPR address system (Fig. 1), which is present in several established FPR ligands of this class (e.g. see structures in panel A, Fig. 1).37–39,43 The chemical modifications performed to introduce EDANS (in 16) and BODIPY (in 17) fluorophores in position C-6 of the pyridazinone ring include the presence of a linker that keeps the optical tags at a distance of six (in 16) or four (in 17) atoms from the targeting module. Thus, it can be assumed that the probes retain the high affinity of previous analogues towards FPRs due to the limited influence of possible steric hindrance induced by the fluorophore, which would then have no detrimental effects on the binding of the targeting module (substantially kept unchanged). Moreover, for probe 16, the EDANS fluorophore in position C-6 can establish a number of additional interactions within FPRs (as detailed in the Molecular Modelling section), which account for the increase of potency (compared to previous series)37–39,43,47 and the overall very high level of activity recorded with this imaging agent.
Biological activity in primary human neutrophils
With the aim of validating the probes as suitable imaging agents for primary phagocytes (i.e. immune cells naturally expressing FPRs, mainly FPR1), we evaluated their activity by measuring Ca2+ flux in treated human neutrophils (hPMNs). For these experiments, we applied the probes with higher FPR1 activity (i.e., 4 and 16) and/or selectivity (over FPR2, i.e., 7), to further verify the results obtained in HL-60 cells. We found that both selective and non-selective ligands identified in HL-60 cell assays also induced intracellular Ca2+ mobilization in human neutrophils (Table 2 and Fig. S3.6 in ESI†). In particular, probe 4 (i.e., moderately potent mixed FPR1/2 partial agonist in FPR-transfected cells) was confirmed to activate neutrophils at micromolar concentrations (∼9 μM) tested as both an agonist and antagonist in hPMNs (Fig. S3.6A in ESI†). In contrast, probe 7 (i.e., FPR1-selective antagonist in transfected cells) produced lower values of EC50 and IC50, although a slightly preferential antagonist effect (IC50 = 19.3 ± 4.98 μM vs. EC50 = 26.0 ± 13.8 μM) was found (Fig. S3.6B in ESI†). More relevant, probe 16 (i.e., potent FPR1/FPR2 mixed partial agonist in transfected cells) demonstrated a very high potency in hPMNs, highlighting a 10-fold increase of activity (i.e., sub-nanomolar) when tested as an antagonist (IC50 = 160 pM vs. EC50 = 1.8 nM) (Fig. S3.6C in ESI†).
Table 2 Summary of biological activity of probes 4, 7, and 16 in human neutrophils. Values are expressed as EC50 and IC50 (μM), as determined through Ca2+ flux assay
| Cmpd |
Human neutrophils |
| EC50, μM |
IC50, μM |
|
4
|
9.2 ± 5.2 |
9.3 ± 1.19 |
|
7
|
26.0 ± 13.8 |
19.3 ± 4.98 |
|
16
|
0.0018 ± 0.00089 |
0.00016 ± 0.000043 |
Molecular modelling
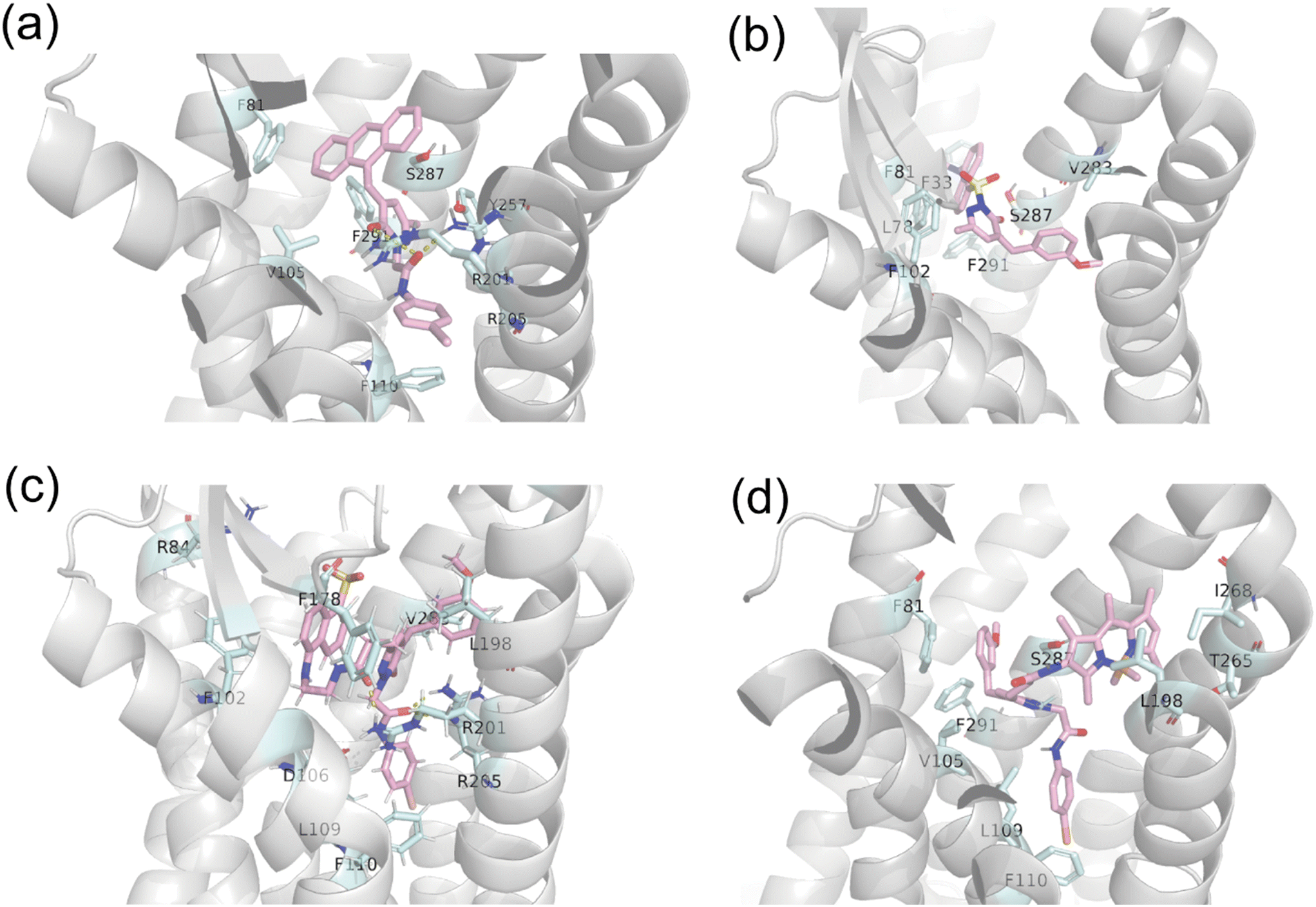
In parallel to the experimental findings, we have conducted molecular modelling simulations to further assess possible binding modes of the optical probes towards FPR1. Initially, the binding free energy was calculated for probes 4, 7, 16, and 17 (Table 3) using the molecular mechanics/general Born surface area (MM/GBSA) method. The contribution of binding free energy of each residue was decomposed from the MM/GBSA free energy, and the top residues are shown in Fig. 2. The predicted energy values (Table 3) showed high match with the experimentally determined IC50 values (Table 1), thus reinforcing reliability of results obtained for predicted binding modes (Fig. 3). Preliminary, the different interaction profiles recorded for the four probes (see Fig. 2, for per-residue decomposition of MM/GBSA free energy) indicated the presence of shifted binding modes between them (when interacting with the receptors), which accounted for the difference in calculated binding free energy. Probe 4 was obtained through modification of C-4 of the pyridazinone by introducing the anthranyl group in the place of a 3-methoxy-phenyl moiety (Fig. 1). As showed in Fig. 2 and 3a, the anthranyl residue forms hydrophobic and pi–pi interaction with F81 (TM2, i.e. transmembrane helix 2, −1.4 kcal mol−1), F110 (TM3, −1.6 kcal mol−1), F291 (TM7, −2.0 kcal mol−1) and S287 (TM7, −1.7 kcal mol−1), as well as H-bonding with R201 (TM5, −3.4 kcal mol−1) and R205 (TM5, −0.6 kcal mol−1), demonstrating a level of affinity for FPR1, which is in line with previously developed pyridazinone-based ligands.37,39,43 Similarly, the dansyl group in probe 7 was coupled to the pyridazin-3(2H)-one core in place of the 4-bromo-phenyl moiety (Fig. 1). The dansyl group shows to interact with F81 (TM2, −4.0 kcal mol−1), F291 (TM7, −1.1 kcal mol−1) and S287 (TM7, −2.0 kcal mol−1) (Fig. 2 and 3b). However, the pi–pi interaction with F110 (TM3, −0.1 kcal mol−1), as well as H-bonds with R201 (TM5, 1.2 kcal mol−1) and R205 (TM5, 0.2 kcal mol−1) are lost. The positive contribution means that the binding of probe 7 is not favoured for R201 and R205, in order to replace the solvent that interact with these residues. These changes in binding mode can explain the decrease in potency values of probe 7 towards FPR1. Differently, probe 16 (i.e. the most active term of the series) is found to bind deeply into the receptor close to TM4, 5 (Fig. 3c). As shown in Fig. 2 and 3c, the 4-bromo-phenyl moiety interacts with L109 (−1.7 kcal mol−1) and F110 (−2.1 kcal mol−1) on TM3 by hydrophobic and pi–pi interactions. The linker that connects the 4-bromo-phenyl residue and the pyridazinone core contains an amide group, which forms H-bonds with D106 (TM3, −4.2 kcal mol−1), R201 (TM5, −0.4 kcal mol−1) and R205 (TM5, −0.8 kcal mol−1). The 3-methoxy-phenyl moiety forms hydrophobic interaction with L198 (TM5, −2.6 kcal mol−1) and V283 (TM7, −1.6 kcal mol−1). The EDANS fluorophore in position C-6 of the pyridazinone scaffold mainly interacts with F102 (TM3, −1.5 kcal mol−1) and F178 (ECL2, i.e. extracellular loop 2, −1.3 kcal mol−1). Besides, the negative sulfonic acid group interacts with the positive guanidyl group in R84 (TM2, −0.8 kcal mol−1). The formation of all these additional interactions, that arise from the presence of the EDANS fluorophore, supports the high level of binding of probe 16 with FPR1. As a result, 16 is more potent than C-6 unmodified 2-arylacetamide-pyridazin-3(2H)-one ligands previously developed (e.g., see panel A in Fig. 1)37,39 and adopted as starting ‘address system’ for the development of the optical agents within this work. Therefore, the strategy of introducing a fluorophore at position C-6 of the pyridazinone scaffold proved a valid approach to access small-molecule fluorescent probes targeted to FPRs and bearing a very high apparent affinity for the receptors. Probe 17 also features C-6 modification in similar fashion to probe 16. However, the binding affinity of 17 (pIC50 = 5.85; Table 3) decreases drastically compared to 16 (pIC50 = 8.70), although it results higher than the other pure antagonist 7 (pIC50 = 4.75). The different length of the linker that connects the EDANS or BODIPY fluorophores in position C-6 of the pyridazin-3(2H)-one core in 16 and 17, respectively, is likely the key factor accounting for such variation of activity. The six-atom-length soft linker in probe 16 made it easier for the ligand to assume a favourable conformation that can be accommodated in the FPR1 binding pocket. For probe 17, the shorter 4-atom linker did not allow that this ligand could assume a pose within the pocket similar to that adopted by 16, thereby directing the BODIPY fluorophore into the space amidst TM1, TM2, and TM7 (Fig. 3d). In the case of 17 in contact with FPR1, the 3-methoxy-phenyl residue is placed near TM1 and the BODIPY stretching towards TM6. The H-bonds with D106 (TM3, 0.05 kcal mol−1), R201 (TM5, 0.4 kcal mol−1), and R205 (TM5, 0.2 kcal mol−1) were absent in comparison with 16, leading to a drastic decrease in binding affinity. Overall, the molecular modelling results highlight that the presence of suitable linkers (of at least six atoms) between the fluorophore and the pyridazinone-based ‘address system’ may represent a crucial requirement to obtain potent FPR-targeted probes based on this class of ligands.
Table 3 Predicted binding free energy (kcal mol−1) by MM/GBSA and pEC50/pIC50 of ligands 4, 7, 16, and 17 on FPR1
|
|
ΔEMM |
ΔGsolv |
−TΔS |
ΔGeffa |
pEC50 |
pIC50 |
|
ΔGeff = ΔEMM − TΔS + ΔGsolv, where ΔEMM was gas-phase interaction energy calculated by molecular mechanics, −TΔS was change of conformational entropy, and ΔGsolv was the change of solvation energy determined by ΔGsolv = ΔGsolv,complex − ΔGsolv,receptor − ΔGsolv,ligand.
|
|
4
|
−98.74 ± 2.06 |
53.70 ± 1.64 |
24.15 ± 1.03 |
−20.88 ± 1.12 |
5.40 |
6.30 |
|
7
|
−62.1 ± 0.403 |
22.94 ± 0.16 |
24.47 ± 0.55 |
−14.75 ± 0.33 |
N.A. |
4.75 |
|
16
|
−310.33 ± 2.94 |
253.10 ± 2.06 |
−30.35 ± 1.44 |
−26.88 ± 2.89 |
8.00 |
8.70 |
|
17
|
−94.73 ± 4.12 |
46.82 ± 3.57 |
−30.31 ± 0.42 |
−17.59 ± 0.44 |
N.A. |
5.85 |
 |
| | Fig. 2 MM/GBSA binding energies decomposition per residue for probes 4, 7, 16, and 17. | |
 |
| | Fig. 3 Binding poses of probes 4 (a), 7 (b), 16 (c), and 17 (d) with FPR1 (PDB ID: 7EUO). | |
Live cell imaging
Based on the high values of activity demonstrated by probe 16 in both FPR1-transfected HL-60 cells and human neutrophils, we have utilized this agent in microscopy experiments to preliminarily assess its efficiency for imaging FPRs within cells, thereby enabling future optical visualisation studies. Probe 16 has a value of absorption max (and, therefore, excitation) of ca. 340 nm (see Experimental section and Fig. S2.1, in ESI†), which is generally lower than the shortest excitation wavelength available on conventional confocal microscopes. For this reason, we opted for the use of a multiphoton microscope to detect the probe interacting with FPR1 in live cells by fixing the excitation wavelength at 760 nm (i.e., the two-photon absorption wavelength of probe 16). Noteworthy, the microscopy experiments highlighted that the fluorescent signal generated by probe 16 was maintained after interaction with the receptors and, to a certain extent, was not only limited to regions of the membrane. For example, we could also observe fluorescence from 16 unevenly distributed in areas of the cytoplasm (for details, see the enlarged view of a single cell in Fig. 4). This result can be explained by taking into consideration the activity profile of probe 16, which is a potent partial agonist for FPR1 (Table 1). Therefore, due to its pronounced agonist effects, 16 is expected to trigger the internalization of FPR1 after activation of the receptor, via a phosphorylation-mediated mechanism,48 in similar fashion to established FPR1 peptide agonists (e.g. fMLF). Taken together, these data suggest that probe 16 represents a valuable optical agent for advanced imaging approaches targeting FPR1, including the possibility of further deciphering how internalization mechanisms may play a role in regulating functions of the receptors upon activation/deactivation signals.
 |
| | Fig. 4 Live-cell multiphoton microscopy imaging of (a) FPR1-transfected HL-60 cells and (b) wild type HL-60 cells. The cells were incubated with probe 16 (1 μM) in HBSS buffer for 10 min on ice. Excess/unreacted probe 16 was removed from the media before imaging. Scale bar = 10 μm. | |
Conclusions
We report the development of a focused series of new small-molecule pyridazinone-based fluorescent probes targeting FPRs. The probes demonstrated affinity for FPR1 and/or FPR2 in functional tests carried out with both FPR-transfected cells and human neutrophils. This validates our molecular design strategy to develop FPR-specific probes focusing on the introduction of fluorophores (having compatible size with small-molecules ligands) in selected positions of an established targeting module (i.e. pyridazinone-based FPR address system). Clearly, the chemical modifications performed on the address system do not hamper the interaction of the molecules with their targets, while, in one case, even determine a substantial increase of the apparent affinity due to the presence of the fluorophore sitting in the binding pocket (i.e. probe 16) and forming multiple interactions with amino acid residues in close proximity. Overall, this work leads to the identification of the first two optically active small-molecules acting as FPR antagonists (i.e., probes 7 and 17) and a highly potent partial agonist for FPR1 and FPR2 (probe 16, EC50 = 1.8 nM and IC50 = 160 pM on human neutrophils), The latter was applied to effectively visualize FPR1 in live cells via multiphoton microscopy. The probes reported here are valid hit candidates to perform further structural modifications, focusing on introduction of fluorophores with enhanced optical properties and efficiency (e.g., for super-resolution and single-molecule localisation microscopy),22 thereby enabling detailed imaging studies to establish the exact roles and functions of FPRs in the activation of immune cells, ultimately leading to the resolution of inflammation.
Experimental section
Chemistry
The synthetic procedures adopted, along with the characterisation analyses and data to confirm purity and formulation of all new intermediates and final optical probes, are reported in Section 1 of ESI.†
Optical characterisation of the probes
The absorption and emission spectra of probe 4, 7 and 16 (10 μM in PBS buffer) were measured in quartz cuvettes with a PerkinElmer UV/VIS Lambda 365 and a Jasco spectrofluorometer FP-8200, respectively. The absorption and emission spectra of probe 17 were measured in methanol at a concentration of 1 μM, using the same instruments. Absorption and emission excited at 337 nm for probe 16 or 500 nm for probe 17 were measured in methanol at six different concentrations to establish the optical properties of the probes, which possess non-conventional fluorophores. The molar extinction coefficient (ε) was determined by the slope of the linear regression equation of concentrations versus absorption according to the equation A = εcl, where A is the maximal absorption, c is the concentration, and l is the length of light path. Meanwhile, the slope of the linear regression equation of peak area of absorption versus peak area of emission was determined. The quantum yield (QY) was calculated by the equation: QYtest = QYreference·Slopetest/Slopereference, using quinine sulphate (QY = 0.52 in 0.05 M of H2SO4) or fluorescein (QY = 0.95 in 0.1 M of NaOH)49 as reference standard.
Molecular modelling
Docking.
The cryo-EM structure of FPR1 in complex with Gi and peptide agonist fMLF (PDB ID: 7EUO)50 was selected as model receptor for docking studies. The receptor protein was prepared in Maestro. Briefly, the N-terminus and C-terminus were capped by acetyl and N-methyl-amino groups, respectively. The bond orders and disulfide bonds were added. Additionally, the protonation states and hydrogen bonds were also optimized. The binding pocket was defined by a cuboid (45 Å × 51 Å × 63 Å) centered around fMLF (i.e., the original ligand in the cryo-EM experimental structure). The initial three-dimensional structures of ligands were prepared by Chem 3D 19.0. The structures were optimized in Gaussian 16 at HF/6-31* basis set. The molecular docking was performed on Autodock GPU.51 The binding poses were visualised with Pymol.
Molecular dynamics (MD) simulations.
The best pose of each ligand from docking was chosen as the initial structure for MD simulations. The complex of receptor and ligand was embedded in a lipid bilayer membrane consisting of DOPC using PACKMOL-Memgen.52 Water molecules, counter ions and salt (0.15 M NaCl) were added to both sides of the bilayer membrane. The thickness of water slat was 17.5 Å over and below the membrane. Default parameters for lipid molecules were applied. The whole solvated system was set as the periodic box for the MD simulation. The force field parameters were assigned by the LEaP module in Amber22.53 The proteins were assigned with ff14SB force field, while the lipids were assigned with the lipid21 force field. TIP3P water model was used. Ligands were assigned with the GAFF2 force field with RESP charges54 calculated in Gaussian 16 at HF/6-31* basis set. MD simulation was performed by Amber22. The energy minimization of the system consisted of 5000 cycles of the steepest descent method and 5000 cycles of the conjugate gradient method. In 1 ns (dt = 0.001 ps), the system was heated from 100 K to 300 K in constant volume (NVT). The system was then released at 300 K and 1 atm for 1 ns (NPT). For all previous steps, all the heavy atoms of the protein (experimental structure) were restrained (restraint_wt = 100). The restraint force was given by k(Δx)2, where k is restraint_wt (kcal mol−1 Å−2), Δx is the difference between one of the Cartesian coordinates of a restrained atom and its reference position. The restraint was decreased gradually (10, 1, 0.1) and, lastly, it was completely removed. The system was equilibrated for 1 ns without the restraint. After the equilibration, productive MD simulation was performed at 300 K and 1 atm for 100 ns. Step size was set to 0.001 ps during the equilibrate stage and switched to 0.002 ps during productive stage. Langevin thermostat (gamma_ln = 1 ps−1) was in use to regulate temperature. SHAKE bond length constrains were used for all bonds involving hydrogen throughout all the MD simulation.
MM/GBSA.
The equilibrated structure from the main productive MD simulation was used as the initial structure of an extra 5 ns MD simulation. Such short MD simulation was repeated three times with randomly assigned velocity. Three trajectories were obtained, and snapshots from these trajectories were used as the conformation assemblage for MM/GBSA calculation. Specifically, 50 even-spaced frames were extracted from each trajectory. The ions, water molecules, and lipid molecules were stripped before the MM/GBSA calculations using the sander program included in Amber22. In the MM/GBSA method, the change of free energy of binding was determined by the equation: ΔGeff = ΔEMM − TΔS + ΔGsolv, where ΔEMM was the molecule energy change, −TΔS was the entropy term and ΔGsolv was the difference of solvation energy between the ligand–receptor complex and sum of unbound receptor and free ligand, ΔΔGsolv = ΔGsolv,complex − ΔGsolv,receptor − ΔGsolv,ligand. Molecule Mechanics method was used to calculate the molecular energy. The molecular energy consisted of electric interaction and Van der Waals interaction, namely EMM = Evdw + Eelec. The change of molecular energy was given by ΔEMM = EMM,complex − EMM,receptor − EMM,ligand. Entropy changes (−TΔS) were estimated by normal mode calculation. GBOBC, a modified generalized Born method was used to calculate the polar solvation free energy (ΔGsolv,polar).55 The salt concentration was set to 0.15 M. Solvent accessible surface area (SASA) of molecules was used to assess the nonpolar solvation free energy (ΔGsolv,nonpolar) using the LCPO algorithm.56 ΔGsolv was given by the sum of polar solvation free energy and nonpolar solvation free energy, ΔGsolv = ΔGsolv,polar + ΔGsolv,nonpolar. Decomposition per residue option was enabled to obtain free energy contribution from each residue. The final results for all energy components were the average of three MM/GBSA calculations based on three independent MD simulations.
Biological assays
Cell culture.
Human promyelocytic leukemia HL-60 cells stably transfected with FPR1 (FPR1-HL60 cells) or FPR2 (FPR2-HL60 cells) were cultured in RPMI-1640 medium supplemented with 10% heat-inactivated fetal calf serum, 10 mM HEPES, 100 μg mL−1 streptomycin, 100 U mL−1 penicillin, and G418 (1 mg mL−1). Although stable cell lines are cultured under G418 selection pressure, G418 may affect some assays, so it was removed in the last round of culture before assays. Wild-type HL-60 cells were cultured under the same conditions, but without G418. FPR surface expression was verified regularly by flow cytometry (FC) using polyclonal FPR antibodies (e.g. for FPR1, from Abcam, Boston, MA, USA, cat# ab113531, or BioLegend, San Diego, CA, USA, cat# 391603, an indicative FC plot of which is provided in Fig. S3.1 in ESI†).
Isolation of human neutrophils.
For isolation of human neutrophils, blood was collected from healthy donors in accordance with a protocol approved by the Institutional Review Board at Montana State University (protocol #MQ041017). Neutrophils were purified from the blood using dextran sedimentation, followed by Histopaque 1077 gradient separation and hypotonic lysis of red blood cells, as described previously.57 Isolated neutrophils were washed twice and resuspended in HBSS−. Neutrophil preparations were routinely >95% pure, as determined by light microscopy, and >98% viable, as determined by trypan blue exclusion. Neutrophils were obtained from multiple different donors; however, the cells from different donors were never pooled during experiments.
Ca2+ mobilization assay (for 4, 7 and 16).
Changes in intracellular Ca2+ concentrations ([Ca2+]i) were measured with a FlexStation 3 scanning fluorometer (Molecular Devices, Sunnyvale, CA, USA). Briefly, FPR1-HL60, FPR2-HL60, or human neutrophils were suspended in HBSS−, loaded with Fluo-4AM at a final concentration of 1.25 μg mL−1, and incubated for 30 min in the dark at 37 °C. After dye loading, the cells were washed with HBSS−, resuspended in HBSS+, separated into aliquots, and loaded into the wells of flat-bottom, half-area-well black microtiter plates (2 × 105 cells per well). To assess direct agonist effects of test compounds on Ca2+ influx, the compounds were added to the wells (final concentration of DMSO was 1%), and changes in fluorescence were monitored (λex = 485 nm, λem = 538 nm) every 5 s for 240 s at room temperature after addition of compound. To evaluate the antagonist effects of the compounds on FPR-dependent Ca2+ influx, the compounds were added to the wells (final concentration of DMSO was 1%) with FPR1-HL60, FPR2-HL60, or human neutrophils. The samples were preincubated for 10 min, followed by the addition of 5 nM fMLF (FPR1-HL60 or neutrophils) or 5 nM WKYMVM (FPR2-HL60). Changes in fluorescence were monitored after addition of fMLF or WKYMVM, as described above. For all assays, the maximum change in fluorescence, expressed in arbitrary units over baseline, was used to determine agonist and antagonist activities. Responses were normalized to the response induced by 5 nM fMLF or 5 nM WKYMVM, which were assigned as 100%. Curve fitting (of at least five or six points) and calculation of median effective concentration values (EC50 or IC50) were performed by nonlinear regression analysis of the dose–response curves generated using Prism 9 (GraphPad Software, Inc., San Diego, CA, USA).
Ca2+ mobilization assay (for 17).
Transfected HL-60 cells were suspended in Ca2+-free HBSS, and the concentration was adjusted to 2 × 106 cells per mL. Fura-2/AM was added to a final concentration of 3 μM, and the cells were incubated for 1 h at 37 °C under darkness. After the incubation, the solution was kept at room temperature for 5–8 min to cool down. Then the cell suspension was centrifuged, and the supernatant was discarded. The cells were washed with Ca2+-free HBSS once, followed by centrifugation and resuspension at a density of 1 × 106 cells per mL. Two mL of cell suspension was added into a cuvette and kept stirred in a purpose built spectrofluorimeter (Cairns Research Ltd., Faversham, Kent, U.K.). The cells were added with 1 mM Ca2+ and warmed up to 37 °C for 3 min. The ratio of emission intensity excited at 340 nm and 380 nm was monitored in real-time. To evaluate the agonist effect of 17, 5 nM fMLF (for FPR1), 5 nM WKYMWm (for FPR2), or 17 were added directly into the cuvette, and the emission response (i.e., ratio of emission values at 340/380 excitation) was recorded consistently. To evaluate the antagonist effect, 17 was preincubated with cells for 30 min before the addition of 5 nM fMLF (for FPR1) or 5 nM WKYMWm (for FPR2). The responses of 5 nM fMLF (for FPR1) or 5 nM WKYMWm were set at 100%, as maximum activity, and the response of 17 at different concentrations was normalized accordingly. EC50 or IC50 values were calculated by non-linear regression analysis of the dose–response curves.
Multiphoton microscopy
Wild type HL-60 cells and FPR1-transfected HL-60 (1.5 × 107) cells were suspended in 1 mL of HBSS buffer. The cell suspensions were treated with 1 μM of probe 16 and incubated for 10 min on ice. Agarose (1%) gel in HBSS was prepared to form a film of 1 mm thickness and, subsequently, pads (1 cm × 1 cm) were trimmed from the agarose film. The cell suspension was added into poly-lysine treated glass bottom dishes (35 mm/20 mm, VWR International). An agarose pad was placed into the dish before the cell media inside the dish was gently removed. Then 200 μL of HBSS buffer was added. Microscopy imaging was conducted on a Leica Stellaris FALCON FLIM Microscope (accessed through Nikon Imaging Centre, KCL) in multiphoton mode. A 63× oil lens was used. Excitation wavelength was set at 760 nm, and emission from 450 nm to 650 nm was detected. The images were analysed by Fiji ImageJ.
Ethical statement
All use of human participants was performed in accordance with the United States Health and Human Services Common Rule for the Protection of Human Subjects and was approved by the Montana State University Institutional Review Board (protocol 2022-168, approved on 23 March 2022). Written informed consent was obtained from all human participants of this study.
Data availability
Supplementary data to this article can be found online. Additional data will be made available upon request as reasonably possible.
Author contributions
QX: experimental work, data curation, methodology, software, formal analysis, writing – original draft, writing – review and editing. KSA: Ca2+ assay supervision, methodology, writing – review and editing. LNK: biology work and cell culture, methodology, writing – review and editing. IAS: experimental work, Ca2+ assay, data curation, methodology, formal analysis, writing – original draft, writing – review and editing. MTQ: formal analysis, funding acquisition, resources, supervision, writing – original draft, writing – review and editing. AC: conceptualization, methodology, data curation, formal analysis, investigation, funding acquisition, project administration, resources, supervision, writing – original draft, writing – review and editing.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
AC acknowledges funding from The Royal Society – Research Grants 2024 (RGS\R2\242316) which provided support to this research. QX is funded by a China Scholarship Council (CSC) International PhD Scholarship (at KCL, AC Lab). QX and AC acknowledge Prof Miraz Rahman (KCL) and Prof Robert Hider (KCL) for useful discussion and inputs on experimental steps. This research was supported in part by National Institutes of Health IDeA Program Grant GM103474 and the Montana State University Agricultural Experiment Station.
References
- E. Weiß and D. Kretschmer, Formyl-peptide receptors in infection, inflammation, and cancer, Trends Immunol., 2018, 39, 815–829, DOI:10.1016/j.it.2018.08.005.
- J. Bylund, M. Gabl, M. Winther, K. Önnheim, C. Dahlgren and H. Forsman, Turning chemoattractant receptors on and off with conventional ligands and allosteric modulators: recent advances in formyl peptide receptor signaling and regulation, Inflammation Cell Signaling, 2014, 1, e73, DOI:10.14800/ics.73.
- J. Mårtensson, M. Sundqvist, A. Manandhar, L. Ieremias, L. Zhang, T. Ulven, X. Xie, L. Björkman and H. Forsman, The two formyl peptide receptors differently regulate GPR84-mediated neutrophil NADPH oxidase activity, J. Innate Immun., 2021, 13, 242–256, DOI:10.1159/000514887.
- M. F. Salamah, D. Ravishankar, R. Vaiyapuri, L. A. Moraes, K. Patel, M. Perretti, J. M. Gibbins and S. Vaiyapuri, The formyl peptide fMLF primes platelet activation and augments thrombus formation, J. Thromb. Haemostasis, 2019, 17, 1120–1133, DOI:10.1111/jth.14466.
- Y. S. Jeong and Y.-S. Bae, Formyl peptide receptors in the mucosal immune system, Exp. Mol. Med., 2020, 52, 1694–1704, DOI:10.1038/s12276-020-00518-2.
- D. A. Dorward, C. D. Lucas, G. B. Chapman, C. Haslett, K. Dhaliwal and A. G. Rossi, The role of formylated peptides and formyl peptide receptor 1 in governing neutrophil function during acute inflammation, Am. J. Pathol., 2015, 185, 1172–1184, DOI:10.1016/j.ajpath.2015.01.020.
- S. L. Skovbakke, A. Holdfeldt, H. Forsman, J. Bylund and H. Franzyk, The role of formyl peptide receptors for immunomodulatory activities of antimicrobial peptides and peptidomimetics, Curr. Pharm. Des., 2018, 24, 1100–1120, DOI:10.2174/1381612824666180403123233.
- J. Zhu, L. Li, J. Ding, J. Huang, A. Shao and B. Tang, The role of formyl peptide receptors in neurological diseases via regulating inflammation, Front. Cell. Neurosci., 2021, 15, 753832, DOI:10.3389/fncel.2021.753832.
- D. A. Dorward, C. D. Lucas, M. K. Doherty, G. B. Chapman, E. J. Scholefield, A. C. Morris, J. M. Felton, T. Kipari, D. C. Humphries, C. T. Robb, A. J. Simpson, P. D. Whitfield, C. Haslett, K. Dhaliwal and A. G. Rossi, Novel role for endogenous mitochondrial formylated peptide-driven formyl peptide receptor 1 signalling in acute respiratory distress syndrome, Thorax, 2017, 72, 928–936, DOI:10.1136/thoraxjnl-2017-210030.
- A. Giebeler, K. L. Streetz, O. Soehnlein, U. Neumann, J. M. Wang and L.-O. Brandenburg, Deficiency of formyl peptide receptor 1 and 2 is associated with increased inflammation and enhanced liver injury after LPS-stimulation, PLoS One, 2014, 9, e100522, DOI:10.1371/journal.pone.0100522.
- M. K. Shin, Y. H. Jang, H. J. Yoo, D. W. Kang, M. H. Park, M. K. Kim, J. H. Song, S. D. Kim, G. Min, H. K. You, K.-Y. Choi, Y.-S. Bae and D. S. Min,
N-formyl-methionyl-leucyl-phenylalanine (fMLP) promotes osteoblast differentiation via the N-formyl peptide receptor 1-mediated signaling pathway in human mesenchymal stem cells from bone marrow, J. Biol. Chem., 2011, 286, 17133–17143, DOI:10.1074/jbc.M110.197772.
- M. Perretti and C. Godson, Formyl peptide receptor type 2 agonists to kick-start resolution pharmacology, Br. J. Pharmacol., 2020, 177, 4595–4600, DOI:10.1111/bph.15212.
- V. M. Caso, V. Manzo, T. Pecchillo Cimmino, V. Conti, P. Caso, G. Esposito, V. Russo, A. Filippelli, R. Ammendola and F. Cattaneo, Regulation of inflammation and oxidative stress by formyl peptide receptors in cardiovascular disease progression, Life, 2021, 11, 243, DOI:10.3390/life11030243.
-
R. H. Ritchie, C. X. Qin, A. Christopoulos, P. M. Sexton and J. B. Baell, WO2016123672A1/US10328075B2, 2019.
- F. Cattaneo, G. Guerra, M. Parisi, A. Lucariello, A. De Luca, N. De Rosa, G. Mazzarella, A. Bianco and R. Ammendola, Expression of formyl-peptide receptors in human lung carcinoma, Anticancer Res., 2015, 35, 2769–2774 CAS , https://oajournals.fupress.net/index.php/ijae/article/view/3991.
- S.-Q. Li, N. Su, P. Gong, H.-B. Zhang, J. Liu, D. Wang, Y.-P. Sun, Y. Zhang, F. Qian, B. Zhao, Y. Yu and R. D. Ye, The expression of formyl peptide receptor 1 is correlated with tumor invasion of human colorectal cancer, Sci. Rep., 2017, 7, 5918, DOI:10.1038/s41598-017-06368-9.
- C. Tian, K. Chen, W. Gong, T. Yoshimura, J. Huang and J. M. Wang, The G-protein coupled formyl peptide receptors and their role in the progression of digestive tract cancer, Technol. Cancer Res. Treat., 2020, 19, 1533033820973280, DOI:10.1177/1533033820973280.
- H. Q. He and R. D. Ye, The formyl peptide receptors: diversity of ligands and mechanism for recognition, Molecules, 2017, 22, 455, DOI:10.3390/molecules22030455.
- M. Maciuszek, A. Cacace, E. Brennan, C. Godson and T. M. Chapman, Recent advances in the design and development of formyl peptide receptor 2 (FPR2/ALX) agonists as pro-resolving agents with diverse therapeutic potential, Eur. J. Med. Chem., 2021, 213, 113167, DOI:10.1016/j.ejmech.2021.113167.
- I. A. Schepetkin, A. I. Khlebnikov, M. P. Giovannoni, L. N. Kirpotina, A. Cilibrizzi and M. T. Quinn, Development of small molecule non-peptide formyl peptide receptor (FPR) ligands and molecular modeling of their recognition, Curr. Med. Chem., 2014, 21, 1478–1504, DOI:10.2174/0929867321666131218095521.
- Y. Zhuang, L. Wang, J. Guo, D. Sun, Y. Wang, W. Liu, H. E. Xu and C. Zhang, Molecular recognition of formylpeptides and diverse agonists by the formylpeptide receptors FPR1 and FPR2, Nat. Commun., 2022, 13, 1054, DOI:10.1038/s41467-022-28586-0.
- C. D. T. Nielsen, D. Dhasmana, G. Floresta, T. Wohland and A. Cilibrizzi, Illuminating the path to target GPCR structures and functions, Biochemistry, 2020, 59, 3783–3795, DOI:10.1021/acs.biochem.0c00606.
- T. Boltersdorf, J. Ansari, E. Y. Senchenkova, J. Groeper, D. Pajonczyk, S. A. Vital, G. Kaur, J. S. Alexander, T. Vogl, U. Rescher, N. J. Long and F. N. E. Gavins, Targeting of formyl peptide receptor 2 for in vivo imaging of acute vascular inflammation, Theranostics, 2020, 10, 6599–6614, DOI:10.7150/thno.44226.
- S. V. Morse, T. Boltersdorf, T. G. Chan, F. N. E. Gavins, J. J. Choi and N. J. Long, In vivo delivery of a fluorescent FPR2/ALX-targeted probe using focused ultrasound and microbubbles to image activated microglia, RSC Chem. Biol., 2020, 1, 385–389, 10.1039/D0CB00140F.
- L. Xiao, Y. Zhang, S. S. Berr, M. D. Chordia, P. Pramoonjago, L. Pu and D. Pan, A novel near-infrared fluorescence imaging probe for in vivo neutrophil tracking, Mol. Imaging, 2012, 11, 372–382, DOI:10.2310/7290.2011.00054.
- J. Zhou, Y.-T. Tsai, H. Weng, E. N. Tang, A. Nair, D. P. Davé and L. Tang, Real-time detection of implant-associated neutrophil responses using a formyl peptide receptor-targeting
NIR nanoprobe, Int. J. Nanomed., 2012, 7, 2057–2068, DOI:10.2147/ijn.S29961.
- B. S. Edwards, S. M. Young, T. I. Oprea, C. G. Bologa, E. R. Prossnitz and L. A. Sklar, Biomolecular screening of formylpeptide receptor ligands with a sensitive, quantitative, high-throughput flow cytometry platform, Nat. Protoc., 2006, 1, 59–66, DOI:10.1038/nprot.2006.9.
- S. M. Young, C. Bologa, E. R. Prossnitz, T. I. Oprea, L. A. Sklar and B. S. Edwards, High-throughput screening with HyperCyt® flow cytometry to detect small molecule formylpeptide receptor ligands, J. Biomol. Screening, 2005, 10, 374–382, DOI:10.1177/1087057105274532.
- C. Pinilla, B. S. Edwards, J. R. Appel, T. Yates-Gibbins, M. A. Giulianotti, J. L. Medina-Franco, S. M. Young, R. G. Santos, L. A. Sklar and R. A. Houghten, Selective agonists and antagonists of formylpeptide receptors: duplex flow cytometry and mixture-based positional scanning libraries, Mol. Pharmacol., 2013, 84, 314–324, DOI:10.1124/mol.113.086595.
- W. Y. Kwon, G. J. Suh, Y. S. Jung, S. M. Park, S. Oh, S. H. Kim, A. R. Lee, J. Y. Kim, H. Kim, K. A. Kim, Y. Kim, B. C. Kim, T. Kim, K. S. Kim, K. Itagaki and C. J. Hauser, Circulating mitochondrial N-formyl peptides contribute to secondary nosocomial infection in patients with septic shock, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2018538118, DOI:10.1073/pnas.2018538118.
- C. N. Jones, F. Ellett, A. L. Robertson, K. M. Forrest, K. Judice, J. M. Balkovec, M. Springer, J. F. Markmann, J. M. Vyas, H. S. Warren and D. Irimia, Bifunctional small molecules enhance neutrophil activities against aspergillus fumigatus in vivo and in vitro, Front. Immunol., 2019, 10, 644, DOI:10.3389/fimmu.2019.00644.
- L. Boneschansker, J. Yan, E. Wong, D. M. Briscoe and D. Irimia, Microfluidic platform for the quantitative analysis of leukocyte migration signatures, Nat. Commun., 2014, 5, 4787, DOI:10.1038/ncomms5787.
- Y. Zhang, B. Kundu, M. Zhong, T. Huang, J. Li, M. D. Chordia, M.-H. Chen, D. Pan, J. He and W. Shi, PET imaging detection of macrophages with a formyl peptide receptor antagonist, Nucl. Med. Biol., 2015, 42, 381–386, DOI:10.1016/j.nucmedbio.2014.12.001.
- E. J. Charles, M. D. Chordia, Y. Zhao, Y. Zhang, J. H. Mehaffey, D. K. Glover, J. Dimastromatteo, W. Z. Chancellor, A. K. Sharma, I. L. Kron, D. Pan and V. E. Laubach, SPECT imaging of lung ischemia-reperfusion injury using [99mTc]cFLFLF for molecular targeting of formyl peptide receptor 1, Am. J. Physiol., 2020, 318, L304–L313, DOI:10.1152/ajplung.00220.2018.
- J. Li, Y. Zhang, M. D. Chordia, H. Wu, L. Shao and D. Pan, Multimodal formyl peptide receptor 1 targeted inflammation imaging probe: cFLFLF-MHI-DOTA, Bioorg. Med. Chem. Lett., 2016, 26, 1052–1055, DOI:10.1016/j.bmcl.2015.12.029.
- T. Boltersdorf, J. Ansari, E. Y. Senchenkova, J. Groeper, D. Pajonczyk, S. A. Vital, G. Kaur, J. S. Alexander, T. Vogl, U. Rescher, N. J. Long and F. N. E. Gavins, Targeting of formyl peptide receptor 2 for in vivo imaging of acute vascular inflammation, Theranostics, 2020, 10, 6599–6614, DOI:10.7150/thno.44226.
- A. Cilibrizzi, M. T. Quinn, L. N. Kirpotina, I. A. Schepetkin, J. Holderness, R. D. Ye, M.-J. Rabiet, C. Biancalani, N. Cesari, A. Graziano, C. Vergelli, S. Pieretti, V. Dal Piaz and M. P. Giovannoni, 6-Methyl-2, 4-disubstituted pyridazin-3 (2H)-ones: a novel class of small-molecule agonists for formyl peptide receptors, J. Med. Chem., 2009, 52, 5044–5057, DOI:10.1021/jm900592h.
- L. Crocetti, C. Vergelli, A. Cilibrizzi, A. Graziano, A. I. Khlebnikov, L. N. Kirpotina, I. A. Schepetkin, M. T. Quinn and M. P. Giovannoni, Synthesis and pharmacological evaluation of new pyridazin-based thioderivatives as formyl peptide receptor (FPR) agonists, Drug Dev. Res., 2013, 74, 259–271, DOI:10.1002/ddr.21076.
- A. Cilibrizzi, I. A. Schepetkin, G. Bartolucci, L. Crocetti, V. Dal Piaz, M. P. Giovannoni, A. Graziano, L. N. Kirpotina, M. T. Quinn and C. Vergelli, Synthesis, enantioresolution, and activity profile of chiral 6-methyl-2, 4-disubstituted pyridazin-3 (2H)-ones as potent N-formyl peptide receptor agonists, Bioorg. Med. Chem., 2012, 20, 3781–3792, DOI:10.1016/j.bmc.2012.04.043.
- A. Cilibrizzi, Correspondence: compound 17b and formyl peptide receptor biased agonism in relation to cardioprotective effects in ischaemia-reperfusion injury, Nat. Commun., 2018, 9, 1–3, DOI:10.1038/s41467-017-02654-2.
- C. Vergelli, I. A. Schepetkin, G. Ciciani, A. Cilibrizzi, L. Crocetti, M. P. Giovannoni, G. Guerrini, A. Iacovone, L. N. Kirpotina, A. I. Khlebnikov, R. D. Ye and M. T. Quinn, 2-Arylacetamido-4-phenylamino-5-substituted pyridazinones as formyl peptide receptors agonists, Bioorg. Med. Chem., 2016, 24, 2530–2543, DOI:10.1016/j.bmc.2016.04.019.
- C. Vergelli, I. A. Schepetkin, G. Ciciani, A. Cilibrizzi, L. Crocetti, M. P. Giovannoni, G. Guerrini, A. Iacovone, L. N. Kirpotina, R. D. Ye and M. T. Quinn, Synthesis of five-and six-membered N-phenylacetamido substituted heterocycles as formyl peptide receptor agonists, Drug Dev. Res., 2017, 78, 49–62, DOI:10.1002/ddr.21370.
- M. P. Giovannoni, I. A. Schepetkin, A. Cilibrizzi, L. Crocetti, A. I. Khlebnikov, C. Dahlgren, A. Graziano, V. Dal Piaz, L. N. Kirpotina, S. Zerbinati, C. Vergelli and M. T. Quinn, Further studies on 2-arylacetamide pyridazin-3 (2H)-ones: design, synthesis and evaluation of 4, 6-disubstituted analogs as formyl peptide receptors (FPRs) agonists, Eur. J. Med. Chem., 2013, 64, 512–528, DOI:10.1016/j.ejmech.2013.03.066.
- B. Camerino and B. Patelli, Some derivatives of 4-keto pimelic acid, and their antituberculotic activities, Farmaco Sci., 1956, 11, 446–450 CAS , https://pubmed.ncbi.nlm.nih.gov/13330869/.
- M. Gupta, S. Mula, M. Tyagi, T. K. Ghanty, S. Murudkar, A. K. Ray and S. Chattopadhyay, Rational design of boradiazaindacene (BODIPY)-based functional molecules, Chem. – Eur. J., 2013, 19, 17766–17772, DOI:10.1002/chem.201302359.
- N. Kassouf, A. Ambily, S. Watson, S. Hassock, H. S. Authi, S. Srivastava, S. P. Watson and K. S. Authi, Phosphatidylinositol-3,4,5-trisphosphate stimulates Ca2+ elevation and Akt phosphorylation to constitute a major mechanism of thromboxane A2 formation in human platelets, Cell. Signalling, 2015, 27, 1488–1498, DOI:10.1016/j.cellsig.2015.03.008.
- A. Cilibrizzi, L. Crocetti, M. P. Giovannoni, A. Graziano, C. Vergelli, G. Bartolucci, G. Soldani, M. T. Quinn, I. A. Schepetkin and C. Faggi, Synthesis, HPLC enantioresolution, and X-ray analysis of a new series of C5-methyl pyridazines as N-formyl peptide receptor (FPR) agonists, Chirality, 2013, 25, 400–408, DOI:10.1002/chir.22162.
- M. H. Hsu, S. C. Chiang, R. D. Ye and E. R. Prossnitz, Phosphorylation of the N-formyl peptide receptor is required for receptor internalization but not chemotaxis, J. Biol. Chem., 1997, 272, 29426–29429, DOI:10.1074/jbc.272.47.29426.
- A. M. Brouwer, Standards for photoluminescence quantum yield measurements in solution (IUPAC Technical Report), Pure Appl. Chem., 2011, 83, 2213–2228, DOI:10.1351/PAC-REP-10-09-31.
- G. Chen, X. Wang, Q. Liao, Y. Ge, H. Jiao, Q. Chen, Y. Liu, W. Lyu, L. Zhu, G. C. P. van Zundert, M. J. Robertson, G. Skiniotis, Y. Du, H. Hu and R. D. Ye, Structural basis for recognition of N-formyl peptides as pathogen-associated molecular patterns, Nat. Commun., 2022, 13, 5232, DOI:10.1038/s41467-022-32822-y.
- D. Santos-Martins, L. Solis-Vasquez, A. F. Tillack, M. F. Sanner, A. Koch and S. Forli, Accelerating AutoDock4 with GPUs and gradient-based local search, J. Chem. Theory Comput., 2021, 17, 1060–1073, DOI:10.1021/acs.jctc.0c01006.
- S. Schott-Verdugo and H. Gohlke, PACKMOL-Memgen: a simple-to-use, generalized workflow for membrane-protein–lipid-bilayer system building, J. Chem. Inf. Model., 2019, 59, 2522–2528, DOI:10.1021/acs.jcim.9b00269.
-
D. A. Case, H. M. Aktulga, K. Belfon, I. Y. Ben-Shalom, J. T. Berryman, S. R. Brozell, D. S. Cerutti, T. E. Cheatham III, G. A. Cisneros, V. W. D. Cruzeiro, T. A. Darden, R. E. Duke, G. Giambasu, M. K. Gilson, H. Gohlke, A. W. Goetz, R. Harris, S. Izadi, S. A. Izmailov, K. Kasavajhala, M. C. Kaymak, E. King, A. Kovalenko, T. Kurtzman, T. S. Lee, S. LeGrand, P. Li, C. Lin, J. Liu, T. Luchko, R. Luo, M. Machado, V. Man, M. Manathunga, K. M. Merz, Y. Miao, O. Mikhailovskii, G. Monard, H. Nguyen, K. A. O'Hearn, A. Onufriev, F. Pan, S. Pantano, R. Qi, A. Rahnamoun, D. R. Roe, A. Roitberg, C. Sagui, S. Schott-Verdugo, A. Shajan, J. Shen, C. L. Simmerling, N. R. Skrynnikov, J. Smith, J. Swails, R. C. Walker, J. Wang, J. Wang, H. Wei, R. M. Wolf, X. Wu, Y. Xiong, Y. Xue, D. M. York, S. Zhao and P. A. Kollman, Amber 2022 [solftware], University of California, San Francisco, 2022 Search PubMed.
- C. I. Bayly, P. Cieplak, W. Cornell and P. A. Kollman, A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model, J. Phys. Chem., 1993, 97, 10269–10280, DOI:10.1021/j100142a004.
- A. Onufriev, D. Bashford and D. A. Case, Exploring protein native states and large-scale conformational changes with a modified generalized born model, Proteins, 2004, 55, 383–394, DOI:10.1002/prot.20033.
- J. Weiser, P. S. Shenkin and W. C. Still, Approximate atomic surfaces from linear combinations of pairwise overlaps (LCPO), J. Comput. Chem., 1999, 20, 217–230, DOI:10.1002/(SICI)1096-987X(19990130)20:2<217::AID-JCC4>3.0.CO;2-A.
- I. A. Schepetkin, L. N. Kirpotina, A. I. Khlebnikov and M. T. Quinn, High-throughput screening for small-molecule activators of neutrophils: identification of novel N-formyl peptide receptor agonists, Mol. Pharmacol., 2007, 71, 1061–1074, DOI:10.1124/mol.106.033100.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Kalwant S.
Authi
b,
Liliya N.
Kirpotina
c,
Igor A.
Schepetkin
c,
Mark T.
Quinn
c and
Agostino
Cilibrizzi
a,
Kalwant S.
Authi
b,
Liliya N.
Kirpotina
c,
Igor A.
Schepetkin
c,
Mark T.
Quinn
c and
Agostino
Cilibrizzi