
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOxidation behavior of iron and binder-mixed iron: insights from TGA–DSC and in situ XRD analysis for field emission application
Supriya E.
More
 *ab,
Suyog A.
Raut
ad,
S.
Premkumar
ac,
Somnath R.
Bhopale
af,
Davy
Deduytsche
e,
Sudha V.
Bhoraskar
a,
Mahendra A.
More
a,
Damien
Thiry
d,
Christophe
Detavernier
e,
Nathalie
De Geyter
b,
Vikas L.
Mathe
*a and
Rino
Morent
b
*ab,
Suyog A.
Raut
ad,
S.
Premkumar
ac,
Somnath R.
Bhopale
af,
Davy
Deduytsche
e,
Sudha V.
Bhoraskar
a,
Mahendra A.
More
a,
Damien
Thiry
d,
Christophe
Detavernier
e,
Nathalie
De Geyter
b,
Vikas L.
Mathe
*a and
Rino
Morent
b
aDepartment of Physics, Savitribai Phule Pune University, Ganeshkhind, Pune 411007, Maharashtra, India. E-mail: vlmathe@physics.unipune.ac.in; Fax: +020-25691684; Tel: +91 9049682053, +020-25692678ext320
bResearch Unit Plasma Technology (RUPT), Department of Applied Physics, Ghent University, Ghent, 9000, Belgium. E-mail: supriya.more@ugent.be; supriyamore89@gmail.com; Tel: +32 472 75 18 68
cArmament Research and Development Establishment (ARDE), Pune 411021, Maharashtra, India
dChimie des Interactions Plasma-Surface (ChIPS), Université de Mons, Mons, 7000, Belgium
eDepartment of Solid States Science, Krijgslaan, Ghent University, Ghent, 9000, Belgium
fSchool of Physics, IISER Thiruvananthapuram, Vithura, Kerala 695551, Thiruvananthapuram, India
First published on 18th November 2025
Abstract
Precise control over the phase composition and surface morphology of materials is crucial for applications in catalysis, sensing, field-emission, and other fields. This study investigates the thermal oxidation of micron-sized iron (M–Fe) both with and without the use of ethyl cellulose as a binder in various oxidation environments. The samples were processed in starving and oxygen-rich conditions with varying heating rates to investigate their impact on oxide formation. A custom-designed radiation heater (RH) was employed in the vacuum system to achieve an ultra-fast heating rate of 12 °C s−1, raising the surface temperature to 750 °C within one minute. Oxidation experiments under reduced oxygen pressure, termed controlled environment thermal oxidation (CETO), were compared to open environment thermal oxidation (OETO), conducted at the same target temperature but with a significantly slower heating rate (2.5 °C min−1) in a muffle furnace. The role of the binder was analyzed using thermogravimetric analysis and differential scanning calorimetry (TGA–DSC), and a deeper understanding was gained through a phase evolution study, as elucidated by in situ X-ray diffraction (XRD) measurements. The phase evolution of iron, both with and without binder, in two distinct surrounding conditions, primarily affects the onset temperature. The findings highlight the critical influence of oxidation conditions, heating rate, and the presence of binder on the surface properties, paving the way for improved design strategies in field emission applications.
1 Introduction
Iron oxides, particularly α-Fe2O3, are among the most studied materials due to their abundance, low cost, environmental friendliness, and remarkable versatility in technological applications.1–3 The three main polymorphs of Fe2O3-hematite (α-Fe2O3), maghemite (γ-Fe2O3), and beta phase (β-Fe2O3, rare high temperature phase) – exhibit unique structural, electronic, and magnetic properties, making them suitable for a broad spectrum of applications. Hematite, the most thermodynamically stable phase, has garnered significant interest due to its applications in photocatalysis, energy conversion, water splitting, sensors, and environmental remediation, primarily because of its narrow band gap (∼2.2 eV), chemical stability, and non-toxic nature.4,5 The versatility of iron oxides is further enhanced by their redox activity, attributed to the variable oxidation states of iron (Fe2+, Fe3+, and Fe0), which enable redox pairs like Fe2+/Fe3+ and Fe3+/Fe0. This property is crucial for catalytic processes, energy storage systems, and biomedicine, where redox reactions drive the performance.6–11Recent advancements in tailoring the surface morphology of iron oxides have significantly improved their functional properties by enhancing surface area, porosity, and the distribution of active sites. Fine control over these parameters has opened new avenues for optimizing performance in supercapacitors, lithium-ion batteries, and heterogeneous catalysis.11–13 The ability to tune surface features according to specific requirements is crucial for maximizing the efficiency of Fe2O3-based materials in various advanced technologies. Thus, the broad applicability, tunable electronic structure, and redox flexibility of iron oxides make them a cornerstone of modern materials research, with hematite (α-Fe2O3) standing out as a prime candidate for innovative solutions to sustainable energy and environmental challenges.3
The growth mechanisms of α-Fe2O3 nanostructures have been extensively explored using a simple thermal oxidation route, particularly in the context of nanowire formation. For instance, Yuan et al.14 demonstrated that heating the iron foil at 400–600 °C with a ramp rate of 20 °C min−1 under controlled oxygen pressure facilitates the formation of nanowires. The oxidation temperature was shown to play a critical role in determining the layered structures, with oxygen-rich environments promoting the growth of longer nanowires. The key mechanism driving this growth was identified as stress-induced surface diffusion, which occurs due to volume expansion at the Fe2O3/Fe3O4 interface. Similarly, Hiralal et al.15 investigated that the resultant morphologies are not affected by oxygen partial pressure, but the formation of oxide thickness directly depends on pressure. These findings underscore the importance of precise control over synthesis parameters, such as temperature, oxygen availability, and processing time, in tailoring the morphology of α-Fe2O3 nanostructures.
Recent research has highlighted the critical role of synthesis methodology and process parameter tuning in controlling the morphology of α-Fe2O3, making it suitable for various applications. Key synthesis methodologies include coprecipitation followed by calcination,16 RF/DC magnetron co-sputtering,17 and atomic layer deposition (ALD),18 which have enabled the creation of surface morphologies such as microspheres, ellipsoids, and nanowires. Advanced approaches, such as microwave-assisted solvothermal synthesis, have enabled the development of hematite half-hexagon nanoplates for battery anodes8 and the mechano-chemical dispersion of platinum on α-Fe2O3 for H2 sensing,9 thereby expanding its potential for applications. Specific studies reveal that calcination temperature and oxygen pressure significantly influence particle size and nanowire growth on iron substrates. Template-assisted synthesis of iron-based metal–organic frameworks (e.g, MIL-100) produces porous structures with superior sensing capabilities,19 while cobalt/copper-doped α-Fe2O3 is used for environmental remediation.20 The choice of precursors, solvents, and synthesis conditions plays a pivotal role in tailoring α-Fe2O3's morphology for targeted functionality.21 Lian et al.,13 synthesized mesoporous hollow microspheres using an ionic liquid-assisted hydrothermal method.
Studies have explored the use of ethyl cellulose (EC) as a useful advanced material, primarily due to its biocompatibility. EC is a biocompatible thermoplastic polymer, widely applied in coatings, pharmaceutical encapsulation, and as a thickener in food and cosmetics.22 Mamata et al.23 synthesized a membrane via electrospinning combining EC and iron nanoparticles for medical applications, while Arias et al.24 developed carbonyl iron/EC core–shell structures for biomedical use. Cordova et al.25 demonstrated in situ synthesis of iron nanoparticles using EC, achieving superparamagnetic properties. Recently, Geetika et al.26 have incorporated α-Fe2O3 nanoparticles into an EC-ethanol nanocomposite for use as antimicrobial coatings, thereby extending the shelf life of capsicum.
Additionally, plasma-assisted thermal oxidation (PATO)27 has been employed to mix nano- or micro- particles of iron with EC, enabling the formation of tunable surface morphologies and use for field emission applications. These findings highlight the versatility of EC in developing functional materials for diverse applications. Table 1 illustrates the impact of synthesis process parameters on phase and morphology, as well as their field emission applications.
| Sr. no. | Synthesis process | Synthesis time | Post-processing | Achieved morphology | Field emission properties (turn on field, MV m−1) | Ref. |
|---|---|---|---|---|---|---|
| 1 | Thermal oxidation | 10 h | X-ray irradiation | Nano-flakes | 10.1–7.8 | 12 |
| 2 | Plasma assisted thermal oxidation | 10 min | NA | Nanowires | 3 | 27 |
| 3 | Thermal oxidation | 15 h | 10 min RF Ar-plasma | Nano-flakes | 8 | 28 |
| 4 | Pulsed laser deposition | 10 min | Annealed in the air for 3 h | Array of nanoparticles | 64–17.8 | 29 |
This study examines the role of binder-mixed iron in influencing phase evolution and morphological changes under various environmental conditions. The workflow begins with thermogravimetric analysis (TGA) of both the raw iron powder and the corresponding slurry. Subsequently, the binder-containing sample (MF) is subjected to thermal treatments under controlled oxygen partial pressures using a rapid heating protocol, referred to as controlled environment thermal oxidation (CETO). In parallel, conventional thermal treatment under ambient conditions is performed on both MF and pure metal powder (MP); this process, termed open environment thermal oxidation (OETO), is carried out with a slower heating rate. Following these treatments, extensive surface and bulk characterizations are conducted to evaluate the resulting structural and morphological transformations. To gain further insights, in situ XRD is employed for both binder-free and binder-containing samples to study phase evolution in two different environments. Finally, samples with distinct morphologies are examined through field electron emission (FEE) measurements, thereby establishing the influence of experimental conditions on material properties and their potential in advanced materials design.
2 Experimental section
2.1 Preparation of M–Fe slurry and thick film
Iron powder (M–Fe, LOBA-Chemie; particle size 25–60 µm, 99.5% purity, electrolytic grade), ethanol (Hayman, premium grade, 100%), and acetylacetone (LOBA-Chemie, 99.5% purity) were used as precursors for preparing the slurry, with ethyl cellulose (EC; Loba-Chemie) as a binder. A detailed preparation procedure is described in a previous report.27 The slurry was applied onto a stainless-steel substrate using the doctor blade method to obtain a film of approximately 0.25 mm thick; named as MF. The coating obtained was subjected to a natural drying process overnight before proceeding with further experimental processes.2.2 Thermal oxidation (TO) process
 | ||
| Fig. 1 Block diagram of the controlled environment thermal oxidation (CETO) process, with a photograph of the radiation heater installed in the chamber. | ||
| Sr. no. | Sample | Reaction system | Process | Pressure | Oxygen Gas flow | Heating time | Heating rate | Sample Name |
|---|---|---|---|---|---|---|---|---|
| 1 | M–Fe film | Controlled | CETO | 10−5 mbar | 0 sccm | 10 min | 12 °C s−1 | MF-0 |
| 2 | M–Fe film | Controlled | CETO | 5 × 10−3 mbar | 30 sccm | 10 min | 12 °C s−1 | MF-30 |
| 3 | M–Fe film | Controlled | CETO | 9 × 10−3 mbar | 100 sccm | 10 min | 12 °C s−1 | MF-100 |
| 4 | M–Fe powder | Open | OETO | Atm. | Atm. air | 300 min | 2.5 °C min−1 | MP-air |
| 5 | M–Fe film | Open | OETO | Atm. | Atm. air | 300 min | 2.5 °C min−1 | MF-air |
2.3 Characterization techniques
Thermogravimetric analysis and differential scanning calorimetry (TGA–DSC) were conducted using a TGA–DSC 1 STARe system to study the oxidation behavior of M–Fe powders and the slurries at elevated temperatures. The samples were heated from 30 °C to 1200 °C at a rate of 10 °C min−1 in a pure air atmosphere with a flow rate of 50 mL min−1. The nitrogen was used as a purge gas at a flow rate of 20 mL min−1 to maintain the surrounding temperature of the micro-furnace. Raman spectroscopic measurements were carried out using a high-resolution (0.1 cm−1) Renishaw inVia Raman Microscope to identify the polymorphs of iron oxide. Laser radiation at 532 nm was used for excitation, and Raman spectra were recorded over a range of 100–3200 cm−1.The structural properties of pre- and post-treated M–Fe films were examined using X-ray diffraction (XRD). XRD patterns were recorded with a Bruker AXS D8 Advance diffractometer equipped with Cu-Kα radiation (λ = 1.5418 Å) to analyze phase changes induced by oxidation processes. Surface morphology of the films and bare M–Fe particles was investigated using scanning electron microscopy (JEOL JSM-6360A and JEOL JSM-6010PLUS/LV).
In situ XRD measurements were conducted using a Bruker D8 Discover XRD system equipped with a Cu-Kα X-ray source (λ = 1.5406 Å) and a linear X-ray detector. The samples were mounted on a heating stage inside a sealed annealing chamber. Samples were annealed from room temperature (RT) to elevated temperature at a heating rate of 10 °C min−1. A K-type thermocouple was used to accurately monitor the temperature. Every 2 seconds, an XRD snapshot was taken over a 2θ range of 29° to 49°, capturing real-time phase transformations during the heating process. The experiments were performed under a controlled gas atmosphere, either compressed air with a continuous flow rate of 50 mL min−1 at atmospheric pressure or pure air.
In situ XRD was further employed to investigate the influence of pure air on selected samples. For this purpose, high-purity N2 and O2 gases were introduced into the chamber and regulated to atmospheric pressure using a mass flow controller, thereby minimizing the effect of moisture. A limitation of the setup was the difficulty in maintaining a precise total gas flow of 50 mL min−1, resulting in minor deviations from the previously established TGA–DSC conditions. To address this, the actual flow rates of individual gases were recalculated using calibration correction factors (1.000 for N2 and 0.988 for O2). For instance, at a target oxygen partial pressure of 30%, by using eqn (1) that the real flow of nitrogen was 61 sccm (70% N2), while the corrected oxygen was 36 sccm (30% O2). Using this methodology, oxygen partial pressures of 10%, 20%, 30%, 50%, and 80% were precisely established for the in situ measurements.
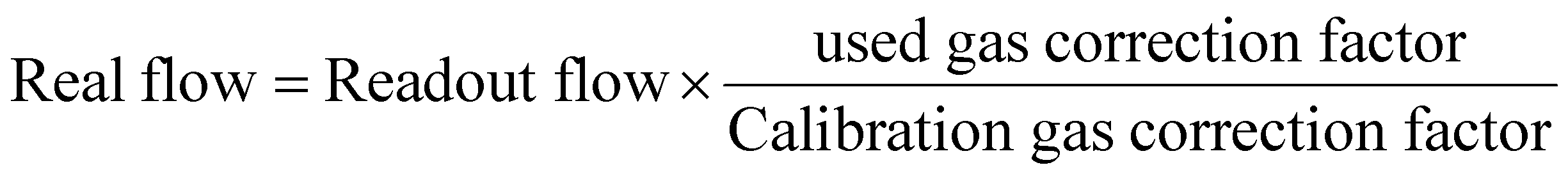 | (1) |
The field electron emission (FEE) properties of morphologically tuned iron oxide films were recorded using a planar diode configuration. In this setup, a semi-transparent cathodoluminescent phosphor screen (diameter ∼50 mm) served as the anode positioned parallel to the cathode. The film under investigation was mounted onto a copper rod using silver paste to secure the electrical contact and acts as the cathode. A copper rod was attached to a linear motion drive, allowing precise adjustment of the cathode–anode separation during FEE measurements. The FEE chamber was sealed and evacuated to a base pressure of 1 × 10−8 mbar using a vacuum system. The field emission measurements were performed with a fixed cathode-anode distance of 1.0 mm (approximately 1000 µm). The emission current was recorded using a Keithley electrometer (Model 6514) while varying the applied voltage from 0 to 40 kV in 40 V increments, using a Spellman high-voltage power source (USA). This configuration enabled a detailed evaluation of the electron emission characteristics of the processed films.
3 Results and discussion
3.1 Thermo-gravimetric and differential scanning calorimetry analysis
The TGA–DSC analysis was conducted to examine the thermal behavior of three different samples: M–Fe only, EC slurry, and M–Fe slurry, and the results are presented in Fig. 2(a)–(c). The thermal oxidation behavior of M–Fe powder and slurries was systematically evaluated. It is well-established that experimental conditions significantly affect TGA–DSC results, including sample size, heating rate, and sample form.30 In the present study, the separate slurry was prepared without Fe powder (only binder EC; Fig. 2(b)) and compared with the M–Fe slurry to investigate oxidation behavior comprehensively, and the thermal profiles of these samples were compared with M–Fe powder (Fig. 2(a)). | ||
| Fig. 2 TGA–DSC recorded for (a) M–Fe powder only, (b) EC slurry only, and (c) M–Fe slurry investigated from temperature 30–1200 °C with a heating rate of 10 °C min−1 and pure airflow 50 mL min−1 used as a reactive gas. | ||
The TGA profiles reveal significant differences between the Fe powder and slurry-based samples. In the slurry samples, the weight loss observed at lower temperatures is attributed to the melting and decomposition of polymeric or organic components, as well as phase transformations within the organic matrix.25,31 Above 400 °C, a noticeable weight gain is observed, corresponding to the oxidation of iron in the case of M–Fe slurry. Sequential thermal decomposition of the binder and subsequent oxidation of metallic iron is expected. The DSC analysis provides complementary insights, with endothermic peaks indicating heat absorption associated with decomposition and melting processes, while exothermic peaks correspond to heat release during oxidation and other exothermic reactions. Together, the TGA–DSC data explain the thermal events and transitions experienced by the materials, elucidate the complex interactions between the ambient environment, binder, and Fe powder, and highlight the critical influence of thermal processing conditions on the overall oxidation and phase evolution of the iron system.
Fig. 2(a) presents the TGA–DSC curves for M–Fe powder. The TGA curve remains stable up to 300 °C, indicating no significant weight change and confirming the material's thermal stability within this temperature range. However, between 300 °C and 900 °C, a substantial weight gain of approximately 41% is observed, corresponding to the oxidation of the M–Fe powder as oxygen reacts with the metallic iron to form iron oxides. The DSC curve reveals a broad exothermic peak between 500 °C and 700 °C, with a maximum at approximately 564 °C, corresponding to phase transformation and crystallization processes. Additionally, minor exothermic features at approximately 600 °C, 730 °C, and 850 °C are linked to solid–solid phase transformations, as reported by Cursaru et al.32
Fig. 2(b) shows the TGA–DSC analysis of the EC slurry without iron powder. The TGA curve exhibits a rapid weight loss from 92 °C to 231 °C, followed by a slower, gradual decline up to 356 °C, culminating in a stabilized plateau. This behavior corresponds to the complete removal of organic components, primarily ethanol and EC. The associated DSC curve features two distinct endothermic peaks between 75 °C and 250 °C. The first peak reflects the evaporation of ethanol, while the second indicates the melting and decomposition of polymeric substances. Subsequently, two exothermic peaks between 350 °C and 450 °C represent crystallization and further solid–solid phase transformations as EC decomposes, leaving behind carbonaceous residues. Heat release during this stage corresponds to the release of aldehydes, carbon dioxide, and water vapor. Up to approximately 457 °C, the organic matrix is entirely decomposed. This analysis provides a detailed understanding of the sequential thermal decomposition and transformation events that occur during thermal treatments applied to EC slurry.
Fig. 2(c) presents the TGA–DSC analysis of the M–Fe slurry. The TGA curve initially shows a minor weight loss near 100 °C, attributed to the evaporation of ethanol, water, and adsorbed species. A more pronounced weight loss is observed up to 550 °C, corresponding to the decomposition and removal of polymeric content, while a slight weight gain is observed at higher temperatures, indicating oxidation of the residual iron. The DSC curve displays a prominent exothermic peak between 400 °C and 550 °C, with a sharp maximum at 490 °C, releasing 1774 J g−1 of thermal energy. This peak is comparable to the broader exothermic peak observed for M–Fe powder (Fig. 2(a)) around 564 °C. Table 3 summarizes the comparison of the TGA–DSC data of M–Fe powder and slurry samples.
Additionally, small humps, like features, are observed at around 600 °C and 800 °C in the DSC curve, indicating iron oxide phase transitions. The complete removal of polymeric content takes place around 550 °C. Slight weight gain above 550 °C indicates oxidation of iron. Here, the role of EC as a binder is evident, as it facilitates the oxidation of iron at a relatively low temperature of 490 °C, likely due to the energy released during EC decomposition, as shown in Fig. 2(b). This localized heat generation creates favourable conditions for initiating oxidation at a relatively lower temperature than that of the M–Fe powder sample. The decreased weight loss in the M–Fe slurry compared to the EC-only sample suggests that heat is more uniformly distributed within the composite system.
3.2 Effect of the CETO and OETO process
Due to the nature of the vacuum system, CETO experiments were restricted to vacuum-based oxidation processes. Key parameters included oxygen pressure, heating rate, and total processing time (summarized in Table 2). These factors collectively govern the structural and morphological transformations during the oxidation process. A comprehensive suite of analytical tools was employed to characterize the samples before and after oxidation, providing insights into the relationship between thermal oxidation conditions, microstructural evolution, and material performance. | ||
| Fig. 3 Pre- and post-CETO processed sample: (a) Raman spectroscopic analysis and (b) corresponding X-ray diffraction (XRD) analysis of MF samples processed under oxygen gas flow conditions of 0, 30 and 100 sccm, referred to as MF-0, MF-30, MF-100, respectively. Here, MF represents untreated micron-sized iron powders mixed with binder and cast into a film on a substrate,while MP denotes untreated micron-sized iron powder. Pre- and post-OETO processed powders (MP/MP-air) and films (MF/MF-air) analysis using (c) Raman spectroscopy and (d) X-ray diffraction analysis, where samples were exposed at 750 °C in a muffle furnace. The corresponding experimental conditions for both processed are summarized in Table 2. (e) Raman spectrum of the post-CETO processed MF-0 sample recorded in the for wavenumber range 3200–100 cm−1. The symbol indicates the respective metal and metal-oxide phases: ■ – metal Fe (JCPDF card no. # 851410), * – Fe3O4/γ-Fe2O3 (JCPDF card no. #861362/#391346), + – α-Fe2O3 (JCPDF card no. 860550), respectively, along with ● – graphitic carbon (JCPDF card no. #752078). | ||
The Raman peaks corresponding to specific vibrational modes presented in iron oxide polymorphs are summarized in Table 4. The characteristic Raman modes observed at 226 cm−1, 245 cm−1, 292 cm−1, 411 cm−1, 491 cm−1, and 612 cm−1 are attributed to the α-Fe2O3 (hematite) phase of iron oxide, based on the reports in the literature.33–35 In Fig. 3(a), the MF-30 sample processed using CETO shows a subtle peak at 292 cm−1 with no significant additional modes. The MF-100 sample, processed with an oxygen flow rate of 100 sccm, displays prominent peaks at 226 cm−1 (A1g) and 292 cm−1 (Eg) with comparable peak intensities, indicating the formation of hematite.
| Iron oxide phases | Wave number in cm−1 |
|---|---|
| Fe3O4 (Magnetite) | 532; 667 |
| γ-Fe2O3 (Maghemite) | 381; 486; 670; 718 |
| α-Fe2O3 (Hematite) | 226; 245; 292; 411; 491; 612 |
Conversely, the MF-0 sample, thermally treated in the absence of oxygen using CETO under vacuum 10−5 mbar, exhibits distinct D and G bands at 1340 cm−1 and 1584 cm−1, respectively, with no detectable Raman peaks corresponding to hematite or magnetite peaks. The intensity ratio of the D to G band (ID/IG) is found to be approximately 0.98, suggesting a high degree of disorder of carbonaceous structures.36,37Fig. 3(e) shows a 2D broad band37 observed in the higher wavenumber region (2500–3200 cm−1), confirming the presence of a multilayer graphene structure at the surface of the MF-0 sample. This result illustrates the influence of oxygen abundance during the oxidation of iron at the surface, which determines the phase composition and structural evolution.38
Fig. 3(c) presents the surface analysis of M–Fe powder and film samples before and after processing with OETO. The pre-processed powder and film exhibit no detectable signature of surface oxidation, indicating that surface oxidation was insignificant during the initial preparation. In contrast, the post-OETO processed powder and film exhibit distinct Raman peaks corresponding to the hematite (α-Fe2O3) phase. All vibrational modes associated with the hematite phase, as listed in Table 4, are observed in the post-OETO processed samples. The most prominent peak observed at 226 cm−1 (A1g) and the second most substantial peak at 292 cm−1 (Eg), both confirm formation of a pure hematite phase at the surface layer through the OETO process. As illustrated in Fig. 3(a) and (c), oxygen-rich environments, regardless of heating rate, promote the formation of hematite as the dominant phase at the surface.
XRD patterns of the M–Fe films and powder samples recorded before and after processing with CETO (Fig. 3(b)) and OETO (Fig. 3(d)) indicate the presence of the (110) peak belonging to metallic iron. This observation confirms that the metallic nature of iron remains intact during slurry formation, with minimal or no oxidation occurring in the initial preparation steps.
In Fig. 3(b), the film processed under vacuum conditions (MF-0) shows no signs of oxidation. Instead, a diffraction peak corresponding to the (111) plane of graphitic carbon having a hexagonal crystal lattice is observed. Notably, no iron-related peaks are present in the MF-0 sample. This absence is attributed to the rapid heating rate and lack of oxygen, which triggers the process of pyrolysis—a thermal decomposition of organic compounds in an oxygen-free environment.30 The rapid heating in a vacuum promotes the formation of a carbonaceous layer on the film surface, effectively obscuring the metallic iron beneath. These findings emphasize the contrasting effects of vacuum (CETO) and atmospheric (OETO) on the phase composition and surface morphology of M–Fe films.
The XRD pattern recorded for the MF-30 film reveals a diffraction peak corresponding to the (311) plane of Fe3O4 (magnetite), indicating its presence at the surface. Additionally, a diffraction line at 2θ = 44.46°, corresponding to the (110) plane, confirms the presence of metallic iron. Notably, no prominent peak corresponding to α-Fe2O3 (hematite) is detected. In contrast, the XRD pattern for the MF-100 film exhibits a characteristic diffraction peak at 2θ = 35.24°, corresponding to the (311) plane of magnetite, with a subtle feature near 2θ = 33° attributed to the (104) plane of hematite. These observations suggest that MF-30 and MF-100 films have mixed phases, including cubic magnetite (Fe3O4), hexagonal hematite, and residual metallic iron.
Surface and bulk characterization together (Fig. 3(a) and (b)) indicate that the MF-100 film develops a structure resembling a core–shell configuration. This structure consists of a surface layer dominated by hematite, underlaid by a magnetite layer, with metallic iron retained beneath. The diffusion dynamics drive the formation of these distinct layers during oxidation: oxygen diffuses inward, while iron diffuses outward. The rapid heating rate and oxygen flow rate of 100 sccm promote the swift oxidation at the surface, forming a passivating oxide layer that inhibits further diffusion of oxygen and iron. Despite achieving complete surface oxidation in MF-100, the bulk iron remains unoxidized due to the limited exposure time and the constraint of fast thermal oxidation processing. These findings underscore the importance of controlling heating rates and oxygen flow rates to tailor the phase at the surface of iron-based films.
Fig. 3(d) presents the structural analysis of pre-and post-OETO processed powder and film samples. The XRD pattern for MP-air shows only the presence of α-Fe2O3 with no detectable traces of metallic iron, confirming complete oxidation of the iron powder. Notably, this process was conducted without a binder, which allowed for uniform oxidation. In contrast, the diffraction pattern for MF-air reveals a mixture of oxide phases and residual metallic iron. The presence of these mixed phases indicates the significant influence of the binder used during film preparation, which appears to hinder complete oxidation by creating a diffusion barrier. This suggests that, while maintaining the same experimental conditions, the resultant phase will differ based on the test sample.
Liang Li29 reported that during the synthesis of hematite nanoplates and particles, variable oxygen abundance significantly influences the resultant phase and morphology. A similar correlation is observed in the present study (Fig. 3), where the relative intensity ratio of the (104) and (110) diffraction planes provides insight into the extent of oxidation and the structural properties of the post-processed films. This correlation reinforces the critical role of oxygen availability, heating profiles, and the presence of binders in determining the final phase composition and microstructure of iron oxide films. A study by Nageshwara et al.8 reported that the parameters of the synthesis process are key factors that determine the shape and size of nanoparticles. Similarly, Panda et al.40 systematically investigated the effect of thermal oxidation temperature on Zn-based thin films. Their findings relate to the correlation between the intensity of specific diffraction planes and corresponding morphologies, providing insights into the possible growth behavior and structural evolution of ZnO under variable thermal treatment. These studies collectively underline the crucial role of synthesis parameters in determining the phase and morphology of metal oxide films.
The OETO process demonstrated that MP-air samples, both at the surface and bulk, give rise to a complete hematite (α-Fe2O3) phase. In contrast, the presence of binders (EC) played a protective role for MF samples, limiting oxidation to the surface while maintaining a metallic iron core. This process led to the creation of a layered structure, where the outermost surface layer was composed of hematite (α-Fe2O3), magnetite (Fe3O4), and the core consisted of metallic iron. Diffraction studies confirmed the phase gradient from the surface to the bulk.
 | ||
Fig. 4 Scanning electron micrographs (SEM) of the CETO processed samples (MF-0, MF-30, MF-100) with magnification of ×3000 are shown in top row. The raw iron powder (MP) and OETO-processed MP-air and MF-air films, recorded at magnification ×3000, ×30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, and ×10000, respectively, are presented in the middle row. The pore size and hair-like morphology distribution for MP-air and MF-air samples are illustrated as histograms in the bottom row. 000, and ×10000, respectively, are presented in the middle row. The pore size and hair-like morphology distribution for MP-air and MF-air samples are illustrated as histograms in the bottom row. | ||
The SEM of post-OETO processed powder and film are presented as MP-air and MF-air, respectively. Surface voids are found to be approximately 150–200 nm in size, as shown in Fig. 4. Similar void formations on micron-sized iron particles have been reported, as shown in cross-sectional views by Lang Qin,41 which reveal a 25% volume expansion and altered pore distribution after oxidation. In the case of MF-air, micrographs reveal a distinct one-dimensional, hair-like morphology with lengths ranging from 0.4 to 0.8 µm.
A comparative structural analysis (Fig. 3) and surface morphology analysis (Fig. 4) of the two different oxidation processes reveal the influence of key factors, including the presence of a binder, processing time, and oxygen abundance, on the phase composition. The use of binders creates a diffusion barrier, slowing the oxidation rate and preventing full-phase transformation. Furthermore, the oxygen flow rate and the distinction between open and controlled environments have a significant impact on oxidation kinetics. High temperatures, combined with controlled oxygen flow, promote faster oxidation, enabling greater precision in controlling the final phase composition and microstructure of iron oxides.
3.2.3.1 Air (moisture present). In situ XRD measurements were performed on MP and MF samples oxidized in an air environment to investigate phase evolution during thermal treatment, as shown in Fig. 5(a) and (b). The blue line in the figure represents the linear temperature increase at a rate of 10 °C per minute, progressing from room temperature to the final set temperature in approximately 530 steps. The in situ XRD results are presented as a color map, where the white denotes the background and the colored lines correspond to diffraction peaks. A prominent diffraction peak at a 2θ value of 44° is attributed to the (110) plane of body-centered cubic (BCC) metallic iron. Additional diffraction peaks at 2θ values of 35° and 33° correspond to the (311) plane of Fe3O4/γ-Fe2O3 and the (104) plane of α-Fe2O3, respectively. For clarity, the identified peaks are labeled P1–P3, from higher to lower 2θ values. To better understand the recorded diffraction pattern shown in Fig. 5(a) and (b), the respective diffracted line intensities were integrated as a function of temperature and are plotted in Fig. 5(a1) and (b1).
 | ||
| Fig. 5 In situ XRD color plots recorded for (a) MP-air and (b) MF-air samples in the temperature range of 30–800 °C with a heating rate of 10 °C min−1 under an airflow (compressed air) of 50 mL min−1. Panels (a1) and (b1) shows the integrated intensities of three diffraction peaks as a function of temperature for samples (a) and (b), respectively. Here, P1 corrosponds to metallic Fe (JCPDF card no. # 851410), P2 to Fe3O4/γ-Fe2O3 (JCPDF card no. #861362/#391346), and P3 to α-Fe2O3 (JCPDF card no. 860550), representing the most intense (hkl) plane (110), (311), and (104), respectively. | ||
Fig. 5(a) and (a1) shows that in the temperature range of 300–800 °C, the oxide phase of iron evolves sequentially. At approximately 400 °C, the diffraction line corresponding to P2 (associated with Fe3O4, magnetite/maghemite), begins to develop. At even higher temperatures, 470 °C, another diffraction line (P3) emerges, indicating the formation of a secondary oxide phase, corresponding to α-Fe2O3 (hematite). From Fig. 5(a1), the sequential phase transition from metallic Fe to FeO, then to magnetite/maghemite, and subsequently to hematite is clearly evident, as indicated by the decreasing intensities of P1 and subsequently P2 at elevated temperatures. The increase in P3 intensity within this range strongly supports the evolution from magnetite/maghemite to pure hematite. This observation aligns with the surface and bulk analysis of the MP-air sample shown in Fig. 3(c) and (d), confirming the complete transformation of Fe to α-Fe2O3 (hematite) under the given thermal conditions.
In the case of MF-air (Fig. 5(b) and (b1)), this provides strong evidence that the presence of a binder results in a different oxidation profile compared to MP-air (Fig. 5(a) and (a1)). The following are the significant differences: (1) the onset temperature of oxide formation shifted to a lower value; (2) the P1 diminishes more rapidly than MP-air, and (3) the P2 and P3 peaks reach higher intensities simultaneously. The P2 intensity profile in Fig. 5(b1) exhibits a distinct behavior, reaching a pronounced maximum that was not observed for MP-air (Fig. 5(a1)).
The variation in the intensities of the P1–P3 peaks provides critical insights into the phase transitions occurring in the samples. The initial oxidation temperature of P2 and P3 indicates that the MF-air sample undergoes oxidation earlier than the MP-air sample. Additionally, the faster decrease in P1 intensity for MF-air compared to MP-air suggests that oxidation is accelerated in the presence of the binder. These observations reveal the significant influence of the binder on oxidation kinetics. The narrower oxidation temperature window for phase evolution suggests that localized heat generated during binder decomposition (Fig. 2(b)) promotes an accelerated phase transition.
3.2.3.2 Pure air (moisture free). Fig. 6(a) and (b) presents the color plots of diffraction patterns for MP-pure and MF-pure samples recorded at 20% oxygen partial pressure, while the corresponding peak intensity profiles are shown in Fig. 6(a1) and (b1). For direct comparison, the TGA–DSC temperature values from (Fig. 2(a) and (c)) are also included to correlate phase transitions of the iron system in the absence of moisture.
 | ||
| Fig. 6 Color maps of in situ XRD recorded for (a) MP-pure and (b) MF-pure samples in the temperature range of 30–920 °C with a heating rate of 10 °C min−1 under a 20% oxygen environment. Panels (a1) and (b1) show the integrated intensities of three diffraction peaks as a function of temperature for samples (a) and (b), respectively. Here, P1 corresponds to metallic Fe (JCPDF #85-1410), P2 to Fe3O4/γ-Fe2O3 (JCPDF #86-1362/#39-1346), and P3 to α-Fe2O3 (JCPDF #86-0550), representing the most intense (hkl) planes (110), (311), and (104), respectively. Additionally, panels (a1) and (b1) highlight characteristic temperature points derived from TGA–DSC measurements (Fig. 2) to provide better correlation and understanding of the phase transitions. | ||
The progressive decrease of P1 intensity, approaching zero, together with the corresponding TGA transition temperature, indicates the complete transformation of the metallic iron precursor into oxide phases. The oxide phase transitions recorded in the DSC can be correlated with the P2 and P3 features in Fig. 6(a1). Broad and well-resolved DSC peaks are observed at 564 °C, 730 °C, and 860 °C, which correspond to the slopes and maxima of P2 and P3. Specifically, tracking the P2 peak over the DSC range of 564–730 °C reveals the following sequence of transitions: Fe → FeO at 564 °C, FeO → Fe3O4/γ-Fe2O3 at 605 °C, and complete formation of Fe3O4/γ-Fe2O3 at 730 °C with maximum P2 intensity. Between 730 °C and 860 °C, the combined DSC signal and changes in P2/P3 intensities indicate the transition from Fe3O4/γ-Fe2O3 to α-Fe2O3, reflected by increasing P3 and decreasing P2 intensities.
Overall, under the given oxygen partial pressure, heating rate, and particle size, the phase transformation sequence is straightforward, like Fe → FeO → Fe3O4/γ-Fe2O3 → α-Fe2O3. Above 860 °C, both DSC signals and P2/P3 intensities saturate, confirming the stabilization of the hematite phase (α-Fe2O3). The oxidation temperature window in this case (295–860 °C) was broader compared to that in Fig. 5(a1) (400–800 °C).
Fig. 6(b) and the corresponding integrated intensities (b1) for the MF-pure sample show that the initial drop in P1 intensity around 400 °C corresponds to binder removal, as confirmed by Fig. 2(b). A subsequent drop in P1 intensity reflects the onset of Fe oxidation. Notably, the onset temperature of P2 is higher than that of MP-pure, suggesting that the binder acts as a protective layer against oxidation. The broad DSC peak at 490 °C (Fig. 2(c)) is associated with the major Fe → FeO phase transition. FeO → Fe3O4/γ-Fe2O3 follows this transition up to ∼600 °C, and the Fe3O4/γ-Fe2O3 → α-Fe2O3 transition up to ∼820 °C. During oxide layer formation, both P2 and P3 phases grow independently and gradually stabilize. For MF-pure, this indicates a layered structure with Fe at the core, Fe3O4/γ-Fe2O3 as an intermediate phase, and α-Fe2O3 as the surface layer.
In contrast, MP-pure exhibits a more direct solid–solid phase transition. The evolution of P2 (intensity drop) and P3 (intensity rise) in the 730–860 °C range reflects a distinct pathway, leading to the complete formation and stabilization of hematite (α-Fe2O3) as a single oxide phase. To further elucidate the phase transitions of MP under varying oxygen partial pressures, additional results are provided in the Fig. S1. The summarized onset temperatures of oxide phase evolution as a function of oxygen partial pressure (in moisture-free conditions) are presented, along with corresponding data for air (moisture-present) conditions (Fig. S2), to highlight the influence of the surrounding environment on the phase transformation behavior.
 | ||
| Fig. 7 XPS survey scan (at left) of MP, MP-pure, MF, and MF-pure. High-resolution XPS Fe 2p spectra (at right) of the MF and MF-pure sample. | ||
The XPS peaks observed at around 710.7 eV and 724.5 eV correspond to Fe 2p3/2 and Fe 2p1/2, respectively,42–45 whereas broad peaks observed at 718.8 eV and 733 eV are the shake-up satellite peaks of Fe 2p3/2 and Fe 2p1/2, respectively. Satellites associated with the Fe 2p core level spectra were used to determine the oxidation states of iron. For the MF-pure sample, the deconvoluted peaks are fitted at 709.45 eV, 710.61 eV, 712.68 eV, 714.69 eV, 718.66 eV, 723.68 eV, 725.65 eV, 727.66 eV, and 732.33 eV. In the current sample, the satellite peak at 718 eV indicates the presence of a maghemite or hematite phase at the surface.46
 | ||
| Fig. 8 Scanning electron micrographs of MP-air and MF 1–3 varying precursor M–Fe: binder ratios—1:0.5 (MF-1), 1:1 (MF-2), and 1:2 (MF-3). All samples processed via the OETO and subjected to a heating rate of 10 °C min−1 under a controlled airflow of 50 mL min−1. | ||
To investigate the effect of binder content on the morphology of iron oxide, additional experiments were conducted in which the M–Fe: binder ratio was varied and thermal oxidation was performed under OETO process parameters, including a heating rate of 10 °C min−1 in an air atmosphere. Fig. 8 suggests that the binder's lower or equal weight ratio to the M–Fe powder yields highly dense NWs, whereas less dense NWs are observed when the binder weight increases. Additionally, Fig. S4 summaries the morphologies obtained for pre and post processed samples under investigation indicates the impact of processing parameters on the obtained surface morphologies. The processes used to obtain distinct α-Fe2O3 morphologies involve careful control of parameters such as heating rate, gaseous environment, and binder usage. The one-dimensional structure of iron oxide with an obtained higher aspect ratio seems to be suitable for field emission applications.11
Fig. 9 provides a comparative overview of phase evolution under thermal treatment for metal Fe (1) only and binder-mixed Fe (2). Under controlled gas conditions, thermal oxidation enables the formation of either a single-phase structure with pore-like morphologies or a layered structure with hair-like surface morphologies. The conventional OETO-processed samples shown in Fig. 3 and 4 (MP-air and MF-air), as well as the respective samples in Fig. 5, exhibit similar phases and morphologies to those summarized in Fig. 9. These observations confirm that the addition of binder, irrespective of environmental conditions, not only narrows the temperature window of oxidation but also promotes the formation of layered structures.
 | ||
| Fig. 9 Graphical representation of the thermal behaviour of (1) MP and (2) MF in a fixed gaseous atmosphere, representing in situ XRD integrated intensity plots for the understanding of phase evolution at given temperature range together with TGA–DSC data; resulting in distinct morphology formation. | ||
Our previous study27 on binder-mixed Fe precursors (micron- and nano-sized) investigated under different oxygen partial pressures and rapid thermal heating demonstrated that surface morphologies could be tuned under plasma conditions, where the processing time was relatively shorter than in existing OETO/CETO processes. Extending this, the present study demonstrates that surface morphologies can also be tailored in conventional processes by adjusting the oxygen partial pressure and binder proportion. Fig. 9 highlights two distinct morphologies that align with the schematic proposed by Lang Qin et al.47 supporting a stress-driven mass transport mechanism for the formation of nanopores and nanowires through mechanical and compressive stresses, respectively.
The layered structures obtained via CETO/OETO processes hold potential for a wide range of applications, including drug delivery, photocatalysis, solid oxide fuel cells,48 volatile organic compound removal,49,50 and as an electrode in lithium- or potassium-ion batteries,51 where iron oxide provides a cost-effective and eco-friendly alternative. In contrast, the oxidation of bare iron particles and film has been extensively studied and reported previously.33,47,52
Overall, the distinct phase evolution and surface morphologies observed for pure and binder-mixed Fe powders highlight the critical role of the binder. Its influence on iron–oxygen diffusion and the formation of distinct oxide layers provides new insights into the mechanisms governing phase transitions and structural evolution during thermal oxidation. A deeper understanding of these dynamics will enable the tailoring of surface and bulk properties through the controlled use of processing parameters.
 | ||
| Fig. 10 (a) Field emission current density versus applied electric field (J–E curve), whereas the inset shows the Fowler–Nordheim (F–N) plot of the corresponding film, (b) photographs of field emission recorded for thermally oxidized M–Fe surface, (c) stability curve showing the continuous electron emission as a function of time. | ||
| Sample name | MF-100 | MF-air |
|---|---|---|
| Low turn-on field 1 µA cm−2 (MV m−1) | 4.4 | 4.4 |
| Threshold field 10 µA cm−2 (MV m−1) | 5.4 | 6.8 |
| Maximum current density (µA cm−2) | 623 | 154 |
Table 5 illustrates the morphology of α-Fe2O3, which significantly influences its field emission properties. The well-defined pyramidal structures in MF-100, characterized by sharp edges and low surface density, provided more effective electron-emitting sites, resulting in superior field emission performance with a low turn-on field of 4.4 MV m−1, a threshold field of 5.4 MV m−1, and a high current density of 623 µA cm−2. In contrast, the high-density nanowires in MF-air exhibited less effective emission sites due to the screening effect, leading to poor electron emission characteristics. As mentioned above, to overcome the screening effect53 achieve better FEE properties, the density of NWs can be tuned by following the initial precursor ratio (EC: M–Fe) using the OETO route; however, this is not within the scope of this article.
Fig. 10(b) shows the field emission electron flux photographs recorded on a phosphor-coated conducting screen (anode) for MF-100 and MF-air. MF-100 demonstrates significantly greater emission sites per unit area than MF-air. The population density of emitting sites strongly influences field emission performance. However, MF-air exhibits a higher density of 1D nanowires with low current density, showcasing stable emission behavior (Fig. 10(c)). These results validate that hematite pyramid morphology could be promising for field emission applications. The enhanced field emission performance of hematite (α-Fe2O3) pyramids is attributed to the rapid heating and oxygen-rich environment created during the CETO process. These results demonstrate that optimizing the morphology through precise control of processing conditions is crucial for enhancing the field-emission performance of α-Fe2O3 nanostructures. Moreover, our reported work based on the advancement of the CETO process27 supports the notion that morphology optimization is a key factor for improved FEE. The presented findings provide a foundation for refining synthesis processes and tailoring surface morphologies to enhance the performance of micron- and nano-iron oxide-based materials in various applications. Additionally, it encourages the use of a similar strategy to study the oxidation behavior of other transition metals (Cu/Ti/Ni).
4 Conclusion
In the present study, the binder was found to serve not only as an inert medium that renders iron powders vacuum-compatible but also as a key factor enabling the investigation of their structural and morphological behavior under rapid heating conditions. The results highlight the significant impact of experimental parameters on surface properties during thermal oxidation. The controlled environmental thermal oxidation process exhibited superior performance compared to the open environmental method, offering enhanced oxygen flow control, reduced processing time, and the formation of well-defined pyramidal morphologies with improved field electron emission characteristics. Furthermore, the combination of thermal analysis and in situ X-ray diffraction revealed that binder incorporation alters the phase evolution pathway, resulting in a narrower temperature window for phase stabilization compared to bare iron powder. Overall, this work demonstrates that the addition of a binder not only facilitates process control but also promotes the formation of stable hematite phases and tailored morphologies advantageous for field electron emission applications.Author contributions
SEM: conceptualization, methodology, formal analysis, investigation, visualization, validation, writing – original draft, writing – review & editing and funding acquisition; SAR: formal analysis, investigation, visualization, writing, review & editing; SP: formal analysis, supervision, writing, review & editing; SRB: investigation, writing – review & editing; DD: methodology, investigation, visualization, formal analysis,; DT: investigation, writing – review & editing; MAM: resources, validation; SVB: formal analysis, writing – review & editing; CD: resources, writing – review and editing, supervision; VLM: resources, writing – review & editing, supervision, funding acquisition; NDG: resources, writing – review & editing, supervision, funding acquisition; RM: resources, writing – review & editing, supervision.Conflicts of interest
There are no conflicts of interest to declare.Data availability
The authors confirmed that the data supporting the findings of this study are available within the articles and in its supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d5ma00591d.Acknowledgements
SEM is thankful to CSIR-SRF (09/137(0575)/2018-EMR-I) for the financial support. NDG wants to acknowledge the financial support of the Research Foundation-Flanders (FWO) for the FWO junior project G060720N. SEM and VLM also gratefully acknowledge the University Grants Commission (UGC) and the Department of Science and Technology (DST), India, for financial support to establish advanced experimental facilities under the UPE, CNQS, PURSE, and FIST Programs. The authors thank Mr Asif Ali from Research Unit Plasma Technology (RUPT), Department of Applied Physics, Ghent University, Ghent 9000 Belgium for their excellent support in acquiring high-quality SEM images used in this study.References
- P. Kumar, N. Thakur, K. Kumar, S. Kumar, A. Dutt, V. K. Thakur, C. Gutiérrez-Rodelo, P. Thakur, A. Navarrete and N. Thakur, Coord. Chem. Rev., 2024, 507, 215750 CrossRef CAS.
- J. Zhao, L. Shi, X. Zhang, Z. Song, H. Lu, A. Abudula, G. Xu and G. Guan, Coord. Chem. Rev., 2024, 518, 216060 CrossRef CAS.
- D. S. Chaudhari, R. P. Upadhyay, G. Y. Shinde, M. B. Gawande, J. Filip, R. S. Varma and R. Zbořil, Green Chem., 2024, 26, 7579–7655 RSC.
- G. S. Parkinson, Surf. Sci. Rep., 2016, 71, 272–365 CrossRef CAS.
- Iron Oxide-Based Nanocomposites and Nanoenzymes: Fundamentals and applications, series ed. D. J. Lockwood, ed. H. Sahoo and J. K. Sahoo, Springer International Publishing, Cham, 2024 Search PubMed.
- U. Singh, V. Ramadesigan, A. K. Chakraborty, S. Shukla and S. Saxena, J. Energy Storage, 2024, 102, 114124 CrossRef.
- M. Zhang, Z. Guan, Y. Qiao, S. Zhou, G. Chen, R. Guo, W. Pan, J. Wu, F. Li and J. Ren, Fuel, 2024, 361, 130541 CrossRef CAS.
- B. Nageswara Rao, J. Pundareekam Goud and N. Satyanarayana, Inorg. Chem. Commun., 2024, 166, 112688 CrossRef CAS.
- M. Šoltić, Z. Klencsár, G. Dražić, M. Gotić, M. Ivanda and N. Baran, Sens. Actuators, A, 2024, 376, 115539 CrossRef.
- X. Zheng and J. Li, Ionics, 2014, 20, 1651–1663 CrossRef CAS.
- L. Junpeng, Y. Tao, Z. Minrui and S. C. Haur, J. Phys. Chem. C, 2011, 115, 8816–8824 CrossRef.
- J. Q. Wu, B. Wang, F. T. Yi, S. Z. Deng, N. S. Xu, J. Chen, J. Q. Wu, B. Wang, F. T. Yi, S. Z. Deng, N. S. Xu and J. Chen, J. Appl. Phys., 2013, 184306, 1–12 Search PubMed.
- J. Lian, X. Duan, J. Ma, P. Peng, T. Kim and W. Zheng, ACS Nano, 2009, 3, 3749–3761 CrossRef CAS.
- L. Yuan, Y. Wang, R. Cai, Q. Jiang, J. Wang, B. Li, A. Sharma and G. Zhou, Mater. Sci. Eng., B, 2012, 177, 327–336 CrossRef CAS.
- P. Hiralal, H. E. Unalan, K. G. U. Wijayantha, A. Kursumovic, D. Jefferson, J. L. MacManus-Driscoll and G. A. J. Amaratunga, Nanotechnology, 2008, 19, 455608 CrossRef CAS.
- X.-M. He, D.-Q. Chen, K.-Y. Su, Z.-F. Yu, Y. Zhang and W. Zhong, Phys. Chem. Chem. Phys., 2024, 26, 2478–2485 RSC.
- S. Sarıtaş, G. Merhan Muğlu, E. Turgut, M. Kundakçı, M. Yıldırım and V. Şenay, Phys. B, 2024, 677, 415704 CrossRef.
- C. Hedrich, N. T. James, L. G. Maragno, V. de Lima, S. Y. G. González, R. H. Blick, R. Zierold and K. P. Furlan, ACS Appl. Mater. Interfaces, 2024, 16, 46964–46974 CrossRef CAS PubMed.
- F. Liu, J. Li, P. Li, J. Shi, X. Gao and H. Xu, Sens. Actuators, B, 2024, 403, 135147 CrossRef CAS.
- P. Kumar, S. Kumar, A. Tapwal and N. Thakur, J. Mater. Res., 2024, 39, 836–849 CrossRef CAS.
- S. Chakrabarty, T. K. Jana, K. De, S. Das, K. Dey and K. Chatterjee, Mater. Res. Express, 2014, 1, 046104 CrossRef CAS.
- H. L. Lai, K. Pitt and D. Q. M. Craig, Int. J. Pharm., 2010, 386, 178–184 CrossRef CAS PubMed.
- M. Shandilya, S. Thakur and S. Thakur, Cellulose, 2020, 27, 10007–10017 CrossRef CAS.
- J. L. Arias, M. López-Viota, M. A. Ruiz, J. López-Viota and A. V. Delgado, Int. J. Pharm., 2007, 339, 237–245 CrossRef CAS PubMed.
- J. G. Puente-Córdova, M. E. Reyes-Melo, B. López-Walle, I. Y. Miranda-Valdez and A. Torres-Castro, Cellulose, 2022, 29, 3845–3857 CrossRef.
- G. Guleria, S. Thakur, M. Shandilya, S. Kumar, P. Kumari, D. K. Sharma and S. Thakur, Mater. Today Proc., 2022, 1–7 Search PubMed.
- S. More, S. Raut, S. Premkumar, S. Bhopale, S. Bhoraskar, M. More and V. Mathe, RSC Adv., 2020, 10, 32088–32101 RSC.
- Z. Zheng, L. Liao, B. Yan, J. X. Zhang, H. Gong, Z. X. Shen and T. Yu, Nanoscale Res. Lett., 2009, 4, 1115–1119 CrossRef CAS PubMed.
- L. Li and N. Koshizaki, J. Mater. Chem., 2010, 20, 2972–2978 RSC.
- P. Arango-Ponton, G. Corjon, J. Dhainaut, S. Heymans, S. Duquesne and J. Lamonier, Adv. Energy Sustainability Res., 2024, 5, 2400072 CrossRef CAS.
- A. Schindler, M. Doedt, Ş. Gezgin, J. Menzel and S. Schmölzer, J. Therm. Anal. Calorim., 2017, 129, 833–842 CrossRef CAS.
- L. M. Cursaru, R. M. Piticescu, D. V. Dragut, I. A. Tudor, V. Kuncser, N. Iacob and F. Stoiciu, Nanomaterials, 2020, 10, 1–22 Search PubMed.
- I. Chamritski and G. Burns, J. Phys. Chem. B, 2005, 109, 4965–4968 CrossRef CAS PubMed.
- D. L. A. De Faria, S. Venâncio Silva and M. T. De Oliveira, J. Raman Spectrosc., 1997, 28, 873–878 CrossRef CAS.
- R. M. Cornell and U. Schwertmann, The Iron Oxides structure, Properties, Reactions, Occurances and Uses, WILEY-VCH GmbH & Co. KGaA, 2003 Search PubMed.
- M. Minakshi, A. Samayamanthry, J. Whale, R. Aughterson, P. A. Shinde, K. Ariga and L. Kumar Shrestha, Chem. – Asian J., 2024, 19, e202400622 CrossRef CAS PubMed.
- A. Rathi and S. I. Kundalwal, J. Compos. Mater., 2022, 56, 1633–1649 CrossRef CAS.
- V. Zólyomi, J. Koltai and J. Kürti, Phys. Status Solidi B, 2011, 248, 2435–2444 CrossRef.
- O. N. Shebanova and P. Lazor, J. Solid State Chem., 2003, 174, 424–430 CrossRef CAS.
- S. A. Panda, S. Choudhary, S. Barala, A. Hazra, S. K. Jena and S. Gangopadhyay, RSC Adv., 2024, 14, 28086–28097 RSC.
- Y. Liu and R. D. L. Smith, Chem. Sci., 2020, 11, 1085–1096 RSC.
- Z. Fishman, Y. He, K. R. Yang, A. Lounsbury, J. Zhu, T. M. Tran, J. B. Zimmerman, V. S. Batista and L. D. Pfefferle, Nanoscale, 2017, 1–13, 10.1039/c7nr03522e.
- M. Fondell, M. Gorgoi, M. Boman and A. Lindblad, J. Electron Spectrosc. Relat. Phenom., 2018, 224, 23–26 CrossRef CAS.
- T. Yamashita and P. Hayes, Appl. Surf. Sci., 2008, 254, 2441–2449 CrossRef CAS.
- A. P. Grosvenor, B. A. Kobe, M. C. Biesinger and N. S. McIntyre, Surf. Interface Anal., 2004, 36, 1564–1574 CrossRef CAS.
- T. Radu, C. Iacovita, D. Benea and R. Turcu, Appl. Surf. Sci., 2017, 405, 337–343 CrossRef CAS.
- L. Qin, A. Mujumder, J. A. Fan, D. Kopechek and L. Fan, J. Mater. Chem. A, 2014, 2, 17511–17520 RSC.
- M. Machado, L. N. Rodrigues, V. B. Vilela, T. S. Moraes, A. S. Ferlauto and F. C. Fonseca, ACS Appl. Energy Mater., 2024, 7, 1766–1776 CrossRef CAS.
- Y. Li, Z. Fan, J. Shi, Z. Liu and W. Shangguan, Chem. Eng. J., 2014, 241, 251–258 CrossRef CAS.
- T. Gelles, A. Krishnamurthy, B. Adebayo, A. Rownaghi and F. Rezaei, Catal. Today, 2020, 350, 3–18 CrossRef CAS.
- M. Valvo, C. Floraki, E. Paillard, K. Edström and D. Vernardou, Nanomaterials, 2022, 12, 1436 CrossRef CAS.
- K. F. Mccarty, M. Monti, S. Nie, D. A. Siegel, E. Starodub, F. El Gabaly, A. H. Mcdaniel, A. Shavorskiy, T. Tyliszczak, H. Bluhm, N. C. Bartelt and J. De Figuera, J. Phys. Chem. C, 2014, 118, 19768–19777 CrossRef CAS.
- L. Wang, F. Gao, S. Chen, C. Li and W. Yang, Appl. Phys. Lett., 2015, 107, 122105 CrossRef.
| This journal is © The Royal Society of Chemistry 2026 |