
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA novel, glutathione-activated prodrug of pimasertib loaded in liposomes for targeted cancer therapy†
Arianna
Amenta‡
 a,
Susanna
Comi‡
b,
Marcelo
Kravicz
b,
Silvia
Sesana
b,
Antonia
Antoniou
c,
Daniele
Passarella
a,
Pierfausto
Seneci
a,
Sara
Pellegrino
c and
Francesca
Re
*b
a,
Susanna
Comi‡
b,
Marcelo
Kravicz
b,
Silvia
Sesana
b,
Antonia
Antoniou
c,
Daniele
Passarella
a,
Pierfausto
Seneci
a,
Sara
Pellegrino
c and
Francesca
Re
*b
aDepartment of Chemistry, University of Milan, Milan, Italy
bSchool of Medicine and Surgery, University of Milano-Bicocca, Monza, Italy. E-mail: francesca.re1@unimib.it
cDepartment of Pharmaceutical Sciences, University of Milan, Milan, Italy
First published on 3rd October 2024
Abstract
Pimasertib, a potent antiproliferative drug, has been extensively studied for treating cancers characterized by dysregulation in the ERK/MAPK signaling pathway, such as melanoma. However, its therapeutic efficacy would greatly benefit from an increased selectivity for tumour cells and a longer half-life. Such improvements may be achieved by combining the rational design of a prodrug with its encapsulation in a potential nanodelivery system. For this reason, we synthesized a glutathione (GSH)-responsive putative prodrug of pimasertib (PROPIMA), which contains a redox-sensitive disulphide linker that can be processed by GSH to activate pimasertib. The synthesis of PROPIMA and its in vitro biological activity on a human melanoma cell line as a model are described. The results showed that PROPIMA, either free or embedded in liposomes, selectively inhibits cell proliferation and cell viability, reducing by about 5-fold the levels of pERK. Additionally, PROPIMA shows stronger inhibition of the cancer cell migration than the parent drug.
Introduction
Pimasertib (MSC1936369B/AS703026) is an investigational drug developed for its potential use in cancer treatment.1 It belongs to a class of drugs known as mitogen-activated protein kinase kinase (MEK) inhibitors, whose target MEK plays a role in cell growth and division. MEK inhibitors such as pimasertib primarily inhibit the growth and spread of cancers with specific mutations in the mitogen-activated protein kinase (MAPK) signaling pathway, such as melanoma with BRAF (V-raf murine sarcoma viral oncogene homolog B1) or NRAS (neuroblastoma RAS viral [V-ras] oncogene homolog) mutations. Such a pathway regulates cell growth and is often dysregulated in cancer, leading to uncontrolled cell division and tumour growth.2Clinical trials have been conducted to evaluate the safety and effectiveness of pimasertib in multiple cancers, including melanoma, non-small cell lung cancer, and ovarian cancer. The most recent phase I study (http://clinicaltrials.gov, NCT00982865) showed its clinical activity in patients with locally advanced/metastatic melanoma, at pharmacologically active doses (>28 mg per day);3 pimasertib has not yet received regulatory approval for any cancer indication, probably due to its short half-life after oral administration and potential toxicity. In such a scenario, prodrugs may offer significant advantages, and recently many efforts have been devoted towards prodrug-based strategies for cancer therapy.4 A prodrug can be defined as a “disguised drug”, i.e. an inactive or minimally active compound that is converted into its active parent drug in vivo when it reaches a cancer-specific microenvironment. Therefore, prodrugs are designed to selectively target and deliver therapeutic agents to tumour cells while minimizing their toxicity to healthy tissues. Several prodrugs have been synthesized, and some of them have been approved by the FDA.5
Considering that the tumour microenvironment is characterized by excessive oxidative stress associated with a 4-fold increase of glutathione (GSH) compared to normal cells,6 higher GSH concentrations can be exploited to activate prodrugs, thus increasing target selectivity. Therefore, we designed a GSH-responsive prodrug of pimasertib (PROPIMA), which is centered on a redox-sensitive disulphide linker that can be processed by GSH to activate pimasertib (Scheme 1).
 | ||
| Scheme 1 GSH-promoted pimasertib release from disulphide-connected cyclic prodrugs. | ||
The mechanism of action and the effect on cell viability of PROPIMA were evaluated in vitro on a melanoma cell line, as a cancer model with dysregulation of the MAPK signalling pathway.7 Moreover, considering that pharmaceutical delivery through liposomes might significantly increase drug pharmacokinetic properties,8,9 we also produced liposomes loaded with PROPIMA to assess their differential effect on healthy and melanoma cell viability in vitro (Scheme 2). The effect of PROPIMA on melanoma cell migration was also evaluated.
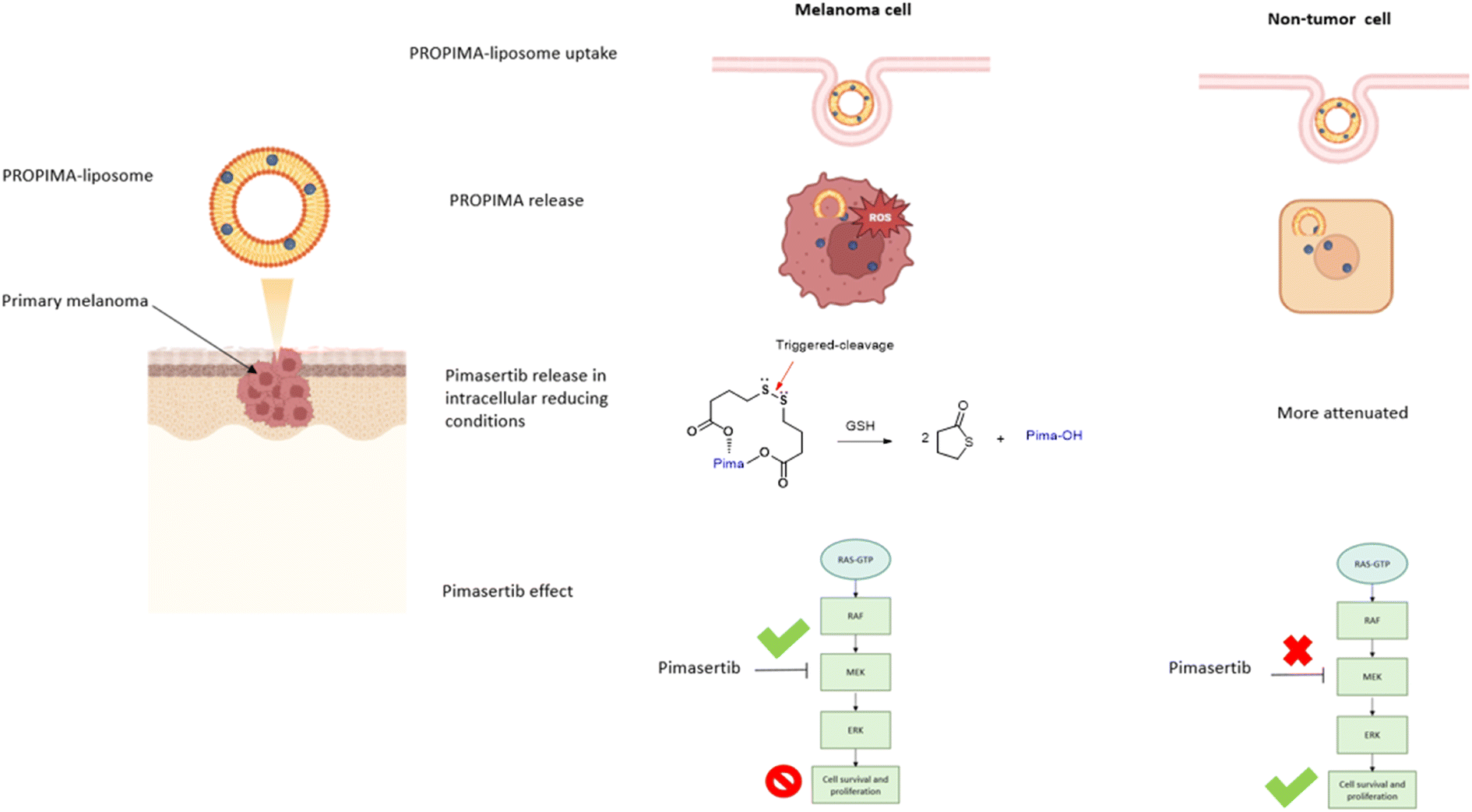 | ||
| Scheme 2 Graphical representation of the experimental plan. | ||
Results and discussion
We have successfully designed and synthesized PROPIMA through an esterification reaction between 4,4′-dithiodibutyric acid and a diol-containing pimasertib (Fig. 1). Its structure was confirmed by 1H and 13C NMR (Fig. S1, ESI†), and its excellent purity (>99%) was determined by LC-MS analysis (Fig. S2, ESI†). | ||
| Fig. 1 Synthesis of GSH-labile pimasertib prodrug, PROPIMA. | ||
The reduction-responsive drug release from PROPIMA was investigated in vitro by incubation at 37 °C with a solution of GSH in sodium phosphate at 10 mM to mimic the intracellular concentration of GSH.10 HPLC analysis, recorded at different times (Fig. 2), revealed a progressive decomposition of the prodrug accompanied by the appearance of a peak corresponding to free pimasertib, therefore showing the drug-release capability of PROPIMA under pathology-related reductive conditions.
 | ||
| Fig. 2 GSH reduction-responsive release of pimasertib from PROPIMA. HPLC chromatograms after incubation of PROPIMA with 10 mM GSH: (A) 1 h; (B) 6 h (≈30% conversion); (C) 12 h (≈70% conversion); (D) 24 h (>95% conversion). | ||
Then, we evaluated the effect of PROPIMA on cell viability compared to free pimasertib. A375 melanoma cells were incubated with different concentrations of the free drug and prodrug dissolved in DMSO for up to 72 h at 37 °C, and cell viability was assessed by MTT assay. The results (Fig. 3A) showed that PROPIMA is cytotoxic, reducing cell viability in a time and dose-dependent manner, similarly to free pimasertib. Differences after 24 or 48 h of incubation between the two tested samples can be ascribed to the time required for the reductive bioactivation of PROPIMA, leading to controlled pimasertib release in situ. Accordingly, by increasing cell treatment time to 72 h, the prodrug became as cytotoxic as free pimasertib. For this reason, the following experiments were performed by incubating A375 cells with PROPIMA for 48 h, to appreciate better the differences with respect to the parental drug. The IC50 values for pimasertib and PROPIMA were calculated (Fig. 3B), confirming a slightly worse IC50 value for PROPIMA. Once more, increasing the incubation time, the IC50 of the prodrug becomes similar to the value of pimasertib. These results confirm that PROPIMA is a prodrug of pimasertib, making it valuable for inhibiting cancer cell growth and spread. The effect of PROPIMA on cell proliferation was assessed by monitoring the cell growth over time with the MTT assay. Results showed that, similarly to pimasertib, PROPIMA slows down cell proliferation in a time and dose-dependent manner (Fig. 3C).
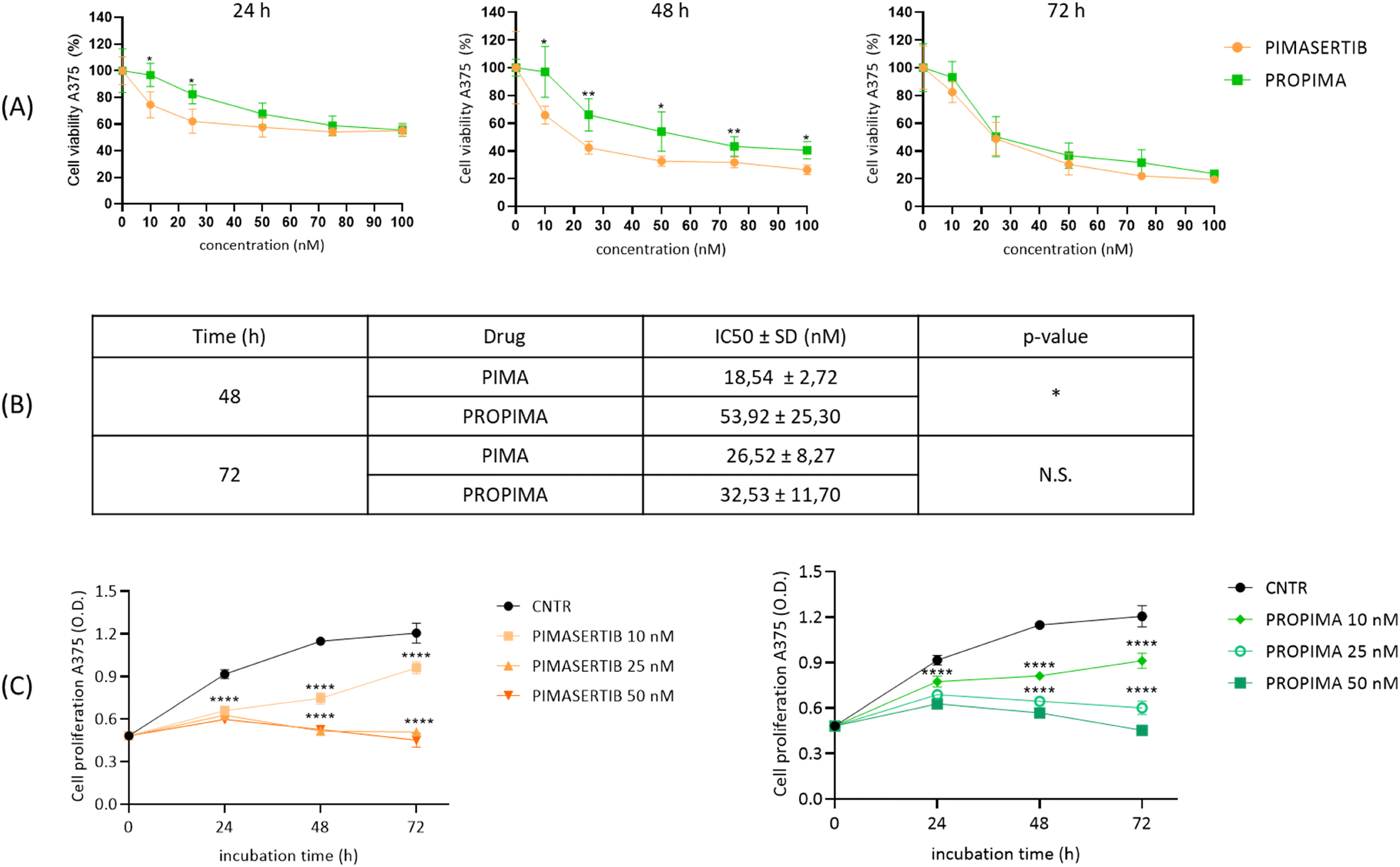 | ||
| Fig. 3 Cytotoxicity of pimasertib and PROPIMA on human A375 melanoma cells. (A) A375 cells were treated with different doses (10–100 nM) of either pimasertib or PROPIMA dissolved in DMSO, and after incubation times up to 72 h, cell viability was assessed by MTT assay. Results are expressed as a percentage of viable cells ± standard deviation. Untreated cells were set as 100% viability. (B) IC50 values for pimasertib and PROPIMA were calculated using GraphPad Prism software with fit spline/LOWESS interpolation function. N.S., not significant; * = p < 0.05; ** = p < 0.01 (two-way ANOVA and Tukey's multiple comparison test). (C) A375 cells were incubated with doses of pimasertib dissolved in DMSO or PROPIMA dissolved in DMSO, at 37 °C for up to 72 h. Cell proliferation at each time point was assessed using the MTT assay. Results are expressed as O.D. (optical density) of viable cells ± standard deviation. Untreated cells were used as the control. **** = p < 0.0001 vs. untreated cells (two-way ANOVA and Tukey's multiple comparison test). | ||
To better discriminate between the toxic effect of PROPIMA on cell viability and the anti-proliferative activity, A375 cells were treated with different doses of the prodrug or pimasertib for 48 h, and LDH release, BrDU incorporation rate and trypan blue uptake were measured. Results showed that treating A375 cells with PROPIMA did not affect the release of LDH and the cellular internalization of trypan blue, indicating no cell membrane damage and, as a consequence, cell viability (Fig. 4A and C). Nevertheless, a dose-dependent effect on BrDU incorporation was observed after treatment with PROPIMA, supporting its anti-proliferative effect (Fig. 4B). Similar results were obtained with the parental drug, thus confirming that the chemical modification of pimasertib did not affect its activity.
 | ||
| Fig. 4 Cytotoxic effect of pimasertib and PROPIMA. A375 were seeded in 96-well plates at a density of 1 × 104, and treated with different doses (10–50 nM) of free pimasertib or PROPIMA for 48 h. (A) % of LDH release was measured after treatment with an LDH assay kit (Roche). Cells treated with lysis buffer were used as a control of maximum LDH release. (B) % of incorporated BrDU determined using a BrDU cell proliferation assay kit #6813 (Cell Signaling). Untreated cells were used as the control. (C) Cell viability was assessed using a trypan blue exclusion assay. Untreated cells were used as the control. Results are expressed as a mean value ± standard deviation. * p < 0.5; ** p < 0.01; *** p < 0.001 (two-way ANOVA test and Tukey's multiple comparison test). | ||
To verify that esterification/cyclization of pimasertib did not affect the drug's mechanism of action, a western blot assay was carried out to confirm the ability of PROPIMA to inhibit the kinase MEK, by measuring the levels of pERK/ERK in treated melanoma cells (Fig. 5A). Treatment of A375 cells with PROPIMA (10 nM) for 48 h reduces the pERK levels by about 5-fold, thus confirming that the prodrug maintains MEK inhibitory activity as pimasertib (Fig. 5B and C).
 | ||
| Fig. 5 Analysis of pERK and ERK 1/2 expression levels in human A375 melanoma cells. (A) Sketched representation of the signaling pathway inhibited by pimasertib and PROPIMA (created using BioRender software). (B) A375 cells were treated with pimasertib (10 nM) or PROPIMA (10 nM), and after 48 h incubation, whole cell lysates were subjected to SDS-PAGE followed by immunoblot analysis. Representative blots are shown, with lanes 1, 2 and 3 representing untreated cells, cells treated with pimasertib and cells treated with PROPIMA, respectively. (C) Semi-quantitative estimation of western blot bands expressed as pERK/ERK ratio ± standard deviation. Cntr, control untreated cells; **** = p < 0.0001 (Student's t-test). | ||
To further rule out any nonspecific effect, either non-tumour cells (i.e. endothelial cells, hCMEC/D3) or tumour cells not presenting dysregulations in the ERK/MAPK signaling pathway (i.e. glioblastoma U87 cells) were also treated. The results (Fig. 6) showed that PROPIMA did not induce any cytotoxicity on both glioblastoma cells (Fig. 6A) and endothelial cells (Fig. 6B) analogously to the parent drug. Moreover, evaluating the effect of these drugs on cell proliferation, the results showed that PROPIMA maintains the specificity for cells bearing specific mutations in the MAPK signaling pathway, as shown by its lower efficacy in inhibiting cell proliferation on U87 (about −20% at 72 h) (Fig. 6C) in comparison to A375 (about −60% at 72 h) (Fig. 3), and an improved selectivity for tumour cells in comparison to healthy cells, as shown by the absence of effect on hCMEC/D3 proliferation in comparison to pimasertib that conversely induced a −25% reduction of proliferation (Fig. 6D). These findings can be attributed to the higher level of glutathione peroxidase 1 (GpX-1), the enzyme involved in the oxidation of GSH and useful to counteract oxidative stress, in A375 cells (9934 ± 593 mU mL−1) compared to hCMEC/D3 cells (4910 ± 959 mU mL−1) (ESI† methods).
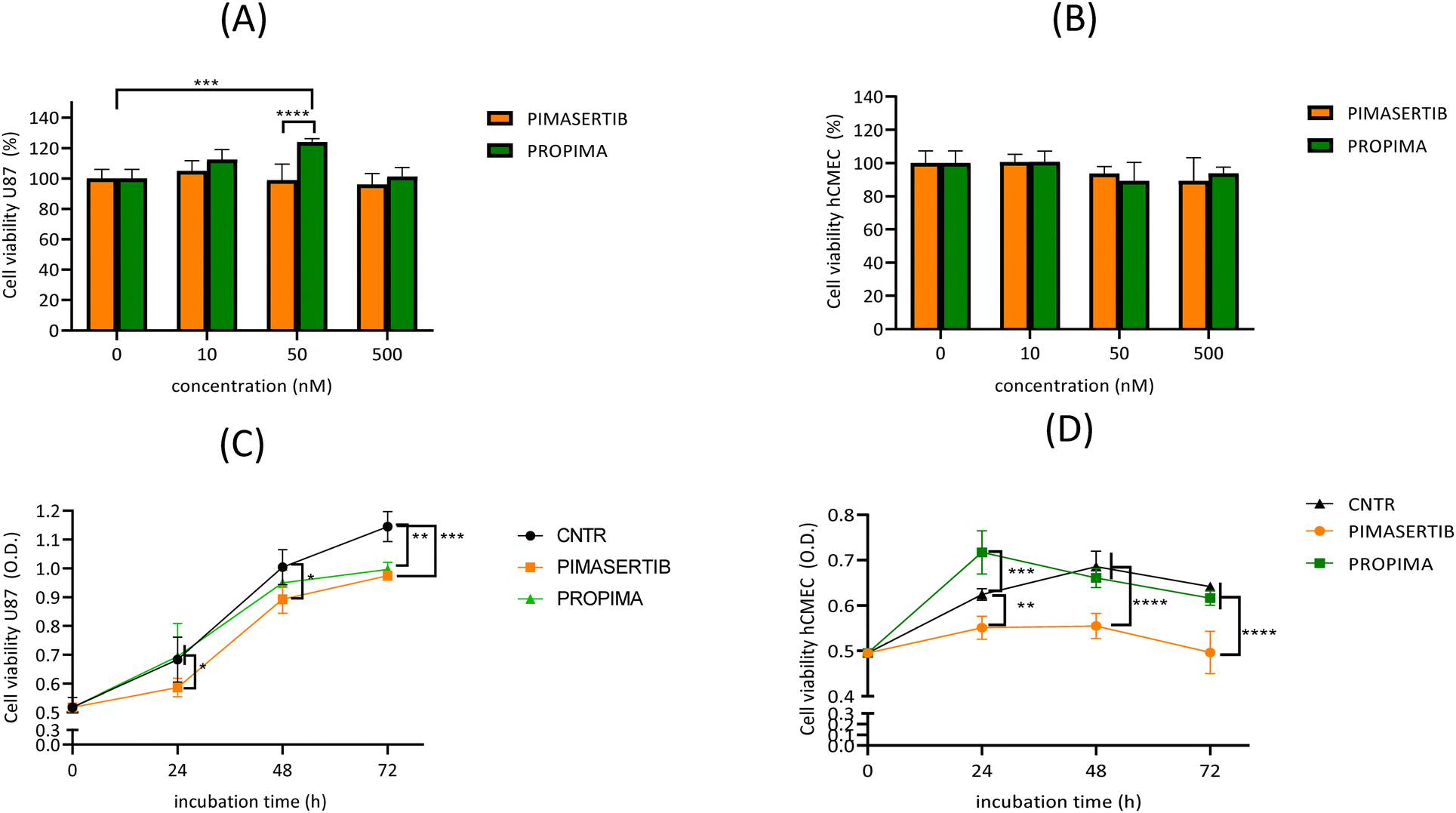 | ||
| Fig. 6 Evaluation of the effect of PROPIMA on U87 and hCMEC/D3 cell viability and proliferation by MTT assay. (A and C) Glioblastoma U87 cells and (B and D) hCMEC/D3 endothelial cells were treated with different doses (10–500 nM) of either pimasertib or PROPIMA, and after different times of incubation, cell viability (A and B) and proliferation (C and D) were characterized by MTT assay. For cell viability, results are expressed as a percentage of viable cells ± standard deviation; untreated cells were set at 100% viability. For cell proliferation, results are expressed as O.D. (optical density) of viable cells ± standard deviation; untreated cells were used as the control. Data were analyzed by two-way ANOVA test: (C) F(2, 36) = 12.21, p < 0.0001; * p < 0.5; ** p < 0.01; *** p < 0.001 vs. CNTR. (D) F(2, 36) = 67.86, p < 0.0001; ** p < 0.01, *** p < 0.001, **** p < 0.0001 vs. CNTR. CNTR = untreated cells. | ||
Considering that the dysregulation of the MAPK pathway in cancer cells (melanoma included) increases the production and secretion of metalloproteinases, which play an essential role in tumour invasion and migration,11 the effect of PROPIMA on A375 migration was assessed by wound healing assay. Distances and width closure were calculated by comparing the images taken between 0 and 48 h (Fig. 7A). We observed that A375 cells migrated and covered approximately 70% to 100% of the wound area within 48 h in the presence of free pimasertib, suggesting that it fails to inhibit cell migration. This is in agreement with the work of Della Corte CM et al.12 showing that an anti-migration effect was obtained only combining pimasertib with another anti-cancer drug. However, a significant lower migration (from 60% to 70% of the wound area within 48 h) was observed when A375 cells were treated with PROPIMA (Fig. 7B). These resulting differences can be due to the different kinetic action profiles between PROPIMA and pimasertib. In support of this, it has been shown that the sustained release of the drug versus drug burst release is favorable for cancer therapy.13 Further investigations are needed to assess this issue, and also the possible effect of PROPIMA on targets other than MEK involved in cell migration, such as PI3K/Akt14 and Rho/ROCK15 pathways. Considering that one of the major concerns of the wound healing assay is the contribution of cell proliferation, the same experiments were performed under 0% FBS conditions to limit cell proliferation, thus promoting cell migration. The results were comparable to those observed with 10% FBS (data shown).
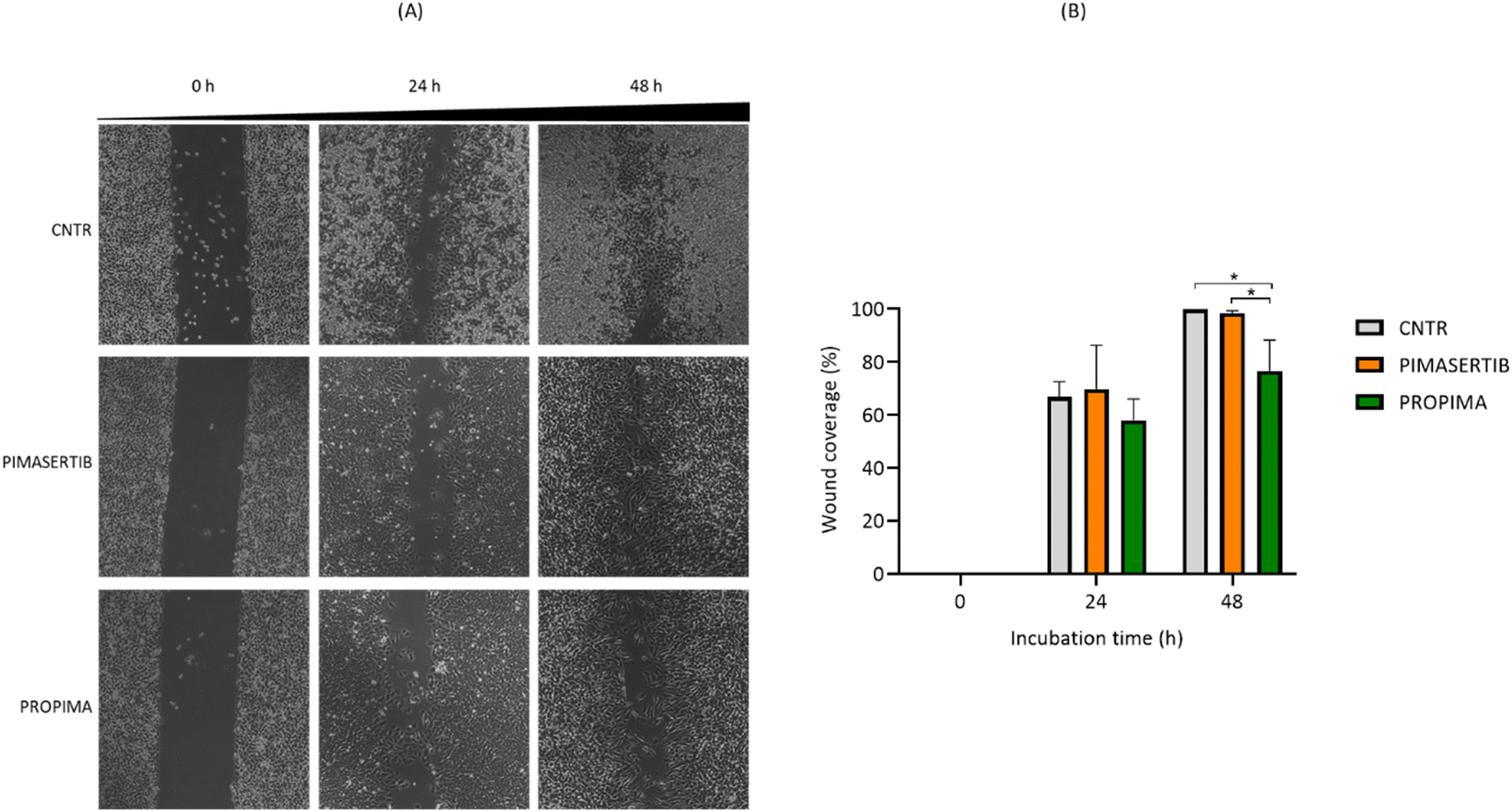 | ||
| Fig. 7 Effects of pimasertib and PROPIMA on A375 migration. A375 cells were seeded in a culture-insert 2-well Petri dish (35 mm, Ibidi). Then, the culture-insert was removed, cells were treated with either 10 nM pimasertib or PROPIMA for 48 h, and images were recorded at different time points. (A) Representative images are shown, and dark bands define the areas lacking cells (wound area, ImageJ). (B) Values of percentage wound closure ± standard deviation are shown. * p < 0.05 (two-way ANOVA and Tukey's multiple comparison test). | ||
Considering the short half-life of pimasertib (3.5 h and 6.3 h after i.v. or oral administration, respectively),1 and predicting similar issues for its release from PROPIMA, we loaded the prodrug in liposomes, as drug delivery systems. Liposomes were chosen because they have been extensively utilized due to their safety and adaptability in delivering both water-soluble and lipid-soluble drugs. Drug delivery via liposomes protects encapsulated drugs by shielding them from catabolic processes, preventing interaction with metabolizing enzymes while in the bloodstream.16,17
Liposomes composed of sphingomyelin/cholesterol (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, M/M) and loaded with pimasertib or PROPIMA were prepared by an ethanol-injection technique.18 Their characterization (Table 1) showed a narrow particle size distribution (PDI < 0.2) with a <200 nm diameter, and an optimal size for passive targeting of liposomes in the tumour through the fenestrations of the endothelium while avoiding excessive phagocytosis by the reticuloendothelial system. Moreover, this size range is more suitable for the accumulation at tumour sites, while larger liposomes with a >300 nm diameter accumulate in the spleen, and smaller, <40 nm ones could be up-taken by the liver or kidneys.17 Their ζ-potential values showed a negative net surface charge, suggesting that their dispersions are stable and not prone to aggregation. Furthermore, negatively charged liposomes should have greater permeability through the skin, and be suitable for the topical treatment of melanoma.17 An increase in their size was detected after drug loading, probably due to their localization across the bilayer and the liposomal core. The encapsulation efficiency (EE%) measured using a spectrophotometer was ∼70% for PROPIMA and ∼30% for pimasertib, probably due to the higher solubility of the prodrug in comparison to pimasertib, which allowed higher drug loading in the liposome core. The drug/lipid ratio (D/L) was 0.06 for PROPIMA and 0.03 for pimasertib. This ratio is a critical process parameter that expresses the capacity of liposomes to accommodate pimasertib or PROPIMA. In particular, a ≤0.95 D/L ratio denotes a high loading efficacy and the effectiveness of the preparation method; a >0.95 D/L ratio would rather indicate a low loading efficiency, with a high probability to damage the liposomal membrane.19 Knowing that the D/L ratio is affected by the liposome composition and the loading method, this parameter could be further optimized, as earlier done for vincristine-loaded liposomes.20
:1, M/M) and loaded with pimasertib or PROPIMA were prepared by an ethanol-injection technique.18 Their characterization (Table 1) showed a narrow particle size distribution (PDI < 0.2) with a <200 nm diameter, and an optimal size for passive targeting of liposomes in the tumour through the fenestrations of the endothelium while avoiding excessive phagocytosis by the reticuloendothelial system. Moreover, this size range is more suitable for the accumulation at tumour sites, while larger liposomes with a >300 nm diameter accumulate in the spleen, and smaller, <40 nm ones could be up-taken by the liver or kidneys.17 Their ζ-potential values showed a negative net surface charge, suggesting that their dispersions are stable and not prone to aggregation. Furthermore, negatively charged liposomes should have greater permeability through the skin, and be suitable for the topical treatment of melanoma.17 An increase in their size was detected after drug loading, probably due to their localization across the bilayer and the liposomal core. The encapsulation efficiency (EE%) measured using a spectrophotometer was ∼70% for PROPIMA and ∼30% for pimasertib, probably due to the higher solubility of the prodrug in comparison to pimasertib, which allowed higher drug loading in the liposome core. The drug/lipid ratio (D/L) was 0.06 for PROPIMA and 0.03 for pimasertib. This ratio is a critical process parameter that expresses the capacity of liposomes to accommodate pimasertib or PROPIMA. In particular, a ≤0.95 D/L ratio denotes a high loading efficacy and the effectiveness of the preparation method; a >0.95 D/L ratio would rather indicate a low loading efficiency, with a high probability to damage the liposomal membrane.19 Knowing that the D/L ratio is affected by the liposome composition and the loading method, this parameter could be further optimized, as earlier done for vincristine-loaded liposomes.20
| Formulation | Diameter (nm) | PDI (polydispersity index) | ζ-Potential (mV) | Encapsulation efficiency% (EE%) |
|---|---|---|---|---|
| Empty liposomes | 118 ± 10 | 0.133 ± 0.001 | −18.34 ± 3.51 | — |
| PIMASERTIB-liposomes | 153 ± 13 | 0.148 ± 0.006 | −11.49 ± 4.50 | 33.32 ± 9.35 |
| PROPIMA-liposomes | 129 ± 14 | 0.138 ± 0.019 | −16.93 ± 2.99 | 71.48 ± 13.17 |
To evaluate if PROPIMA after loading in liposomes maintains the pharmacological activity of the free prodrug, A375 cells were treated with PROPIMA-liposomes for 48 h. This time point has been selected to maintain consistency with the previous experiments, and because the blood circulation of liposomes in vivo is typically no longer than 48 hours.21 The results (Fig. 8A) showed that PROPIMA-liposomes reduce cell viability similarly to free PROPIMA, suggesting that its incorporation in liposomes did not affect its cytotoxicity. Moreover, we observed reduced pERK levels after treatment with PROPIMA-liposomes (Fig. 8B and C), confirming their efficacy through the same mechanism of action. These findings suggest that the liposomal formulation of PROPIMA represents a promising approach against melanoma, similar to other prodrugs that have shown enhanced metabolic stability, improved intracellular cancer cell delivery and release, and increased efficacy after being incorporated into liposomes.22,23 Additionally, this strategy can be further optimized by modifying the liposome surface with ligands that facilitate the delivery of prodrugs across biological barriers, such as the blood–brain barrier,24 where cancer metastasis, including melanoma, can occur.25
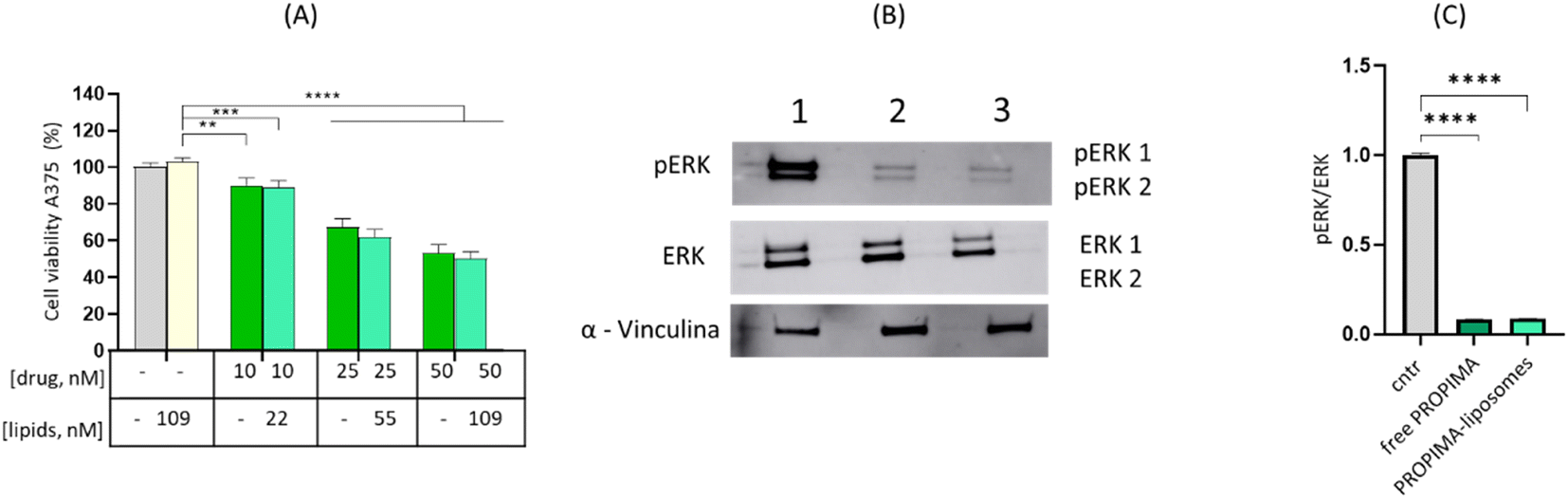 | ||
| Fig. 8 Effects of PROPIMA-liposomes on A375 cells. (A) A375 cells were treated with either free PROPIMA (10–50 nM, dark green), or the same embedded in liposomes (light green), and after 48 h, cell viability was assessed by MTT assay. Results are expressed as a percentage of viable cells ± standard deviation. Untreated cells were set at 100% viability. Empty liposomes and the highest lipid dose were also tested as blanks (yellow bar). **** = p < 0.0001 (two-way ANOVA and Tukey's multiple comparison test). (B) A375 cells were treated with PROPIMA (10 nM), either free or embedded in liposomes, and after 48 h incubation, whole cell lysates were subjected to SDS-PAGE followed by immunoblot analysis. Representative blots are shown, where lanes 1, 2 and 3 represent untreated cells, cells treated with free PROPIMA and cells treated with PROPIMA-liposomes, respectively. (C) Semi-quantitative estimation of bands expressed as pERK/ERK ratio ± standard deviation; cntr: control untreated cells; **** = p < 0.0001 (Student's t-test). | ||
Experimental procedures
General
Reagents and solvents were purchased from Sigma-Aldrich and used without further purification. Pimasertib was purchased from Advanced ChemBlocks Inc. Chemical reactions were carried out in oven-dried glassware and with dry solvents, under a nitrogen atmosphere, and were monitored by TLC on silica gel (Merck precoated 60F254 plates), with detection by UV light (254 nm) or by permanganate. HPLC was performed on an Agilent 1100 Series System, using a Gemini 5 μM C18 110 Å LC column (150 × 3 mm) and a H2O/ACN gradient from 5% ACN to 100% ACN in 40 min, 1.0 mL min−1 flux and 20 μL sample injection. 1H NMR and 13C NMR spectra were recorded on a Bruker Advance spectrometer (400 MHz), using commercially available deuterated chloroform (chloroform-d) solvent at room temperature.Synthesis of PROPIMA, (S)-N-((5,14-dioxo-1,4-dioxa-9,10-dithiacyclotetradecan-2-yl)methyl)-3-((2-fluoro-4-iodophenyl)amino) isonicotinamide
EDC (74 mg, 0.38 mmol) and DMAP (46 mg, 0.38 mmol) were added under stirring at room temperature to a solution of 4,4′-dithiodibutyric acid (39 mg, 0.16 mmol) in dry DMF. After 30 min, pimasertib (70 mg, 0.16 mmol) was added, and the mixture was stirred at room temperature overnight. The reaction mixture was then diluted with ethyl acetate (10 mL) and washed with saturated NH4Cl (3 × 10 mL). The organic layer was dried over sodium sulfate and the solvent was removed under reduced pressure. The crude product was purified by column chromatography on silica gel (98:2 DCM:MeOH) to give pure PROPIMA (80 mg, 79%) as a yellowish oil. 1H NMR (400 MHz, CDCl3): δ (ppm) = 9.02 (s, 1H), 8.58 (s, 1H), 8.16 (s, 1H), 7.48 (d, J = 10.0 Hz, 1H), 7.42 (d, J = 8.5 Hz, 1H), 7.24 (d, J = 4.5 Hz, 1H), 7.13 (t, J = 8.4 Hz, 1H), 6.84 (bs, 1H), 5.36–5.27 (m, 1H), 4.52 (dd, J = 12.2, 2.1 Hz, 1H), 4.21 (dd, J = 12.2, 7.2 Hz, 1H), 3.78–3.64 (m, 2H), 2.86–2.68 (m, 4H), 2.62–2.45 (m, 4H), 2.00 (m, 4H). 13C NMR (100 MHz, CDCl3): δ (ppm) = 173.1 (C), 172.5 (C), 167.7 (C), 154.8 (d, J = 258.5 Hz, C), 140.4 (CH), 139.5 (CH), 133.8 (d, J = 3.7 Hz, CH), 128.7 (d, J = 11.3 Hz, C), 125.6 (d, J = 21.9 Hz, CH), 132.5 (C), 123.0 (CH), 84.7 (d, J = 7.4 Hz, C), 70.8 (CH), 63.0 (CH2), 40.6 (CH2), 39.0 (CH2), 38.9 (CH2), 33.0 (CH2), 32.9 (CH2), 24.7 (CH2), 24.5 (CH2).
LC-MS: m/z = 634.532 [M+ H]+, purity >99%.
In vitro GSH-mediated release of pimasertib from PROPIMA
The release profile of PROPIMA by GSH was investigated by incubation of a 1 mg mL−1 prodrug solution at 37 °C in 10 mM sodium phosphate in the presence of 10 mM GSH. After 12 h and 24 h, respectively, 200 μL of the solution were taken out to run an HPLC and quantify the release of pimasertib. The same experiment was carried out in the absence of 10 mM GSH as a negative control.Cell cultures
The epithelial cell line A375 (ATCC® CRL-1619, ATCC, Manassas, MD, USA), originally isolated from the skin of a 54 year-old patient, was used as an in vitro model of malignant melanoma. Cells were maintained in culture in complete DMEM medium supplemented with 10% FBS (fetal bovine serum), 4 mM L-glutamine, 1% penicillin–streptomycin (P/S) and 1 mM sodium pyruvate. Cultured cells were maintained at 37 °C in a humidified environment and 5% CO2, changing the medium every 2 to 3 days.21 Immortalized human cerebral microvascular endothelial cells (hCMEC/D3) were provided by Prof. Bourdoulous (Institut Cochin, Inserm, Paris, France) and used as a non-tumour cellular model.26 Cells between 25 and 35 passages were seeded on tissue culture flasks pre-treated with rat tail collagen type I (0.05 mg mL−1). Cells were grown in a complete culture medium (endothelial basal medium, EBM-2 supplemented with 10% FBS, 1% chemically defined lipid concentrate, 1% P/S, 10 mM HEPES, 5 μg mL−1 ascorbic acid, 1 ng mL−1 basic fibroblast growth factor, and 1.4 μM hydrocortisone) and maintained at 37 °C and 5% CO2. The culture medium was changed every 2 days.27 U87-MG glioblastoma cells were purchased from the American Type Culture Collection (ATCC, VA, USA) and used as an in vitro cancer model without alterations in the MAPK pathway. Cells were grown in culture in a complete DMEM medium supplemented with 10% FBS, 4 mM L-glutamine, 1% P/S and 1 mM sodium pyruvate and maintained at 37 °C and 5% CO2. The culture medium was changed every 2 days.Cell viability and proliferation assay
Cell viability and proliferation were determined by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay.28 Cells were seeded in 96-well plates at densities of 1 × 104 (A375), 3 × 104 (hCMEC/D3) and 1.2 × 104 (U87 cells) cells per well.21 Different doses (10–500 nM) of either free pimasertib or free PROPIMA (or PROPIMA-liposomes) were added to the culture medium and incubated for up to 72 h. Free drugs were dissolved in DMSO; PROPIMA-liposomes were dissolved in phosphate buffered saline (PBS). Untreated cells or cells treated with empty liposomes (109 nM total lipids, corresponding to the maximum dose of PROPIMA-liposomes used) were used as a control. Each experiment was carried out at least in triplicate. IC50 values were calculated using GraphPad Prism software with the fit spline/LOWESS interpolation function.LDH assay
The cytotoxic effect of pimasertib and PROPIMA was evaluated by measuring the LDH release using an LDH assay kit (Roche). The assay is based on the measurement of cytoplasmic LDH activity released from damaged cells after exposure to treatments. A375 cells were seeded in 96-well plates at a density of 1 × 104, and treated with different doses (10–50 nM) of either pimasertib or PROPIMA for 48 h. Free drugs were dissolved in DMSO. After treatment, LDH released in the culture media was detected following the manufacturer's protocol (Roche). Briefly, 100 μL of cell medium was collected from each well, and then 100 μL of the reaction mixture (freshly prepared) was added and incubated for 30 min at RT. Finally, 50 μL of STOP solution was added to each well on the 96-well plate. Absorbance was measured at two different wavelengths, one being the “measurement wavelength” (492 nm) and the other being the “reference wavelength” (690 nm) using a microplate spectrophotometer. Maximum LDH release (positive control) was obtained by adding 5 μL lysis buffer to untreated control cells. The average values of the culture medium background were subtracted from all values of experimental wells and the percentage of death cells was calculated in relation to the maximum LDH release. Each experiment was carried out at least in triplicate.BrDU cell proliferation assay
Cell proliferation was determined using a BrDU cell proliferation assay kit #6813 (Cell Signaling). A375 cells were seeded in 96-well plates at a density of 1 × 104, and treated with different doses (10–50 nM) of either pimasertib or PROPIMA for 48 h. Free drugs were dissolved in DMSO. Briefly, 10 μL per well BrdU labeling solution (10×) was added and cells were re-incubated for an additional 4 h at 37 °C. Then, the culture medium was removed and cells were fixed in fixing/denaturing solution, at 100 μL per well for 30 min at RT. The working solution of anti-BrdU antibody diluted in detection antibody diluent was subsequently added (100 μL per well for 60 min, RT), and following this, the working solution of anti-mouse IgG coupled with horseradish peroxidase diluted in HRP-linked antibody diluent was added (100 μL per well for 30 min, RT). Incorporated BrdU was detected using tetramethylbenzidine substrate (TMB) (100 μL per well for 30 min at RT). A total of 100 μL per well of STOP solution was added to stop the enzymatic reaction, and the quantification was performed with a microplate spectrophotometer at 450 nm. Untreated cells were used as 100% of incorporated BrDU. Each experiment was carried out at least in triplicate.Trypan blue exclusion assay
A375 cells were seeded in 96-well plates at a density of 1 × 104, and treated with different doses (10–50 nM) of either pimasertib or PROPIMA for 48 h. Free drugs were dissolved in DMSO. After treatment, cells were washed with PBS 1× and 50 μL of 0.25% trypsin/EDTA for 7 min at 37 °C was added to detach the cells. Trypsin activity was stopped adding 50 μL of DMEM, 10% FBS and the cell suspension was collected in Eppendorf tubes. The cells were diluted with a 1:1 ratio of 0.4% trypan blue staining solution (Sigma-Aldrich) and added to a Burker chamber to count the vital cells under a light microscope. Untreated cells were set as 100% viability. Each experiment was carried out at least in triplicate.
Immunoblotting
A375 cells were plated in 6-well plates (3 × 105 cells per well) and incubated with 10 nM of free pimasertib, free PROPIMA or PROPIMA-liposomes for 48 h. Whole cell lysates were obtained and analyzed as previously described.20 Anti-ERK1/2 (1:1000), anti-p-ERK1/2 (1:2000) and anti-α vinculin (1:1000) antibodies from Cell Signaling (Beverly, MA) were used. Mouse anti-rabbit IgG secondary antibodies conjugated with horseradish peroxidase (1:5000) from Biorad were used for 1 h at room temperature. Immunoreactive proteins were visualized by enhanced chemiluminescence (ECL plus, Thermo Fisher Scientific (Rockford, IL) with an Amersham Imager 600 (GE Healthcare)), and the band intensity was quantified using ImageLab software. Each experiment was carried out at least in triplicate.
Cell migration assay
Cell migration assay was performed as described,29 with modifications. Briefly, A375 cells were seeded in a culture-insert 2-well Petri dish (35 mm, Ibidi) at a density of 2 × 103 cells per well and after an overnight incubation. The culture-insert was removed with sterile tweezers, and cells were washed twice with PBS. Cells were then treated with 10 nM of free pimasertib or PROPIMA for 48 h. Pictures of wounds were taken with an optical microscope at time points of 0, 24 h and 48 h. Untreated cells were used as the control. The percentage of the covered area at each time was calculated using ImageJ software.Preparation and characterization of pimasertib and PROPIMA-liposomes
Cholesterol and sphingomyelin were purchased from Avanti Polar Lipids Inc., Alabaster, AL, USA. Sphingomyelin and cholesterol in a 1:1 molar ratio, M/M (8 μmol of total lipids), were solubilized in ethanol together with pimasertib (0.27 mg mL−1) or PROPIMA (0.4 mg mL−1). This solution (lipid phase) was dropped in 3 mL of PBS (pH 7.4) under stirring. Ethanol was evaporated at 40 °C under reduced pressure for 5 minutes, and the formulation was sonicated for 12 minutes at 65 °C. Any residual free drug or prodrug was removed by ultracentrifugation using an Amicon®Ultra 10 kDa filter (Millipore, Sigma Aldrich). Free pimasertib or PROPIMA was quantified using a spectrophotometer at 340 nm, and lipid recovery was measured by Stewart's assay.30 The encapsulation efficiency (EE%) and drug-to-lipid mass ratio (D/L, μmol μmol−1) were calculated as described.31 The size, polydispersity index (PDI) and ζ-potential were analysed by dynamic light scattering (DLS) with an instrument made by Brookhaven Instruments Corporation, Holtsville, NY, USA, equipped with a ZetaPALS device, as previously described.32,33
Conclusions
We herein report the successful design and synthesis of a novel pimasertib prodrug with potential to compete with present treatments against various types of cancers characterized by dysregulation in the ERK/MAPK signaling pathway. The in vitro selectivity of PROPIMA for cancer cells outlines an efficient approach to managing the lack of selectivity of standard chemotherapy drugs. Moreover, the combined and innovative strategy to encapsulate the prodrug in nanocarriers without loss of pharmacological activity is a powerful tool to also improve the pharmacokinetic profile of the active compound. This issue deserves further investigation. Therefore, both PROPIMA and its liposome-loaded version represent two pharmacological entities with putative broad applicability against various types of cancer. However, increasing knowledge of prodrug specificity toward different substrates and mechanisms of action, alongside further liposome functionalization, could lead to the future development of successful targeted prodrug delivery.Data availability
The data supporting the article titled: “A novel, glutathione-activated prodrug of pimasertib loaded in liposomes for targeted cancer therapy” by Arianna Amenta, Susanna Comi, Marcelo Kravicz, Silvia Sesana, Antonia Antoniou, Daniele Passarella, Pierfausto Seneci, Sara Pellegrino and Francesca Re have been included as part of the ESI† and are available in the interactive notebook [Susanna Comi_PROPIMA, Google Drive Folder] at [https://drive.google.com/drive/folders/1JvKAYpYzIEIcYYXSGPIhYITu-Qz55--9?usp=sharing].Author contributions
All authors have approved the final version of the manuscript. These authors contributed equally.A. Amenta and A. Antoniou synthesized and characterized PROPIMA and performed the in vitro GSH sensitivity assay. S. C. performed all the in vitro experiments. A. A. and S. C. prepared the first draft of the manuscript. M. K. conducted the preparation of liposomes using ethanol-injection techniques. M. K. and S. C. carried out the PROPIMA loading in liposomes. S. S. performed the physicochemical characterization of liposomes. F. R., S. P. and P. S. conceived the project. F. R. and P. S. supervised the research and revised the manuscript with contributions from all co-authors.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Fondazione Regionale per la Ricerca Biomedica, research project “New frontiers of engineered nanovectors to improve treatment efficacy and safety in neurological–NEVERMIND Project”, project nr. CP2_16/2018, to F. R. and P. S. We thank Prof. Bulbarelli A. and Dr. Lonati E. of the School of Medicine and Surgery, University of Milano-Bicocca, for suggestions on biochemical experimental sections.References
- O. von Richter, G. Massimini, H. Scheible, I. Udvaros and A. Johne, Pimasertib, a Selective Oral MEK1/2 Inhibitor: Absolute Bioavailability, Mass Balance, Elimination Route, and Metabolite Profile in Cancer Patients, Br. J. Clin. Pharmacol., 2016, 82(6), 1498–1508, DOI:10.1111/bcp.13078.
- U. Degirmenci, M. Wang and J. Hu, Targeting Aberrant RAS/RAF/MEK/ERK Signaling for Cancer Therapy, Cell, 2020, 9(1), 198, DOI:10.3390/cells9010198.
- C. Lebbé, A. Italiano, N. Houédé, A. Awada, P. Aftimos, T. Lesimple, M. Dinulescu, J. H. M. Schellens, S. Leijen, S. Rottey, V. Kruse, R. Kefford, E. Raymond, S. Faivre, C. Pages, C. Gomez-Roca, A. Schueler, S. Goodstal, G. Massimini and J.-P. Delord, Selective Oral MEK1/2 Inhibitor Pimasertib in Metastatic Melanoma: Antitumor Activity in a Phase I, Dose-Escalation Trial, Target Oncol., 2021, 16(1), 47–57, DOI:10.1007/s11523-020-00767-1.
- J. Rautio, N. A. Meanwell, L. Di and M. J. Hageman, The Expanding Role of Prodrugs in Contemporary Drug Design and Development, Nat. Rev. Drug Discovery, 2018, 17(8), 559–587, DOI:10.1038/nrd.2018.46.
- G. Lemercier, P. Carpentier, H. Sentenac-Roumanou and P. Morelis, Histological and Histochemical Changes in the Central Nervous System of the Rat Poisoned by an Irreversible Anticholinesterase Organophosphorus Compound, Acta Neuropathol., 1983, 61(2), 123–129, DOI:10.1007/BF00697391.
- E. E. Ramsay and P. J. Dilda, Glutathione S-Conjugates as Prodrugs to Target Drug-Resistant Tumors, Front. Pharmacol., 2014, 5, 181, DOI:10.3389/fphar.2014.00181.
- G. S. Inamdar, S. V. Madhunapantula and G. P. Robertson, Targeting the MAPK Pathway in Melanoma: Why Some Approaches Succeed and Other Fail, Biochem. Pharmacol., 2010, 80(5), 624–637, DOI:10.1016/j.bcp.2010.04.029.
- D. Guimarães, A. Cavaco-Paulo and E. Nogueira, Design of Liposomes as Drug Delivery System for Therapeutic Applications, Int. J. Pharm., 2021, 601, 120571, DOI:10.1016/j.ijpharm.2021.120571.
- S. Sesana, F. Re, A. Bulbarelli, D. Salerno, E. Cazzaniga and M. Masserini, Membrane Features and Activity of GPI-Anchored Enzymes: Alkaline Phosphatase Reconstituted in Model Membranes, Biochemistry, 2008, 47(19), 5433–5440, DOI:10.1021/bi800005s.
- C. A. Labarrere and G. S. Kassab, Glutathione: A Samsonian Life-Sustaining Small Molecule That Protects against Oxidative Stress, Ageing and Damaging Inflammation, Front. Nutr., 2022, 9, 1007816, DOI:10.3389/fnut.2022.1007816.
- T.-Y. Yang, M.-L. Wu, C.-I. Chang, C.-I. Liu, T.-C. Cheng and Y.-J. Wu, Bornyl Cis-4-Hydroxycinnamate Suppresses Cell Metastasis of Melanoma through FAK/PI3K/Akt/MTOR and MAPK Signaling Pathways and Inhibition of the Epithelial-to-Mesenchymal Transition, Int. J. Mol. Sci., 2018, 19(8), 2152, DOI:10.3390/ijms19082152.
- C. M. Della Corte, V. Ciaramella, C. Di Mauro, M. D. Castellone, F. Papaccio, M. Fasano, F. C. Sasso, E. Martinelli, T. Troiani, F. De Vita, M. Orditura, R. Bianco, F. Ciardiello and F. Morgillo, Metformin Increases Antitumor Activity of MEK Inhibitors through GLI1 Downregulation in LKB1 Positive Human NSCLC Cancer Cells, Onco Targets Ther, 2016, 7(4), 4265–4278, DOI:10.18632/oncotarget.6559.
- X. Bai, Z. L. Smith, Y. Wang, S. Butterworth and A. Tirella, Sustained Drug Release from Smart Nanoparticles in Cancer Therapy: A Comprehensive Review, Micromachines, 2022, 13(10), 1623, DOI:10.3390/mi13101623.
- X. Peng, Z. Wang, Y. Liu, X. Peng, Y. Liu, S. Zhu, Z. Zhang, Y. Qiu, M. Jin, R. Wang, Q. Zhang and D. Kong, Oxyfadichalcone C Inhibits Melanoma A375 Cell Proliferation and Metastasis via Suppressing PI3K/Akt and MAPK/ERK Pathways, Life Sci., 2018, 206, 35–44, DOI:10.1016/j.lfs.2018.05.032.
- M.-C. Hsieh, W.-P. Hu, H.-S. Yu, W.-C. Wu, L.-S. Chang, Y.-H. Kao and J.-J. Wang, A DC-81-Indole Conjugate Agent Suppresses Melanoma A375 Cell Migration Partially via Interrupting VEGF Production and Stromal Cell-Derived Factor-1α-Mediated Signaling, Toxicol. Appl. Pharmacol., 2011, 255(2), 150–159, DOI:10.1016/j.taap.2011.06.008.
- C. Zylberberg and S. Matosevic, Pharmaceutical Liposomal Drug Delivery: A Review of New Delivery Systems and a Look at the Regulatory Landscape, Drug Delivery, 2016, 23(9), 3319–3329, DOI:10.1080/10717544.2016.1177136.
- G. Bozzuto and A. Molinari, Liposomes as Nanomedical Devices, Int. J. Nanomed., 2015, 10, 975–999, DOI:10.2147/IJN.S68861.
- Z. Hammoud, M. Kayouka, A. Trifan, E. Sieniawska, J. M. B. Jemâa, A. Elaissari and H. Greige-Gerges, Encapsulation of α-Pinene in Delivery Systems Based on Liposomes and Cyclodextrins, Molecules, 2021, 26(22), 6840, DOI:10.3390/molecules26226840.
- N. T. T. Le, V. Cao, T. N. Q. Nguyen, T. T. H. Le, T. T. Tran and T. T. Hoang Thi, Soy Lecithin-Derived Liposomal Delivery Systems: Surface Modification and Current Applications, Int. J. Mol. Sci., 2019, 20(19), 4706, DOI:10.3390/ijms20194706.
- M. Chountoulesi, N. Naziris, N. Pippa and C. Demetzos, The Significance of Drug-to-Lipid Ratio to the Development of Optimized Liposomal Formulation, J. Liposome Res., 2018, 28(3), 249–258, DOI:10.1080/08982104.2017.1343836.
- L. Taiarol, C. Bigogno, S. Sesana, M. Kravicz, F. Viale, E. Pozzi, L. Monza, V. A. Carozzi, C. Meregalli, S. Valtorta, R. M. Moresco, M. Koch, F. Barbugian, L. Russo, G. Dondio, C. Steinkühler and F. Re, Givinostat-Liposomes: Anti-Tumor Effect on 2D and 3D Glioblastoma Models and Pharmacokinetics, Cancers, 2022, 14(12), 2978, DOI:10.3390/cancers14122978.
- C. Gao, L. Zhang, M. Xu, Y. Luo, B. Wang, M. Kuang, X. Liu, M. Sun, Y. Guo, L. Teng, C. Wang, Y. Zhang and J. Xie, Pulmonary Delivery of Liposomes Co-Loaded with SN38 Prodrug and Curcumin for the Treatment of Lung Cancer, Eur. J. Pharm. Biopharm., 2022, 179, 156–165, DOI:10.1016/j.ejpb.2022.08.021.
- S. Salmaso, F. Mastrotto, M. Roverso, V. Gandin, S. De Martin, D. Gabbia, M. De Franco, C. Vaccarin, M. Verona, A. Chilin, P. Caliceti, S. Bogialli and G. Marzaro, Tyrosine Kinase Inhibitor Prodrug-Loaded Liposomes for Controlled Release at Tumor Microenvironment, J. Controlled Release, 2021, 340, 318–330, DOI:10.1016/j.jconrel.2021.11.006.
- N. Wang, W. Zhang, D. Hu, L. Jiang, X. Liu, S. Tang, X. Zhou, T. Liu, X. Tang, Y. Chai, M. Li, H. Peng and Z. Du, “Prodrug-Like” Acetylmannosamine Modified Liposomes Loaded With Arsenic Trioxide for the Treatment of Orthotopic Glioma in Mice, J. Pharm. Sci., 2020, 109(9), 2861–2873, DOI:10.1016/j.xphs.2020.06.001.
- J. I. Caulfield and H. M. Kluger, Emerging Studies of Melanoma Brain Metastasis, Curr. Oncol. Rep., 2022, 24(5), 585–594, DOI:10.1007/s11912-022-01237-9.
- T. Mantso, I. Anestopoulos, E. Lamprianidou, I. Kotsianidis, A. Pappa and M. I. Panayiotidis, Isothiocyanate-Induced Cell Cycle Arrest in a Novel In Vitro Exposure Protocol of Human Malignant Melanoma (A375) Cells, Anticancer Res., 2019, 39(2), 591–596, DOI:10.21873/anticanres.13152.
- B. Weksler, I. A. Romero and P.-O. Couraud, The HCMEC/D3 Cell Line as a Model of the Human Blood Brain Barrier, Fluids Barriers CNS, 2013, 10(1), 16, DOI:10.1186/2045-8118-10-16.
- R. Dal Magro, A. Vitali, S. Fagioli, A. Casu, A. Falqui, B. Formicola, L. Taiarol, V. Cassina, C. A. Marrano, F. Mantegazza, U. Anselmi-Tamburini, P. Sommi and F. Re, Oxidative Stress Boosts the Uptake of Cerium Oxide Nanoparticles by Changing Brain Endothelium Microvilli Pattern, Antioxidants, 2021, 10(2), 266, DOI:10.3390/antiox10020266.
- L. L. Ramenzoni, T. Attin and P. R. Schmidlin, In Vitro Effect of Modified Polyetheretherketone (PEEK) Implant Abutments on Human Gingival Epithelial Keratinocytes Migration and Proliferation, Materials, 2019, 12(9), 1401, DOI:10.3390/ma12091401.
- J. C. Stewart, Colorimetric Determination of Phospholipids with Ammonium Ferrothiocyanate, Anal. Biochem., 1980, 104(1), 10–14, DOI:10.1016/0003-2697(80)90269-9.
- M. T. Vu, D. T. D. Nguyen, N. H. Nguyen, V. T. Le, T. N. Dao, T. H. Nguyen, T. D. Cong, T. L.-B. Pham, T. D. Lam and N. T. T. Le, Development, Characterization and In Vitro Evaluation of Paclitaxel and Anastrozole Co-Loaded Liposome, Processes, 2020, 8(9), 1110, DOI:10.3390/pr8091110.
- S. Giofrè, A. Renda, S. Sesana, B. Formicola, B. Vergani, B. E. Leone, V. Denti, G. Paglia, S. Groppuso, V. Romeo, L. Muzio, A. Balboni, A. Menegon, A. Antoniou, A. Amenta, D. Passarella, P. Seneci, S. Pellegrino and F. Re, Dual Functionalized Liposomes for Selective Delivery of Poorly Soluble Drugs to Inflamed Brain Regions, Pharmaceutics, 2022, 14(11), 2402, DOI:10.3390/pharmaceutics14112402.
- B. Formicola, Differential Exchange of Multifunctional Liposomes Between Glioblastoma Cells and Healthy Astrocytes via Tunneling Nanotubes, Frontiers in Bioengineering and Biotechnology, 2019, 7, 403, DOI:10.3389/fbioe.2019.00403.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4md00517a |
| ‡ These authors have contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |