
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The synthesis and investigation of novel 3-benzoylbenzofurans and pyrazole derivatives for anti-HIV activity†
Sinothile S.
Khuzwayo
 a,
Mamoalosi A.
Selepe
b,
Debra
Meyer
c and
Ntombenhle H.
Gama
*a
a,
Mamoalosi A.
Selepe
b,
Debra
Meyer
c and
Ntombenhle H.
Gama
*a
aDepartment of Biochemistry, Genetics and Microbiology, University of Pretoria, 2 Lynnwood Road, Pretoria, 0002, South Africa. E-mail: snothilesementha81@gmail.com
bChemistry Department, University of Pretoria, 2 Lynnwood Road, Pretoria, 0002, South Africa
cSchool of Natural and Applied Sciences, Sol Plaatje University, Kimberley, 8300, South Africa
First published on 5th February 2025
Abstract
The emergence of drug-resistant viruses continues to be an obstacle to effectively controlling the HIV/AIDS pandemic. The development of novel drugs with high potency and the ability to fully eradicate HIV-1 infections is therefore of critical importance. Novel pyrazole derivatives were synthesized from 3-benzoylbenzofurans and characterized by mass spectrometry (MS) and nuclear magnetic resonance (NMR) spectroscopy. The 3-benzoyl benzofurans were determined to be highly cytotoxic in TZM-bl cells, while their pyrazole derivatives were mild to non-cytotoxic. Evaluation of anti-HIV activities in pseudoviruses revealed two 3-benzoyl benzofurans (3g and 4b) and pyrazoles (5f and 5h) as the most potent inhibitors. The IC50 values of 4b and 5f were 0.49 ± 0.11 μM and 0.39 ± 0.13 μM in Q23 and 0.12 ± 0.05 μM and 1.00 ± 0.15 μM in the CAP210 pseudovirus, respectively. Further evaluations for mechanism of action involved the time of addition assay and direct enzyme inhibition, which showed that 3g and 4b were non-nucleotide reverse transcriptase inhibitors while 5f and 5h inhibited HIV entry. Additionally, 3g, 4b, and 5h were found to be mild inhibitors of HIV-1 protease, while 5f was the most active protease inhibitor. The IC50 value of 5f was 31.59 ± 3.83 μM, and it displayed interactions with the active site of HIV-1 PR, suggesting competitive inhibition using molecular docking. The promising anti-HIV activities of 5f in pseudoviruses and HIV-PR motivate its further development for antiretroviral drugs.
1. Introduction
Four decades after its discovery, infection by the human immunodeficiency virus (HIV-1) remains one of the world's leading global epidemics. UNAIDS estimated 39 million people were living with HIV in 2022, with 630![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 AIDS-related deaths reported globally in that same year.1,2 Eastern and southern Africa have been most severely affected by the HIV-1 epidemic, with 53.3% of HIV-infected people and 38.5% of new infections reported in 2022.1 South Africa reported 7.6 million people living with HIV in 2022, which makes up a total of 17.8% of adults.3 The number of newly infected people was 160000, and a total of 45000 people died from AIDS-related illnesses in South Africa alone in 2022.3
000 AIDS-related deaths reported globally in that same year.1,2 Eastern and southern Africa have been most severely affected by the HIV-1 epidemic, with 53.3% of HIV-infected people and 38.5% of new infections reported in 2022.1 South Africa reported 7.6 million people living with HIV in 2022, which makes up a total of 17.8% of adults.3 The number of newly infected people was 160000, and a total of 45000 people died from AIDS-related illnesses in South Africa alone in 2022.3
Rapid HIV replication and the virus' propensity for replication errors, resulting in numerous genetic variations in its offspring, provide the greatest challenges to eradicating HIV. Candidate HIV-1 vaccines have shown considerable promise, including ALVAC-HIV, a DNA vaccine made up of gp120 plasmids from five different HIV-1 subtypes;4 VRC01, an anti-gp120 monoclonal antibody now undergoing phase II clinical trials;5 and PDPHV, an envelope and gp120 vaccine.6 However, none of these vaccines have been approved. Therefore, the use of combination antiretroviral therapy (cART) for controlling HIV-1 infections remains a significant necessity7 for disease management. Approximately 78% of HIV-1-infected people in sub-Saharan Africa were receiving treatment in 2022.8 Unfortunately, cART also has drawbacks, including drug toxicity, which leads to treatment non-adherence, and the emergence of drug resistance.9
Recently, new drug targets for HIV have been discovered, including the inhibition of the HIV-1 capsid.10 The capsid protein has been identified as a promising target for HIV treatment because it not only houses the HIV genome but also aids in virus uncoating in the host cytoplasm, nuclear export, and virion assembly.11 Compounds GS-CA1 and its analogue GS-6207 (Fig. 1) are capsid inhibitors that have been shown to have a better potency than all the existing HIV drugs,12 while lenacapavir (GS-6207) was the first capsid inhibitor to be FDA-approved for HIV treatment in 2022.13 Additionally, lenacapavir is the first pyrazole-based drug to be approved for the treatment of HIV-1 multi-drug-resistant infections.13 Numerous experimental pyrazole compounds are potent inhibitors of HIV infections and HIV-1 enzymes.14–16 Lersivirine, a pyrazole derivative, was demonstrated to be a potent inhibitor of HIV-1 reverse transcriptase (RT). However, it was discontinued in 2013 during its phase III clinical trials due to its lack of improved efficacy over already available drugs.17 A pyrazole-piperidine was shown to inhibit both HIV-RT and viral entry with IC50 of 9 μM, 0.8 μM, and 3.8 μM for HIV-RT, CCR5, and CXCR4 co-receptors, respectively.18 More potent pyrazole compounds have been shown to inhibit HIV-1 RT with IC50 values of 0.93 and 1.28 μM.19 In addition to the potent anti-HIV activity, the minimal cytotoxicity of the pyrazoles makes these compounds attractive candidates for the development of antiretrovirals.20
 | ||
| Fig. 1 Comparison of the structures of potent HIV inhibitors with the generic structures of benzoylbenzofurans and pyrazole derivatives prepared in the current study. The pyrazole scaffold found in lenacapavir, the most potent HIV drug, was mimicked with the derivatives synthesized in this study. The benzoylbenzofurans contain acetyl and methoxyphenyl groups found in atervidine. | ||
Benzofurans are well-studied due to their distinguished pharmacological applications, and some of these compounds are FDA-approved drugs against various human diseases, including cancer, malaria, and various bacterial infections.21 Similarly to the pyrazoles, benzofuran compounds were shown to be potent inhibitors of HIV-1 RT, and their activity was higher compared to atervidine, an HIV drug that closely resembles benzofuran compounds as shown in Fig. 1.22 More benzofuran derivatives have been identified as HIV-1 protease (PR) inhibitors,23,24 These compounds include benzofuran-carboxamide derivatives, which demonstrated dual activities against both RT and PR, which further motivates the potential of the benzofuran scaffold for exploration for antiretroviral development.25
In the current study, the pyrazoles were synthesized from the 3-benzoyl benzofuran precursors, and both compound classes were investigated for cytotoxicity and their inhibitory effects on the HIV life cycle.
2. Results and discussion
2.1 Synthesis of 3-benzoylbenzofurans and pyrazole derivatives
The synthesis of 3-benzoylbenzofurans 3a–h, 4a–e, and their pyrazole derivatives 5a–h is demonstrated in Scheme 1. The synthesis of 3-benzoylbenzofurans involved the condensation of 2-methoxyacetophenones 1a–d and N,N-dimethylformamide dimethyl acetal (DMF-DMA) producing enaminoketones 2a–d, which were reacted without further purification with 1,4-benzoquinone or 1,4-naphthoquinone derivatives to form the 3-benzoyl benzofurans 3a–h.26,27 The 3-benzoylbenzofurans were obtained in moderate to low percentage yields over two steps. Numerous methods for the synthesis of benzofurans have been reported in the literature.28,29 The current study produced the benzofurans using the most common method called the Nenitzescu reaction, which involves the condensation of an enaminoketone and 1,4-benzoquinone.30 The reaction produces two common products, the 5-hydroxyindoles and benzofuran derivatives, depending on the reaction conditions.30,31 The first step of the reaction is the Michael Addition of an enaminoketone to the benzoquinone, which is followed by the production of a zwitter ion, which is then O-protonated and results in a hydroquinone adduct, which under acidic conditions (acetic acid) forms a benzofuran.30,31 However, oxidation could also occur in less or non-acidic conditions, which results in the formation of the 5-hydroxyindole.30,31 The low yields of 3-benzoylbenzofurans obtained in the present study could be attributed to the production of byproducts (observed from the thin layer chromatography); however, the byproducts were not characterized. | ||
| Scheme 1 Synthesis of 3-benzoylbenzofurans and pyrazole derivatives. Reagents and conditions: (a) DMF-DMA, 90–190 °C, Ar, 12–72 h. (b) 1,4-benzoquinone/1,4-naphthoquinone, acetic acid, rt, 12 h. (c) CH3I, K2CO3, DMF, 100 °C, Ar, 5–12 h. (d) Hydrazine hydrate, methanol, rt, 2–5 h. | ||
The pyrazole derivatives were synthesized from the 3-benzoylbenzofurans using a method described by Abdel-Aziz et al.,32 which treated the 3-benzoylbenzofurans with hydrazine hydrate in the presence of methanol, forming the pyrazole derivatives. The 3-benzoylbenzofurans 3f, 3g, and 3h spontaneously reacted with hydrazine hydrate to form the pyrazole derivatives 5f, 5g, and 5h in percentage yields of 31, 69, and 16%, respectively. However, the substituted 3-benzoylbenzofurans presented difficulties when synthesizing the pyrazole derivatives. Multiple byproducts were observed when reacting these 3-benzoylbenzofurans (3a–3e) with hydrazine hydrate, and stringent purification methods were applied; unfortunately, the products were not free of impurities. The pyrazole derivatives 5a–5e were rather formed smoothly from the 5-methoxybenzoylbenzofurans 4a–4e. The compounds 4a–4e were prepared by treatment of 3-benzoylbenzofurans 3a–3e with methyl iodide (CH3I) in the presence of potassium carbonate (K2CO3) and dry DMF (Scheme 1). Methylation reactions have been reported to occur rapidly in the presence of K2CO3 as a base and dry DMF at high temperatures.33 The methylation reactions were conducted at 100 °C and the compounds were obtained in high percentage yields ranging from 71 to 96%. The methylated 3-benzoylbenzofurans (4a–4e) readily reacted with hydrazine hydrate to produce the pyrazole derivatives (5a–5e) which did not require further chromatographic purification steps. The pyrazole derivatives (5a–5e) were obtained at yields of 72–95% (Scheme 1). The obtained percentage yields and the rapidness of the reactions corresponded to other studies that obtained high yields of pyrazole compounds under similar conditions.32,34
The current study synthesized the pyrazole derivatives from the α,β-unsaturated ketone of 3-benzoylbenzofurans and hydrazine hydrate in methanol, which produced the 3,4-substituted pyrazole derivatives at room temperature. Numerous methods have been reported for the synthesis of pyrazoles.35–37 Examples include refluxing chromones and chalcones with hydrazine derivatives in ethanol.35,36 The 1, 3, and 5 substituted pyrazoles36 and the 3,5-substituted pyrazoles35 were obtained at high percentage yields ranging from 51–98% after recrystallization from ethanol. Contrary to other researchers,34–36 the current study followed a method that transformed the benzofuran moiety into the pyrazole scaffold.32 In this study, the yields of the pyrazole compounds depended largely on the benzofuran precursors, as it has been shown that 5-methoxybenzoylbenzofurans resulted in high yields of pyrazole products that did not require further chromatographic purification.
2.2 Cytotoxicity evaluations
The synthesized compounds were evaluated for cytotoxicity in TZM-bl cells using an MTT assay. Investigating the cytotoxicity of compounds in this cell line was crucial since these cells were further used for infections with pseudoviruses in the evaluation of potential anti-HIV activity. The current study demonstrated that the derivatization of cytotoxic 3-benzoylbenzofurans to pyrazoles resulted in a general decrease in cytotoxicity, as shown in Table 1. Only the pyrazole derivative 5f demonstrated increased cytotoxicity from its precursor 3f; however, this difference was not significant with a P value of 0.9918. There was a significant decrease in cytotoxicity of the pyrazole derivatives 5g and 5h compared to their precursors 3g and 3h respectively (P = 0.0005). The methylated 3-benzoylbenzofurans 4a–4e had the highest cytotoxicity, with CC50 values ranging from 7.77 to 59.15 μM. There was a significant decrease in the cytotoxicity upon derivatization of 4b, 4c, 4d, and 4e to pyrazoles 5b, 5c, 5d, and 5e. The decrease in the cytotoxicity for pyrazole derivatives 5b, 5c, 5d, and 5e ranged from 3 to 10-fold. The cytotoxicity of 5a was only 1-fold lower than that of the precursor 4a; this was not significantly different. Overall, these results show that converting the benzofuran scaffold to a pyrazole decreases the cytotoxicity of the compounds which is an excellent characteristic of potential HIV treatment.| Benzoylbenzofurans | CC50 (μM) ± SEM (TZM-bl cells) | Pyrazole derivatives | CC50 (μM) ± SEM (TZM-bl cells) |
|---|---|---|---|
| CC50 represents the concentration of compounds that cause a 50% decrease in TZM-bl cell viability. The superscript (*) represents the pyrazoles that have significantly low cytotoxicity compared to their 3-benzoylbenzofuran precursors with P < 0.05. | |||
| 4a | 59.15 ± 6.56 | 5a | 88.85 ± 22.10 |
| 4b | 8.42 ± 1.16 | 5b | 46.45 ± 6.66* |
| 4c | 9.65 ± 2.37 | 5c | 47.69 ± 17.65* |
| 4d | 7.77 ± 1.54 | 5d | 74.96 ± 6.40 |
| 4e | 35.19 ± 2.32 | 5e | 103.40 ± 11.14* |
| 3f | 41.94 ± 6.33 | 5f | 36.07 ± 7.95 |
| 3g | 79.85 ± 7.25 | 5g | 140.10 ± 17.13* |
| 3h | 49.98 ± 10.47 | 5h | 142.30 ± 13.24* |
| Control (cisplatin) | 56.67 ± 5.20 | ||
Pyrazole compounds are known for their minimal cytotoxicity in human cells,19,20 while benzofurans are well known for their cytotoxicity and are more common in cancer treatment.38,39 The compound cytotoxicity data obtained in this study agreed with available literature about benzofuran and pyrazole cytotoxicity, except for the fluorinated pyrazole derivative (5f) which was more cytotoxic than its benzofuran precursor. However, fluorine has been well-documented to increase the toxicity of benzofuran compounds.40 Similar to benzofurans, the addition of single or multiple fluorine atoms to the pyrazole derivatives has been shown to increase the lipophilicity of the drugs, thus allowing for better absorption, but unfortunately also increasing cytotoxicity.41 The current study observed that methylating 3-benzoylbenzofurans increased their lipophilicity (MlogP) by a value greater than 0.2 (Table 6). This was expected, given that the methoxy functional group has previously been demonstrated to contribute to lipophilicity more than the hydroxy group.42
The mechanisms of apoptosis for chemotherapy drugs, including cisplatin, involve the induction of reactive oxygen species,43 DNA intercalation,44 and increased expression of caspases 3/7 and p53, which are tumour suppressor proteins.45 Benzofurans46,47 and pyrazoles48,49 have been demonstrated to cause cytotoxicity through similar mechanisms. The low cytotoxicity of pyrazole derivatives has been reported by many researchers16 and this may be attributed to the physicochemical properties of the pyrazole scaffold. Researchers demonstrated that drugs with topological polar surface area (TPSA) values lower than 75 Å have significantly higher toxicity than drugs with larger TPSA values.50 Similar phenomena applied to the 3-benzoylbenzofurans, and their pyrazole derivatives were investigated in this study. The 3-benzoylbenzofurans had lower TPSA values, with the most toxic compound 4b having a TPSA of 57.90 Å and its pyrazole derivative 5b having a TPSA value of 76.60 Å (Table S1†). All the pyrazole derivatives had TPSA values that were higher by 18.70 Å compared to their precursors, showing the modification caused by the pyrazole scaffold. Therefore, the remarkably low cytotoxicity observed with the pyrazole derivatives prepared in this study was due to their physicochemical properties. Additionally, the mechanisms of apoptosis were not explored in this study. Therefore, it is inconclusive that mechanism of apoptosis for 3-benzoylbenzofurans may be altered because of derivatization to pyrazoles.
2.3 Inhibition of HIV-1 infections by 3-benzoylbenzofurans and pyrazole derivatives
The 3-benzoylbenzofurans were investigated for the inhibition of HIV-1 Q23 and CAP210 pseudoviruses, and as can be seen in Fig. 2, both Q23 and the CAP210 pseudoviruses were inhibited by the 3-benzoylbenzofurans. The IC50 values of the 3-benzoylbenzofurans in the Q23 pseudovirus ranged from 0.49 to 40.58 μM, as shown in Table 2. The 3-benzoylbenzofurans demonstrated higher inhibitory activity to the CAP210 pseudovirus with the IC50 values ranging from 0.12 to 11.16 μM (Table 2). 4b, which is a (2,5-dimethoxybenzoyl)-4,7-dimethylbenzofuran, was the most potent inhibitor with IC50 values of 0.49 ± 0.11 μM and 0.12 ± 0.05 μM in Q23 and CAP210 viruses, respectively. 3g, a (2,3,4-trimethoxybenzoyl)benzofuran, was the second active 3-benzoylbenzofuran in the Q23 pseudovirus with an IC50 of 3.90 ± 1.63 μM. Compound 3f, which is the 4-fluoro-2-methoxybenzoylbenzofuran, was the least active with IC50 values of 40.58 ± 7.85 μM in Q23 and 11.16 ± 0.31 μM in the CAP210 pseudovirus. | ||
| Fig. 2 Inhibition of HIV-1 infections by the 3-benzoylbenzofurans. The 3-benzoylbenzofurans were evaluated against Q23 (A) and CAP210 (B). Nevirapine (NVP) was used as a control at similar concentrations, and 99% inhibition was obtained in all tested concentrations. The bars represent the mean ± SEM obtained from three independent experiments. | ||
| Compounds | IC50 (μM) Q23 | SI (Q23) | IC50 (μM) CAP210 | SI (CAP210) |
|---|---|---|---|---|
| IC50 represents the concentration of compounds that inhibit 50% of HIV-1 infections by pseudoviruses. Q23 and CAP210 belong to HIV-1 subtypes A and C, respectively. | ||||
| 4a | 18.12 ± 1.78 | 3.26 | 8.12 ± 2.56 | 7.26 |
| 4b | 0.49 ± 0.11 | 17.18 | 0.12 ± 0.05 | 70.17 |
| 4c | 12.56 ± 0.53 | 0.76 | 2.43 ± 1.28 | 3.97 |
| 4d | 22.44 ± 3.47 | 0.35 | 3.28 ± 1.48 | 2.36 |
| 4e | 20.95 ± 2.20 | 1.68 | 4.27 ± 0.19 | 8.24 |
| 3f | 40.58 ± 7.85 | 1.03 | 11.16 ± 0.31 | 3.76 |
| 3g | 3.90 ± 1.63 | 20.47 | 4.91 ± 1.18 | 16.26 |
| 3h | 5.74 ± 1.51 | 8.71 | 4.45 ± 0.82 | 11.23 |
| NVP | 0.11 ± 0.004 | >500 | 0.10 ± 0.002 | >500 |
Selectivity indices (SI) represent the ratio of the CC50 of drugs against healthy cells to the IC50 of compounds against pathogens, and an SI value higher than 1 suggests favourable inhibition.51 In Q23, the 3-benzoylbenzofurans 4c, 4d, and 3f displayed low SI values ranging from 0.35 to 1.03, indicating general toxicity and non-specificity. However, in CAP210 pseudoviruses, these 3-benzoylbenzofurans had higher SI values ranging from 2.36 to 3.97 (Table 2). The 3-benzoylbenzofurans 4a, 4b, 3h, and 3g had high SI values that ranged from 3.26 to 20.47 and 7.26 to 70.17 in Q23 and CAP210 pseudoviruses, respectively (Table 2). Therefore, only compounds 4a, 4b, 3g, and 3h were considered to be inhibitors of the Q23 pseudovirus, while all the 3-benzoylbenzofurans inhibited the CAP210 pseudovirus, with SI values of above 1 for all the compounds. The CAP210 pseudovirus belongs to the HIV-1 subtype C strain, which accounts for 46.6% of infections globally and 98% of HIV infections in South Africa.52 Research has shown that the subtype C HIV viruses maintain intact proviruses, which gives them an advantage for better adaptation in a new host and, therefore, increased transmission.53 This study reported 3-benzoylbenzofurans with potent inhibitory activities against HIV subtype C, the most prevalent HI-virus in South Africa. Additionally, while the 3-benzoylbenzofurans are active against subtype C HIV infections, they represent medication that potentially won't be effective against the other eight HIV subtypes of group M, which are prevalent throughout Europe, Asia, East and West Africa, and the Caribbean. In contrast to subtype C, these HIV subtypes are less virulent.54 Among the 3-benzoylbenzofurans, 4b was the most effective inhibitor found; however, it was still far less potent than the common non-nucleotide reverse transcriptase inhibitors (NNRTIs) zidovudine (DZV) and nevirapine (NVP), which have been demonstrated to inhibit HIV-1 subtype C pseudoviruses with IC50 values of 0.05 uM and 0.03 uM, respectively.55
Pyrazole derivatives of the 3-benzylbenzofurans were successfully synthesized and these were also evaluated against Q23 and CAP210 pseudoviruses, as shown in Fig. 3. The pyrazole derivatives inhibited HIV infections by both pseudoviruses. Compound 5f was the most potent pyrazole derivative, with IC50 values of 0.39 ± 0.13 μM in Q23 and 1.00 ± 0.15 μM in CAP210 pseudovirus, as shown in Table 3. The pyrazoles 5g and 5h also demonstrated good activities with both compounds having IC50 values lower than 10 μM in Q23 and 13.80 ± 1.70 μM and 6.31 ± 0.31 μM in CAP210, respectively. As shown in Fig. 3, the pyrazole derivative 5f exhibited over 90% anti-HIV activity at the lowest concentrations tested, similar to NVP. However, the current study did not test lower concentrations of NVP to determine its actual IC50. Therefore, it can be assumed that the IC50 for NVP is much lower than 0.11 μM. Literature reports typically show IC50 values around 0.03 μM or lower against various HIV-1 subtypes.55 Thus, NVP and other HIV drugs are significantly more potent (in the nano and picomolar range) than 5f. Nonetheless, compound 5f remains the most potent inhibitor among the pyrazoles investigated in this study. Due to the pyrazole scaffold in 5f, which has been shown to reduce toxicity, 5f is the most promising compound identified in this study for further development as an antiretroviral agent.
 | ||
| Fig. 3 Inhibition of HIV-1 infections by the pyrazole derivatives. The pyrazoles were evaluated against Q23 (A) and CAP210 (B). NVP was used as a control at similar concentrations, and 99% inhibition was obtained in all tested concentrations. The bars represent the mean ± SEM obtained from three independent experiments. | ||
| Compounds | IC50 (μM) Q23 | SI (Q23) | IC50 (μM) CAP210 | SI (CAP210) |
|---|---|---|---|---|
| IC50 represents the concentration of compounds that inhibit 50% of HIV-1 infections by pseudoviruses. Q23 and CAP210 belong to HIV-1 subtypes A and C, respectively. SI = CC50 (TZM-bl)/IC50 (virus), SI greater than 1 indicates a favorable inhibition of HIV-1 infections. | ||||
| 5a | 13.32 ± 1.12 | 6.67 | 18.38 ± 3.20 | 4.83 |
| 5b | 10.09 ± 1.66 | 4.60 | 7.87 ± 0.91 | 5.90 |
| 5c | 12.96 ± 3.75 | 3.68 | 20.49 ± 6.80 | 2.33 |
| 5d | 16.21 ± 2.55 | 4.62 | 20.77 ± 1.00 | 3.61 |
| 5e | 15.68 ± 8.17 | 6.60 | 29.09 ± 0.18 | 3.55 |
| 5f | 0.39 ± 0.13 | 92.49 | 1.00 ± 0.15 | 36.43 |
| 5g | 9.93 ± 0.69 | 14.11 | 13.80 ± 1.70 | 10.15 |
| 5h | 8.14 ± 2.63 | 17.48 | 6.31 ± 0.31 | 22.55 |
| NVP | 0.11 ± 0.04 | >500 | 0.10 ± 0.02 | >500 |
Pyrazoles 5f, 5g, and 5h have hydroquinone substituents on carbon 4 but differ in the carbon 3 substituents which are 4-fluoro-2-methoxyphenyl, 2,3,4-trimethoxyphenyl and 2,5-dimethyphenyl respectively. Amongst these compounds, 5f, which is a fluorinated pyrazole, had the highest anti-HIV activities (Fig. 3). 5g and 5h had activities that were significantly lower than NVP. Fluorinated pyrazoles have been well documented for their high potency against HIV;4,56,57 therefore, 5f was expected to demonstrate the highest activity. Based on these findings, it can be deduced that the fluorine of 5f influenced the anti-HIV activities more than the hydroquinone substituent, which is also found in 5g and 5h, which had lower anti-HIV activities. Additionally, these derivatives (5f, 5g, and 5h) demonstrated high selectivity indices, which ranged from 3.68 to 92.49 in Q23 and 2.33 to 36.43 in the CAP210 pseudovirus, making them all favourable inhibitors of HIV-1 infections (Table 3). The difference in IC50 values of pyrazole derivatives in Q23 and CAP210 viruses was not significant; therefore, these compounds could potentially be developed for the treatment of both subtype A and C infections. Subtypes A and C have been documented to have approximately 15 to 20% differences in their genetic sequences; therefore, it is plausible that similar drugs would inhibit both viruses.58
The anti-HIV activities of 3-benzoylbenzofurans and their pyrazole derivatives were compared in Q23 and CAP210, as shown in Fig. 4. There was no significant difference between the anti-HIV activity of 3-benzoylbenzofurans (4a, 4c, 4d, 4e, 3g, and 3h) and their pyrazole derivatives (5a, 5c, 5d, 5e, 5g, and 5h) in Q23 (Fig. 4A), while the anti-HIV activity of 3-benzoylbenzofuran 4b was significantly higher than that of its pyrazole derivative 5b. The pyrazole derivative 5f also had a significantly higher activity than its precursor 3f. There was no significant difference between the IC50 values of NVP, 4b, and 5f, which were 0.11 ± 0.04, 0.49 ± 0.11, and 0.39 ± 0.13 μM, respectively. Even though derivatization to pyrazoles did not significantly increase the anti-HIV activities of the majority of the compounds, there were noticeable differences in specificity for HIV strains. The 3-benzoylbenzofurans were more specific for CAP210, while the pyrazoles had general activity against both subtype A and C viruses (Fig. 4). Therefore, cART formulated from these pyrazole derivatives would be equally effective against various HIV strains, while 3-benzoylbenzofurans would produce cART that could only be effective in certain strains, thus impending efforts to eradicate the HIV epidemic.
 | ||
| Fig. 4 Comparison of inhibition of Q23 (A) and CAP210 (B) infections by 3-benzoylbenzofurans and pyrazole derivatives. The superscripts *, **, ***, and **** represent the compound pair with significantly higher activity than their precursors with P ≤ 0.05, P ≤ 0.01, P ≤ 0.001, and P ≤ 0.0001, respectively. 4b and 5f had significantly higher activity than their derivative and precursor in Q23, respectively. Only 5f had a significantly higher activity in CAP210, while all benzofurans except 3h were significantly more active than their pyrazole derivatives in CAP210. The bars represent the mean ± SEM obtained from three independent experiments. | ||
A time of addition assay was conducted in an attempt to elucidate the target of the most potent 3-benzoylbenzofurans (3g and 4b) and pyrazoles (5f and 5h). The compounds were added from the onset of infections and every two hours for 8 hours. 3g and 4b were observed to inhibit HIV infections for up to 8 hours similarly to NVP, a reverse transcriptase inhibitor (Fig. 5). 5f and 5h only inhibited HIV infections when added on the onset of infection, then the inhibition significantly decreased from 2 hours post-infection. These findings suggest that the pyrazoles target the entry stage of infections, while the 3-benzoylbenzofurans potentially target reverse transcription, which occurs 5 to 8 hours post-infection.59 Since the structure of the 3-benzoylbenzofurans does not closely resemble the DNA nucleotides, which are substrates for HIV RT, it can be deduced that these compounds are NNRTIs like NVP.
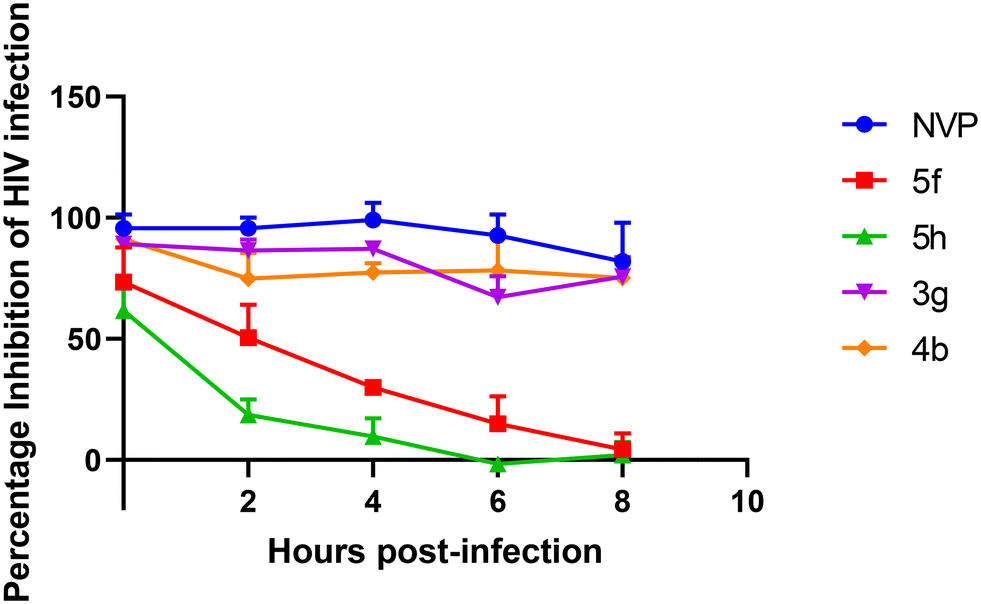 | ||
| Fig. 5 Time of addition assay of CAP210 HIV infection treated with active compounds. TZM-bl cells were infected with CAP210 pseudoviruses and treated with compounds at various hours post-infection. NVP, 3g, and 4b were used at 3 μM, while 5f and 5h were used at 25 μM. The bars represent the mean ± SEM obtained from three independent experiments. | ||
Pyrazoles 5f and 5h were demonstrated to exhibit inhibition against the early stages of infection; therefore, they could either target the viral receptors gp120 and CD4, host cell receptors like CD4 and CCR5, or the fusion, which all occur within the first two hours of HIV infection.59 Previous studies have identified pyrazole derivatives that inhibit HIV entry through binding to CCR5 and CXCR4, which are potential targets for 5f and 5h.18 Pyrazole 5f was six times more active than this pyrazole-piperidine compound at inhibiting HIV infections. Moreover, the pyrazole-piperidine compounds had dual targets, including HIV entry and HIV RT, which was not observed with 5f. 5f is a fluorophenylpyrazole, and fluorinated pyrazoles are more potent inhibitors of HIV-1 infection.57 Evidence of this is the recently approved long-acting capsid inhibitor lenacapavir, which inhibits more than 23 HIV-1 isolates with an IC50 ranging from 20 to 160 pM,12 making it significantly more potent compared to 5f, which has an IC50 ranging from 0.39 to 1.00 μM. Furthermore, 5f has demonstrated no selectivity for HIV subtypes A and C, suggesting that it may inhibit a variety of HIV isolates. Therefore, 5f holds potential for development as an anti-HIV drug. Future studies could focus on performing structural modifications to enhance the anti-HIV activity of compound 5f. Modifications to 5f, such as incorporating active functional groups like trifluoromethyl (CF3) and sulfonyl (SO2) groups, may prove beneficial. These groups are known to enhance lipophilicity, thereby improving membrane absorption; increase stability, leading to a longer half-life and potential for long-acting formulations; and enhance binding affinity to viral targets. Furthermore, these functional groups are present in several HIV drugs, including protease inhibitors (darunavir, atazanavir), integrase inhibitors (raltegravir, dolutegravir), and capsid inhibitors (lenacapavir), and their inclusion in 5f could enhance its anti-HIV activity.60–62 However, these modifications would need to be validated in lead optimization studies, which would include target-based validation.
2.4 Inhibition of HIV-1 protease (PR) inhibition
Previous studies reported 3-benzoylbenzofurans with dual inhibitory activities against HIV RT and PR.25 The current study screened the most potent 3-benzoylbenzofurans and pyrazoles in a direct enzyme inhibition assay against HIV PR. Dose-dependent inhibition of the enzyme was observed against PR (Fig. 6). All the compounds demonstrated more than 50% inhibition of HIV-1 PR at the highest tested concentration. | ||
| Fig. 6 Inhibition of HIV-1 PR by active compounds. All the compounds resulted in percentage inhibition above 50% at the highest concentration with 5f demonstrating the highest activity at most of the tested concentrations. Acetyl pepstatin (AP) was used at 1.6 μM and afforded 99.99% PR inhibition. The four compounds were screened at four decreasing concentrations. The bars represent the mean ± SEM obtained from three independent experiments. | ||
The pyrazole derivatives were the most potent inhibitors of PR, with 5f having the highest IC50 value of 31.59 μM and 5h having an IC50 value of 50.68 μM (Table 4). The cytotoxicity of pyrazoles was evaluated in HIV-negative peripheral blood mononuclear cells (PBMCs) and demonstrated low to mild cytotoxicity as demonstrated in Table 4. The selectivity indices of 5f and 5h were 3.1 and 1.6 respectively, demonstrating that they are true inhibitors of HIV-1 PR.
| Compound | IC50 (μM) HIV-1 PR | CC50 (μM) PBMCs | SI (HIV-1 PR) |
|---|---|---|---|
| SI = CC50 (PBMCs)/IC50 (HIV-1 PR), SI greater than 1 indicates a favorable inhibition of HIV-1 protease. | |||
| 3g | 70.90 ± 12.41 | 89.54 ± 11.60 | 1.3 |
| 4b | 57.48 ± 10.13 | 41.29 ± 6.78 | 0.7 |
| 5f | 31.59 ± 3.83 | 98.21 ± 1.30 | 3.1 |
| 5h | 50.68 ± 0.78 | 83.22 ± 9.70 | 1.6 |
| AP | >1.6 | — | — |
Among the 3-benzoylbenzofurans, only 3g inhibited HIV PR activity with an IC50 of 70.90 μM, while 4b did not inhibit HIV-1 PR due to its low selectivity index, which demonstrated its lack of selectivity for the virus against healthy human cells (Table 4). These results demonstrate that the pyrazole derivatives inhibit HIV PR activity, while the 3-benzoylbenzofurans show mild to no inhibition of the enzyme.
The compounds were docked against HIV-1 PR to evaluate whether the observed inhibition was attributed to the competitive interactions of the compounds with the active site of the enzyme. Docking of compounds allows for the identification of the potential mechanism of action as well as identifying biological targets. Adeniyi et al. (2013) reported that docking scores lower than −5 kcal mol−1 represent the highest binding energies, making them acceptable docking scores for potential inhibitors.63 HIV-1 PR is a homodimer, and between these two subunits is a hydrophobic core and two Asp25 residues, which make up the active site of the enzyme (Fig. S2†). For the inhibition to take place, the drug binds to the active site, and the flaps enclose, trapping the inhibitor inside the active site interface, which consequently prevents further incoming and exiting of substrates.64 HIV-1 PR active site residues include Arg8, Asp25, Gly27, Asp29, Asp30, Gly48, Ile50, and Thr80, and protease Inhibitors (PIs) have been demonstrated to interact with these residues.65 The compounds were docked into the active site, with saquinavir (SQV), a potent PI, and acetyl pepstatin (AP) as docking controls to confirm the docking method. Docking scores of −10.06 and −9.42 were obtained for SQV and AP, respectively; and as expected, these ligands were shown to interact with active site residues, including Asp25, from both subunits, which are critical for enzyme activity (Fig. 6).
Only the pyrazole derivatives (5f and 5h) demonstrated good docking scores of −5.62 and −6.18 kcal mol−1 (Table 5). Compounds 5f and 5h interacted with two active site residues (Asp30 and Ile50) (Fig. 6). However, unlike SQV, AP and the 3-benzoylbenzofurans, the pyrazoles did not interact with Asp25 in the enzyme's active site. Importantly, like all the competitive inhibitors of HIV-1 PR, both pyrazoles interacted with Ile50, which is the flap residue, and at least one active site residue, which suggests these compounds would still occupy the active site, enclose the flaps and prevent the incoming protease substrate.66 Han et al. (1998) reported a pyrazole derivative that interacted with Asp30 through the NH group of the pyrazole scaffold.67 The pyrazoles in the current study interacted with Asp30 through the hydroxyl group of the hydroquinone substituents (Fig. 6). Additional interaction of another OH group from the hydroquinone with Ile50 residues and H2O in the active site was also observed (Fig. 6).
| Compounds | Docking scores (kcal mol−1) |
|---|---|
| 3g | −4.87 (Asp25 (A), Asp25 (B), Ile50) |
| 4b | −4.35 (Asp25 (A), Ile50) |
| 5f | −5.62 (Asp30 (B), Ile50) |
| 5h | −6.18 (Asp30 (B), Ile50) |
| Saquinavir | −10.06 (Asp25 (A), Asp25 (B), Gly27 (A), Arg8 (A), Ile50) |
| AP | −9.42 (Asp25 (A), Asp25 (B), Gly27(A), Gly48 (B), Ile50) |
The 3-benzoylbenzofurans showed poor docking scores (−4.35 and −4.87 kcal mol−1 for 4b and 3g), indicating weak interactions with the active site (Table 5). Surprisingly, the 3-benzoylbenzofurans interacted with Asp25 through the carbonyl oxygen, and the OCH3 of the benzoyl group interacted with H2O and Ile50 of the enzyme (Fig. 7). Therefore, the 3-benzoylbenzofurans competitively inhibit HIV-1 PR, even though they are mild inhibitors due to their smaller size, which limits their interactions with other active site residues.
 | ||
| Fig. 7 Comparison of the interactions of active compounds and controls with the active site of HIV-1 PR crystal structure (PDB: 1HIV). The figure was generated using Maestro 13.1 ligand interaction. | ||
The 3-benzoylbenzofurans and pyrazole derivatives investigated in this study exhibited mild HIV protease inhibition, with acceptable selectivity, as shown in Table 5. These compounds could be further modified to enhance their inhibition of HIV protease, thereby improving both their potency and selectivity. Potential modifications could include the incorporation of functional groups that form key interactions with the protease active site. Such groups include hydroxy, amine, and amide, which can form hydrogen bonds with the Asp25 and Asp30 residues. Additionally, crucial π–π interactions between the aromatic groups and the flap residues (Ile50 of both subunits) could further strengthen binding. These modifications would enhance the compounds' binding affinity for the HIV protease active site, reducing off-target interactions and minimizing potential toxicity.
2.5 Analysis of drug-likeness of active compounds
Numerous drug compounds have been observed to fail during drug development due to poor absorbed, distributed, metabolized, excreted, and toxic (ADMET) profiles. Excellent oral bioavailability is crucial for the drug to proceed with clinical trials. Lipinski's rule of five describes the lead compounds' physicochemical properties, which determine their bioavailability potential.68,69 These physicochemical properties include a molecular weight of less than 500 g mol−1, the number of H-bond donors (OH and NH) of less than 5, the number of H-bond acceptors (O and N atoms) of less than 10, and a partition coefficient (log P) of less than 5.68 Additionally, the Pfizer 3/75 rule predicts the toxicity of drug compounds with constraints of log P greater than 3 and TPSA lower than 75 Å2.70All the 3-benzoylbenzofurans and pyrazole derivatives analyzed in this study were within the parameters of Lipinski's rule of five, as shown in Table 6. Surprisingly, lenacapavir and SQV did not obey Lipinski's rule, although the drugs are FDA-approved for HIV treatment. Additionally, adherence to the Pfizer rule was investigated, 3-benzoylbenzofuran (4b) was the only compound that did not obey the Pfizer rule, which was attributed to its low TPSA of 57.90 Å2. This also proves the validity of the in vitro cytotoxicity results, which demonstrated 4b as the most toxic compound (Table 1). Compound 3g and the pyrazole derivatives obeyed the Pfizer rule and were demonstrated to be mild to non-cytotoxic in vitro. Lenacapavir and saquinavir agreed with the Pfizer rule, which was attributed to their large TPSA, which makes these drugs non-cytotoxic. The lack of cytotoxicity is important for novel HIV drugs since toxicity poses a threat to adherence to treatment and, consequently, the emergence of drug resistance.71 The pyrazole derivatives gained popularity in HIV research due to their low cytotoxicity in human cells.16 This study has confirmed both in vitro and in silico that derivatizing benzofurans to pyrazoles indeed decreases the toxicity of compounds, thus making them better HIV drug candidates. No PAINS alerts were predicted with both the 3-benzoylbenzofurans and pyrazoles investigated in this study, which suggests the compounds are not promiscuous and are suitable to be in a drug discovery pipeline.72
| Compound | MW (g mol−1) | MLogP |
nHA | nHD | TPSA Å2 | Lipinski rule | Pfizer rule | PAINS alerts |
|---|---|---|---|---|---|---|---|---|
| Molecular weight; MW; g mol−1, lipophilicity; Mlog P, number of hydrogen bond acceptors; nHA, number of hydrogen bond donors; nHD. | ||||||||
| 3g | 328.32 | 0.74 | 6 | 1 | 78.13 | Yes | Yes | 0 |
| 4b | 340.37 | 1.75 | 5 | 0 | 57.90 | Yes | No | 0 |
| 5f | 300.28 | 1.78 | 5 | 3 | 78.37 | Yes | Yes | 0 |
| 5h | 342.35 | 0.79 | 6 | 3 | 96.83 | Yes | Yes | 0 |
| Saquinavir | 670.84 | 1.40 | 7 | 5 | 166.75 | No | Yes | 0 |
| Lenacapavir | 968.30 | 4.86 | 12 | 2 | 174.70 | No | Yes | 0 |
This study synthesized novel 3-benzoylbenzofurans and their pyrazole derivatives that inhibited HIV-1 pseudoviruses and HIV-1 proteases and exhibited good physiochemical properties, including low cytotoxicity. These novel compounds, therefore, have the potential to be further investigated for development into antiretroviral treatments. Further studies on the active compounds could include investigation for additional targets such as screening for their potential as HIV entry inhibitors through a time-of-addition study and inhibition of HIV capsid, especially pyrazole derivatives since their scaffold is similar to that of lenacapavir. Since the pseudovirus experiments are based on the triggering of the luciferase gene following integration,73 and HIV RT inhibition has been ruled out (Fig. S1†), the compounds could potentially inhibit HIV integration, which could be further investigated.
3. Conclusion
Overall, novel 3-benzoylbenzofurans and pyrazole derivatives were successfully synthesized. Four compounds, including two 3-benzoylbenzofurans (3g and 4b) and two pyrazole derivatives (5f and 5h), were the most potent inhibitors of HIV-1 pseudovirus infections (Q23 and CAP210) with IC50 values less than 10 μM. Compounds 4b and 5f were the most potent inhibitors among the 3-benzoylbenzofurans and pyrazole derivatives, respectively. Compounds 3g and 5h were mild inhibitors of HIV-1 protease, while pyrazole (5f) was the most potent HIV PR inhibitor. The 3-benzoylbenzofurans produced in this study were shown to be cytotoxic, and derivatizing them to pyrazoles decreased their cytotoxicity while maintaining their potent anti-HIV activity. Additionally, there was no significant difference in anti-HIV activities against subtype A and C infections, which makes pyrazoles ideal HIV inhibitor since they have the potential to be effective against various HIV isolates. Together, these observations are compelling for the further development of these compounds for HIV management.4. Experimental procedure
4.1 General procedures
The chemicals and reagents used for synthesis were bought from Sigma-Aldrich/Merck (Pty) Ltd. Radchem (Pty) Ltd. was used to source all the solvents. Ethyl acetate and hexanes, the solvents employed for chromatographic purification, were first distilled before use. Both the deuterated solvents for NMR and the reagent-grade solvents for synthesis were bought from Sigma-Aldrich. All reactions that required inert conditions were carried out under a nitrogen or argon atmosphere. Using suitable solvent mixtures, the progression of the reactions was monitored on the aluminium-backed silica gel 60 F254 TLC plates (Sigma Aldrich/Merck). The UV lamp (254 nm) and fluorescent lamp (365 nm) were used to view the TLC plates. The purification of the major intermediates and final products was conducted by column chromatography using silica gel (230–400 mesh with a particle size of 40–63 μM) and hexanes/ethyl acetate mixtures as eluent. The intermediates and final products were characterized using techniques that included mass spectrometry (MS), and Nuclear Magnetic Resonance (NMR) spectroscopy. The HRMS (ESI-QTOF) analysis was carried out using a Waters UPLC coupled to a QTOF Synapt G2 mass spectrometry. LRMS and the purity of the final compounds were measured on an Agilent 1260 Infinity HPLC with a PDA detector coupled to a Bruker AmaZon SL ion trap mass spectrometry. The NMR spectra of the compounds were recorded at room temperature using Bruker Avance III and Avance III HD spectrometers operating at 300, 400 and 500 MHz for 1H and 75, 100 and 125 MHz for 13C analyses. The spectra were processed with Bruker TopSpin 3.6.5 software and the chemical shifts were referenced against solvent peaks at δH 2.50 and δC 39.5 for DMSO-d6, δH 3.31 and δC 49.05 for methanol-d4 and, δH 7.26 and δC 77.16 for chloroform-d1.4.1.1.1 (5-Hydroxybenzofuran-3-yl)(2,3,4-trimethoxyphenyl)methanone (3g). The benzofuran 3g was synthesized from 2,3,4-trimethoxyacetophenone (0.61 g, 2.90 mmol) and 1,4-benzoquinone (0.38 g, 3.48 mmol) following GP1. Compound 3g was purified with column chromatography using hexanes/ethyl acetate (7
:3). It was obtained as a white powder with a yield of 0.37 g (39%); mp 218–225 °C. 1H NMR (400 MHz, DMSO-d6) δ 3.74 (s, 3H), 3.79 (s, 3H), 3.87 (s, 3H), 6.85 (dd, J = 2.6 and 8.9 Hz, 1H), 6.92 (d, J = 8.7 Hz, 1H), 7.23 (d, J = 8.7 Hz, 1H), 7.47 (d, J = 2.6 Hz, 1H), 7.51 (d, J = 8.9 Hz, 1H), 8.36 (s, 1H), and 9.50 (s, 1H). 13C NMR (125 MHz, DMSO-d6) δ 56.0, 60.5, 61.7, 106.4, 107.4, 112.1, 114.3, 121.8, 124.0, 125.2, 127.2, 141.7, 149.2, 151.4, 154.8, 155.4, 155.6, and 188.5. HRMS (ESI/Q-TOF) m/z calcd. for C18H17O6 329.1020 [M + H]+, found 329.1020.
4.1.1.2 (5-Hydroxybenzofuran-3-yl)(2,5-dimethoxyphenyl)methanone (3h). The benzofuran 3h was synthesized from 2,5-dimethoxyacetophenone (3.00 g, 16.65 mmol) and 1,4-benzoquinone (2.16 g, 19.98 mmol) following GP1. Compound 3h was purified with column chromatography using hexanes/ethyl acetate (7
:3). It was obtained as a white powder with a yield of 0.61 g (12%); mp 173–180 °C. The 1H NMR (400 MHz, DMSO-d6) δ 3.69 (s, 3H), 3.74 (s, 3H), 6.85 (dd, J = 2.6 and 8.9 Hz, 1H), 6.97 (d, J = 2.9 Hz, 1H), 7.08 (dd, J = 2.9 and 9.0 Hz, 1H) 7.12 (d, J = 9.0 Hz, 1H), 7.46 (d, J = 2.6 Hz, 1H), 7.49 (d, J = 8.9 Hz, 1H), 8.33 (s, 1H) and 9.50 (s, 1H). The 13C NMR (125 MHz, DMSO-d6) δ 55.5, 56.0, 106.3, 112.0, 113.5, 113.6, 114.3, 116.8, 121.6, 124.9, 129.9, 149.1, 150.2, 152.8, 154.8, 155.9, and 189.3. HRMS (ESI/Q-TOF) m/z calcd. for C17H15O5 299.0914 [M + H]+, found 299.0916.
4.1.2.1 (2,5-Dimethoxyphenyl)(5-methoxy-4,6-dimethyl-1-benzofuran-3-yl)methanone (4a). Compound 4a was synthesized by methylating (2,5-dimethoxyphenyl)(5-hydroxy-4,6-dimethyl-1-benzofuran-3-yl)methanone (3a) (0.37 g, 1.15 mmol) according to GP2. Compound 4a was purified using column chromatography using hexanes/ethyl acetate (7
:3). It was obtained as a yellow solid with a yield of 0.28 g (71%); mp 80–85 °C. 1H NMR (400 MHz, methanol-d4), δ 2.39 (s, 3H), 2.62 (s, 3H), 3.70 (s, 3H), 3.74 (s, 3H), 3.78 (s, 3H), 7.01 (d, J = 3.2 Hz, 1H), 7.06–7.11 (m, 2H), 7.23 (s, 1H), and 7.87 (s, 1H). 13C NMR (125 MHz, methanol-d4), δ 15.2, 17.5, 56.8, 57.3, 61.2, 112.1, 115.1, 116.3, 119.3, 124.3, 126.9, 127.1, 131.7, 132.5, 153.6, 154.6, 155.4, 155.9, 156.7, and 192.0. HRMS (ESI/Q-TOF) m/z calcd. for C20H21O5 341.1384 [M + H]+, found m/z was 341.1389.
4.1.2.2 (2,5-Dimethoxyphenyl)(5-methoxy-4,7-dimethyl-1-benzofuran-3-yl)methanone (4b). The benzofuran 4b was synthesized by methylating (2,5-dimethoxyphenyl)(5-hydroxy-4,7-dimethyl-1-benzofuran-3-yl)methanone (3b) (0.30 g, 0.92 mmol) according to GP2. Compound 4b was purified using column chromatography using hexanes/ethyl acetate (7
:3). It was obtained as a yellow sticky solid with a yield of 0.30 g (96%); mp 59–61 °C. 1H NMR (400 MHz, methanol-d4), δ 2.44 (s, 3H), 2.49 (s, 3H), 3.67(s, 3H), 3.76 (s, 3H), 3.84 (s, 3H), 6.87 (s, 1H), 6.99 (d, J = 2.8 Hz, 1H), 7.02–7.08 (m, 2H) and 7.88 (s, 1H). The 13C NMR (125 MHz, methanol-d4), δ 13.9, 14.9, 56.3, 56.8, 57.5, 112.9, 114.6, 115.9, 118.8, 119.1, 120.2, 125.2, 126.6, 132.0, 151.5, 153.1, 154.8, 156.0, 156.8, and 191.7. HRMS (ESI/Q-TOF) m/z calcd. for C20H21O5 341.1384 [M + H] +, found 341.1385.
4.1.2.3 (2,5-Dimethoxyphenyl)(5-methoxynaphtho[1,2-b]furan-3-yl)methanone (4c). The benzofuran 4c was synthesized by methylating compound 3c (0.30 g, 0.86 mmol) according to GP2. Compound 4c was purified using column chromatography using hexanes/ethyl acetate (7
:3). It was obtained as a cream white solid with a yield of 0.29 g (94%); mp 140–149 °C. 1H NMR (400 MHz, methanol-d4), δ 3.76 (s, 3H), 3.81 (s, 3H), 4.07 (s, 3H), 7.03–7.04 (m, 1H), 7.09–7.13 (m, 2H), 7.53–7.57 (m, 2H), 7.66 (brt, J = 7.7 Hz, 1H), 8.19–8.22 (m, 2H), and 8.34 (brd, J = 8.3 Hz, 1H). 13C NMR (125 MHz, methanol-d4), δ 56.36, 56.42, 56.8, 97.9, 114.6, 115.4, 118.3, 120.6, 121.5, 122.7, 124.2, 125.0, 125.9, 126.5, 128.6, 131.7, 147.6, 152.5, 154.9, 155.0, 155.1, and 192.6. HRMS (ESI/Q-TOF) m/z calcd. for C22H19O5 363.1227 [M + H] +, found 363.1249.
4.1.2.4 (5-Bromo-2-methoxyphenyl)(5-methoxynaphtho[1,2-b]furan-3-yl)methanone (4d). Compound 4d was synthesized by methylating (5-bromo-2-methoxyphenyl)(5-hydroxynaphtho[1,2-b]furan-3-yl)methanone (3d) (0.09 g, 0.227 mmol) according to GP2. Compound 4d was purified using column chromatography using hexanes/ethyl acetate (7
:3). It was obtained as white crystals with a yield of 0.09 g (96%); mp 156–162 °C. 1H NMR (400 MHz, methanol-d4), δ 3.81 (s, 3H), 4.08 (s, 3H), 7.16 (d, J = 8.8 Hz, 1H), 7.56–7.58 (m, 3H), 7.65–7.69 (m, 2H), 8.21–8.24 (m, 2H) and 8.33 (d, J = 8.3 Hz, 1H). 13C NMR (125 MHz, methanol-d4), δ 56.4, 56.5, 97.8, 113.6, 115.2, 120.6, 121.4, 122.6, 124.2, 124.8, 125.9, 126.6, 128.6, 132.4, 132.8, 135.9, 147.7, 155.0, 155.1, 157.6, and 191.0. HRMS (ESI/Q-TOF) m/z calcd. for C21H16BrO4 411.0227 [M + H]+, found 411.0239.
4.1.2.5 (4-Fluoro-2-methoxyphenyl)(5-methoxynaphtho[1,2-b]furan-3-yl)methanone (4e). Compound 4e was synthesized by methylating (4-fluoro-2-methoxyphenyl)(5-hydroxynaphtho[1,2-b]furan-3-yl)methanone (3e) (0.15 g, 0.45 mmol) according to GP2. Compound 4e was purified using column chromatography using hexanes/ethyl acetate (7
:3). It was obtained as white crystals with a yield of 0.15 g (95%); mp 133–138 °C. 1H NMR (300 MHz, methanol-d4), δ 3.78 (s, 3H), 4.04 (s, 3H), 6.93 (td, J = 8.4, 2.2 Hz, 1H), 7.14 (dd, J = 11.6, 2.2 Hz, 1H), 7.52–7.57 (m, 2H), 7.59–7.64 (m, 1H), 7.70–7.76 (m, 1H), and 8.21 (brd, J = 8.4 Hz, 1H), 8.28 (brd, J = 8.5 Hz, 1H), 8.53 (s, 1H). 13C NMR (75 MHz, methanol-d4), δ 56.0, 56.2, 96.9, 100.5–100.9 (d, 2JCF = 26.1 Hz), 107.0–107.2 (d, 2JCF = 21.8 Hz), 119.5, 120.2, 120.7, 122.9, 123.1, 123.7, 125.8, 125.9 (d, 4JCF = 3.1 Hz), 128.0, 130.7–130.8 (d, 3JCF = 11.0 Hz), 145.4, 152.9, 154.4, 158.6–158.7 (d, 3JCF = 10.8 Hz), 162.9–166.1 (d, 1JCF = 247.2 Hz), and 189.0. HRMS (ESI/Q-TOF) m/z calcd. for C21H16FO4 351.1027 [M + H]+, found 351.1029.
4.1.3.1 2-[3-(2,5-Dimethoxyphenyl)-1H-pyrazol-4-yl]-4-methoxy-3,5-dimethylphenol (5a). The pyrazole 5a was synthesized by reacting compound 4a (0.20 g, 0.59 mmol) with hydrazine hydrate (0.12 g, 2.35 mmol) according to GP3. Compound 5a was obtained as white crystals with a yield of 0.15 g (72%); mp 111–123 °C. 1H NMR (400 MHz, methanol-d4), δ 1.81 (s, 3H), 2.22 (s, 3H), 3.46 (s, 3H), 3.57 (s, 3H), 3.74 (s, 3H), 6.57 (s, 1H), 6.76 (d, J = 3.2 Hz, 1H), 6.79 (dd, J = 3.2 and 8.9 Hz, 2H), 6.93 (d, J = 8.9 Hz, 1H) and 7.45 (s, 1H). 13C NMR (100 MHz, methanol-d4), δ 13.8, 16.3, 55.7, 56.4, 60.4, 113.6, 114.8, 115.7, 115.8, 116.0, 116.2, 119.6, 120.5, 131.7, 132.4, 141.3, 151.3, 152.1, 152.6, and 154.8. HRMS (ESI/Q-TOF) m/z calcd. for C20H23N2O4 was 355.1653 [M + H]+, found 355.1655.
4.1.3.2 2-[3-(2,5-Dimethoxyphenyl)-1H-pyrazol-4-yl]-4-methoxy-3,6-dimethylphenol (5b). The pyrazole 5b was synthesized by reacting benzofuran 4b (0.20 g, 0.59 mmol) with hydrazine hydrate (0.12 g, 2.35 mmol) according to GP3. Compound 5b was obtained as white crystals with a yield of 0.18 g (86%); mp 133–146 °C. 1H NMR (300 MHz, methanol-d4), δ 1.72 (s, 3H), 2.22 (s, 3H), 3.45 (brs, 3H), 3.72 (s, 6H), 6.70 (s, 1H), 6.76 (brs, 1H), 6.80 (dd, J = 8.9, 3.0 Hz, 1H), 6.94 (brd, J = 8.9 Hz, 1H) and 7.45 (brs, 1H). 13C NMR (75 MHz, methanol-d4), δ 13.3, 16.8, 55.7, 56.4, 56.6, 113.6, 114.2, 114.6, 115.2, 116.6, 119.9, 122.9, 123.2, 125.4, 138.9, 141.4, 148.1, 152.0, 152.5, and 154.8. HRMS (ESI/Q-TOF) m/z calcd. for C20H23N2O4 was 355.1653 [M + H]+, found 355.1653.
4.1.3.3 2-[3-(2,5-Dimethoxyphenyl)-1H-pyrazol-4-yl]-4-methoxynaphthalen-1-ol (5c). The pyrazole 5c was synthesized by reacting benzofuran 4c (0.20 g, 0.55 mmol) with hydrazine hydrate (0.11 g, 2.21 mmol) according to GP3. Compound 5c was obtained as brownish-red crystals with a yield of 0.18 g (88%); mp 175–179 °C. 1H NMR (400 MHz, methanol-d4), δ 3.49 (brs, 3H), 3.56 (brs, 3H), 3.64 (s, 3H), 6.47 (s, 1H), 6.85–6.95 (m, 3H), 7.39–7.48 (m, 2H), 7.89 (brs, 1H), 8.09 (brd, J = 8.3 Hz, 1H), and 8.18 (brd, J = 8.3 Hz, 1H). 13C NMR (100 MHz, methanol-d4), δ 55.8, 55.9, 56.3, 107.6, 113.7, 116.0, 116.2, 117.0, 118.0, 122.6, 123.1, 126.0, 126.7, 126.8, 128.2, 133.3, 134.9, 139.4, 144.0, 150.0, 152.7, and 154.9. HRMS (ESI/Q-TOF) m/z calcd. for C22H21N2O4 377.1496 [M + H]+, found 377.1496.
4.1.3.4 2-[3-(5-Bromo-2-methoxyphenyl)-1H-pyrazol-4-yl]-4-methoxynaphthalen-1-ol (5d). The pyrazole 5d was synthesized by reacting compound 4d (0.06 g, 0.15 mmol) with hydrazine hydrate (0.03 g, 0.58 mmol) according to GP3. The product was obtained as a brownish-red powder with a yield of 0.06 g (91%); mp 155–161 °C. 1H NMR (400 MHz, methanol-d4), δ 3.48 (s, 3H), 3.63 (s, 3H), 6.42 (s, 1H), 6.91 (d, J = 8.7 Hz, 1H), 7.37–7.49 (m, 4H), 7.95 (s, 1H), 8.07 (brd, J = 8.3 Hz, 1H) and 8.17 (brd, J = 8.7 Hz, 1H). 13C NMR (75 MHz, methanol-d4), δ 55.9, 56.0, 107.3, 113.4, 114.3, 115.9, 118.5, 122.7, 123.1, 126.0, 126.8, 128.3, 133.3, 133.5, 133.6, 134.4, 134.6, 143.8, 150.1, and 158.0. HRMS (ESI/Q-TOF) m/z calcd. for C20H18BrN2O3, 425.0494 [M + H]+, found 425.0493.
4.1.3.5 2-[3-(4-Fluoro-2-methoxyphenyl)-1H-pyrazol-4-yl]-4-methoxynaphthalen-1-ol (5e). The pyrazole 5e was synthesized by reacting compound 4e (0.10 g, 0.29 mmol) with hydrazine hydrate (0.12 g, 2.35 mmol) according to GP3. Compound 5e was washed with hexanes and recrystallized from methanol and the pure product was obtained as a yellow powder with a yield of 0.10 g (95%); mp 123–127 °C. 1H NMR (300 MHz, chloroform-d1), δ 3.84 (s, 3H), 3.86 (s, 3H), 6.48 (td, J = 8.1, 2.5 Hz, 1H), 6.53 (s, 1H), 6.69 (dd, J = 10.2, 2.5 Hz, 1H), 6.98 (brs, 2H), 7.51–7.52 (m, 2H), 7.72 (s, 1H) and 8.18–8.22 (m, 2H). 13C NMR (125 MHz, chloroform-d1), δ 55.9, 56.2, 99.8–100.0 (d, 2JCF = 26.4 Hz), 106.2, 108.1–108.2 (d, 2JCF = 21.4 Hz), 111.9, 114.0–114.1 (d, 4JCF = 3.4 Hz), 114.6, 121.9, 122.4, 125.4, 126.0, 126.1, 126.2, 130.6 (d, 3JCF = 10.2 Hz), 137.8, 139.8, 143.0, 149.2, 157.6–157.7 (d, 2JCF = 9.8 Hz) and 162.5–164.5 (d, 1JCF = 249.0 Hz). HRMS (ESI/Q-TOF) m/z calcd. for C21H18FN2O3 365.1295 [M + H]+, found 365.1297.
4.1.3.6 2-[3-(4-Fluoro-2-methoxyphenyl)-1H-pyrazol-4-yl]benzene-1,4-diol (5f). The pyrazole 5f was synthesized by reacting (4-fluoro-2-methoxyphenyl)(5-hydroxy-1-benzofuran-3-yl)methanone (3f) (0.2 g, 0.70 mmol) with hydrazine hydrate (0.14 g, 2.79 mmol) according to GP3. The product was obtained as cream white crystals with a yield of 0.13 g (61%); mp 255–258 °C. 1H NMR (400 MHz, chloroform-d1), δ 3.96 (s, 3H), 6.65 (ddd, J = 2.4, 7.7 and 8.8 Hz, 1H), 6.65–6.76 (m, 1H), 6.82 (dd, J = 2.4 and 10.4 Hz, 1H), 6.83–6.93 (m, 2H), 7.29 (dd, J = 6.4 and 8.9, 1H), and 8.14 (brs, 1H). 13C NMR (100 MHz, chloroform-d1), δ 56.3, 100.4–100.7 (d, 2JCF = 26.6 Hz), 108.8–109.0 (d, 2JCF = 22.1 Hz), 109.6 (d, 4JCF = 4.2 Hz), 117.0, 117.4, 118.4, 118.6, 131.7–131.8 (d, 3JCF = 10.9 Hz), 134.5, 141.2, 147.4, 148.4, 148.8, 158.8–158.9 (d, 3JCF = 10.3 Hz), and 164.7–167.7 (d, 1JCF = 255 Hz). HRMS (ESI/Q-TOF) m/z calcd. for C16H14FN2O3 301.0983 [M + H]+, found 301.0984.
4.1.3.7 2-[3-(2,3,4-Trimethoxyphenyl)-1H-pyrazol-4-yl]benzene-1,4-diol (5g). The pyrazole (5g) was synthesized by reacting compound 3g (0.10 g, 0.31 mmol) with hydrazine hydrate (0.06 g, 1.22 mmol) according to GP3. Compound 5g was purified using column chromatography using hexanes/ethyl acetate (4
:6). It was obtained as pale pink crystals with a yield of 0.02 g (16%); 141–148 °C. 1H NMR (400 MHz, methanol-d4), δ 3.57 (s, 3H), 3.81 (s, 3H), 3.85 (s, 3H), 6.46 (d, J = 3.0 Hz, 1H), 6.52 (dd, J = 8.7 and 3.0 Hz, 1H), 6.67 (d, J = 8.7 Hz, 1H), 6.76 (d, J = 8.7 Hz, 1H), 6.97 (d, J = 8.7 Hz, 1H) and 7.80 (s, 1H). 13C NMR (125 MHz, methanol-d4), δ 56.5, 61.2, 61.3, 108.6, 115.3, 117.3, 117.6, 118.0, 122.5, 126.8, 137.8, 141.4, 143.5, 148.8, 150.9, 153.1, and 155.5. HRMS (ESI/Q-TOF) m/z calcd. for C18H19N2O5 343.1289 [M + H]+, found 343.1287.
4.1.3.8 2-[3-(2,5-Dimethoxyphenyl)-1H-pyrazol-4-yl]benzene-1,4-diol (5h). Compound 5h was synthesized by reacting compound 3h (0.20 g, 0.67 mmol) with hydrazine hydrate (0.13 g, 2.68 mmol) according to GP3. 5h was purified and obtained as white crystals with a yield of 0.15 g (69%); mp 210–213 °C. 1H NMR (300 MHz, chloroform-d1), δ 3.51 (s, 3H), 3.93 (s, 3H), 6.72–6.73 (m, 1H), 6.86–6.87 (m, 2H), 6.92 (d, J = 1.7 Hz, 1H), 7.02–7.03 (m, 2H), and 8.11 (s, 1H). 13C NMR (75 MHz, chloroform-d1), δ 55.8, 56.3, 113.0, 113.6, 114.4, 117.1, 117.3, 117.8, 117.9, 118.2, 119.5, 134.2, 140.9, 147.4, 149.4, 151.4 and 153.2. HRMS (ESI/Q-TOF) m/z calcd. for C17H17N2O4 313.1183 [M + H]+, found 313.1188.
4.2 Biological assays
:9 dilution MTS prepared in RPMI-1640) was added to the wells and incubated for 4 to 24 hours. MTT samples were solubilized with 100 μL solubilization buffer (1 mL HCl: 9 mL isopropanol) for 30 minutes, and the absorbance reading was taken at 550 nm and 690 nm wavelengths. MTS sample readings were taken at 490 nm without a solubilization step using the SpectraMaxR ParadigmR Multi-Mode Detection Platform plate reader (Molecular Devices, California, USA).
4.3 In silico analysis of 3-benzoylbenzofurans and pyrazole derivatives
4.4 Data analysis and statistics
The graphs were plotted using GraphPad Prism 7.0 software, as was the statistical analysis. The student t-test and one-way ANOVA were used for statistical analysis. A p-value of <0.05 is considered significantly different.Data availability
Data will be made available on request.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was supported by the Technology Innovation Agency, the National Research Foundation of South Africa (grant numbers 121996 and 138404), the University of Pretoria, and the Council for Scientific and Industrial Research.References
- AIDS Tpte, UNAIDS Global AIDS Update 2023 [Available from: https://www.thepath.unaids.org/wp-content/themes/unaids2023/assets/files/2023_report.pdf.
- UNAIDS, Fact sheets 2022: Global HIV statistics 2023 [Available from: https://www.unaids.org/en/regionscountries/countries/southafrica.
- UNAIDS, Country factsheets: South Africa 2022 [Available from: https://www.unaids.org/en/regionscountries/countries/southafrica.
- G. E. Gray, L. G. Bekker, F. Laher, M. Malahleha, M. Allen and Z. Moodie, et al. Vaccine Efficacy of ALVAC-HIV and Bivalent Subtype C gp120-MF59 in Adults, N. Engl. J. Med., 2021, 384(12), 1089–1100 CrossRef CAS PubMed.
- L. Corey, P. B. Gilbert, M. Juraska, D. C. Montefiori, L. Morris and S. T. Karuna, et al. Two randomized trials of neutralizing antibodies to prevent HIV-1 acquisition, N. Engl. J. Med., 2021, 384(11), 1003–1014 CrossRef CAS PubMed.
- S. Wang, N. L. Yates, J. Pollara, Y. Voronin, S. Stanfield-Oakley and D. Han, et al. Broadly binding and functional antibodies and persisting memory B cells elicited by HIV vaccine PDPHV, npj Vaccines, 2022, 7(1), 18 CrossRef CAS PubMed.
- T. Cihlar and M. Fordyce, Current status and prospects of HIV treatment, Curr. Opin. Virol., 2016, 18, 50–56 CrossRef CAS PubMed.
- UNAIDS, Danger: UNAIDS global AIDS update 2022, UN, 2022 Search PubMed.
- E. Venanzi Rullo, M. Ceccarelli, F. Condorelli, A. Facciolà, G. Visalli and F. D'Aleo, et al. Investigational drugs in HIV: Pros and cons of entry and fusion inhibitors, Mol. Med. Rep., 2019, 19(3), 1987–1995 Search PubMed.
- R. Bocanegra, A. Rodríguez-Huete, M. Fuertes, M. Del Álamo and M. G. Mateu, Molecular recognition in the human immunodeficiency virus capsid and antiviral design, Virus Res., 2012, 169(2), 388–410 CrossRef CAS PubMed.
- M. Cevik and C. Orkin, Insights into HIV-1 capsid inhibitors in preclinical and early clinical development as antiretroviral agents, Expert Opin. Invest. Drugs, 2019, 28(12), 1021–1024 CrossRef CAS PubMed.
- K. Singh, F. Gallazzi, K. J. Hill, D. H. Burke, M. J. Lange and T. P. Quinn, et al. GS-CA Compounds: First-In-Class HIV-1 Capsid Inhibitors Covering Multiple Grounds, Front. Microbiol., 2019, 10, 1227 CrossRef PubMed.
- J. Paik, Lenacapavir: first approval, Drugs, 2022, 82(14), 1499–1504 CrossRef CAS PubMed.
- Z. K. Sweeney, S. F. Harris, N. Arora, H. Javanbakht, Y. Li and J. Fretland, et al. Design of annulated pyrazoles as inhibitors of HIV-1 reverse transcriptase, J. Med. Chem., 2008, 51(23), 7449–7458 CrossRef CAS.
- J. Fichez, C. Soulie, L. Le Corre, S. Sayon, S. Priet and K. Alvarez, et al. Discovery, SAR study and ADME properties of methyl 4-amino-3-cyano-1-(2-benzyloxyphenyl)-1 H-pyrazole-5-carboxylate as an HIV-1 replication inhibitor, RSC Med. Chem., 2020, 11(5), 577–582 RSC.
- S. Kumar, S. Gupta, V. Rani and P. Sharma, Pyrazole containing anti-HIV agents: An update, Med. Chem., 2022, 18(8), 831–846 CrossRef CAS PubMed.
- M. Platten and G. Fätkenheuer, Lersivirine–a new drug for HIV infection therapy, Expert Opin. Invest. Drugs, 2013, 22(12), 1687–1694 CrossRef CAS PubMed.
- B. D. Cox, A. R. Prosser, Y. Sun, Z. Li, S. Lee and M. B. Huang, et al. Pyrazolo-piperidines exhibit dual inhibition of CCR5/CXCR4 HIV entry and reverse transcriptase, ACS Med. Chem. Lett., 2015, 6(7), 753–757 CrossRef CAS PubMed.
- R.-M. Rui, C.-R. Tang, C.-T. Zhang, W.-K. Pan, K. Gan and R.-H. Luo, et al. C6-structural optimizations of 2-aryl-1H-pyrazole-S-DABOs: From anti-HIV to anti-DENV activity, Bioorg. Chem., 2022, 119, 105494 CrossRef CAS.
- M. D. Cherne, J. Hall, A. Kellner, C. F. Chong, A. L. Cole and A. M. Cole, Avirulins, a novel class of HIV-1 reverse transcriptase inhibitors effective in the female reproductive tract mucosa, Viruses, 2019, 11(5), 408 CrossRef CAS PubMed.
- Y.-H. Miao, Y.-H. Hu, J. Yang, T. Liu, J. Sun and X.-J. Wang, Natural source, bioactivity and synthesis of benzofuran derivatives, RSC Adv., 2019, 9(47), 27510–27540 RSC.
- S. A. Galal, A. S. Abd El-All, K. H. Hegab, A. A. Magd-El-Din, N. S. Youssef and H. I. El-Diwani, Novel antiviral benzofuran-transition metal complexes, Eur. J. Med. Chem., 2010, 45(7), 3035–3046 CrossRef CAS PubMed.
- M. F. Armentano, P. Lupattelli, F. Bisaccia, R. D'Orsi, R. Miglionico and I. Nigro, et al. Novel wild type and mutate HIV-1 protease inhibitors containing heteroaryl carboxamides in P2: Synthesis, biological evaluations and in silico ADME prediction, Results Chem., 2023, 6, 101165 CrossRef.
- F. Tramutola, M. F. Armentano, F. Berti, L. Chiummiento, P. Lupattelli and R. D'Orsi, et al. New heteroaryl carbamates: Synthesis and biological screening in vitro and in mammalian cells of wild-type and mutant HIV-protease inhibitors, Bioorg. Med. Chem., 2019, 27(9), 1863–1870 CrossRef CAS.
- M. Zhu, Q. Shan, L. Ma, B. Dong, J. Wang and G. Zhang, et al. Structure based design and evaluation of benzoheterocycle derivatives as potential dual HIV-1 protease and reverse transcriptase inhibitors, Eur. J. Med. Chem., 2023, 246, 114981 CrossRef CAS PubMed.
- P. Kunyane, M. S. Sonopo and M. A. Selepe, Synthesis of isoflavones by tandem demethylation and ring-opening/cyclization of methoxybenzoylbenzofurans, J. Nat. Prod., 2019, 82(11), 3074–3082 CrossRef CAS.
- M. A. Selepe, P. Kunyane, P. Seboletswe, S. Nair, N. Cele and M. Engelbrecht, et al. Synthesis and evaluation of benzoylbenzofurans and isoflavone derivatives as sirtuin 1 inhibitors with antiproliferative effects on cancer cells, Bioorg. Chem., 2022, 128, 106101 CrossRef CAS PubMed.
- L. Chiummiento, R. D'Orsi, M. Funicello and P. Lupattelli, Last decade of unconventional methodologies for the synthesis of substituted benzofurans, Molecules, 2020, 25(10), 2327 CrossRef CAS PubMed.
- M. M. Heravi and V. Zadsirjan, Recent advances in the synthesis of benzo [b] furans, Adv. Heterocycl. Chem., 2015, 117, 261–376 CrossRef CAS.
- T. Mukhanova, L. Alekseeva, E. Kuleshova, Y. N. Sheinker and V. Granik, Synthesis of 3-acyl-5-hydroxyindoles and 3-acyl-5-hydroxybenzofurans. Influence of solvent on the course of the Nenitzescu reaction, Pharm. Chem. J., 1993, 27(2), 136–142 CrossRef.
- R. Hermans, M. Van Hoof, L. Van Meervelt and W. Dehaen, Exploration of the Divergent Outcomes for the Nenitzescu Reaction of Piperazinone Enaminoesters, Organics, 2023, 4(2), 146–163 CrossRef CAS.
- H. A. Abdel-Aziz, P. Ahmad, A. Kadi, K. A. Al-Rashood, H. A. Ghabbour and H.-K. Fun, Unexpected ring-opening of 3-aroylbenzo [b] furans at room temperature: a new route for the construction of phenol-substituted pyrazoles, Tetrahedron Lett., 2013, 54(26), 3424–3426 CrossRef CAS.
- L. Cai, R. Xu, X. Guo and V. W. Pike, Rapid Room-Temperature 11C-Methylation of Arylamines with [11C]Methyl Iodide Promoted by Solid Inorganic-Bases in DMF, Eur. J. Org. Chem., 2012, 2012(7), 1303–1310 CrossRef CAS.
- S. S. Abd El-Karim, M. M. Anwar, N. A. Mohamed, T. Nasr and S. A. Elseginy, Design, synthesis, biological evaluation and molecular docking studies of novel benzofuran–pyrazole derivatives as anticancer agents, Bioorg. Chem., 2015, 63, 1–12 CrossRef CAS PubMed.
- A. Amr, M. Abdalla, S. Essaouy, M. Areef, M. Elgamal and T. Nassear, et al. Synthesis of some substituted 5 H-furo [3, 2-g] chromene and benzofuran sulfonate derivatives as potent anti-HIV agents, Russ. J. Gen. Chem., 2017, 87, 1591–1600 CrossRef CAS.
- J. Rangaswamy, H. V. Kumar, S. T. Harini and N. Naik, Synthesis of benzofuran based 1,3,5-substituted pyrazole derivatives: as a new class of potent antioxidants and antimicrobials--a novel accost to amend biocompatibility, Bioorg. Med. Chem. Lett., 2012, 22(14), 4773–4777 CrossRef CAS PubMed.
- K. Karrouchi, S. Radi, Y. Ramli, J. Taoufik, Y. N. Mabkhot and F. A. Al-Aizari, et al. Synthesis and pharmacological activities of pyrazole derivatives: A review, Molecules, 2018, 23(1), 134 CrossRef PubMed.
- J. Farhat, L. Alzyoud, M. Alwahsh and B. Al-Omari, Structure–Activity Relationship of Benzofuran Derivatives with Potential Anticancer Activity, Cancers, 2022, 14(9), 2196 CrossRef CAS PubMed.
- A. A. Abbas and K. M. Dawood, Anticancer therapeutic potential of benzofuran scaffolds, RSC Adv., 2023, 13(16), 11096–11120 RSC.
- I. Hayakawa, R. Shioya, T. Agatsuma, H. Furukawa, S. Naruto and Y. Sugano, 4-Hydroxy-3-methyl-6-phenylbenzofuran-2-carboxylic acid ethyl ester derivatives as potent anti-tumor agents, Bioorg. Med. Chem. Lett., 2004, 14(2), 455–458 CrossRef CAS PubMed.
- R. J. Glyn and G. Pattison, The Effects on Lipophilicity of Replacing Oxygenated Functionality with Their Fluorinated Bioisosteres, 2021 Search PubMed.
- M. Maximo da Silva, M. Comin, T. Santos Duarte, M. A. Foglio, J. E. De Carvalho and M. do Carmo Vieira, et al. Synthesis, antiproliferative activity and molecular properties predictions of galloyl derivatives, Molecules, 2015, 20(4), 5360–5373 CrossRef CAS PubMed.
- S. Mirzaei, K. Hushmandi, A. Zabolian, H. Saleki, S. M. R. Torabi and A. Ranjbar, et al. Elucidating role of reactive oxygen species (ROS) in cisplatin chemotherapy: a focus on molecular pathways and possible therapeutic strategies, Molecules, 2021, 26(8), 2382 CrossRef CAS PubMed.
- P. Jordan and M. Carmo-Fonseca, Molecular mechanisms involved in cisplatin cytotoxicity, Cell. Mol. Life Sci., 2000, 57, 1229–1235 CrossRef CAS PubMed.
- C. Yang, V. Kaushal, R. Haun, R. Seth, S. Shah and G. Kaushal, Transcriptional activation of caspase-6 and-7 genes by cisplatin-induced p53 and its functional significance in cisplatin nephrotoxicity, Cell Death Differ., 2008, 15(3), 530–544 CrossRef CAS PubMed.
- M. Napiórkowska, M. Cieślak, J. Kaźmierczak-Barańska, K. Królewska-Golińska and B. Nawrot, Synthesis of new derivatives of benzofuran as potential anticancer agents, Molecules, 2019, 24(8), 1529 CrossRef.
- D. Coskun, M. Erkisa, E. Ulukaya, M. F. Coskun and F. Ari, Novel 1-(7-ethoxy-1-benzofuran-2-yl) substituted chalcone derivatives: Synthesis, characterization and anticancer activity, Eur. J. Med. Chem., 2017, 136, 212–222 CrossRef CAS PubMed.
- V. Pandey, G. Tripathi, D. Kumar, A. Kumar and P. K. Dubey, Novel 3, 4-diarylpyrazole as prospective anti-cancerous agents, Heliyon, 2020, 6(7), e04397 CrossRef PubMed.
- P. A. Halim, S. M. Z. Sharkawi and M. B. Labib, Novel pyrazole-based COX-2 inhibitors as potential anticancer agents: Design, synthesis, cytotoxic effect against resistant cancer cells, cell cycle arrest, apoptosis induction and dual EGFR/Topo-1 inhibition, Bioorg. Chem., 2023, 131, 106273 CrossRef CAS PubMed.
- T. Yukawa and R. Naven, Utility of physicochemical properties for the prediction of toxicological outcomes: Takeda perspective, ACS Med. Chem. Lett., 2020, 11(2), 203–209 CrossRef CAS PubMed.
- J. J. Lica, M. Wieczór, G. J. Grabe, M. Heldt, M. Jancz and M. Misiak, et al. Effective Drug Concentration and Selectivity Depends on Fraction of Primitive Cells, Int. J. Mol. Sci., 2021, 22(9), 4931 CrossRef CAS PubMed.
- M. J. Gartner, M. Roche, M. J. Churchill, P. R. Gorry and J. K. Flynn, Understanding the mechanisms driving the spread of subtype C HIV-1, EBioMedicine, 2020, 53, 102682 CrossRef PubMed.
- G. Q. Lee, K. Reddy, K. B. Einkauf, K. Gounder, J. M. Chevalier and K. L. Dong, et al. HIV-1 DNA sequence diversity and evolution during acute subtype C infection, Nat. Commun., 2019, 10(1), 2737 CrossRef PubMed.
- V. Madhavi, A. Kulkarni, A. Shete, W. S. Lee, M. R. Mclean and A. B. Kristensen, et al. Effect of combination antiretroviral therapy on HIV-1-specific antibody-dependent cellular cytotoxicity responses in subtype B-and subtype C-infected cohorts, J. Acquir. Immune Defic. Syndr., 2017, 75(3), 345–353 CrossRef CAS.
- L. G. Mavhandu, H. Cheng, Y.-C. Bor, D. M. Tebit, M.-L. Hammarskjold and D. Rekosh, et al. Development of a pseudovirus assay and evaluation to screen natural products for inhibition of HIV-1 subtype C reverse transcriptase, J. Ethnopharmacol., 2020, 258, 112931 CrossRef CAS PubMed.
- G. Di Perri, Pharmacological outlook of Lenacapavir: a novel first-in-class Long-Acting HIV-1 Capsid Inhibitor, Infez. Med., 2023, 31(4), 495 CAS.
- P. K. Mykhailiuk, Fluorinated pyrazoles: From synthesis to applications, Chem. Rev., 2020, 121(3), 1670–1715 CrossRef PubMed.
- B. S. Taylor, M. E. Sobieszczyk, F. E. McCutchan and S. M. Hammer, The challenge of HIV-1 subtype diversity, N. Engl. J. Med., 2008, 358(15), 1590–1602 CrossRef CAS PubMed.
- M. Van Loock, C. Van den, J. Hansen, P. Geluykens, T. Ivens and S. Sauviller, et al. An automated time-of-drug-addition assay to routinely determine the mode of action of HIV-1 inhibitors, Assay Drug Dev. Technol., 2013, 11(8), 489–500 CrossRef CAS PubMed.
- A. Hirata, K. Sugimoto, T. Konno and T. Morii, Amyloid-forming propensity of the hydrophobic non-natural amino acid on the fibril-forming core peptide of human tau, Bioorg. Med. Chem. Lett., 2007, 17(11), 2971–2974 CrossRef CAS PubMed.
- R. Subramanian, J. Tang, J. Zheng, B. Lu, K. Wang and S. R. Yant, et al. Lenacapavir: A Novel, Potent, and Selective First-in-Class Inhibitor of HIV-1 Capsid Function Exhibits Optimal Pharmacokinetic Properties for a Long-Acting Injectable Antiretroviral Agent, Mol. Pharmaceutics, 2023, 20(12), 6213–6225 CrossRef CAS PubMed.
- J. Zeuli, S. Rizza, R. Bhatia and Z. Temesgen, Bictegravir, a novel integrase inhibitor for the treatment of HIV infection, Drugs Today, 2019, 55(11), 669–682 CrossRef CAS PubMed.
- A. A. Adeniyi and P. A. Ajibade, Comparing the suitability of autodock, gold and glide for the docking and predicting the possible targets of Ru (II)-based complexes as anticancer agents, Molecules, 2013, 18(4), 3760–3778 CrossRef CAS PubMed.
- A. Badaya and Y. U. Sasidhar, Inhibition of the activity of HIV-1 protease through antibody binding and mutations probed by molecular dynamics simulations, Sci. Rep., 2020, 10(1), 5501 CrossRef PubMed.
- M. S. Ellithey, N. Lall, A. A. Hussein and D. Meyer, Cytotoxic, cytostatic and HIV-1 PR inhibitory activities of the soft coral Litophyton arboreum, Mar. Drugs, 2013, 11(12), 4917–4936 CrossRef CAS PubMed.
- I. T. Weber and J. Agniswamy, HIV-1 Protease: Structural Perspectives on Drug Resistance, Viruses, 2009, 1(3), 1110–1136 CrossRef CAS PubMed.
- Q. Han, C.-H. Chang, R. Li, Y. Ru, P. K. Jadhav and P. Y. Lam, Cyclic HIV protease inhibitors: Design and synthesis of orally bioavailable, pyrazole P2/P2 ‘cyclic ureas with improved potency, J. Med. Chem., 1998, 41(12), 2019–2028 CrossRef CAS PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings, Adv. Drug Delivery Rev., 1997, 23(1–3), 3–25 CrossRef CAS.
- C. A. Lipinski, Lead-and drug-like compounds: the rule-of-five revolution, Drug Discovery Today:Technol., 2004, 1(4), 337–341 CrossRef CAS PubMed.
- T. Yukawa and R. Naven, Utility of Physicochemical Properties for the Prediction of Toxicological Outcomes: Takeda Perspective, ACS Med. Chem. Lett., 2020, 11(2), 203–209 CrossRef CAS PubMed.
- D. S. Clutter, M. R. Jordan, S. Bertagnolio and R. W. Shafer, HIV-1 drug resistance and resistance testing, Infect., Genet. Evol., 2016, 46, 292–307 CrossRef CAS PubMed.
- S. Babalola, N. Igie and I. Odeyemi, Structure-based Discovery of Multitarget Directed Anti-inflammatory p-nitrophenyl hydrazones; Molecular Docking, Drug-likeness, in-silico Pharmacokinetics, and Toxicity Studies, 2022 Search PubMed.
- Q. Xiang, L. Li, J. Wu, M. Tian and Y. Fu, Application of pseudovirus system in the development of vaccine, antiviral-drugs, and neutralizing antibodies, Microbiol. Res., 2022, 258, 126993 CrossRef CAS PubMed.
- M. Y. Lugongolo, et al. The combination of low level laser therapy and efavirenz drastically reduces HIV infection in TZM-bl cells, Biomed. J., 2021, 44(6), 37 CrossRef PubMed.
- M. Ghasemi, T. Turnbull, S. Sebastian and I. Kempson, The MTT Assay: Utility, Limitations, Pitfalls, and Interpretation in Bulk and Single-Cell Analysis, Int. J. Mol. Sci., 2021, 22(23), 12827 CrossRef CAS PubMed.
- P. Kapewangolo, A. A. Hussein and D. Meyer, Inhibition of HIV-1 enzymes, antioxidant and anti-inflammatory activities of Plectranthus barbatus, J. Ethnopharmacol., 2013, 149(1), 184–190 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4md00844h |
| This journal is © The Royal Society of Chemistry 2025 |