DOI:
10.1039/D4MD00645C
(Research Article)
RSC Med. Chem., 2025,
16, 296-311
Breaking boundaries in diabetic nephropathy treatment: design and synthesis of novel steroidal SGLT2 inhibitors†
Received
20th August 2024
, Accepted 13th October 2024
First published on 15th October 2024
Abstract
The activity of sodium glucose co-transporter 2 (SGLT2) has always been an important parameter influencing chronic kidney disease in type-2 diabetic patients. Herein, we have meticulously designed, synthesized, and evaluated several novel steroidal pyrimidine molecules that possess the capability to successfully bind to the SGLT2 protein and inhibit its activity, thereby remedying kidney-related ailments in diabetic patients. The lead steroidal pyrimidine compounds were selected after virtually screening from a library of probable N-heterocyclic steroidal scaffolds. A nano-catalyzed synthetic route was also explored for the synthesis of the steroidal pyrimidine analogs demonstrating an environmentally benign protocol. Extensive in vitro investigations encompassing SGLT2 screening assays and cell viability assessments were conducted on the synthesized compounds. Among the steroidal pyrimidine derivatives evaluated, compound 9a exhibited the highest SGLT2 inhibition activity and underwent further scrutiny. Western blot analysis was employed to determine the impact of 9a on inflammatory and fibrotic proteins, aiming to elucidate its mechanism of action. Additionally, in silico analyses were performed to illuminate the structural dynamics and molecular interaction mechanism of 9a. The overall investigation is crucial for advancing the development of the next generation of anti-diabetic drugs.
Introduction
Diabetic nephropathy (DN) stands as a significant microvascular complication of diabetes, often progressing to end-stage renal disease (ESRD) with a mortality rate 3 to 12 times higher than in patients without such complications.1 Chronic hyperglycemia deteriorates pancreatic β-cells,2 leading to a spectrum of macro and microvascular complications including nephropathy, neuropathy, retinopathy, and cardiovascular issues.3 Presently, DN ranks among the leading causes of kidney dialysis and transplantation. However, treatments specifically targeting diabetic kidney disease (DKD) remain elusive. Current therapeutic approaches focus on glycemic control, blood pressure regulation, and lipid management. Yet, it's recognized that these interventions alone may fall short in effectively managing the occurrence and progression of DKD.4 In this context, SGLT2 inhibitors are emerging as a novel therapeutic class with nephroprotective potential.
Sodium glucose co-transporters located in the proximal tubule of the kidneys have the ability to absorb all the glucose from the glomerular filtrate. Specifically, SGLT2 can absorb 90% of the total filtered glucose. It is found on the luminal surface of proximal tubule epithelial cells. Sodium glucose co-transporter 1 (SGLT1) is responsible for absorbing the remaining 10% and is situated on the luminal surface at the end of the proximal tubule.4–6 During chronic hyperglycemia, there is an increase in SGLT2 expression, causing the renal threshold of blood glucose to rise from a normal level of 10 mmol L−1 to 11–13 mmol L−1. This ultimately leads to heightened reabsorption of glucose and elevation in blood sugar levels.4,6 In recent years, promising research has enhanced our comprehension of the fundamental mechanisms of action of SGLT2 inhibitors. Randomized clinical trials involving diabetic patients at increased risk of vascular diseases have been conducted, supporting the beneficial use of SGLT2 inhibitors.7 Moreover, various reports suggest that SGLT2 inhibitors can effectively manage inflammation and fibrosis during diabetic nephropathy through different mechanisms.4,8,9
For instance, canagliflozin, an FDA-approved SGLT2 inhibitor, can directly or indirectly phosphorylate and activate AMP-activated protein kinase (AMPK).10 This enzyme serves as a key energy sensor in the body and mitigates oxidative stress by boosting the expression of antioxidants, thereby controlling multiple downstream effects.11 Oxidative stress plays a pivotal role in the pathogenesis of diabetic nephropathy. Chronic hyperglycemia triggers the activation of the reduced nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX) enzyme, leading to increased production of reactive oxygen species (ROS) and a state of oxidative stress. Over time, oxidative stress leads to renal inflammation, fibrosis, and vascular dysfunction.12–15 NOX-4, notably expressed in renal cells, emerges as the primary ROS source during diabetes, contributing to significant renal injury.12 The heightened production of ROS induces kidney inflammation and fibrosis, resulting in glomerular hypertrophy, mesangial expansion, and mitochondrial dysfunction.16
Additionally, numerous studies have highlighted the association between nuclear factor kappa B (NF-κB) activation and the progression of kidney inflammation, inducing various inflammatory and fibrotic target genes.8,17,18 NF-κB activation triggers the stimulation of several downstream targets and proinflammatory genes, including tumor necrosis factor-alpha (TNF-α) and interleukin 6 (IL-6), all implicated in diabetic nephropathy development.12,19 IL-6 plays a crucial role in promoting mesangial cell proliferation, expanding the extracellular matrix, and increasing endothelial cell permeability. Meanwhile, TNF-α has been observed to stimulate ROS production through a positive feedback loop.12,20 Consequently, NOX-4-induced ROS lead to the uncoupling of endothelial nitric oxide synthase (eNOS), reducing nitric oxide (NO) bioavailability in diabetes and initiating kidney fibrosis.12 ROS play a pivotal role in profibrotic pathways, including the stimulation of growth factors like transforming growth factor-beta-1 (TGF-β1). Elevated TGF-β1 expression, in conjunction with ROS, results in excessive extracellular matrix (ECM) build-up and the accumulation of proteins such as collagen IV and fibronectin, ultimately contributing to kidney dysfunction.21,22
The structures of several commercially available SGLT2 inhibitors, along with their brand names, are depicted in Fig. 1. Despite the availability of numerous commercial SGLT2 inhibitors, the search for enhanced and alternative medications persists. Notably, most SGLT2 inhibitors are primarily based on a glucitol backbone, with some variations incorporating a thiophene ring or a phenyl ring. Leveraging our prior experience in synthesizing biologically relevant steroidal derivatives from 16-dehydropregnenolone acetate,23,24 a key natural product derived from Dioscorea (i.e. wild yam), prompted us to explore the potential of steroidal scaffolds as SGLT2 inhibitors. The novel approach of developing an SGLT2 inhibitor without a glucitol backbone, sourced from a natural product, served as the central impetus for our investigation.
 |
| | Fig. 1 SGLT2 inhibition mechanism in the kidneys and some commercially available SGLT2 inhibitors along with the synthesized target drug (9a). | |
Results and discussion
Design and virtual screening of viable steroidal SGLT2 inhibitors
Discovering active compounds marks the pivotal outset for any drug discovery endeavor. These compounds ideally exhibit or have the capacity to exhibit upon refinement favorable physical chemistry and properties akin to drugs. Screening, encompassing both physical and virtual methods, entails probing a protein target extensively with numerous compounds to pinpoint promising starting points for drug development. Crafting an effective screening strategy necessitates a thorough understanding of the particular protein target.25–27 The expansive realm of steroidal chemistry motivated our exploration into virtually screening numerous promising heterocycles featuring the steroidal framework for potential binding to the SGLT2 protein. While there exists a multitude of heterocyclic compounds with a steroid core, our focus primarily centred on integrating pregnenolone as the foundation structure. As such, all structures considered for virtual screening shared the pregnenolone core. We endeavoured to introduce a nitrogen-containing heterocyclic group to mimic the function of glucitol in facilitating SGLT2 inhibition. Following an extensive literature review and conceptualization, a diverse library of steroidal N-heterocyclic compounds was subjected to virtual screening by high-precision molecular docking (Table S1†). Among these, six distinct classes of derivatives were identified based on their docking scores, and subsequent synthetic routes were devised accordingly. The central theme guiding the retrosynthesis pathway was rooted in 16-dehydropregnenolone acetate being the primary starting material as it can be directly sourced in bulk from a natural product. Our prior familiarity with synthesizing steroidal derivatives28 from 16-dehydropregnenolone acetate facilitated the retrosynthetic analysis for the mentioned compounds. Feasible pathways were consequently mapped out for the synthesis of the six intended SGLT2 inhibitor candidates (Fig. 2).
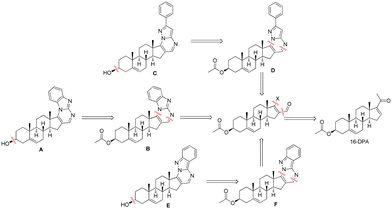 |
| | Fig. 2 Retrosynthesis of the six targeted SGLT2 inhibitors (screened by molecular docking). | |
Synthesis of the screened SGLT2 inhibitors
The commonality for all the intended target molecules was their starting steroidal precursor, 16-dehydropregnenolone acetate, and the synthesis process commenced with oximation, followed by Vilsmeier–Haack formylation, resulting in a distinctive β-formylenamide intermediate.29 This intermediate was then coupled with specific aminopyrazole (4a–e) or aminobenzimidazole (7a–c) units to yield steroidal pyrimidine derivatives 5a–e and 8a–d, respectively. Efforts were also made to incorporate an environment benign catalytic system for this particular synthetic step, by utilizing freshly prepared nickel(0) nanoparticles.30 Upon completion, the highly superparamagnetic nanocatalyst facilitated easy recovery via an external magnet, allowing for its reuse multiple times in the subsequent synthesis of other derivatives.
The remaining target molecules (6a–d and 9a–c) were obtained through mild base hydrolysis of their respective precursors (Scheme 1). Subsequently, the synthesized compounds underwent meticulous purification via column chromatography, with certain samples undergoing additional refinement through HPLC techniques to further improve their purity. The structure of the synthesized molecules was characterized and confirmed through 1HNMR, 13C{1H}NMR, and HRMS data before advancing to in vitro SGLT2 screening and cytotoxicity assays (Fig. 3).
 |
| | Scheme 1 Synthesis of targeted steroidal N-heterocyclic compounds. | |
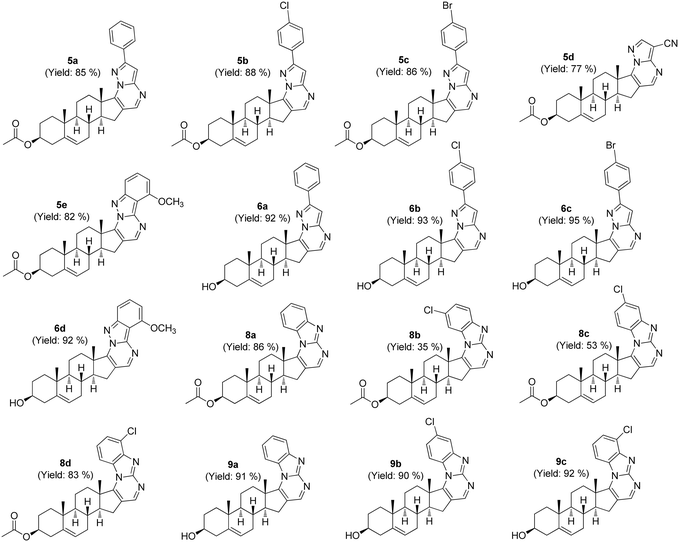 |
| | Fig. 3 Synthesized library of steroidal N-heterocycles. | |
In vitro screening of the synthesized steroidal compounds for SGLT2 and SGLT1 activity
Cytotoxicity assay of 5a–9c in NRK-52E kidney proximal tubular cells.
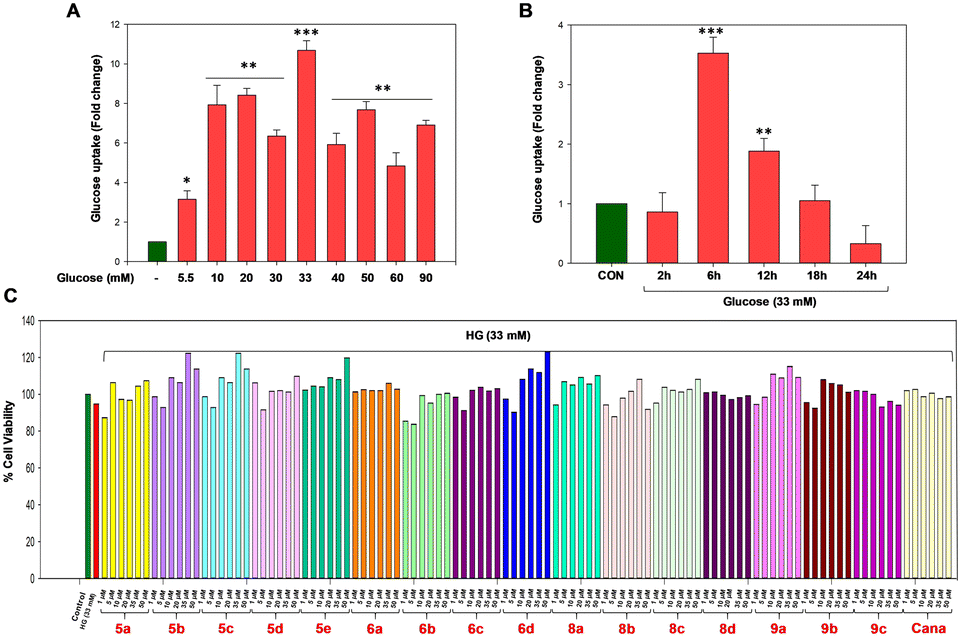
In the preliminary screening, the cytotoxic effects of compounds 5a–9c and the standard drug canagliflozin were assessed at various concentrations (1, 5, 10, 20, 35, and 50 μM) against NRK-52E kidney proximal tubular cells using the Alamar blue cell viability assay. The concentration and time-dependent studies pertaining to the high glucose treatment were also conducted and the results are shown in Fig. 4A and B. In NRK-52E cells cultured in 33 mM D-glucose medium for 6 hours, a significant increase in glucose uptake was observed (p < 0.001) and the results indicated that compounds 5a–9c exhibited no toxic effects on NRK-52E kidney proximal tubular cells, as evidenced by the lack of significant cell death compared to the control group (Fig. 4C).
 |
| | Fig. 4 Dose and time dependent glucose uptake analysis and cell viability analysis in renal NRK-52E proximal tubular cells. (A and B) Representative images of the effect of varying concentrations and times of HG on SGLT2 mediated glucose uptake. (C) Cell viability of NRK-52E cells by the Alamar blue assay upon 5a–9c treatment. Synthetic compound concentrations: 1, 5, 10, 20, 35 and 50 μM, respectively. Results are expressed as mean ± SEM. (***) p < 0.001, (**) p < 0.01 and (**) p < 0.05 vs. control group. n = 3 independent experiments for each group. | |
Glucose uptake inhibition analysis of 5a–9c in NRK-52E kidney proximal tubular cells.
6-NBDG, a fluorescent glucose analog, was employed for screening the glucose uptake mediated by SGLT2 receptors in kidney proximal tubular cells. Proximal tubular cells are known to express two types of glucose carriers i.e. SGLT2 and facilitative glucose transporters 2 (GLUT2).31 SGLT2 receptor mediated glucose uptake is an electrogenic process which involves coupling with Na+ transport in a stoichiometric ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.4 Therefore, Na+ transport occurs simultaneously along with glucose transport via SGLT2, whereas GLUT2 receptors transport glucose via facilitative diffusion. In our study, we used both sodium buffer and sodium-free buffer during cell incubation to measure glucose uptake. 6-NBDG tagged glucose transported in the presence of Na+ buffer is considered as the total uptake, which is the sum of contributions from SGLT2 and GLUT2, whereas the glucose uptake through GLUT2 alone was determined by incubating the cells in Na+-free buffer. By subtracting the fluorescence intensity of the Na+-free buffer from that of the Na+ buffer, we obtain the specific value of glucose uptake mediated by the SGLT2 receptor. The study was performed to evaluate the effect of the compounds on high glucose (HG)-treated NRK-52E cells which in turn helps to determine the glucose uptake by SGLT2 receptors. The optimised concentration of 33 mM of high glucose (Fig. 4A and B) was employed for 6 h to induce diabetic nephropathy. The FDA approved drug canagliflozin was implemented as a standard for the experiments.32,33 For the screening of glucose uptake, 1, 5 and 10 μM concentrations of the compounds and canagliflozin were considered. In the overall study, 5e and 6d were found to be ineffective in reducing glucose uptake compared to the HG-induced group (Fig. 5A); however, among the effective compounds, 9a was found to be the most potent candidate in reducing glucose uptake with a 2-fold decrease at 1 μM compared to the HG-induced group.
:1.4 Therefore, Na+ transport occurs simultaneously along with glucose transport via SGLT2, whereas GLUT2 receptors transport glucose via facilitative diffusion. In our study, we used both sodium buffer and sodium-free buffer during cell incubation to measure glucose uptake. 6-NBDG tagged glucose transported in the presence of Na+ buffer is considered as the total uptake, which is the sum of contributions from SGLT2 and GLUT2, whereas the glucose uptake through GLUT2 alone was determined by incubating the cells in Na+-free buffer. By subtracting the fluorescence intensity of the Na+-free buffer from that of the Na+ buffer, we obtain the specific value of glucose uptake mediated by the SGLT2 receptor. The study was performed to evaluate the effect of the compounds on high glucose (HG)-treated NRK-52E cells which in turn helps to determine the glucose uptake by SGLT2 receptors. The optimised concentration of 33 mM of high glucose (Fig. 4A and B) was employed for 6 h to induce diabetic nephropathy. The FDA approved drug canagliflozin was implemented as a standard for the experiments.32,33 For the screening of glucose uptake, 1, 5 and 10 μM concentrations of the compounds and canagliflozin were considered. In the overall study, 5e and 6d were found to be ineffective in reducing glucose uptake compared to the HG-induced group (Fig. 5A); however, among the effective compounds, 9a was found to be the most potent candidate in reducing glucose uptake with a 2-fold decrease at 1 μM compared to the HG-induced group.
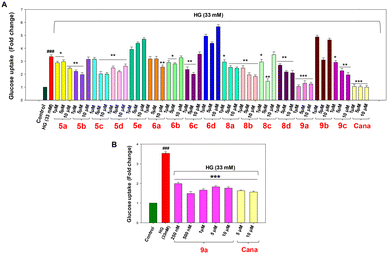 |
| | Fig. 5 (A) SGLT2 mediated glucose uptake analysis on NRK-52E cells upon treatment with compounds 5a–9c in HG-treated NRK-52E cells. HG-treated (33 mM); Cana, canagliflozin (1, 5 and 10 μM in HG medium); 5a–9c (1, 5 and 10 μM in HG medium). (B) SGLT2 mediated glucose uptake analysis and inhibition of SGLT2 protein expression on NRK-52E cells and HG-treated NRK-52E cells upon 9a treatment. Results are expressed as mean ± SEM. (###) p < 0.001 vs. control group; (***) p < 0.001, (**) p < 0.01 and (*) p < 0.05 vs. HG group. n = 3 independent experiments for each group. | |
To determine the half-maximal inhibitory concentration (IC50) values of canagliflozin and 9a in SGLT2-mediated glucose uptake inhibition, we exposed HG-induced NRK-52E cells to a range of concentrations from 1 nM to 20 μM for 24 hours (Fig. S50†). The results indicated that the IC50 values for glucose uptake inhibition were 2.35 nM for canagliflozin and 2.91 nM for 9a.
Glucose uptake inhibition & SGLT2 expression of 9a in NRK-52E kidney proximal tubular cells.
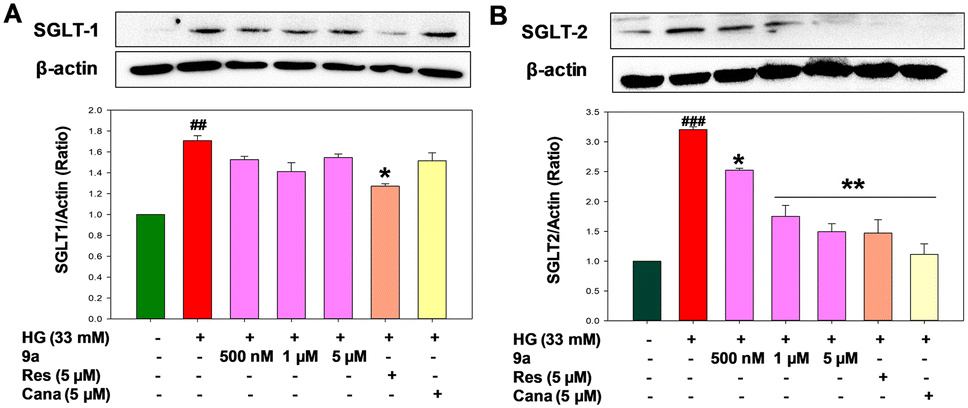
Experimental evidence from studies on humans and rodents with diabetes and related complications indicates that the threshold for glucose reabsorption through renal SGLT2 is elevated under hyperglycemic conditions.31 Additionally, increased glucose uptake in diabetic kidneys is primarily responsible for metabolic adjustments, leading to enhanced gluconeogenesis, aberrant fatty acid oxidation, and lipid accumulation in the kidneys.34 Since compound 9a demonstrated the most significant activity at the lowest concentration (1 μM), we conducted a dose-dependent inhibition study with concentrations ranging from 100 nM to 10 μM. Among these, 500 nM showed the most significant inhibition of glucose uptake (by 1.4-fold), followed by 1 μM and 5 μM (Fig. 5B). We investigated the expression levels of SGLT2 in 9a-treated cells under high glucose conditions. A significant increase in SGLT2 expression was observed in high glucose-treated cells (p < 0.001). However, treatment with 9a at concentrations of 1 μM and 5 μM significantly reduced SGLT2 expression in these cells (p < 0.01). Conversely, 9a did not significantly affect SGLT1 expression in high glucose-induced cells (p < 0.05). These results demonstrate that 9a inhibits SGLT2 expression in NRK-52E cells without affecting SGLT1, ultimately reducing glucose uptake from the glomerular filtrate (Fig. 6).4
 |
| | Fig. 6 Representative western blot images of (A) SGLT1 and (B) SGLT2 proteins and levels of expression. HG (33 mM) treated NRK-52E cells; Cana, canagliflozin (5 and 10 μM in HG medium); Res, resveratrol (5 μM in HG medium); 9a (500 nM, 1 and 5 μM in HG medium). Results are expressed as mean ± SEM. (###) p < 0.001 vs. control group; (***) p < 0.001, (**) p < 0.01 and (*) p < 0.05 vs. HG group. n = 3 independent experiments for each group. | |
Intracellular ROS generation and MMP studies of 9a in NRK-52E kidney proximal tubular cells.
Several reports indicate that SGLT2 inhibitors can control inflammation and fibrosis in diabetic nephropathy by activating various pathways. Oxidative stress is also closely linked to the pathogenesis of DN. Chronic hyperglycemia, a hallmark of diabetes mellitus, activates the intra-renal renin-angiotensin system (RAS) and increases the production of reactive oxygen species, leading to renal cell injury and fibrosis.12,17 Excessive ROS production causes kidney inflammation and fibrosis, resulting in significant injuries through lipid peroxidation, DNA damage, protein modification, and mitochondrial dysfunction.12 The ROS generation inhibition activity of 9a was investigated at concentrations ranging from 500 nM to 10 μM using the fluorescent DCFDA dye. Spectrofluorometric analysis showed that 9a reduced intracellular ROS generation in NRK-52E cells compared to the HG group. High glucose (33 mM) led to an increase in ROS production compared to the control cells; however, a significant decrease in ROS levels was observed at all concentrations of 9a in NRK-52E cells pre-treated with high glucose (Fig. 7).
 |
| | Fig. 7 Analysis of ROS generation upon treatment with 9a in HG-treated NRK-52E cells. HG (33 mM) treated NRK-52E cells; Cana, canagliflozin (500 nM, 5 and 10 μM in HG medium); Res, resveratrol (500 nM, 5 and 10 μM in HG medium); 9a (100–500 nM and 1–10 μM in HG medium). Results are expressed as mean ± SEM. (###) p < 0.001 vs. control group; (**) p < 0.01 and (*) p < 0.05 vs. HG group. n = 3 independent experiments for each group. | |
Flow cytometry analysis was used to determine the reduction in intracellular ROS generation in NRK-52E cells. The data revealed a significant increase (p < 0.001) in intracellular ROS production (2-fold change) in NRK-52E cells treated with high glucose (Fig. 8B) compared to control cells (Fig. 8A). However, treatment with 500 nM, 1 μM, and 5 μM compound 9a significantly reduced ROS production in NRK-52E cells (Fig. 8). Moreover, rhodamine-123 (RH-123) staining was employed to investigate whether 9a could reverse the loss of mitochondrial membrane potential (MMP) induced by high glucose in NRK-52E cells. MMP was quantified via flow cytometry analysis using RH-123 staining, where high green fluorescence indicated high MMP. HG-treated (33 mM) NRK-52E cells exhibited reduced green fluorescence compared to untreated NRK-52E cells, suggesting disrupted MMP (Fig. 9). At concentrations of 500 nM, 1 μM, and 5 μM, compound 9a led to a notable increase (p < 0.01) in green fluorescence intensity compared to NRK-52E cells treated only with high glucose. The data revealed a significant increase (p < 0.001) in mitochondrial membrane depolarization in HG-treated NRK-52E cells compared to control cells; however, treatment with 9a restored HG-induced mitochondrial membrane depolarization in NRK-52E cells (Fig. 9).
 |
| | Fig. 8 Flow cytometric determination of intracellular ROS in HG-induced NRK-52E and its reversal. NRK-52E cells were cultured in (A) control, (B) 33 mM HG, (C–E) 33 mM HG with 500 nM, 1 μM and 5 μM 9a respectively, (F) 33 mM HG with 5 μM resveratrol, (G) 33 mM HG with 5 μM canagliflozin. The ROS generation was determined by using an H2DCFDA fluorescence probe with the help of flow cytometry. Bar graph representing quantitative analysis of the intracellular ROS generation (expressed by a fold change value with respect to the control) determined by flow cytometry analysis. Results are expressed as mean ± SEM. (###) p < 0.001 vs. control group; (**) p < 0.01 and (**) p < 0.05 vs. HG group. n = 3 independent experiments for each group. | |
 |
| | Fig. 9 Compound 9a inhibited HG-induced mitochondrial membrane depolarization in NRK-52E cells. NRK-52E cells were cultured in (A) control, (B) 33 mM HG, (C–E) 33 mM HG with 500 nM, 1 μM and 5 μM 9a, (F) 33 mM HG with 5 μM resveratrol and (G) 33 mM HG with 5 μM canagliflozin. Mitochondrial energization induces quenching of RH-123 fluorescence and the rate of fluorescence decay is proportional to the mitochondrial membrane potential. The bar diagram represents the level of fluorescence decay. The RH-123 fluorescence decay is maximum under HG conditions and it decreases upon treatment with 9a. Results are expressed as mean ± SEM. (###) p < 0.001 vs. control group; (**) p < 0.01 and (*) p < 0.05 vs. HG group. n = 3 independent experiments for each group. | |
Western blot analysis of AMPK, NOX-4, NF-κB, IL-6, TGF-β1, fibronectin and collagen-IV.
Multiple studies have demonstrated that inhibiting SGLT2 exerts anti-inflammatory and anti-fibrotic effects, guarding against extracellular matrix dysregulation and oxidative damage in the kidneys. Inflammatory pathways in diabetic nephropathy involve various factors including transcriptional factors, cytokines, adhesion molecules, chemokines, nuclear receptors, and others, all of which are pivotal in DN treatment.12 Hence, delving into these pathways and understanding the molecular mechanisms behind inflammation and fibrosis could significantly slow down or halt the advancement of DN and aid in the creation of therapeutic targets with anti-inflammatory and anti-fibrotic properties. AMPK, an energy sensor found in all eukaryotic cells, forms a heterotrimeric complex comprising α, β, and γ subunits.33,35 Significantly, abundant evidence has established a strong association between NOX activity and AMPK in diabetes and diabetic nephropathy. Of the various NOX isoforms, NOX-4 is notably expressed and activated in the kidneys, particularly in the proximal tubular compartment during DN. Several studies have demonstrated that AMPK activation could reduce HG-induced up-regulation of NOX-4.36 In our current investigation, we discovered that 9a notably enhanced AMPK phosphorylation and notably reduced NOX-4 overexpression, particularly at concentrations of 1 and 5 μM. This implies that 9a likely exerts its anti-inflammatory and anti-fibrotic effects against renal inflammation and fibroblast proliferation by stimulating AMPK activity and inhibiting NOX-4 (Fig. 10B and C). Inflammatory and pro-inflammatory cytokines are consistently pivotal in the onset and advancement of diabetic nephropathy. NF-κB stands out as a critical transcription factor activated by the generation of free radicals and the production of cytokines resulting from prolonged hyperglycemia.17 Multiple studies have highlighted the heightened phosphorylation of NF-κB associated with the onset of DN.18 Under normal conditions, NF-κB exists in the cytoplasm as a heterodimer of p50 and p65 units bound to an inhibitory protein, IκB. Upon stimulation by oxidative stress, inactive NF-κB dissociates from IκB, allowing the active p65-NF-κB complex to translocate into the nucleus and regulate the expression of pro-inflammatory cytokines such as TNF-α, IL-1β, IL-6, and others.12,19
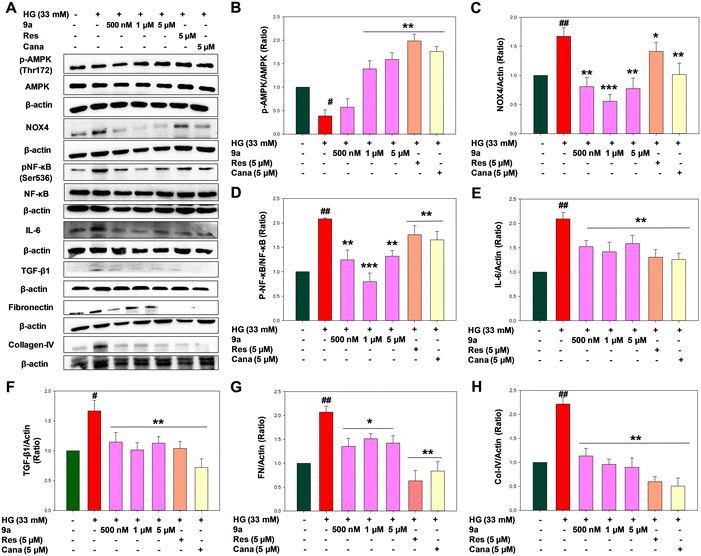 |
| | Fig. 10 Compound 9a ameliorates AMPK-mediated ROS signalling in HG-treated NRK-52E cells. (A) Representative western blot expression bands for proteins. Representative western blot level of expression of (B) AMPK, (C) NOX-4, (D) NF-κB, (E) IL-6, (F) TGF-β1, (G) fibronectin, and (H) collagen-IV. HG (33 mM) treated NRK-52E cells; Cana, canagliflozin (5 μM in HG medium); Res, resveratrol (5 μM in HG medium); 9a (500 nM, 1 and 5 μM in HG medium). Results are expressed as mean ± SEM. (##) p < 0.01 and (#) p < 0.05 vs. control group; (***) p < 0.001, (**) p < 0.01 and (*) p < 0.05 vs. HG group. n = 3 independent experiments for each group. | |
Our findings revealed elevated phosphorylation of p65-NF-κB units in the high glucose-treated group, indicating oxidative stress conditions in renal cells. Moreover, treatment with 9a notably suppressed NF-κB activation and the subsequent generation of cytokines (Fig. 10D). Therefore, treatment with 9a at concentrations of 500 nM, 1 μM, and 5 μM appears to disrupt NF-κB activation, potentially serving as one of the mechanisms for inflammation control in diabetic nephropathy. IL-6 expression correlates positively with NF-κB activation. NF-κB-induced IL-6 expression, in turn, contributes to cell proliferation, extracellular matrix expansion, and increased expression of fibronectin and collagen, resulting in thickening of the glomerular basement membrane in renal cells. The study demonstrated that treatment with 9a at concentrations of 500 nM, 1 μM, and 5 μM significantly reduces the elevated level of renal IL-6 expression in HG-induced NRK-52E cells, thereby alleviating renal inflammation (Fig. 10E). Moreover, heightened ROS production triggers the activation of profibrotic growth factors like TGF-β1, stimulating the production and accumulation of extracellular matrix proteins such as collagen IV and fibronectin. This cascade ultimately leads to the development of renal fibrosis and sclerosis.12,37 At the same time, reports suggest that the interaction between TGF-β1 and NOX-4 also contributes to tubular injury and renal fibrosis. It was evident from this study that 9a reduced the expression of TGF-β1 in kidney cells compared to NRK-52E cells treated with high glucose (Fig. 10F). Fibronectin and collagen-IV are crucial fibrotic factors of the extracellular matrix. The upregulation of TGF-β1 enhances extracellular matrix formation, leading to kidney fibrosis by increasing the expression of fibronectin and collagen-IV.21 The elevated levels of these proteins contribute to the thickening of the basement membrane in kidney cells, resulting in tubular injury and fibrosis.12 Notably, compound 9a significantly inhibited the expression of fibronectin and collagen-IV at concentrations of 500 nM, 1 μM, and 5 μM (p < 0.01 & p < 0.05) (Fig. 10G and H). These findings suggest that 9a exerts an overall positive effect in ameliorating kidney fibrosis to regulate diabetic nephropathy.
In silico studies of SGLT2 interaction with 9a.
Molecular docking and molecular dynamics simulations revealed that 9a can exhibit strong and stable interactions with human SGLT2, especially in the glucose entry site. After MD simulation, the MMGBSA binding energy of compound 9a was found to be −51.38 kcal mol−1. The interaction and binding pose of 9a with SGLT2 are represented in Fig. 11A and B. The analysed RMSD revealed the formation of a stable complex and reached a stable plateau region after the first few nanoseconds (Fig. 11C). Similarly, the RMSD of the protein was nominal resulting in a stable protein–ligand complex throughout the simulation. Following the molecular docking, the binding pose of 9a exhibited a minor deviation of 4 Å within the early 1 to 2 ns of simulation. Subsequently, it remained in the same binding position, effectively blocking the glucose transport channel of SGLT2. The RMSD remained below 1.6 Å from the initial 5 ns of the simulation until the final frame of the simulation (Fig. 11D). The solvent accessible surface area (SASA) of SGLT2 exhibited a considerable decrease throughout the simulation period (Fig. 11E) after interacting with 9a. This suggests the inhibition of the glucose transport channel in SGLT2. The analysis of protein–ligand interaction fractions revealed that 9a was held at the glucose binding positions of SGLT2 majorly by hydrogen bonding, hydrophobic interactions and ionic interactions (Fig. 11F). The SGLT2 residues viz. GLU 99 and ASP 454 exhibited strong hydrogen bonding with 9a within 2.5 Å for up to 97% of the simulation time (Fig. 11G). Moreover, PHE 453 interacted with 9a by cation–π interaction for over 69% of the simulation time.
 |
| | Fig. 11 Interaction of 9a with SGLT2 (A) represented within the transmembrane; (B) cartoon representation of the 9a binding site in SGLT2 (in both the subfigures, the 9a ligand is highlighted within a yellow circle); (C) RMSD analysis of the MD simulation trajectory of the 9a–SGLT2 complex with reference to the docked pose; (D) RMSD of the MD simulation trajectory with reference to the equilibrated pose after 5 ns; (E) SASA of the 9a–SGLT2 complex throughout the simulation time; (F) interaction fraction of different non-covalent interactions; (G) 2D interaction diagram depicting interaction of amino acid residues of SGLT2 and different moieties of 9a. | |
ADMET properties.
The predicted physicochemical properties, medicinal chemistry parameters and ADMET (absorption, distribution, metabolism, excretion and toxicity) properties of 9a are listed in Tables S2–S11.†9a exhibited promising drug-likeliness parameters as represented by Lipinski's rule38 and the golden triangle.39 Various molecular descriptors of 9a, along with their corresponding upper and lower limit values for drug likeliness properties, were represented by a radar plot in Fig. 12. 9a exhibited over 90% probability of human intestinal absorption. The compound revealed a low probability of plasma protein binding and a higher value of fraction unbound in plasma. 9a has exhibited optimal values of volume distribution and no probability of crossing the blood brain barrier. Moreover, 9a can be easily metabolized and excreted based on different metabolism and excretion parameters computed. The prediction of different toxicity parameters revealed negative results for all the TOX21 pathway parameters and lower probability values for most of the toxicity parameters evaluated. However, significant probability values were observed for some toxicity parameters such as skin sensitization and respiratory toxicity.
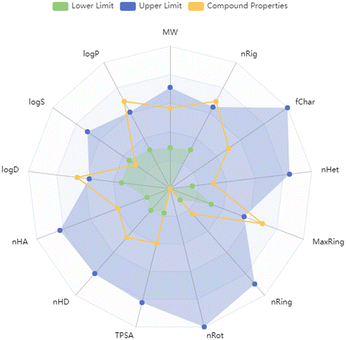 |
| | Fig. 12 Radar plot showing the drug likeliness of 9a by comparing different values of its molecular descriptors and their acceptable ranges. | |
Conclusions
In summary, among the synthesized target compounds, 9a displayed significant potential against diabetic nephropathy by inhibiting SGLT2-mediated glucose uptake in kidney cells and inhibiting SGLT2 expression. Overall, compound 9a alleviated inflammation and fibrosis in high glucose-induced human proximal tubular cells through the AMPK/NOX-4/NF-κB signalling cascade. Additionally, our study revealed that treatment with 9a reduced intracellular ROS generation and mitochondrial membrane depolarization in the presence of high glucose. These findings contribute to the progress in anti-diabetic drug development, highlighting the promise of steroidal pyrimidine compounds as potential candidates for the next generation of therapies targeting chronic kidney disease in type-2 diabetic patients.
Experimental
General
Commercial reagents were purchased from Sigma-Aldrich, Merck and TCI and were used without further purification. The 1H and 13C{1H}NMR spectra were recorded at ambient temperature on a 400 MHz (100 MHz for 13C{1H}) NMR spectrometer with CDCl3 or DMSO-d6 as the solvent. Chemical shifts were referenced to the residual peaks of the solvent [CDCl3: δ = 7.26 (1H), 77.16 ppm (13C); DMSO-d6: δ = 2.50 (1H), 39.52 ppm (13C)]. Column chromatography was performed using EM silica gel 60 (100–200 mesh). HRMS spectra were obtained using a Xevo XS QT mass spectrometer, Waters ACQUITY UHPLC. A Shimadzu Shimpack GIS C16 analytical column (5 μm, 250 × 4.6 mm) was used for HPLC analysis with a mobile phase of water (pump A) and acetonitrile–methanol (50:50) (pump B). An isocratic method with 70% acetonitrile–methanol (50:50) with a flow rate of 1 ml min−1 for 20 minutes was used to run a sample injection volume of 20 μl. The HPLC chromatogram trace and purity of our main target synthesized compound (9a) for which the detailed in vitro study was conducted are included in the ESI† (Table S12) (purity > 96%) and the purity for all the synthesized compounds was also verified by NMR and HRMS to be >95%. NRK-52E cells were purchased from NCCS, Pune. The culture medium comprised high glucose DMEM with 10% FBS. Trypsin (cat# 25200072), antibiotic antimycotic (cat# 15240062), DPBS (cat# 21300025), and 6-NBDG (cat# N23106) were purchased from Invitrogen. Sodium buffer (Na+ buffer): 140 mM NaCl, 5 mM KCl, 2.5 mM CaCl2, 1 mM MgSO4, 1 mM KH2PO4, 10 mM HEPES (pH 7.4). Sodium free buffer (Na+ free buffer): 140 mM N-methyl-D-glucamine, 5 mM KCl, 2.5 mM CaCl2, 1 mM MgSO4, 1 mM KH2PO4, 10 mM HEPES (pH 7.4). 33 mM high glucose solution, 6-NBDG solution, Triton X (0.1%) pH 10 in 1× PBS, DCFDA solution, DMSO. PMSF (Roche, 10837091001), Halt protease and phosphatase inhibitor cocktail (Biorad 78441) were supplied by Thermo Scientific. Sodium dodecyl sulphate (SDS, cat# L3771), skim milk (cat# GRM1254), Tris base (cat# TC072) and sodium chloride (cat# TC046) were purchased from HiMedia. SGLT2 and anti-beta actin (HRP) (cat# 49900) antibodies were bought from Abcam. AMPKα (cat# 2532), phospho-AMPKα (Thr172) rabbit mAb (cat# 2535), NF-κB (cat# 4882P), pNF-κB p65 (Ser536) (cat# 3031), anti-rabbit IgG HRP linked secondary antibodies (cat# 7074), and SGLT1 (cat# 5042S) were obtained from Cell Signaling Technology (Beverly, MA, USA). NOX-4 (cat# MA5-32090) and IL-6 (cat# M620) were purchased from Invitrogen. TGF beta 1 (cat# A16640), fibronectin (cat# A12977) and collagen-IV (cat# A10710) were purchased from Abclonal.
Molecular docking and molecular dynamics simulation
The crystal structure of human SGLT2 complexed with empagliflozin was obtained from the PDB (7VSI) and processed by removing water molecules, adding hydrogen atoms, filling in missing side chains, generating alternate residue positions and restraining minimization in Modeller.40 The docking grid was generated near the glucose entry site of SGLT2 based on the binding position of cocrystallized empagliflozin. The docking grid x, y and z coordinates were −2.68, 2.14 and −10.95, respectively. The docking method was validated by docking empagliflozin to the binding site and comparing its root mean square deviation (RMSD) to the cocrystallized empagliflozin, where the RMSD value was less than 1 Å. The ligand library was meticulously compiled using a diverse range of biologically potent N-heterocyclic scaffolds integrated with a steroid core. The structure of each of the steroidal heterocyclic compounds was generated using Marvin (Chemaxon, academic version 2024), followed by energy minimization and geometry optimization to generate the 3D conformation.
The binding poses of each ligand with SGLT2 were derived by molecular docking using SeeSAR (BioSolveIt suite 2019). The compounds with the highest docking score and binding energies were subjected to chemical synthesis and subsequent analysis. The selected ligand bound SGLT2 complex with the highest score was extracted in PDB format and subjected to molecular dynamics simulation. The MD simulation was carried out in Desmond academic version (Schrödinger suite 2024). The simulation system was generated within an orthorhombic periodic boundary with a minimum distance of 10 Å between the solute and edge of the boundary. The system comprised TIP3P water, 0.15 M NaCl and a docked SGLT2–9a complex in a POPC (300 K) membrane model. The membrane was placed within the transmembrane domains of SGLT2 as outlined in the UniProt database (UniProt ID: P31639). The system was neutralized with the required number of Na+/Cl− ions and minimized with 10000 steps of the steepest descent algorithm followed by equilibration with NVT and NPγT ensembles at 300 K and 1 bar for 100 ps. The reversible reference system propagator algorithms (r-RESPA)41 were used for trajectory integration with a 2 fs time step. The Coulomb cut off and van der Waals radius were 1.5 Å and 10 Å. The system was thermostatically maintained at 300 K and 1 bar using the Nosé–Hoover chain thermostat and Martyna Tobias Klein barostat using isotropic coupling with a relaxation time of 2 ps for each. The production MD run was carried out for 100 ns using the NPγT ensemble and the trajectory was recorded within 10 ps interval. After completion of the MD simulation run, the trajectory was analysed for root mean square deviations (RMSDs) and interactions of the ligand with SGLT2 residues throughout the simulation time.
Determination of ADMET properties
The ADMET properties were determined using the ADMETlab 3.0 server.42 The three dimensional structure of 9a interacting with SGLT2 was obtained in SDF format after the MD simulation. The ADMET attributes were then computed using prediction models trained by the directed message passing neural network (DMPNN) framework in the ADMETlab 3.0 server.
Statistical analysis
The results were presented as mean ± standard deviation (SD). Student's unpaired t-test was performed to analyse individual group statistical comparisons, and multiple-group comparisons between the diabetic control group and test groups were evaluated by performing one-way ANOVA and the Student–Newman–Keuls test in proprietary scientific graphing and data analysis software. Values of p < 0.05, 0.01 and 0.001 were considered statistically significant.
Chemistry
Synthesis of target compounds 5, 6, 8, and 9. General procedure for the synthesis: a 25 mL Schlenk tube equipped with a stirring bar was charged with a β-formyl enamide derivative of 16-DPA (1 mmol), aminopyrazole/aminopyridine/benzamidine/aminobenzimidazole (1.2 mmol), Ni nanoparticles (5 mol%) and DMF (5 mL). The reaction mixture was stirred under 110 °C heating conditions in an oil bath. Upon completion, the tube was cooled to room temperature, the catalyst was recovered using a magnetic rod and the solution was poured into ice-cold water. The mixture was extracted with EtOAc (3 × 10 mL) and washed with water. The organic layer was collected and concentrated in a vacuum. The residue was purified by column chromatography on silica gel with hexane–EtOAc as the eluent to afford 5a–e and 8a–d. Some of the compounds were subjected to HPLC purification. The aforementioned compounds were hydrolysed using 10% KOH to obtain the target molecules 6a–d and 9a–c.
Spectral data of synthesised compounds
3β-Acetoxy-2′-phenyl-androst[16,17-e]pyrazolo(1′,5′-a)pyrimidin-5-ene (5a).
White solid (yield: 85%). 1HNMR (400 MHz, CDCl3) δ 8.34 (s, 1H), 7.96 (d, J = 6.9 Hz, 2H), 7.45–7.32 (m, 3H), 7.03 (s, 1H), 5.39 (d, J = 5.2 Hz, 1H), 2.99 (d, J = 15.4 Hz, 1H), 2.91–2.82 (m, 1H), 2.59–2.54 (m, 1H), 2.36–2.25 (m, 2H), 2.10 (d, J = 17.1 Hz, 1H), 1.99 (s, 3H), 1.88–1.69 (m, 8H), 1.61–1.53 (m, 2H), 1.22 (s, 3H), 1.18–1.10 (m, 2H), 1.08 (s, 3H) ppm. 13C{1H}NMR (100 MHz, CDCl3) δ 170.6, 156.9, 145.7, 140.3, 132.8, 129.2, 128.8, 126.7, 121.7, 120.2, 92.2, 73.7, 56.17, 50.1, 47.1, 38.1, 36.9, 36.8, 33.4, 31.3, 30.3, 29.7, 28.8, 27.7, 21.5, 20.3, 19.3, 14.4 ppm. HRMS (ESI) m/z calcd. for C31H36N3O2 (M + H)+ 482.2808, found 482.2889.
3β-Acetoxy-2′-(p-chlorophenyl)-androst[16,17-e]pyrazolo(1′,5′-a)pyrimidin-5-ene (5b).
White solid (yield: 88%). 1HNMR (400 MHz, CDCl3) δ 8.31 (s, 1H), 7.89 (d, J = 8.5 Hz, 2H), 7.36 (d, J = 8.5 Hz, 2H), 6.90 (s, 1H), 5.38 (d, J = 4.6 Hz, 1H), 4.60–4.52 (m, 1H), 2.96 (d, J = 12.8 Hz, 1H), 2.85 (dd, J = 14.5, 5.2 Hz, 1H), 2.56–2.50 (m, 1H), 2.36–2.25 (m, 2H), 2.09 (d, J = 14.8 Hz, 1H), 1.99 (s, 3H), 1.88–1.68 (m, 8H), 1.62–1.51 (m, 1H), 1.19 (s, 3H), 1.16–1.10 (m, 2H), 1.07 (s, 3H) ppm. 13C{1H}NMR (100 MHz, CDCl3) δ 170.6, 157.4, 155.1, 149.8, 146.7, 140.3, 134.7, 131.7, 128.9, 127.8, 121.7, 120.6, 92.50, 73.7, 56.2, 50.1, 46.9, 38.1, 36.9, 36.8, 33.5, 31.3, 30.2, 28.8, 27.7, 21.5, 20.3, 19.3, 14.4 ppm. HRMS (ESI) m/z calcd. for C31H35ClN3O2 (M + H)+ 516.2418, found 516.2526.
3β-Acetoxy-2′-(p-bromophenyl)-androst[16,17-e]pyrazolo(1′,5′-a)pyrimidin-5-ene (5c).
White solid, (yield: 86%). 1HNMR (400 MHz, CDCl3) δ 8.31 (s, 1H), 7.82 (d, J = 8.6 Hz, 2H), 7.51 (d, J = 8.6 Hz, 2H), 6.88 (s, 1H), 5.38 (d, J = 4.9 Hz, 1H), 4.59–4.52 (m, 1H), 2.95 (d, J = 12.6 Hz, 1H), 2.89–2.81 (m, 2H), 2.55–2.49 (m, 1H), 2.35–2.25 (m, 2H), 2.10–2.06 (m, 1H), 1.98 (s, 3H), 1.87–1.82 (m, 4H), 1.78–1.67 (m, 3H), 1.61–1.52 (m, 1H), 1.19 (s, 3H), 1.16–1.09 (m, 2H), 1.07 (s, 3H) ppm. 13C{1H}NMR (100 MHz, CDCl3) δ 170.6, 157.2, 155.0, 150.0, 146.9, 140.3, 132.2, 131.9, 128.1, 122.9, 121.7, 120.7, 92.6, 73.7, 56.2, 50.1, 46.8, 38.1, 36.9, 36.8, 33.5, 31.3, 30.2, 28.8, 27.7, 21.5, 20.3, 19.3, 14.4 ppm. HRMS (ESI) m/z calcd. for C31H35BrN3O2 (M + 2H)2+ 561.1991, found 561.2067.
3β-Acetoxy-3′-cyano-androst[16,17-e]pyrazolo(1′,5′-a)pyrimidin-5-ene (5d).
White solid (yield: 77%). 1HNMR (400 MHz, CDCl3) δ 8.56 (s, 1H), 8.30 (s, 1H), 5.37 (d, J = 5.4 Hz, 1H), 4.62–4.49 (m, 1H), 2.93 (dd, J = 14.7, 5.7 Hz, 1H), 2.79–2.76 (m, 1H), 2.62–2.56 (m, 1H), 2.35–2.23 (m, 1H), 2.11–2.07 (m, 2H), 1.98 (s, 3H), 1.88–1.67 (m, 7H), 1.60–1.50 (m, 2H), 1.17 (s, 3H), 1.15–1.08 (m, 2H), 1.06 (s, 3H) ppm. 13C{1H}NMR (100 MHz, CDCl3) δ 170.6, 158.9, 150.4, 150.3, 147.6, 140.3, 124.1, 121.5, 113.3, 82.2, 73.6, 56.2, 49.9, 47.1, 38.1, 36.8, 36.8, 33.2, 31.2, 30.2, 28.8, 27.7, 21.5, 20.2, 19.3, 14.7 ppm. HRMS (ESI) m/z calcd. for C26H31N4O2 (M + H)+ 431.2447, found 431.2509.
3β-Acetoxy-5′-methoxy-phenyl-androst[16,17-e]indazolo(7′,6′-a)pyrimidin-5-ene (5e).
Yellow solid (yield: 82%). 1HNMR (400 MHz, CDCl3) δ 8.57 (s, 1H), 7.48–7.44 (m, 1H), 7.37 (d, J = 8.5 Hz, 1H), 6.52 (d, J = 7.5 Hz, 1H), 5.38 (d, J = 5.4 Hz, 1H), 4.61–4.53 (m, 1H), 4.07 (s, 3H), 3.07 (d, J = 12.8 Hz, 1H), 2.97 (dd, J = 14.7, 5.6 Hz, 1H), 2.64 (dd, J = 14.6, 3.2 Hz, 1H), 2.35–2.25 (m, 2H), 2.10 (d, J = 14.6 Hz, 1H), 1.98 (s, 3H), 1.88–1.69 (m, 8H), 1.62–1.51 (m, 1H), 1.21 (s, 3H), 1.18–1.09 (m, 2H), 1.07 (s, 3H) ppm. 13C{1H}NMR (100 MHz, CDCl3) δ 170.6, 155.7, 155.5, 153.2, 143.8, 143.6, 140.3, 130.4, 124.5, 121.7, 108.6, 104.5, 98.7, 73.7, 56.2, 56.0, 50.1, 47.1, 38.1, 36.9, 36.8, 33.5, 31.2, 30.3, 29.1, 27.7, 21.5, 20.3, 19.3, 14.0 ppm. HRMS (ESI) m/z calcd. for C30H36N3O3 (M + H)+ 486.2757, found 486.2895.
3β-Hydroxy-2′-phenyl-androst[16,17-e]pyrazolo(1′,5′-a)pyrimidin-5-ene (6a).
White solid (yield: 92%). 1HNMR (400 MHz, CDCl3) δ 8.32 (s, 1H), 7.95 (d, J = 7.2 Hz, 2H), 7.42–7.31 (m, 3H), 6.96 (s, 1H), 5.36 (d, J = 5 Hz, 1H), 3.54–3.46 (m, 1H), 3.01 (d, J = 12.8 Hz, 1H), 2.88 (dd, J = 14.3, 5.4 Hz, 1H), 2.58–2.52 (m, 1H), 2.31–2.22 (m, 2H), 2.12–2.07 (m, 1H), 1.89–1.68 (m, 7H), 1.57–1.43 (m, 2H), 1.21 (s, 3H), 1.16–1.08 (m, 2H), 1.06 (s, 3H) ppm. 13C{1H}NMR (100 MHz, CDCl3) δ 156.5, 149.7, 146.2, 141.3, 132.9, 129.0, 128.8, 126.6, 120.8, 120.3, 92.4, 71.6, 56.3, 50.2, 46.9, 42.2, 37.1, 36.8, 33.5, 31.6, 31.3, 30.3, 29.7, 28.8, 20.4, 19.4, 14.4 ppm. HRMS (ESI) m/z calcd. for C29H34N3O (M + H)+ 440.2624, found 440.2628.
3β-Hydroxy-2′-(p-chlorophenyl)-androst[16,17-e]pyrazolo(1′,5′-a)pyrimidin-5-ene (6b).
White solid (yield: 93%). 1HNMR (400 MHz, CDCl3) δ 8.33 (s, 1H), 7.89 (d, J = 8.3 Hz, 2H), 7.37 (d, J = 8.2 Hz, 2H), 6.95 (s, 1H), 5.36 (d, J = 5.0 Hz, 1H), 3.54–3.46 (m, 1H), 2.97 (d, J = 11.9 Hz, 1H), 2.87 (dd, J = 14.5, 5.5 Hz, 1H), 2.59–2.53 (m, 1H), 2.32–2.19 (m, 2H), 2.12–2.07 (m, 1H), 1.86–1.71 (m, 8H), 1.54–1.43 (m, 1H), 1.21 (s, 3H), 1.18–1.08 (m, 2H), 1.06 (s, 3H) ppm. 13C{1H}NMR (100 MHz, DMSO-D6) δ 156.3, 154.1, 150.4, 148.3, 142.2, 133.9, 132.1, 129.4, 128.3, 121.7, 120.4, 93.1, 70.4, 55.7, 50.1, 46.6, 42.7, 37.2, 36.8, 33.5, 31.8, 31.1, 30.2, 28.7, 20.3, 19.5, 14.5 ppm. HRMS (ESI) m/z calcd. for C29H33ClN3O (M + H)+ 474.2312, found 474.2347.
3β-Hydroxy-2′-(p-bromophenyl)-androst[16,17-e]pyrazolo(1′,5′-a)pyrimidin-5-ene (6c).
White solid, (yield: 95%). 1HNMR (400 MHz, CDCl3) δ 8.32 (s, 1H), 7.83 (d, J = 8.5 Hz, 2H), 7.52 (d, J = 8.5 Hz, 2H), 6.91 (s, 1H), 5.35 (d, J = 5.3 Hz, 1H), 3.54–3.46 (m, 1H), 2.96 (d, J = 12.7 Hz, 1H), 2.85 (dd, J = 14.4, 5.8 Hz, 1H), 2.54 (dd, J = 14.3, 11.0 Hz, 1H), 2.31–2.18 (m, 2H), 2.12–2.07 (m, 1H), 1.89–1.67 (m, 8H), 1.54–1.43 (m, 1H), 1.19 (s, 3H), 1.15–1.08 (m, 2H), 1.06 (s, 3H) ppm. 13C{1H}NMR (100 MHz, CDCl3) δ 157.6, 155.2, 149.8, 146.7, 141.3, 132.1, 131.9, 128.1, 123.0, 120.8, 120.7, 92.5, 71.6, 56.3, 50.2, 46.9, 42.2, 37.1, 36.78, 33.5, 31.6, 31.3, 30.3, 28.9, 20.4, 19.4, 14.4 ppm. HRMS (ESI) m/z calcd. for C29H33BrN3O (M + H)+ 518.1807, found 518.1883.
3β-Hydroxy-5′-methoxy-phenyl-androst[16,17-e]indazolo(7′,6′-a)pyrimidin-5-ene (6d).
Yellow solid (yield: 92%). 1HNMR (400 MHz, CDCl3) δ 8.56 (s, 1H), 7.49–7.41 (m, 1H), 7.37 (d, J = 8.5 Hz, 1H), 6.52 (d, J = 7.5 Hz, 1H), 5.34 (d, J = 5.3 Hz, 1H), 4.07 (s, 3H), 3.53–3.45 (m, 1H), 3.09–3.04 (m, 1H), 2.96 (dd, J = 14.7, 5.9 Hz, 1H), 2.64 (dd, J = 14.6, 11.3 Hz, 1H), 2.31–2.17 (m, 2H), 2.12–2.06 (m, 1H), 1.89–1.69 (m, 8H), 1.53–1.42 (m, 1H), 1.21 (s, 3H), 1.14–1.08 (m, 2H), 1.06 (s, 3H) ppm. 13C{1H}NMR (100 MHz, CDCl3) δ 155.7, 155.5, 153.2, 143.8, 143.7, 141.4, 130.4, 124.5, 120.8, 108.6, 104.5, 98.7, 71.6, 56.3, 56.0, 50.2, 47.1, 42.2, 37.1, 36.8, 33.5, 31.6, 31.3, 30.3, 29.1, 20.3, 19.4, 14.0 ppm. HRMS (ESI) m/z calcd. for C28H34N3O2 (M + H)+ 444.2651, found 444.2646.
3β-Acetoxy-androst[16,17-e]benzimidazolo(1′,2′-a)pyrimidin-5-ene (8a).
Yellow solid (yield: 86%). 1HNMR (400 MHz, CDCl3) δ 8.63 (s, 1H), 8.09 (d, J = 8.5 Hz, 1H), 7.97 (d, J = 8.2 Hz, 1H), 7.50 (t, J = 7.7 Hz, 1H), 7.32 (t, J = 7.8 Hz, 1H), 5.40 (d, J = 5.0 Hz, 1H), 4.60–4.52 (m, 1H), 2.98 (d, J = 12.0 Hz, 1H), 2.89 (dd, J = 14.2, 5.5 Hz, 1H), 2.63–2.57 (m, 1H), 2.37–2.25 (m, 2H), 2.10 (d, J = 19.2 Hz, 1H), 1.99 (s, 3H), 1.92–1.71 (m, 8H), 1.62–1.53 (m, 1H), 1.38 (s, 3H), 1.18–1.12 (m, 2H), 1.09 (s, 3H) ppm. 13C{1H}NMR (100 MHz, CDCl3) δ 170.6, 159.7, 153.2, 152.1, 144.6, 140.1, 126.0, 126.0, 121.8, 121.4, 120.7, 120.5, 115.4, 73.6, 57.4, 49.8, 48.5, 38.0, 36.8, 36.8, 34.2, 30.9, 30.3, 28.6, 27.7, 21.5, 20.6, 19.3, 13.7 ppm. HRMS (ESI) m/z calcd. for C29H34N3O2 (M + H)+ 456.2651, found 456.2646.
3β-Acetoxy-6′-chloro-androst[16,17-e]benzimidazolo(1′,2′a)pyrimidin-5-ene (8b).
Pale yellow solid (yield: 35%). 1HNMR (400 MHz, CDCl3) δ 8.63 (s, 1H), 7.99 (d, J = 9 Hz, 1H), 7.91 (s, 1H), 7.26 (dd, J = 8.9, 2.0 Hz, 1H), 5.39 (d, J = 4.2 Hz, 1H), 4.60–4.52 (m, 1H), 2.89 (d, J = 13.3 Hz, 2H), 2.60 (t, J = 12.7 Hz, 1H), 2.35–2.26 (m, 2H), 2.10 (d, J = 17.6 Hz, 1H), 1.99 (s, 3H), 1.89–1.70 (m, 8H), 1.62–1.53 (m, 1H), 1.36 (s, 3H), 1.18–1.13 (m, 2H), 1.09 (s, 3H) ppm. 13C{1H}NMR (100 MHz, CDCl3) δ 170.6, 159.7, 153.5, 152.9, 145.8, 140.1, 131.7, 124.6, 121.8, 121.7, 121.1, 119.9, 116.2, 73.6, 57.4, 49.8, 48.5, 38.03, 36.8, 34.1, 30.9, 30.3, 28.6, 27.7, 21.5, 20.6, 19.3, 13.7 ppm. HRMS (ESI) m/z calcd. for C29H33ClN3O2 (M + H)+ 490.2261, found 490.2297.
3β-Acetoxy-5′-chloro-androst[16,17-e]benzimidazolo(1′,2′a)pyrimidin-5-ene (8c).
Pale yellow solid (yield: 53%). 1HNMR (400 MHz, CDCl3) δ 8.63 (s, 1H), 8.06 (s, 1H), 7.88 (d, J = 8.7 Hz, 1H), 7.46 (dd, J = 8.8, 1.8 Hz, 1H), 5.40 (d, J = 5.4 Hz, 1H), 4.60–4.52 (m, 1H), 2.90 (dd, J = 13.5, 6.4 Hz, 2H), 2.60 (t, J = 11.6 Hz, 1H), 2.37–2.26 (m, 2H), 2.13–2.08 (m, 1H), 1.99 (s, 3H), 1.92–1.72 (m, 8H), 1.62–1.53 (m, 1H), 1.37 (s, 3H), 1.18–1.15 (m, 2H), 1.10 (s, 3H) ppm. 13C{1H}NMR (100 MHz, CDCl3) δ 170.6, 159.6, 153.6, 152.6, 143.3, 140.2, 126.8, 126.7, 126.3, 121.7, 121.5, 121.1, 115.1, 73.6, 57.4, 49.8, 48.6, 38.0, 36.8, 36.8, 34.4, 30.9, 30.3, 28.6, 27.7, 21.5, 20.7, 19.3, 13.9 ppm. HRMS (ESI) m/z calcd. for C29H33ClN3O2 (M + H)+ 490.2261, found 490.2297.
3β-Acetoxy-4′-chloro-androst[16,17-e]benzimidazolo(1′,2′a)pyrimidin-5-ene (8d).
Yellow solid (yield: 83%). 1HNMR (400 MHz, CDCl3) δ 8.63 (s, 1H), 7.98 (d, J = 8.4 Hz, 1H), 7.51 (d, J = 7.7 Hz, 1H), 7.24–7.22 (m, 1H), 5.39 (d, J = 5.0 Hz, 1H), 4.59–4.52 (m, 1H), 2.93–2.86 (m, 2H), 2.62–2.56 (m, 1H), 2.33–2.28 (m, 2H), 2.09 (d, J = 14.5 Hz, 1H), 1.98 (s, 3H), 1.86–1.69 (m, 8H), 1.61–1.51 (m, 1H), 1.36 (s, 3H), 1.15–1.11 (m, 2H), 1.08 (s, 3H) ppm. 13C{1H}NMR (100 MHz, CDCl3) δ 170.6, 159.8, 153.8, 152.2, 142.2, 140.1, 126.9, 125.6, 125.2, 121.7, 121.4, 121.3, 113.9, 73.6, 57.3, 49.8, 48.5, 38.0, 36.8, 34.2, 30.9, 30.3, 29.7, 28.6, 27.7, 21.5, 20.6, 19.3, 13.7 ppm. HRMS (ESI) m/z calcd. for C29H33ClN3O2 (M + H)+ 490.2261, found 490.2297.
3β-Hydroxy-androst[16,17-e]benzimidazolo(1′,2′-a)pyrimidin-5-ene (9a).
Pale yellow solid (yield: 91%). 1HNMR (400 MHz, CDCl3) δ 8.58 (s, 1H), 8.07 (d, J = 8.6 Hz, 1H), 7.93 (d, J = 8.3 Hz, 1H), 7.47 (t, J = 7.7 Hz, 1H), 7.30 (t, J = 7.8 Hz, 1H), 5.35 (d, J = 5.5 Hz, 1H), 3.56–3.48 (m, 1H), 3.42 (s, 1H), 2.95 (d, J = 12.4 Hz, 1H), 2.85 (dd, J = 14.1, 6.1 Hz, 1H), 2.60–2.54 (m, 2H), 2.34–2.24 (m, 2H), 2.07 (d, J = 12.4 Hz, 1H), 1.90–1.77 (m, 5H), 1.73–1.66 (m, 2H), 1.55–1.46 (m, 1H), 1.35 (s, 3H), 1.22–1.17 (m, 2H), 1.06 (s, 3H) ppm. 13C{1H}NMR (100 MHz, CDCl3) δ 159.7, 153.1, 152.1, 144.8, 141.4, 126.0, 125.9, 121.4, 120.7, 120.5, 115.4, 71.4, 57.5, 49.9, 48.5, 42.2, 37.1, 36.7, 34.2, 31.5, 31.0, 30.3, 28.6, 20.6, 19.3, 13.6 ppm. HRMS (ESI) m/z calcd. for C27H32N3O (M + H)+ 414.2545, found 414.2543.
3β-Hydroxy-5′-chloro-androst[16,17-e]benzimidazolo(1′,2′a)pyrimidin-5-ene (9b).
Pale yellow solid (yield: 90%). 1HNMR (400 MHz, DMSO-D6) δ 7.78 (s, 1H), 7.30 (s, 1H), 7.25 (d, J = 8.4 Hz, 1H), 6.97 (d, J = 8.3 Hz, 1H), 5.26 (d, J = 4.8 Hz, 1H), 4.60 (d, J = 4.5 Hz, 1H), 3.25–3.16 (m, 1H), 2.56 (dd, J = 8.8, 6.4 Hz, 1H), 2.15–1.96 (m, 3H), 1.72–1.53 (m, 7H), 1.41–1.15 (m, 4H), 0.92 (s, 3H), 0.90–0.79 (m, 2H), 0.76 (s, 3H) ppm. 13C{1H}NMR (100 MHz, DMSO-D6) δ 151.3, 150.8, 142.1, 141.9, 134.5, 131.3, 125.4, 121.0, 120.5, 113.9, 70.4, 50.3, 49.9, 47.6, 42.7, 37.3, 36.8, 31.9, 31.8, 31.0, 30.9, 26.2, 20.5, 19.6, 14.7 ppm. HRMS (ESI) m/z calcd. for C27H31ClN3O (M + H)+ 448.2156, found 448.2155.
3β-Hydroxy-4′-chloro-androst[16,17-e]benzimidazolo(1′,2′a)pyrimidin-5-ene (9c).
Yellow solid (yield: 92%). 1HNMR (400 MHz, CDCl3) δ 8.67 (s, 1H), 8.00 (d, J = 8.4 Hz, 1H), 7.53 (d, J = 7.7 Hz, 1H), 7.23 (t, J = 8.1 Hz, 1H), 5.37 (d, J = 5.3 Hz, 1H), 3.55–3.47 (m, 1H), 2.95–2.88 (m, 2H), 2.64–2.58 (m, 1H), 2.34–2.19 (m, 2H), 2.10 (dd, J = 16.9, 2.5 Hz, 1H), 1.92–1.71 (m, 7H), 1.59–1.48 (m, 2H), 1.37 (s, 3H), 1.14–1.11 (m, 2H), 1.08 (s, 3H) ppm. 13C{1H}NMR (100 MHz, CDCl3) δ 159.9, 153.9, 152.2, 142.2, 141.2, 126.9, 125.7, 121.4, 120.8, 113.9, 71.5, 57.5, 49.9, 48.6, 42.1, 37.0, 36.7, 34.2, 31.9, 31.5, 30.3, 29.7, 28.6, 22.7, 20.6, 19.3, 13.7 ppm. HRMS (ESI) m/z calcd. for C27H31ClN3O (M + H)+ 448.2156, found 448.2150.
Cell culture and treatment with high glucose and synthetic compounds
NRK-52E cells were cultured in high glucose DMEM medium with 10% FBS and 1% antibiotic-antimycotic at 37 °C in a humidified atmosphere containing 5% (v/v) CO2. NRK-52E cells ranging in passages from 10–30 were used. Various concentrations of D-glucose (5.5 to 90 mM) and 24 h time were used for the investigation of concentration dependence. Further, the application of D-glucose (33 mM) for various times (0–24 h) was used for the investigation of time dependence. After obtaining the results, NRK-52E cells were exposed to high (33 mM D-glucose) glucose for 6 h, followed by a 24 h treatment with or without synthetic compounds (100 nM to 10 μM). Control cells were treated with media only. The cells were also treated with two standard drugs, canagliflozin (500 nM to 10 μM) and resveratrol (500 nM to 10 μM). Upon termination of incubation, the cells were lysed in radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris pH 8, 150 mM NaCl, 1% NP-40, 0.5% deoxycholic acid, 0.1% SDS) supplemented with protease and phosphatase inhibitors (1 mM PMSF, 5 μg mL−1 leupeptin, 2 μg mL−1 aprotinin, 1 mM EDTA, 10 mM NaF, and 1 mM NaVO4). Lysates were cleared by centrifugation and total protein concentrations were determined by the bicinchoninic acid (BCA) assay (Pierce/Thermo Scientific, Rockford, IL). Each experiment was repeated three times, and the average of the three values was used.
Cell viability assay
The cell toxicity test of synthetic compounds 5a–9c was carried out in NRK-52E murine proximal tubular cells using the Alamar blue reduction bioassay as described previously.43 When the cells reach 80–90% confluency, the cells were treated with 33 mM high glucose except for the control group and after 6 h, they were treated with compounds 5a–9c with doses ranging from 1–50 μM (0.1% DMSO + 1× PBS as a vehicle) and the control group was treated with the vehicle for the next 24 h. The original medium was removed after 24 h and incubated with Alamar blue dye for 4 h. After 4 h, OD values were assayed at two different wavelengths of 570 nm and 600 nm in a microplate reader. Cell viability was expressed as the ratio of the absorbance to that of the control group.
Glucose uptake assay
The glucose uptake assay was performed using 6-[N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino]-2-deoxy-D-glucose (6-NBDG) as described previously with slight modifications.44,45 In brief, NRK-52E cells were evenly plated in a 96 well plate at a density of 1 × 104 cells per well. After 24 h of sub-confluence, the cells were treated with HG (33 mM made in 1× PBS) for the next 6 h followed by treatment with 5a–9c compounds and the standard drug, canagliflozin, with doses of 1, 5 and 10 μM (made in 0.1% DMSO + 1× PBS as a vehicle), and the control cells were treated with the vehicle for the next 24 h. Sodium buffer (Na+ buffer), prepared for cell incubation in the presence of sodium conditions, contained 140 mM NaCl, 5 mM KCl, 2.5 mM CaCl2, 1 mM MgSO4, 1 mM KH2–PO4, and 10 mM HEPES (pH 7.4). Sodium free buffer (Na+-free buffer) contained 140 mM N-methyl-D-glucamine instead of NaCl and was used for cell incubation to measure the 6-NBDG uptake in the absence of sodium. For experiments, all culture medium was removed from each well and rinsed in Na+-free buffer two times. NRK-52E cells were then incubated in 100 μL Na+ or Na+-free buffer in the presence of (6-NBDG; 20 μM) fluorescent nonhydrolyzable glucose for 20 min in a humidified chamber with 5% CO2 at 37 °C. The 6-NBDG uptake reaction was stopped by removing the incubation medium and washing the cells two times with pre-cold Na+-free buffer. The cells were washed two times with 1× PBS after incubation and lysed with 70 μL PBS containing 1% Triton X (pH 10) and kept in the dark for 10 min. After 10 min, 30 μL of DMSO was added and the mixture was homogenized by pipetting up and down. Fluorescence was measured immediately with a microplate reader at emission and excitation wavelengths of 470 nm and 540 nm, respectively. Results were reported as fold change with respect to the control. Further, dose-dependent glucose uptake analysis of compound 9a was carried out at doses ranging from 100 nM to 10 μM with the same protocol as described.
ROS scavenging assay
The ROS level in the NRK-52E cells post-HG treatment was determined using the fluorogenic probe 2′,7′-dichlorofluorescein diacetate (H2DCFDA) according to a reported procedure.45 Briefly, NRK-52E cells (1 × 106) at 80% confluence were cultured in 24-well plates and kept overnight. The cells were treated with HG (33 mM made in 1× PBS) for 6 h followed by different concentrations of compound 9a (100 nM, 250 nM, 500 nM, 1 μM, 5 μM, 10 μM), and the control cells were treated with the vehicle, along with canagliflozin (500 nM, 5 and 10 μM) and resveratrol (500 nM, 5 and 10 μM) (0.1% DMSO + 1× PBS as the vehicle) for the next 24 h. The medium was aspirated out and the cells were then washed with 1× PBS and incubated with 10 μM H2DCFDA in the dark for 30 min at 37 °C. The cells were then washed twice with 200 μL 1× PBS and incubated with 1% Triton X-100 (170 μL). The reaction was stopped by adding DMSO (130 μL). The cells were scraped and transferred to a black plate. Absorbance was measured at excitation and emission wavelengths of 480 and 530 nm, respectively, using a microplate reader (in triplicate). The ROS production by HG-treated cells was considered 100% (baseline), to which other activities were compared. In another set of experiments, intracellular ROS levels generated post-HG exposure in NRK-52E cells were measured by flow cytometry (BD-FACS-Melody, USA) analysis using H2DCFDA, according to a reported procedure.45 Briefly, NRK-52E cells were treated with compound 9a after exposure with HG (33 mM) for 6 h at different concentrations (500 nM, 1 μM and 5 μM), canagliflozin (5 μM, as the positive control) and resveratrol (5 μM, as the positive control) for the next 24 h. The adherent and non-adherent cells were collected and washed twice with 1× PBS (pH 7.4). The cells were then incubated with 10 μM H2DCFDA at 37 °C for 30 min in the dark, which was followed by washing twice with chilled 1× PBS. The fluorescence intensities of 2′,7′-dichlorodihydrofluorescein (DCF) produced by intracellular ROS were analysed by flow cytometry with excitation and emission at 480 nm and 530 nm, respectively.
Measurement of mitochondrial membrane potential in HG-induced NRK-52E cells
Rhodamine 123 (Invitrogen, Life Technologies, Carlsbad, CA, USA) was used to measure MMP, as previously described.46 Briefly, 2 × 105 cells were plated in each well of a 6-well plate and allowed to attach for 22–24 h. After treatment for 24 h, the cells were treated with HG (33 mM) treatment for 6 h followed by the treatment of the best concentrations based on the glucose uptake analysis and ROS analysis of compound 9a, i.e. 500 nM, 1 μM and 5 μM for the next 24 h. The standard drugs, canagliflozin (5 μM) and resveratrol (5 μM), were used. After 24 h, the cells were washed with 1× PBS and incubated with 200 ng mL−1 Rhodamine 123. After incubation for 30 min at 37 °C, the cells were washed thrice and resuspended in 1 mL of 1× PBS. Cytofluorimetric analysis was performed using flow cytometry (BD-FACS-Melody, USA) and FACS Chorus software (Beckman Coulter, Brea, CA, USA) using ∼655–730 nm emission for PI.
Western blot analysis
The total protein from NRK-52E cells was lysed in RIPA buffer as previously described.47–49 All samples consisting of approximately the same amount of protein (∼20–30 μg) were subjected to electrophoresis using 10% sodium dodecyl polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to nitrocellulose membranes. Subsequently, the membranes were blocked in 1% bovine serum albumin (BSA) for 40 min and incubated with the respective primary antibody overnight at 4 °C. Primary antibodies against SGLT2 (1:1000), AMPKα (1:1000), phospho-AMPKα (Thr172) rabbit mAb (1:1000), NF-κB (1:1000), pNF-κB p65 (Ser536) (1:1000), NOX-4 (1:1000), TGF beta 1 (1:1000), fibronectin (1:1000), collagen-IV (1:1000), IL-6 (1:1000), and SGLT1 (1:1000) were used. After overnight incubation, TBS-T (50 mmol L−1 Tris-HCl, pH 7.6, 150 mmol L−1 NaCl, 0.1% Tween 20) was used to wash the membranes for 30 min and then incubated at room temperature with HRP-conjugated goat anti-rabbit secondary antibodies (1:1000) for 2 h. The blotted membrane was detected by an enhanced chemiluminescence method with an ECL western blotting detection substrate and the intensity of each immunoblotting band was measured using the histogram tool of Adobe Photoshop CS5. HRP conjugated β-actin was used as an internal control.
Abbreviation used
| SGLT2 | Sodium glucose co-transporter 2 |
| DN | Diabetic nephropathy |
| ESRD | End-stage renal disease |
| NRK-52E | Normal rat kidney-52E |
| DKD | Diabetic kidney disease |
| SGLT1 | Sodium glucose co-transporter 1 |
| NF-κB | Nuclear factor kappa B |
| TNF-α | Tumor necrosis factor-alpha |
| IL-6 | Interleukin 6 |
| ECM | Extracellular membrane |
| ROS | Reactive oxygen species |
| NOX-4 | NADPH oxidase-4 |
| eNOS | Endothelial nitric oxide synthase |
| NO | Nitric oxide |
| TGF-β1 | Transforming growth factor-beta-1 |
| NMR | Nuclear magnetic resonance |
| HRMS | High-resolution mass spectrometry |
| HG | High Glucose |
| 6-NBDG | 6-[N-(7-Nitrobenz-2-oxa-1,3-diazol-4-yl)amino]-2-deoxy-D-glucose |
| GLUT2 | Glucose transporters 2 |
| FDA | Food and Drug Administration |
| Cana | Canagliflozin |
| Res | Resveratrol |
| SEM | Structural equation modeling |
| RAS | Renin angiotensin system |
| DNA | Deoxyribonucleic acid |
| DCFDA | 2′,7′-Dichlorodihydrofluorescein diacetate |
| H2DCFDA | 2′,7′-Dichlorofluorescein diacetate |
| RH-123 | Rhodamine-123 |
| MMP | Mitochondrial membrane potential |
| AMPK | Adenosine monophosphate-activated protein kinase |
| RMSD | Root mean square deviation |
| MD | Molecular dynamics |
| SASA | Solvent accessible surface area |
| CDCl3 | Deuterated chloroform |
| DMSO-d6 | Deuterated dimethyl sulfoxide |
| HPLC | High performance liquid chromatography |
| DMEM | Dulbecco's modified Eagle medium |
| FBS | Fetal bovine serum |
| DPBS | Dulbecco's phosphate-buffered saline |
| HEPES | 4-(2-Hydroxyethyl)-1-piperazineethanesulfonic acid |
| PBS | Phosphate-buffered Saline |
| OD | Optical density |
| SDS | Sodium dodecyl sulphate |
| HRP | Horseradish peroxidase |
| RIPA | Radioimmunoprecipitation assay |
| PMSF | Phenylmethylsulfonyl fluoride |
| EDTA | Ethylenediaminetetraacetic acid |
| BCA | Bicinchoninic acid |
| DCF | 2′,7′-Dichlorodihydrofluorescein |
| SDS-PAGE | Sodium dodecyl polyacrylamide gel electrophoresis |
| BSA | Bovine serum albumin |
| TBS-T | Tris buffered saline with Tween 20 |
| ECL | Enhanced chemiluminescence |
| PDB | Protein Data Bank |
| TIP3P | Transferable intermolecular potential with 3 points |
| POPC | 1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine |
| r-RESPA | Reversible reference system propagator algorithms |
| SD | Standard deviation |
| 16-DPA | 16-Dehydropregnenolone acetate |
| DMF |
N,N-Dimethylformamide |
| EtOAc | Ethyl acetate |
| ADMET | Absorption, distribution, metabolism, excretion and toxicity |
| DMPNN | Directed message passing neural network |
Data availability
The manuscript titled “Breaking boundaries in diabetic nephropathy treatment: design and synthesis of novel steroidal SGLT2 inhibitors” contains the primary research data, including the NMR spectra, HRMS spectra, HPLC chromatogram, and western blot raw data. These details are included in the ESI† provided along with the research article.
Author contributions
GSN and SAA contributed equally to this work. The manuscript was written and contributed by all authors.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors are thankful to the Director, IASST (DST, Govt. of India) for providing the necessary facilities for conducting the research and the Sophisticated Analytical Instrument Centre (SAIC) for providing us with the necessary infrastructure facilities. GSN, SAA and KS acknowledge IASST (DST, Govt. of India) for fellowship grants. IASST in-house core funding (research project no. IASST/R&D/LSD/IHP-4/2023-24/1275-1283) is also acknowledged. NRK-52E cells were purchased from NCCS, Pune.
References
- Y. S. Kanwar, L. Sun, P. Xie, F. Liu and S. Chen, Annu. Rev. Pathol.: Mech. Dis., 2011, 6, 395–423 CrossRef CAS PubMed.
- T. Wu, L. Ding, V. Andoh, J. Zhang and L. Chen, Life, 2023, 13, 539 CrossRef CAS PubMed.
- W. T. Cade, Phys. Ther., 2008, 88, 1322–1335 CrossRef PubMed.
- R.-F. Jorge, D.-A. Rodrigo, C.-B. Maria Ximena, L.-M. Victor, A.-F. Emilio, P.-G. Nehomar, C. Jose, C. José, C. Manuel, D. Amable, C. Alejandro, E. Javier, B. José, U. Isabella, Z. Julio, C. Abraham, R. Alfonso and L. Luis, J. Clin. Nephrol., 2020, 4, 044–055 CrossRef.
- T. Rieg and V. Vallon, Diabetologia, 2018, 61, 2079–2086 CrossRef CAS.
- E. C. Chao and R. R. Henry, Nat. Rev. Drug Discovery, 2010, 9, 551–559 CrossRef CAS PubMed.
-
I. S. Padda, A. U. Mahtani and M. Parmar, Sodium-Glucose Transport Protein 2 (SGLT2) Inhibitors, 2022 Search PubMed.
- D. Kawanami, K. Matoba, Y. Takeda, Y. Nagai, T. Akamine, T. Yokota, K. Sango and K. Utsunomiya, Int. J. Mol. Sci., 2017, 18, 1083 CrossRef PubMed.
- H. Yaribeygi, L. E. Simental-Mendía, M. Banach, S. Bo and A. Sahebkar, Biomed. Pharmacother., 2019, 120, 109526 CrossRef CAS.
- S. A. Hawley, R. J. Ford, B. K. Smith, G. J. Gowans, S. J. Mancini, R. D. Pitt, E. A. Day, I. P. Salt, G. R. Steinberg and D. G. Hardie, Diabetes, 2016, 65, 2784–2794 CrossRef CAS.
- T. Moore, R. E. Yanes, M. A. Calton, D. Vollrath, G. M. Enns and T. M. Cowan, PLoS One, 2020, 15, e0240517 CrossRef CAS.
- J. C. Jha, C. Banal, B. S. M. Chow, M. E. Cooper and K. Jandeleit-Dahm, Antioxid. Redox Signaling, 2016, 25, 657–684 CrossRef CAS.
- Y. Gorin and F. Wauquier, Mol. Cells, 2015, 38, 285–296 CrossRef CAS.
- M. Li, H. Li, X. Min, J. Sun, B. Liang, L. Xu, J. Li, S.-H. Wang and X. Xu, J. Med. Chem., 2024, 67, 8406–8419 CrossRef CAS PubMed.
- X. Min, S. Guo, Y. Lu and X. Xu, J. Lumin., 2024, 269, 120437 CAS.
- M. T. Coughlan and K. Sharma, Kidney Int., 2016, 90, 272–279 CAS.
- J. Donate-Correa, D. Luis-Rodríguez, E. Martín-Núñez, V. G. Tagua, C. Hernández-Carballo, C. Ferri, A. E. Rodríguez-Rodríguez, C. Mora-Fernández and J. F. Navarro-González, J. Clin. Med., 2020, 9, 458 CAS.
- M. A. El-Rous, S. Saber, E. M. Raafat and A. A. E. Ahmed, Inflammopharmacology, 2021, 29, 1169–1185 CAS.
- H. Zhang and S.-C. Sun, Cell Biosci., 2015, 5, 63 Search PubMed.
- H. Su, C.-T. Lei and C. Zhang, Front. Immunol., 2017, 8, 1–10 Search PubMed.
- R. D. Bülow and P. Boor, J. Histochem. Cytochem., 2019, 67, 643–661 CrossRef PubMed.
- L. Fan, Q. Xiao, L. Zhang, X. Wang, Q. Huang, S. Li, X. Zhao and Z. Li, Biomed. Pharmacother., 2018, 108, 1640–1650 CAS.
- G. S. Nongthombam, K. Borah, T. Muinao, Y. Silla, M. Pal, H. P. Deka Boruah and R. C. Boruah, ACS Comb. Sci., 2019, 21, 11–27 CAS.
- G. S. Nongthombam and R. C. Boruah, Tetrahedron Lett., 2021, 67, 152893 CAS.
- M. Feng, B. Liang, J. Sun, X. Min, S.-H. Wang, Y. Lu and X. Xu, J. Mol. Struct., 2024, 1310, 138311 CrossRef CAS.
- S. Lang and M. J. Slater, J. Med. Chem., 2024, 67, 6897–6898 CrossRef CAS PubMed.
- J. Lin, D. Xiao, L. Lu, B. Liang, Z. Xiong and X. Xu, J. Mol. Struct., 2023, 1283, 135279 CrossRef CAS.
- G. S. Nongthombam and R. C. Boruah, Heterocycles, 2019, 98, 19 CrossRef CAS.
- G. S. Nongthombam and R. C. Boruah, Synth. Commun., 2021, 51, 2853–2861 CrossRef CAS.
- N. G. Singh, R. Nagarajaprakash, J. W. S. Rani, C. Kathing, R. Nongrum and R. Nongkhlaw, New J. Chem., 2015, 39, 3908–3915 RSC.
- E. Ferrannini, S. A. Veltkamp, R. A. Smulders and T. Kadokura, Diabetes Care, 2013, 36, 1260–1265 CrossRef CAS.
- P. Chonlaket, T. Wongwan and S. Soodvilai, Exp. Physiol., 2018, 103, 250–260 CAS.
- Y. Guo, Z. Ran, Y. Zhang, Z. Song, L. Wang, L. Yao, M. Zhang, J. Xin and X. Mao, Biomed. Pharmacother., 2020, 131, 110684 CrossRef CAS.
- J. E. Gerich, Diabetic Med., 2010, 27, 136–142 CrossRef CAS PubMed.
- K.-F. Tsai, Y.-L. Chen, T. T.-Y. Chiou, T.-H. Chu, L.-C. Li, H.-Y. Ng, W.-C. Lee and C.-T. Lee, Antioxidants, 2021, 10, 1166 CrossRef CAS PubMed.
- Q. Yang, F. Wu, J. Wang, L. Gao, L. Jiang, H.-D. Li, Q. Ma, X. Liu, B. Wei, L. Zhou, J. Wen, T. t. Ma, J. Li and X. Meng, Free Radical Biol. Med., 2018, 124, 466–472 CrossRef CAS.
- L. Wang, H.-L. Wang, T.-T. Liu and H.-Y. Lan, Int. J. Mol. Sci., 2021, 22, 7881 CrossRef CAS.
- C. A. Lipinski, Drug Discovery Today: Technol., 2004, 1, 337–341 CrossRef CAS.
- T. W. Johnson, K. R. Dress and M. Edwards, Bioorg. Med. Chem. Lett., 2009, 19, 5560–5564 CrossRef CAS PubMed.
- B. Webb and A. Sali, Curr. Protoc. Bioinf., 2016, 54, 5.6.1–5.6.37 Search PubMed.
- M. Tuckerman, B. J. Berne and G. J. Martyna, J. Chem. Phys., 1992, 97, 1990–2001 CrossRef CAS.
- L. Fu, S. Shi, J. Yi, N. Wang, Y. He, Z. Wu, J. Peng, Y. Deng, W. Wang, C. Wu, A. Lyu, X. Zeng, W. Zhao, T. Hou and D. Cao, Nucleic Acids Res., 2024, 1–10 Search PubMed.
- S. A. Ahmed, P. Sarma, S. R. Barge, D. Swargiary, G. S. Devi and J. C. Borah, Chem.-Biol. Interact., 2023, 371, 110347 CrossRef CAS PubMed.
- Y.-T. Lu, X.-L. Ma, Y.-H. Xu, J. Hu, F. Wang, W.-Y. Qin and W.-Y. Xiong, Nat. Prod. Bioprospect., 2019, 9, 13–21 CrossRef CAS.
- D. Swargiary, B. Kashyap, P. Sarma, S. A. Ahmed, S. Gurumayum, S. R. Barge, D. Basumatary and J. C. Borah, Fitoterapia, 2024, 173, 105803 CrossRef CAS.
- M.-M. Tiao, C.-W. Liou, L.-T. Huang, P.-W. Wang, T.-K. Lin, J.-B. Chen, Y.-M. Chou, Y.-H. Huang, H.-Y. Lin, C.-L. Chen and J.-H. Chuang, PLoS Genet., 2013, 9, e1003696 CrossRef CAS.
- S. Gurumayum, S. Bharadwaj, Y. Sheikh, S. R. Barge, K. Saikia, D. Swargiary, S. A. Ahmed, D. Thakur and J. C. Borah, J. Ethnopharmacol., 2023, 303, 115936 CrossRef CAS.
- X.-T. Xu, J. Chen, X. Ren, Y.-R. Ma, X. Wang, Y.-Y. Ma, D.-G. Zhao, R.-P. Zhou, K. Zhang, S. Goodin, D.-L. Li and X. Zheng, Eur. J. Pharmacol., 2021, 893, 173840 CrossRef CAS.
- J. Chen, D.-L. Li, L.-N. Xie, Y. Ma, P.-P. Wu, C. Li, W.-F. Liu, K. Zhang, R.-P. Zhou, X.-T. Xu, X. Zheng and X. Liu, Phytomedicine, 2020, 78, 153309 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Semim Akhtar
Ahmed‡
ab,
Kangkon
Saikia
a,
Sanjib
Gogoi
a,
Semim Akhtar
Ahmed‡
ab,
Kangkon
Saikia
a,
Sanjib
Gogoi