
Small molecule targeted protein degradation via the UPS: venturing beyond E3 substrate receptors
Renyu
Guo
 ,
Fukang
Yang
and
Emily C.
Cherney
*
,
Fukang
Yang
and
Emily C.
Cherney
*
Discovery & Development Sciences, Bristol Myers Squibb Co, Princeton, New Jersey 08543, USA. E-mail: emily.cherney@bms.com
First published on 12th February 2025
Abstract
The ubiquitin proteasome system (UPS) has been successfully hi-jacked by both bifunctional and monovalent small molecules to affect the degradation of proteins that were once considered undruggable. This field has primarily focused on the targeted recruitment of proteins to substrate receptors on E3 ubiquitin ligases, which are only one part of the UPS. More recently, the field has begun to explore recruitment to other types of UPS proteins including E2 ubiquitin-conjugating enzymes, substrate adaptor proteins within the E3 complex, chaperone proteins that associate with E3s, proteasomal subunits, and proteasome-associated proteins. While these approaches are relatively nascent compared to more traditional E3 substrate receptor-based degradation, these approaches are starting to show promise and could offer unique advantages. This review will cover key findings in small molecule UPS-mediated targeted protein degradation (TPD) affected by co-opting proteins beyond traditional E3 substrate receptors.
 Renyu Guo | Renyu Guo grew up in Chengdu, China. He obtained his B.S. degree at Nankai University in 2016 with Prof. Liangfu Tang. He was a visiting student in the laboratories of Prof. David Sarlah (University of Illinois Urbana-Champaign) and Prof. Zhen Yang (Peking University), before joining Prof. Kevin Brown's research group at Indiana University Bloomington for his doctoral training. After obtaining his Ph.D. degree in 2022, he started his career as a medicinal chemist at Bristol Myers Squibb and is currently working on protein homeostasis chemistry. |
 Fukang Yang | Fukang Yang is a principal scientist at Bristol Myers Squibb. Prior to joining the protein homeostasis team, he was a medicinal chemist pursuing new medicines for CNS and infectious disease indications at BMS for over 20 years. He was a key contributor to the discoveries of the HCV drug Daklinza as well as a mGluR5 positive allosteric modulator clinical candidate. He graduated from East China University of Science & Technology with B.S. in Chemistry and subsequently obtained his M.S. in Organic University of Wisconsin-Milwaukee. |
 Emily Cherney | Emily Cherney is a scientific associate director at Bristol Myers Squibb where she is a medicinal chemist responsible for supporting protein degradation programs across the portfolio and expanding the protein homeostasis platform at BMS. Emily received her PhD from the Scripps Research Institute in La Jolla, California where she studied natural product total synthesis in the laboratory of Phil S. Baran. Since joining BMS, she's had the privilege of contributing to several programs including IDO1 inhibitors, STING agonists, and Helios degraders. Her key areas of interest include synthetic organic chemistry, immuno-oncology, and protein homeostasis. |
Introduction
Over the past two decades, the field of targeted protein degradation (TPD) has expanded exponentially driven by the promise of event-driven pharmacology.1,2 This field aims to target proteins that were previously deemed undruggable due to lack of enzymatic function, scaffolding effects, and/or lack of druggable binding sites by either marking them for degradation or recruiting to native degradation machinery. Proteins can be marked for degradation with small molecules via a variety of approaches including hydrophobic tagging (HyT), intercepting partially folded proteins to mimic misfolding, and triggering oligomerization.3–5 Proteins can also be recruited to myriad degradation machinery spanning the ubiquitin proteasome system (UPS) and autophagy-lysosome system. Targeted recruitment can be achieved using bifunctional molecules (e.g. targeting chimeras (TACs)) encompassing PROTACs, LYTACs, MoDE-As, AUTACs, AUTOTACs, ATTECs, AbTACs, KineTACs, RIPTACs, DENTACs, CHAMPs, IFLDs, CIDEs, IUDs, and more.6–8 Targeted recruitment can also be achieved with monovalent small molecules, typically referred to as molecule glue degraders (MGDs), that facilitate protein–protein interactions (PPIs) between a protein of interest and degradation machinery. While MGDs most frequently co-opt cereblon with immunomodulatory imide drugs (IMiDs) or cereblon E3 ligase modulatory drugs (CELMoDs), MGDs have also been found to facilitate PPIs with other E3s.9The UPS involves a complex cascade (Fig. 1A). The fundamental components of this system include ubiquitin, E1 ubiquitin-activating enzymes, E2 ubiquitin-conjugating enzymes, E3 ubiquitin ligases, and the proteasome itself, which can exist in several assembly states and degrade ubiquitinated targets. Within these components lie individual subunits. For instance, the Cullin RING family of E3 ligases (CRLs) can consist of RING-box (Rbx), Cullin (Cul), substrate adaptor, and substrate receptor proteins (Fig. 1B). Additionally, this RING complex requires neddylation with NEDD8 for activation.10,11 The 26S proteasome is also a multi-protein complex that consists of two 19S regulatory particles and one 20S core particle which contain dozens of subunits between them (Fig. 1C). Beyond these, there are a range of proteins that associate with these UPS components including deubiquitinating enzymes (DUBs) and chaperone proteins. The complexity of this system as well as its interplay with other biological pathways is seemingly endless, but advances are being made in our understanding of the UPS constantly. These advances in understanding, coupled with the drive to push the boundaries of TPD, are the sources of inspiration for the novel TPD approaches discussed in this review.
 | ||
| Fig. 1 A. The UPS pathway. B. E3 ligase families and modes of endogenous substrate recognition. C. The 26S proteasome machinery. D. Overview of non-substrate receptor UPS components targeted with small molecules (SMs) for TPD that will be discussed in this review. | ||
Within the UPS-directing bifunctional degrader space (i.e. PROTACs), the vast majority of focus has been on recruitment to proteins within E3 complexes that directly interact with substrates endogenously (yellow proteins in Fig. 1). These types of targets include substrate receptors like CRBN and VHL, as well as monomeric E3s like cIAP and MDM2. Recruitment to substrate receptors has also been the most successful and validated approach for degradation via bifunctional recruitment with multiple assets in clinical trials.12 Despite this success, only a small handful of the hundreds of substrate receptors within E3 ubiquitin ligases have been co-opted. This has created intense interest in attempting to expand the chemical toolbox of E3 ligase substrate receptor ligands. Several different approaches have been implemented to prioritize which of the hundreds of potential substrate receptor targets to pursue, and these approaches have been thoroughly reviewed elsewhere.13 Considerations include essentiality to address resistance mechanisms, tissue or cell type specificity for context-specific degradation, sub-cellular localization, induction within a specific disease context, confirmed ability to degrade endogenous substrates as opposed to facilitating a translocation or other alternative events, and more general assessments for target tractability such as druggability.14–16 While most efforts focus on these substrate receptors based on their known propensity to form PPIs and target proteins for degradation, they represent only one of many types of proteins that work in concert within the UPS.
Herein, we will explore TPD efforts to co-opt UPS proteins beyond E3 substrate receptors. These efforts will be discussed in the order in which the UPS targets appear in the pathway (Fig. 1D): E2 ubiquitin-conjugating enzymes, non-substrate receptor E3 components, E3-associated proteins, the proteasome itself, and, lastly, proteasome associated proteins. While the details of co-opting these novel UPS targets may be less understood, many of the chemical tools and approaches established for validating degrader targets and interrogating mechanism of action (MoA) for substrate receptor-based degraders translate.17,18 The assays developed for assessing degradation of both tagged and endogenous proteins of interest (e.g. BRD4 and AR) are still foundational for profiling novel degraders. The chemical tools for interrogating neddylation dependence and proteasome dependence are still critical for validating a proposed MoA.19 Demonstrating target engagement, ternary complex formation, genetic validation through knock-down/out, and reconstituting ubiquitination in an in vitro setting build confidence that a degrader is working through the implicated UPS machinery.20–22 Proteomics is still key for assessing degradation selectivity.23–25 Despite the preliminary nature of some of the studies reviewed below, they still provide a window into how innovative leaders in TPD are looking to push the boundaries of the field into new space and inspire others in the field to do the same in unique, unprecedented ways.
Co-opting E2 ubiquitin-conjugating enzymes
Humans have ∼40 ubiquitin-conjugating enzymes (E2s), which function to transfer ubiquitin (Ub) or ubiquitin-like (Ubl) proteins from an ubiquitin-activating enzyme (E1) to a substrate protein on E3 ubiquitin ligase (Fig. 1A) either directly or via the E3 (Fig. 1B).26 Additionally, E2s can directly engage substrate proteins in the absence of E3s.27–29 All E2s contain one, highly conserved core catalytic (UBC) domain that contains an active site cysteine. The C-terminal carboxylate of Ub is conjugated to this E2 active site cysteine in an E1-catalyzed, ATP-driven transthiolation reaction.30 Subsequently, Ub can be transferred to a nucleophilic residue (typically lysine, though transfer to non-lysine residues has been observed) on a target protein through aminolysis.31E2s have been targeted with both large and small molecule strategies. While somewhat outside of the scope of this review, BioPROTACs, which replace a natural substrate recognition domain with a peptide that recognizes a protein of interest, have recently been extended to work through not only E3s, but also E2s.32 Small molecule E2 binders typically function via covalent modification of the catalytic cysteine. Two examples of inhibitors that covalently target the catalytic Cys85 in UBE2N through a conjugate addition reaction are BAY 11-7082 and NSC697923 (Fig. 2A).33BAY 11-7082 was shown to inhibit not only UBE2N but also other E2 enzymes as well as the proteasome.34 In contrast, NSC697923 was found to be specific for UBE2N because of additional occupancy of a cleft in the active site that is not accessible in other E2s.35 These types of E2 ligands, however, cause E2 loss of function (as evidenced by NF-κB downregulation), and prevent Ub transfer onto the E2. Therefore, these ligands presumably could not be leveraged for the targeted recruitment and degradation of a protein of interest.
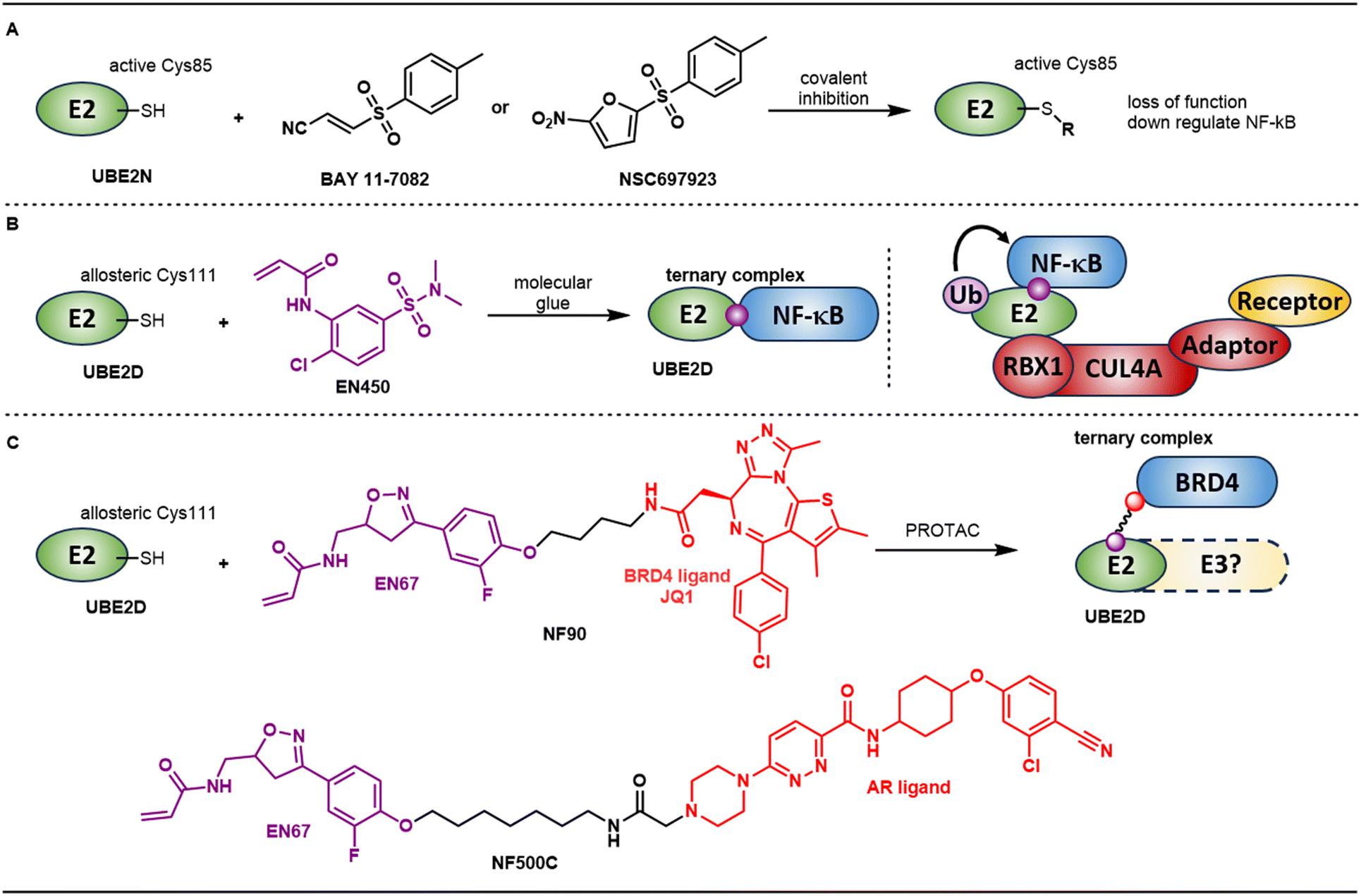 | ||
| Fig. 2 A. BAY 11-7082 and NSC697923 are covalent inhibitors of UBE2N that modify the catalytically active Cys85 within the UBC domain. B. EN450 is a covalent ligand that modifies allosteric Cys111 on UBE2D and induces degradation of NF-κB via a molecule glue mechanism. C. EN67 also modifies Cys111 on UBE2D. This ligand was incorporated into PROTACs NF90 and NF500C targeting BRD4 and AR respectively. | ||
Instead of targeting E2s through covalent modification of the active cysteine on UBE2N to downregulate NF-κB, the Nomura group recently reported a covalent molecular glue degrader that directly recruits NF-κB through allosteric cysteines on UBE2D.36 In this study, they identified a cysteine reactive covalent ligand EN450 through a phenotypic screen based on impairment of HAP1 cell viability. The authors conjectured that EN450 induces the proximity of a target protein with a component of the CUL4A/RBX1/NEDD8 ubiquitin-proteasome system because the inhibition of HAP1 proliferation was found to be both neddylation and proteasome dependent. Further chemoproteomic profiling revealed covalent interaction of EN450 with 81 cysteines including a single UPS target cysteine: the allosteric Cys111 in UBE2D. Quantitative proteomics profiling with tandem mass tagging (TMT)-based upon treatment with EN450 showed only NF-κB1 was significantly downregulated. Pulldown studies demonstrated that EN450 promoted ternary complex formation between UBE2D1 and NF-κB1. Ubiquitination of NF-κB1 could be reconstituted in a biochemical setting, but only in the presence of a CUL4A/RBX1/NEDD8 complex. One possible explanation for this is the need for the E2 to be in complex with E3 machinery to be activated for Ub transfer. While authors proposed NF-κB1 as the putative degradation target via a molecule glue mechanism, they recognize several limitations in their studies. Their studies do not definitively confirm the mechanism is via direct recruitment of the substrate to the E2 bypassing the E3. EN450 contains a potentially promiscuous covalent warhead and it is unclear from a structural standpoint why this warhead would have good selectivity against the catalytic Cys85 vs. allosteric cysteines on E2 enzymes.37 Inhibition via covalent modification of the catalytic Cys85 on E2s, such as UBE2N, (vide infra) has previously been shown to impact NF-κB signaling. This precedent further complicates the interpretation of the observed results.
Following this study, the Nomura group also incorporated UBE2D covalent modifiers into bifunctional degraders to investigate their ability to recruit and degrade target proteins.38 The authors initially prepared bifunctional degraders linking EN450 and the BRD4 inhibitor JQ1, but observed very weak degradation of only the short form of BRD4. Considering EN450 was discovered through a phenotypic screening as opposed to a targeted screen, they performed a direct covalent binder screening against recombinant human UBE2D C85S protein and identified EN67 (Fig. 2C). Similarly, EN67 also targeted Cys111 according to LCMS/MS experiments and did not inhibit functional activity of UBE2D to ubiquitinate TP53 in a reconstituted ubiquitination assay. PROTACs were prepared by linking EN67 and JQ1 with different linkers, and 4-carbon linked NF90 selectively degraded short isoform BRD4 over long isoform in both HEK293T and MDA-MB-231 cells. To further support on-target degradation effects, knockdown of all four UBE2D family members ablated degradation. However, knockdown could also be affecting the activity of the E3s that UBE2D family members interact with, so the observed degradation rescue effects could still potentially be indirect/off-target. Additionally, 7-carbon linked linker androgen accepter (AR) degrader NF500C was synthesized by linking EN67 with the ARV-110 AR ligand.39 In AR-positive LNCaP prostate cancer cells, NF500C showed reasonable degradation and the result was further supported by TMT-based quantitative proteomic profiling. Most surprisingly, non-reactive analogs of the above PROTACs that lacked the covalent warhead maintained measurable activity in both UBE2D binding and degradation assays. This result is difficult to rationalize in the context of the originally proposed mechanism which outlines recruitment via covalent modification of Cys111 on UBE2D.
Co-opting E3 substrate adaptors
Cullin-RING ligases (CRLs) consist of four main components: a RING-box (RBX) that binds to an E2, a scaffolding cullin, a substrate adaptor that connects the substate receptor to the cullin, and a substrate receptor that binds directly to a substrate protein (Fig. 1B). Adaptor proteins for different cullins include SKP1 (Cul1), EloB/C (Cul2/5), BTB (Cul3), and DDB1 (Cul4). Each adaptor can host dozens of different substrate receptors, some of which can form protein–protein interactions (PPIs) with multiple substrate proteins. These adaptor proteins are generally essential and do not have redundancy with other adaptors the way that some non-essential substrate receptors do. Co-opting substrate adaptors not only has the advantage of bypassing substrate receptors, but also can avoid resistance mechanisms of non-essential substrate receptors like CRBN.40,41 To date, two of these adaptor proteins have been targeted for TPD: DDB1 and SKP1.Damage specific DNA binding protein 1 (DDB1), as its name suggests, was initially found to be involved in the nucleotide excision repair pathway.42 Besides several other functions,43 DDB1 is a core component of a ubiquitin–E3 ligase complex and serves as an adapter protein between Cullin 4A (Cul4A) and over 20 CUL4-associated factors (DCAFs) to target substrates for ubiquitination.44 It should be noted that adaptor proteins like DDB1 and EloB/C are known to directly interact with viral proteins to trigger ubiquitination and degradation.45
In a systematic drug sensitivity study of 4518 clinical and pre-clinical drugs across 578 cell lines and E3 ligase mRNA level correlations, CR8, a cyclin-dependent kinase (CDK) inhibitor, was found to be dependent upon DDB1 based on CRISPR-mediated inactivation (Fig. 3A).46 Quantitative proteomic profiling showed cyclin K was the only protein consistently and significantly downregulated with CR8 treatment. The observed down regulation was determined to be NEDDylation and proteasome dependent which indicated cullin ring ligase (CRL) involvement, as the process was rescued by MLN4924 (a neddylation inhibitor) and MG132 (a proteasome inhibitor). However, no DCAFs were implicated in genetic screens. Indeed, only other components of the Cul4 ligase assembly were identified. Co-immunoprecipitation (IP) and in vitro ubiquitination assays suggested that the RBX1-CUL4A-DDB1 ligase core alone is sufficient to drive robust ubiquitination of cyclin K, adding support to the CRISPR screen results. Finally, a ternary X-ray structure confirmed that the CDK12-bound form of CR8 induces the formation of a complex between CDK12 and DDB1 (Fig. 3A). It also confirmed direct binding of cyclin K to CDK12, which would then be poised for ubiquitination and degradation. This structure shows CR8 can bypass the need for an endogenous substrate receptor and instead co-opt CDK12 to act as a “neosubstrate receptor”. Around this time, related structures, such as dCeMM3 (Fig. 3A), were uncovered utilizing comparative profiling of drugs in hyponeddylated cells.47 In this report, authors demonstrate the ability of dCeMM3 to bypass a substrate receptor and induce dimerization between CDK12 and DDB1, confirm that UBE2Z and UBE2G1 are the necessary priming and extending E2s, and show that UBA6 is the main E1 responsible for degradation.
 | ||
| Fig. 3 A. CR8, a CDK inhibitor, leads to the degradation of cyclin K via a molecular glue degrader mechanism through the recruitment of CDK12 directly to DDB1. B. Other reported reversible and covalent ligands of DDB1 were subsequently incorporated into bifunctional degraders. | ||
Since these initial DDB1-CDK12 molecular glues were reported, various structural changes on the same pyrazolo[4,3-d]pyrimidine core that preserve degradation activity have also been reported.48,49 In addition, modifications on other pan-CDK inhibitors such as dinaciclib and AT-7519 revealed that they can also function as cyclin K molecular glue degraders.50 Other groups have found 5-methylthiazol-2-amine derivatives also behave as glue degraders of cyclin K through the same recruitment mechanism.51,52 With the understanding of the binding models, an evaluation of 91 candidate degraders in structural, biophysical and cellular studies reveal all compounds acquire glue activity via simultaneous CDK12 binding and engagement of DDB1 interfacial residues.53 All molecular glues maintained hinge binding interactions with CDK12 and interacted with Arg928 on DDB1.
The extension of DDB1-based MGDs to bifunctional PROTACs has also been explored (Fig. 3B). In 2020, Winter and coworkers filed a patent containing potential DDB1 ligands to recruit neosubstrates.54 Bifunctional degraders with BRD4 ligands or CDK ligands were prepared and degradation was observed. However, it is not clear exactly where the ligands are binding (DDB1, CUL4, or the interface between the two) based on chemoproteomics, pulldown studies, and mTurbo. In the same year, a patent from Liu and coworkers also claimed nitrothiazole-based DDB1 ligands that demonstrated weak binding by SPR (KD = 5–60 μM).55 These were also incorporated into PROTACs that demonstrated degradation against several therapeutic targets, including P300 and CBP, in a UPS-dependent manner. The Nomura group identified a covalent binder MM-02-57 that targets Cys173 on DDB1 using activity-based protein profiling (ABPP) and cysteine chemoproteomic screening.56 PROTACs against BRD4 were prepared and MM-02-08 selectively degraded short BRD4 isoform over long isoform in a proteasome, NEDDylation, and DDB1-dependent manner. MM-03-73, an AR-DDB1 based bifunctional degrader, could similarly degrade AR in corresponding cell lines.
S-phase kinase associated protein 1 (SKP1) is the adaptor protein of the SCF (RBX1-CUL1-Skp1-F box protein) ligase complex (Fig. 4A).57 Early studies showed CRL1-SKP1 auto-degradation of F-box substrate receptor proteins, suggesting the potential of SKP1 to directly recruit neosubstrates and bypass F-box proteins.58 Further support for bypassing F-box proteins was provided by Thoma and co-workers who applied covalent functionalization followed by E3 electroporation (COFFEE) in live cells.59 They directly attached the kinase inhibitor dasatinib onto SKP1 (at both Cys120 and Cys159) via covalent maleimide-thiol chemistry (Fig. 4B). Degradation of multiple kinase targets of dasatinib was then demonstrated after introducing the modified SKP1-dasatinib chimera into live cells with electroporation. The Nomura group discovered another cysteine-reactive covalent recruiter, EN884, that targets Cys159 of SKP1 using covalent chemoproteomic approaches (Fig. 4C).60 Encountering synthetic challenges, the pyridine nitrogen atom of EN884 was replaced with CH for PROTAC preparation. With linker and corresponding TBM attached on the meta position, the authors observed desired degradation against BRD4 and AR. As was observed in the E2 example highlighted in Fig. 2C (vide supra), non-covalent versions of these SKP1-based PROTACs maintained SKP1 binding and degradation activity, which is, again, difficult to rationalize with the proposed mechanism. Two other small-molecule ligands have been reported to inhibit SKP1, which may provide opportunities for TPD purposes.61,62
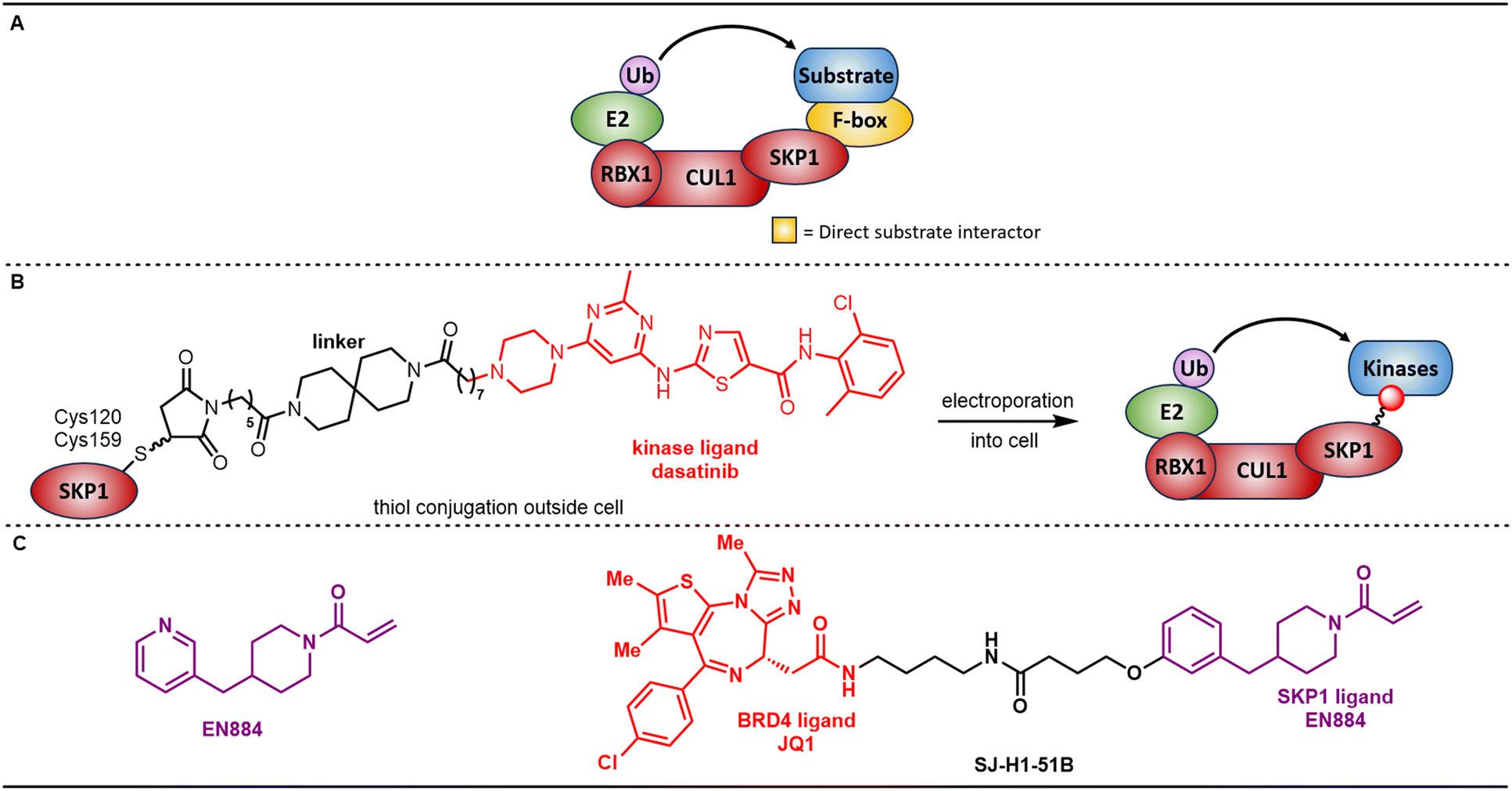 | ||
| Fig. 4 A. Canonical SKP1, Cullin, F-box containing (SCF) complex for SKP1-containing E3 ligases. B. Covalent functionalization followed by E3 electroporation (COFFEE) of SKP1 with dasatinib-derived electrophiles demonstrates the ability to directly recruit substrates to SKP1 for degradation bypassing the need for F-box substrate receptor proteins. C. A covalent ligand that modifies Cys160 on SKP1, which was incorporated into bifunctional degraders of BRD4 and AR. | ||
Co-opting E3-associated proteins
Heat shock proteins (HSPs) function as chaperone proteins to protect cells from outside stress, including exposure to cold, UV light, and during wound healing or tissue remodeling.63 In particular, they can stabilize new proteins to ensure correct folding and fold proteins that were damaged due to cell stress. In addition to participating in protein folding, HSPs can also recognize misfolded proteins and accelerate their degradation through the UPS. About 30% of human E3 ubiquitin ligases were found to bind to HSP90, thus recruiting neosubstrates to HSP90 could be an alternative approach to indirectly recruit to E3s to cause TPD.64 Beyond HSP90, other E3-associated HSPs like HSP70 may be amenable to hi-jacking, though no reports of HSP70-based bifunctional degraders have emerged at this time.65–67As opposed to E3 ligands, which are relatively scarce, over 20 clinical and pre-clinical HSP90 inhibitors have been reported representing a wide range of chemotypes.68 Ying and coworkers were the first to exploit these ligands in their CHAperone-mediated protein degradation (CHAMP) platform. They selected several different reported HSP90 ligands including SNX2112 (ref. 69) and ganetespib (STA-9090)70 for the HSP90 binding motif and prepared CHAMPs against various targets including BRD4,71,72 KRAS-G12C,73 KRAS-G12D74 and ERK5 (ref. 75) (Fig. 5A).
 | ||
| Fig. 5 A. Schematic of how E3 associated chaperone proteins could be exploited for TPD along with structures of HSP90 inhibitors exploited for TPD efforts. B. Bifunctional degraders reported subsequently for the degradation of CDK4/6 and AR (respectively) with demonstrated in vivo efficacy. | ||
Other groups in academia have since reported on leveraging known HSP90 inhibitors to co-opt HSP90 for TPD (Fig. 5B). Li and co-workers used EC144 (a derivative of BIIB021) as the HSP90 ligand and incorporated it into bifunctional molecules, which they dubbed heat shock protein 90 (HSP90)–mediated targeting chimeras (HEMTACs), with the clinical CDK4/6 inhibitor palbociclib.76 They observed CDK4/6 degradation in B16F10 cells and substantial tumor growth inhibition (TGI) in a B16F10 xenograft model in 57BL/6J mice with both 20 and 40 mg kg−1 IP QD dosing of HEMTAC 26. While efficacy was superior compared to the HPS90 inhibitor BIIB021 alone, the bifunctional degraders were not benchmarked to palbociclib in the efficacy study. Notably, the authors also demonstrated preliminary support for potential tumor-selective effects due to the overexpression of both HSP90 and CDK4/6 in tumors. Qin and co-workers attached various AR ligands to ganetespib-derived HSP90 ligands with a variety of linkers and observed antiproliferative activity in both LNCaP and 22RV1 cells, the latter of which is Enzalutamide resistant and expresses a resistance-associated truncated variant of full-length AR: AR-V7.77 One key finding was that extracellular HSP90 seemed to facilitate the uptake of these bifunctional degraders. SQA-710 showed ∼50% TGI in a LNCap xenograft model in Balb/c nude mice with 5 mg kg−1 IP QoD dosing. Intriguingly, both AR-FL and AR-V7 downregulation was observed. It is unclear how the V7 form, which lacks the canonical binding site for AR antagonists, would be recruited directly to HSP90 with these bifunctional molecules, so perhaps an alternative mechanism is responsible.
Direct recruitment to the proteasome
The proteasome is the most essential and complex component of the UPS. It is also the machinery that ultimately causes protein degradation.78 It is ubiquitously expressed and consists of a barrel-shaped proteolytic core (20S) which can be capped by two 19S regulatory particles to form the 26S proteasome (Fig. 6A).79,80 The 19S recognizes ubiquitinated proteins, deubiquitinates them, and starts to unfold them to feed them into 20S for degradation. The 19S consists of two subcomplexes: a ring base that contains six ATPase subunits (Rpt1 to Rpt6) which connect the 19S to the 20S and a lid of non-ATPase subunits (Rpn1 to Rpn13) involved in substrate recognition and deubiquitination. The 20S contains four stacked rings: two outer rings comprised of α subunits (α1 to α7) that create a “gate” for protein entry and two inner rings comprised of β subunits (β1 to β7) that form the proteolytically active sites. | ||
| Fig. 6 A. The 26S proteasome assembly with subunits targeted to establish POC for ubiquitin-independent degradation via induced proximity to the proteasome highlighted in purple. B. Rapamycin-induced heterodimerization of proteasomal subunits Rpn2, Rpn10, and Rpt5 with His3 reveals degradation upon recruitment to Rpn10. C. Induced dimerization of using HaloTagging to recruit BRD2 to Rpn13 with JQ1 leads degradation of BRD2. | ||
While the canonical mechanism for proteasomal degradation involves recognition of a ubiquitinated substrate, evidence of ubiquitin-independent degradation in endogenous settings has been presented.81–85 The ability for a protein to be degraded in a ubiquitin-independent manner is hypothesized to potentially be reliant upon intrinsically disordered regions (IDRs) as well as patches of hydrophobicity. Indeed, direct recruitment to the 20S is one mechanism by which hydrophobic tag-based protein degradation (HyT-PD) is reported to occur.3,86 Several examples of artificially induced proximity to the proteasome have also demonstrated ubiquitin-independent proteasomal degradation. Wilmington and Matouschek collaborated to discover that chemical inducers of dimerization (CIDs) can induce proteasomal degradation in the absence of ubiquitin by inducing proximity to the proteasome with a Ubl domain.87 Janse and coworkers also demonstrated ubiquitin-independent degradation through induced proximity (Fig. 6B).88 By fusing Fpr1 with proteasomal subunits Rpn2, Rpn10, and Rpt5, and fusing Tor with the target protein His3, rapamycin can induce dimerization. Authors found that induced proximity with Rpn10 caused His3 degradation, but induced proximity with Rpn2 or Rpt5 did not. Authors also attempted fusions with several other subunits, but found them to be lethal presumably because they inhibited functional proteasome assembly. Rpn13 is a ubiquitin receptor within the lid of the 19S that recognizes ubiquitinated proteins marked for degradation. More recently, induced proximity to the proteasomal subunit Rpn13 via a HaloTagging approach also led to the successful degradation of BRD2 upon surveying a small set of PEG-based linker lengths (Fig. 6C).89
Recruiting directly to the proteasome to affect TPD would bypass several of the complex cascades required of more traditional PROTACs that target E3 substrate receptor proteins (see Fig. 7 inset). E3 ligase assembly, substrate receptor occupancy of the necessary adaptors, Ub transfer geometry, the requirement of a suitable lysine on the protein of interest, and competing de-ubiquitination from deubiquitinases (DUBs) could, in theory, become non-issues. Tissue and sub-cellular co-localization between the E3 and the protein of interest would likely also be non-issues due to the ubiquitous expression of proteasomes. While efforts to recruit to the proteasome are comparatively less developed than recruitment to E3 substrate receptors, several groups have recognized the potential of this approach and have demonstrated it is feasible.
 | ||
| Fig. 7 A. Peptidic macrocycle (MC1) is a ligand for Rpn1 that, when connected to a BRD4 (BETi) ligand through a PEG linker installed via click chemistry, can degrade BRD4. B. Two RPN11 ligand-based bifunctional molecules can successfully degrade BRD4 in a proteasome dependent manner. C. Non-covalent ligand for Rpn13 (TCL-1) was also elaborated into a BRD4 bifunctional degrader. D. A, B, and C all aim to affect degradation via direct recruitment to the proteasome and bypass the need for E3 recruitment and ubiquitination. | ||
Bashore and coworkers discovered a potent, cell permeable macrocyclic peptide MC1 that binds to Rpn1 (PSMD2) (Fig. 7).90 Cryo-EM guided the incorporation of MC1 into BRD4 ligand-linked Chemical Inducers of Degradation (CIDEs) that maintained both Rpn1 and BRD4 binding as well as cellular permeability. Degradation of BRD4 in HEK293 cells was found to be proteasome dependent as well as dependent upon Rpn1 binding based on a negative control for MC1 possessing D-amino acids and BRD4 binding via competition experiments conducted in the presence of a BET inhibitor.
Rpn11 (PSMD14) is an essential DUB in the lid subcomplex of the 19S.91 It is a zinc-based metalloprotease responsible for cleaving ubiquitin from polyubiquitinated proteins before they enter the 20S. Orthosteric inhibitors for Rpn11 were identified by Deshaies and Cohen by screening a metal-binding pharmacophore library and subsequent optimization of an 8-thioquinoline hit leading to the identification of capzimin (Fig. 7).92,93 Ciulli and coworkers then incorporated these bidentate ligands into bifunctional molecules targeting proteins including BRD4 (11JQ15 and 11JQ16) and kinases while capping the thiol as a thioester prodrug, presumably to enhance cellular permeability.94 Degradation of BRD2, 3, and 4 was observed to be proteasome dependent upon co-treatment with bortezomib, and competition studies with JQ1 and capzimin provided further support to the proposed mechanism. Finally, they demonstrated that the bifunctional degraders maintained binding to Rpn11 by isothermal calorimetry (ITC). Ternary complex formation to validate a direct recruitment event between Rpn11 and BRD4 was not reported.
Rpn13 (ADMR1) is a non-essential ubiquitin receptor on the 19S.95 While several covalent or peptidic Rpn13 inhibitors have been previously reported, an acid-containing non-covalent ligand TCL-1 which binds weakly to Rpn13's Pru domain was recently disclosed (NMR KD ∼ 26 μM).96–99 Interestingly, the acid did not prove essential for binding as the ester was reported to retain binding. Using this acid as a functional group for elaboration, Rpn13-based bypassing E3 targeting chimeras (ByeTACs) containing JQ1 (TEC3 and TEC4) were prepared and evaluated (Fig. 7).100 Mechanistic studies show degradation is proteasome dependent, ubiquitin-independent, and Rpn 13 dependent. Once again, however, ternary complex formation is not shown.
In addition to the dozens of essential subunits within the 26S proteasome, there are also multiple types of proteasome-associated proteins.101 Among these are proteasome-associated DUBs including USP14 and UCH37 (UCHL5).102 USP14 and UCH37 are both cysteine-based DUBs associated with the 19S: USP14 associates with proteasomal subunit Rpn1 (PSMD2) and UCH37 associates with proteasomal subunit Rpn13. Interests in DUBs as therapeutic targets implicated in human cancers and neurodegenerative diseases have motivated the pursuit of DUB inhibitors.103 The roughly 100 DUBs present in humans can be broadly categorized into two families: cysteine proteases and metalloproteases. In contrast to Rpn 11, (vide supra) which is a zinc-containing metalloprotease, USP14 and UCH37 are both cysteine protease DUBs. While non-covalent inhibitors of USP14 have been reported (see IU-1 in Fig. 8A), most inhibitors that are reported to target UCH37 are promiscuous DUB inhibitors that rely on the modification of the active site cysteine with reversible or irreversible covalent warheads like b-AP15 and WP1130 (Fig. 8A).104–109
 | ||
| Fig. 8 A. DUB inhibitors known to inhibit the function of proteasome associated DUBs USP14 and UCH37. B. Bifunctional degraders against a variety of targets featuring a similar reversible covalent warhead. Compound A39 notable demonstrates both plasma and brain exposure after i.v. administration. Later compounds (below) showcase a further truncated reversible covalent warhead. | ||
A series of patents from Testa and coworkers appear to be leveraging similar warheads in bifunctional degraders against a variety of targets including BRD4, CDK9, PARP1, Kras G12C, mutant EGFR, ER, AR, and SMARCA (Fig. 8B).110–113 While a specific UPS target is not implicated in the patent, the inventors state that these degraders may cause degradation “by acting to bring the target protein into proximity with a proteasome”.92 Many of these bifunctional degraders are small in size compared to more typical CRBN- and VHL-based bifunctional degraders. In addition to demonstrating the ability to degrade target proteins, inventors demonstrated exposure of A39 in both plasma and brain. This is in contrast to CRBN- and VHL-based bifunctional degraders, which typically do not allow for CNS penetration.92 Subsequent applications have disclosed a truncated warhead with a simple t-butyl terminus (Fig. 8B), which may further improve the physicochemical properties of these streamlined bifunctional degraders. While the exact mechanism for these degraders has not been reported, they could, in theory, work by modifying the catalytic cysteine in a proteasome-associated DUB, thereby leading to direct recruitment of a target to the proteasome for degradation. Based on the simple structure of these warheads it seems unlikely that these would be highly selective for proteasome associated DUBs. Perhaps, due to this presumed promiscuity, the reversible nature of these covalent warheads is essential to allow the warhead to reversibly modify unproductive cysteines before eventually binding to a productive (e.g. proteasome-associated DUB) cysteine.
Conclusion
As the field of TPD has grown over the past several years, a wide array of degradation approaches have blossomed that co-opt both the autophagy-lysosome system and the UPS. Within the UPS, small molecule degrader efforts have focused on E3 ligases, and, most intensely, on the specific E3 ligase components known to interact with endogenous substrates (e.g. substrate receptor proteins). While recruitment to these types of UPS components is the most advanced and characterized approach, limitations to its utility have been revealed. Certain targets may not be amenable to TPD with a specific E3 substrate receptor due to lack of co-localization either within the cell or more broadly in terms of cell or tissues type. Recruitment to chaperones that associate with multiple E3s, such as HSP90, can potentially avoid such E3-substrate mismatches. E3s can have redundant functions, and, if an E3 is not essential, degraders co-opting them may be more susceptible to a range of resistance from mutations up to and including entire deletion. Recruitment to essential UPS components including E2s and substrate adaptors like SKP1 and DDB1 can potentially address these mechanisms of resistance. When a target is recruited to an E3, a complex series of steps must still occur to lead to productive degradation: functionally active E3 assembly, efficient poly-ubiquitination, and successful recruitment to the proteasome before de-ubiquitination occurs. All of these steps could be bypassed by direct recruitment to the proteasome or proteasome-associated proteins.Limitations presented by E3 substrate receptors have driven pioneers on the TPD space to explore alternative methods of recruiting to the UPS for TPD beyond E3 substrate receptors. These efforts to recruit to other UPS target classes is comparatively nascent with each class presenting its own unique set of challenges and opportunities. First and foremost, the identification of selective, drug-like ligands for these UPS components is required. Much like E3 substrate receptors, many of these alternative UPS components serve scaffolding functions and lack highly druggable pockets. For families of targets that serve a distinct catalytic function, such as E2s or DUBs, which can possess conserved catalytic machinery, achieving suitable selectivity is both challenging and crucial. Of particular concern for direct-to-proteasome approaches, ligand binding needs to preserve proteasome assembly and function. Historically, small molecule chemists have encountered challenges of comparable difficulty and succeeded in identifying drug-like, selective chemical matter. Given the high potential value presented by hijacking these alternative UPS components and the intense investment in protein homeostasis research across academic and industrial communities, it is likely that chemists will succeed again. As our understanding of these targets increases and the chemical tools available to interrogate them improve, they may well be representative of the next frontier for TPD.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review. The primary data for all results discussed has been properly cited.Conflicts of interest
There is no conflict of interest to declare.References
- M. Békés, D. R. Langley and C. M. Crews, Nat. Rev. Drug Discovery, 2022, 21, 181–200 CrossRef PubMed.
- K. T. G. Samarasinghe and C. M. Crews, Cell Chem. Biol., 2021, 28, 934–951 CrossRef CAS PubMed.
- Q. He, X. Zhao, D. Wu, S. Jia, C. Liu, Z. Cheng, F. Huang, Y. Chen, T. Lu and S. Lu, Eur. J. Med. Chem., 2023, 260, 115741 CrossRef CAS PubMed.
- G. Spagnolli, T. Massignan, A. Astolfi, S. Biggi, M. Rigoli, P. Brunelli, M. Libergoli, A. Ianeselli, S. Orioli, A. Boldrini, L. Terruzzi, V. Bonaldo, G. Maietta, N. L. Lorenzo, L. C. Fernandez, Y. B. Codeseira, L. Tosatto, L. Linsenmeier, B. Vignoli, G. Petris, D. Gasparotto, M. Pennuto, G. Guella, M. Canossa, H. C. Altmeppen, G. Lolli, S. Biressi, M. M. Pastor, J. R. Requena, I. Mancini, M. L. Barreca, P. Faccioli and E. Biasini, Commun. Biol., 2021, 4, 62 CrossRef CAS PubMed.
- M. Słabicki, H. Yoon, J. Koeppel, L. Nitsch, S. S. Roy Burman, C. Di Genua, K. A. Donovan, A. S. Sperling, M. Hunkeler, J. M. Tsai, R. Sharma, A. Guirguis, C. Zou, P. Chudasama, J. A. Gasser, P. G. Miller, C. Scholl, S. Fröhling, R. P. Nowak, E. S. Fischer and B. L. Ebert, Nature, 2020, 588, 164–168 CrossRef PubMed.
- J. Kim, I. Byun, D. Y. Kim, H. Joh, H. J. Kim and M. J. Lee, Chem. Soc. Rev., 2024, 53, 3253–3272 RSC.
- S. Ghosh, B. Ramadas and D. Manna, RSC Med. Chem., 2022, 13, 1476–1494 RSC.
- D. VanDyke, J. D. Taylor, K. J. Kaeo, J. Hunt and J. B. Spangler, Curr. Opin. Biotechnol., 2022, 78, 102807 CrossRef CAS PubMed.
- J. M. Sasso, R. Tenchov, D. Wang, L. S. Johnson, X. Wang and Q. A. Zhou, Biochemistry, 2023, 62, 601–623 CrossRef CAS PubMed.
- D. C. Scott, V. O. Sviderskiy, J. K. Monda, J. R. Lydeard, S. Ei Cho, J. W. Harper and B. A. Schulman, Cell, 2014, 157, 1671–1684 CrossRef CAS PubMed.
- K. Baek, D. C. Scott and B. A. Schulman, Curr. Opin. Struct. Biol., 2021, 67, 101–109 CrossRef CAS PubMed.
- D. Chirnomas, K. R. Hornberger and C. M. Crews, Nat. Rev. Clin. Oncol., 2023, 20, 265–278 CrossRef CAS PubMed.
- P. Jevtić, D. L. Haakonsen and M. Rapé, Cell Chem. Biol., 2021, 28, 1000–1013 CrossRef PubMed.
- P. Ottis, C. Palladino, P. Thienger, A. Britschgi, C. Heichinger, M. Berrera, A. Julin-Laferriere, F. Roudnicky, T. Kam-Thong, J. R. Bischoff, B. Martoglio and P. Pettazzoni, ACS Chem. Biol., 2019, 14, 2215–2223 CAS.
- L. T. Kramer and X. Zhang, Curr. Res. Chem. Biol., 2022, 2, 100020 CrossRef CAS.
- S. Tateno, M. Iida, S. Fujii, T. Suwa, M. Katayama, H. Tokuyama, J. Yamamoto, T. Ito, S. Sakamoto, H. Handa and Y. Yamaguchi, Sci. Rep., 2020, 10, 4012 CrossRef CAS PubMed.
- Y. Prozzillo, G. Fattorini, M. V. Santopietro, L. Suglia, A. Ruggiero, D. Ferreri and G. Messina, Biology, 2020, 9, 421–436 CrossRef CAS PubMed.
- J. O. Cresser-Brown, G. P. Marsh and H. J. Maple, Curr Opin Pharmacol., 2021, 59, 43–51 CrossRef CAS PubMed.
- G. M. Burslem and C. M. Crews, Cell, 2020, 181, 102–114 CrossRef CAS PubMed.
- D. L. Daniels, K. M. Riching and M. Urh, Drug Discovery Today:Technol., 2019, 31, 61–68 CrossRef PubMed.
- K. M. Riching, S. Mahan, C. R. Corona, M. McDougall, J. D. Vasta, M. B. Robers, M. Urh and D. L. Daniels, ACS Chem. Biol., 2018, 13, 2758–2770 CrossRef CAS PubMed.
- R. Hjerpe, F. Aillet, F. Lopitz-Otsoa, V. Lang, P. England and M. S. Rodriguez, EMBO Rep., 2009, 10, 1250–1258 CrossRef CAS PubMed.
- H.-T. Huang, D. Dobrovolsky, J. Paulk, G. Yang, E. L. Weisberg, Z. M. Doctor, D. L. Buckley, J.-H. Cho, E. Ko, J. Jang, K. Shi, H. G. Choi, J. D. Griffin, Y. Li, S. P. Treon, E. S. Fischer, J. E. Bradner, L. Tan and N. S. Gray, Cell Chem. Biol., 2018, 25, 88–99 CrossRef CAS PubMed.
- D. P. Bondeson, B. E. Smith, G. M. Burslem, A. D. Buhimschi, J. Hines, S. Jaime-Figueroa, J. Wang, B. D. Hamman, A. Ishchenko and C. M. Crews, Cell. Chem. Biol., 2018, 25, 78–87 CrossRef CAS PubMed.
- G. Sathe and G. P. Sapkota, Trends Pharm. Sci., 2023, 44, 786–801 CrossRef CAS PubMed.
- M. D. Stewart, T. Ritterhoff, R. E. Klevit and P. S. Brzovic, Cell Res., 2016, 26, 423–440 CrossRef CAS PubMed.
- R. K. McGinty, R. C. Henrici and S. Tan, Nature, 2014, 514, 591–596 CrossRef CAS PubMed.
- V. Bernier-Villamor, D. A. Sampson, M. J. Matunis and C. D. Lima, Cell, 2002, 108, 345–356 CrossRef CAS PubMed.
- K.-S. Ryu, Y.-S. Choi, J. Ko, S.-O. Kim, H. J. Kim, H.-K. Cheong, Y. H. Jeon, B.-S. Choi and C. Cheong, BMB Rep., 2008, 41, 852857 Search PubMed.
- A. Hershko, H. Heller, S. Elias and A. Ciechanover, J. Biol. Chem., 1983, 258, 8206–8214 CrossRef CAS PubMed.
- G. S. McDowell and A. Philpott, Int. J. Biochem. Cell Biol., 2013, 45, 1833–1842 CrossRef CAS PubMed.
- J. D. Taylor, N. Barrett, S. M. Cuesta, K. Cassidy, F. Pachl, J. Dodgson, R. Patel, T. M. Eriksson, A. Riley, M. Burrell, C. Bauer, D. G. Rees, R. Cimbro, A. X. Zhang, R. R. Minter, J. Hunt and S. Legg, Commun. Biol., 2024, 7, 1179 CrossRef CAS PubMed.
- C. D. Hodge, R. A. Edwards, C. J. Markin, D. McDonald, M. Pulvino, M. S. Y. Huen, J. Zhao, L. Spyracopoulos, M. J. Hendzel and J. N. M. Glover, ACS Chem. Biol., 2015, 10, 1718–1728 CrossRef CAS PubMed.
- S. Strickson, D. G. Campbell, C. H. Emmerich, A. Knebel, L. Plater, M. S. Ritorto, N. Shpiro and P. Cohen, Biochem. J., 2013, 451, 427–437 CrossRef CAS PubMed.
- M. Pulvino, Y. Liang, D. Oleksyn, M. DeRan, E. Van Pelt, J. Shapiro, I. Sanz, L. Chen and J. Zhao, Blood, 2012, 120, 1668–1677 CrossRef CAS PubMed.
- E. A. King, Y. Cho, N. S. Hsu, D. Dovala, J. M. McKenna, J. A. Tallarico, M. Schire and D. K. Nomura, Cell Chem. Biol., 2023, 30, 394–402 CrossRef CAS PubMed.
- Y.-D. Li, M. W. Ma, M. M. Hassan, M. Hunkeler, M. Teng, K. Puvar, J. C. Rutter, R. J. Lumpkin, B. Sandoval, C. Y. Jin, A. M. Schmoker, S. B. Ficarro, H. Cheong, R. J. Metivier, M. Y. Wang, S. Xu, W. S. Byun, B. J. Groendyke, I. You, L. H. Sigua, I. Tavares, C. Zou, J. M. Tsai, P. M. C. Park, H. Yoon, F. C. Majewski, H. T. Sperling, J. A. Marto, J. Qi, R. P. Nowak, K. A. Donovan, M. Słabicki, N. S. Gray, E. S. Fischer and B. L. Ebert, Nat. Chem. Biol., 2024, 10, 1640–1649 CrossRef PubMed.
- N. Forte, D. Dovala, M. J. Hesse, J. M. McKenna, J. A. Tallarico, M. Schirle and D. K. Nomura, ACS Chem. Biol., 2023, 18, 897–904 CrossRef CAS PubMed.
- S. Ponnusamy, Y. He, D.-J. Hwang, T. Thiyagarajan, R. Houtman, V. Bocharova, B. G. Sumpter, E. Fernandez, D. Johnson, Z. Du, L. M. Pfeffer, R. H. Getzenberg, I. J. McEwan, D. D. Miller and R. Narayanan, Clin. Cancer Res., 2019, 25, 6764–6780 CrossRef CAS PubMed.
- S. Gooding, N. Ansari-Pour, F. Towfic, M. Ortiz Estévez, P. P. Chamberlain, K.-T. Tsai, E. Flynt, M. Hirst, D. Rozelle, P. Dhiman, P. Neri, K. Ramasamy, N. Bahlis, P. Vyas and A. Thakurta, Blood, 2021, 137, 232–237 CrossRef CAS PubMed.
- A. Hanzl, R. Casement, H. Imrichova, S. J. Hughes, E. Barone, A. Testa, S. Bauer, J. Wright, M. Brand, A. Ciulli and G. E. Winter, Nat. Chem. Biol., 2023, 19, 323–333 CrossRef CAS PubMed.
- A. Scrima, R. Koníčková, B. K. Czyzewski, Y. Kawasaki, P. D. Jeffrey, R. Groisman, Y. Nakatani, S. Iwai, N. P. Pavletich and N. H. Thomä, Cell, 2008, 135, 1213–1223 CrossRef CAS PubMed.
- B. Iovine, M. L. Iannella and M. A. Bevilacqua, Int. J. Biochem. Cell Biol., 2011, 43, 1664–1667 CrossRef CAS PubMed.
- J. Lee and P. Zhou, Mol. Cell, 2007, 26, 775–780 CrossRef CAS PubMed.
- C. Mahon, N. J. Krogan, C. S. Craik and E. Pick, Biomolecules, 2014, 4, 897–930 CrossRef PubMed.
- M. S. Łabicki, Z. Kozicka, G. Petzold, Y.-D. Li, M. Manojkumar, R. D. Bunker, K. A. Donovan, Q. L. Sievers, J. Koeppel, D. Suchyta, A. S. Sperling, E. C. Fink, J. A. Gasser, L. R. Wang, S. M. Corsello, R. S. Sellar, M. Jan, D. Gillingham, C. Scholl, S. Fröhling, T. R. Golub, E. S. Fischer, N. H. Thomä and B. L. Ebert, Nature, 2020, 585, 293–297 CrossRef PubMed.
- C. Mayor-Ruiz, S. Bauer, M. Brand, Z. Kozicka, M. Siklos, H. Imrichova, I. H. Kaltheuner, E. Hahn, K. Seiler, A. Koren, G. Petzold, M. Fellner, C. Bock, A. C. Müller, J. Zuber, M. Geyer, N. H. Thomä, S. Kubicek and G. E. Winter, Nat. Chem. Biol., 2020, 16, 1199–1207 CrossRef CAS PubMed.
- T. Houles, J. Boucher, G. Lavoie, G. MacLeod, S. Lin, S. Angers and P. P. Roux, Cell Death Discovery, 2023, 9, 459–467 CrossRef CAS PubMed.
- R. Jorda, L. Havlíček, M. Peřina, V. Vojácková, T. Pospíšil, S. Djukic, J. Skerlová, J. Grúz, N. Renešová, P. Řezáčová, M. Strnad and V. Kryštof, J. Med. Chem., 2022, 65, 8881–8896 CrossRef CAS PubMed.
- K. L. Thomas, H. Bouguenina, D. S. J. Miller, F. J. Sialana, T. G. Hayhow, J. S. Choudhary, O. W. Rossanese and B. R. Bellenie, ACS Chem. Biol., 2024, 19, 173–184 CrossRef CAS PubMed.
- L. Lv, P. Chen, L. Cao, Y. Li, Z. Zeng, Y. Cui, Q. Wu, J. Li, J.-H. Wang, M.-Q. Dong, X. Qi and T. Han, eLife, 2022, 9, e599994 Search PubMed.
- S. M. Dieter, C. Siegl, P. L. Codó, M. Huerta, A. L. Ostermann-Parucha, E. Schulz, M. K. Zowada, S. Marin, K. Laaber, A. Nowrouzi, M. Blatter, S. Kreth, F. Westermann, A. Benner, U. Uhrig, K. Putzker, J. Lewis, A. Haegebarth, D. Mumberg, S. J. Holton, J. Weiske, L.-M. Toepper, U. Scheib, G. Siemeister, C. R. Ball, B. Kuster, G. Stoehr, H. Hahne, S. Johannes, M. Lange, F. Herbst and H. Glimm, Cell Rep., 2021, 36, 109394 CrossRef CAS PubMed.
- Z. Kozicka, D. J. Suchyta, V. Focht, G. Kempf, G. Petzold, M. Jentzsch, C. Zou, C. Di Genua, K. A. Donovan, S. Coomar, M. Cigler, C. Mayor-Ruiz, J. L. Schmid-Burgk, D. Häussinger, G. E. Winter, E. S. Fischer, M. Slabicki, D. Gillingham, B. L. Ebert and N. H. Thomä, Nat. Chem. Biol., 2024, 20, 93–102 CrossRef CAS PubMed.
- G. Winter, C. Mayor Ruiz and S. Kubicek, WO Pat., 2021074414A1, 2020 Search PubMed.
- J. Liu, M. B. Plewe, M. R. Lee, X. Han, L. Chen, C. Zhang and J. Wang, WO Pat., 2021239117A1, 2021 Search PubMed.
- M. Meyers, S. Cismoski, A. Panidapu, B. Chie-Leon and D. K. Nomura, ACS Chem. Biol., 2024, 19, 58–68 CrossRef CAS PubMed.
- C. Bai, P. Sen, K. Hofmann, L. Ma, M. Goebl, J. W. Harper and S. J. Elledge, Cell, 1996, 86, 263–274 CrossRef CAS PubMed.
- G. A. Cope and R. J. Deshaies, BMC Biochem., 2006, 7, 1 CrossRef PubMed.
- B. J. Pinch, D. L. Buckley, S. Gleim, S. M. Brittan, L. Tandeske, P. L. D'Alessandro, Z. J. Hauseman, J. Lipps, L. Xu, E. P. Harvey, M. Schirle, E. R. Sprague, W. C. Forrester, D. Dovala, L. M. McGregor and C. R. Thoma, Cell Chem. Biol., 2022, 29, 57–66 CrossRef CAS PubMed.
- S. H. Hong, A. Divakaran, A. Osa, O. W. Huang, I. E. Wertz, D. K. Nomura and A. C. S. Chem, Biologia, 2024, 19, 442–450 CAS.
- Y.-Q. Liu, X.-L. Wang, X. Cheng, Y.-Z. Lu, G.-Z. Wang, X.-C. Li, J. Zhang, Z.-S. Wen, Z.-L. Huang, Q.-L. Gao, L.-N. Yang, Y.-X. Cheng, S.-C. Tao, J. Liu and G.-B. Zhou, Onco Targets Ther, 2015, 6, 34953–34967 Search PubMed.
- M. Hussain, Y. Lu, M. Tariq, H. Jiang, Y. Shu, S. Luo, Q. Zhu, J. Zhang and J. Liu, iScience, 2022, 25, 104591 CrossRef CAS PubMed.
- B. J. Lang, T. L. Prince, Y. Okusha, H. Bunch and S. K. Calderwood, Biochim. Biophys. Acta, Mol. Cell Res., 2022, 1896, 119187 CrossRef PubMed.
- M. Taipale, I. Krykbaeva, M. Koeva, C. Kayatekin, K. D. Westover, G. I. Karras and S. Lindquist, Cell, 2012, 150, 9871001 CrossRef PubMed.
- R. Rosenzweig, N. B. Nillegoda, M. P. Mayer and B. Bukau, Nat. Rev. Mol. Cell Biol., 2019, 20, 665–680 CrossRef CAS PubMed.
- M. R. Fernández-Fernández, M. Gragera, L. Ochoa-Ibarrola, L. Quintana-Gallardo and J. María Valpuesta, FEBS Lett., 2017, 591, 2648–2660 CrossRef PubMed.
- S. Reeg, T. Jung, J. P. Castro, K. J. A. Davies, A. Henze and T. Grune, Free Radical Biol. Med., 2016, 99, 153–166 CrossRef CAS PubMed.
- Z.-N. Li and Y. Luo, Oncol. Rep., 2022, 49, 6 CrossRef PubMed.
- S. Chandarlapaty, A. Sawai, Q. Ye, A. Scott, M. Silinski, K. Huang, P. Fadden, J. Partdrige, S. Hall, P. Steed, L. Norton, N. Rosen and D. B. Solit, Clin. Cancer Res., 2008, 14, 240–248 CrossRef CAS PubMed.
- T. Shimamura, S. A. Perera, K. P. Foley, J. Sang, S. J. Rodig, T. Inoue, L. Chen, D. Li, J. Carretero, Y.-C. Li, P. Sinha, D. C. Carey, C. L. Borgman, J.-P. Jimenez, M. Meyerson, W. Ying, J. Barsoum, K.-K. Wong and G. I. Shapiro, Clin. Cancer Res., 2012, 18, 4973–4985 CrossRef CAS PubMed.
- W. Ying, L. Ye and K. P. Forley, WO Pat., 2020207395, 2020 Search PubMed.
- W. Ying, L. Ye and K. P. Forley, WO Pat., 2020207396, 2020 Search PubMed.
- W. Ying, K. P. Foley, M. Wang, C. Ying, L. Ye, W. Yin and L. Zhang, WO Pat., 2022, 2022078414 Search PubMed.
- W. Ying, K. P. Foley, C. Ying, Y. Wang, Y. Dai, G. Wang, W. Yin, J. Li, T. Prince and Z. Wang, WO Pat., 2024044334, 2024 Search PubMed.
- W. Ying, K. P. Foley, M. Wang, C. Ying, L. Ye, W. Yin and L. Zhang, WO Pat., 2022078350, 2022 Search PubMed.
- Z. Li, S. Ma, L. Zhang, S. Zhang, Z. Ma, L. Du and M. Li, J. Med. Chem., 2023, 66, 733–751 CrossRef CAS PubMed.
- S. Zhang, X. Meng, S. Zhang, B. Li, W. Jin, C. Zhang, Z. Liu, X. Hu, L. Ge, Z. Yu, Z. Li, S. Ma, X. Wang, L. Li and C. Qin, J. Med. Chem., 2023, 66, 4784–4801 CrossRef CAS PubMed.
- C. Arkinson, K. C. Dong, C. L. Gee and A. Martin, Nat. Rev. Mol. Cell Biol., 2024 DOI:10.1038/s41580-024-00778-0.
- G. C. Lander, E. Estrin, M. E. Matyskiela, C. Bashore, E. Nogales and A. Martin, Nature, 2012, 482, 186–193 CrossRef CAS PubMed.
- T. A. Thibaudeau and D. M. Smith, Pharmacol. Rev., 2019, 71, 170–197 CrossRef CAS PubMed.
- P. Coffino, Nat. Rev. Mol. Cell Biol., 2001, 2, 188–194 CrossRef CAS PubMed.
- I. Jariel-Encontre, G. Bossis and M. Piechaczyk, Biochim. Biophys. Acta, Gen. Subj., 2008, 1786, 153–177 CAS.
- G. Ben-Nissan and M. Sharon, Biomolecules, 2014, 4, 862–884 CrossRef PubMed.
- L.-S. Hsieh, W.-M. Su, G.-S. Han and G. M. Carman, J. Biol. Chem., 2015, 290, 11467–11478 CrossRef CAS PubMed.
- W. He, H. P. Rao, S. Tang, N. Bhagwat, D. S. Kulkarni, Y. Ma, M. A. W. Chang, C. Hall, J. W. Bragg, H. S. Manasca, C. Baker, G. F. Verhees, L. Ranjha, X. Chen, N. M. Hollingsworth, P. Cejka and N. Hunter, Mol. Cell, 2020, 78, 168–183 CrossRef CAS PubMed.
- Y. Shi, M. J. Long, M. M. Rosenberg, S. Li, A. Kobjack, P. Lessans, R. T. Coffey and L. Hedstrom, ACS Chem. Biol., 2016, 11, 3328–3337 CrossRef CAS PubMed.
- S. R. Wilmington and A. Matouschek, PLoS One, 2016, 11, e0152679 CrossRef PubMed.
- D. M. Janse, B. Crosas, D. Finley and G. M. Church, J. Biol. Chem., 2004, 279, 21415–21420 CrossRef CAS PubMed.
- M. Balzarini, W. Gui, I. M. Jayalath, B.-B. Schell, J. Tong and T. Kodadek, bioRxiv, 2023, preprint, DOI:10.1101/2023.07.19.549534.
- C. Bashore, S. Prakash, M. C. Johnson, R. J. Conrad, I. A. Kekessie, S. J. Scales, N. Ishisoko, T. Kleinheinz, P. S. Liu, N. Popovych, A. T. Weckler, L. Zhou, C. Tam, I. Zilberleyb, R. Srinivasan, R. A. Blake, A. Song, S. T. Staben, Y. Zhang, D. Arnott, W. J. Fairbrother, S. A. Foster, I. E. Wertz, C. Ciferri and E. C. Dueber, Nat. Chem. Biol., 2022, 19, 55–63 CrossRef PubMed.
- R. Verma, L. Arvind, R. Oania, W. H. McDonald, J. R. Yates III, E. V. Koonin and R. J. Deshaies, Science, 2002, 298, 611–615 CrossRef CAS PubMed.
- C. Perez, J. Li, F. Parlati, M. Rouffet, Y. Ma, A. L. Mackinnon, T.-F. Chou, R. J. Deshaies and S. M. Cohen, J. Med. Chem., 2017, 60, 1343–1361 CrossRef CAS PubMed.
- J. Li, T. Yakushi, F. Parlati, A. L. Mackinnon, C. Perez, Y. Ma, K. P. Carter, S. Colayco, G. Magnuson, B. Brown, K. Nguyen, S. Vasile, E. Suyama, L. H. Smith, E. Sergienko, A. B. Pinkerton, T. D. Y. Chung, A. E. Palmer, I. Pass, S. Hess, S. M. Cohen and R. J. Deshaies, Nat. Chem. Biol., 2017, 13, 486–493 CrossRef CAS PubMed.
- A. Ciulli, A. Testa, S. Hughes and S. P. Butcher, WO Pat., 2019238817, 2019 Search PubMed.
- K. Husnjak, S. Elsasser, N. Zhang, X. Chen, L. Randles, Y. Shi, K. Hofmann, K. J. Walters, D. Finley and I. Dikic, Nature, 2008, 453, 481–488 CrossRef CAS PubMed.
- R. K. Anchoori, B. Karanam, S. Peng, J. W. Wang, R. Jiang, T. Tanno, R. Z. Orlowski, W. Matsui, M. Zhao, M. A. Rudek, C.-F. Hung, X. Chen, K. J. Walters and R. B. S. Roden, Cancer Cell, 2013, 24, 791–805 CrossRef CAS PubMed.
- D. J. Trader, S. Simanski and T. Kodadek, J. Am. Chem. Soc., 2015, 137, 6312–6319 CrossRef CAS PubMed.
- X. Lu, V. R. Sabbasani, V. Osei-Amponsa, C. N. Evans, J. C. King, S. G. Tarasov, M. Dyba, S. Das, K. C. Chan, C. D. Schwieters, S. Choudhari, C. Fromont, Y. Zhao, B. Tran, X. Chen, H. Matsuo, T. Andresson, R. Chari, R. E. Swenson, N. I. Tarasova and K. J. Walters, Nat. Commun., 2021, 12, 7318 CrossRef CAS PubMed.
- C. A. Loy, C. S. Muli, E. M. H. Ali, D. Xie, M. H. Ahmed, C. B. Post and D. J. Trader, Bioorg. Med. Chem. Lett., 2023, 95, 129485 CrossRef CAS PubMed.
- E. M. H. Ali, C. A. Loy and D. J. Trader, bioRxiv, 2024, preprint, DOI:10.1101/2024.01.20.576376.
- D. S. Leggett, J. Hanna, A. Borodovsky, B. Crosas, M. Schmidt, R. T. Baker, T. Walz, H. Ploegh and D. Finley, Mol. Cell., 2002, 10, 495–507 CrossRef CAS PubMed.
- C. A. Loy, C. S. Muli, E. M. H. Ali, D. Xie, M. H. Ahmed, C. B. Post and D. J. Trader, Int. J. Mol. Sci., 2021, 22, 6213 CrossRef PubMed.
- J. A. Harrigan, X. Jacq, N. M. Martin and S. P. Jackson, Nat. Rev. Drug Discovery, 2018, 17, 57–78 CrossRef CAS PubMed.
- B.-H. Lee, M. J. Lee, S. Park, D.-C. Oh, S. Elsasser, P.-C. Chen, C. Gartner, N. Dimova, J. Hanna, S. P. Gygi, S. M. Wilson, R. W. King and D. Finley, Nature, 2010, 467, 179–184 CrossRef CAS PubMed.
- M. Boselli, B.-H. Lee, J. Robert, M. A. Prado, S.-W. Min, C. Cheng, M. C. Silva, C. Seong, S. Elsasser, K. M. Hatle, T. C. Gahman, S. P. Gygi, S. J. Haggarty, L. Gan, R. W. King and D. Finley, J. Biol. Chem., 2017, 292, 19209–19225 CrossRef CAS PubMed.
- Y. Wang, Y. Jiang, S. Ding, J. Li, N. Song, Y. Ren, D. Hong, C. Wu, B. Li, F. Wang, W. He, J. Wang and Z. Mei, Cell Res., 2018, 28, 1186–1194 CrossRef CAS PubMed.
- P. D'Arcy, S. Brnjic, M. H. Olofsson, M. Fryknäs, K. Lindsten, M. De Cesare, P. Perego, B. Sadeghi, M. Hassan, R. Larsson and S. Linder, Nat. Med., 2011, 17, 1636–1640 CrossRef PubMed.
- X. Wang, P. D'Arcy, T. R. Caulfield, A. Paulus, K. Chitta, C. Mohanty, J. Gullbo, A. Chanan-Khan and S. Linder, Chem. Biol. Drug Des., 2015, 86, 1036–1048 CrossRef CAS PubMed.
- V. Kapuria, L. F. Peterson, D. Fang, W. G. Bornmann, M. Talpaz and N. J. Donato, Cancer Res., 2010, 70, 9265–9276 CrossRef CAS PubMed.
- A. Testa, C. MacGregor, D. McGarry, G. Meier, I. Churcher and M. Mathieson, WO Pat., 2022129925, 2022 Search PubMed.
- A. Testa, C. MacGregor, G. Meier and D. McGarry, WO Pat., 2023242597, 2023 Search PubMed.
- A. Testa, C. MacGregor, G. Meier, C. Hamby and C. Fallan, WO Pat., 2023242598, 2023 Search PubMed.
- C. Hamby, G. Meier, J. Osborne, A. Testa, G. A. Brown, M. Ambler and C. Fallan, WO Pat., 2024457021, 2024 Search PubMed.
| This journal is © The Royal Society of Chemistry 2025 |