
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNatural peptides and their synthetic congeners acting against Acinetobacter baumannii through the membrane and cell wall: latest progress
Gautam
Kumar
 *
*
Department of Pharmacy, Birla Institute of Technology and Science Pilani, Pilani Campus, Rajasthan 333031, India. E-mail: gautam.kumar@pilani.bits-pilani.ac.in
First published on 20th November 2024
Abstract
Acinetobacter baumannii is one of the deadliest Gram-negative bacteria (GNB), responsible for 2–10% of hospital-acquired infections. Several antibiotics are used to control the growth of A. baumannii. However, in recent decades, the abuse and misuse of antibiotics to treat non-microbial diseases have led to the emergence of multidrug-resistant A. baumannii strains. A. baumannii possesses a complex cell wall structure. Cell wall-targeting agents remain the center of antibiotic drug discovery. Notably, the antibacterial drug discovery intends to target the membrane of the bacteria, offering several advantages over antibiotics targeting intracellular systems, as membrane-targeting agents do not have to travel through the plasma membrane to reach the cytoplasmic targets. Microorganisms, insects, and mammals produce antimicrobial peptides as their first line of defense to protect themselves from pathogens and predators. Importantly, antimicrobial peptides are considered potential alternatives to antibiotics. This communication summarises the recently identified peptides of natural origin and their synthetic congeners acting against the A. baumannii membrane by cell wall disruption.
 Gautam Kumar | Gautam Kumar is an Assistant Professor at the Department of Pharmacy, Birla Institute of Technology and Science Pilani, Pilani Campus, Rajasthan, India. He received his B. Pharm. and M. Pharm. degrees from Jamia Hamdard, Hamdard University, India. He did his PhD dissertation at the Department of Natural Products, National Institute of Pharmaceutical Education and Research (NIPER) S.A.S Nagar, Punjab, India. During his PhD, he was involved in synthesizing heterocyclic compounds, including furan, thiophene, dihydroquinoline, and coumarin derivatives as potential anti-tubercular agents. He conducted post-doctoral research at the Indian Institute of Technology Bombay, India. During his post-doctoral research work, he was involved in designing and synthesising small bioactive molecules and chemical probes inspired by lipid molecules from Mycobacterium tuberculosis (M.tb), the causative agent of tuberculosis. He has developed “clickable” chemical probes based on the M.tb virulence-associated glycolipid, trehalose dimycolate, for real-time proteomics and imaging. Currently, he leads a lab in the area of Natural Products at the Department of Pharmacy, Birla Institute of Technology and Science Pilani, Pilani Campus, with a focus on bioassay-guided fractionation of extracts, isolation of natural products, phytopharmaceutical formulation development, and synthesis of biologically active molecules. |
1. Introduction
Acinetobacter baumannii is a Gram-negative aerobic coccobacilli bacterium belonging to the Moraxellaceae family and Acinetobacter genus. The genus Acinetobacter came from the Greek word “akinetos”, meaning non-motile. It has characteristic features including non-motile, non-fastidious, and non-fermentative.1A. baumannii has a vast habitat, including soil, water, and food. There are more than 50 recognized Acinetobacter species, and most of them are non-pathogenic. The Acinetobacter calcoaceticus–Acinetobacter baumannii (Acb) complex comprises the phylogenetically most closely clinically relevant members of the Acinetobacter species, including A. calcoaceticus, A. baumannii, A. nosocomialis, A. pittii, A. seifertii, and A. lactucae.2 Nevertheless, many Acinetobacter species are opportunistic human pathogens responsible for nosocomial infections. A. baumannii is responsible for 2–10% of hospital-acquired infections, including skin and soft tissue infections, pneumonia, urinary tract infection, bacteremia, endocarditis, and meningitis. On the other hand, A. calcoaceticus and A. lwoffii are responsible for fewer human-associated infections.3A. baumannii nosocomial infections are responsible for severe life-threatening complications and high morbidity and death rates. Patients with previous illnesses such as diabetes, kidney disease, cancer, and chronic obstructive pulmonary disease are more prone to A. baumannii infections.4 Also, a person with a habit of heavy smoking and excessive alcohol consumption frequently suffers from A. baumannii infections.5 The most common mode of nosocomial infection transmission is frequently encountered due to medical devices used during surgical operations. A. baumannii colonizes medical surgical tools through biofilm formation over the surface of Foley catheters, cerebrospinal fluid shunts, and vascular catheters and becomes a source of new infections.61.1. A. baumannii virulence factors
The genotypic and phenotypic analysis revealed A. baumannii virulence factors, including outer membrane porins, biofilm-associated protein (Bap), phospholipases, capsules, lipopolysaccharides (LPS), proteases, iron-chelating systems, penicillin-binding protein, and protein secretion systems.7 The genomic studies of Acinetobacter baylyi led to identifying genes responsible for pilus biogenesis, iron scavenging, quorum sensing, and the type IV secretion system.8 The virulence factors of A. baumannii allow it to survive under adverse environmental conditions and protect itself from being exposed to antimicrobials.8 The virulence factors of A. baumannii are discussed below.Porins present in the outer membrane of A. baumannii are responsible for cellular permeability in the bacteria. OmpA is a β-barrel porin abundantly present in the outer membrane of A. baumannii. In mammals, OmpA is responsible for the induction of apoptosis of epithelial cells by activating caspase 8 and 9. It also causes changes in cytochrome c and modulates mitochondrial activity in humans.8,9 Meanwhile, the porins Omp34 and Ompw are highly immunogenic. Omp34 interacts with eukaryotic cells and induces apoptosis through a caspase-dependent mechanism. Moreover, Omp34 inhibits autophagy and promotes bacterial persistence in autophagosomes.10 The concentration of glycosyltransferase (LpsB) increases during lipopolysaccharide biosynthesis in A. baumannii and is responsible for pathogenicity in mammals. The lipid anchor of LPS is hepta-acylated lipid A. Lipid A is the immunostimulatory component that stimulates toll-like receptor 4 (TLR4) in human cells. Also, lipid A is a crucial virulence component of the bacterial membrane and induces proinflammatory cytokine expression in humans.10,11
A. baumannii encodes two phospholipase C and three phospholipase D enzymes substrate-specific to the eukaryotic membrane component phosphatidylcholine. The enzymes phospholipase C and D displayed hemolytic activity against human erythrocytes. The phospholipase D genes are also associated with serum resistance, epithelial cell invasion, and in vivo, pathogenesis in humans.10 The A. baumannii capsule comprises L-rhamnose, D-glucose, D-glucuronic acid, and D-mannose, which promote its adhesion to epithelial cells and protect itself against phagocytosis.8 A recent study revealed that A. baumannii's thicker capsule is responsible for increased pathogenicity and antibiotic resistance. Also, thicker capsules are responsible for antimicrobial and disinfectant resistance in A. baumannii.12 Moreover, A. baumannii pathogenicity depends upon the specific iron acquisition proteins, siderophores, and hemophores, which compete with host binding proteins for essential iron and are necessary for bacterial survival and growth under limited low-iron conditions.8 A recent study demonstrated from their work that polymicrobial infections of A. baumannii and K. pneumoniae promote their co-existence and provide cross-protection against antibiotics.13A. baumannii virulence and its host response are depicted in Fig. 1.
 | ||
| Fig. 1 A diagrammatic representation depicting A. baumannii virulence and its host response. A. A. baumannii activates the host cell response by binding to plasma membrane-bound TLR4 and endosome-bound TLR9. Activating the TLR signaling activates the nuclear translocation of the NF-kB transcription factor. Also, NF-kB activates pattern recognition receptors NOD1 and NOD2. Further, the activated NF-kB leads to the enhanced expressions of proinflammatory cytokines, including TNF, IL-6, and IL-8. Furthermore, NF-kB also increases the expressions of antimicrobial peptides (AMPs) like β-defensins, which inhibit the growth of A. baumannii. B. A. baumannii secretes outer membrane vesicles (OMVs) with outer membrane proteins that interact with the DRP1 host protein, causing the release of mitochondrial protein cytochrome C and fragmentation of mitochondria. OmpA enhances the release of ROS levels, decreases the ATP release, and increases the mitochondrial depolarization, and cytochrome C induces apoptotic cell death.10,11 | ||
1.2. Acinetobacter baumannii biofilm role in pathogenesis
The exopolysaccharide poly-β-(1-6)-N-acetylglucosamine is responsible for biofilm formation in A. baumannii. A population of A. baumannii bacteria encased in the extracellular matrix on biotic or abiotic surfaces is called a biofilm, which plays a critical role in pathogenesis and provides protection from antimicrobials. The exopolysaccharide poly-β-(1-6)-N-acetylglucosamine is also responsible for eukaryotic cell adhesion.14 Biofilm-associated protein (BAP) significantly contributes to biofilm formation and is responsible for increased resistance to the immune host response. A. baumannii, despite lacking flagella, possesses uncharacterized surface-associated motility. The Acinetobacter genus produces polyamine 1,3-diamino propane, which is responsible for surface-associated motility and virulence. A GNAT-family acetyltransferase is conserved in A. baumannii and is responsible for regulating polyamines. Diaminopropane acetyltransferase (Dpa) performs the acetylation of polyamine 1,3-diaminopropane. It has been found that Dpa expression is upregulated in the bacteria, forming a pellicle, which helps in adhering to eukaryotic epithelial cells more than planktonic bacterial cells. Notably, deleting 1,3-diaminopropane acetyltransferase gene dpa enhances motility and negatively affects biofilm formation on plastic.15 Moreover, the colonized A. baumannii biofilm is reported to cause post-neurosurgical meningitis and surgical wound infections.16 The biofilm formation in A. baumannii is mediated by Csu pili, assembled through the “archaic” chaperone–usher pathway. The CsuC–CsuE chaperone–adhesin X-ray structure revealed hydrophobic finger-like loops at the tip of the pilus, which form attachments to the hydrophobic surface and, thus, biofilm formation can be a source of new infections.6 Several findings have demonstrated the role of Csu pili in the spread of A. baumannii through biofilm formation over the surface of medical surgical instruments.61.3. A. baumannii diagnosis
The conventional diagnosis of A. baumannii in most laboratories is carried out by cultures of pathogens. Additionally, biochemical assays and Gram staining techniques are used to identify A. baumannii. The detection of A. baumannii using a cultural method is time-consuming and laborious. Moreover, the polymerase chain reaction can identify A. baumannii, depending on DNA or complementary DNA amplification.17 Also, antibody-based assays such as enzyme-linked immunosorbent assays (ELISA) are more specific in detecting pathogens.18 In recent years, several techniques have been evaluated to identify A. baumannii, including a phage-based real-time quantitative polymerase chain reaction (qPCR), dual aptamer-based.18,19 A most important reliable biomarker is the blaOXA-51 gene, which is responsible for the weak hydrolysis of carbapenems used to identify A. baumannii.201.4. A. baumannii cell wall
The membrane of the bacteria is composed of complex conserved structures. Membrane-targeting antibiotics are effective in killing slow-growing or dormant bacteria. Also, membrane-targeting antibiotics have pronounced and rapid bactericidal effects on bacteria.21 Moreover, membrane-targeting antibiotics do not require to cross the membrane to reach the targets, which is a significant hurdle in developing antibiotics. However, there is always a challenge associated with conventional antibiotics to cross the plasma membrane and reach intracellular targets. Also, antibacterial agents targeting the membrane are less likely to acquire resistance than those targeting intracellular systems, and thus, antibiotics will have prolonged clinical efficacy.22 Membrane-targeting antibiotics must selectively target bacterial membranes, not mammalian cell membranes, as both membranes comprise lipid bilayers.22 A diagrammatic representation of the A. baumannii cell wall is shown in Fig. 2. | ||
| Fig. 2 A diagrammatic representation depicting the A. baumannii cell wall. | ||
A. baumannii is intrinsically resistant to toxic compounds and antibiotics because of the low permeability of its outer membrane. Notably, the outer membrane of A. baumannii is ∼100-fold less permeable than that of E. coli.23A. baumannii's outer membrane comprises an asymmetric bilayer with glycerophospholipids in the inner leaflet. The glycolipids are mainly constricted in the outer leaflet of the outer membrane and, depending upon the type of polysaccharide attached to glycolipids, are referred to as lipopolysaccharides and lipooligosaccharides (LOS).24 Lipooligosaccharides are acylated disaccharides (lipid A) connected to an oligosaccharide. Also, lipopolysaccharides are acylated disaccharides linked to variable amounts of repeat units of polysaccharides called O-antigen.25 The outer membrane proteins comprising β-barrel structures formed by 8 to 26 strands are anchored to the outer membrane. These large extended loops are present on the extracellular side and shorter loops on the periplasmic side. Due to this complex structure, the outer membrane proteins give high stability to the membrane, thus making it capable of tolerating harsh environments.26 The LPS provides structural roles such as a decrease in the membrane permeability and an increase in the rigidity of the bacterial cells.27
Next to the outer membrane, well-defined components of peptidoglycan layers are present. They comprise glycan layers of alternating N-acetylglucosamine (GlcNAc) and N-acetylmuramic acid (MurNAc) connected through MurNAc-attached peptides. Peptidoglycan provides cell-shape rigidity and mechanical strength to counteract osmotic stress. Also, the outer membrane asymmetry provides the membrane's impermeability due to the presence of LPS occupying the outer leaflet of the bacteria, as well as glycerophospholipids in the inner leaflet. The LPS comprises the lipid A anchor, core oligosaccharide, and O-antigen. Lipid A is a hydrophobic glycolipid and is a glucosamine-based phospholipid phosphorylated at the 1 and 4′ positions. Notably, A. baumannii biosynthesizes hepta-acylated lipid A, while other GNBs, such as E. coli, produce hexa-acylated lipid A. Two 3-deoxy-D-manno-oct-2-ulosonic acid and oligomers of sugars form the core of LPS. Also, the antigens are attached to the core oligosaccharide LPS, generating an intact LPS. Notably, the lipid A and core moieties in A. baumannii are phosphorylated, generating an overall negative charge for the endotoxin molecules. The final step in the biosynthesis of lipid A involves the secondary acylation of lipid A precursors and is catalyzed by members of the superfamily enzymes known as lysophospholipid acyltransferases (LPLATs).28,29 Also, the LPS strengthens the outer membrane by balancing the electrostatic net charge.29 It protects the cell from cationic peptides and cell lysis. Moreover, hepta-acylated lipid A is accountable for polymyxin resistance.
BamA, LptD, Omp33-36, and OmpW are outer membrane proteins identified in A. baumannii. BamA is responsible for allowing the other outer membrane proteins to be assembled. LptD enables the transportation of LPS to the outer membrane, while Omp33-36 is a porin channel for the passage of water. OmpW is a hydrophobic channel in the outer membrane and cytoplasm responsible for iron homeostasis.30 Chemical structures of lipid A present in wild-type A. baumannii and colistin-resistant A. baumannii are shown in Fig. 3.
 | ||
| Fig. 3 Chemical structures of lipid A present in wild-type A. baumannii and colistin-resistant A. baumannii. | ||
1.5. Conventional treatment of A. baumannii infections
A. baumannii survives for extended periods in the environment and thus can spread bacilli in the surroundings. The World Health Organization (WHO) has categorized and placed A. baumannii on the list of nosocomial ESKAPE (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter spp.) pathogens and mentioned an urgent need for research and development of antibiotics to tackle A. baumannii.31A. baumannii infections can be treated by several classes of antibiotics including carbapenems, aminoglycosides, fluoroquinolones, cephalosporins, and β-lactamase inhibitors. In recent decades, there has been a decrease in the susceptibility of A. baumannii to antibiotics, leading to an increase in the minimum inhibitory concentrations (MICs) of the prescribed antibiotics.5Sulbactam, clavulanic acid, and tazobactam belong to class A β-lactamase inhibitors. Sulbactam is a semisynthetic penicillanic acid sulfone derivative with intrinsic activity against carbapenem-resistant Acinetobacter strains.32 It showed antibacterial activity through inhibition of the penicillin-binding protein (PBP) types 1a and 2, which results in the interference of the cell wall synthesis. Sulbactam is given in combination with cefoperazone and ampicillin to prevent the hydrolysis of the β-lactam ring by β-lactamase enzymes.33 Sulbactam, in combination with cefoperazone and ampicillin, has been approved for treating skin and soft tissue intra-abdominal and gynaecological infections caused by A. baumannii.34 In a study, it has been found that spontaneous resistance to sulbactam in susceptible A. baumannii strains is rare. On the other hand, low-frequency and high-level resistance was observed in sulbactam and was associated with pbp3 mutants.32 Avibactam, relebactam, and vaborbactam inhibit clinically relevant class A and C β-lactamases and a limited number of class D enzymes. ETX2514 is a broad-spectrum β-lactamase inhibitor. ETX2514, in combination with sulbactam, has shown potent in vitro and in vivo efficacy against MDR A. baumannii.35
Imipenem, meropenem, and doripenem belong to the carbapenem class and are considered the first-line treatment for the infection caused by A. baumannii.36 Carbapenem antibiotics pass through porins to reach the targets and bind to the penicillin-binding proteins. PBPs, including PBP1a, 1b, 2, and 3, are responsible for peptidoglycan biosynthesis in the bacteria's cell wall. Notably, carbapenems structurally resemble acylated D-alanyl-D-alanine (the building block of peptidoglycan) and bind irreversibly to the PBP active sites. When β-lactam, like carbapenem, binds to the PBPs, it cannot further participate in the transpeptidation, preventing the cell wall formation and resulting in bactericidal activity.37
However, the rise in multidrug-resistant (MDR) A. baumannii strains reduced the effectiveness of β-lactams.38 AmpC β-lactamase, known as Acinetobacter-derived cephalosporinase (ADC), is naturally produced by A. baumannii.39 Carbapenemase enzymes, especially oxacillinases, are responsible for the neutralization of the carbapenem and are the major contributor to carbapenem resistance.39 OXA-23 is the most common class D carbapenemase reported to be present in all countries' strains and is associated with endemic clones CC113/CC79, CC104/CC15, CC110/ST25, and CC109/CC1.40 The recent research suggested that there is a near-complete absence of therapeutic options to treat infections caused by carbapenem-resistant A. baumannii (CRAB).41
Moreover, most of the clinical isolates of A. baumannii are resistant to cephalosporins, including third and fourth-generation (like ceftazidime and cefepime) agents. Cefiderocol is a novel siderophore cephalosporin that exhibits potent activity against MDR A. baumannii. Cefiderocol, in combination with antibiotics, is used to treat GNB infections. Time-kill analysis revealed that cefiderocol demonstrated synergistic activity with β-lactamase inhibitors. Also, cefiderocol, in combination with β-lactamase inhibitors, showed increased antibacterial activity against A. baumannii isolates.42
Tigecycline is a semisynthetic 9-t-butylglycylamido derivative of minocycline, belonging to a glycylcycline class. It is bacteriostatic and exhibits antibacterial activity by binding to the 30S ribosomal subunit. Tigecycline has been recommended for treating infections such as complicated skin and intra-abdominal infections and community-acquired pneumonia.43 Tigecycline showed resilience to several resistance mechanisms and demonstrated broad-spectrum antibacterial activity compared to its parent congener tetracyclines. Minocycline regimen therapy in combination with colistin or carbapenems showed clinical and microbiological success rates of 72.6% and 60.2% in patients suffering from pneumonia caused by A. baumannii.44
Aminoglycosides such as tobramycin and amikacin are used to treat MDR A. baumannii. Aminoglycosides are generally used in combination with other antimicrobials. Recent studies demonstrated that L-lysine potentiated the activity of aminoglycosides and killed clinically resistant MDR A. baumannii. The exogenous use of lysine increases the proton motive force through the transmembrane and causes the accumulation of aminoglycosides in the bacterial cells.45
Rifampin acts by binding to the bacterial RNA polymerase and prevents transcription in the bacteria. Rifampin demonstrated partial and synergistic effects in MDR A. baumannii in combination with biapenem, colistin, and tigecycline. Rifabutin, a semisynthetic derivative of rifampin, effectively cures patients suffering from A. baumannii infections.46
Fosfomycin showed antimicrobial activity by interfering with peptidoglycan biosynthesis. Fosfomycin, upon entering the bacterial cells, irreversibly binds to MurA (a UDP-GlcNAc enolpyruvyl transferase), which catalyzes the first step in peptidoglycan via the synthesis of UDP-GlcNAc-3-O-enolpyruvate from UDP-GlcNAc and phosphoenolpyruvate. A recent study suggested that peptidoglycan remodelling by the AmpD and Anmk pathway contributes to intrinsic resistance to fosfomycin in A. baumannii.47
Bacteriophages are made up of proteins and nucleic acids. They are amplified in the presence of susceptible bacteria and cause lysis of the cells. Bacteriophages inhibit biofilm formation and are active against resistant bacterial strains.3 ISTD and NOVI bacteriophages were assessed against A. baumannii isolates.3 In another study, phage øFG02, in combination with ceftazidime, was evaluated in an in vivo model of A. baumannii AB900 infection. The phage øFG02 treated A. baumannii AB900 showed a loss of genes involved in the capsule biosynthesis, increasing the sensitivity to ceftazidime of A. baumannii. This finding suggests that phage therapy, in combination with antibiotics, can restore the activity in resistant A. baumannii strains.48
Drug repurposing is one of the effective strategies for developing new antimicrobials.
A study assessed anticancer drugs, including 5-fluorouracil, cisplatin, mitomycin C, and melphalan, against A. baumannii ATTC BAA-747. Among these drugs, mitomycin C effectively inhibited A. baumannii ATTC BAA-747 with a MIC50 value of 7 μg mL−1 and completely inhibited the growth at a concentration of 25 μg mL−1. Moreover, mitomycin C increased the survival of the insect larvae Galleria mellonella infected with sensitive A. baumannii ATTC BAA-747 and MDR A560 and A578 strains, respectively.49
Auranofin is an anti-rheumatic drug that exhibits antibacterial activity in GPB with a MIC value of 0.0625 μg mL−1 against methicillin-resistant Staphylococcus aureus MRSA.50 It shows multiple modes of antibacterial action, including DNA, cell wall, and protein synthesis inhibition. Moreover, auranofin inhibits GNB, including E. coli, A. baumannii, and K. pneumoniae, with higher MIC values of 125, 15.6, and 125 μg mL−1, respectively. pentamidine is an antiprotozoal drug that inhibits GNB by interacting with lipopolysaccharides, disrupting the outer membrane and increasing the membrane permeability. pentamidine inhibits E. coli, A. baumannii, and K. pneumoniae with higher MIC values of 125, 250, and 500 μg mL−1, respectively. Yu et al. in their study demonstrated that the combination of both auranofin and pentamidine displayed a strong synergistic effect against GNB (E. coli, A. baumannii, and K. pneumoniae) with fraction inhibitory concentration index (FIC) values ranging from 0.094–0.506. Also, the combination decreased the MIC of auranofin in sensitive A. baumannii and MDR A. baumannii up to 1 μg mL−1. The mode of action of the combination is through disruption of the membrane, leading to an increase in the permeability of auranofin inside the bacterial cell. This study demonstrates that combining non-antibiotic drugs with complementary antibacterial mechanisms can lead to the discovery of new medications that can delay drug resistance.50
1.6. Antibiotic resistance in Acinetobacter baumannii spp.
A. baumannii is intrinsically resistant to several classes of antibiotics. A. baumannii has environmental persistence ability and follows a “persist and resist strategy” in which bacteria keep off oxidative stress and resist complement-mediated killing.31A. baumannii has developed several modes of antibiotic resistance, including enzyme-mediated metabolism that leads to inactivation of the drug, reduction in the access of the drug to the target site, overexpression of efflux pumps, and change in the drug permeability through the membrane.9 For instance, the aminoglycoside class of antibiotics is reported to acquire resistance to all these mechanisms.51 The community-acquired A. baumannii is more susceptible than hospital-acquired A. baumannii strains. The genome sequence of community-acquired A. baumannii revealed that it lacks antibiotic resistance islands (AbaR), which encode genes that make it resistant to multiple antibiotics, including aminoglycosides, β-lactams, sulphonamides, and tetracyclines. It could be reasoned that community-acquired A. baumannii is more susceptible to prescribed antibiotics.52Recent studies suggested that there is an increase in the progression of A. baumannii-resistant strains, including multidrug-resistant (MDR), extensively drug-resistant (XDR), and pan-drug-resistant (PDR) A. baumannii. MDR A. baumannii strains are non-sensitive to any three classes of antibiotics prescribed for A. baumannii. In contrast, XDR A. baumannii strains resist three classes of antibiotics and carbapenems. Meanwhile, PDR A. baumannii strains showed resistance to three classes of antibiotics: beta-lactams, fluoroquinolones, and aminoglycosides.53 The pan β-lactam-resistant bacteria are only sensitive to one or two potential active drugs, while they resist all antibiotics.
The porin channel and outer membrane of A. baumannii are essential for transporting antimicrobial agents to access bacterial targets. Several studies confirmed that the loss of protein and porin channels led to the development of carbapenem resistance in A. baumannii.54 The point mutations could be responsible for antibiotic resistance in A. baumannii. For example, mutations in the bacterial targets gyrA and parC topoisomerase enzymes are responsible for quinolone resistance in A. baumannii.54 Also, A. baumannii showed improved expression of chromosomal genes for efflux systems, which played a significant role in extruding a range of structurally dissimilar antibiotics. These pumps are responsible for reduced susceptibility to antibiotics, which leads to the generation of MDR.55
The use of β-lactam antibiotics has mainly contributed to the emergence and rapid progression of A. baumannii-resistant strains. A. baumannii possesses a series of β-lactamases that hydrolyze and acquire resistance to broad-spectrum β-lactam penicillins, cephalosporins, and carbapenems. It has been found that the use of colistin methanesulfonate to treat carbapenem-resistant A. baumannii led to the emergence of A. baumannii-resistant strains.56 Importantly, colistin-resistant isolates have emerged and restricted chemotherapy choices.57
One of the most common resistance mechanisms to β-lactam antibiotics is enzymatic hydrolysis of antibiotics by β-lactamase enzymes. Based on their amino acid sequences, β-lactamases are classified into four ambler classes (A–D). The β-lactamase enzymes A, C, and D types perform hydrolysis of β-lactam antibiotics by forming an intermediate acyl–enzyme complex with the serine residue, and class B carries out the hydrolysis of β-lactam antibiotics by using one or more zinc ions.53 Also, the association of OXA-23 with OM porins is responsible for carbapenemase activity. The topology of the OXA-23 with OM porin complex revealed that catalytic site Oxa-23-K82 concentrates near the inner surface of OM porins. Notably, this orientation of Oxa-23-K82 near the porins would benefit the hydrolysis of antibiotics immediately entering the cells before their diffusion in the cellular matrix. The increased concentration of OXA-23 in the periplasm causes increased carbapenem resistance.58 Also, the increased overexpression of OXA-51, OXA-23, and metallo-β-lactamases in A. baumannii is responsible for carbapenem resistance. While OXA-51 and OXA-40 belong to the D β-lactamase class, their overexpression is responsible for ceftazidime resistance.52 Oxacillinases are specific β-lactamases in Acinetobacter spp. and Pseudomonas aeruginosa. In contrast, β-lactamases such as blaOXA-23-like, blaOXA-51-like, and blaOXA-134 enzymes are identified in A. radioresistens, A. baumannii, and A. lwoffi. Oxacillinases are responsible for the hydrolysis of oxacillin, benzylpenicillin, methicillin, amoxicillin, and some antibiotics of the cephalosporin class, respectively.57 Specifically, the blaOXA-23-like oxacillinase is responsible for carbapenem resistance, OXA-type β-lactamases and blaOXA-40-like β-lactamases can hydrolyze penicillin but showed weak hydrolytic activity against cephalosporins and carbapenems (imipenem and ceftazidime), and blaOXA-51-like enzymes can inactivate clavulanic acid, tazobactam, and NaCl, respectively. In contrast, blaOXA-58-like enzymes can hydrolyze penicillin, imipenem, and oxacillin, while they are not active against extended-spectrum cephalosporins.57
Moreover, the polymyxin-dependent resistant A. baumannii strains possess mutations in the lpxC (lipopolysaccharide biosynthesis) and katG (reactive oxygen species scavenging) genes. The multi-omics study revealed that there is a remarkably abundant increased proportion of phosphatidylglycerol in the outer membrane of polymyxin-dependent resistant A. baumannii.59
The above reports suggest the loss of antibacterial activity of conventional antibiotics in A. baumannii; thus, there is a need for novel alternative treatments to address A. baumannii. Antibiotics with unique modes of action can overcome the existing resistance mechanisms in A. baumannii. This review discusses natural peptides, their synthetic congeners acting against A. baumannii, their modes of action, and their potential benefits and shortcomings.
2. Antimicrobial peptides (AMPs)
AMPs are produced from multiple organisms, including bacteria, reptiles, plants, and mammals, as part of their defense mechanism to protect themselves from pathogens and predators. AMPs are short-chain peptides with amino acids ranging from 10 to 100. AMPs are generally amphiphilic and positively charged (usually +2 to +9). The hydrophobic domains acquired approximately 50% of the amino acid residues of the AMP. In addition to positive charges, AMPs can have neutral or negative charges.60 AMPs' physicochemical properties and antimicrobial activity depend upon the length of the peptide, amino acid composition, cationic charge residues, hydrophobicity of the lipid constituents, overall net charge on the molecule, and the helicity of the spatial arrangement.61,62 Importantly, AMPs display remarkable diversity in their structure and are considered potential alternatives to antibiotics. It is generally believed that developing microbial resistance against AMPs is unlikely since AMPs attack multiple low-affinity targets rather than a single high-affinity target like conventional antibiotics.63 AMPs display various modes of action through interactions with intracellular targets and disruption of critical cellular processes.64 Most of the AMPs have membrane-permeabilizing and disrupting abilities. Moreover, they inhibit the cytoplasmic membrane septum formation and cell wall synthesis. At the same time, some AMPs inhibit DNA and protein synthesis, chaperone-assisted protein folding, and enzymatic activity.64,65 The nucleic acids, DNA, and RNA are considered intracellular targets of AMPs.66 Mostly, AMPs are cationic and preferentially bind to the negatively charged bacterial cytoplasmic membranes rather than the zwitterionic membrane of mammalian cells. The peptide-to-lipid ratio influences the AMP interaction with the cell membrane. When the peptide-to-lipid ratio is low, the peptide orients parallel to the plasma membrane and vertical to the hydrophobic center of the membrane as the peptide-to-lipid ratio rises. The binding of AMPs to the bacterial membrane causes leakage of intracellular ions, metabolites, and cytoplasmic contents, leading to bacterial cell death.67Despite continuous efforts to combat MDR pathogens, only a few compounds entered the clinical evaluation and received approval for treating human MDR infections. At the same time, drug repurposing and antibiotic adjuvant combination therapy have received attention for treating MDR infections.68,69 The AMPs are considered one of the promising alternatives to antibiotics, and they showed activity against both susceptible and MDR bacterial strains. Moreover, several studies have shown that AMPs as adjuvants exhibited potent synergistic activity with antibiotics. The synergistic combination of AMPs and antibiotics increases the therapeutic effects of the antibiotics, and it further allows the reduction of antibiotic dosage and toxicity effects, preventing antibiotic resistance development. Moreover, combining AMPs with antibiotics can help tackle MDR bacteria and prevent the further development of antibiotic-resistant strains.70
The AMP activity has been defined by theoretical models for the membrane-centric activity, which include barrel-stave, toroidal-pore, carpet, and aggregate models. In the barrel-stave model, the peptides with a specific orientation are sandwiched between membranes. Then, peptide monomers are aggregated to form transmembrane pores within the hydrophobic membrane core. In the barrel stave pore model, the AMPs will have a linear structure aligned with the shape of the pore to maximize their interaction with the membrane.71 In the toroidal pore model, the AMPs are adsorbed to the bilayer, and after internalizing between the lipid bilayer, the phospholipid molecules bend inward to form pores. In this model, the hydrophilic domain of the AMP orients to the bilayer membrane to form the outer part of the pore, and the hydrophobic domain acts as the internal part. The peptide chain is positioned within the lipid bilayer, heads inside the pore, and embeds in the hydrophilic and hydrophobic interface. Notably, the transmembrane pore is broadened by the interaction between lipid head groups and the AMPs, allowing the water core to be lined. Toroidal pores are more common and are formed by a broader range of peptides than barrel-stave pores.71 In the carpet model, the AMPs are electrostatically attached to the anionic phospholipid head groups and carpet the surface of the membrane. When the concentration of AMPs exceeds a certain threshold, they induce the formation of pores, causing severe membrane perturbation following the release of mixed peptide–lipid complexes.72 The carpet mechanism, or detergent model, impacts the integrity of the membrane molecular structure.71 In the aggregate model, the AMPs bind to the anionic cytoplasmic membrane, leading to transient peptide–lipid micelles. The AMPs, lipids, and water channels allow ions and cellular contents to leak, leading to cell death.72 The AMP modes of action with their types of models are given in Fig. 4.
 | ||
| Fig. 4 A schematic representation depicting the AMP modes of action, i.e., detergent, carpet, toroidal-pore, and barrel-stave models.71 | ||
The pharmacodynamics of AMPs is complex, and several AMPs demonstrate multiple effects on the body, such as immunomodulatory activities, including enhanced chemotaxis of immune cells, activation of immune cells, and their cell differentiation and maturation.73 Cationic host defense peptides exhibit various immune functions and modulate innate and adaptive immune responses. For example, IDR-1002 is bovine bactenecin, a host defense peptide that decreases TNF-α, IL-6, IL-8, and nitric oxide production triggered by toll-like receptor ligands and increases the expression of the anti-inflammatory cytokine IL-10.63 The human cathelicidin LL-37 exhibited multiple immunomodulatory activities, including macrophage activation, upregulation of chemokines and chemokine receptor production, and promotion of wound healing.74 Additionally, LL-37 acts on monocytes and induces chemokines such as CXCL8, CCL2, and CCL7 and anti-inflammatory cytokine expression, promoting differentiation to proinflammatory macrophages.75 Host defense peptides are short-cationic peptides with diverse sequences produced by several cells and tissues in all complex life forms and play an essential role in the body's response to infection and inflammation.76 α-Defensins, including HNP1, HNP2, HNP3, and HNP4, act on lung epithelial cells and stimulate cytokine release such as CXCL8, CCL2, and GM-CSF. In contrast, α-defensins such as HD5 and HD6 act on intestinal epithelial cells, activate NF-kB, and induce chemokine production such as CXCL8. β-Defensins HBD1, HBD2, HBD3, and HBD4 act on intestinal epithelial cells and promote migration and wound healing, and through epidermal keratinocytes stimulate chemokine and cytokine production such as IL-6, IL-10, and CCL2, and CCL20.76 AMPs used for treating bacterial, viral, and fungal infections are given in Table 1.
| S. no. | Peptide | Target | Therapeutic indication | Ref. |
|---|---|---|---|---|
| 1. | Colistin | Colistin binds to LPS and phospholipids in the outer cell membrane of GNB | GNB infections | 77 |
| 2. | Polymyxins | They interact with the lipid A component of LPS | GNB infections | 78 |
| 3. | Bacitracin | It inhibits dephosphorylation of the lipid carrier and obstructs the process of cell wall synthesis | GPB infections | 79 |
| 4. | Gramicidin D | It increases the permeability and disrupts the inner cellular content | GPB infections | 80 |
| 5. | Daptomycin | It disrupts the multiple aspects of the cell membrane function | GPB infections | 81 |
| 6. | Teicoplanin | It inhibits the cell wall peptidoglycan synthesis | GPB infections | 82 |
| 7. | Telavancin | It disrupts the peptidoglycan synthesis and cell membrane function | GPB infections | 83 |
| 8. | Oritavancin | It binds to the D-alanyl-D-alanine terminus of the peptidoglycan precursor linked to the C55-lipid transporter (collectively referred to as lipid II) | GPB infections | 84 |
| 9. | Dalbavancin | Dalbavancin interacts with terminal D-alanyl-D-alanine residues of peptidoglycan precursors. It prevents peptidoglycan cross-linking, thereby destroying the integrity of the cell wall | GPB infections | 85 |
| 10. | Vancomycin | It inhibits the peptidoglycan synthesis of the cell wall | GPB infections | 86 |
| 11. | Micafungin | It is a non-competitive inhibitor of 1,3-β-D-glucan synthesis, an essential fungal cell wall component | Fungicidal activity against Candida species | 87 |
| 12. | Caspofungin | It is a non-competitive inhibitor of 1,3-β-D-glucan synthesis | Fungicidal activity against Candida species | 88 |
| 13. | Enfuvirtide | T-20 effectively impedes the gp41-mediated fusion between the viral and host cell membranes | It is used for treating HIV-1 infections | 89 |
| 14. | Cobicistat | It is inactive and selectively inhibits CYP3A isoenzymes and the number of transmembrane drug transporters at different blood–target tissue barriers | It is used in combination with other anti-retroviral drugs as a pharmacoenhancer | 90 |
The efficacy of AMPs is often questionable due to their limited contribution as clinical agents.91 Most of the AMPs' experimental in vitro and in vivo results correlate little. Also, AMPs are sensitive to environmental conditions, including pH, serum, and salt. Moreover, some AMPs lost their activities under physiological salt conditions due to electrostatic attraction between peptides and the cell membrane, and some of the AMPs bind to serum protein, including albumin and lipoproteins, which leads to a decrease in their activity.91
2.1. Natural peptides and their analogs
 | ||
| Fig. 5 A diagrammatic representation depicting the polymyxins' mode of action. | ||
Roberts et al. evaluated polymyxin B (1), polymyxin B1 (2, Fig. 6), polymyxin B2 (3, Fig. 6), colistin (4), colistin A (5, Fig. 6), and colistin B (6, Fig. 6) against a panel of GNB, including P. aeruginosa, A. baumannii, K. pneumoniae, and E. cloacae. Here, polymyxin B represents 53% of the content of polymyxin B1 and 23% of polymyxin B2, and colistin represents 58% of the content of colistin A and 19% of the content of colistin B, respectively. Currently, colistin and polymyxin B are the only AMPs used in the clinic for treating infections caused by GNB. Polymyxin B, polymyxin B1, B2, colistin, colistin A, and colistin B showed antibacterial activity against sensitive and resistant A. baumannii strains. Also, these compounds were found to be active in the in vivo model of GNB infections compared to the commercial polymyxin and colistin.99 To retain essential interactions of polymyxins with lipid A and to reduce their toxicity, Roberts et al. synthesized polymyxin analogs in that all Dab1, Dab5, Dab8, and Dab9 residues were conserved in the native polymyxins. At the same time, the modifications were carried out in the N-terminus and in positions 6 and 7 to reduce the hydrophobicity at the binding site. The removal of positive charge, the change in stereochemistry and the increase or reduction in the side chain length were carried out at position 3. This work led to the identification of potent polymyxin compound F365 (QPX9003) (7, Fig. 6), which inhibits GNB, including A. baumannii ATCC19606, A. baumannii FADDI-AB030*, and A. baumannii FADDI-AB034* with MIC values of 0.25, 0.25, and 2 μg mL−1, respectively. Moreover, F365 was superior to polymyxin B and colistin in terms of therapeutic efficacy and safety. Additionally, it showed superior safety and effectiveness in lung infections caused by MDR pathogens, including A. baumannii, P. aeruginosa, and K. pneumoniae.95
 | ||
| Fig. 6 Chemical structures of lipopeptides. | ||
Polymyxin S2 (ASK0912) (8, Fig. 6) is a cyclic peptide derived from polymyxin B2 by substituting basic amino acid L-Dab3 with polar amino acid D-Ser3 and hydrophobic amino acid L-Leu7 with polar amino acid L-Thr7. Polymyxin S2 demonstrated broad-spectrum antibacterial activity against susceptible and resistant E. coli, K. pneumoniae, and A. baumannii. Polymyxin S2 inhibited A. baumannii with a MIC value of 0.25 μg mL−1 and was nontoxic to Vero cells up to the concentration of >500 μg mL−1.100,101 Polymyxin S2 increased the survival rate of mice infected with sepsis (induced by A. baumannii) by lowering the bacterial loads in organs and blood.101
NAB739 (9, Fig. 6) is a polymyxin derivative with three positive amino acid residues. It showed potential efficacy in treating E. coli in a murine model of infections. Also, NAB739 was less nephrotoxic to cynomolgus monkeys than polymyxin B. It has been established in several studies that polymyxins increase the permeability of the outer membrane and allow the entry of other drugs. Considering the importance of NAB739, Tyrrell et al. evaluated NAB739 in combination with several antibiotics against polymyxin-resistant (PMR) strains. Notably, NAB739 demonstrated synergistic activity with antibiotics against most GNB strains. For instance, the combination of NAB739 with rifampin or retapamulin demonstrated synergistic activity against polymyxin-resistant strains, including E. coli and K. pneumoniae. Also, NAB739, in combination with rifampin, demonstrated synergistic activity against A. baumannii. Notably, the antibiotics acting on GPB are generally ineffective on GNB. However, this study suggested that polymyxin and its derivatives, combined with other antibiotics (acting against GPB), can treat GNB infections.102
The plasmid-borne mobilized colistin-resistance (mcr-1) gene and its relatives significantly contribute to colistin-resistance in A. baumannii. The gene mcr-1 encodes a phosphoethanolamine transferase that ligates phosphoethanolamine to a phosphate group in lipid A, thus reducing the electrostatic interactions between colistin and lipopolysaccharides, responsible for bacterial resistance to colistin.103 Wang et al. performed the bioinformatic analysis of the sequenced bacterial genomes and used biosynthetic gene clusters to identify and predict a structurally divergent colistin congener macolacin (10, Fig. 6). Macolacin inhibits K. pneumonia 10031 and A. baumannii 17978, with the same MIC value of 1 μg mL−1. Colistin and polymyxin B showed a 32-fold higher MIC to K. pneumonia and A. baumannii expressed with the mcr-1 gene. In contrast, macolacin showed only a two- to four-fold increase in MIC against these strains. Further optimization of the macolacin structure generated a biphenyl analog of macolacin (11, Fig. 6) that was more effective in treating a mouse neutropenic infection model of extensively drug-resistant A. baumannii. The mode of action of 10 and 11 is by binding to the lipid A of A. baumannii.103
Arylomycins belong to macrocyclic lipopeptides obtained from Streptomyces culture, which inhibit the bacterial type I signal peptidase (SPase), an essential membrane-bound protease. The signal peptidase is present in the periplasmic space between the cytoplasm and outer membranes. Arylomycins do not exhibit antibacterial activity against ESKAPE pathogens because of their high molecular weight and lipophilicity. Recent research revealed that arylomycins are capable of penetrating the outer membrane. Also, they are inactive against GNB due to naturally occurring mutations in Gram-negative SPase LepB, which reduces the binding affinity of arylomycins in the membrane. The chemical optimization of arylomycins A–C16 (12, Fig. 7) led to the identification of compound G0775 (13, Fig. 7) with increased target affinity and improved penetration in the outer membrane. The access of G0775 to LepB in the bacteria does not require a porin mode of transportation. Arylomycins A-C16 and G0775 inhibited A. baumannii ATCC 17978, with MIC values of >64 and 1 μg mL−1, respectively. Moreover, G0775 showed potential efficacy in treating several GNB infection in vivo models of mice infected with E. coli, P. aeruginosa, K. pneumoniae, and A. baumannii. Also, it was nontoxic to mammalian cells.104
 | ||
| Fig. 7 Chemical structures of arylomycins A–C16, G0775, tridecaptin A1, Oct-TriA1, and tridecaptin B1 analogs. | ||
Tridecaptins are produced from the Bacillus and Paenibacillus species belonging to the lipopeptide class. Tridecaptins exhibit antibacterial activity against GNB.105 Tridecaptins are linear non-ribosomal peptides comprising 13 amino acids, acylated N-terminal fatty acid chains, and more than half of non-proteinogenic amino acid residues. Tridecaptins kill GNB by binding to lipid II and disrupting the inner membrane.106 Tridecaptin A1 (14, Fig. 7) is a member of the tridecaptin family and demonstrated potent antimicrobial activity against A. baumannii ATCC19606, with a MIC value of 12.5 μg mL−1.107,108 The structure–activity relationship suggests that the tridecaptin stereochemistry and structure of the lipid tail are essential for antibacterial activity.109 The removal of the N-terminal lipid from tridecaptin abolishes its antimicrobial activity. At the same time, replacing an octanol chain gives Oct-TriA1 (15, Fig. 7) that can kill GNB.110
SRCM 37 was derived from Paenibacillus NRRL B-30507, and to confirm the chemical structure, Cochrane et al. performed its synthesis. Also, they synthesized new variants of tridecaptin B1, including (6′S)-TriB1 (16, Fig. 7), (6′R)-TriB1 (17, Fig. 7), and Oct-TriB1 (18, Fig. 7), and assessed them for antimicrobial activities. (6′S)-TriB1, (6′R)-TriB1, and Oct-TriB1 showed broad-spectrum antibacterial activity and inhibited E. coli, Salmonella enterica, Pseudomonas aeruginosa, and Klebsiella pneumoniae. Moreover, (6′S)-TriB1, (6′R)-TriB1 and Oct-TriB1 inhibited A. baumannii ATCC19606 and A. baumannii ATCC BAA 1605 with MIC values of 25, 50, and 50 μg mL−1 and 25, 25, and 50 μg mL−1, respectively.109
Oct-TriA1 is a tridecaptin analog consisting of D/L-diaminobutyric acid and D-allo-isoleucine. It is synthesized from costlier amino acid residues. Ballantine et al. synthesized eight tridecaptin analogs that were cheaper in synthetic cost and evaluated them against MDR GNB. Tridecaptin analogs 1–8 (19–26, Fig. 8) demonstrated activity against sensitive and resistant A. baumannii strains. Among the synthesized compounds, tridecaptin-7 (25) displayed potent activity against MDR strains (clinical and environmental isolates) of A. baumannii, K. pneumoniae, and E. cloacae. Also, the tridecaptin-7 was four-fold more active against P. pseudoalcaligenes than canonical Oct-TriA1 and was least in causing hemolysis to RBC.111
 | ||
| Fig. 8 Chemical structures of tridecaptin and its analogs 1–8, tridecaptin M, nisin, and isopedopeptins A–H. | ||
Tridecaptin M (27, Fig. 8) was isolated from the mud bacterium Paenibacillus jamilae.106 Tridecaptin M is in preclinical development for treating XDR Enterobacteriaceae infections. In a study, Jangra et al. evaluated tridecaptin M in combination with other antibiotics against A. baumannii strains. Tridecaptin M sensitizes A. baumannii to vancomycin, ceftazidime, and rifampicin. The tridecaptin M and rifampicin combination was the most promising against A. baumannii. Tridecaptin M killed the bacteria in an ex vivo model of A. baumannii and was superior to rifampicin monotherapy.112
Lactococcus lactis and some Streptococcus spp. produce nisin (28, Fig. 8). It belongs to the bacteriocin class and displayed antimicrobial activity in GPB by binding to the lipid II complex. This increases membrane permeability through pore formation and by inhibiting peptidoglycan synthesis. It is mainly used in the food industry as a natural preservative because of its low toxicity to humans and high effectiveness against GPB. However, it is inactive against GNB. A recent study revealed that nisin, in combination with polymyxin B, demonstrated synergistic activity against pan-drug-resistant and extensively resistant A. baumannii. Thus, these findings suggest that nisin can be combined to decrease the adverse effects of polymyxin B by lowering its dose.113
Synthetic modifications of nisin were carried out to enhance its antibacterial activity against GNB. Several short peptides active against GNB were fused as a tail to the C terminus of either the entire or truncated nisin species. Among these, tail T2 [DKYLPRPRPV], T6 [NGVQPKY], and T8 [KIAKVALKAL] were attached to nisin, which displayed improved activity against GNB. Further, work on this led to the identification of T16m2 (29, Fig. 8) (ABCDE rings + SV+ PRPPHPRLK), which inhibits A. baumannii with a MIC value of 0.5 μM.114
Lipodepsipeptides contain a peptide skeleton with one or more additional ester bonds. Pedobacter cryoconitis UP508 bacterial strain was isolated from the soil samples. Nord et al. isolated isopedopeptins A–H (30–37, Fig. 8) by bioassay-guided fractionation of Pedobacter cryoconitis UP508 extracts. Among the isolated compounds, isopedopeptins B showed promising antibacterial activity against Gram-negative carbapenem-resistant A. baumannii with a MIC value of 1 μg mL−1, and against colistin-resistant strains A. baumannii, with a MIC value of 8 μg mL−1, respectively. The isopedopeptins B exhibited an antibacterial mode of action in the E. coli liposome system by causing a significant leakage and disruption in the bacterial membrane.115
Brevicidine (38, Fig. 9) is a non-ribosomal cyclic lipopeptide isolated from the fermentation of Brevibacillus laterosporus DSM25. Earlier studies revealed that brevicidine exhibits broad-spectrum antibacterial activity against Enterobacter cloacae, Escherichia coli, Pseudomonas aeruginosa, and Klebsiella pneumoniae. Brevicidine demonstrated potent antibacterial activity against Escherichia coli by interacting with lipopolysaccharides on the outer membrane and phosphatidylglycerol and cardiolipin on the inner membrane, resulting in the dissipation of proton motive force and membrane disruption. Zhong et al. evaluated brevicidine with antibiotics for combination antibacterial studies. Notably, brevicidine (1 μM) decreased the MIC of several antibiotics, including erythromycin, azithromycin, and rifampicin, against A. baumannii strains by 32–128-fold. Among them, the potent synergistic activity was achieved for brevicidine and erythromycin combination against A. baumannii. Moreover, this combination showed potent antibacterial activity in the A. baumannii-induced mouse peritonitis–sepsis models. Brevicidine demonstrated antibacterial activity against A. baumannii by disrupting the bacterial membrane. Also, the brevicidine and erythromycin combination showed enhanced killing of A. baumannii by adenosine triphosphate biosynthesis inhibition and by accumulation of reactive oxygen species, resulting in the pronounced death of A. baumannii.116
 | ||
| Fig. 9 Chemical structures of brevicidine B and its analogs, laterocidine and darobactins. | ||
Brevicidine B contains a single substitution from D-Tyr to D-Phe in the exocyclic linear segment compared to brevicidine. In another study, brevicidine analogs (39–45, Fig. 9) were assessed for antibacterial activity. Interestingly, a single amino acid substitution led to a broader spectrum antimicrobial activity toward E. coli, P. aeruginosa, and K. pneumoniae, with MIC values ranging from 4–8 μg mL−1. Brevicidine B inhibits A. baumannii ATCC 19606 and A. baumannii AB5075 with the same MIC value of 16 μg mL−1. Brevicidine B was inactive against GPB, including MRSA strains.117 The Li et al. group analyzed 7395 bacterial genomes and assessed their potential to synthesize cationic non-ribosomal peptides considering activity against GNB. This work identified two novel compounds, brevicidine and laterocidine (46, Fig. 9), which exhibit broad-spectrum activity against GNB. Laterocidine inhibited A. baumannii, with a MIC value of 4 μg mL−1. Moreover, brevicidine and laterocidine showed efficacy in mouse models of E. coli infections.118
Darobactins are produced by entomopathogenic Photorhabdus khaini. Darobactins are ribosomal synthesized and post-translationally modified peptides. They mimic the outer membrane protein (OMP) signaling peptide. They bind to the β-barrel of the outer membrane protein BamA, interfering with the integration of the OMP into the membrane. Due to this, BamA cannot close the lateral gate, affecting the entire complex's mechanical, kinetic, and energetic properties in the membrane of the bacteria. Using a biosynthetic engineering pathway, Seyfert et al. synthesized analogs of darobactin (DA) (47, Fig. 9). Among the synthesized analogs, DA9 (48, Fig. 9) and DA22 (49, Fig. 9) were the most superior analogs exhibiting antibacterial activity. DA and its analog DA22 demonstrated broad-spectrum antibacterial activity against A. baumannii DSM-30007 and A. baumannii DSM-30008 with MIC values of 16 and 4 μg mL−1 and 2 and 0.25 μg mL−1, respectively. D22 was nontoxic to HepG2 cells and the zebrafish larvae model.119
Specific outer membrane receptor (OMR) transporters in the GNB recognize the siderophore complex and internalize the iron complex in the periplasm. Natural sideromycins are the covalent combination of siderophores and antibiotics, which are recognized by the OMR and allow antibiotics to be delivered inside the cells. In recent years, synthetic sideromycins have been synthesized to mimic the Trojan horse, allowing it to deliver antibiotics inside the cells. Daptomycin is a cyclic lipopeptide produced by the fermentation of Streptomyces roseosporus.120,121 Daptomycin belongs to the class of calcium-dependent antibiotics and has an anionic charge. It is active against broad-spectrum GPB, including Staphylococcus aureus, Streptococcus pyogenes, Streptococcus agalactiae, Streptococcus dysgalactiae subsp. equisimilis and Enterococcus faecalis.122 It acts against GPB by bacterial membrane disruption, causing rapid membrane depolarization and leaking of ions from the cell.123 However, it is inactive against GNB because of its polyanionic character and its considerable size, thus it can't penetrate the outer membrane of GNB. In a study by Ghosh et al., they synthesized a hybrid of siderophores and daptomycin, which can solve the issue of daptomycin membrane permeabilization in GNB. The resultant mixed-ligand daptomycin conjugate (50, Fig. 10) was active against GNB and GPB strains. Notably, the mixed-ligand daptomycin conjugate inhibited susceptible and MDR strains, including A. baumannii ATCC 17961, with a MIC value of 0.4 μM. Moreover, the daptomycin conjugate increases the survival rate of mice infected with the A. baumannii ATCC 17961 sepsis model.124 In contrast, other siderophore-based conjugates, including bis-catechol daptomycin (51, Fig. 10) and tri-catechol daptomycin conjugates (52, Fig. 10) showed potent antibacterial activity against A. baumannii strains. The above study suggests that combining siderophores with daptomycin facilitates the entry of conjugates into A. baumannii demonstrating its potential antibacterial activity against GNB.124,125
 | ||
| Fig. 10 Chemical structures of daptomycin–siderophore, daptomycin–catechol, vancomycin analogs, paenimucillin A, and paenimucillin C. | ||
Vancomycin belongs to a glycopeptide obtained from Amycolatopsis orientalis. It possesses a sugar-containing heptapeptide structure.126 The long-chain fatty acid of vancomycin is responsible for its lipophilicity character. Vancomycin and its derivatives showed antibacterial activity by acting on the cell wall precursors containing the D-Ala–D-Ala sequence.127 It is active against GPB but does not possess activity against GNB. Recent work revealed that a vancomycin lipophilic cationic derivative can eradicate GNB in vitro and in vivo models.128 Sarkar et al. synthesized vancomycin derivative VanQAmC10 (53, Fig. 10), and its efficacy was assessed in GPB and GNB. VanQAmC10 demonstrated activity against A. baumannii strains with MIC values ranging from 3.9 to 15.5 μM. It demonstrated bactericidal activity against the carbapenem-resistant A. baumannii. It exhibited antibacterial activity by disruption of biofilm formation in A. baumannii. Moreover, VanQAmC10 promoted the intracellular degradative mechanism autophagy in mammalian cells, which helps in the clearance of intracellular pathogens such as S. aureus and Salmonella typhimurium.129 In another study by Guan et al., they modified vancomycin with sulfonium ions. The synthesized sulfonium–vancomycin analogs were assessed for their activity in GPB and GNB. The sulfonium–vancomycin analogs showed enhanced antibacterial activity, a good pharmacokinetic profile, stability, and toxicity. The sulfonium–vancomycin conjugate demonstrated antibacterial activity by disrupting the bacterial membrane. The representative vancomycin sulfonium conjugate (54, Fig. 10) inhibited A. baumannii with MIC values ranging from 4–8 μg mL−1. This finding suggests that vancomycin having sulfonium modification enhanced the interaction with the bacterial membrane and promoted the disruption of membrane integrity in GNB.130
Sanderink et al. evaluated a combination of colistin with glycopeptide antibiotics vancomycin or teicoplanin against A. baumannii. The colistin–teicoplanin combination increased the survival of mice infected with carbapenem-resistant A. baumannii pneumonia by 74%. However, colistin in combination with vancomycin showed similar efficacy compared to colistin alone. This finding suggests that teicoplanin can be combined with colistin to lower the dose of colistin and can decrease the adverse effects associated with colistin.131
Paenimucillin A (55, Fig. 10) was initially identified using a culture-independent synthetic-bioinformatic natural product (syn-BNP). It was synthesized based on the bioinformatic prediction of the NRPS gene cluster of Paenibacillus mucilaginosus K02. Importantly, paenimucillin A shares similarities in five of their 13 residues and an acyl substituent at the N-terminal of tridecaptin. Paenimucillin A exhibited broad-spectrum antibacterial activity against GPB and mild antibacterial activity against A. baumannii. Vila-Farres et al. carried out the bioinformatics-guided synthesis of paenimucillin A analogs. Among the synthesized compounds, paenimucillin C (56, Fig. 10) was cationic and inhibited the growth of MDR A. baumannii 1788 with a MIC value of 4 μg mL−1. Moreover, in rat cutaneous wound models infected with A. baumannii, paenimucillin C sterilized the wound infections completely, with no sign of rebound infection. Paenimucillin C demonstrated antibacterial activity against A. baumannii by pore formation and promoting cytoplasmic contents' leakage. In contrast, the closest similar structure, tridecaptin, showed antibacterial activity by binding to the target lipid II. Furthermore, paenimucillin C was the least toxic to HT-29 cells.132
X33 (57) is an oligopeptide isolated from Streptomyces lavendulae X33. It is active against Penicillium digitatum and Candida albicans. X33 showed moderate antibacterial activity against A. baumannii with MIC and MBC values of 78.13 and 312.50 μg mL−1, respectively. It causes changes in the cell wall permeability, induces changes in the cell morphology, and promotes leakage of cell contents. Moreover, X33 interfered with the tricarboxylic acid (TCA) cycle, glycolysis, oxidative phosphorylation, and pyruvate metabolism and induced oxidative stress in the bacterial cells.133
Lynronne-1 (58) is a 19 amino acid cationic peptide isolated from the bovine rumen microbiome. It inhibited A. baumannii ATCC 19606 with MIC values ranging from 16–8 μg mL−1, respectively. It displayed more selectivity to bacterial cells and exhibited the least hemolysis of the RBC cells. The biophysical models confirmed that the lynronne-1 mode of action is by membrane lysis. However, this has to be confirmed on the bacterial cells.134
HLF (1–11) (63) belongs to the cathelicidin family and is derived from an N-terminal fragment of human lactoferrin. HLF showed promising antibacterial activity against MDR bacteria, including Staphylococcus aureus and A. baumannii, in vitro and in vivo models of infections. HLF (1–11) (GRRRRSVQWCA, C-terminal amide) contains N-terminal arginine 2 and 3, essential for bactericidal activity. Additionally, HLF (1–11) improves the survival of mice infected with A. baumannii-induced pneumonia by suppressing the pyroptosis of macrophages and inflammatory cytokine production of activated macrophages.140 In a study, cathelicidin-related antimicrobial peptide (CRAMP) was evaluated in a murine model for immune response to multidrug-resistant A. baumannii. Wild-type and CRAMP knockout mice were infected intranasally with the multidrug-resistant Acinetobacter baumannii. These studies demonstrated that CRAMP−/− mice exhibited an increase in the colony-forming units of the bacteria, and levels of IL-6 and CXCL1 were lower, and IL-10 was more in the BAL fluid compared to wild-type mice. The above results suggest that the cathelicidin-related antimicrobial peptide is essential in providing host defense against pulmonary infection with bacteria by activating neutrophils and the innate immune response.141
Defensins are derived from human peptides. They are cationic amphipathic with a β-sheet-rich group. They form a network of disulfide bridges leading to a conserved structural fold, an important characteristic of defensins. Defensins exhibit antibacterial activity by creating pores, leading to the loss of cellular contents. They have three types, i.e., α-, β-, and θ-defensins. The α-defensin and β-defensin peptides can be differentiated based on their disulfide bridge. In α-defensin peptides, the intramolecular bonding occurs between cysteines 1–6, 2–4, and 3–5, whereas in β-defensins, intramolecular bonding occurs between 1–5, 2–4, and 3–6 cysteine residues, respectively.142 Further, β-defensins are sub-classified into β-defensin-1 (hBD-1), β-defensin-2 (hBD-2), β-defensin-3 (hBD-3) and β-defensin-4 (hBD-4). Among them, β-defensin-2 comprises 41 amino acids. The synthetic β-defensin-2 (64) (GIGDPVTCLKSGAICHPVFCPRRYKQIGTCGLPGTKCCKKP) showed bactericidal activity against A. baumannii, Pseudomonas aeruginosa, Enterococcus faecalis, Enterococcus faecium and Staphylococcus aureus. MDR A. baumannii (vLD90 = 3.25–4.5 μg mL−1) was more susceptible to β-defensin-2 than wild-type A. baumannii (vLD90 = 3.90–9.35 μg mL−1). Interestingly, β-defensin-2 does not exhibit promising bactericidal activity in Escherichia coli, Klebsiella pneumoniae, and Proteus mirabilis. In contrast, β-defensin-2 showed enhanced activity in the bacterial strains resistant to β-lactam antibiotics.143
Apolipoprotein E (ApoE) is a glycosylated protein in humans. It regulates microglial activation and inflammatory response during acute and chronic nerve injury. The proteolytic fragments of apolipoprotein E demonstrated antimicrobial and other biological activities. For instance, ApoE (130–162) showed antibacterial activity against Escherichia coli and Pseudomonas aeruginosa. Notably, ApoE23 is active in A. baumannii. Wang et al. evaluated the apolipoprotein E mimetic COG1410 (65) peptide for antibacterial activity. COG1410 mimics the residues located between 138 and 149 of the ApoE N-terminal domain and has substitutions at positions 140 and 145 with aminoisobutyric acid. COG1410 inhibited A. baumannii ATCC19606 and PDR A. baumannii YQ4 with the same MIC value of 16 μg mL−1 and quickly eliminated a large inoculum of A. baumannii. COG1410 demonstrated antibacterial activity by disrupting the plasma membrane, leaking the cytoplasmic contents, and inhibiting biofilm formation. COG1410 exhibited synergistic activity with polymyxin B and cured C. elegans infected with PDR A. baumannii. This finding suggests that COG1410 can be combined with polymyxin B to reduce the dose of polymyxins.144
Proline-rich peptides (Pr-AMPs) possess potent antimicrobial activities against bacterial species, including K. pneumoniae, A. baumannii, and E. coli. Pr-AMPs showed antimicrobial activity by acting on targets other than bacterial membranes. Bac7 (1–35) (71) is a proline-rich peptide that exhibits potent antimicrobial activity against many GNB species, including A. baumannii, with a MIC value of ≤8 μM. Notably, at the sub-MIC values of Bac7, it decreased the biofilm formation and motility.146 Bac5 (1–17) is a bovine proline-rich cathelicidin, and its fragments possess antibacterial activity against GNB, including E. coli, A. baumannii, and K. pneumoniae. Notably, the N-terminal 1–17 fragment of Bac 5 is mainly responsible for antimicrobial activity and protein synthesis inhibition. Mardirossian et al. modified Bac5 to identify the critical residues for antimicrobial activity. Among the synthesized compounds, the representative derivatives Bac5-258 (72) (RFRPPIRRPPIRPPFYR), Bac5-272 (73) (RFRWPIRRPPIRPPFYR), Bac5-278 (74) (RWRWPIRRPPIRPPFYR), Bac5-281 (75) (RWRRPIRRRPIRPPFYW), and Bac5-291 (76) (RWRRPIRRRPIRPPFWR) showed broad spectrum activity against GNB. Bac5-258, Bac5-272, Bac5-278, Bac5-281, and Bac5-291 inhibited A. baumannii ATCC19606 with MIC values of >64, 64, 8, 4, 2, and 8 μM, and MBC values of >64, 64, 16, 8, 8, and 16 μM, respectively. Only Bac5-281 demonstrated protein synthesis inhibition among these peptides, mainly due to its membrane permeabilization. The other peptides showed dual modes of action, including protein synthesis inhibition and bacterial membrane destabilization. Notably, the Bac5 derivatives, which have more arginine and tryptophan residues, demonstrated enhanced antimicrobial activity and broader spectrum activity while retaining low cytotoxicity to eukaryotic cells.147
Barksdale et al. characterized and evaluated the antimicrobial activity of peptides Apo5 (87), Apo6 (88), and A1P (89) derived from Alligator mississippiensis. Apo5 and Apo6 are related peptides, nested fragments of a purported apolipoprotein, and A1P is the C-terminal fragment of the alpha-1-proteinase inhibitor of the serpin family. Apo5 (FSTKTRNWFSEHFKKVKEKLKDTFA), Apo6 (KTRNWFSEHFKKVKEKLKDTFA), and A1P (PPPVIKFNRPFLMWIVERDTRSILFMGKIVNPKAP) exhibited broad-spectrum antibacterial activity. They inhibited A. baumannii ATCC 9955 and A. baumannii ATCC BAA-1794 with EC50 values of 0.644 and 0.234 μg mL−1, 0.233 and 0.126 μg mL−1, and 224.0 and 23.6 μg mL−1, respectively. Additionally, these peptides inhibited the growth of S. aureus, Escherichia coli, and P. aeruginosa. The mode of action of Apo5 and Apo6 is by depolarisation of the bacterial membrane. However, A1P was unable to depolarise the bacterial membrane at low concentrations. This suggests that Apo5 and Apo6 modes of antibacterial action are by membrane disruption. Importantly, these peptides do not cause hemolysis of sheep red blood cells.151
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 933 with MIC values of 6.25 and 3.13 μM and 25 and 12.5 μM, respectively. 2K2L and 2K4L have the same net charge, with high mean hydrophobic values of 14.94 and 17.14, respectively, suggesting that the peptide's hydrophobicity plays a vital role in antibacterial activity. The increase in the hydrophobicity remarkably increases the permeability of the peptides in the membrane. Due to its strong positive charge on the 2K4L peptide, it promotes interaction with lipopolysaccharides and disrupts membrane integration, leading to leakage of cell contents. Moreover, 2K4L inhibited biofilm formation. Interestingly, 2K4L showed protection activity in mice infected with A. baumannii-induced sepsis.152
933 with MIC values of 6.25 and 3.13 μM and 25 and 12.5 μM, respectively. 2K2L and 2K4L have the same net charge, with high mean hydrophobic values of 14.94 and 17.14, respectively, suggesting that the peptide's hydrophobicity plays a vital role in antibacterial activity. The increase in the hydrophobicity remarkably increases the permeability of the peptides in the membrane. Due to its strong positive charge on the 2K4L peptide, it promotes interaction with lipopolysaccharides and disrupts membrane integration, leading to leakage of cell contents. Moreover, 2K4L inhibited biofilm formation. Interestingly, 2K4L showed protection activity in mice infected with A. baumannii-induced sepsis.152
Hylin a1 (92) (IFGAILPLALGALKNLIK-NH2) was obtained from the arboreal South American frog Hypsiboas albopunctatus. It is an eighteen-amino acid peptide and demonstrates antimicrobial activity by acting on the bacterial membrane. However, hylin a1 exhibits hemolytic activity against red blood cells. Park et al. designed and synthesized hylin a1 analog peptides to decrease hemolytic activity. The hylin a1 analogs, including hylin a1-11K (93) (IAKAILPLALKALKNLIK-NH2) and hylin a1-15K (94) (IAKAILPLALKALKKLIK-NH2), exhibited broad-spectrum antimicrobial and anti-biofilm activity against carbapenem-resistant A. baumannii and showed lower hemolytic effects and lower selectivity to mammalian cells. Hylin a1-11K and hylin a1-15K showed antibacterial activity against A. baumannii by binding to lipopolysaccharides and causing the disruption of the bacterial membrane. Moreover, these analogs decreased the inflammation by suppressing the proinflammatory response generated due to A. baumannii infections and also ameliorated carbapenem resistance in mice infected with A. baumannii.153
Aurein is an antibacterial peptide secreted from the granular dorsal gland of Litoria aurea. P0 is a 45 amino acid α-helical peptide belonging to the cathelicidin class, and AO is an 11 amino acid α-helical aurein derived peptide. P0 and A0 were modified to P7 and A3 derivatives by truncating the peptide chain and substituting amino acids with hydrophobic and positively charged amino acids tryptophan and arginine. From these two parent peptides, α-helical cathelicidin (P7) and aurein (A3), a hybrid peptide was synthesized and designated as A3P7. Jariyarattanarach et al. modified the hybrid peptide A3P7 by flipping methodology. The parent compound A3P7 exhibits potent antibacterial activity against GNB and GPB but has hemolytic activity. To reduce the toxicity of A3P7, a series of eight compounds (AP12-AP19) were synthesized by truncating the C-terminal amino acids. Among them, AP19 (RLFRRVKKVAGKIAKRIWK-NH2) (95) demonstrated potent activity against A. baumannii with minimal hemolytic and cytotoxic activities against RBCs and mammalian cells. Further, to enhance the stability of AP19, the D-form of amino acids was incorporated to give D-AP19, which demonstrated antibacterial activity against MDR and XDR clinical isolates of A. baumannii and rapidly killed the bacteria. The mode of action of D-AP19 is by membrane disruption and leakage of the cytoplasmic intracellular contents. The bacteria treated with D-AP19 showed rough and shrunk surfaces, with blebbing and distortion of the membrane.154
Esculentin-1a (Esc) (96) is a 21-amino residue peptide from frog skin. It is a broad-spectrum AMP active against GPB and GNB. Amp Esc (1–21), in combination with colistin, showed synergistic activity against MDR A. baumannii clinical isolates. Esc (1–21), in combination with colistin, demonstrated antibacterial action by potentiating the membrane perturbation effects.155
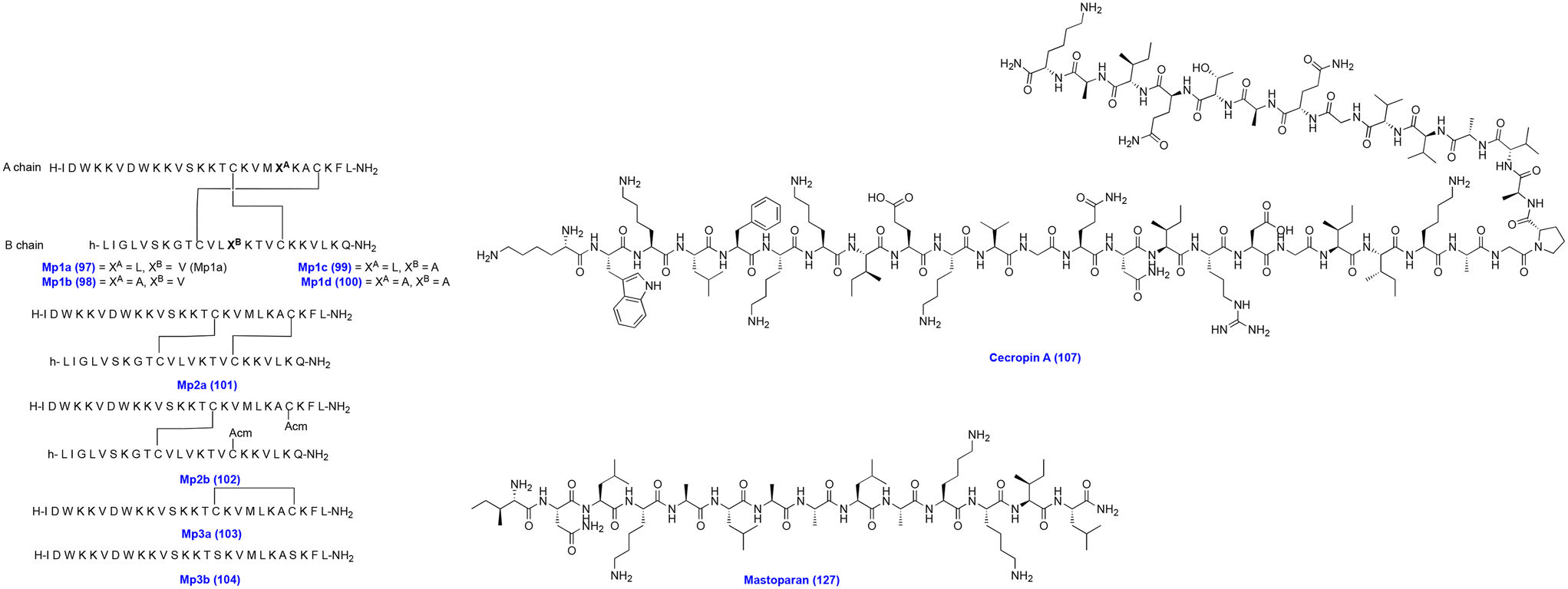 | ||
| Fig. 11 Chemical structures of Mp1a analogs, cecropin A, and mastoparan. | ||
LyeTx I (105) (H-IWLTALKFLGKNLGKHLAKQQLAKL-NH2) is an AMP obtained from Lycosa erythrognatha spider venom and exhibits broad-spectrum antimicrobial activity, including GPB and GNB, and fungi. Lima et al. modified the C-terminal portion of the LyeTx I peptide to generate LyeTx I mnΔK (106) (H-IWLTKALKFLGKNLGK-NH2), a short peptide comprising 16 amino acids. LyeTx I mnΔK showed antibacterial activity against carbapenem-resistant A. baumannii AC10, A. baumannii AC37, A. baumannii AC30, A. baumannii AC31, A. baumannii AC35, and A. baumannii AC03, with MIC and MBC values of 2 and 2 μM, 4 and 4 μM, 8 and 8 μM, 16 and 16 μM, 4 and 4 μM, and 4 and 4 μM, respectively. Moreover, intranasal dosing of LyeTx I mnΔK reduced the bacterial load in the mouse model of carbapenem-resistant A. baumannii. Also, it showed synergistic activity with meropenem and colistin against carbapenem-resistant A. baumannii. It demonstrated significantly low cytotoxicity and hemolytic activity in Vero cells and RBCs. LyeTx I mnΔK exhibits an antibacterial mode of action by disrupting the bacterial cell membrane, causing leakage of cell contents and reducing the preformed biofilms.157
Cecropins are amphipathic peptides secreted by insects. Cecropin A (107, Fig. 11) was first isolated from the giant silk moth Hyalophora cecropia. Cecropin A is a hydrophobic cationic AMP, which exhibits antibacterial activity by facilitating membrane permeabilization and inhibiting proton gradient formation. However, it suffers from stability issues due to abundant lysine and arginine in the structure, a common recognition sequence for protease enzymes. Several synthetic modifications were carried out to address the inherent instability issue of cecropin A.158,159 The insect Musca domestica cecropin showed antibacterial activity by acting on the cell membrane integrity of Escherichia coli and A. baumannii. Cec4 (108) (GWLKKIGKKIERVGQNTRDATIQAIGVAQQAANVAATLKG) is an α-helix peptide, which exhibited antibacterial activity against A. baumannii (ATCC 19606), MDR A. baumannii (ID: 4367661), and extensive drug-resistant A. baumannii (PRAB) with the same MIC value of 4 μg mL−1 and showed minimum biofilm inhibition concentration (MBIC90) values of 64, 64 and 128 μg mL−1 and minimum biofilm eradication concentration (MBEC) values of 128, 128, and 256 μg mL−1, respectively. Importantly, Cec4 showed no toxicity to hemolytic effects on human cells. Cec4 treatment of A. baumannii led to the destruction of the bacterial membrane and caused leakage of the cytoplasmic contents.160 In another study, Cec4 demonstrated activity against carbapenem-resistant A. baumannii with a MIC value of 4 μg mL−1, a MBIC value of 64–128 μg mL−1, and a MBEC value of 256–512 μg mL−1, respectively. Scanning electron microscopy (SEM) and confocal laser scanning microscopy (CLSM) revealed that Cec4-treated A. baumannii cells showed biofilm disruption. Meanwhile, transcriptome analysis revealed that multiple metabolic pathways were affected in A. baumannii.161
Papiliocin is an antimicrobial peptide of 37 amino acid residues belonging to a class of cecropins. It is present in the swallowtail butterfly larvae. It is active against MDR GNB. However, it is responsible for immunogenic potential. Kim et al. designed a short 12-meric antimicrobial peptide by substituting 12 papiliocin (pap12-1) peptide residues at the N-terminal end to increase the antibacterial activity and bacterial membrane interactions. Notably, the designed 12-meric peptides displayed enhanced antibacterial activity by destabilizing and increasing the membrane's permeability and causing leakage of cellular contents. Among these peptides, Pap12-6 (109) (RWKIFKKVVKKW-NH2) inhibited A. baumannii with a MIC value of 4 μM. Moreover, Pap12-6 significantly reduced the bacterial growth in a mouse sepsis model. Also, it significantly reduced inflammatory cytokines.162
Mandel et al. synthesized OMN6 (110, Fig. 11), a cyclic peptide of 40 amino acids. It displayed improved stability and a significant decrease in proteolytic degradation. OMN6 showed potent inhibitory activity against GNB, including A. baumannii, Klebsiella pneumoniae, and Escherichia coli. OMN6 showed antibacterial activity against A. baumannii ACC 00445, A. baumannii ACC 00527, A. baumannii ACC 00535, and A. baumannii ACC 01077 with the same MIC value of 4 μg mL−1, respectively. The mode of action of OMN6 was determined using E. coli. It disrupted the membrane and caused leakage of the cell contents. Moreover, OMN6 did not cause hemolysis of the red blood cells and was nontoxic to the HEK293T cells.163
Peptides D6 and D8 are derived from cecropin–melittin hybrids. D6 and D8 showed antimicrobial activity by binding to the LPS of the bacterial membrane. In combination with antibiotics, these peptides potentiated the effects of vancomycin against a murine model for abscess formation by P. aeruginosa. Also, D6 and D8 comprise D-amino acid residues, which are costlier and are not recognized by mammalian proteases, making them more stable in vivo models. Keeping this in view, Schouten et al. synthesized L forms of peptides L6 (111) (RRLFRRILRWL) and L8 (112) (KRIVQRIKKWLR). The peptides L6 and L8 demonstrated synergistic activity with vancomycin against E. coli, A. baumannii, and K. pneumoniae. Further, L6 potentiated the action of vancomycin in A. baumannii. However, the peptides L6 and L8 were unstable in human serum. Stapled peptides were generated by introducing hydrocarbons, which reduce flexibility in the peptides. The stapled peptide L8S1 potentiated the activity of vancomycin in the in vivo model of A. baumannii infections.164
Protaetiamycine (113) is an AMP present in insects belonging to the defensin class. Earlier studies have identified Pro9-3 (114) (RLWLAIWRR-NH2) and Pro9-3D (rlwlairwrr-NH2) (115) peptides based on the chemical structure of defensins. Further, based on the defensin, 10-meric peptide Pro10-1 and its enantiomer, Pro10-1D, were synthesized by including an Arg residue at the N-terminus of Pro9-3. Krishnan et al. synthesized 9-meric peptides R-Pro9-3 (116) (RRWIALWLR-NH2) and R-Pro9-3D (117) (rrwialwlr-NH2). Pro9-3, Pro9-3D, R-Pro9-3, and R-Pro9-3D inhibited A. baumannii with MIC values of 16, 8, 16, and 8 μM, respectively. Moreover, these peptides showed inhibitory action against CRAB. Also, R-Pro9-3D protected mice infected with the A. baumannii-induced sepsis model. These peptides showed antibacterial activity by interacting with LPS in the outer membrane, causing permeabilization. Furthermore, R-Pro9-3D demonstrated good proteolytic stability and low toxicity.165
CM11 (118) (COOH-WKLFKKILKVL-NH2) is a cecropin A–melittin peptide hybrid. The modification of CM11 led to peptide mCM11 (119) (NH2-WRLFRRILRVL-NH2). The mCM11 peptide showed antibacterial and antibiofilm formation activity. It inhibited MDR A. baumannii 31, PDR A. baumannii 71, XDR A. baumannii 23, and XDR A. baumannii 59, with MIC values of <4, <4, 4, and 4 μg mL−1, respectively. Moreover, mCM11 disrupted the bacterial membrane due to its high arginine content, which enhanced its antimicrobial activity. Furthermore, it demonstrated low hemolytic activity on human RBCs.166
Eales et al. assessed the antibacterial activity of bicarinalin (120) and BP100 (121) against A. baumannii strains. Bicarinalin (120) (KIKIPWGKVKDFLVGGMKAV) is an amphipathic obtained from the venom of the myrmicine ant Tetramorium bicarinatum. It exhibits antibacterial activity against GPB and GNB. Melittin is an alpha-helical peptide isolated from the European honey bee, which is cytotoxic to the cells. To overcome the proteolytic degradation and melittin's toxicity, a peptide BP100 (121) (KKLFKKILKYL) was synthesized. BP100 is a hybrid of cecropin A and melittin. BP100 showed potent antibacterial activity against GNB. Bicarinalin and BP100 inhibited A. baumannii with MIC and MBC values of 4 and 4 μg mL−1, respectively. Bicarinalin and BP100 inhibited biofilm formation in A. baumannii. Notably, A. baumannii treated with bicarinalin and BP100 showed morphological changes in the cells, with pronounced blebbing with variable cell shape, shrinkage, and membrane disruption. Further liposome assays confirmed that these peptides cause membrane disruption by pore-formation.167
HP (2–20) (122) is a protein obtained from the N-terminal region of Helicobacter pylori ribosomal protein L1 (RPL1). The peptides cecropin A, melittin, magainin 2, and HP (2–20) are known for their antibacterial activity. Gopal et al. synthesized four chimeric peptides HPME (123) (AKKVFKRLGIGAVLKVLTTG, a combination of HP 2–9 and ME 1–12), HPMA (124) (AKKVF KRLGIGKFLHSAKKF-NH2, a combination of HP 2–9 and MA 1–12), CAME (125) (KWKLFKKI GIGAVLKVLTTG-NH2, a combination of CA 1–8 and ME 1–12), and CAMA (126) (KWKLFKK IGIGKFLHSAKKF-NH2, a combination of CA 1–8 and MA 1–12). Interestingly, all these four peptides, HPME, HPMA, CAME, and CAMA, exhibited antibacterial activity with MIC values ranging from 3.12–12.5 μM in A. baumannii strains that were resistant to antibiotics, including ampicillin, cefotaxime, ciprofloxacin, tobramycin, and erythromycin. Importantly, these peptides exhibited synergistic activity with the commercial antibiotics and inhibited biofilm formation.168
Mastoparan (127, Fig. 11) (H-INLKALAALAKKIL-NH2) is an AMP that showed potent antibacterial activity against colistin-susceptible and colistin-resistant A. baumannii. However, mastoparan is unstable in serum. To enhance the stability and maintain the antimicrobial activity, Vila-Farrés et al. synthesized mastoparan analogs. Among the synthesized mastoparan analogs, H-inlkalaalakkil-NH2 (128) and Gu-INLKALAALAKKIL-NH2 (129) showed activity against A. baumannii Ab113 with MIC values of 2.7 and 2.6 μM, respectively. In contrast, mastoparan inhibited A. baumannii Ab113 with a MIC value of 2.7 μM. Importantly, mastoparan analogs H-inlkalaalakkil-NH2 and Gu-INLKALAALAKKIL-NH2 were stable for 24 h in human serum, whereas mastoparan was stable for six hours. Also, A. baumannii treated cells with mastoparan and its analogs showed structural damage to the membrane, depicting fractures and causing dye leakage in the dye-assay experiment. This finding suggests that the mode of action of these peptides is by disruption of the bacterial membrane.169
Lycosin-II (130) (VWLSALKFIGKHLAKHQLSKL) is a peptide isolated from the venom of the spider Lycosa singoriensis. It is a linear cationic amphipathic α-helical peptide. Lycosin-II inhibited A. baumannii with MIC values ranging from 3.1–6.3 μM. In the presence of magnesium ions, there is an increase in the MIC of lycosin-II against A. baumannii, which suggests that magnesium might compete with the bacterial cell membrane. However, lycosin-II is unstable in vivo, which limits further development.170
DvAMP (131) (EKKPWARLRFKFKLLKGLAKKMK) is a novel antimicrobial peptide identified by pre-screening the uniport database. A typical α-helical peptide showed 100% homology to Drosophila virilis. It exhibits antifungal activity against Cryptococcus neoformans.171 DvAMP inhibited A. baumannii ATCC19606, A. baumannii CRAB 01, A. baumannii CRAB 02, A. baumannii CRAB 03, A. baumannii CRAB 04, and A. baumannii CRAB 05, with the same MIC value of 8 μg mL−1. DvAMP binds to exogenous lipopolysaccharides and phospholipids of A. baumannii, which results in enhanced membrane permeability, causing the dissipation of proton motive force, reducing intracellular ATP, and increasing ROS levels, ultimately leading to bacterial death. Additionally, it showed antibiofilm activity in A. baumannii. Moreover, it increases the survival rates of mice infected with the A. baumannii pneumonia model by decreasing the bacterial load.172
Luo et al. screened peptides from different species of scorpion, including VsCT1 from Vaejovis subcristatus, BmKn1 from Mesobuthus martensii Karsch, spiniferin from Heterometrus spinifer, Hp1404 from Heterometrus petersii, ctriporin from Chaerilus tricostatus, and Im4 and Im5 from Isometrus maculatus, respectively. Peptides VsCT1 (141) (FLKGIIDTVSNWL), BmKn1 (142) (FIGAVAGLLSKIF), spiniferin (143) (ILGEIWKGIKDIL), Hp1404 (134) (GILGKLWEGVKSIF), ctriporin (144) (FLWGLIPGAISAVTSLIKK), Im4 (145) (FIGMIPGLIGGLISAIK) and Im5 (146) (FLGSLFSIGSKLLPGVIKLFQRKKQ) showed activity against Acinetobacter baumannii ATCC19606 with MIC values of >40, >40, >40, 5, 20, >40, and 2.5 μg mL−1, respectively. Hp1404, ctriporin, and Im5 showed antibacterial activity against a series of A. baumannii clinical isolates resistant to carbapenem. Hp1404, Im5, and ctriporin showed activity against CRAB strains with MIC values of 6.4, 5.0, and 29.1 μg mL−1, respectively. Among these, Hp1404 was better compared to Im5 and ctriporin due to its extensive therapeutic index. It caused hemolysis of red blood cells at very high concentrations. Hp1404 caused dose-dependent disruption of the bacterial cell membrane and lysis of the bacterial membrane.175
Octominin is a defensin protein-3-based AMP obtained from Octopus minor. In O. minor, the prohibitin-2 gene is responsible for the biosynthesis of prohibitin, which inhibits cellular proliferation. Jayathilaka et al. used the prohibitin-2 gene to synthesize octoprohibitin using charged amino acid residues, including arginine and lysine. Octoprohibitin (148) exhibited moderate antibacterial activity against A. baumannii with MIC and MBC values of 50 and 100 μg mL−1, respectively. The field emission electron microscopy analysis of A. baumannii cells treated with octoprohibitin showed prominent and severe damage with opening and shrinkage of the cells. It also inhibited the biofilm formation in A. baumannii and increased the survival of zebrafish infected with A. baumannii.178
The cecropin A (CecA) N terminus is responsible for antibacterial activity against A. baumannii. LysMK34 is prepared from the lysin of A. baumannii phage MK34. Notably, LysMK34 showed a turgor pressure-dependent intrinsic antibacterial activity. Abdelkader et al. prepared a chimeric protein by fusing lysin (LysMK34) with the N-terminus of cecropin A via a linker of three Ala-Gly repeats to give engineered LysMK34 (eLysMK34) (152). eLysMK34 showed antibacterial activity against sensitive and resistant A. baumannii strains. Also, eLysMK34 was active against other GNB, including P. aeruginosa, E. coli, and colistin-resistant strains. Moreover, eLysMK34 was nontoxic to the HaCaT human epithelial cell line. The eLysMK34 antibacterial activity does not depend on high intracellular osmotic pressure. The mode of action of eLysMK34 is due to its combined actions of peptidoglycan degradation and pressure-induced osmotic lysis, eventually killing bacterial cells.183
Yang et al. prepared a peptide-modified lysin PlyA (153) using cecropin A peptide residues 1–8 (KWKLFKKI) and OBPgp279. Importantly, PlyA showed good antibacterial activity against A. baumannii and P. aeruginosa but reduced activity against bacterial cells in their stationary phase. It has been found that the addition of the outer membrane permeabilizers EDTA and citric acid enhances the antibacterial activity against stationary phase cells. However, PlyA gets deactivated in complicated media, including bio-matrices, milk, and serum. This could limit its further development as a therapeutic drug candidate.184
Earlier studies demonstrated that AbEndolysin of phage Ab1656-2 (154) consists of N-terminal N-acetylmuramidase and C-terminal peptidoglycan binding domains. AbEndolysin showed antibacterial activity against clinical A. baumannii strains. Islam et al. synthesized a chimeric protein eAbEndolysin (155) by combining cecropin A with the N-terminus of AbEndolysin. Interestingly, cecropin A-fused AbEndolysin exhibited enhanced bactericidal activity by 2–8 fold against various MDR A. baumannii clinical isolates and more than its parent lysin. Moreover, eAbEndolysin showed synergistic activity with β-lactam antibiotics, including cefotaxime, ceftazidime, and aztreonam, and an additive effect with meropenem and imipenem against A. baumannii. Importantly, eAbEndolysin did not show any toxicity to A549 cell lines and cured mice infected with A. baumannii.185
LysAB2 (156) is endolysin obtained from A. baumannii phage ΦAB2, which showed antibacterial activity against GPB and GNB. Notably, the detailed assessment of LysAB2 revealed that the sequence between 113 and 145 is essential for antimicrobial activity. Peng et al. synthesized four peptides (LysAB2 P0-P3) based on the amino acid sequence present between 113 and 145 of LysAB2. Among them, the representative LysAB2 P3 (157) (NPEKALEKLIAIQKAIKGMLNGWFTGVGFRRKR) showed antibacterial activity against MDR and colistin-resistant A. baumannii strains. LysAB2 P3 caused damage to the bacterial cell membrane, with the collapse of the cellular structure. Furthermore, it decreased the bacterial load in a mouse model infected with A. baumannii infections, and it cured 60% of the mice infected with bacteremia induced by A. baumannii. However, it caused moderate hemolysis of RBCs.186
The C-terminal region of A. baumannii phage lysin LysAB2 has a cationic character and is involved in the permeabilization to the outer membrane. A. baumannii phage lysin PlyF307 and its amino acids between 108 to 138 partly resembled the LysAB2 peptide named P307 (158). Further, P307 was modified with eight amino acids, with a complete C-terminal part of native PlyF307 to give a peptide P307AE-8 (159). Additionally, the fusion of eight amino acids gave P307SQ-8. The derivatives P307 and P307SQ-8 exhibited antibacterial activity against A. baumannii and inhibited biofilm formation. Moreover, P307AE-8 killed the bacteria by disrupting the bacterial plasma membrane. It did not cause hemolysis in human red blood cells.187
The lysin LysP53 (160) is derived from A. baumannii phage 53. It showed the highest similarity of 87% with the endopeptidase derived from Acinetobacter phage DMU1. The time-kill kinetic studies revealed that LysP53 (100 μg mL−1) caused a 4-log reduction in A. baumannii in 15 minutes of exposure. Additionally, it is active against P. aeruginosa, K. pneumoniae, and E. coli. It contained an N-terminal domain of α-helical fragments in the N-terminus of LysP53, especially the N-terminal 33 amino acid sequence. Further, two peptides were generated from LysP53, i.e., P103 (1–17 aa) and P104 (1–33 aa), and these peptides did not show bactericidal activity compared to whole-length peptides. Notably, LysP53 showed no toxicity to A549 cells even at a very high concentration of 500 μg mL−1. Moreover, LysP53 (57.6 μM) upon topical application to a mouse model infected with A. baumannii-associated burn reduced the bacterial loads better than minocycline.182
Artilysins are prepared by an engineered fusion of endolysins with membrane-destabilizing peptides. Using a specific lipopolysaccharide destabilizing peptide facilitates the translocation of the endolysin moiety, which eventually leads to enzymatic degradation of peptidoglycan, resulting in the lysis of the cells. Artilysin Art-175 (161) comprises a modified variant of endolysin KZ144 and a giant P. aeruginosa bacteriophage ΦKZ, linking to the N-terminal of SMAP-29, an antibacterial peptide derived from the sheep myeloid. Art-175 showed bactericidal activity against A. baumannii and P. aeruginosa. It inhibited MDR A. baumannii with MIC values ranging between 4 and 20 μg mL−1 and eliminated a large inoculant of A. baumannii ≥108 CFU mL−1. It also killed the actively dividing and persister cells. The antibacterial mode of action of Art-175 is by osmotic lysis and peptidoglycan degradation.188
2.2. Synthetics and peptidomimetics
 | ||
| Fig. 12 Chemical structure of G3KL and T7 dendrimers. | ||
SA4 (167) (IOWAGOLFOLFO-NH2) is a cationic amphiphilic peptide.198 It inhibits GPB and GNB strains with MIC values ranging between 1 and 32 μg mL−1. The calcein dye leakage and transmission electron microscopy studies confirmed that SA-4 acts on the bacterial membrane and causes perturbation in the membrane.198 Sharma et al. synthesized a poly-N-substituted glycine congener of SA4 and named it SPO (168, Fig. 13). SA4 and SPO exhibited antibacterial activity against A. baumannii strains with MIC values ranging between 50 and 100 μg mL−1 and demonstrated biofilm inhibition activity. Even SPO at a higher concentration of 500 μg mL−1 did not cause hemolysis of the erythrocytes. A. baumannii cells treated with SA4 and SPO showed abnormalities like disrupted cell structures and cellular debris.199
 | ||
| Fig. 13 Chemical structures of SPO, paenipeptin hexanoyl and carboxybenzyl analogs, MSI-78-ADCA, MSI-78-ACA, and macrocyclic tethered peptides. | ||
Paenipeptin is a synthetically derived lipopeptide AMP, consisting of an N-terminal C8 fatty acyl chain with three positive charge amino acids. From the initial study of paenipeptin analogs, two compounds, paenipeptin A and paenipeptin B (169 and 170, Fig. 13), were found to be non-hemolytic. Analog A has a shorter lipid chain of C6 than paenipeptin, whereas analog B has a lipid chain modified with the carboxybenzyl group. Importantly, these two analogs potentiated the activity of clarithromycin and rifampin against carbapenem-resistant K. pneumoniae, A. baumannii, and A. baumannii FDA-CDC AR 0063 strains. Both paenipeptin analogs A and B were stable in 95% human serum for 24 hours. Moreover, 169 and 170 analogs were nontoxic to kidney cells.200
Nagarajan et al. designed AMPs using computational graph optimization and a maximum common subgraph approach. This work led to the design and identification of five peptides, Ω03, Ω13, Ω17, Ω76, and Ω93. Ω76 (171) (FLKAIKKFGKEFKKIGAKLK) displayed antibacterial activity against carbapenem and tigecycline-resistant microorganisms. Ω76 displayed antibacterial activity against E. coli and A. baumannii with the same MBC50 value of 4 mg per liter. In infected mice models of A. baumannii, Ω76 displayed high efficacy against carbapenem and tigecycline-resistant A. baumannii. Interestingly, Ω76 adopts an α-helical structure in the membrane, causing rapid membrane disruption, leakage, and bacterial death.201
β-Lactam antibiotics showed antibacterial activity by inhibiting the bacterial cell wall synthesis. However, the β-lactam antibiotics are deactivated by bacterial β-lactamases. In recent years, linking the core structure of the β-lactam ring and its synthetic modification allows the escape from the β-lactamase, and thus it retains antibacterial activity. 7-Aminodesacetoxycephalosporanic acid is a precursor to β-lactam and cephalosporin antibiotics. MSI-78, CA(1–7)M(2–9)NH2, and des-Chex1-Arg20 are cationic AMPs. Li et al. carried out the chemical modification of 7-aminodesacetoxycephalosporanic acid by linking it to MSI-78, CA(1–7)M(2–9)NH2, and des-Chex1-Arg20. MSI-78 is a modified AMP from the magainin family. Among these, β-lactam antibiotics linked to MSI-78 conjugates MSI-78-ADCA (172Fig. 13) and MSI-78-ACA (173, Fig. 13) have shown significant antibacterial activity against GNB, including K. pneumoniae, A. baumannii, and P. aeruginosa. Moreover, these peptides were found to be nontoxic to mammalian cell lines.202
In a study, He et al. screened cyclic peptides acting against Staphylococcus aureus, E. coli, and A. baumannii. Among the screened peptides, the cyclic peptide B2 (174) (cyclo-RRWWRRW) showed activity against A. baumannii and MDR A. baumannii with the same MIC value of 6 μg mL−1. The antibacterial activity of peptide B2 was retained in salt, including NaCl and CaCl2. However, the peptide B2 exhibited slight toxicity to sheep red blood cells and human lung fibroblast HLF-1 cells. The MDR A. baumannii treated with peptide B2 displayed membrane rupture, full perforations, and the leakage of cellular contents from lysed cells. Moreover, the peptide B2 showed significant therapeutic effects on lung infections in mice infected with MDR A. baumannii. Further analysis suggests that the mode of binding of peptide B2 is by forming a complex with BamA.203
| Category | Compound name | Source | Mode of action | Morphological changes | Targeted A. baumannii | Other targeted bacteria | Ref. |
|---|---|---|---|---|---|---|---|
| Lipopeptides | Polymyxin B (1) | The polymyxin molecule binds to lipid A | Polymyxin and colistin disrupt membrane lipid packing and make the outer membrane permeable | Polymyxin B (1) MIC A. baumannii ATCC19606 = 1 μg mL−1 | P. aeruginosa, K. pneumoniae, E. cloacae | 99 | |
| Polymyxin B (1) MIC A. baumannii FADDI-AB03 = 0.5 μg mL−1 | |||||||
| Polymyxin B (1) MIC A. baumannii ATCC17978 = 1 μg mL−1 | |||||||
| Polymyxin B1 (2) | Polymyxin B1 (2) MIC A. baumannii ATCC19606 = 0.5 μg mL−1 | ||||||
| Polymyxin B1 (2) MIC A. baumannii FADDI-AB03 = 0.5 μg mL−1 | |||||||
| Polymyxin B1 (2) MIC A. baumannii ATCC17978 = 1 μg mL−1 | |||||||
| Polymyxin B2 (3) | Polymyxin B2 (3) MIC A. baumannii ATCC19606 = 1 μg mL−1 | ||||||
| Polymyxin B2 (3) MIC A. baumannii FADDI-AB03 = 1 μg mL−1 | |||||||
| Polymyxin B2 (3) MIC A. baumannii ATCC17978 = 0.5 μg mL−1 | |||||||
| Colistin (4) | Colistin (4) MIC A. baumannii ATCC19606 = 1 μg mL−1 | ||||||
| Colistin (4) MIC A. baumannii FADDI-AB03 = 1 μg mL−1 | |||||||
| Colistin (4) MIC A. baumannii ATCC17978 = 0.5 μg mL−1 | |||||||
| Colistin A (5) | Colistin A (5) MIC A. baumannii ATCC19606 = 2 μg mL−1 | ||||||
| Colistin A (5) MIC A. baumannii FADDI-AB03 = 1 μg mL−1 | |||||||
| Colistin A (5) MIC A. baumannii ATCC17978 = 1 μg mL−1 | |||||||
| Colistin B (6) | Colistin B (6) MIC A. baumannii ATCC19606 = 2 μg mL−1 | ||||||
| Colistin B (6) MIC A. baumannii FADDI-AB03 = 1 μg mL−1 | |||||||
| Colistin B (6) MIC A. baumannii ATCC17978 = 1 μg mL−1 | |||||||
| Lipopeptides | F365 (QPX9003) (7) | Synthetic polymyxin analogs | F365 (QPX9003) (7) MIC A. baumannii ATCC19606 = 0.25 μg mL−1 | P. aeruginosa ATCC 27853, P. aeruginosa FADDI-PA025, P. aeruginosa FADDI-PA038*, K. pneumoniae FADDI-KP032*, K. pneumoniae FADDI-KP065* | 95 | ||
| F365 (QPX9003) (7) MIC A. baumannii FADDI-AB030* = 0.25 μg mL−1 | |||||||
| F365 (QPX9003) (7) MIC A. baumannii FADDI-AB034* = 2 μg mL−1 | |||||||
| Lipopeptides | Polymyxin S2 (ASK0912) (8) | Synthetic polymyxin analogs | Polymyxin S2 (ASK0912) (8) MIC A. baumannii = 0.25 μg mL−1 | 101 | |||
| Lipopeptides | NAB739 (9) | Synthetic polymyxin analogs | NAB739 (9), in combination with rifampin, demonstrated synergistic activity against A. baumannii | E. coli | 102 | ||
| Lipopeptides | Macolacin (10) | Colistin congener | It binds to lipid A | Macolacin (10) MIC A. baumannii 17978 = 1 μg mL−1 |
P. aeruginosa PA01, E. cloacae 0150 | 103 | |
| Lipopeptides | Arylomycins A-C16 (12) | Streptomyces culture | It inhibits essential bacterial type I signal peptidase | Arylomycins A-C16 (12) MIC A. baumannii ATCC 17978 = >64 μg mL−1 | E. coli ATCC 25922, K. pneumoniae ATCC 43816, Enterobacter cloacae ATCC 13407, Enterobacter aerogenes ATCC 13408, Citrobacter werkmanii ATCC 51114, Serratia marcescens CDC4385-74, and P. aeruginosa ATCC 27853 | 104 | |
| Lipopeptides | G0775 (13) | Arylomycins A-C16 derivative | They inhibit essential bacterial type I signal peptidase | G0775 (13) MIC A. baumannii ATCC 17978 = 1 μg mL−1 | E. coli ATCC 25922, K. pneumoniae ATCC 43816, Enterobacter cloacae ATCC 13407, Enterobacter aerogenes ATCC 13408, Citrobacter werkmanii ATCC 51114, Serratia marcescens CDC4385-74, and P. aeruginosa ATCC 27853 | 104 | |
| Lipopeptides | Tridecaptin A1 (14) | It binds to lipid II | It disrupts the inner membrane | Tridecaptin A1 (14) MIC A. baumannii ATCC19606 = 12.5 μg mL−1 | Klebsiella pneumoniae ATCC 13883, E. coli ATCC 25922, S. enterica ATCC 13311, P. aeruginosa ATCC 27853, C. jejuni NCTC 11168 | 107, 108 | |
| Lipopeptides | Tridecaptin analogs 1–8 (19–26) | Tridecaptin analogs 1–8 demonstrated activity against A. baumannii, A. baumannii ACM 11, and A. baumannii ACM 29 with MIC values of 12.5, 25, and 25 μg mL−1, and 25, 100, and 50 μg mL−1, and 100, 50, and 50 μg mL−1, and 50, 25, and 50 μg mL−1, and 12.5, 50, and 50, μg mL−1, and 6.25, 25, and 25, μg mL−1, and 25, 25, and 50 μg mL−1, and >100, >100> and >100 μg mL−1, respectively | E. cloacae, K. pneumoniae, K. pneumoniae IMP 170, and P. pseudoalcaligenes | 111 | |||
| Lipopeptides | Tridecaptin M (27) | Tridecaptin M (27) sensitizes A. baumannii to vancomycin, ceftazidime, and rifampicin against A. baumannii strains | 112 | ||||
| Bacteriocins | Nisin (28) | Lactococcus lactis | It binds to the lipid-II complex | It increases the membrane permeability through pore formation and inhibits peptidoglycan synthesis | Nisin (28), in combination with polymyxin B, demonstrated synergistic activity against pan-drug-resistant and extensively resistant A. baumannii | 114 | |
| Bacteriocins | T16m2 (29) | Nisin derivative | T16m2 (29) MIC A. baumannii LMG01041 = 0.5 μM | E. coli LMG15862, K. pneumoniae LMG20218, P. aeruginosa LMG 6395, E. aerogenes LMG02094, L. lactis MG1363 | 113 | ||
| Lipodepsipeptides | Isopedopeptins B (31) | They cause a significant leakage of cytoplasmic contents and disruption in the bacterial membrane | Isopedopeptins B (31) MIC carbapenem-resistant A. baumannii = 1 μg mL−1 | E. coli, and Klebsiella pneumoniae | 115 | ||
| Lipopeptides | Brevicidine (38) | Brevibacillus laterosporus DSM25 | It interacts with lipopolysaccharides on the outer membrane and phosphatidylglycerol and cardiolipin on the inner membrane of E. coli | It causes the dissipation of proton motive force and membrane disruption | Brevicidine (38) demonstrated synergistic activity with erythromycin against A. baumannii | 116 | |
| Lipopeptides | Brevicidine B (38) | Brevicidine B (38) MIC A. baumannii ATCC 19606 = 16 μg mL−1 | E. coli, P. aeruginosa, and K. pneumoniae | 117 | |||
| Lipopeptides | Laterocidine (46) | Laterocidine (46) MIC A. baumannii = 4 μg mL−1 | E. coli TOP10, P. aeruginosa, K. pneumoniae NRRL-B-408, Enterobacter cloacae NRRL-B-413 | 118 | |||
| Cyclopeptide | DA (47) | It binds to the β-barrel of the outer membrane protein (OMP) BamA | It interfered with the integration of the OMP into the membrane. Thus, BamA cannot close the lateral gate, affecting the entire complex's mechanical, kinetic, and energetic properties | DA (47), D9 (48), and D22 (49) inhibited CRAB isolates A. baumannii 038 (OXA-23), 045 (OXA-58), 046 (OXA-40), 047 (OXA-235), 070 (NDM-1), 054 (OXA-51-ISAba1) with MIC values of 16, 16–32, 8–16, 16–32, 8, and 16–32 μg mL−1, and 4, 8, 4, 4–8, 2, and 8 μg mL−1, and 0.5, 0.5, 0.5, 0.25, 0.0625, and 0.125 μg mL−1, respectively | Citrobacter freundii DSM-30009, E. coli ATC-25922, K. pneumoniae DSM-30104, P. aeruginosa | 119 | |
| Darobactin analogue DA 9 (48) and DA22 (49) | |||||||
| Cyclic lipopeptide | Mixed-ligand daptomycin conjugate (50) | Daptomycin derivative | These conjugates allow membrane permeabilization in GNB | Mixed-ligand daptomycin conjugate (50) MIC A. baumannii ATCC 17961 = 0.4 μM | 124 | ||
| Bis-catechol daptomycin conjugate (51) | Daptomycin derivative | Bis-catechol daptomycin conjugate (51) and Tri-catechol daptomycin conjugate (52) inhibit A. baumannii ATCC 17961, A. baumannii ARC 3484, A. baumannii ARC 3486, A. baumannii ARC 5079, A. baumannii ARC 5081, A. baumannii ATCC 17978, A. baumannii ATCC 17978 pNT165b, A. baumannii ATCC 17978 pNT221, A. baumannii ATCC 17978 pNT255, A. baumannii ATCC 17978 pNT320c with MIC values of 0.4 and 0.2 μM, 1.6 and 3 μM, 0.8 and 3 μM, 3 and 12.5 μM, 50 and 12.5 μM, 1.6 and 0.8 μM, 1.6 and 0.4 μM, 0.8 and 0.4 μM, 1.6 and 0.8 μM, and 1.6 and 0.4 μM, respectively | 124, 125 | ||||
| Tri-catechol daptomycin conjugate (52) | |||||||
| Glycopeptide | VanQAmC10 (53) | Vancomycin derivative | Disruption of biofilm formation | VanQAmC10 (53) MIC A. baumannii = 3.9 to 15.5 μM | S. aureus and Salmonella typhimurium | 129 | |
| Glycopeptide | Vancomycin sulfonium conjugates (54) | Vancomycin derivative | Disruption of the bacterial membrane | Vancomycin sulfonium conjugates (54) MIC A. baumannii = 4–8 μg mL−1 | E. coli, K. pneumoniae, P. aeruginosa, and M. catarrhalis | 130 | |
| Linear peptide | Paenimucillin C (56) | Paenibacillus mucilaginosus K02 | It showed antibacterial activity through pore formation and caused leakage of cytoplasmic contents | Paenimucillin C (56) inhibited MDR A. baumannii 1788, A. baumannii 1790, A. baumannii 1791, A. baumannii 1795, A. baumannii 1797, A. baumannii S3, and A. baumannii S5 with MIC values of 4, 4, 2, 4, 4, 4, and 4 μg mL−1, respectively | 132 | ||
| Oligopeptide | X33 (57) | Streptomyces lavendulae X33 | It causes cell wall permeability and morphology changes and promotes cell content leakage | X33 (57) MIC A. baumannii 1788 = 78.13 μg mL−1 | 133 | ||
| Cationic peptide | Lynronne-1 (58) | Bovine rumen microbiome | It causes bacterial membrane lysis | Lynronne-1 (58) MIC A. baumannii ATCC 19606 = 16–8 μg mL−1 | S. aureus ATCC 29213, and S. aureus ATCC 6538 | 134 | |
| Cathelicidin | LL-37 (59) | Produced from hCAP-18 | LL-37 binds to OmpA (AbOmpA) protein | LL-37 (59) MIC pan-drug-resistant A. baumannii = 32–64 μg mL−1 | 135–138 | ||
| LL-37, in combination with oncorhyncin II, showed synergistic activity and demonstrated rapid killing of A. baumannii | |||||||
| Cathelicidin | LL/CAP18 (60) | LL-37 derivative | LL/CAP18 (60) MIC pan-drug-resistant A. baumannii = 32–64 μg mL−1 | 136, 137 | |||
| Cathelicidin | FF/CAP18 (61) | LL-37 derivative | FF/CAP18 (61) MIC pan-drug-resistant A. baumannii = 8–16 μg mL−1 | 136, 137 | |||
| Cathelicidin | P-10 (62) | LL-37 derivative | P-10 (62), in combination with nisin, showed synergistic activity against extensively drug-resistant A. baumannii | P-10, in combination with nisin, is also active against colistin-resistant P. aeruginosa | 139 | ||
| Cathelicidin | HLF (1–11) (63) | Produced from human lactoferrin | HLF (63) is active against in vitro and in vivo models of A. baumannii infections | MDR Staphylococcus aureus | 140 | ||
| Defensins | Synthetic β-defensin-2 (64) | Synthetic β-defensin-2 (64) MIC MDR A. baumannii vLD90 = 3.25–4.5 μg mL−1 | Pseudomonas aeruginosa, Enterococcus faecalis, Enterococcus faecium and Staphylococcus aureus | 143 | |||
| Synthetic β-defensin-2 (64) MIC wild-type A. baumannii vLD90 = 3.90–9.35 μg mL−1 | |||||||
| Apolipoprotein E (ApoE) | COG1410 (65) | Apolipoprotein E mimetic | It disrupts the plasma membrane, leaking the cytoplasmic contents and inhibiting biofilm formation | COG1410 (65) MIC A. baumannii ATCC19606 = 16 μg mL−1. | 144 | ||
| COG1410 (65) MIC PDR A. baumannii YQ4 = 16 μg mL−1 | |||||||
| COG1410 exhibited synergistic activity with polymyxin B and cured C. elegans infected with PDR Acinetobacter baumannii | |||||||
| Cathelicidin | BMAP-28 (66) | BMAP-28 (66) MIC pan-drug-resistant A. baumannii ATCC19606 = 5 μg mL−1 | 145 | ||||
| Cathelicidin | A837 (67) | BMAP-28 and its analogs bind to the outer membrane protein OmpA (AbOmpA) of A. baumannii | 67, 68, 69, and 70 inhibited pan-drug-resistant A. baumannii ATCC19606 10, 5–10, 5–10, 5–10 μg mL−1, and clinical isolates of pan-drug-resistant A. baumannii strains with MIC values of 5–10, 20, 5–220, 5–20, and 5–20 μg mL−1, respectively | 145 | |||
| A838 (68) | |||||||
| A839 (69) and A840 (70) | |||||||
| Cathelicidin | Bac7 (71) | Bac7 (71) MIC A. baumannii ATCC19606 = ≤8 μM | 146 | ||||
| Cathelicidin | Bac5-258 (72),Bac5-272 (73), Bac5-278 (74), Bac5-281 (75), and Bac5-291 (76) |
Bac5 analogues | Peptides cause protein synthesis inhibition and bacterial membrane destabilization | 72, 73, 74, 75 and 76 MIC A. baumannii ATCC19606 = >64, 64, 8, 4, 2, and 8 μM, respectively | 147 | ||
| Cathelicidin | Cathelicidin-BF-a4 (ZY4) (81) | Cathelicidin-BF derivative | It induces membrane permeabilization and disruption of the membrane, causing pore formation | Cathelicidin-BF-a4 (81) demonstrated antibacterial activity against GNB, including P. aeruginosa strains and A. baumannii isolates, with MIC ranging between 2.0 and 4.5 μg mL−1 and 4.6 and 9.4 μg mL−1, respectively | 149 | ||
| Defensin | Am23SK (85) | Crocodylian β-defensin synthetic analogue | Am23SK (85) displayed antibacterial activity against Salmonella enterica serovar Typhimurium ATCC 14028, Staphylococcus aureus SAP0017, Enterobacter cloacae 218R1, and A. baumannii Ab5075 with MIC values of 16, 16, 8, and 2 μg mL−1, respectively | Salmonella enterica serovar Typhimurium ATCC 14028, Staphylococcus aureus SAP0017, and Enterobacter cloacae 218R1 | 150 | ||
| Apolipoprotein | Apo5 (87), Apo6 (88), and A1P (89) | Alligator mississippiensis | Apo5 and Apo6 induced membrane disruption | 87, 88, and 89 inhibited A. baumannii ATCC 9955 and A. baumannii ATCC BAA-1794 with EC50 values of 0.644 and 0.234 μg mL−1, 0.233 and 0.126 μg mL−1, and 224.0 and 23.6 μg mL−1, respectively | S. aureus, Escherichia coli, and P. aeruginosa | 151 | |
| Linear peptide | 2K4L (90) | Temporin-1CEc analogs derived from Rana chensinensis skin secretion | 2K4L demonstrated interactions with lipopolysaccharides | They cause membrane disruption | 2K4L(90) and 2K2L (91) showed activity against A. baumannii MRAB 0227 and A. baumannii 22933 with MIC values of 6.25 and 3.13 μM and 25 and 12.5 μM, respectively |
152 | |
| 2K2L (91) | |||||||
| Hylin a1-11K (93) | Hylin a1 analogue | Hylin a1-11K and Hylin a1-15K bind to the lipopolysaccharide | These peptides disrupt the membrane | Hylin a1-11K (93) and hylin a1-15K (94) exhibited broad-spectrum antimicrobial activity and anti-biofilm activity against carbapenem-resistant A. baumannii | 153 | ||
| Hylin a1-15K (94) | |||||||
| AP19 (95) | A hybrid of cathelicidin (P7) and aurein (A3) | The D-form of AP19 causes membrane disruption and leakage of cytoplasmic contents | The AP19 (95) D-form exhibited antibacterial activity against MDR and XDR clinical isolates of A. baumannii | 154 | |||
| Esculentin-1a (96) | Esculentin-1a, in combination with colistin, enhanced the membrane perturbation effects | Esculentin-1a, in combination with colistin, demonstrated synergistic activity against MDR A. baumannii clinical isolates | 155 | ||||
| Δ-Myrtoxin-Mp1a (97) and its analogues (98–104) | Mp1a is obtained from the venom of Myrmecia pilosula | Mp1a and its derivatives demonstrated potent cell membrane binding and disrupted the cytoplasmic membrane | 97–104 MIC A. baumannii ATCC 19606 = 0.025, 0.015–0.03, 0.015–0.03, 0.015–0.03, 0.05–0.1, 0.025–0.1, 0.2, and 0.4–0.8 μM, respectively | 156 | |||
| LyeTx I mnΔK (106) | LyeTx I mnΔK is derived from LyeTx I AMP | LyeTx I mnΔK disrupts the bacterial cell membrane and causes leakage of cell contents, reducing the preformed biofilms. | LyeTx I mnΔK (106) MIC carbapenem-resistant A. baumannii AC10 = 2 μM | 157 | |||
| Cecropin | Cec4 (108) | Cec4 is derived from cecropin, a peptide from Musca domestica | Cec4 treatment of A. baumannii led to the destruction of the bacterial membrane and caused leakage of the cytoplasmic contents | Cec4 (108) demonstrated activity against A. baumannii (ATCC 19606), Multidrug-resistance A. baumannii and extensive drug-resistant A. baumannii (PRAB) with the same MIC value of 4 μg mL−1 | 160, 161 | ||
| Cecropin | Pap12-6 (109) | Pap12-6 derived from papiliocin | It destabilizes and increases the membrane's permeability. Also, it causes leakage of cellular contents | Pap12-6 (109) inhibited E. coli, P. aeruginosa, A. baumannii, and S. typhimurium, with MIC values of 4, 4, 4, and 8 μM, respectively | E. coli, P. aeruginosa, S. typhimurium | 162 | |
| OMN6 (110) | OMN6 causes membrane disruption and leakage of cellular contents in E. coli | OMN6 (110) inhibited A. baumannii with a 4 μg mL−1 MIC value | Klebsiella pneumoniae and Escherichia coli | 163 | |||
| L6 (111) and L8 (112) | L6 and L8 are formed from the L-form of D6 and D8. Notably, D6 and D8 are derived from cecropin–melittin hybrids | D6 and D8 bind to the LPS of the bacterial membrane | L6 (111) and L8 (112) synergized with vancomycin in A. baumannii | 164 | |||
| Defensin | Pro9-3 (114), Pro9-3D (115), R-Pro9-3 (116), and R-Pro9-3D (117) | Pro9-3, Pro9-3D, R-Pro9-3, and R-Pro9-3D are derived from protaetiamycine | These peptides interact with LPS in the outer membrane, causing permeabilization | 114–117 MIC A. baumannii = 16, 8, 16, and 8 μM, respectively | 165 | ||
| mCM11 (119) | mCM11 is derived from CM11 | mCM11 disrupts the membrane | mCM11 (119) inhibited MDR A. baumannii 31, PDR A. baumannii 71, XDR A. baumannii 23, and XDR A. baumannii 59, with MIC values of <4, <4, 4, and 4 μg mL−1, respectively | 166 | |||
| Bicarinalin (120) | Bicarinalin is obtained from the venom of Tetramorium bicarinatum | Bicarinalin and BP100 demonstrated morphological changes in the cells, with pronounced blebbing, variable cell shape, and shrinkage. Also, thy induces membrane disruption | Bicarinalin (120) and BP100 (121) inhibited the A. baumannii with MIC and MBC values of 4 and 4 μg mL−1, respectively | 167 | |||
| BP100 (121) | |||||||
| BP100 is a hybrid of cecropin A and melittin | |||||||
| HPME (123) | HPME is derived from HP 2–9, and ME 1–12, HPMA is from HP 2–9 and MA 1–12, CAME is from CA 1–8 and ME 1–12, and CAMA is from CA 1–8 and MA 1–12, respectively | HPME (123), HPMA (124), CAME (125), and CAMA (126) exhibited antibacterial activity with MIC values ranging from 3.12–12.5 μM in A. baumannii strains | 168 | ||||
| HPMA (124) | |||||||
| CAME (125) | |||||||
| CAMA (126) | |||||||
| Mastoparan (127) and its analogues | Mastoparan and its analogs cause structural damage to the bacterial membrane and cause dye leakage in dye-assay experiments | Mastoparan (127), H-inlkalaalakkil-NH2 (128), and Gu-INLKALAALAKKIL-NH2 (129) inhibit A. baumannii Ab113 with MIC values of 2.7, 2.7, and 2.6 μM, respectively | 169 | ||||
| H-inlkalaalakkil-NH2 (128) and Gu-INLKALAALAKKIL-NH2 (129) | |||||||
| Linear peptide | Lycosin-II (130) | From venom of Lycosa singoriensis | Lycosin-II (140) competes with the bacterial membrane | Lycosin-II (130) showed broad-spectrum antibacterial activity against E. coli, K. pneumoniae, Streptococcus pyogenes, Staphylococcus saprophyticus, Staphylococcus epidermidis, Pseudomonas aeruginosa, Viridans streptococci, S. aureus, and A. baumannii with MIC values of 12.5, 50, 50, 3.1, 3.1, 12.5, 3.1, 3.1, and 3.1–6.3 μM, respectively | E. coli, K. pneumoniae, Streptococcus pyogenes, Staphylococcus saprophyticus, Staphylococcus epidermidis, Pseudomonas aeruginosa, Viridans streptococci, and S. aureus | 170 | |
| DvAMP (131) | It showed 100% homology to drosophila virilis | It binds to exogenous lipopolysaccharides and phospholipids of A. baumannii | It enhanced membrane permeability, causing the dissipation of proton motive force, reducing intracellular ATP, and increasing ROS levels, ultimately leading to bacterial death | DvAMP (131) showed broad-spectrum antibacterial activity and inhibited E. coli ATCC25922, P. aeruginosa CMCC10104, K. pneumoniae ATCC70060, S. aureus ATCC25923, A. baumannii ATCC19606, A. baumannii CRAB 01, A. baumannii CRAB 02, A. baumannii CRAB 03, A. baumannii CRAB 04, and A. baumannii CRAB 05, with MIC values of 8, 16, 16, 64, 8, 8, 8, 8, 8, and 8 μg mL−1, respectively | 172 | ||
| Arenicin | AA139 (133) | Derived from arenicin-3 | It causes membrane disruption and leakage of cytoplasmic contents in P. aeruginosa | AA139 (133) inhibited E. coli ATCC 25922, K. pneumoniae BAA-2146 (NDM-1), non-MDR A. baumannii, MDR A. baumannii, and XDR A. baumannii with MIC values of 0.125, 1, 2, 1, and 0.5 μg mL−1, respectively | E. coli ATCC 25922, K. pneumoniae BAA-2146 (NDM-1) | 173 | |
| Hp1404 (134), and its analogs Hp1404-V (137), Hp1404-L (138), Hp1404-I (139), Hp1404-W (140) | These peptides cause bacterial membrane destruction and inhibition of biofilm formation. | Hp1404 (134), and its analogs Hp1404-V (137), Hp1404-L (138), Hp1404-I (139), and Hp1404-W (140) inhibited A. baumannii 409081, A. baumannii 719705, and A. baumannii 907233 with MIC values of 3.13, 6.25, 3.13, 3.13, and 3.13 μg mL−1, and 3.13, 12.5, 3.13, 6.25, and 3.13, and 3.13, 6.25, 3.13, and 3.13 μg mL−1, respectively |
Hp1404 (134) also inhibits the growth of Staphylococcus aureus | 174, 175 | |||
| Im5 (146) | Isometrus maculatus | Im5 (146) MIC Acinetobacter baumannii ATCC19606 = 2.5 μg mL−1 | 175 | ||||
| Octopromycin (147) | Octopus minor | Octopromycin causes severe cell wall damage. It forms irregularly shaped cells and increases ROS generation inside the cells | Octopromycin (147) MIC Acinetobacter baumannii = 50 μg mL−1 | 177 | |||
| Octoprohibitin (148) | Synthesized from the prohibitin-2 gene | It causes severe damage with opening and shrinkage of cells | Octoprohibitin (148) MIC Acinetobacter baumannii = 50 μg mL−1 | 178 | |||
| TP2-5 (150) and TP2-6 (151) | Derived from tilapia piscidin 2 (TP2) | These peptides cause membrane depolarization with deformed cells | TP2-5 (150) and TP2-6 (151) inhibited sensitive and MDR isolates, including A. baumannii 10591, A. baumannii 14B0091, A. baumannii 2088, A. baumannii 921, A. baumannii 1019, and A. baumannii 1033 with the same MIC value of 3.125 μg mL−1 |
179 | |||
| LysMK34 (eLysMK34) (152) | A chimeric protein having a fusion of lysin and cecropin A | It causes peptidoglycan degradation and pressure-induced osmotic lysis | eLysMK34 (152) inhibited sensitive and resistant strains A. baumannii MK34, A. baumannii RUH134, A. baumannii Greek46, A. baumannii Greek47, and A. baumannii NCTC13423 with MIC values of 0.76, 0.76, 0.76, 1.2, and 0.45 μM, respectively | P. aeruginosa, E. coli | 183 | ||
| PlyA (153) | It is derived from cecropin A, and OBPgp279 | PlyA (153) showed potent antibacterial activities against A. baumannii | P. aeruginosa | 184 | |||
| eAbEndolysin (155) | A chimeric protein derived from cecropin A fusion with the AbEndolysin N-terminus | eAbEndolysin (154) showed potent antibacterial activity against MDR A. baumannii | 185 | ||||
| LysAB2 P3 (156) | Derived from LysAB2 | LysAB2 P3 causes damage to bacterial cell walls and the collapse of the cellular structure | LysAB2 P3 (156) showed antibacterial activity against MDR and colistin-resistant strains. It inhibited A. baumannii ATCCC17978, A. baumannii ATCCC19606, A. baumannii M3237, A. baumannii M6337, A. baumannii M105656, A. baumannii M2925, and A. baumannii M17720, with MIC and MBC values of 4 and 8, 8 and 8, 16 and 16, 4 and 4, 8 and 8, 8 and 8, and 8 and 8 μM, respectively | 186 | |||
| P307 (158), and P307AE-8 (159) | Derived from LysAB2 | P307 (158) and P307SQ-8 (159) exhibited antibacterial activity against A. baumannii | 187 | ||||
| LysP53 (160) | Derived from A. baumannii phage 53 | LysP53 (100 μg mL−1) caused a 4-log reduction in A. baumannii in 15 minutes of exposure | P. aeruginosa, K. pneumoniae, and E. coli | 182 | |||
| Artilysin Art-175 (161) | This is an engineered fusion of endolysin KZ144 linked to SMAP-29 | It causes enzymatic degradation of the peptidoglycan and osmotic lysis | Artilysin Art-175 (161) inhibits MDR A. baumannii with MIC values ranging between 4 and 20 μg mL−1 | P. aeruginosa | 188 | ||
| Lys1S-L9P (162) | Lys1S-L9P (162) showed potent activity against MDR A. baumannii | E. coli, and P. aeruginosa | 190 | ||||
| Guavanin 2 (163) | Derived from glycine-rich peptide Pg-AMP1 | It triggers hyperpolarization and causes membrane disruption in the bacterial cells | Guavanin 2 (163) inhibited Escherichia coli ATCC 25922, Pseudomonas aeruginosa ATCC 27853, Acinetobacter baumannii ATCC 19606, and Klebsiella pneumoniae with 6.25, 25, 6.25, and 80 μg mL−1, respectively | Escherichia coli ATCC 25922, Pseudomonas aeruginosa ATCC 27853, and Klebsiella pneumoniae | 191 | ||
| Dendrimer | G3KL (164) | T7 causes membrane disruption activity in bacterial cells | G3KL (164) inhibited A. baumannii (10 OXA-23, 7 OXA-24, and 11 OXA-58 carbapenemase producers) and P. aeruginosa (18 VIM and 3 IMP carbapenemase producers) strains with MIC values ranging from 4–16 and 4–32 μg mL−1, and MBC values ranging from 4–16 and 4–128 μg mL−1, respectively | P. aeruginosa | 194–196 | ||
| T7 (165) | |||||||
| T7 (165) showed broad-spectrum activity against Gram-negative MDR strains. It inhibited A. baumannii ATCC 19606, E. coli bla OXA-48, E. coli W3110, P. aeruginosa ZEM-9A, P. aeruginosa ZEM-1A, E. cloacae bla NDM, K. pneumoniae NCTC 418, and K. pneumoniae bla OXA-48 with MIC values of 8, 8–16, 8, 8, 8, 8, 8–16, 16, and 16 μg mL−1 | E. coli bla OXA-48, E. coli W3110, P. aeruginosa ZEM-9A, P. aeruginosa ZEM-1A, E. cloacae Bla NDM, K. pneumoniae NCTC 418, K. pneumoniae Bla OXA-48 | ||||||
| Chimeric peptide | TAT-RasGAP317–326 (166) | A hybrid of TAT48–57 and Src homology 3 domain | Biofilm inhibition | TAT-RasGAP317–326 (166) inhibited A. baumannii ATCC 19606 with a MIC value of 8 μg mL−1 | P. aeruginosa PA14 | 197 | |
| SA4 (167) | Membrane perturbation | SA4 (167) and SPO (168) inhibited A. baumannii strains with MIC values ranging between 50 and 100 μg mL−1 | 198, 199 | ||||
| SPO (168) | |||||||
| Paenipeptin hexanoyl (169) | Derived from paenipeptin | 169 and 170 potentiated the activity of clarithromycin and rifampin against carbapenem-resistant A. baumannii and K. pneumoniae strains | 200 | ||||
| Paenipeptin carboxybenzyl (170) | |||||||
| Ω76 (171) | It causes rapid membrane disruption, leakage, and bacterial death | Ω76 (171) MIC A. baumannii = 4 mg per liter | E. coli | 201 | |||
| MSI-78-ADCA (172) and MSI-78-ACA (173) | MSI-78-ADCA (172) and MSI-78-ACA (173) showed significant antibacterial activity against K. pneumoniae, A. baumannii, P. aeruginosa, and A. baumannii 156 with MIC values of 4.6 and 3.4, 3.6 and 3.7, 2.9 and 3.1, and 4.2 and 9.6 μM, respectively | K. pneumoniae and P. aeruginosa | 202 | ||||
| B2 (174) | It causes membrane rupture, perforations, and the leakage of cellular contents from lysed cells | B2 (174) inhibited A. baumannii and MDR A. baumannii with the same MIC value of 6 μg mL−1 | 203 | ||||
| RO7075573 (176), RO7055137 (177), and zosurabalpin (179) | Zosurabalpin inhibited the inner membrane LptB2FGC complex to block the LPS transport in A. baylyi | RO7075573 (176), RO7055137 (177), and zosurabalpin (179) inhibited A. baumannii ATCC 17978, A. baumannii ATCC 19606, and A. baumannii (10 MDR isolates) with MIC values of ≤0.06, 0.12, and 0.5 (≤0.06–0.5) mg L−1, ≤0.06, ≤0.06, and 0.12 (≤0.06–0.12) mg L−1, and ≤0.06, 0.25, and 0.25 (0.12–1) mg L−1, respectively | 204 |
3. Conclusion
A. baumannii is on the list of nosocomial ESKAPE pathogens of the WHO, and there is an urgent need for research to discover antibiotics to tackle its infections. Several antibiotics, including carbapenems, aminoglycosides, fluoroquinolones, cephalosporins, and β-lactamase inhibitors, are used to treat A. baumannii infections. However, in recent decades, abuse and misuse of antibiotics to treat other diseases other than the mentioned use led to the rapid emergence of MDR A. baumannii, consequently leading to an increase in the MICs of prescribed antibiotics. The emergence of MDR A. baumannii, XDR A. baumannii, and PDR A. baumannii strains significantly affect health, and it is a threat to human life and health and is responsible for a significant economic burden to society. Several targets, including ribosomes, DNA gyrase, and the cell wall, have already been explored in A. baumannii to control its growth. However, targeting the cell wall and its components is one of the primary focuses of antibiotics research, and it continues to hold the center stage in antibiotics discovery, as cell wall targeting antibiotics are less likely to acquire mutation-based resistance. Moreover, membrane-targeting antibiotics do not have to cross the plasma membrane to exhibit their activity. Among them, AMPs are next-generation membrane-targeting antibiotics and exhibit broad-spectrum activity against GNB, GPB, and related clinical-resistant strains. Moreover, most AMPs act on the membrane targets and other generalized cytoplasmic targets, and some of them activate the human immune response for the effective eradication of pathogens from the body. This paper discussed AMPs from natural or synthetic origins and their congeners acting through membrane disruption of A. baumannii.Abbreviations
| AbaR | Antibiotic resistance island |
| Acb | Acinetobacter calcoaceticus–Acinetobacter baumannii |
| ADC | Acinetobacter-derived cephalosporinase |
| AMPs | Antimicrobial peptides |
| ApoE | Apolipoprotein E |
| BAP | Biofilm-associated protein |
| CecA | Cecropin A |
| CLSM | Confocal laser scanning microscopy |
| CRAB | Carbapenem-resistant A. baumannii |
| Dab, L-2 | 4-Diaminobutyric acid |
| Dpa | Diaminopropane acetyltransferase |
| ELISA | Enzyme-linked immunosorbent assays |
| ESKAPE | (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter spp.) |
| GlcNAc | N-Acetylglucosamine |
| hBD-1 | Human β-defensin-1 |
| hBD-2 | Human β-defensin-2 |
| hBD-3 | Human β-defensin-3 |
| hBD-4 | Human β-defensin-4 |
| HD5 | Human defensin 5 |
| Hst5 | Histatin 5 |
| Kdo | Ketodeoxyoctanoic acid |
| LPS | Lipopolysaccharides |
| LpsB | Glycosyltransferase |
| LOS | Lipooligosaccharides |
| LPLATs | Lysophospholipid acyltransferase |
| MBEC | Minimum biofilm eradication concentration |
| MBIC90 | Minimum biofilm inhibition concentration |
| MCP | Tethered macrocyclic peptide |
| MDR | Multidrug-resistant |
| MICs | Minimum inhibitory concentrations |
| MurNAc | N-Acetylmuramic acid |
| OMP | Outer membrane protein |
| OMR | Outer membrane receptors |
| OMVs | Outer membrane vesicles |
| PBPs | Penicillin-binding proteins |
| PDR | Pan-drug-resistant |
| PMR | Polymyxin-resistant |
| Pr-AMPs | Proline-rich peptides |
| qPCR | Quantitative polymerase chain reaction |
| SEM | Scanning electron microscopy |
| SPase | Signal peptidase |
| syn-BNP | Synthetic-bioinformatic natural product |
| TCA | Tricarboxylic acid cycle |
| TLR4 | Toll-like receptor 4 |
| WHO | World Health Organization |
| XDR | Extensively drug-resistant |
| XDR-ABC | Drug-resistant Acinetobacter baumannii–calcoaceticus complex |
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
GK thanks the Director of Birla Institute of Technology and Science Pilani, Pilani Campus, Rajasthan for providing the necessary facilities to carry out the research work.References
- A. Gedefie, W. Demsis, M. Ashagrie, Y. Kassa, M. Tesfaye, M. Tilahun, H. Bisetegn and Z. Sahle, Infect. Drug Resist., 2021, 14, 3711–3719 CrossRef PubMed.
- A. Nemec, L. Krizova, M. Maixnerova, O. Sedo, S. Brisse and P. G. Higgins, Int. J. Syst. Evol. Microbiol., 2015, 65, 934–942 CrossRef CAS PubMed.
- N. Bagińska, M. Cieślik, A. Górski and E. Jończyk-Matysiak, Antibiotics, 2021, 10, 1–15 CrossRef.
- T. Ballouz, J. Aridi, C. Afif, J. Irani, C. Lakis, R. Nasreddine and E. Azar, Front. Cell. Infect. Microbiol., 2017, 7, 1–8 CrossRef.
- M. Asif, I. A. Alvi and S. U. Rehman, Infect. Drug Resist., 2018, 11, 1249–1260 CrossRef CAS PubMed.
- N. Pakharukova, M. Tuittila, S. Paavilainen, H. Malmi, O. Parilova, S. Teneberg, S. D. Knight and A. V. Zavialov, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 5558–5563 CrossRef CAS.
- C. M. Harding, S. W. Hennon and M. F. Feldman, Nat. Rev. Microbiol., 2018, 16, 91–102 CrossRef CAS PubMed.
- R. Vázquez-López, S. G. Solano-Gálvez, J. J. Juárez Vignon-Whaley, J. A. Abello Vaamonde, L. A. Padró Alonzo, A. Rivera Reséndiz, M. Muleiro Álvarez, E. N. Vega López, G. Franyuti-Kelly, D. A. Álvarez-Hernández, V. Moncaleano Guzmán, J. E. Juárez Bañuelos, J. Marcos Felix, J. A. González Barrios and T. Barrientos Fortes, Antibiotics, 2020, 9, 1–22 CrossRef PubMed.
- C.-R. Lee, J. H. Lee, M. Park, K. S. Park, I. K. Bae, Y. B. Kim, C.-J. Cha, B. C. Jeong and S. H. Lee, Front. Cell. Infect. Microbiol., 2017, 7, 1–35 Search PubMed.
- F. C. Morris, C. Dexter, X. Kostoulias, M. I. Uddin and A. Y. Peleg, Front. Microbiol., 2019, 10, 1–20 Search PubMed.
- J. M. Boll, A. T. Tucker, D. R. Klein, A. M. Beltran, J. S. Brodbelt, B. W. Davies and M. S. Trent, MBio, 2015, 6, 1–11 Search PubMed.
- C. Y. Chin, K. A. Tipton, M. Farokhyfar, E. M. Burd, D. S. Weiss and P. N. Rather, Nat. Microbiol., 2018, 3, 563–569 Search PubMed.
- L. Semenec, A. K. Cain, C. J. Dawson, Q. Liu, H. Dinh, H. Lott, A. Penesyan, R. Maharjan, F. L. Short, K. A. Hassan and I. T. Paulsen, Nat. Commun., 2023, 14, 1–18 Search PubMed.
- B. Shin, C. Park and W. Park, Appl. Microbiol. Biotechnol., 2020, 104, 1259–1271 Search PubMed.
- J. Armalytė, A. Čepauskas, G. Šakalytė, J. Martinkus, J. Skerniškytė, C. Martens, E. Sužiedėlienė, A. Garcia-Pino and D. Jurėnas, Nat. Commun., 2023, 14, 1–11 Search PubMed.
- S. Roy, G. Chowdhury, A. K. Mukhopadhyay, S. Dutta and S. Basu, Front. Med., 2022, 9, 1–32 Search PubMed.
- E. De Gregorio, E. Roscetto, V. D. Iula, M. Martinucci, R. Zarrilli, P. P. Di Nocera and M. R. Catania, New Microbiol., 2015, 38, 251–257 CAS.
- C.-H. Su, M.-H. Tsai, C.-Y. Lin, Y.-D. Ma, C.-H. Wang, Y.-D. Chung and G.-B. Lee, Biosens. Bioelectron., 2020, 159, 1–7 CrossRef PubMed.
- J. Luo, M. Jiang, J. Xiong, J. Li, X. Zhang, H. Wei and J. Yu, Anal. Chim. Acta, 2018, 1044, 147–153 CrossRef CAS PubMed.
- C. Ayoub Moubareck and D. Hammoudi Halat, Antibiotics, 2020, 9, 1–29 CrossRef.
- G. Kumar and S. Kapoor, Bioorg. Med. Chem., 2023, 81, 117212 CrossRef CAS.
- K. B. Steinbuch and M. Fridman, MedChemComm, 2016, 7, 86–102 RSC.
- M. Zahn, S. P. Bhamidimarri, A. Baslé, M. Winterhalter and B. van den Berg, Structure, 2016, 24, 221–231 CrossRef CAS.
- L. M. VanOtterloo, L. A. Macias, M. J. Powers, J. S. Brodbelt and M. S. Trent, MBio, 2024, 15, 1–20 CrossRef CAS PubMed.
- E. Geisinger, W. Huo, J. Hernandez-Bird and R. R. Isberg, Annu. Rev. Microbiol., 2019, 73, 481–506 CrossRef CAS.
- J. M. Boll, A. A. Crofts, K. Peters, V. Cattoir, W. Vollmer, B. W. Davies and M. S. Trent, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E6228–E6237 CrossRef CAS.
- B. W. Simpson, M. Nieckarz, V. Pinedo, A. B. McLean, F. Cava and M. S. Trent, MBio, 2021, 12, 1–19 CrossRef.
- D. Dovala, C. M. Rath, Q. Hu, W. S. Sawyer, S. Shia, R. A. Elling, M. S. Knapp and L. E. Metzger, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E6064–E6071 CrossRef CAS PubMed.
- M. J. Powers and M. S. Trent, Mol. Microbiol., 2018, 107, 47–56 CrossRef CAS PubMed.
- D. Nie, Y. Hu, Z. Chen, M. Li, Z. Hou, X. Luo, X. Mao and X. Xue, J. Biomed. Sci., 2020, 27, 1–8 CrossRef PubMed.
- C. Whiteway, A. Breine, C. Philippe and C. Van der Henst, Trends Microbiol., 2022, 30, 199–200 CrossRef CAS PubMed.
- W. F. Penwell, A. B. Shapiro, R. A. Giacobbe, R.-F. Gu, N. Gao, J. Thresher, R. E. McLaughlin, M. D. Huband, B. L. M. DeJonge, D. E. Ehmann and A. A. Miller, Antimicrob. Agents Chemother., 2015, 59, 1680–1689 CrossRef PubMed.
- Y. Doi, G. Murray and A. Peleg, Semin. Respir. Crit. Care Med., 2015, 36, 085–098 CrossRef PubMed.
- H. Chen, Q. Liu, Z. Chen and C. Li, J. Infect. Chemother., 2017, 23, 278–285 CrossRef CAS PubMed.
- T. F. Durand-Réville, S. Guler, J. Comita-Prevoir, B. Chen, N. Bifulco, H. Huynh, S. Lahiri, A. B. Shapiro, S. M. McLeod, N. M. Carter, S. H. Moussa, C. Velez-Vega, N. B. Olivier, R. McLaughlin, N. Gao, J. Thresher, T. Palmer, B. Andrews, R. A. Giacobbe, J. V. Newman, D. E. Ehmann, B. de Jonge, J. O'Donnell, J. P. Mueller, R. A. Tommasi and A. A. Miller, Nat. Microbiol., 2017, 2, 1–10 Search PubMed.
- M. I. El-Gamal, I. Brahim, N. Hisham, R. Aladdin, H. Mohammed and A. Bahaaeldin, Eur. J. Med. Chem., 2017, 131, 185–195 CrossRef CAS PubMed.
- M. Nguyen and S. G. Joshi, J. Appl. Microbiol., 2021, 131, 2715–2738 CrossRef CAS PubMed.
- J. Fishbain and A. Y. Peleg, Clin. Infect. Dis., 2010, 51, 79–84 CrossRef.
- E.-T. Piperaki, L. S. Tzouvelekis, V. Miriagou and G. L. Daikos, Clin. Microbiol. Infect., 2019, 25, 951–957 CrossRef CAS PubMed.
- C. H. Rodríguez, M. Nastro and A. Famiglietti, Rev. Argent. Microbiol., 2018, 50, 327–333 Search PubMed.
- M. R. Galac, E. Snesrud, F. Lebreton, J. Stam, M. Julius, A. C. Ong, R. Maybank, A. R. Jones, Y. I. Kwak, K. Hinkle, P. E. Waterman, E. P. Lesho, J. W. Bennett and P. Mc Gann, Antimicrob. Agents Chemother., 2020, 64, 1–11 CrossRef PubMed.
- E. Rando, S. L. Cutuli, F. Sangiorgi, E. S. Tanzarella, F. Giovannenze, G. De Angelis, R. Murri, M. Antonelli, M. Fantoni and G. De Pascale, JAC Antimicrob. Resist., 2023, 5, 1–9 Search PubMed.
- W. Ni, Y. Han, J. Zhao, C. Wei, J. Cui, R. Wang and Y. Liu, Int. J. Antimicrob. Agents, 2016, 47, 107–116 CrossRef CAS PubMed.
- P. Fragkou, G. Poulakou, A. Blizou, M. Blizou, V. Rapti, D. Karageorgopoulos, D. Koulenti, A. Papadopoulos, D. Matthaiou and S. Tsiodras, Microorganisms, 2019, 7, 1–20 Search PubMed.
- W. Deng, T. Fu, Z. Zhang, X. Jiang, J. Xie, H. Sun, P. Hu, H. Ren, P. Zhou, Q. Liu and Q. Long, Emerging Microbes Infect., 2020, 9, 639–650 Search PubMed.
- M. C. Phillips, N. Wald-Dickler, K. Loomis, B. M. Luna and B. Spellberg, Open Forum Infect. Dis., 2020, 7, 1–9 Search PubMed.
- M. L. Gil-Marqués, P. Moreno-Martínez, C. Costas, J. Pachón, J. Blázquez and M. J. McConnell, J. Antimicrob. Chemother., 2018, 73, 2960–2968 Search PubMed.
- F. L. Gordillo Altamirano, X. Kostoulias, D. Subedi, D. Korneev, A. Y. Peleg and J. J. Barr, EBioMedicine, 2022, 80, 104045 Search PubMed.
- M. Y. Cruz-Muñiz, L. E. López-Jacome, M. Hernández-Durán, R. Franco-Cendejas, P. Licona-Limón, J. L. Ramos-Balderas, M. Martinéz-Vázquez, J. A. Belmont-Díaz, T. K. Wood and R. García-Contreras, Int. J. Antimicrob. Agents, 2017, 49, 88–92 Search PubMed.
- Y. Yu, H. Zhao, J. Lin, Z. Li, G. Tian, Y. Y. Yang, P. Yuan and X. Ding, Int. J. Antimicrob. Agents, 2022, 59, 1–6 Search PubMed.
- S. Garneau-Tsodikova and K. J. Labby, MedChemComm, 2016, 7, 11–27 CAS.
- C. O. Vrancianu, I. Gheorghe, I. B. Czobor and M. C. Chifiriuc, Microorganisms, 2020, 8, 1–40 Search PubMed.
- M. S. Ramirez, R. A. Bonomo and M. E. Tolmasky, Biomolecules, 2020, 10, 1–32 Search PubMed.
- L. L. Maragakis and T. M. Perl, Clin. Infect. Dis., 2008, 46, 1254–1263 CAS.
- S. N. Abdi, R. Ghotaslou, K. Ganbarov, A. Mobed, A. Tanomand, M. Yousefi, M. Asgharzadeh and H. S. Kafil, Infect. Drug Resist., 2020, 13, 423–434 CrossRef CAS.
- Z. A. Qureshi, L. E. Hittle, J. A. O'Hara, J. I. Rivera, A. Syed, R. K. Shields, A. W. Pasculle, R. K. Ernst and Y. Doi, Clin. Infect. Dis., 2015, 60, 1295–1303 CrossRef.
- S. Ibrahim, N. Al-Saryi, I. M. S. Al-Kadmy and S. N. Aziz, Mol. Biol. Rep., 2021, 48, 6987–6998 CrossRef CAS PubMed.
- X. Wu, J. D. Chavez, D. K. Schweppe, C. Zheng, C. R. Weisbrod, J. K. Eng, A. Murali, S. A. Lee, E. Ramage, L. A. Gallagher, H. D. Kulasekara, M. E. Edrozo, C. N. Kamischke, M. J. Brittnacher, S. I. Miller, P. K. Singh, C. Manoil and J. E. Bruce, Nat. Commun., 2016, 7, 1–14 Search PubMed.
- Y. Zhu, J. Lu, M. Han, X. Jiang, M. A. K. Azad, N. A. Patil, Y. Lin, J. Zhao, Y. Hu, H. H. Yu, K. Chen, J. D. Boyce, R. A. Dunstan, T. Lithgow, C. K. Barlow, W. Li, E. K. Schneider-Futschik, J. Wang, B. Gong, B. Sommer, D. J. Creek, J. Fu, L. Wang, F. Schreiber, T. Velkov and J. Li, Adv. Sci., 2020, 7, 1–13 Search PubMed.
- L. Zhang and R. L. Gallo, Curr. Biol., 2016, 26, R14–R19 CrossRef CAS PubMed.
- S. Li, Y. Wang, Z. Xue, Y. Jia, R. Li, C. He and H. Chen, Trends Food Sci. Technol., 2021, 109, 103–115 CrossRef CAS.
- Q. Wu, J. Patočka and K. Kuča, Toxins, 2018, 10, 1–17 Search PubMed.
- M. Mahlapuu, C. Björn and J. Ekblom, Crit. Rev. Biotechnol., 2020, 40, 978–992 CrossRef CAS PubMed.
- A. Peschel and H. G. Sahl, Nat. Rev. Microbiol., 2006, 4, 529–536 CrossRef CAS.
- H. Q. Ning, Y. Q. Li, Q. W. Tian, Z. S. Wang and H. Z. Mo, LWT--Food Sci. Technol., 2019, 99, 62–68 CrossRef CAS.
- M. Magana, M. Pushpanathan, A. L. Santos, L. Leanse, M. Fernandez, A. Ioannidis, M. A. Giulianotti, Y. Apidianakis, S. Bradfute, A. L. Ferguson, A. Cherkasov, M. N. Seleem, C. Pinilla, C. de la Fuente-Nunez, T. Lazaridis, T. Dai, R. A. Houghten, R. E. W. Hancock and G. P. Tegos, Lancet Infect. Dis., 2020, 20, 1–15 CrossRef PubMed.
- E. F. Haney, S. C. Mansour and R. E. W. Hancock, in Methods in Molecular Biology, 2017, vol. 1548, pp. 3–22 Search PubMed.
- A. Panjla, G. Kaul, S. Chopra, A. Titz and S. Verma, ACS Chem. Biol., 2021, 16(12), 2731–2745 CrossRef CAS PubMed.
- C. Chen, J. Shi, D. Wang, P. Kong, Z. Wang and Y. Liu, Crit. Rev. Microbiol., 2024, 50, 267–284 CrossRef CAS.
- J. M. Sierra, E. Fusté, F. Rabanal, T. Vinuesa and M. Viñas, Expert Opin. Biol. Ther., 2017, 17, 663–676 CrossRef.
- W. C. Wimley and K. Hristova, J. Membr. Biol., 2011, 239, 27–34 CrossRef CAS PubMed.
- Q.-Y. Zhang, Z.-B. Yan, Y.-M. Meng, X.-Y. Hong, G. Shao, J.-J. Ma, X.-R. Cheng, J. Liu, J. Kang and C.-Y. Fu, Mil. Med. Res., 2021, 8, 1–25 Search PubMed.
- H. X. Luong, T. T. Thanh and T. H. Tran, Life Sci., 2020, 260, 118407 CrossRef CAS.
- A. M. van der Does, P. S. Hiemstra and N. Mookherjee, in Advances in Experimental Medicine and Biology, Springer New York LLC, 2019, vol. 1117, pp. 149–171 Search PubMed.
- N. Mookherjee, M. A. Anderson, H. P. Haagsman and D. J. Davidson, Nat. Rev. Drug Discovery, 2020, 19, 311–332 CrossRef CAS PubMed.
- R. E. W. Hancock, E. F. Haney and E. E. Gill, Nat. Rev. Microbiol., 2016, 16, 321–334 CAS.
- S. Biswas, J. M. Brunel, J. C. Dubus, M. Reynaud-Gaubert and J. M. Rolain, Expert Rev. Anti-Infect. Ther., 2012, 10, 917–934 CrossRef CAS.
- J. H. Moffatt, M. Harper and J. D. Boyce, in Advances in Experimental Medicine and Biology, Springer New York LLC, 2019, vol. 1145, pp. 55–71 Search PubMed.
- J. Zhu, S. Wang, C. Wang, Z. Wang, G. Luo, J. Li, Y. Zhan, D. Cai and S. Chen, Synth. Syst. Biotechnol., 2023, 8, 314–322 CrossRef CAS.
- G. Pavithrra and R. Rajasekaran, Int. J. Pept. Res. Ther., 2020, 26, 191–199 CrossRef CAS.
- M. Heidary, A. D. Khosravi, S. Khoshnood, M. J. Nasiri, S. Soleimani and M. Goudarzi, Daptomycin, J. Antimicrob. Chemother., 2018, 73, 1–11 CrossRef CAS.
- C. H. Lee, C. Y. Tsai, C. C. Li, C. C. Chien and J. W. Liu, J. Antimicrob. Chemother., 2015, 70, 257–263 CrossRef CAS.
- J. A. Karlowsky, K. Nichol and G. G. Zhanel, Clin. Infect. Dis., 2015, 61, S58–S68 CrossRef CAS PubMed.
- K. D. Brade, J. M. Rybak and M. J. Rybak, Infect. Dis. Ther., 2016, 5, 1–15 CrossRef PubMed.
- J. R. Smith, K. D. Roberts and M. J. Rybak, s, Infect. Dis. Ther., 2015, 4, 245–258 CrossRef PubMed.
- R. Álvarez, L. E. L. Cortés, J. Molina, J. M. Cisneros and J. Pachón, Antimicrob. Agents Chemother., 2016, 60, 2601–2609 CrossRef PubMed.
- G. D. Marena, M. A. dos Santos Ramos, T. M. Bauab and M. Chorilli, Crit. Rev. Anal. Chem., 2021, 51, 312–328 CrossRef CAS.
- J. C. Song and D. A. Stevens, Crit. Rev. Microbiol., 2016, 42, 813–846 CrossRef CAS.
- H. Pang, Z. Qu, V. Kumar, Y. Wang, Y. Wu, M. H. Lin, D. Harrich and F. Y. Han, Adv. Ther., 2024, 7, 2300439 CrossRef CAS.
- N. V. Hentig, HIV/AIDS, 2016, 8, 1–16 Search PubMed.
- H. B. Koo and J. Seo, Pept. Sci., 2019, 111, e24122 CrossRef.
- S. Baron, L. Hadjadj, J.-M. Rolain and A. O. Olaitan, Expert Rev. Anti-Infect. Ther., 2012, 10, 917–934 CrossRef.
- M. H. Rigatto, D. R. Falci and A. P. Zavascki, in Advances in Experimental Medicine and Biology, 2019, vol. 1145, pp. 197–218 Search PubMed.
- J. H. Moffatt, M. Harper and J. D. Boyce, in Advances in Experimental Medicine and Biology, 2019, vol. 1145, pp. 55–71 Search PubMed.
- K. D. Roberts, Y. Zhu, M. A. K. Azad, M. Han, J. Wang, L. Wang, H. H. Yu, A. S. Horne, J. Pinson, D. Rudd, N. H. Voelcker, N. A. Patil, J. Zhao, X. Jiang, J. Lu, K. Chen, O. Lomovskaya, S. J. Hecker, P. E. Thompson, R. L. Nation, M. N. Dudley, D. C. Griffith, T. Velkov and J. Li, Nat. Commun., 2022, 13, 1–15 Search PubMed.
- P. Nasr, J. Hosp. Infect., 2020, 104, 4–11 CAS.
- T. C. Menegucci, J. Albiero, L. B. Migliorini, J. L. B. Alves, G. F. Viana, J. Mazucheli, F. E. Carrara-Marroni, C. L. Cardoso and M. C. B. Tognim, Int. J. Antimicrob. Agents, 2016, 47, 380–385 CrossRef CAS.
- K. Kengkla, K. Kongpakwattana, S. Saokaew, A. Apisarnthanarak and N. Chaiyakunapruk, J. Antimicrob. Chemother., 2018, 73, 22–32 CrossRef CAS PubMed.
- K. D. Roberts, M. A. K. Azad, J. Wang, A. S. Horne, P. E. Thompson, R. L. Nation, T. Velkov and J. Li, ACS Infect. Dis., 2015, 1, 568–575 CrossRef CAS PubMed.
- A.-L. Cui, X.-X. Hu, Y. Gao, J. Jin, H. Yi, X.-K. Wang, T.-Y. Nie, Y. Chen, Q.-Y. He, H.-F. Guo, J.-D. Jiang, X.-F. You and Z.-R. Li, J. Med. Chem., 2018, 61, 1845–1857 CrossRef CAS PubMed.
- C. Xie, P. Wang, H. Wu, X. Hu, T. Nie, X. Li, P. Pang, G. Li, Y. Lu, X. Yang, X. Wang, C. Li and X. You, Biomed. Pharmacother., 2023, 164, 1–11 CrossRef.
- J. M. Tyrrell, A. F. Aboklaish, T. R. Walsh, T. Vaara and M. Vaara, Peptides, 2019, 112, 149–153 CrossRef CAS PubMed.
- Z. Wang, B. Koirala, Y. Hernandez, M. Zimmerman, S. Park, D. S. Perlin and S. F. Brady, Nature, 2022, 601, 606–611 CrossRef CAS.
- P. A. Smith, M. F. T. Koehler, H. S. Girgis, D. Yan, Y. Chen, Y. Chen, J. J. Crawford, M. R. Durk, R. I. Higuchi, J. Kang, J. Murray, P. Paraselli, S. Park, W. Phung, J. G. Quinn, T. C. Roberts, L. Rougé, J. B. Schwarz, E. Skippington, J. Wai, M. Xu, Z. Yu, H. Zhang, M.-W. Tan and C. E. Heise, Nature, 2018, 561, 189–194 CrossRef CAS PubMed.
- M. Jangra, M. Kaur, R. Tambat, R. Rana, S. K. Maurya, N. Khatri, A. Ghafur and H. Nandanwar, Antimicrob. Agents Chemother., 2019, 63, 1–16 CrossRef PubMed.
- S. J. Bann, R. D. Ballantine and S. A. Cochrane, RSC Med. Chem., 2021, 12, 538–551 RSC.
- S. A. Cochrane, C. T. Lohans, J. R. Brandelli, G. Mulvey, G. D. Armstrong and J. C. Vederas, J. Med. Chem., 2014, 57, 1127–1131 CrossRef CAS PubMed.
- S. A. Cochrane and J. C. Vederas, Int. J. Antimicrob. Agents, 2014, 44, 493–499 CrossRef CAS.
- S. A. Cochrane, C. T. Lohans, M. J. van Belkum, M. A. Bels and J. C. Vederas, Org. Biomol. Chem., 2015, 13, 6073–6081 RSC.
- S. A. Cochrane, B. Findlay, A. Bakhtiary, J. Z. Acedo, E. M. Rodriguez-Lopez, P. Mercier and J. C. Vederas, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 11561–11566 CrossRef CAS.
- R. D. Ballantine, C. E. McCallion, E. Nassour, S. Tokajian and S. A. Cochrane, MedChemComm, 2019, 10, 484–487 RSC.
- M. Jangra, V. Raka and H. Nandanwar, Molecules, 2020, 25, 3255 CrossRef CAS.
- V. M. Thomas, R. M. Brown, D. S. Ashcraft and G. A. Pankey, Int. J. Antimicrob. Agents, 2019, 53, 663–668 CrossRef CAS PubMed.
- Q. Li, M. Montalban-Lopez and O. P. Kuipers, Appl. Environ. Microbiol., 2018, 84, 1–15 CAS.
- C. Nord, J. Bjerketorp, J. J. Levenfors, S. Cao, A. A. Strömstedt, B. Guss, R. Larsson, D. Hughes, B. Öberg and A. Broberg, ACS Chem. Biol., 2020, 15, 2937–2944 CrossRef CAS.
- X. Zhong, K. Deng, X. Yang, X. Song, Y. Zou, X. Zhou, H. Tang, L. Li, Y. Fu, Z. Yin, H. Wan and X. Zhao, Front. Microbiol., 2023, 14, 1–14 Search PubMed.
- D. Palpal-latoc, A. J. Horsfall, A. J. Cameron, G. Campbell, S. A. Ferguson, G. M. Cook, V. Sander, A. J. Davidson, P. W. R. Harris and M. A. Brimble, J. Nat. Prod., 2024, 87, 764–773 CrossRef CAS.
- Y.-X. Li, Z. Zhong, W.-P. Zhang and P.-Y. Qian, Nat. Commun., 2018, 9, 1–9 CrossRef.
- C. E. Seyfert, C. Porten, B. Yuan, S. Deckarm, F. Panter, C. D. Bader, J. Coetzee, F. Deschner, K. H. M. E. Tehrani, P. G. Higgins, H. Seifert, T. C. Marlovits, J. Herrmann and R. Müller, Angew. Chem., Int. Ed., 2023, 62, 1–11 Search PubMed.
- S. Patel, S. Ahmed and J. S. Eswari, World J. Microbiol. Biotechnol., 2015, 31, 1177–1193 CrossRef CAS PubMed.
- M. Heidary, A. D. Khosravi, S. Khoshnood, M. J. Nasiri, S. Soleimani and M. Goudarzi, J. Antimicrob. Chemother., 2018, 73, 1–11 CrossRef CAS PubMed.
- A. Raja, J. LaBonte, J. Lebbos and P. Kirkpatrick, Nat. Rev. Drug Discovery, 2003, 2, 943–944 CAS.
- J. E. Page and S. Walker, Curr. Opin. Microbiol., 2021, 61, 16–24 CrossRef CAS.
- M. Ghosh, P. A. Miller, U. Möllmann, W. D. Claypool, V. A. Schroeder, W. R. Wolter, M. Suckow, H. Yu, S. Li, W. Huang, J. Zajicek and M. J. Miller, J. Med. Chem., 2017, 60, 4577–4583 CrossRef CAS.
- M. Ghosh, Y.-M. Lin, P. A. Miller, U. Möllmann, W. C. Boggess and M. J. Miller, ACS Infect. Dis., 2018, 4, 1529–1535 CrossRef CAS PubMed.
- A. S. Lee, H. De Lencastre, J. Garau, J. Kluytmans, S. Malhotra-Kumar, A. Peschel and S. Harbarth, Nat. Rev. Dis. Primers, 2018, 4, 1–23 CrossRef PubMed.
- E. Breukink and B. de Kruijff, Nat. Rev. Drug Discovery, 2006, 5, 321–323 CrossRef CAS PubMed.
- V. Yarlagadda, G. B. Manjunath, P. Sarkar, P. Akkapeddi, K. Paramanandham, B. R. Shome, R. Ravikumar and J. Haldar, ACS Infect. Dis., 2016, 2, 132–139 CrossRef CAS.
- P. Sarkar, S. Samaddar, V. Ammanathan, V. Yarlagadda, C. Ghosh, M. Shukla, G. Kaul, R. Manjithaya, S. Chopra and J. Haldar, ACS Chem. Biol., 2020, 15, 884–889 CrossRef CAS PubMed.
- D. Guan, F. Chen, Y. Qiu, B. Jiang, L. Gong, L. Lan and W. Huang, Am. Ethnol., 2019, 131, 6750–6754 Search PubMed.
- D. Sanderink, V. Cassisa, R. Chenouard, R. Mahieu, M. Kempf, V. Dubée and M. Eveillard, Future Microbiol., 2019, 14, 581–586 CAS.
- X. Vila-Farres, J. Chu, M. A. Ternei, C. Lemetre, S. Park, D. S. Perlin and S. F. Brady, mSphere, 2018, 3, 1–10 CrossRef.
- Q. Lu, X. Wu, Y. Fang, Y. Wang and B. Zhang, Synth. Syst. Biotechnol., 2024, 9, 312–321 CAS.
- E. S. Jayawant, J. Hutchinson, D. Gašparíková, C. Lockey, L. Pruñonosa Lara, C. Guy, R. L. Brooks and A. M. Dixon, ChemBioChem, 2021, 22, 2430–2439 CAS.
- M. F. Lin, P. W. Tsai, J. Y. Chen, Y. Y. Lin and C. Y. Lan, PLoS One, 2015, 10, 1–19 Search PubMed.
- Y. Guo, L. Wang, J. Lei, J. Xu and L. Han, Jundishapur J. Microbiol., 2017, 10, 1–7 CrossRef.
- H. Memariani and M. Memariani, World J. Microbiol. Biotechnol., 2023, 39, 1–28 CrossRef.
- M. Safari, R. Rafiei Tabatabaei, H. Abtahi, S. Fahimirad and A. Alimoradian, Jundishapur J. Microbiol., 2023, 15, 1–8 Search PubMed.
- A. Jahangiri, A. Neshani, S. A. Mirhosseini, K. Ghazvini, H. Zare and H. Sedighian, Microb. Pathog., 2021, 150, 1–7 CrossRef.
- M. Dai, P. Pan, H. Li, S. Liu, L. Zhang, C. Song, Y. Li, Q. Li, Z. Mao, Y. Long, X. Su and C. Hu, Exp. Cell Res., 2018, 364, 95–103 CrossRef CAS PubMed.
- M.-J. Kang, A.-R. Jang, J.-Y. Park, J.-H. Ahn, T.-S. Lee, D.-Y. Kim, D.-H. Jung, E.-J. Song, J. J. Hong and J.-H. Park, Immune Netw., 2020, 20, 1–13 CrossRef PubMed.
- M. Amerikova, I. Pencheva El-Tibi, V. Maslarska, S. Bozhanov and K. Tachkov, Biotechnol. Biotechnol. Equip., 2019, 33, 671–682 CAS.
- J. G. Routsias, P. Karagounis, G. Parvulesku, N. J. Legakis and A. Tsakris, Peptides, 2010, 31, 1654–1660 CAS.
- B. Wang, F.-W. Zhang, W.-X. Wang, Y.-Y. Zhao, S.-Y. Sun, J.-H. Yu, M. P. Vitek, G. F. Li, R. Ma, S. Wang, Z. Hu and W. Chen, Front. Microbiol., 2022, 13, 1–16 Search PubMed.
- Y. Guo, M. Xun and J. Han, Medicine, 2018, 97, 1–7 Search PubMed.
- L. Dolzani, A. Milan, M. Scocchi, C. Lagatolla, R. Bressan and M. Benincasa, J. Med. Microbiol., 2019, 68, 1253–1265 CAS.
- M. Mardirossian, R. Sola, B. Beckert, D. W. P. Collis, A. Di Stasi, F. Armas, K. Hilpert, D. N. Wilson and M. Scocchi, ChemMedChem, 2019, 14, 2025–2033 CAS.
- C. Liu, B. Shan, J. Qi and Y. Ma, Front. Cell. Infect. Microbiol., 2017, 7, 1–11 Search PubMed.
- J. Mwangi, Y. Yin, G. Wang, M. Yang, Y. Li, Z. Zhang and R. Lai, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 26516–26522 CrossRef CAS.
- F. L. Santana, I. Arenas, E. F. Haney, K. Estrada, R. E. W. Hancock and G. Corzo, Peptides, 2021, 141, 1–11 CrossRef.
- S. M. Barksdale, E. J. Hrifko, E. M.-C. Chung and M. L. van Hoek, BMC Microbiol., 2016, 16, 189 CrossRef.
- F. Ji, G. Tian, D. Shang and F. Jiang, Front. Microbiol., 2023, 14, 1–18 Search PubMed.
- H. J. Park, H. K. Kang, E. Park, M. K. Kim and Y. Park, Eur. J. Pharm. Sci., 2022, 175, 1–13 CrossRef.
- P. Jariyarattanarach, N. Klubthawee, M. Wongchai, S. Roytrakul and R. Aunpad, Sci. Rep., 2022, 12, 15852 CrossRef CAS PubMed.
- F. Sacco, C. Bitossi, B. Casciaro, M. R. Loffredo, G. Fabiano, L. Torrini, F. Raponi, G. Raponi and M. L. Mangoni, Antibiotics, 2022, 11, 1–12 CrossRef.
- Z. Dekan, S. J. Headey, M. Scanlon, B. A. Baldo, T. Lee, M. Aguilar, J. R. Deuis, I. Vetter, A. G. Elliott, M. Amado, M. A. Cooper, D. Alewood and P. F. Alewood, Angew. Chem., Int. Ed., 2017, 56, 8495–8499 CrossRef CAS.
- W. G. Lima, J. C. M. Brito, M. E. de Lima, A. C. S. T. Pizarro, M. A. M. de M. Vianna, M. C. de Paiva, D. C. S. de Assis, V. N. Cardoso and S. O. A. Fernandes, J Antibiot, 2021, 74, 425–434 CrossRef CAS.
- A. L. Carboni, M. A. Hanson, S. A. Lindsay, S. A. Wasserman and B. Lemaitre, Genetics, 2022, 220, 1–10 CrossRef PubMed.
- M. Wang, J. Lin, Q. Sun, K. Zheng, Y. Ma and J. Wang, Appl. Microbiol. Biotechnol., 2019, 103, 1765–1775 CrossRef CAS.
- J. Peng, H. Long, W. Liu, Z. Wu, T. Wang, Z. Zeng, G. Guo and J. Wu, Infect. Drug Resist., 2019, 12, 2417–2428 CrossRef CAS.
- W. Liu, Z. Wu, C. Mao, G. Guo, Z. Zeng, Y. Fei, S. Wan, J. Peng and J. Wu, Front. Microbiol., 2020, 11, 1–13 CrossRef.
- J. Kim, B. Jacob, M. Jang, C. Kwak, Y. Lee, K. Son, S. Lee, I. D. Jung, M. S. Jeong, S.-H. Kwon and Y. Kim, Sci. Rep., 2019, 9, 1–13 CrossRef.
- S. Mandel, J. Michaeli, N. Nur, I. Erbetti, J. Zazoun, L. Ferrari, A. Felici, M. Cohen-Kutner and N. Bachnoff, Sci. Rep., 2021, 11, 1–22 CrossRef.
- G. Schouten, F. Paulussen, O. Kuipers, W. Bitter, T. Grossmann and P. van Ulsen, Antibiotics, 2022, 11, 1–16 CrossRef.
- M. Krishnan, J. Choi, A. Jang, Y. K. Yoon and Y. Kim, Int. J. Mol. Sci., 2021, 22, 1–22 Search PubMed.
- S. Eshtiaghi, R. Nazari and M. Fasihi-Ramandi, Curr. Microbiol., 2023, 80, 1–13 CrossRef PubMed.
- M. G. Eales, E. Ferrari, A. D. Goddard, L. Lancaster, P. Sanderson and C. Miller, Res. Microbiol., 2018, 169, 296–302 CrossRef CAS PubMed.
- R. Gopal, Y. G. Kim, J. H. Lee, S. K. Lee, J. D. Chae, B. K. Son, C. H. Seo and Y. Park, Antimicrob. Agents Chemother., 2014, 58, 1622–1629 CrossRef.
- X. Vila-Farrés, R. López-Rojas, M. E. Pachón-Ibáñez, M. Teixidó, J. Pachón, J. Vila and E. Giralt, Eur. J. Med. Chem., 2015, 101, 34–40 CrossRef.
- Y. Wang, L. Wang, H. Yang, H. Xiao, A. Farooq, Z. Liu, M. Hu and X. Shi, Toxins, 2016, 8, 1–9 CrossRef.
- L. Yang, Z. Tian, W. Zhao, J. Zhang, C. Tian, L. Zhou, Z. Jiao, J. Peng and G. Guo, Bioorg. Chem., 2023, 138, 1–16 Search PubMed.
- L. Yang, Y. Gao, J. Zhang, C. Tian, F. Lin, D. Song, L. Zhou, J. Peng and G. Guo, Int. J. Antimicrob. Agents, 2024, 63, 1–12 Search PubMed.
- A. G. Elliott, J. X. Huang, S. Neve, J. Zuegg, I. A. Edwards, A. K. Cain, C. J. Boinett, L. Barquist, C. V. Lundberg, J. Steen, M. S. Butler, M. Mobli, K. M. Porter, M. A. T. Blaskovich, S. Lociuro, M. Strandh and M. A. Cooper, Nat. Commun., 2020, 11, 1–13 CrossRef.
- M. J. Hong, M. K. Kim and Y. Park, Int. J. Mol. Sci., 2021, 22, 1–18 Search PubMed.
- X. Luo, X. Ye, L. Ding, W. Zhu, Z. Zhao, D. Luo, N. Liu, L. Sun and Z. Chen, Microb. Pathog., 2021, 157, 1–7 CrossRef.
- D. C. Rajapaksha, E. H. T. T. Jayathilaka, S. L. Edirisinghe, C. Nikapitiya, J. Lee, I. Whang and M. De Zoysa, Fish Shellfish Immunol., 2021, 117, 82–94 CrossRef CAS.
- D. C. Rajapaksha, S. L. Edirisinghe, C. Nikapitiya, I. Whang and M. De Zoysa, Antibiotics, 2023, 12, 1–13 CrossRef PubMed.
- E. H. T. T. Jayathilaka, D. C. Rajapaksha, C. Nikapitiya, J. Lee, M. De Zoysa and I. Whang, Pharmaceuticals, 2022, 15, 1–19 CrossRef PubMed.
- P. K. Hazam, C.-C. Cheng, C.-Y. Hsieh, W.-C. Lin, P.-H. Hsu, T.-L. Chen, Y.-T. Lee and J.-Y. Chen, Int. J. Mol. Sci., 2022, 23, 1–12 Search PubMed.
- M. T. P. Gontijo, P. M. P. Vidigal, M. E. S. Lopez and M. Brocchi, Res. Microbiol., 2021, 172, 1–9 CrossRef PubMed.
- W. C. B. Lai, X. Chen, M. K. Y. Ho, J. Xia and S. S. Y. Leung, Int. J. Pharm., 2020, 589, 1–17 CrossRef PubMed.
- C. Li, M. Jiang, F. M. Khan, X. Zhao, G. Wang, W. Zhou, J. Li, J. Yu, Y. Li, H. Wei and H. Yang, ACS Infect. Dis., 2021, 7, 3336–3344 CrossRef CAS PubMed.
- K. Abdelkader, D. Gutiérrez, H. Tamés-Caunedo, P. Ruas-Madiedo, A. Safaan, A. S. Khairalla, Y. Gaber, T. Dishisha and Y. Briers, Appl. Environ. Microbiol., 2022, 88, 1–15 CrossRef PubMed.
- H. Yang, M. Wang, J. Yu and H. Wei, Front. Microbiol., 2015, 6, 1–9 Search PubMed.
- M. M. Islam, D. Kim, K. Kim, S.-J. Park, S. Akter, J. Kim, S. Bang, S. Kim, J. Kim, J. C. Lee, C.-W. Hong and M. Shin, Front. Microbiol., 2022, 13, 1–13 Search PubMed.
- S. Y. Peng, R. I. You, M. J. Lai, N. T. Lin, L. K. Chen and K. C. Chang, Sci. Rep., 2017, 7, 1–12 CrossRef.
- M. Thandar, R. Lood, B. Y. Winer, D. R. Deutsch, C. W. Euler and V. A. Fischetti, Antimicrob. Agents Chemother., 2016, 60, 2671–2679 CrossRef CAS PubMed.
- V. Defraine, J. Schuermans, B. Grymonprez, S. K. Govers, A. Aertsen, M. Fauvart, J. Michiels, R. Lavigne and Y. Briers, Antimicrob. Agents Chemother., 2016, 60, 3480–3488 CrossRef CAS PubMed.
- I. Rázquin-Olazarán, H. Shahrour and G. Martínez-De-Tejada, J. Biomed. Sci., 2020, 27, 1–19 CrossRef PubMed.
- S. M. Son, J. Kim and S. Ryu, Front. Microbiol., 2023, 14, 1–11 CAS.
- W. F. Porto, L. Irazazabal, E. S. F. Alves, S. M. Ribeiro, C. O. Matos, Á. S. Pires, I. C. M. Fensterseifer, V. J. Miranda, E. F. Haney, V. Humblot, M. D. T. Torres, R. E. W. Hancock, L. M. Liao, A. Ladram, T. K. Lu, C. de la Fuente-Nunez and O. L. Franco, Nat. Commun., 2018, 9, 1–12 CrossRef CAS PubMed.
- S. Paul, S. Verma and Y.-C. Chen, ACS Infect. Dis., 2024, 10, 1034–1055 CAS.
- M. Stach, T. N. Siriwardena, T. Köhler, C. van Delden, T. Darbre and J. Reymond, Angew. Chem., 2014, 126, 13041–13045 Search PubMed.
- J. Pires, T. N. Siriwardena, M. Stach, R. Tinguely, S. Kasraian, F. Luzzaro, S. L. Leib, T. Darbre, J.-L. Reymond and A. Endimiani, Antimicrob. Agents Chemother., 2015, 59, 7915–7918 CAS.
- T. N. Siriwardena, A. Lüscher, T. Köhler, C. van Delden, S. Javor and J. Reymond, Helv. Chim. Acta, 2019, 102, 1–11 CrossRef.
- T. N. Siriwardena, A. Capecchi, B. Gan, X. Jin, R. He, D. Wei, L. Ma, T. Köhler, C. van Delden, S. Javor and J. Reymond, Angew. Chem., 2018, 130, 8619–8623 CrossRef.
- T. Heinonen, S. Hargraves, M. Georgieva, C. Widmann and N. Jacquier, J. Global Antimicrob. Resist., 2021, 25, 227–231 CAS.
- S. Joshi, G. S. Bisht, D. S. Rawat, A. Kumar, R. Kumar, S. Maiti and S. Pasha, Biochim. Biophys. Acta, Biomembr., 2010, 1798, 1864–1875 CrossRef CAS.
- D. Sharma, M. Choudhary, J. Vashistt, R. Shrivastava and G. S. Bisht, Biochem. Biophys. Res. Commun., 2019, 518, 472–478 CrossRef CAS.
- S. H. Moon and E. Huang, Antimicrob. Agents Chemother., 2018, 62, 1–5 CrossRef.
- D. Nagarajan, N. Roy, O. Kulkarni, N. Nanajkar, A. Datey, S. Ravichandran, C. Thakur, S. T, I. V. Aprameya, S. P. Sarma, D. Chakravortty and N. Chandra, Sci. Adv., 2019, 5, 1–19 Search PubMed.
- W. Li, N. M. O'Brien-Simpson, J. A. Holden, L. Otvos, E. C. Reynolds, F. Separovic, M. A. Hossain and J. D. Wade, Pept. Sci., 2018, 110, 1–9 Search PubMed.
- Q. He, L. Zhao, G. Li, Y. Shen, Y. Hu and Y. Wang, New J. Chem., 2022, 46, 6577–6586 RSC.
- C. Zampaloni, P. Mattei, K. Bleicher, L. Winther, C. Thäte, C. Bucher, J.-M. Adam, A. Alanine, K. E. Amrein, V. Baidin, C. Bieniossek, C. Bissantz, F. Boess, C. Cantrill, T. Clairfeuille, F. Dey, P. Di Giorgio, P. du Castel, D. Dylus, P. Dzygiel, A. Felici, F. García-Alcalde, A. Haldimann, M. Leipner, S. Leyn, S. Louvel, P. Misson, A. Osterman, K. Pahil, S. Rigo, A. Schäublin, S. Scharf, P. Schmitz, T. Stoll, A. Trauner, S. Zoffmann, D. Kahne, J. A. T. Young, M. A. Lobritz and K. A. Bradley, Nature, 2024, 625, 566–571 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |