
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNovel sigma 1-antagonists with cis-(+)-normetazocine scaffold: synthesis, molecular modeling, and antinociceptive effect†
Giuliana
Costanzo
 a,
Giuseppe
Cosentino
a,
Margherita
Grasso
b,
Vincenzo
Patamia
a,
Sara
Zuccalà
a,
Alessandro
Coco
a,
Elisabetta
Novello
c,
Mahmoud
Al-Khrasani
d,
Raffaele
Morrone
e,
Giovanni Mario
Pitari
c,
Emanuele
Amata
a,
Agostino
Marrazzo
a,
Antonio
Rescifina
a,
Lorella
Pasquinucci‡
*a and
Carmela
Parenti‡
a
a,
Giuseppe
Cosentino
a,
Margherita
Grasso
b,
Vincenzo
Patamia
a,
Sara
Zuccalà
a,
Alessandro
Coco
a,
Elisabetta
Novello
c,
Mahmoud
Al-Khrasani
d,
Raffaele
Morrone
e,
Giovanni Mario
Pitari
c,
Emanuele
Amata
a,
Agostino
Marrazzo
a,
Antonio
Rescifina
a,
Lorella
Pasquinucci‡
*a and
Carmela
Parenti‡
a
aDepartment of Drug and Health Sciences, University of Catania, Viale A. Doria 6, 95125 Catania, Italy. E-mail: lpasquin@unict.it; Tel: (+39) 095 7384273
bOasi Research Institute – IRCCS, Troina, Italy
cVera Salus Ricerca S.r.l., Via Sigmund Freud 62/B, 96100 Siracusa, Italy
dDepartment of Pharmacology and Pharmacotherapy, Faculty of Medicine, Semmelweis University, Nagyvárad tér 4, H-1445 Budapest, Hungary
eCNR – Istituto di Chimica Biomolecolare, Sezione Catania Via del Santuario 110, 95028 Valverde Catania, Italy
First published on 29th November 2024
Abstract
Inflammatory pain represents one of the unmet clinical needs for patients, as conventional therapies cause several side effects. Recently, new targets involved in inflammatory pain modulation have been identified, including the sigma-1 receptor (σ1R). Selective σ1R antagonists have demonstrated analgesic efficacy in acute and chronic inflammatory pain models. Considering these findings, a series of novel N-normetazocine derivatives has been designed and synthesized to investigate the pivotal role of N-normetazocine stereochemistry in their pharmacological fingerprint. The affinity profile of new ligands versus sigma receptors and opioid receptors was evaluated in vitro, and compound 7 showed a relevant σ1R affinity, with Kiσ1 = 27.5 ± 8.1 nM, and selectivity over sigma-2 receptor (σ2R) and opioid receptors. Furthermore, in vivo, compound 7 significantly reduced inflammatory pain in the second phase of the formalin test. Molecular modeling studies were also performed to analyze the binding mode and the key interactions between the new ligands and σ1R.
1. Introduction
Nowadays, sigma receptors (σRs) are considered promising therapeutic targets for the management of various debilitating disorders, including pain, depression, and neurodegenerative diseases.1,2 Initially, σRs were presented as subtypes of the opioid receptors; now, they are being reclassified as a unique receptor type.3 Two subtypes are identified and termed sigma-1 receptor (σ1R) and sigma-2 receptor (σ2R), different in structure, biological functions, and pharmacological profile. In particular, σ1R is a unique ligand-regulated chaperone membrane receptor, considered a regulatory subunit that modulates target protein functions, thus playing a crucial role in various biological pathways.4 For instance, it can modulate opioid receptors,5 voltage-gated calcium channels,6 and the NMDA receptor activity.7 In particular, σ1R activation is considered a valid strategy to treat neurodegenerative disorders,8 while σ1R blockage is evaluated as an innovative approach in the oncology area for managing different cancers such as glioblastoma, pancreas, breast, and prostate tumors.9 Moreover, owing to its distribution in key regions of pain control, such as dorsal root ganglia, spinal dorsal horn, periaqueductal grey matter, locus coeruleus, and rostroventral medulla, σ1R antagonists have a central role in pain modulation.10 In fact, σ1R agonists counteract opioid receptor-mediated analgesia, acting as an endogenous anti-opioid system, whereas σ1R antagonists determined a marked morphine-induced antinociception enhancement, both at central and peripheral levels.11 This observation is supported by further preclinical studies showing that σ1R antagonists produced antinociceptive effects in persistent pain conditions.12 In this regard, it has been reported that the intrathecal blockade of σ1R prevents nerve injury-induced hyperalgesia, and the σ1R knockout (KO) mice and the treatment of wild-type animals with σ1R antagonists can counteract inflammatory pain behaviors evoked by carrageenan13,14 or complete Freund's adjuvant.15 Furthermore, σ1R antagonists showed antiallodynic and antihyperalgesic effects in sensitizing conditions without modifying the normal pain behavior.10The role of σ1R activation in inflammatory pain could be linked to its capability to enhance Ca2+ release, induced by bradykinin and nitric oxide (NO) signaling with a reinforcement of the inflammatory process.16 Pain sensitization after peripheral inflammation also involves plastic changes defined by increased spinal excitatory neurotransmission and activation of different kinases, like ERK1/2, modulated by σ1R.17 Moreover, σ1R plays a role in pain modulation at the spinal level by activating neurosteroids.18 In light of this, developing novel σ1R antagonists is challenging in medicinal chemistry and is considered a good strategy to counteract persistent pain.
To develop new σ1R ligands, we evaluated the profile of newly synthesized N-substituted normetazocine compounds 6–15 (Fig. 1). In particular, the (+)-cis-normetazocine scaffold was chosen due to its affinity for σ1R, and it is known that (+)-SKF-10,047 was the first drug discovered to interact with σ1R.19 Carrol et al. developed numerous analogs of (+)-SKF10,047, and the impact of altering its N-substituent with respect to the ligand's affinity and selectivity was evaluated. In particular, it has emerged that lipophilic N-substituents may improve σ1R selectivity and potency, and the N-benzyl compound showed the best σ1R affinity.20 In a successive study,21 the ortho-, meta-, and para-substituted N-benzyl-analogues of (+)-cis-normetazocine were synthesized, and this study highlighted both the influence of substituent position and substituent volume. For instance, the analog with the F-atom in para-position retained the high σ1R affinity profile of the lead compound. Thus, para-position and low steric hindrance analogs with N-benzyl-substituent were preferred. To obtain new insights into the structural requirements for σ1R interaction, a series of 2-aminoethyl derivatives of cis-(+)-N-normetazocine was synthesized.22 Bearing a second basic nitrogen, these compounds displayed a good affinity for σ1R while showing a reduced σ2R/σ1R selectivity. Moreover, a compound with 2-(N-phenylcarbamoyl)ethyl substituent, (+)-LP1, was also synthesized, and it demonstrated a nanomolar σ1R affinity and a potent analgesic effect in a mouse model of inflammatory pain. However, a low selectivity ratio to σ2R was highlighted (σ2R/σ1R = 10).23 Subsequently, we synthesized the cis-(+)-N-normetazocine derivative of (+)-LP2 (Fig. 1), bearing an N-phenethylnormetazocine and a methoxy group at the benzylic carbon. (+)-LP2 showed high affinity for σ1R and selectivity versus σ2R. Moreover, (+)-LP2 has shown a σ1R antagonist profile with analgesic effects in both inflammatory and neuropathic animal pain models.24,25 (+)-LP2 was used as a diastereomeric mixture of (+)-(2′R)-LP2 and (+)-(2′S)-LP2.
 | ||
| Fig. 1 Chemical structures of (+)-LP2 and the newly synthesized compounds. | ||
Our new compounds 6–15 were designed as derivatives of (+)-LP2 and here we investigated the influence of various structural modifications. In particular, we examined the impact of altering the chain length of the N-substituent from 2 to 4 carbon atoms, assessed the importance of the phenyl ring by substituting it with a proton, and explored the effects of replacing the methoxy group with either an ester or carboxylic acid function. With respect to the methoxy group substitution of the (+)-LP2, we aimed to examine its impact on σ1R affinity by introducing ester and acid functionalities that show different hydrogen bonding capabilities. A molecular modeling analysis was also performed to analyze the binding mode and the key interactions between the new ligands and σ1R.
2. Results and discussion
2.1. Synthesis
The new compounds 6–15 were synthesized according to Scheme 1. Compounds 1–3 were obtained as previously described.26 Briefly, the compound 1 was prepared by primary alcohol sulfonylation of ethyl tropate with methanesulfonyl chloride and triethylamine in CH2Cl2. The ethyl 4-chloro-2-(R/S)-phenylbutanoate and the ethyl 5-chloro-2-(R/S)-phenylpentanoate were synthesized starting with a suspension of ethyl phenyl cyanoacetate in potassium t-butoxide in anhydrous DMF by adding 1-bromo-2-chloroethane and 1-chloro-3-iodopropane, respectively. The subsequent decarboxylation reaction on relative intermediate compounds in a saturated aqueous solution of K2CO3 and CH3OH, followed by hydrolysis of their cyano group in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of HCl (12 N) and CH3CH2OH, allowed the compounds 2 and 3 to be obtained. Target esters 6–10 were prepared by alkylation of (+)-cis-normetazocine with the alkylation agents 1–5, respectively. The acid derivatives 11–15 were synthesized by ester saponification. Compounds 6–15 possess a chiral stereocenter at the N-substituent of (+)-cis-normetazocine scaffold and exist as a mixture of diastereoisomers, which could not be separated using our purification procedures. An HPLC analysis on diastereomeric mixture of final compound 7 was performed with a Chiralcel OJ column to verify the diastereoisomers ratio. HPLC analysis highlighted the separation of the diastereoisomers of compound 7 with a peak at tR = 12.3 min (55.6%) and a peak at tR = 14.6 min (44.4%) (Fig. S1†). All compounds were characterized by 1H NMR, 13C NMR, attached proton test (APT), and elemental analysis.
:1 mixture of HCl (12 N) and CH3CH2OH, allowed the compounds 2 and 3 to be obtained. Target esters 6–10 were prepared by alkylation of (+)-cis-normetazocine with the alkylation agents 1–5, respectively. The acid derivatives 11–15 were synthesized by ester saponification. Compounds 6–15 possess a chiral stereocenter at the N-substituent of (+)-cis-normetazocine scaffold and exist as a mixture of diastereoisomers, which could not be separated using our purification procedures. An HPLC analysis on diastereomeric mixture of final compound 7 was performed with a Chiralcel OJ column to verify the diastereoisomers ratio. HPLC analysis highlighted the separation of the diastereoisomers of compound 7 with a peak at tR = 12.3 min (55.6%) and a peak at tR = 14.6 min (44.4%) (Fig. S1†). All compounds were characterized by 1H NMR, 13C NMR, attached proton test (APT), and elemental analysis.
 | ||
| Scheme 1 Synthesis of target compounds 6–15. Reagents and conditions: a) NaHCO3, KI, DMF, 55 °C, 24 h; b) NaOH 1 N, 110 °C, 5 h. | ||
2.2. Binding properties on σRs and opioid receptors
Target compounds 6–15 were tested versus σ1R and σ2R with the [3H]-(+)-pentazocine and [3H]-1,3-di-ortho-tolylguanidine ([3H]-DTG) radio probes, respectively (Table 1). Competitive inhibition curves of compounds 7, 8, and 10–11 at 10−10–10−5 M concentration range were reported in Fig. S2.† Inhibition constant (Ki) values were obtained using nonlinear regression analysis GraphPad Prism, version 7.0 (GraphPad Software In., San Diego, USA). Compared to (+)-LP2 (Kiσ1 = 26.6 ± 2.4 nM), only compound 7 maintained high σ1R affinity with a Ki value of 27.5 ± 28.1 nM. Shortening or increasing spacer length in compounds 6 and 8 significantly increased their Ki values versus σ1R. Compound 10, with a similar distance between normetazocine scaffold and ester functionality but lacking the phenyl ring in the N-substituent, exhibited a moderate σ1R affinity (Ki = 159 ± 23 nM). Instead, compound 9, with a methylene deletion in the N-substituent spacer, showed negligible affinity for σ1R. Replacing the ester group with the corresponding carboxylic acid group in compounds 11–15 was not tolerated, and all compounds showed negligible σ1R affinity. Regarding σ2R, all new compounds have shown negligible binding affinity. Moreover, compounds 6–15 were tested versus mu-opioid receptor (MOR), delta-opioid receptor (DOR), and kappa-opioid receptor (KOR) with the [3H]-DAMGO, [3H]-Deltorphin and [3H]-U69,593 radio probes, respectively, with Ki values >5000.| Compound | N-Substituent | K i (nM) ± SDa | |
|---|---|---|---|
| σ1R | σ2R | ||
| a Each value is the mean ± SD of at least two experiments performed in duplicate. Reference compounds were tested with the same membrane homogenates. b Ref. 24. c 1,3-Di-ortho-tolylguanidine. | |||
| (+)-LP2b |

|
26.6 ± 2.4 | 2393 ± 514 |
| 6 |

|
>10000 |
>10000 |
| 7 |

|
27.5 ± 8.1 | >1000 |
| 8 |
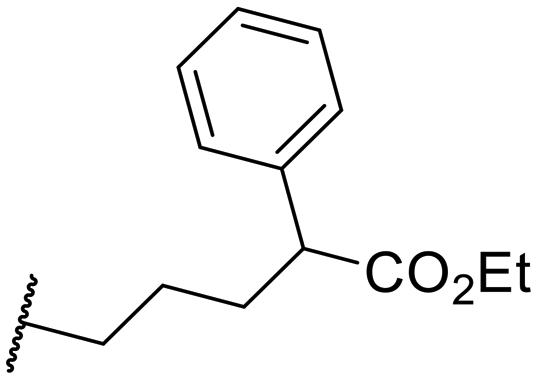
|
594.0 ± 168 | >10000 |
| 9 |

|
>10000 |
>10000 |
| 10 |

|
159.0 ± 23 | >10000 |
| 11 |

|
664.0 ± 200 | >10000 |
| 12 |

|
>10000 |
>10000 |
| 13 |

|
>10000 |
>10000 |
| 14 |
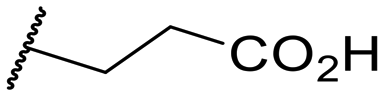
|
>10000 |
>10000 |
| 15 |

|
>10000 |
>10000 |
| (+)-Pentazocine | 4.3 ± 0.5 | 1465 ± 224 | |
| DTGc | 124.0 ± 19 | 18.0 ± 1 | |
2.3. Molecular modeling studies
To rationalize ligand-receptor interactions within the receptor site, in silico and molecular modeling studies have been performed, thanks to which we are better able to understand how the nature of the substituents of each ligand may influence the interaction with the various residues in the receptor pocket. In silico studies were carried out with AutoDock, integrated into the YASARA program, on compounds that demonstrated noteworthy inhibitory activity experimentally, i.e., 7, 8, 10, and 11. At physiological pH (7.4), all the compounds had positively charged nitrogen, and for compound 11, the acidic function is deprotonated. As in our previous study, LP2 was used as a reference compound.21 Considering the protonated nitrogen atom as an asymmetric center, we first docked the four (+)-LP2 stereoisomers to identify the most suitable ones for further molecular modeling studies. The (2′S,3R)-(+)-LP2 and (2′R,3R)-(+)-LP2 stereoisomers had the computed Ki value closest to the observed one, with (2′S,3R)-(+)-LP2 being the more active of the two. Therefore, subsequent docking experiments focused on the (2′S,3R)- and (2′R,3R)-stereoisomers, maintaining the nitrogen stereocenter as 3R. The stereoisomers with lower predicted Ki values are listed in Table 2. However, the inhibitory activity between the (2′S,3R)- and (2′R,3R)-stereoisomers did not differ significantly across most compounds. For σ1R, a detailed analysis of the docking poses within the receptor site has been performed for compounds 7 and 10, which possess an ΔG of −10.21 and −9.05 kcal mol−1, respectively.| Compound | Calcd ΔG σ1R | Calcd Ki σ1R | Exp Kiσ1Ra | Calcd ΔG σ2R | Calcd Kiσ2R | Exp Kiσ2Ra |
|---|---|---|---|---|---|---|
| a The experimental values of Ki refer to the mixture of diastereoisomers. | ||||||
| (2′S,3R)-(+)-LP2 | −9.70 | 77.0 | 26.6 ± 2.4 | −7.40 | 3741 | 2393 ± 514 |
| (2′R,3R)-(+)-LP2 | −9.67 | 81.0 | −7.38 | 3870 | ||
| (3′R,3R)-7 | −10.21 | 32.5 | 27.5 ± 8.1 | −7.14 | 5803 | >1000 |
| (3′S,3R)-7 | −10.14 | 36.6 | −7.14 | 5803 | ||
| (4′S,3R)-8 | −8.48 | 603.9 | 594.0 ± 168 | — | — | >10000 |
| (4′R,3R)-8 | −8.46 | 624.7 | ||||
| (3R)-10 | −9.05 | 230.6 | 159.0 ± 23 | — | — | >10000 |
| (2′R,3R)-11 | −8.38 | 715.0 | 664.0 ± 200 | — | — | >10000 |
| (2′S,3R)-11 | −8.37 | 727.2 | ||||
Fig. 2 illustrates the 3D and 2D poses of the two most active compounds within the receptor site of σ1R. Compound 7 (Fig. 2a) establishes a dense network of interactions within the receptor cavity, similar to typical σ1R inhibitors.27,28 It forms hydrogen bonds with the Asp126 residue and electrostatic interactions with Glu172. Additionally, the non-polar portion of compound 7 engages in hydrophobic interactions with various amino acid residues. In contrast, compound 10 (Fig. 2b) demonstrates lower inhibitory activity, lacking the typical interaction with Asp126. However, it forms a hydrogen bond and an electrostatic interaction with Glu172. Furthermore, compound 10 establishes an unconventional hydrogen bond and an electrostatic-type interaction with Tyr103, π–π interactions with Trp89 and Phe107, and several hydrophobic interactions with other residues.
 | ||
| Fig. 2 3D and 2D poses of compounds 7 (a) and 10 (b) within the σ1R. | ||
Molecular modeling studies support that these N-substituents can establish interactions with key amino acid residues of the σ1R receptor site. As shown in Fig. 2, which presents the poses of the two ligands within the receptor site, the presence of the phenyl group in compound 7 increases the number of hydrophobic interactions. Specifically, compound 7 can interact with residues Leu182 and Met93 through its phenyl group, interactions that do not occur with compound 10. Additionally, the presence of the aromatic group allows ligand 7 to adopt a conformation within the receptor site that enables it to form a crucial interaction with the residue Asp126, an interaction absent in compound 10.
2.4. Chemical stability of compound 7
Compound 7's chemical stability was evaluated in vitro at 37 °C in aqueous phosphate buffer (PBS) at pH 7.4 using HPLC with a diode array detector (DAD). Across all incubation times, the concentration of compound 7 remained constant (Fig. S3 and S4†), indicating optimal stability under physiological conditions. It has a half-life (t1/2) of 300 min.2.5. Plasma stability assay of compound 7
Stability studies were performed to determine the decay over time of compound 7 in mouse plasma. As indicated by correspondent chromatograms (Fig. S5†), compound 7 in mouse plasma was clearly identifiable as a sharp peak at tR = 10.52 min (in red box). Compound concentrations remained stable up to 5 min, starting to decline at times ≥20 min. Half-life estimations (Fig. S6†) indicated that compound 7 was reduced to 50% of its initial concentrations at 31.07 min, supporting the temporal pharmacology of compound 7 in the inflammatory pain model examined in the present studies.2.6. ADME profile of compound 7
A comprehensive computational prediction of the pharmacokinetic profile and brain/plasma distribution of compound 7 was conducted using the Calculator Plugins of ChemAxon and the SwissADME web tool.29 Notably, with high accuracy, the SwissADME platform incorporates advanced algorithms capable of identifying potential false positive results, a common issue in biochemical assays for small molecules. This provides valuable insights into compound 7's reliability in terms of pharmacological profiling.Several key descriptors essential for determining bioactivity parameters were utilized in these predictions, as summarized in Table 3.
| Descriptor | Value |
|---|---|
| MW | 407.55 |
| LogP |
4.57 |
| LogD |
2.90 |
| Topological polar surface area | 49.77 A2 |
| Number of hydrogen bond donors | 1 |
| Number of hydrogen bond acceptors | 4 |
| Number of nitrogen atom count | 1 |
| Oxygen atom count | 3 |
| Nitrogen-oxygen atoms count | 4 |
| Number of aromatic rings | 2 |
| Number of heavy atoms | 30 |
| MWHBN | 0.20 |
| pKa | 9.37 |
| Solubility | 5.23 × 10−5 mg mL−1; 1.28 × 10−7 mol L−1 |
| Chemaxon BBB score | 5.04 |
| Chemaxon CNS MPO score | 3.49 |
Compound 7 was evaluated for drug-likeness according to Lipinski's rule of five, a widely accepted guideline for predicting oral bioavailability. The analysis confirmed compound 7 meets all criteria for orally active drugs without any violations, reinforcing its suitability for development. It also adheres to Lipinski's30 and Veber's31 rules, highlighting its drug-like properties. Veber's rule, in particular, emphasizes the importance of molecular flexibility and polar surface area in drug absorption, parameters in which compound 7 excelled. The compound's bioavailability score of 0.55 indicates moderate oral bioavailability, suggesting it may achieve sufficient systemic exposure when administered orally.
Crucially, compound 7 passed the PAINS (pan assay interference compounds) model,32 which screens for chemical structures likely to interfere with high-throughput biological assays and produce false positive results. The absence of PAINS alerts underscores its potential as a reliable lead compound for further development, free from problematic structures that often complicate drug discovery efforts. These assessments position compound 7 as a promising candidate for drug development, with minimal risks associated with common small-molecule pitfalls.
Moreover, compound 7 was predicted to have moderate water solubility, a favorable attribute for oral delivery, as sufficient solubility is essential for absorption through the gastrointestinal tract. The absorption and distribution characteristics were further analyzed using the extended BOILED-Egg model (Brain Or IntestinaL EstimateD). This computational tool visually predicts a drug's capacity to permeate biological barriers. As depicted in Fig. 3, compound 7 resides within the “yellow yolk” region, suggesting a high probability of passive diffusion across the blood–brain barrier (BBB). This indicates strong potential for targeted therapies for the central nervous system (CNS), where BBB penetration is a critical challenge. Simultaneously, the compound is predicted to exhibit high gastrointestinal absorption, further enhancing its feasibility for oral administration. Interestingly, despite its BBB permeability, a blue dot marker in the model indicates that compound 7 is likely a substrate for P-glycoprotein (P-gp), a transporter protein responsible for pumping drugs out of the brain. This suggests that while compound 7 can cross into the CNS, it may be subject to efflux from the brain via P-gp, an essential consideration for CNS drug development. This efflux mechanism could limit its retention within the brain, and strategies to modulate or circumvent this effect may be necessary for the future development of CNS-targeted therapies.
 | ||
| Fig. 3 BOILED-Egg plot for compound 7. | ||
The Chemaxon BBB permeability score of 5.04 and CNS MPO score of 3.49 validate the predictions obtained using the SwissADME platform, confirming compound 7's potential for CNS drug development. A BBB score of 5.04 indicates a strong likelihood of the compound crossing the blood–brain barrier, which is critical for CNS-targeted therapies. This aligns with the results from SwissADME, which suggested high BBB permeability. The CNS MPO score of 3.49, while slightly below the ideal threshold of 4, still indicates reasonable suitability for CNS activity. Although this score suggests room for optimization in specific molecular properties, it confirms that compound 7 is a promising candidate for CNS applications, consistent with the SwissADME predictions of its drug-like behavior. Overall, the combination of these scores supports the viability of compound 7 for further development, particularly in therapies aimed at the brain, with potential adjustments to improve its CNS MPO score in future optimizations.
Metabolic stability and interaction with cytochrome P450 (CYP) enzymes were also explored via in silico predictions. Compound 7 was identified as an inhibitor of two key CYP isoforms: CYP2D6 and CYP3A4. These isoforms play significant roles in the metabolism of many drugs, so inhibition could lead to potential drug–drug interactions (DDIs) if compound 7 is co-administered with other medications metabolized by these enzymes. However, compound 7 showed no significant inhibition of CYP1A2, CYP2C19, or CYP2C9, indicating a more selective interaction with the CYP system. While the inhibition of CYP2D6 and CYP3A4 suggests that compound 7 could pose DDI risks, this can be managed through careful dosing strategies and further optimization. Understanding these inhibitory interactions is critical for anticipating side effects, managing metabolic liabilities, and ensuring the compound's safety profile in clinical applications.
2.7. In vivo formalin test
Based on competition binding assays and molecular modeling studies, we evaluated the antinociceptive efficacy of compound 7 in the mouse formalin test. The intraplantar (i.pl.) injection of a dilute solution of formalin (5%) into the plantar surface of the mouse hind paw induces a biphasic response characterized by an early nociceptive phase (phase I) and a tonic, inflammatory pain phase (phase II). Phase I is induced by the direct activation of peripheral nerve terminals, while phase II is sustained by mechanisms of central sensitization in the dorsal horn of the spinal cord.To assess the systemic antinociceptive effect of compound 7 in both phases of the formalin test, vehicle or compound 7 (3, 5, and 7 mg kg−1) was administrated intraperitoneally (i.p.) 20 minutes before formalin injection. As shown in Fig. 4, pretreatment with compound 7 at the dose of 3 mg kg−1 i.p. did not significantly modify the nociceptive behavior (licking and flicking or shanking the injected paw) in either phase of the formalin test. On the other hand, pretreatment with 5 or 7 mg kg−1 dose did not alter the pain behaviors of the first phase but significantly decreased the nociceptive behavior during the second phase. This reduction was evidenced by decreased flinches and the lifting/licking time compared to vehicle-treated mice, suggesting a σ1R antagonist profile for compound 7. Compound 7 showed the same pharmacological profile as (+)-LP2, which evidentiated an antinociceptive effect in the second phase of the formalin test.24
 | ||
| Fig. 4 Effects of compound 7 at the doses of 3.0, 5.0, and 7.0 mg kg−1 i.p. in phase I (0–10) and phase II (10–60) of formalin test. Results are reported as MEAN ± SEM (n = 4–11 per group) of the percentage of inhibition of pain responses of the respective vehicle group. *p < 0.05, versus vehicle, two-way ANOVA followed by Bonferroni's post hoc test. | ||
3. Conclusions
In summary, we investigated the influence of the N-substituent on the affinity profile and selectivity profile of (+)-(2S,6S,11S)-normetazocine-based compounds, known for their capacity to bind σ1R.33 Among the newly synthesized ligands, compound 7 exhibited high σ1R affinity, while compound 10 displayed moderate affinity. Both compounds showed negligible affinity for σ2R and opioid receptors. These findings underscore the crucial role of normetazocine stereochemistry in achieving σ1R affinity. Regarding the N-substituent structure, compounds 7 and 10 possess an ester functional group positioned at an optimal distance from the nitrogen atom of the normetazocine scaffold despite the absence of a phenyl ring in compound 10. The higher σ1R affinity of compound 7 compared to compound 10 aligns with the σ1R pharmacophore model proposed by Glennon et al.,34 which features two hydrophobic regions at an appropriate distance from the nitrogen atom.35Moreover, compound 7 produced a potent antinociceptive effect in a mouse model of inflammatory pain, suggesting that it acts as a σ1R antagonist. These in vivo results, consistent with literature reports,36 support the potential of compound 7 as a therapeutic agent for managing inflammatory pain through σ1R antagonism. Notably, the i.p. administration of compound 7 reduces formalin-induced nociception associated with the second phase of the formalin test without affecting the first phase. The injection of formalin induces a biphasic pain response reflecting two different forms of pain. Phase I is a rapid pain response occurring immediately after formalin injection, lasting about 10 minutes, representing acute phasic pain due to direct activation of nociceptors. Phase II is a late pain response starting after 10–15 minutes and lasting up to 50–60 minutes, representing more persistent, tonic, inflammatory pain. This second phase is clinically significant as it reflects mechanisms of nociceptive sensitization, particularly in the dorsal horn of the spinal cord,37 where σ1Rs play a crucial role in pain transmission and modulation.38 Given its ability to reduce pain during the second phase, compound 7 could represent a promising drug for treating persistent pain conditions, such as neuropathic pain, characterized by central sensitization. Previously reported σ1R compounds with a similar profile in formalin-induced inflammatory pain have shown efficacy in counteracting neuropathic pain.39 In particular, (+)-LP2, which exhibits a comparable profile to compound 7 during the second phase of the formalin test, demonstrated inhibition of mechanical allodynia and modulation of neuroinflammation in models of chronic neuropathic pain.25
In silico ADME studies demonstrate compound 7 has a favorable balance of pharmacokinetic and pharmacodynamic properties, meeting critical criteria for oral bioavailability, solubility, absorption, and CNS penetration. While its interaction with CYP enzymes requires consideration, the compound holds significant potential for further development, particularly in CNS-targeted therapies. The absence of PAINS liabilities and compliance with major drug-likeness rules enhance its prospects as a lead candidate. Stability of compound 7 was also evaluated. While compound 7's chemical stability in vitro at 37 °C in aqueous phosphate buffer (PBS) at pH 7.4 was established and a half-life (t1/2) of 300 min was measured, the plasma stability assay on compound 7 showed its progressive hydrolysis with a half-life (t1/2) of 31.07 min. However, its high BBB permeability score measured and the in vivo evidence, as compound 7 demonstrated pain modulation in the inflammatory pain model, support the pharmacological profile of compound 7. Therefore, future studies are required to improve stability of compound 7 with the aim to obtain its analogues useful in treating neuropathic pain.
4. Experiment section
4.1. Chemistry
The intermediates 1–3 were obtained as previously reported.26
:5) to give a white solid. Yield: 88%; m.p.: 183–186 °C. TLC CH2Cl2/MeOH (95:5 v/v) Rf = 0.49. 1H NMR (500 MHz, CDCl3): δ = 7.83–7.79 (m, 1H, CH-aryl), 7.32–7.26 (m, 4H, CH-aryl), 6.86 (d, 1H, J = 8.3 Hz, CH-aryl benzomorphan), 6.70 (d, 1H, J = 3.4 Hz, CH-aryl benzomorphan), 6.58 (dd, 1H, J = 8.3 and 3.4 Hz, CH-aryl benzomorphan), 3.82–3.79 (m, 1H, CH–CO), 3.68 (s, 3H, OCH2CH3), 2.99–2.97 (m, 2H, CH2-benzomorphan), 2.91–2.88 (m, 2H, CH2), 2.89 (d, 2H, J = 15.0 Hz, CH2–CH3), 2.56–2.67 (m, 4H, CH2-benzomorphan), 1.79–1.70 (m, 2H, CH2-benzomorphan), 1.32 (s, 3H, CH3-benzomorphan), 0.79 (d, 3H, J = 7.0, CH3-benzomporphan). The –OH signal of benzomorphan is not observed (Fig. S7†). APT NMR (126 MHz, CDCl3) δ = 174.11 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 154.62 (C-aryl–OH benzomorphan), 137.83 (C-aryl benzomorphan), 137.57 (C-aryl), 128.74 (CH-aryl), 128.25 (CH-aryl benzomorphan), 128.32 (CH-aryl), 128.16 (CH-aryl), 128.10 (C-aryl benzomorphan), 112.99 (CH-aryl benzomorphan), 112.39 (CH-aryl benzomorphan), 59.95 (O–CH2CH3), 58.28 (CH piperidine benzomorphan), 57.34 (N–CH2), 46.40 (CH2 piperidine benzomorphan), 42.36 (CH2 piperidine benzomorphan), 41.72 (CH piperidine benzomorphan), 41.62 (CH2–CH–aryl/COOCH2CH3), 36.56 (C-piperidine benzomorphan), 25.71 (CH3 benzomorphan), 24.95 (CH2 aliphatic benzomorphan), 14.33 (O–CH2CH3), 13.71 (CH3 benzomorphan) (Fig. S17†). Anal. (C25H31NO3) C, H, N (Table S1†).
:5) to give an orange solid. Yield: 25%; m.p.: 158–161 °C; TLC CH2Cl2/MeOH (95:5 v/v) Rf = 0.36. Chiral HPLC: 55.6%, tR = 12.3 min; 44.4%, and tR = 14.6 min (Fig. S1†). 1H NMR (200 MHz, CDCl3): δ = 7.40–7.32 (m, 5H, CH-aryl), 6.87 (d, 1H, J = 8.0 Hz, CH-aryl benzomorphan), 6.95 (d,1H, J = 2.2 Hz, CH-aryl benzomorphan), 6.71 (dd, 1H, J = 8.0 and 2.2 Hz, CH-aryl benzomorphan), 4.21 (q, 2H, J = 7.2 Hz, O–CH2–CH3), 3.30–3.08 (m, 1H, CH–CO), 3.01–2.93 (m, 7H, CH-benzomorphan, CH2-benzomporphan, CH2, CH2), 2.66–2.46 (m, 4H, CH2-benzomorphan), 1.46–1.44 (m, 1H, CH-benzomorphan), 1.20 (s, 3H, CH3-benzomoprphan), 1.12 (t, 3H, J = 7.2 Hz, O–CH2–CH3), 0.89 (d, 3H, J = 7.0 Hz, CH3-benzomorphan). The –OH signal of benzomorphan was not observed (Fig. S8†). 13C NMR (126 MHz, CDCl3) δ = 167.36 (CO), 155.66 (C-aryl–OH benzomorphan), 133.45 (C-aryl benzomorphan), 133.06 (C-aryl), 129.25 (CH-aryl), 129.20 (CH-aryl benzomorphan), 129.11 (CH-aryl), 129.08 (C-aryl benzomorphan), 128.28 (CH-aryl), 114.19 (CH-aryl benzomorphan), 112.43 (CH-aryl benzomorphan), 54.04 (O–CH2CH3), 54.01 (CH piperidine benzomorphan), 54.00 (N–CH2–CH2), 53.37 (CH2 piperidine benzomorphan), 53.22 (CH2 piperidine benzomorphan), 35.24 (CH piperidine benzomorphan), 34.84 (CH2–CH–(aryl/COOCH2CH3), 34.85 (C-piperidine benzomorphan), 34.55 (N–CH2–CH2), 29.48 (CH3 benzomorphan), 23.84 (CH2 aliphatic benzomorphan), 13.53 (O–CH2CH3), 13.50 (CH3 benzomorphan) (Fig. S18†). Anal. (C26H33NO3) C, H, N (Table S1†).
:5) to give an orange solid. Yield: 40%; m.p.: 160–163 °C; TLC CH2Cl2/MeOH (95:5 v/v) Rf = 0.47. 1H NMR (500 MHz, CDCl3): δ = 7.48–7.31 (m, 4H, CH-aryl), 7.28–7.27 (m, 1H, CH-aryl), 7.01 (d, 1H, J = 10.0 Hz, CH-benzomorphan), 6.76 (d, 1H, J = 10 Hz, CH-aryl benzomorphan), 6.69 (d, 1H, J = 10.0 Hz, CH-aryl benzomorphan), 3.47–3.24 (m, 2H, CH2–CH3), 3.17–3.06 (m, 3H, CH2-benzomorphan, CH-CO), 2.14–1.97 (m, 6H, CH2, CH2, CH2), 1.66–1.65 (m, 3H, CH, CH2-benzomorphan), 1.55–1.40 (m, 2H, CH2-benzomorphan),1.30 (s, 3H, CH3-benzomorphan), 1.27 (s, 3H, CH2–CH3), 1.21–1.24 (m, 1H, CH-benzomorphan), 0.93 (d, 3H, J = 7.2 Hz, CH3-benzomorphan). The –OH signal of benzomorphan is not observed (Fig. S9†). 13C NMR (126 MHz, CDCl3) δ = 167.34 (CO), 155.65 (C-aryl–OH benzomorphan), 133.44 (C-aryl benzomorphan), 133.05 (C-aryl), 129.23 (CH-aryl), 129.19 (CH-aryl benzomorphan), 129.10 (CH-aryl), 129.07 (C-aryl benzomorphan), 128.27 (CH-aryl), 114.17 (CH-aryl benzomorphan), 112.42 (CH-aryl benzomorphan), 63.30 (O–CH2CH3), 54.03 (CH piperidine benzomorphan), 54.02 (N–CH2–CH2–CH2), 54.00 (CH2 piperidine benzomorphan), 53.36 (CH2 piperidine benzomorphan), 53.35 (CH piperidine benzomorphan), 53.21 (N–CH2–CH2–CH2), 35.23 (CH2–CH–(aryl/COOCH2CH3), 34.84 (C-piperidine benzomorphan), 34.53 (N–CH2–CH2–CH2), 29.47 (CH3 benzomorphan), 24.01 (CH2 aliphatic benzomorphan), 13.65 (O–CH2CH3), 13.49 (CH3 benzomorphan) (Fig. S19†). Anal. (C27H35NO3) C, H, N (Table S1†).
:7) to give a white solid. Yield: 78%; m.p.: 139–142 °C. TLC CH2Cl2/MeOH (93:7 v/v) Rf = 0.49. 1H NMR (200 MHz, CDCl3): δ = 6.83 (d, 1H, J = 8.0 Hz, CH-aryl benzomorphan), 6.63 (m, 1H, CH-aryl benzomorphan), 6.55 (d, 1H, J = 8.0 Hz, CH-aryl benzomorphan), 3.58 (s, 3H, O–CH3), 2.98–2.91 (m, 4H, CH2-benzomorphan, CH2–CH2–CO), 2.82–2.57 (m, 4H, CH2-benzomorphan, CH2–CH2–CO), 2.21–2.09 (m, 1H, CH-benzomorphan), 1.90–1.76 (m, 2H, CH2-benzomorphan), 1.29–1.44 (m, 1H, CH-benzomorphan), 1.21 (s, 3H, CH3-benzomoprphan), 0.75 (d, 3H, 8.0 Hz, CH3-benzomorphan). The –OH signal of benzomorphan was not observed (Fig. S10†). APT NMR (126 MHz, DMSO-d6) δ = 174.63 (CO), 155.67 (C-aryl–OH benzomorphan), 141.26 (C-aryl benzomorphan), 127.62 (CH-aryl benzomorphan), 124.96 (C-aryl benzomorphan), 113.21 (CH-aryl benzomorphan), 111.72 (CH-aryl benzomorphan), 57.11 (O–CH3), 54.06 (N–CH2–CH2), 44.26 (CH2 piperidine benzomorphan), 40.81 (CH2 piperidine benzomorphan), 40.80 (CH piperidine benzomorphan), 40.79 (CH piperidine benzomorphan), 35.88 (N–CH2–CH2), 34.54 (C-aliphatic benzomorphan), 25.30 (CH3 benzomorphan), 23.43 (CH2 aliphatic benzomorphan), 13.94 (CH3 benzomorphan) (Fig. S20†). Anal. (C18H25NO3) C, H, N (Table S1†).
:7) to give a white solid. Yield: 75%; m.p.: 143–146 °C. TLC CH2Cl2/MeOH (93:7 v/v) Rf = 0.50. 1H NMR (200 MHz, CDCl3): δ = 6.61 (d, 1H, J = 10.0 Hz, CH-aryl benzomorphan), 6.38 (m, 1H, CH-aryl benzomorphan), 6.34 (d, 1H, J = 10.0 Hz, CH-aryl benzomorphan), 3.38 (s, 3H, O–CH3), 2.66–2.57 (m, 4H, CH2-benzomorphan, CH2–CH2–CH2–CO), 2.04–1.96 (m, 1H, CH-benzomorphan), 1.96–1.90 (m, 2H, CH2-benzomorphan), 1.90–1.85 (m, 2H, CH2–CH2–CH2–CO), 1.85–1.62 (m, 4H, CH2-benzomorphan, CH2–CH2–CH2–CO), 1.02–0.95 (m, 1H, CH-benzomorphan), 0.91 (s, 3H, CH3-benzomorphan), 0.54 (d, 3H, 10.0 Hz, CH3-benzomorphan). The –OH signal of benzomorphan was not observed (Fig. S11†). APT NMR (126 MHz, DMSO-d6) δ = 174.65 (CO), 155.68 (C-aryl–OH benzomorphan), 141.27 (C-aryl benzomorphan), 127.64 (CH-aryl benzomorphan), 124.97 (C-aryl benzomorphan), 113.22 (CH-aryl benzomorphan), 111.73 (CH-aryl benzomorphan), 57.12 (O–CH3), 54.07 (N–CH2–CH2–CH2), 44.27 (CH2 piperidine benzomorphan), 40.83 (CH2 piperidine benzomorphan), 40.80 (CH piperidine benzomorphan), 40.79 (CH piperidine benzomorphan), 38.02 (N–CH2–CH2–CH2), 35.90 (N–CH2–CH2–CH2), 34.40 (C-aliphatic benzomorphan), 25.31 (CH3 benzomorphan), 23.44 (CH2 aliphatic benzomorphan), 13.95 (CH3 benzomorphan) (Fig. S21†). Anal. (C19H27NO3) C, H, N (Table S1†).
:5 v/v): Rf = 0.32. 1H NMR (500 MHz, DMSO-d6): δ = 9.08 (br, 1H, OH), 7.28–7.24 (m, 5H, CH-aryl benzomorphan), 6.83–6.82 (m, 1H, CH-aryl benzomorphan), 6.81–6.80 (m, 1H, CH-aryl benzomorphan), 6.56–6.50 (m, 1H, CH-aryl benzomorphan), 4.07–4.02 (m, 1H, CH–CO), 3.12–3.08 (m, 4H, CH2-benzomorphan, CH2), 2.80–2.75 (m, 3H, CH, CH2-benzomorphan), 2.41–2.42 (m, 1H, CH2-benzomorphan), 1.99–1.76 (m, 2H, CH-benzomorphan), 1.19 (s, 3H, CH3-benzomorphan), 0.68 (d, 3H, CH3-benzomorphan). The –OH signal of benzomorphan was not observed (Fig. S12†). APT NMR (126 MHz, DMSO-d6) δ = 174.44 (CO), 156.95 (C-aryl–OH benzomorphan), 141.65 (C-aryl benzomorphan), 138.81 (C-aryl), 129.54 (CH-aryl), 129.48 (CH-aryl benzomorphan), 129.16 (CH-aryl), 129.02 (CH-aryl), 128.94 (C-aryl benzomorphan), 114.50 (CH-aryl benzomorphan), 112.83 (CH-aryl benzomorphan), 60.52 (CH piperidine benzomorphan), 56.26 (N–CH2), 52.00 (CH piperidine benzomorphan), 47.74 (CH2 piperidine benzomorphan), 46.08 (CH2–CH–(aryl/COOH), 41.00 (CH2 piperidine benzomorphan), 36.17 (C-piperidine benzomorphan), 27.58 (CH3 benzomorphan), 26.39 (CH2 aliphatic benzomorphan), 13.93 (CH3 benzomorphan) (Fig. S22†). Anal. (C23H27NO3) C, H, N (Table S1†).
:5 v/v) Rf = 0.30. 1H NMR (500 MHz, DMSO-d6): δ = 8.43 (br, 1H, OH), 7.22–7.20 (m, 2H, CH-aryl), 7.13–7.10 (m, 2H, CH-aryl), 7.03–7.01 (m, 1H, CH-aryl), 6.74 (d, 1H, J = 10.0 Hz, CH-aryl benzomorphan), 6.52 (d, 1H, J = 10.0 Hz, CH-aryl benzomorphan), 6.43–6.41 (dd, 1H, J = 10.0 and 3.4 Hz, CH-aryl benzomorphan), 3.13–3.10 (m, 2H, CH–CO, CH-benzomorphan), 2.68–2.62 (m, 2H, CH2-benzomorphan), 2.38–2.21 (m, 4H, CH2, CH2), 1.80–1.77 (m, 2H, CH2-benzomorphan), 1.80–1.77 (m, 2H, CH2-benzomorphan), 1.64–1.60 (m, 1H, CH-benzomorphan), 1.16 (s, 3H, CH3-benzomorphan), 0.66 (d, 3H, J = 7.2 Hz, CH3-benzomorphan). The –OH signal of benzomorphan was not observed (Fig. S13†). APT NMR (126 MHz, DMSO-d6) δ = 176.93 (CO), 155.91 (C-aryl–OH benzomorphan), 144.63 (C-aryl benzomorphan), 142.17 (C-aryl), 127.89 (CH-aryl), 127.64 (CH-aryl benzomorphan), 127.51 (CH-aryl), 126.59 (C-aryl benzomorphan), 125.43 (CH-aryl), 113.12 (CH-aryl benzomorphan), 111.92 (CH-aryl benzomorphan), 56.43 (CH piperidine benzomorphan), 52.75 (N–CH2–CH2), 51.75 (CH piperidine benzomorphan), 44.80 (CH2 piperidine benzomorphan), 41.78 (CH2 piperidine benzomorphan), 36.21 (CH2–CH–aryl/COOH), 32.91 (C-piperidine benzomorphan), 28.89 (N–CH2–CH2), 25.28 (CH3 benzomorphan), 22.43 (CH2 aliphatic benzomorphan), 13.44 (CH3 benzomorphan) (Fig. S23†). Anal. (C24H29NO3) C, H, N (Table S1†).
:5 v/v) Rf = 0.28. 1H NMR (500 MHz, DMSO-d6): δ = 7.48–7.31 (m, 5H, CH-aryl), 7.28–7.27 (m, 1H, CH-aryl), 7.01 (d, 1H, J = 10.0 Hz, CH-aryl benzomorphan), 6.76 (d, 1H, J = 10 Hz, CH-aryl benzomorphan), 6.69 (dd, 1H, J = 10.0 and 3.4 Hz, CH-aryl benzomorphan), 3.47–3.24 (m, 2H, CH–CO, CH-benzomorphan), 3.17–3.06 (m, 2H, CH2-benzomorphan), 2.14–1.97 (m, 4H, CH2, CH2), 1.66–1.65 (m, 4H, CH2-benzomorphan), 1. 23 (s, 3H, CH3-benzomorphan), 1.21–1.22 (m, 1H, CH-benzomorphan), 0.93 (d, 3H, J = 7.2 Hz, CH3-benzomorphan). The –OH signals of benzomorphan and carboxylic acid moiety were not observed (Fig. S14†). APT NMR (126 MHz, DMSO-d6) δ = 174.50 (CO), 156.10 (C-aryl–OH benzomorphan), 143.39 (C-aryl benzomorphan), 139.79 (C-aryl), 128.68 (CH-aryl), 128.39 (CH-aryl benzomorphan), 128.05 (CH-aryl), 127.88 (CH-aryl), 127.14 (C-aryl benzomorphan), 113.76 (CH-aryl benzomorphan), 112.50 (CH-aryl benzomorphan), 57.76 (CH piperidine benzomorphan), 51.24 (CH piperidine benzomorphan), 50.79 (N–CH2–CH2–CH2), 39.62 (CH2 piperidine benzomorphan), 35.10 (CH2 piperidine benzomorphan), 33.57 (C-piperidine benzomorphan), 31.22 (N–CH2–CH2–CH2), 29.09 (CH2–CH–(aryl/COOH), 25.25 (N–CH2–CH2–CH2), 24.86 (CH2 aliphatic benzomorphan), 19.73 (CH3 benzomorphan), 13.74 (CH3 benzomorphan) (Fig. S24†). Anal. (C25H31NO3) C, H, N (Table S1†).
:5 v/v) Rf = 0.28. 1H NMR (500 MHz, DMSO-d6): δ = 6.82 (d, 1H, J = 10.0 Hz, CH-aryl benzomorphan), 6.60 (d, 1H, J = 5.0 Hz, CH-aryl benzomorphan), 6.50 (dd, 1H, J = 10.0 and 5.0 Hz, CH-aryl benzomorphan), 2.76–2.81 (m, 2H, CH2-benzomorphan), 2.65–2.60 (m, 4H, CH2-benzomorphan, CH2–CH2–CO), 2.42–2.39 (m, 2H, m, CH2-benzomorphan), 2.07–2.05 (m, 2H, CH2–CH2–CO), 1.92–1.70 (m, 1H, CH-benzomorphan), 1.22 (s, 3H, CH3-benzomorphan), 1.18–1.15 (m, 1H, CH-benzomorphan), 0.72 (d, 3H, J = 10.0 Hz, CH3-benzomorphan). The –OH signals of benzomorphan and carboxylic acid moiety were not observed (Fig. S15†). APT NMR (126 MHz, DMSO-d6) δ = 171.47 (CO), 156.01 (C-aryl–OH benzomorphan), 139.84 (C-aryl benzomorphan), 128.20 (CH-aryl benzomorphan), 122.98 (C-aryl benzomorphan), 113.74 (CH-aryl benzomorphan), 111.62 (CH-aryl benzomorphan), 57.90 (N–CH2–CH2), 48.70 (CH2 piperidine benzomorphan), 44.98 (CH2 piperidine benzomorphan), 40.80 (CH piperidine benzomorphan), 35.72 (CH2 aliphatic benzomorphan), 34.58 (N–CH2–CH2), 33.46 (CH piperidine benzomorphan), 29.54 (C-aliphatic benzomorphan), 24.08 (CH3 benzomorphan), 12.84 (CH3 benzomorphan) (Fig. S25†). Anal. (C17H23NO3) C, H, N (Table S1†).
:5 v/v) Rf = 0.30. 1H NMR (500 MHz, DMSO-d6): δ = 7.09 (d, 1H, J = 4.0 Hz, CH-aryl benzomorphan), 6.85 (d, 1H, J = 2.0 Hz, CH-aryl benzomorphan), 6.77 (dd, 1H, J = 4.0 and 2.0 Hz, CH-aryl benzomorphan), 3.38–3.34 (m, 1H, CH-benzomorphan), 2.95–2.88 (m, 4H, CH2-benzomorphan, CH2–CH2–CH2–CO), 2.36–2.33 (m, 2H, CH2-benzomorphan), 2.16–2.15 (m, 2H, CH2–CH2–CH2–CO), 1.97–1.92 (m, 4H, CH2-benzomorphan, CH2–CH2–CH2–CO), 1.59–1.50 (m, 1H, CH-benzomorphan), 1.46 (s, 3H, CH3-benzomorphan), 0.96 (d, 3H, J = 4.0 Hz, CH3-benzomorphan). The –OH signals of benzomorphan and carboxylic acid moiety were not observed (Fig. S16†). APT NMR (126 MHz, DMSO-d6) δ = 173.44 (CO), 154.48 (C-aryl–OH benzomorphan), 140.07 (C-aryl benzomorphan), 126.43 (CH-aryl benzomorphan), 123.77 (C-aryl benzomorphan), 112.02 (CH-aryl benzomorphan), 110.52 (CH-aryl benzomorphan), 52.87 (N–CH2–CH2–CH2), 43.07 (CH2 piperidine benzomorphan), 39.62 (CH2 piperidine benzomorphan), 39.50 (CH piperidine benzomorphan), 39.48 (CH piperidine benzomorphan), 34.89 (N–CH2–CH2–CH2), 33.35 (C-aliphatic benzomorphan), 24.11 (CH3 benzomorphan), 22.24 (N–CH2–CH2–CH2), 20.27 (CH2 aliphatic benzomorphan), 12.75 (CH3 benzomorphan) (Fig. S26†). Anal. (C18H25NO3) C, H, N (Table S1†).
O), 154.62 (C-aryl–OH benzomorphan), 137.83 (C-aryl benzomorphan), 137.57 (C-aryl), 128.74 (CH-aryl), 128.25 (CH-aryl benzomorphan), 128.32 (CH-aryl), 128.16 (CH-aryl), 128.10 (C-aryl benzomorphan), 112.99 (CH-aryl benzomorphan), 112.39 (CH-aryl benzomorphan), 59.95 (O–CH2CH3), 58.28 (CH piperidine benzomorphan), 57.34 (N–CH2), 46.40 (CH2 piperidine benzomorphan), 42.36 (CH2 piperidine benzomorphan), 41.72 (CH piperidine benzomorphan), 41.62 (CH2–CH–aryl/COOCH2CH3), 36.56 (C-piperidine benzomorphan), 25.71 (CH3 benzomorphan), 24.95 (CH2 aliphatic benzomorphan), 14.33 (O–CH2CH3), 13.71 (CH3 benzomorphan) (Fig. S17†). Anal. (C25H31NO3) C, H, N (Table S1†).
:5) to give an orange solid. Yield: 25%; m.p.: 158–161 °C; TLC CH2Cl2/MeOH (95:5 v/v) Rf = 0.36. Chiral HPLC: 55.6%, tR = 12.3 min; 44.4%, and tR = 14.6 min (Fig. S1†). 1H NMR (200 MHz, CDCl3): δ = 7.40–7.32 (m, 5H, CH-aryl), 6.87 (d, 1H, J = 8.0 Hz, CH-aryl benzomorphan), 6.95 (d,1H, J = 2.2 Hz, CH-aryl benzomorphan), 6.71 (dd, 1H, J = 8.0 and 2.2 Hz, CH-aryl benzomorphan), 4.21 (q, 2H, J = 7.2 Hz, O–CH2–CH3), 3.30–3.08 (m, 1H, CH–CO), 3.01–2.93 (m, 7H, CH-benzomorphan, CH2-benzomporphan, CH2, CH2), 2.66–2.46 (m, 4H, CH2-benzomorphan), 1.46–1.44 (m, 1H, CH-benzomorphan), 1.20 (s, 3H, CH3-benzomoprphan), 1.12 (t, 3H, J = 7.2 Hz, O–CH2–CH3), 0.89 (d, 3H, J = 7.0 Hz, CH3-benzomorphan). The –OH signal of benzomorphan was not observed (Fig. S8†). 13C NMR (126 MHz, CDCl3) δ = 167.36 (CO), 155.66 (C-aryl–OH benzomorphan), 133.45 (C-aryl benzomorphan), 133.06 (C-aryl), 129.25 (CH-aryl), 129.20 (CH-aryl benzomorphan), 129.11 (CH-aryl), 129.08 (C-aryl benzomorphan), 128.28 (CH-aryl), 114.19 (CH-aryl benzomorphan), 112.43 (CH-aryl benzomorphan), 54.04 (O–CH2CH3), 54.01 (CH piperidine benzomorphan), 54.00 (N–CH2–CH2), 53.37 (CH2 piperidine benzomorphan), 53.22 (CH2 piperidine benzomorphan), 35.24 (CH piperidine benzomorphan), 34.84 (CH2–CH–(aryl/COOCH2CH3), 34.85 (C-piperidine benzomorphan), 34.55 (N–CH2–CH2), 29.48 (CH3 benzomorphan), 23.84 (CH2 aliphatic benzomorphan), 13.53 (O–CH2CH3), 13.50 (CH3 benzomorphan) (Fig. S18†). Anal. (C26H33NO3) C, H, N (Table S1†).
:5) to give an orange solid. Yield: 40%; m.p.: 160–163 °C; TLC CH2Cl2/MeOH (95:5 v/v) Rf = 0.47. 1H NMR (500 MHz, CDCl3): δ = 7.48–7.31 (m, 4H, CH-aryl), 7.28–7.27 (m, 1H, CH-aryl), 7.01 (d, 1H, J = 10.0 Hz, CH-benzomorphan), 6.76 (d, 1H, J = 10 Hz, CH-aryl benzomorphan), 6.69 (d, 1H, J = 10.0 Hz, CH-aryl benzomorphan), 3.47–3.24 (m, 2H, CH2–CH3), 3.17–3.06 (m, 3H, CH2-benzomorphan, CH-CO), 2.14–1.97 (m, 6H, CH2, CH2, CH2), 1.66–1.65 (m, 3H, CH, CH2-benzomorphan), 1.55–1.40 (m, 2H, CH2-benzomorphan),1.30 (s, 3H, CH3-benzomorphan), 1.27 (s, 3H, CH2–CH3), 1.21–1.24 (m, 1H, CH-benzomorphan), 0.93 (d, 3H, J = 7.2 Hz, CH3-benzomorphan). The –OH signal of benzomorphan is not observed (Fig. S9†). 13C NMR (126 MHz, CDCl3) δ = 167.34 (CO), 155.65 (C-aryl–OH benzomorphan), 133.44 (C-aryl benzomorphan), 133.05 (C-aryl), 129.23 (CH-aryl), 129.19 (CH-aryl benzomorphan), 129.10 (CH-aryl), 129.07 (C-aryl benzomorphan), 128.27 (CH-aryl), 114.17 (CH-aryl benzomorphan), 112.42 (CH-aryl benzomorphan), 63.30 (O–CH2CH3), 54.03 (CH piperidine benzomorphan), 54.02 (N–CH2–CH2–CH2), 54.00 (CH2 piperidine benzomorphan), 53.36 (CH2 piperidine benzomorphan), 53.35 (CH piperidine benzomorphan), 53.21 (N–CH2–CH2–CH2), 35.23 (CH2–CH–(aryl/COOCH2CH3), 34.84 (C-piperidine benzomorphan), 34.53 (N–CH2–CH2–CH2), 29.47 (CH3 benzomorphan), 24.01 (CH2 aliphatic benzomorphan), 13.65 (O–CH2CH3), 13.49 (CH3 benzomorphan) (Fig. S19†). Anal. (C27H35NO3) C, H, N (Table S1†).
:7) to give a white solid. Yield: 78%; m.p.: 139–142 °C. TLC CH2Cl2/MeOH (93:7 v/v) Rf = 0.49. 1H NMR (200 MHz, CDCl3): δ = 6.83 (d, 1H, J = 8.0 Hz, CH-aryl benzomorphan), 6.63 (m, 1H, CH-aryl benzomorphan), 6.55 (d, 1H, J = 8.0 Hz, CH-aryl benzomorphan), 3.58 (s, 3H, O–CH3), 2.98–2.91 (m, 4H, CH2-benzomorphan, CH2–CH2–CO), 2.82–2.57 (m, 4H, CH2-benzomorphan, CH2–CH2–CO), 2.21–2.09 (m, 1H, CH-benzomorphan), 1.90–1.76 (m, 2H, CH2-benzomorphan), 1.29–1.44 (m, 1H, CH-benzomorphan), 1.21 (s, 3H, CH3-benzomoprphan), 0.75 (d, 3H, 8.0 Hz, CH3-benzomorphan). The –OH signal of benzomorphan was not observed (Fig. S10†). APT NMR (126 MHz, DMSO-d6) δ = 174.63 (CO), 155.67 (C-aryl–OH benzomorphan), 141.26 (C-aryl benzomorphan), 127.62 (CH-aryl benzomorphan), 124.96 (C-aryl benzomorphan), 113.21 (CH-aryl benzomorphan), 111.72 (CH-aryl benzomorphan), 57.11 (O–CH3), 54.06 (N–CH2–CH2), 44.26 (CH2 piperidine benzomorphan), 40.81 (CH2 piperidine benzomorphan), 40.80 (CH piperidine benzomorphan), 40.79 (CH piperidine benzomorphan), 35.88 (N–CH2–CH2), 34.54 (C-aliphatic benzomorphan), 25.30 (CH3 benzomorphan), 23.43 (CH2 aliphatic benzomorphan), 13.94 (CH3 benzomorphan) (Fig. S20†). Anal. (C18H25NO3) C, H, N (Table S1†).
:7) to give a white solid. Yield: 75%; m.p.: 143–146 °C. TLC CH2Cl2/MeOH (93:7 v/v) Rf = 0.50. 1H NMR (200 MHz, CDCl3): δ = 6.61 (d, 1H, J = 10.0 Hz, CH-aryl benzomorphan), 6.38 (m, 1H, CH-aryl benzomorphan), 6.34 (d, 1H, J = 10.0 Hz, CH-aryl benzomorphan), 3.38 (s, 3H, O–CH3), 2.66–2.57 (m, 4H, CH2-benzomorphan, CH2–CH2–CH2–CO), 2.04–1.96 (m, 1H, CH-benzomorphan), 1.96–1.90 (m, 2H, CH2-benzomorphan), 1.90–1.85 (m, 2H, CH2–CH2–CH2–CO), 1.85–1.62 (m, 4H, CH2-benzomorphan, CH2–CH2–CH2–CO), 1.02–0.95 (m, 1H, CH-benzomorphan), 0.91 (s, 3H, CH3-benzomorphan), 0.54 (d, 3H, 10.0 Hz, CH3-benzomorphan). The –OH signal of benzomorphan was not observed (Fig. S11†). APT NMR (126 MHz, DMSO-d6) δ = 174.65 (CO), 155.68 (C-aryl–OH benzomorphan), 141.27 (C-aryl benzomorphan), 127.64 (CH-aryl benzomorphan), 124.97 (C-aryl benzomorphan), 113.22 (CH-aryl benzomorphan), 111.73 (CH-aryl benzomorphan), 57.12 (O–CH3), 54.07 (N–CH2–CH2–CH2), 44.27 (CH2 piperidine benzomorphan), 40.83 (CH2 piperidine benzomorphan), 40.80 (CH piperidine benzomorphan), 40.79 (CH piperidine benzomorphan), 38.02 (N–CH2–CH2–CH2), 35.90 (N–CH2–CH2–CH2), 34.40 (C-aliphatic benzomorphan), 25.31 (CH3 benzomorphan), 23.44 (CH2 aliphatic benzomorphan), 13.95 (CH3 benzomorphan) (Fig. S21†). Anal. (C19H27NO3) C, H, N (Table S1†).
:5 v/v): Rf = 0.32. 1H NMR (500 MHz, DMSO-d6): δ = 9.08 (br, 1H, OH), 7.28–7.24 (m, 5H, CH-aryl benzomorphan), 6.83–6.82 (m, 1H, CH-aryl benzomorphan), 6.81–6.80 (m, 1H, CH-aryl benzomorphan), 6.56–6.50 (m, 1H, CH-aryl benzomorphan), 4.07–4.02 (m, 1H, CH–CO), 3.12–3.08 (m, 4H, CH2-benzomorphan, CH2), 2.80–2.75 (m, 3H, CH, CH2-benzomorphan), 2.41–2.42 (m, 1H, CH2-benzomorphan), 1.99–1.76 (m, 2H, CH-benzomorphan), 1.19 (s, 3H, CH3-benzomorphan), 0.68 (d, 3H, CH3-benzomorphan). The –OH signal of benzomorphan was not observed (Fig. S12†). APT NMR (126 MHz, DMSO-d6) δ = 174.44 (CO), 156.95 (C-aryl–OH benzomorphan), 141.65 (C-aryl benzomorphan), 138.81 (C-aryl), 129.54 (CH-aryl), 129.48 (CH-aryl benzomorphan), 129.16 (CH-aryl), 129.02 (CH-aryl), 128.94 (C-aryl benzomorphan), 114.50 (CH-aryl benzomorphan), 112.83 (CH-aryl benzomorphan), 60.52 (CH piperidine benzomorphan), 56.26 (N–CH2), 52.00 (CH piperidine benzomorphan), 47.74 (CH2 piperidine benzomorphan), 46.08 (CH2–CH–(aryl/COOH), 41.00 (CH2 piperidine benzomorphan), 36.17 (C-piperidine benzomorphan), 27.58 (CH3 benzomorphan), 26.39 (CH2 aliphatic benzomorphan), 13.93 (CH3 benzomorphan) (Fig. S22†). Anal. (C23H27NO3) C, H, N (Table S1†).
:5 v/v) Rf = 0.30. 1H NMR (500 MHz, DMSO-d6): δ = 8.43 (br, 1H, OH), 7.22–7.20 (m, 2H, CH-aryl), 7.13–7.10 (m, 2H, CH-aryl), 7.03–7.01 (m, 1H, CH-aryl), 6.74 (d, 1H, J = 10.0 Hz, CH-aryl benzomorphan), 6.52 (d, 1H, J = 10.0 Hz, CH-aryl benzomorphan), 6.43–6.41 (dd, 1H, J = 10.0 and 3.4 Hz, CH-aryl benzomorphan), 3.13–3.10 (m, 2H, CH–CO, CH-benzomorphan), 2.68–2.62 (m, 2H, CH2-benzomorphan), 2.38–2.21 (m, 4H, CH2, CH2), 1.80–1.77 (m, 2H, CH2-benzomorphan), 1.80–1.77 (m, 2H, CH2-benzomorphan), 1.64–1.60 (m, 1H, CH-benzomorphan), 1.16 (s, 3H, CH3-benzomorphan), 0.66 (d, 3H, J = 7.2 Hz, CH3-benzomorphan). The –OH signal of benzomorphan was not observed (Fig. S13†). APT NMR (126 MHz, DMSO-d6) δ = 176.93 (CO), 155.91 (C-aryl–OH benzomorphan), 144.63 (C-aryl benzomorphan), 142.17 (C-aryl), 127.89 (CH-aryl), 127.64 (CH-aryl benzomorphan), 127.51 (CH-aryl), 126.59 (C-aryl benzomorphan), 125.43 (CH-aryl), 113.12 (CH-aryl benzomorphan), 111.92 (CH-aryl benzomorphan), 56.43 (CH piperidine benzomorphan), 52.75 (N–CH2–CH2), 51.75 (CH piperidine benzomorphan), 44.80 (CH2 piperidine benzomorphan), 41.78 (CH2 piperidine benzomorphan), 36.21 (CH2–CH–aryl/COOH), 32.91 (C-piperidine benzomorphan), 28.89 (N–CH2–CH2), 25.28 (CH3 benzomorphan), 22.43 (CH2 aliphatic benzomorphan), 13.44 (CH3 benzomorphan) (Fig. S23†). Anal. (C24H29NO3) C, H, N (Table S1†).
:5 v/v) Rf = 0.28. 1H NMR (500 MHz, DMSO-d6): δ = 7.48–7.31 (m, 5H, CH-aryl), 7.28–7.27 (m, 1H, CH-aryl), 7.01 (d, 1H, J = 10.0 Hz, CH-aryl benzomorphan), 6.76 (d, 1H, J = 10 Hz, CH-aryl benzomorphan), 6.69 (dd, 1H, J = 10.0 and 3.4 Hz, CH-aryl benzomorphan), 3.47–3.24 (m, 2H, CH–CO, CH-benzomorphan), 3.17–3.06 (m, 2H, CH2-benzomorphan), 2.14–1.97 (m, 4H, CH2, CH2), 1.66–1.65 (m, 4H, CH2-benzomorphan), 1. 23 (s, 3H, CH3-benzomorphan), 1.21–1.22 (m, 1H, CH-benzomorphan), 0.93 (d, 3H, J = 7.2 Hz, CH3-benzomorphan). The –OH signals of benzomorphan and carboxylic acid moiety were not observed (Fig. S14†). APT NMR (126 MHz, DMSO-d6) δ = 174.50 (CO), 156.10 (C-aryl–OH benzomorphan), 143.39 (C-aryl benzomorphan), 139.79 (C-aryl), 128.68 (CH-aryl), 128.39 (CH-aryl benzomorphan), 128.05 (CH-aryl), 127.88 (CH-aryl), 127.14 (C-aryl benzomorphan), 113.76 (CH-aryl benzomorphan), 112.50 (CH-aryl benzomorphan), 57.76 (CH piperidine benzomorphan), 51.24 (CH piperidine benzomorphan), 50.79 (N–CH2–CH2–CH2), 39.62 (CH2 piperidine benzomorphan), 35.10 (CH2 piperidine benzomorphan), 33.57 (C-piperidine benzomorphan), 31.22 (N–CH2–CH2–CH2), 29.09 (CH2–CH–(aryl/COOH), 25.25 (N–CH2–CH2–CH2), 24.86 (CH2 aliphatic benzomorphan), 19.73 (CH3 benzomorphan), 13.74 (CH3 benzomorphan) (Fig. S24†). Anal. (C25H31NO3) C, H, N (Table S1†).
:5 v/v) Rf = 0.28. 1H NMR (500 MHz, DMSO-d6): δ = 6.82 (d, 1H, J = 10.0 Hz, CH-aryl benzomorphan), 6.60 (d, 1H, J = 5.0 Hz, CH-aryl benzomorphan), 6.50 (dd, 1H, J = 10.0 and 5.0 Hz, CH-aryl benzomorphan), 2.76–2.81 (m, 2H, CH2-benzomorphan), 2.65–2.60 (m, 4H, CH2-benzomorphan, CH2–CH2–CO), 2.42–2.39 (m, 2H, m, CH2-benzomorphan), 2.07–2.05 (m, 2H, CH2–CH2–CO), 1.92–1.70 (m, 1H, CH-benzomorphan), 1.22 (s, 3H, CH3-benzomorphan), 1.18–1.15 (m, 1H, CH-benzomorphan), 0.72 (d, 3H, J = 10.0 Hz, CH3-benzomorphan). The –OH signals of benzomorphan and carboxylic acid moiety were not observed (Fig. S15†). APT NMR (126 MHz, DMSO-d6) δ = 171.47 (CO), 156.01 (C-aryl–OH benzomorphan), 139.84 (C-aryl benzomorphan), 128.20 (CH-aryl benzomorphan), 122.98 (C-aryl benzomorphan), 113.74 (CH-aryl benzomorphan), 111.62 (CH-aryl benzomorphan), 57.90 (N–CH2–CH2), 48.70 (CH2 piperidine benzomorphan), 44.98 (CH2 piperidine benzomorphan), 40.80 (CH piperidine benzomorphan), 35.72 (CH2 aliphatic benzomorphan), 34.58 (N–CH2–CH2), 33.46 (CH piperidine benzomorphan), 29.54 (C-aliphatic benzomorphan), 24.08 (CH3 benzomorphan), 12.84 (CH3 benzomorphan) (Fig. S25†). Anal. (C17H23NO3) C, H, N (Table S1†).
:5 v/v) Rf = 0.30. 1H NMR (500 MHz, DMSO-d6): δ = 7.09 (d, 1H, J = 4.0 Hz, CH-aryl benzomorphan), 6.85 (d, 1H, J = 2.0 Hz, CH-aryl benzomorphan), 6.77 (dd, 1H, J = 4.0 and 2.0 Hz, CH-aryl benzomorphan), 3.38–3.34 (m, 1H, CH-benzomorphan), 2.95–2.88 (m, 4H, CH2-benzomorphan, CH2–CH2–CH2–CO), 2.36–2.33 (m, 2H, CH2-benzomorphan), 2.16–2.15 (m, 2H, CH2–CH2–CH2–CO), 1.97–1.92 (m, 4H, CH2-benzomorphan, CH2–CH2–CH2–CO), 1.59–1.50 (m, 1H, CH-benzomorphan), 1.46 (s, 3H, CH3-benzomorphan), 0.96 (d, 3H, J = 4.0 Hz, CH3-benzomorphan). The –OH signals of benzomorphan and carboxylic acid moiety were not observed (Fig. S16†). APT NMR (126 MHz, DMSO-d6) δ = 173.44 (CO), 154.48 (C-aryl–OH benzomorphan), 140.07 (C-aryl benzomorphan), 126.43 (CH-aryl benzomorphan), 123.77 (C-aryl benzomorphan), 112.02 (CH-aryl benzomorphan), 110.52 (CH-aryl benzomorphan), 52.87 (N–CH2–CH2–CH2), 43.07 (CH2 piperidine benzomorphan), 39.62 (CH2 piperidine benzomorphan), 39.50 (CH piperidine benzomorphan), 39.48 (CH piperidine benzomorphan), 34.89 (N–CH2–CH2–CH2), 33.35 (C-aliphatic benzomorphan), 24.11 (CH3 benzomorphan), 22.24 (N–CH2–CH2–CH2), 20.27 (CH2 aliphatic benzomorphan), 12.75 (CH3 benzomorphan) (Fig. S26†). Anal. (C18H25NO3) C, H, N (Table S1†).
4.2. Radioligand binding assays
4.2.1.1. Materials. [3H](+)-Pentazocine (26.9 Ci mmol−1) and [3H]1,3-di-o-tolylguanidine ([3H]DTG, 35.5 Ci mmol−1) were purchased from PerkinElmer (Zaventem, Belgium). Ultima Gold MV Scintillation cocktail was from PerkinElmer (Milan, Italy). All the other materials were obtained from Merck Life Science S.r.l. (Milan, Italy). The test compound solutions were prepared by dissolving approximately 10 μmol of the test compound in DMSO to obtain a 10−2 M stock solution. The required test concentrations for the assay (from 10−5 to 10−10 M) have been prepared by diluting the DMSO stock solution with the respective assay buffer. All experiments were performed using ultrapure water obtained with a Millipore Milli-Q Reference Ultrapure Water Purification System. All the laboratory glassware was washed with 6 M HCl water solution and then rinsed with ultrapure water.
4.2.1.2. Preparation of membrane homogenates from rat liver. Rat livers (∼21 g, male Sprague Dawley rats – ENVIGO RMS S.R.L., Udine, Italy) were cut into small pieces with a scalpel and homogenized in two portions with 6 volumes of cold 0.32 M sucrose with a Potter–Elvehjem glass homogenizer. The suspension was centrifuged at 1030 × g for 10 min at 4 °C. The supernatant was separated and centrifuged at 31
100 × g for 20 min at 4 °C. The pellet was resuspended with 6 volumes of ice-cold Tris buffer (50 mM, pH 8) and incubated at rt for 30 min. Then, the suspension was centrifuged at 31100 × g for 20 min at 4 °C. The final pellet was resuspended with 6 volumes of ice-cold Tris buffer and stored at −80 °C in ∼1 mL portions containing about 6 mg protein mL−1.41,42
4.2.1.3. Protein determination. The protein concentration was determined using Bradford's method. The Bradford solution was prepared by dissolving 10 mg of Coomassie Brilliant Blue G 250 in 5 mL of 95% ethanol. To this solution, 10 mL of 85% phosphoric acid were added, and the mixture was stirred and filled to a total volume of 100 mL with ultrapure water. The calibration was carried out with bovine serum albumin as a standard at different concentrations. In a 96-well plate, 30 μL of the calibration solution or 30 μL of the membrane receptor preparation were mixed with 240 μL of the Bradford solution, respectively. After 5 min of incubation at rt, the UV absorbance was measured at λ = 595 nm using a microplate spectrophotometer reader (Synergy HT, BioTek).
4.2.1.4. σ1R ligand binding assays. In vitro σ1R ligand binding assays were carried out in Tris buffer (50 mM, pH 8.0) for 150 min at 37 °C. The thawed membrane preparation 8 (250 μg per sample) was incubated with increasing concentrations of test compounds and [3H](+)-pentazocine (2 nM) in a final volume of 0.5 mL. The Kd value of [3H](+)-pentazocine was 2.9 nM. Unlabeled (+)-pentazocine (10 μM) was used to measure non-specific binding. Bound and free radioligand were separated by fast filtration under reduced pressure using a Millipore filter apparatus through Whatman GF/6 glass fiber filters, presoaked in a 0.5% poly(ethyleneimine) water solution for 120 min. Each filter paper was rinsed three times with 3 mL ice-cold Tris buffer (50 mM, pH 7.4), dried at r.t., and incubated overnight with 3 mL scintillation cocktail into pony vials. The bound radioactivity has been determined using a liquid scintillation counter (Beckman LS 6500).
4.2.1.5. σ2R ligand binding assays. In vitro σ2R ligand binding assays were carried out in Tris buffer (50 mM, pH 8.0) for 120 min at rt. The thawed membrane preparation of rat liver (250 μg per sample) (∼21 g, male Sprague Dawley rats – ENVIGO RMS S.R.L., Udine, Italy) was incubated with increasing concentrations of test compounds and [3H]DTG (2 nM) in the presence of (+)-pentazocine (5 μM) as σ1R masking agent in a final volume of 0.5 mL. The Kd value of [3H]DTG was 17.9 nM. Non-specific binding was evaluated with unlabeled DTG (10 μM). Bound and free radioligand were separated by fast filtration under reduced pressure using a Millipore filter apparatus through Whatman GF/6 glass fiber filters, presoaked in a 0.5% poly(ethyleneimine) water solution for 120 min. Each filter paper was rinsed three times with 3 mL ice-cold Tris buffer (10 mM, pH 8), dried at r.t., and incubated overnight with 3 mL scintillation cocktail into pony vials. The bound radioactivity has been determined using a liquid scintillation counter (Beckman LS 6500).
4.2.1.6. Data analysis. The Ki-values were calculated using GraphPad Prism® 7.0 (GraphPad Software, San Diego, CA, USA). The Ki-values are given as mean value ± SD from at least two independent experiments performed in duplicate.
4.2.2.1. Materials. [3H]-DAMGO (48.4 Ci mmol−1), [3H]-(2-D-Ala)-[tyrosyl-3,5-] DELTORPHIN II (54.7 Ci mmol−1), and [3H]-U69,593 (49.3 Ci mmol−1) were purchased from PerkinElmer (Zaventem, Belgium). Unlabeled naloxone hydrochloride, DAMGO, (−)-U50,488, and naltrindole hydrochloride were purchased from Sigma-Aldrich (St. Louis, MO, USA). The Ultima Gold MV Scintillation cocktail was from PerkinElmer (Milano, Italy). A 10 mM stock solution was obtained by dissolving the test compound in DMSO and then diluting it with the assay buffer to obtain the required test concentrations for the assay (from 10−5 to 10−9 M). All experiments were performed using ultrapure water obtained with a Millipore Milli-Q Reference Ultrapure Water Purification System (Millipore, Burlington, MA, USA). The bound radioactivity has been determined using a Beckman LS 6500 liquid scintillation counter (Beckman Coulter, Brea, CA, USA).
4.2.2.2. Preparation of membrane homogenates from Sprague–Dawley rat brains for MOR and DOR binding assays or guinea pig brains for KOR binding assays. Sprague–Dawley rat brains (for MOR and DOR binding assays – ENVIGO RMS S.R.L., Udine, Italy) or guinea pig brains (for KOR binding assays – 200–300 g, male Dunkin–Hartley guinea pigs – Harlan Laboratories, S.Pietro al Natisone (UD)) were homogenized in ice-cold Tris buffer (50 mM, pH 7.4) by using a Dounce glass homogenizer (Wheaton, Millville, NJ, USA) with a loose inner tolerance pestle first and a tight inner tolerance pestle later in a cylindrical glass tube of 40 mL volume. The suspension was centrifuged at 40
000 × g for 20 min at 4 °C (Beckmann J2-20 centrifuge and a JA-21 rotor). The pellet was resuspended in ice-cold Tris buffer and then incubated at 37 °C for 30 min to remove endogenous ligands. After incubation, the suspension was centrifuged at 40000 × g for 20 min at 4 °C, and the final pellet was resuspended in ice-cold Tris buffer and frozen at −80 °C in ∼1 mL portions containing about 10 mg protein mL−1.
4.2.2.3. Protein determination. The protein concentration was determined using the Bradford method. The calibration curve was built with bovine serum albumin as the standard compound at 7 concentrations ranging from 60 μg mL−1 to 210 μg mL−1 with blank correction. In a 96-well plate, 30 μL of the calibration solution or 30 μL of the membrane receptor preparation were mixed with 240 μL of the Bradford solution (10 mg of Coomassie Brilliant Blue G 250 in 5 mL of 95% ethanol, 10 mL of 85% phosphoric acid, and water up to 100 mL; with ultrapure water). After 5 min of incubation at RT, the UV absorbance was measured at λ = 595 nm using a microplate spectrophotometer reader (Synergy HT, BioTek, Winooski, VT, USA).
4.2.2.4. Opioid receptor ligand binding assays. MOR and DOR binding experiments were carried out by incubating 400 μg per sample and 500 μg per sample of rat brain membranes, respectively, for 45 min at 35 °C with 1 nM [3H]-DAMGO or 2 nM [3H]-Deltorphin II (2-D-Ala)-[tyrosyl-3,5-3H] in 50 mM Tris-HCl (pH 7.4). For KOR binding assays, guinea pig brain membranes (400 μg per sample) were incubated for 30 min at 30 °C with 1 nM [3H]-U69,593. Test compounds were added to a final volume of 1 mL. The Kd values of [3H]DAMGO, [3H]-Deltorphin II (2-D-Ala)-[tyrosyl-3,5-3H] and [3H]-U69,593 were 1.0, 1.5, and 2.3 nM, respectively. Nonspecific binding was assessed in the presence of 10 μM unlabelled naloxone. The reaction was terminated by filtering the solution under reduced pressure using a Millipore filter apparatus through Whatman glass fiber filters, GF/C for MOR and DOR and GF/B for KOR, presoaked for 1 h in a 0.1% poly(ethyleneimine) solution. Filters were washed with 50 mM ice-cold Tris-HCl buffer (2 × 4 mL), dried at r.t., soaked overnight in 4 mL of scintillation cocktail into 6 mL pony vials, and counted on a liquid scintillation counter.
4.2.2.5. Data analysis. K i values were calculated using nonlinear regression analysis to fit a logistic equation to the competition data using GraphPad Prism 9.3.0 (GraphPad Software Inc., San Diego, CA, USA).
4.3. Molecular modeling studies
4.4. Chemical stability. In vitro assay
First, a standard calibration curve (concentration range, 10–250 μM; in methanol) was performed to calculate unknown sample concentrations (Fig. S3†). Samples were prepared as 10 mM solution in DMSO of compound 7, added to the medium previously heated at 37 °C to have a final concentration of 200 μM. The resultant solution was incubated at 37 ± 0.5 °C, and at proper time intervals, an amount of 150 μL of the reaction mixture was withdrawn and added to the same amount of acetonitrile to obtain a final concentration of 100 μM. The sample was vortexed, filtered by 0.22 μm filters, and analyzed by HPLC. The experiment was performed in duplicate. The stability of compound 7 was evaluated over 300 minutes (Fig. S4†). The data were acquired with a HITA903-0337 Chromaster System, equipped with an autosampler for direct injection, a column oven, a quaternary gradient pump, and a diode array detector. The system was equipped with a Kinetex C18 5μ (4 × 3.0 mm ID) column, maintained at 25 °C and connected to a SecurityGuard Cartridge. A 30 min gradient run was performed, with a mobile phase A (acetonitrile 0.1% in TFA), a mobile phase B (H2O 0.1% in TFA), a work flow at 1 ml min−1, an injection volume of 10 μl and the detector wavelength of λ = 285 nm. The half-life (t1/2) of compound 7 was calculated by matching the data with one phase exponential decay equation using Prism software 9.3.0 (Graph Pad, San Diego, CA, USA).4.5. Stability in mouse plasma
The stability of compound 7 to the esterase was evaluated by incubating it in mouse C57 plasma. The solution of compound 7 (10 mM in DMSO) was added to plasma preheated at 37 °C; thus, obtaining a final concentration of the compound of 200 μM. The solution was incubated at 37° ± 0.5 °C, and at appropriate time intervals (0, 1, 5, 20, and 60 minutes) an aliquot of 200 μL of reaction mixture was withdrawn and added to 200 μL of acetonitrile containing 0.1% HCOOH, to deproteinize the plasma. The final concentration was of 100 μM. The sample was vortexed and centrifuged for 15 minutes 13000 rpm. The supernatant was filtered through 0.22 μm filters and analyzed by RP-HPLC. All experiments were performed in duplicate. The HPLC analyses were performed as described above. The half-life (t1/2) in mouse plasma of compound 7 was calculated by matching the data with one phase exponential decay equation with semi-log plots. The plateau was fixed to a constant value of ‘0’. All statistical analyses were performed with GraphPad Prism 8.0 (GraphPad Software, Inc., San Diego, CA).
4.6. In vivo pharmacology
Data availability
The data supporting my article have been included as part of the ESI† that I included in my manuscript (see file attached).Author contributions
G. C., L. P. and C. P. were responsible for the study design, analysis, and interpretation of the data. G. C. synthesized, purified, and characterized the compounds. G. C. and G. Co. performed in vitro binding experiments. E. N. and G. M. P. performed HPLC analysis. R. M. performed chiral HPLC analysis. V. P., A. C., and A. R. conceived the computational molecular modeling experiments. M. G., S. Z., and C. P. conducted in vivo pharmacology experiments and described and discussed the relative results. G. C., L. P., C. P., V. P., and A. R. drafted the main text. M. A., E. A., and A. M. contributed to the writing of the manuscript. All authors have participated in the refinement of the writing and have approved the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the University of Catania, PIA.CE.RI. 2020–2022 Linea di intervento 2, project DETTAGLI (grant 57722172125). The authors gratefully acknowledge Fabbrica Italiana Sintetici (Italy) for providing cis-(+)-normetazocine. The authors thank Prof. Giuseppe Politi and Mr. Salvatore Leotta from the Department of Physics and Astronomy, University of Catania, and Mr. Emanuele Bonanno for technical and instrumental support of the Beckman LS6500 liquid scintillation counter. The authors acknowledge the Center for Advanced Preclinical in vivo Research (CAPiR) of the University of Catania for its technical contribution. Free academic license from ChemAxon for its suite of programs is gratefully acknowledged.References
- L. Nguyen, B. P. Lucke-Wold, S. A. Mookerjee, J. Z. Cavendish, M. J. Robson, A. L. Scandinaro and R. R. Matsumoto, Role of sigma-1 receptors in neurodegenerative diseases, J. Pharmacol. Sci., 2015, 127, 17–29, DOI:10.1016/j.jphs.2014.12.005.
- C. G. Rousseaux and S. F. Greene, Sigma receptors [σRs]: biology in normal and diseased states, J. Recept. Signal Transduction, 2016, 36(4), 327–388, DOI:10.3109/10799893.2015,1015737.
- F. J. Kim, Introduction to Sigma Proteins: Evolution of the Concept of Sigma Receptors, Handb. Exp. Pharmacol., 2017, 244, 1–11, DOI:10.1007/164_2017_41.
- C. Sanchez-Fernandez, J. M. Entrena, J. M. Baeyens and J. C. Cobos, Sigma-1 Receptor Antagonists: A New Class of Neuromodulatory Analgesics, Adv. Exp. Med. Biol., 2017, 964, 109–132, DOI:10.1007/978-3-319-50174-1_9.
- F. J. Kim, I. Kovalyshyn, M. Burgman, C. Neilan, C.-C. Chien and G. W. Pasternak, Sigma 1 receptor modulation of G-protein coupled receptor signaling: potentiation of opioid transduction independent from receptor binding, Mol. Pharmacol., 2010, 77, 695–703, DOI:10.1124/mol.109.057083.
- H. Zhang and J. Cuevas, Sigma receptors inhibit high-voltageactivated calcium channels in rat sympathetic and parasympathetic neurons, J. Neurophysiol., 2002, 87, 2867–2879, DOI:10.1152/jn.2002.87.6.2867.
- M. Rodríguez-Muñoz, P. Sánchez-Blázquez, R. Herrero- Labrador, R. Martínez-Murillo, M. Merlos, J. M. Vela and J. Garzón, The sigma1 receptor engages the redox-regulated HINT1 protein to bring opioid analgesia under NMDA receptor negative control, Antioxid. Redox Signaling, 2015, 22, 799–818, DOI:10.1089/ars.2014.5993.
- A. V. Bolshakova, E. O. Kukanova, A. N. Gainullina, V. A. Zhemkov, S. A. Korban and I. B. Bezprozvanny, Sigma-1 receptor as a potential pharmacological target for the treatment of neuropathology, St. Petersbg. State Polytech. Univ. J.: Phys. Math., 2016, 237(1), 48–65, DOI:10.1016/j.spjpm.2016.03.003.
- M. Rui, D. Rossi, A. Marra, M. Paolillo, S. Schinelli, D. Curti, A. Tesei, M. Cortesi, A. Zamagni, E. Laurini, S. Pricl, D. Schepmann, B. Wünsch, E. Urban, V. Pace and S. Collina, Synthesis and biological evaluation of new aryl-alkyl(alkenyl)-4-benzylpiperidines, novel Sigma Receptor (SR) modulators, as potential anticancer-agents, Eur. J. Med. Chem., 2016, 124, 649–665, DOI:10.1016/j.ejmech.2016.08.067.
- P. Linciano, G. Rossino, R. Listro, D. Rossi and S. Collina, Sigma-1 receptor antagonists: promising players in fighting neuropathic pain, Pharm. Pat. Anal., 2020, 9(3), 77–85, DOI:10.4155/ppa-2020-0007.
- J. Mei and G. W. Pasternak, Sigma1 receptor modulation of opioid analgesia in the mouse, J. Pharmacol. Exp. Ther., 2002, 300(3), 1070–1074, DOI:10.1124/jpet.300.3.1070.
- M. Merlos, L. Romero, D. Zamanillo, C. Plata-Salamán and J. M. Vela, Sigma-1 Receptor and Pain, Handb. Exp. Pharmacol., 2017, 244, 131–161, DOI:10.1007/164_2017_9.
- T. J. Cirino, S. O. Eans, J. M. Medina, L. L. Wilson, L. Mottinelli, S. Intagliata, C. R. McCurdy and J. P. McLaughlin, Characterization of sigma 1 receptor antagonist CM-304 and its analog, AZ-66: novel therapeutics against allodynia and induced pain, Front. Pharmacol., 2019, 10, 678, DOI:10.3389/fphar.2019.00678.
- J. M. Vela, M. Merlos and C. Almansa, Investigational sigma-1 receptor antagonists for the treatment of pain, Expert Opin. Invest. Drugs, 2015, 24(7), 883–896, DOI:10.1517/13543784.2015.1048334.
- G. Gris, M. Merlos, J. M. Vela, D. Zamanillo and E. Portillo-Salido, S1RA, a selective sigma-1 receptor antagonist, inhibits inflammatory pain in the carrageenan and complete Freund's adjuvant models in mice, Behav. Pharmacol., 2014, 25(3), 226–235, DOI:10.1097/FBP.0000000000000038.
- T. Hayashi, T. Maurice and T. P. Su, Ca(2+) signaling via sigma(1)-receptors: Novel regulatory mechanism affecting intracellular Ca(2+) concentration, J. Pharmacol. Exp. Ther., 2000, 293, 788–798 CrossRef CAS PubMed.
- M. A. Tejada, A. Montilla-García, C. Sánchez-Fernández, J. M. Entrena, C. Perazzoli, J. M. Baeyens and E. J. Cobos, Sigma-1 receptor inhibition reverses acute inflammatory hyperalgesia in mice: role of peripheral sigma-1 receptors, Psychopharmacology, 2014, 231(19), 3855–3869, DOI:10.1007/s00213-014-3524-3.
- S. M. Wang, N. Goguadze, Y. Kimura, Y. Yasui, B. Pan, T. Y. Wang, Y. Nakamura, Y. T. Lin, Q. H. Hogan, K. L. Wilson, T. P. Su and H. E. Wu, Genomic Action of Sigma-1 Receptor Chaperone Relates to Neuropathic Pain, Mol. Neurobiol., 2021, 58(6), 2523–2541, DOI:10.1007/s12035-020-02276-8.
- R. Turnaturi, L. Montenegro, A. Marrazzo, R. Parenti, L. Pasquinucci and C. Parenti, Benzomorphan skeleton, a versatile scaffold for different targets: A comprehensive review, Eur. J. Med. Chem., 2018, 155, 492–502, DOI:10.1016/j.ejmech.2018.06.017.
- F. I. Carroll, P. Abraham, K. Parham, X. Bai, X. Zhang, G. A. Brine, S. W. Mascarella, B. R. Martin, E. L. May, C. Sauss, L. Di Paolo, P. Wallace, J. M. Walker and W. D. Bowen, Enantiomeric N-substituted N-normetazocines: a comparative study of affinities at sigma, PCP, and mu opioid receptors, J. Med. Chem., 1992, 35(15), 2812–2818, DOI:10.1021/jm00093a014.
- S. W. Mascarella, X. Bai, W. Williams, B. Sine, W. D. Bowen and F. I. Carroll, (+)-cis- N-(para-, meta-, and ortho-substituted benzyl)-N-normetazocines: synthesis and binding affinity at the [3H]-(+)-pentazocine-labeled (sigma 1) site and quantitative structure-affinity relationship studies, J. Med. Chem., 1995, 38(3), 565–569, DOI:10.1021/jm00003a019.
- G. Ronsisvalle, A. Marrazzo, O. Prezzavento, L. Pasquinucci, F. Vittorio, V. Pittalà, M. S. Pappalardo, S. Cacciaguerra and S. Spampinato, (+)-cis-N-ethyl-eneamino-N-normetazocine derivatives. Novel and selective sigma ligands with antagonist properties, J. Med. Chem., 1998, 41(10), 1574–1580, DOI:10.1021/jm970333f.
- R. Turnaturi, L. Pasquinucci, S. Chiechio, M. Grasso, A. Marrazzo, E. Amata, M. Dichiara, O. Prezzavento and C. Parenti, Exploiting the Power of Stereochemistry in Drug Action: 3-[(2S,6S,11S)-8-Hydroxy-6,11-dimethyl-1,4,5,6-tetrahydro-2,6-methano-3-benzazocin-3(2H)-yl]-N-phenylpropanamide as Potent Sigma-1 Receptor Antagonist, ACS Chem. Neurosci., 2020, 11(7), 999–1005, DOI:10.1021/acschemneuro.9b00688.
- R. Turnaturi, S. Chiechio, L. Pasquinucci, S. Spoto, G. Costanzo, M. Dichiara, S. Piana, M. Grasso, E. Amata, A. Marrazzo and C. Parenti, Novel N-normetazocine Derivatives with Opioid Agonist/Sigma-1 Receptor Antagonist Profile as Potential Analgesics in Inflammatory Pain, Molecules, 2022, 27, 5135, DOI:10.3390/molecules27165135.
- S. Denaro, L. Pasquinucci, R. Turnaturi, C. Alberghina, L. Longhitano, S. Giallongo, G. Costanzo, S. Spoto, M. Grasso, A. Zappalà, G. Li Volti, D. Tibullo, N. Vicario, R. Parenti and C. Parenti, Sigma-1 Receptor Inhibition Reduces Mechanical Allodynia and Modulate Neuroinflammation in Chronic Neuropathic Pain, Mol. Neurobiol., 2024, 61(5), 2672–2685, DOI:10.1007/s12035-023-03717-w.
- G. Costanzo, V. Patamia, R. Turnaturi, C. Parenti, C. Zagni, J. Lombino, E. Amata, A. Marrazzo, L. Pasquinucci and A. Rescifina, Design, synthesis, in vitro evaluation, and molecular modeling studies of N-substituted benzomorphans, analogs of LP2, as novel MOR ligands, Chem. Biol. Drug Des., 2023, 101(6), 1382–1392, DOI:10.1111/cbdd.14220.
- K. Szczepańska, S. Podlewska, M. Dichiara, D. Gentile, V. Patamia, N. Rosier, D. Mönnich, M. C. Ruiz Cantero, T. Karcz, D. Łażewska, A. Siwek, S. Pockes, E. J. Cobos, A. Marrazzo, H. Stark, A. Rescifina, A. J. Bojarski, E. Amata and K. Kieć-Kononowicz, Structural and Molecular Insight into Piperazine and Piperidine Derivatives as Histamine H3 and Sigma-1 Receptor Antagonists with Promising Antinociceptive Properties, ACS Chem. Neurosci., 2022, 13(1), 1–15, DOI:10.1021/acschemneuro.1c00435.
- C. Barbaraci, V. Di Giacomo, A. Maruca, V. Patamia, R. Rocca, M. Dichiara, A. Di Rienzo, I. Cacciatore, A. Cataldi, M. Balaha, M. Rapino, C. Zagni, D. Zampieri, L. Pasquinucci, C. Parenti, E. Amata, A. Rescifina, S. Alcaro and A. Marrazzo, Discovery of first novel sigma/HDACi dual-ligands with a potent in vitro antiproliferative activity, Bioorg. Chem., 2023, 140, 106794, DOI:10.1016/j.bioorg.2023.106794.
- A. Daina, O. Michielin and V. Zoete, SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules, Sci. Rep., 2017, 7, 42717, DOI:10.1038/srep42717.
- A. K. Ghose, V. N. Viswanadhan and J. J. Wendoloski, A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases, J. Comb. Chem., 1999, 1(1), 55–68, DOI:10.1021/cc9800071.
- D. F. Veber, S. R. Johnson, H. Y. Cheng, B. R. Smith, K. W. Ward and K. D. Kopple, Molecular properties that influence the oral bioavailability of drug candidates, J. Med. Chem., 2002, 45(12), 2615–2623, DOI:10.1021/jm020017n.
- J. B. Baell and G. A. Holloway, New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays, J. Med. Chem., 2010, 53(7), 2719–2740, DOI:10.1021/jm901137j.
- G. W. Pasternak, M. Carroll-Buatti and K. Spiegel, The binding and analgesic properties of a sigma opiate, SKF 10,047, J. Pharmacol. Exp. Ther., 1981, 219(1), 192–198 CAS.
- R. A. Glennon, S. Y. Ablordeppey, A. M. Ismaiel, M. B. El-Ashmawy, J. B. Fischer and K. B. Howie, Structural Features Important for sigma1 Receptor Binding, J. Med. Chem., 1994, 37, 1214–1219 CAS.
- B. Wunsch, Pharmacophore models and development of spirocyclic ligands for σ1 receptors, Curr. Pharm. Des., 2012, 18(7), 930–937, DOI:10.2174/138161212799436548.
- G. Gris, E. J. Cobos, D. Zamanillo and E. Portillo-Salido, Sigma-1 receptor and inflammatory pain, Inflammation Res., 2015, 64(6), 377–381, DOI:10.1007/s00011-015-0819-8.
- T. J. Coderre and R. Melzack, The contribution of excitatory amino acids to central sensitization and persistent nociception after formalin-induced tissue injury, J. Neurosci., 1992, 12(9), 3665–3670, DOI:10.1523/JNEUROSCI.12-09-03665.1992.
- H. W. Kim, Y. B. Kwon, D. H. Roh, S. Y. Yoon, H. J. Han, K. W. Kim, A. J. Beitz and J. H. Lee, Intrathecal treatment with sigma1 receptor antagonists reduces formalin-induced phosphorylation of NMDA receptor subunit 1 and the second phase of formalin test in mice, Br. J. Pharmacol., 2006, 148(4), 490–498, DOI:10.1038/sj.bjp.0706764.
- Y. Peng, Q. Zhang and W. J. Welsh, Novel Sigma 1 Receptor Antagonists as Potential Therapeutics for Pain Management, J. Med. Chem., 2021, 64(1), 890–904, DOI:10.1021/acs.jmedchem.0c01964.
- M. Dichiara, F. A. Ambrosio, C. Barbaraci, R. González-Cano, G. Costa, C. Parenti, A. Marrazzo, L. Pasquinucci, E. J. Cobos, S. Alcaro and E. Amata, Synthesis, Computational Insights, and Evaluation of Novel Sigma Receptors Ligands, ACS Chem. Neurosci., 2023, 14(10), 1845–1858, DOI:10.1021/acschemneuro.3c00074.
- N. Elkholy, A. Abdelwaly, K. Mohamed, E. Amata, J. Lombino, G. Cosentino, S. Intagliata and M. A. Helal, Discovery of 3-(2- aminoethyl)-thiazolidine-2,4-diones as a novel chemotype of sigma-1 receptor ligands, Chem. Biol. Drug Des., 2022, 100, 25–40, DOI:10.1111/cbdd.14047.
- M. Dichiara, A. Artacho-Cordón, R. Turnaturi, M. Santos- Caballero, R. González-Cano, L. Pasquinucci, C. Barbaraci, I. Rodríguez-Gómez, M. Gómez-Guzmán, A. Marrazzo, E. J. Cobos and E. Amata, Dual Sigma-1 receptor antagonists and hydrogen sulfide- releasing compounds for pain treatment: Design, synthesis, and pharmacological evaluation, Eur. J. Med. Chem., 2022, 230, 114091, DOI:10.1016/j.ejmech.2021.114091.
- D. Gentile, A. Coco, V. Patamia, C. Zagni, G. Floresta and A. Rescifina, Targeting the SARS-CoV-2 HR1 with Small Molecules as Inhibitors of the Fusion Process, Int. J. Mol. Sci., 2022, 23(17), 10067, DOI:10.3390/ijms231710067.
- M. Zammataro, S. Merlo, M. Barresi, C. Parenti, H. Hu, M. A. Sortino and S. Chiechio, Chronic Treatment with Fluoxetine Induces Sex-Dependent Analgesic Effects and Modulates HDAC2 and mGlu2 Expression in Female Mice, Front. Pharmacol., 2017, 8, 743, DOI:10.3389/fphar.2017.00743.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4md00397g |
| ‡ These authors share the last authorship. |
| This journal is © The Royal Society of Chemistry 2025 |