
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnraveling the electronic control of hydride-ion diffusivity in oxyhydrides from model studies on BaTiO3−2xHx□x
Lucas
Fine
 ab,
Maths
Karlsson
a,
Itai
Panas
a and
Michael Marek
Koza
*b
ab,
Maths
Karlsson
a,
Itai
Panas
a and
Michael Marek
Koza
*b
aDepartment of Chemistry and Chemical Engineering, Chalmers University of Technology, Göteborg, 412 96, Sweden
bInstitut Laue Langevin, 71 avenue des Martyrs, Grenoble, 38042, France. E-mail: koza@ill.fr
First published on 24th September 2025
Abstract
Mixed hydride–electronic conductors are technologically important materials, but the mechanism of hydride-ion diffusivity is generally not fully understood. The diffusivities of hydride-ions and oxygen vacancies are closely related because hydride-ions are accommodated in oxygen vacancies, and a neighbouring oxygen vacancy is required for the inter-site migration of a hydride-ion. Here, we investigate the impact of electron localization in the hydride-ion-accepting oxygen vacancy on the inter-site hydride-ion migration dynamics in the perovskite-type oxyhydride BaTiO3−2xHx□x (where □ denotes oxygen vacancies) using density functional theory (DFT). Supercell calculations were designed to model two ( ), one
), one  , and zero
, and zero  electrons localized in the hydride-ion-accepting site and correspondingly, zero, one, and two electrons delocalized in the conduction band formed from the Ti4+ 3d orbitals. It is found that the trapping of electrons causes the activation energy for inter-site migration to increase from 0.29 eV for
electrons localized in the hydride-ion-accepting site and correspondingly, zero, one, and two electrons delocalized in the conduction band formed from the Ti4+ 3d orbitals. It is found that the trapping of electrons causes the activation energy for inter-site migration to increase from 0.29 eV for 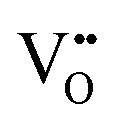 , to 0.39 eV for
, to 0.39 eV for  , to 0.60 eV when
, to 0.60 eV when 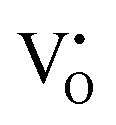 turns into
turns into  during the migration, and to 0.83 eV for
during the migration, and to 0.83 eV for  . In an analogous way, the mobility of oxygen vacancies becomes increasingly hindered with increased electron occupation in the vacancies. This suggests that the tailoring of the degree of electron localization by, e.g., bandgap engineering or the introduction of electron trapping impurity states may be effective in tuning the hydride-ion conductivity in oxyhydrides, not limited to BaTiO3−2xHx□x.
. In an analogous way, the mobility of oxygen vacancies becomes increasingly hindered with increased electron occupation in the vacancies. This suggests that the tailoring of the degree of electron localization by, e.g., bandgap engineering or the introduction of electron trapping impurity states may be effective in tuning the hydride-ion conductivity in oxyhydrides, not limited to BaTiO3−2xHx□x.
1 Introduction
Perovskite-type oxyhydrides (BaTiO3−x−yHx□y, x < 0.6) represent a novel class of mixed-anion materials in which hydride-ions (H−) and oxide-ions (O2−) share the same anion sub-lattice.1 BaTiO3−x−yHx□y (x < 0.6) displays a mixed electron and H− conductivity, which may be exploited for diverse applications in e.g., catalysis, topochemical synthesis, and electrochemistry.2–5 A striking example is the high catalytic activity for ammonia synthesis when using BaTiO3−x−yHx□y as a support for catalytic Fe and Ru nanoparticles.2,6 This high catalytic activity is attributed to the H− mobility through the BaTiO3−x−yHx□y support as well as the donation of electrons from this support to the catalytic nanoparticles.2 Therefore, both H− and electronic conductivities are key to the performance of BaTiO3−x−yHx□y in catalytic applications, and potentially in many other applications as well.The H− conductivity in BaTiO3−x−yHx□y occurs, in part, through the hopping (migration) of H− into adjacent vacant sites in the O2− sub-lattice (VO), as demonstrated by both experimental7 and theoretical studies.4,8 Therefore, the formation of percolation pathways enabling H− conduction throughout the material inherently depends on the diffusion of  within the O2− sub-lattice, which occurs via the migration of adjacent O or H− into the
within the O2− sub-lattice, which occurs via the migration of adjacent O or H− into the  . The electron conductivity, on the other hand, is due to the presence of H− and/or
. The electron conductivity, on the other hand, is due to the presence of H− and/or  in the
in the  sub-lattice, which act as n-type dopants of the empty 3d states of titanium (Ti).9,10 When O2− is replaced by H−
sub-lattice, which act as n-type dopants of the empty 3d states of titanium (Ti).9,10 When O2− is replaced by H− or when a
or when a  forms, the valence electrons originally associated with
forms, the valence electrons originally associated with  , referred to as doping electrons, can adopt two possible states. They may either localize in the
, referred to as doping electrons, can adopt two possible states. They may either localize in the  or near a Ti4+ ion, as an electron polaron, which results in a singly charged defect
or near a Ti4+ ion, as an electron polaron, which results in a singly charged defect  or a neutral defect (
or a neutral defect ( ). This implies a semiconductor-like behavior of the material. Alternatively, they may delocalize across the Ti4+ sub-lattice and populate the conduction band (CB) as a band state, which results in a doubly charged defect
). This implies a semiconductor-like behavior of the material. Alternatively, they may delocalize across the Ti4+ sub-lattice and populate the conduction band (CB) as a band state, which results in a doubly charged defect  . This implies a metal-like behaviour of the material.11–14
. This implies a metal-like behaviour of the material.11–14
Previous studies of BaTiO3−x−yHx□y with y in the range of 0.1–0.31, combining DFT with experimental techniques such as inelastic neutron scattering (INS)15 and nuclear magnetic resonance (NMR),16 showed that the doping electrons form a band state. However, in ref. 15 it is shown that the energy difference between the two scenarios, electron polaron vs. band state, is low and lies in the range of −0.15 to 0.12 eV, depending on the H− concentration and the DFT settings used. Similarly, in ref. 16 it is concluded that band states are prevalent but the existence of polarons cannot be ruled out. Therefore, the conclusion that the doping electrons always form band states cannot simply be generalized to all compositions of BaTiO3−x−yHx□y (x, y < 0.6). In fact, both metal-like and semiconductor-like behaviors have been reported in various experimental studies.1,9,11,17
Crucially, as observed in various transition metal oxide cathode materials, such as LiV3O8,18 LiV2O5,19 and LiFePO4,20 electron polarons can significantly influence the activation energy for ion conductivity. Nonetheless, for BaTiO3−x−yHx□y, the H− and electron conductivities have, to the best of our knowledge, been studied only independently, and the localization effects of the doping electrons were not explicitly taken into account.4,8,21,22 As a result, the influence of electron localization on H− conductivity remains unclear. Yet, elucidating the mechanisms underlying mixed hydride–electronic conductivity in BaTiO3−x−yHx□y is essential for the rational design of new oxyhydrides and related technologies. Notably, electron localization engineering has been used to enhance catalytic activity, such as in CO2 reduction, through dopant incorporation and nanostructuring.23–25 These studies highlight the potential of controlling electron localization in the design of advanced catalysts.
Towards this aim, we build upon previous DFT studies by explicitly modeling the localization of doping electrons in BaTiO3−2xHx□x and assessing its effect on H− migration. We consider three cases with two ( ), one
), one  , and zero
, and zero  electrons localized in the hydride-ion-accepting site and correspondingly, zero, one, and two delocalized electrons in the conduction band. For each, we calculate the activation energy (Ea) for H− migration to a neighboring oxygen vacancy. Rather than attempting to predict whether the doping electrons are localized or delocalized in BaTiO3−2xHx□x, our aim is to establish a physical understanding of how their degree of localization influences H− transport behavior.
electrons localized in the hydride-ion-accepting site and correspondingly, zero, one, and two delocalized electrons in the conduction band. For each, we calculate the activation energy (Ea) for H− migration to a neighboring oxygen vacancy. Rather than attempting to predict whether the doping electrons are localized or delocalized in BaTiO3−2xHx□x, our aim is to establish a physical understanding of how their degree of localization influences H− transport behavior.
2 Methodology
The DFT calculations were conducted using a plane wave basis set as implemented in the Vienna ab initio simulation package (VASP)26–28, used with the projector augmented-wave method.29 We used supercells containing 2 × 2 × 2 and 3 × 3 × 3 conventional unit cells of BaTiO3 (39 and 134 atoms, respectively) and one H− and one inserted next to each other, which correspond to BaTiO2.75H0.125□0.125 and BaTiO2.93H0.037□0.037. Another 2 × 2 × 2 supercell of composition BaTiO2.75H0.125□0.125 was arranged in a channel geometry in such a way that the H− and the
inserted next to each other, which correspond to BaTiO2.75H0.125□0.125 and BaTiO2.93H0.037□0.037. Another 2 × 2 × 2 supercell of composition BaTiO2.75H0.125□0.125 was arranged in a channel geometry in such a way that the H− and the  form a linear chain along the crystallographic a axis. Additionally, the hydrogen-free but oxygen-vacancy containing material BaTiO2.75□0.25 and the fully stoichiometric material BaTiO3 were modeled using a 2 × 2 × 2 supercell. All supercells are depicted in Fig. S1 of the Supporting Information (SI). Brillouin zones were sampled over Monkhorst–Pack grids of 5 × 5 × 5 points for the 2 × 2 × 2 supercells and 3 × 3 × 3 points for the 3 × 3 × 3 supercell. Spin-polarized calculations were conducted, where a cut-off energy of 500 eV was applied to the plane wave basis set and electronic structures were converged to energy changes less than 10−7 eV. Prior to calculating the electronic properties and Ea, every structure was optimized to relax inter-atomic forces below 10−4 eV Å−1. Minimum energy paths and Ea were calculated using the nudged elastic band with a climbing image method.30 The doping electrons are introduced in the system as a consequence of
form a linear chain along the crystallographic a axis. Additionally, the hydrogen-free but oxygen-vacancy containing material BaTiO2.75□0.25 and the fully stoichiometric material BaTiO3 were modeled using a 2 × 2 × 2 supercell. All supercells are depicted in Fig. S1 of the Supporting Information (SI). Brillouin zones were sampled over Monkhorst–Pack grids of 5 × 5 × 5 points for the 2 × 2 × 2 supercells and 3 × 3 × 3 points for the 3 × 3 × 3 supercell. Spin-polarized calculations were conducted, where a cut-off energy of 500 eV was applied to the plane wave basis set and electronic structures were converged to energy changes less than 10−7 eV. Prior to calculating the electronic properties and Ea, every structure was optimized to relax inter-atomic forces below 10−4 eV Å−1. Minimum energy paths and Ea were calculated using the nudged elastic band with a climbing image method.30 The doping electrons are introduced in the system as a consequence of  , as confirmed by calculations on BaTiO3 and BaTiO2.75□0.25 (see the SI). Specifically, since charge neutrality is maintained in the calculations, introducing one
, as confirmed by calculations on BaTiO3 and BaTiO2.75□0.25 (see the SI). Specifically, since charge neutrality is maintained in the calculations, introducing one  in the supercell provides two doping electrons, while substituting one oxygen atom with a hydrogen atom yields one doping electron and one H− ion
in the supercell provides two doping electrons, while substituting one oxygen atom with a hydrogen atom yields one doping electron and one H− ion  . This results in a net total of three doping electrons, which can be found either localized near the
. This results in a net total of three doping electrons, which can be found either localized near the  , forming
, forming  (high localization) or
(high localization) or  (partial localization), or delocalized over the Ti sub-lattice, forming
(partial localization), or delocalized over the Ti sub-lattice, forming 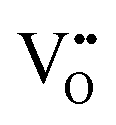 (full delocalization).
(full delocalization).
Different combinations of exchange–correlation (XC) functionals and supercells are employed to investigate the impact of varying degrees of electron localization on H− mobility. The functionals employed include the Perdew–Burke–Ernzerhof (PBEsol) functional, suitable for metallic bulk and surface systems,31 and the PBE functional32 within the DFT+U framework (further referred to as PBE+U).33 They are summarized in Table 1 and further described in the following.
 . The charge state of
. The charge state of  was monitored at the initial state (I.S.) or at the transition state (T.S.)
was monitored at the initial state (I.S.) or at the transition state (T.S.)
| Label | Setting | Composition | State of
|
E a (eV) | |
|---|---|---|---|---|---|
| I.S. | T.S. | ||||
| a E a is given without zero-point vibrational energy correction. | |||||
| (1) High localization | 2 × 2 × 2 + PBE+U | BaTiO2.75H0.125□0.125 |

|

|
0.83 |
| (2) Partial localization | 2 × 2 × 2 + PBEsol | BaTiO2.75H0.125□0.125 |

|

|
0.60 |
| (3) Partial localization | Channel + PBEsol | BaTiO2.75H0.125□0.125 |

|

|
0.39 |
| (4) Full delocalization | 3 × 3 × 3 + PBEsol | BaTiO2.93H0.037□0.037 |

|

|
0.29 |

(1) High localization ( ) of the doping electrons was modeled using the PBE+U functional with the 2 × 2 × 2 supercell. The Hubbard correction (U = 3.3 eV) is applied here as a means to localize the doping electrons near Ti4+ ions, thereby compensating for the over-delocalization that is characteristic of standard XC functionals.34 This approach has previously proven successful in modeling polarons in BaTiO2.875H0.125,15 a composition closely matching that of the 2 × 2 × 2 supercell used in this study, i.e., BaTiO2.75H0.125□0.125. In ref. 15, the U parameter was tuned to reproduce a linear dependence of the total energy on the occupation of the polaronic level – a known property of the exact XC functional35 – resulting in a calibrated value of U = 3.3 eV.
) of the doping electrons was modeled using the PBE+U functional with the 2 × 2 × 2 supercell. The Hubbard correction (U = 3.3 eV) is applied here as a means to localize the doping electrons near Ti4+ ions, thereby compensating for the over-delocalization that is characteristic of standard XC functionals.34 This approach has previously proven successful in modeling polarons in BaTiO2.875H0.125,15 a composition closely matching that of the 2 × 2 × 2 supercell used in this study, i.e., BaTiO2.75H0.125□0.125. In ref. 15, the U parameter was tuned to reproduce a linear dependence of the total energy on the occupation of the polaronic level – a known property of the exact XC functional35 – resulting in a calibrated value of U = 3.3 eV.
Fig. 1(a) shows the electronic band structure and the partial density of states (PDOS), revealing two defect (localized) states in the bandgap, close to the CB edge. The Fermi level is found above the CB edge, which indicates the presence of delocalized electrons in CB states, referred to as band states.
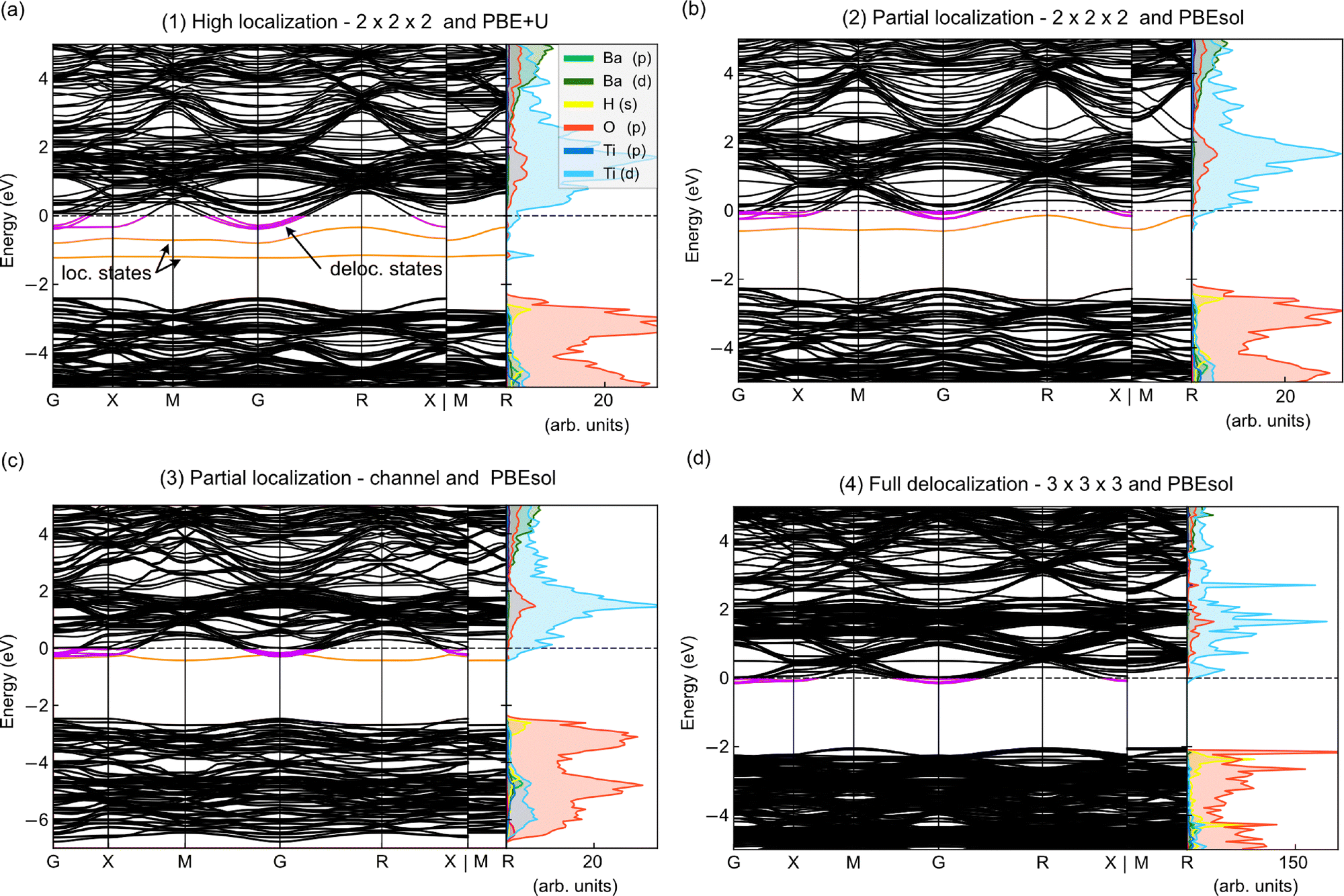 | ||
| Fig. 1 Electronic band structure and orbital-decomposed (s, p, d) PDOS for each calculation setup. (a) High localization of doping electrons, (b) partial localization of doping electrons, (c) partial localization of doping electrons in the channel geometry, and (d) full delocalization of doping electrons. The PDOS of H is scaled 10× in (a)–(c) and 100× in (d). The dashed line represents the Fermi level. The orange and purple colors in the electronic band structures indicate localized (loc.) and delocalized (deloc.) states, respectively. | ||
Fig. 2(a) shows real-space charge densities in the energy range of −2 to 0 eV, which, when integrated, account for a total of three doping electrons. One doping electron is delocalized over the whole Ti sub-lattice, occupying Ti orbitals of local t2g symmetry, i.e. 3dxz, 3dxy, and 3dyz orbitals [Fig. 2(a3)], which correspond to the occupied band states in the PDOS. Two doping electrons are localized near the  [Fig. 2(a1 and a2)], which correspond to the two defect states in the PDOS. The defect state of lowest energy is localized around Ti3 and occupies a Ti orbital of t2g symmetry, i.e. 3dyz [Fig. 2(a1)], while the defect state of highest energy is shared between Ti1 and Ti3 orbitals of eg symmetry, i.e. 3dz2 [Fig. 2(a2)]. Overall, two doping electrons reside near the
[Fig. 2(a1 and a2)], which correspond to the two defect states in the PDOS. The defect state of lowest energy is localized around Ti3 and occupies a Ti orbital of t2g symmetry, i.e. 3dyz [Fig. 2(a1)], while the defect state of highest energy is shared between Ti1 and Ti3 orbitals of eg symmetry, i.e. 3dz2 [Fig. 2(a2)]. Overall, two doping electrons reside near the  , which results in
, which results in  . We note that H contributes in the valence band (VB) through its 1s orbital [Fig. 1(a) and Fig. S2]. Its hydridic character is confirmed by a Bader charge analysis,36 showing an excess of ∼0.6 electrons, in agreement with a previous study on BaTiO2.82H0.1□0.08.15
. We note that H contributes in the valence band (VB) through its 1s orbital [Fig. 1(a) and Fig. S2]. Its hydridic character is confirmed by a Bader charge analysis,36 showing an excess of ∼0.6 electrons, in agreement with a previous study on BaTiO2.82H0.1□0.08.15
 | ||
| Fig. 2 Band-decomposed electron density isosurfaces at 10% for each calculation setup. (1) High localization of doping electrons, (2) partial localization of doping electrons, (3) partial localization of doping electrons in the channel geometry, and (4) full delocalization of doping electrons. Each row represents the three most populated bands in the energy range of −2 to 0 eV. | ||
(2) Partial localization of the doping electrons was modeled using the PBEsol functional with a 2 × 2 × 2 supercell. Removing the Hubbard correction restores the over-delocalization characteristic of the XC functional and, consequently, more doping electrons are expected to form band states. However, one localized defect state remains in the bandgap, making this setting useful for studying a partially localized case [Fig. 1(b)]. This unexpected defect state is attributed to the periodic boundary conditions used in our supercell calculations, which result in the close ordering of
of the doping electrons was modeled using the PBEsol functional with a 2 × 2 × 2 supercell. Removing the Hubbard correction restores the over-delocalization characteristic of the XC functional and, consequently, more doping electrons are expected to form band states. However, one localized defect state remains in the bandgap, making this setting useful for studying a partially localized case [Fig. 1(b)]. This unexpected defect state is attributed to the periodic boundary conditions used in our supercell calculations, which result in the close ordering of  along the Ti–□–Ti direction, leading to spurious □–□ interactions and causing electron localization. The real-space charge density shows two delocalized doping electrons occupying Ti t2g orbitals [Fig. 2(b2 and b3)], which correspond to band states in the PDOS [Fig. 1(b)], and one localized doping electron occupying Ti1 and Ti3 eg orbitals, corresponding to the defect state in the PDOS. Overall, only one doping electron resides near the
along the Ti–□–Ti direction, leading to spurious □–□ interactions and causing electron localization. The real-space charge density shows two delocalized doping electrons occupying Ti t2g orbitals [Fig. 2(b2 and b3)], which correspond to band states in the PDOS [Fig. 1(b)], and one localized doping electron occupying Ti1 and Ti3 eg orbitals, corresponding to the defect state in the PDOS. Overall, only one doping electron resides near the  , which results in
, which results in  .
.
(3) Partial localization in a channel. In the 2 × 2 × 2 supercell with PBEsol (2), the partial localization is due to spurious □–□ interactions. In the channel geometry, the  are more closely packed along the □–H–□ direction, with a separation of 5.6 Å compared to 8 Å in the 2 × 2 × 2 supercell, thereby exacerbating the spurious interaction effects. At the initial state, the electronic structure in the channel geometry is similar to that of the 2 × 2 × 2 supercell with PBEsol (2). We observe one defect state in the band gap touching the CB edge, as well as occupied band states. Overall, one doping electron resides near the
are more closely packed along the □–H–□ direction, with a separation of 5.6 Å compared to 8 Å in the 2 × 2 × 2 supercell, thereby exacerbating the spurious interaction effects. At the initial state, the electronic structure in the channel geometry is similar to that of the 2 × 2 × 2 supercell with PBEsol (2). We observe one defect state in the band gap touching the CB edge, as well as occupied band states. Overall, one doping electron resides near the  , resulting in
, resulting in  [Fig. 1(c) and Fig. 2(c)]. However, due to the channel geometry, the degree of localization may differ from that of the 2 × 2 × 2 supercell during H− migration, making this geometry an additional case of electron localization.
[Fig. 1(c) and Fig. 2(c)]. However, due to the channel geometry, the degree of localization may differ from that of the 2 × 2 × 2 supercell during H− migration, making this geometry an additional case of electron localization.
(4) Full delocalization of the doping electrons was modeled using the PBEsol functional with a 3 × 3 × 3 supercell. Given that the 2 × 2 × 2 supercell and the channel geometry cause electron localization due to spurious □–□ interactions, the 3 × 3 × 3 supercell was employed in order to mitigate the effect of periodic boundary conditions and obtain only delocalized electrons. Consequently, only band states are observed, as indicated by the band structure [Fig. 1(d)] and the real-space charge density [Fig. 2(d1–d3)]. Overall, no doping electron resides near the
of the doping electrons was modeled using the PBEsol functional with a 3 × 3 × 3 supercell. Given that the 2 × 2 × 2 supercell and the channel geometry cause electron localization due to spurious □–□ interactions, the 3 × 3 × 3 supercell was employed in order to mitigate the effect of periodic boundary conditions and obtain only delocalized electrons. Consequently, only band states are observed, as indicated by the band structure [Fig. 1(d)] and the real-space charge density [Fig. 2(d1–d3)]. Overall, no doping electron resides near the  , which results in
, which results in 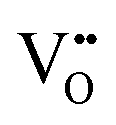 .
.
3 Results and discussion
After having established different degrees of electron localization, we now turn to investigate how this impacts the inter-site migration of a H− with a neighbouring , which is also commonly referred to as the localized diffusion of H−. In previous studies, two H− migration pathways were proposed. The first involves protonic species diffusing through interstitial sites and subsequently swapping positions with neighboring H−.21,22 The second consists of H− hopping into an adjacent
, which is also commonly referred to as the localized diffusion of H−. In previous studies, two H− migration pathways were proposed. The first involves protonic species diffusing through interstitial sites and subsequently swapping positions with neighboring H−.21,22 The second consists of H− hopping into an adjacent  while preserving its hydridic character.4,7,8,16 Experimental studies have indicated that H− migration occurs via the second pathway,7,16 as no interstitial protonic species have been observed,37 whereas the presence of
while preserving its hydridic character.4,7,8,16 Experimental studies have indicated that H− migration occurs via the second pathway,7,16 as no interstitial protonic species have been observed,37 whereas the presence of  has been confirmed.10,37 Based on this, we modeled H− migration according to the second pathway, where a H− undergoes vacancy-mediated hopping. In order to identify the saddle point for the inter-site migration, the minimum energy path (MEP) was first calculated with 5 intermediate positions of the H− in addition to the initial and final positions. This was done using the less computationally demanding setup PBEsol with a 2 × 2 × 2 supercell. Importantly, a Bader charge analysis at each configuration revealed an excess charge of approximately 0.6 electrons around the hydrogen atom, consistent with the value at the initial state, indicating that the hydrogen does not dissociate into a proton but retains its hydridic character throughout the migration process. The saddle point, further referred to as the transition state, is found exactly in the middle of the migration path, and the energy barrier is found to be symmetric around the transition state (Fig. 3). In our further calculations, only the transition state was thus calculated to save computational time.
has been confirmed.10,37 Based on this, we modeled H− migration according to the second pathway, where a H− undergoes vacancy-mediated hopping. In order to identify the saddle point for the inter-site migration, the minimum energy path (MEP) was first calculated with 5 intermediate positions of the H− in addition to the initial and final positions. This was done using the less computationally demanding setup PBEsol with a 2 × 2 × 2 supercell. Importantly, a Bader charge analysis at each configuration revealed an excess charge of approximately 0.6 electrons around the hydrogen atom, consistent with the value at the initial state, indicating that the hydrogen does not dissociate into a proton but retains its hydridic character throughout the migration process. The saddle point, further referred to as the transition state, is found exactly in the middle of the migration path, and the energy barrier is found to be symmetric around the transition state (Fig. 3). In our further calculations, only the transition state was thus calculated to save computational time.
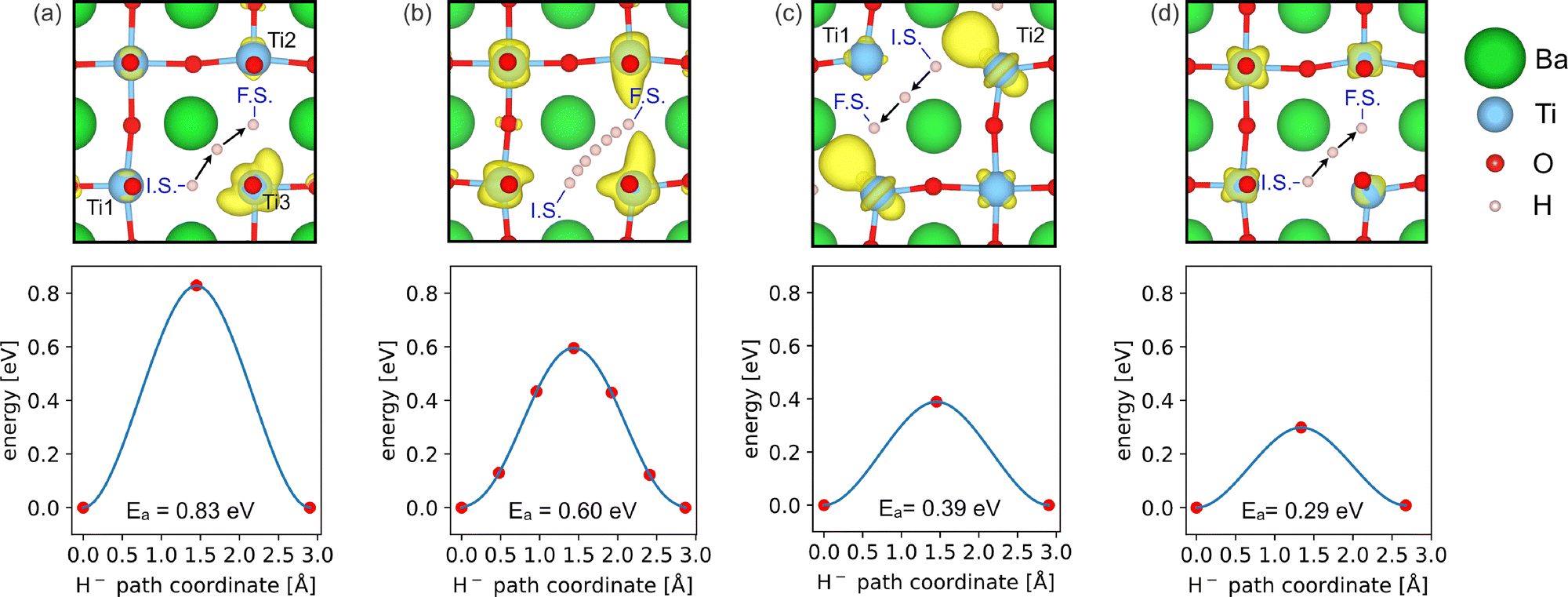 | ||
Fig. 3 Schematic illustration of the H− migration between two neighbouring  together with the energy barrier for each calculation setup (the red points represent the calculated energies, and blue lines are polynomial fits). (a) High localization of doping electrons, (b) partial localization of doping electrons, (c) partial localization of doping electrons in the channel geometry, and (d) full delocalization of doping electrons. H− is shown at the initial state (I.S.), transition state, and final state (F.S.), and part (b) shows, additionally, H− at 5 intermediate states used for the MEP calculation. In yellow are overplotted isosurfaces at 10% of the maximum electronic density for bands in the energy range of −2 to 0 eV at the transition state. together with the energy barrier for each calculation setup (the red points represent the calculated energies, and blue lines are polynomial fits). (a) High localization of doping electrons, (b) partial localization of doping electrons, (c) partial localization of doping electrons in the channel geometry, and (d) full delocalization of doping electrons. H− is shown at the initial state (I.S.), transition state, and final state (F.S.), and part (b) shows, additionally, H− at 5 intermediate states used for the MEP calculation. In yellow are overplotted isosurfaces at 10% of the maximum electronic density for bands in the energy range of −2 to 0 eV at the transition state. | ||
At the transition state, two  are symmetrically arranged around Ti3, thus constraining a doping electron localized in the initial
are symmetrically arranged around Ti3, thus constraining a doping electron localized in the initial  to delocalize over both
to delocalize over both  . As a result, the symmetry of the Ti3 3dz2 orbital is violated and cannot handle the doping electron at the transition state. Instead, the Ti3 3dx2−y2 and 3dyz orbitals, initially well separated in the VB and CB, respectively, become mixed and accommodate the doping electron near Ti3. This band mixing affects the band structure and raises the energy levels of the defect states (see the SI). The position of the mixed bands, either in the band gap or in the CB, defines the degree of localization at the transition state, and therefore Ea. The main results are compiled in Table 1 and summarized in the following.
. As a result, the symmetry of the Ti3 3dz2 orbital is violated and cannot handle the doping electron at the transition state. Instead, the Ti3 3dx2−y2 and 3dyz orbitals, initially well separated in the VB and CB, respectively, become mixed and accommodate the doping electron near Ti3. This band mixing affects the band structure and raises the energy levels of the defect states (see the SI). The position of the mixed bands, either in the band gap or in the CB, defines the degree of localization at the transition state, and therefore Ea. The main results are compiled in Table 1 and summarized in the following.
(1) For high localization of the doping electrons, the mixed states are located within the bandgap and are localized near Ti3 [Fig. 3(a)]. Because the electrons are confined near the jump path of H−, Ea increases as a result of an increase in Pauli repulsion between e− and H−, and takes the value Ea = 0.83 eV.
(2) For partial localization of the doping electrons, the mixed states are located in the CB as band states (see the SI). In other words,  at the initial state becomes
at the initial state becomes  at the transition state [Fig. 3(b)], which indicates that the localized electron uses the CB to swap position with H−. In this case, we calculate Ea = 0.60 eV, which reflects the energy cost to pump the doping electron to the CB.
at the transition state [Fig. 3(b)], which indicates that the localized electron uses the CB to swap position with H−. In this case, we calculate Ea = 0.60 eV, which reflects the energy cost to pump the doping electron to the CB.
(3) For partial localization in a channel geometry, one mixed state is a defect state, maintaining  at the transition state, whereas the others are band states (see the SI). Interestingly, the doping electron, which was initially localized in the
at the transition state, whereas the others are band states (see the SI). Interestingly, the doping electron, which was initially localized in the  , relocates near Ti2 at the transition state [Fig. 3(c)]. This relocation is facilitated by the close periodic arrangement of
, relocates near Ti2 at the transition state [Fig. 3(c)]. This relocation is facilitated by the close periodic arrangement of  along the □–H–□ direction, resulting in a lower Ea of 0.39 eV compared to (2) partial localization. Moreover, Ea is also found to be significantly lower than that of (1) high localization, at Ea = 0.83 eV, despite the presence of a localized doping electron. This reduction is due to the H− and the localized electron migrating together in the same direction, whereas for (1) high localization, they must move against each other, intensifying their interaction.
along the □–H–□ direction, resulting in a lower Ea of 0.39 eV compared to (2) partial localization. Moreover, Ea is also found to be significantly lower than that of (1) high localization, at Ea = 0.83 eV, despite the presence of a localized doping electron. This reduction is due to the H− and the localized electron migrating together in the same direction, whereas for (1) high localization, they must move against each other, intensifying their interaction.
(4) For full delocalization of the doping electrons, the electronic structure remains unchanged, that is, the three doping electrons form band states and  is conserved during the H− migration, which results in Ea = 0.29 eV [Fig. 3(d)]. Note that this value is very similar to Ea = 0.28 eV, as previously calculated8 in an equally large supercell, but using DFT+U instead of PBEsol. Since we find the same value with PBEsol, we conclude that in such low concentrations of H− and
is conserved during the H− migration, which results in Ea = 0.29 eV [Fig. 3(d)]. Note that this value is very similar to Ea = 0.28 eV, as previously calculated8 in an equally large supercell, but using DFT+U instead of PBEsol. Since we find the same value with PBEsol, we conclude that in such low concentrations of H− and  , in spite of the Hubbard correction, doping electrons always tend to delocalize or are readily displaced. Such consistency among different calculation settings strongly indicates that we are accurately modeling the actual states of doping electrons in BaTiO2.93H0.037□0.037, namely, band states. Another important observation is that the hydridic character is preserved at the transition state, as indicated by the Bader charge analysis showing an excess charge of ∼0.55 electrons, and that only the electrons forming H− are displaced during the migration, with the rest of the doping electrons residing in the CB.
, in spite of the Hubbard correction, doping electrons always tend to delocalize or are readily displaced. Such consistency among different calculation settings strongly indicates that we are accurately modeling the actual states of doping electrons in BaTiO2.93H0.037□0.037, namely, band states. Another important observation is that the hydridic character is preserved at the transition state, as indicated by the Bader charge analysis showing an excess charge of ∼0.55 electrons, and that only the electrons forming H− are displaced during the migration, with the rest of the doping electrons residing in the CB.
By combining the results from the four different degrees of localization, we observe that when a doping electron forming  or
or  must migrate against H−, it entails an additional cost in energy. This cost in energy depends on the degree of localization of the doping electron. When the electron is delocalized as a band state forming
must migrate against H−, it entails an additional cost in energy. This cost in energy depends on the degree of localization of the doping electron. When the electron is delocalized as a band state forming  , which is favored by a lower concentration of H− and
, which is favored by a lower concentration of H− and  , Ea is reduced. Conversely, when the doping electron remains localized during migration, as favored by the presence of an electron polaron, Ea increases. Interestingly, even in the presence of a localized doping electron, we find that Ea can still be reduced if the electron migrates in the same direction as H−, as enabled by the periodic arrangement of
, Ea is reduced. Conversely, when the doping electron remains localized during migration, as favored by the presence of an electron polaron, Ea increases. Interestingly, even in the presence of a localized doping electron, we find that Ea can still be reduced if the electron migrates in the same direction as H−, as enabled by the periodic arrangement of  within the channel geometry.
within the channel geometry.
To further assess the role of electron localization in H− migration, we systematically depleted the doping electrons in our calculations and monitored the resulting changes in Ea. For both the PBEsol and PBE+U functionals, a consistent decrease in Ea was observed with increasing electron depletion. When all localized doping electrons were removed, Ea reached a minimum of approximately 0.4 eV, closely approaching the value of Ea = 0.28 eV obtained for case (4). These results confirm that the presence of localized electrons significantly hinders H− migration (see details in the SI).
To verify that these results are not accidental nor dependent on the choice of the XC functional, we also carried out calculations using the hybrid functional HSE06.38 The HSE06 calculations yielded one localized electron at both the initial and transition states, similar to case (1), and a comparable migration barrier of Ea = 0.89 eV (see details in the SI). The agreement in both activation energies and localization behavior confirms that this correspondence reflects the similar impact of both approaches on the system. This suggests that U parametrization offers a robust and transferable description of localization effects, providing a reliable framework for this material. This understanding provides an explanation for variations in Ea derived in previous DFT studies on H− migration in BaTiO3−x−yHx□y. On one hand, Ea was found to have a value of 0.28 eV,8 which approximates our case (4) of full electron delocalization. On the other hand, an Ea of 1 eV was derived4 close to our case (1) of high localization of the doping electrons. These varying results should be due to differences in the computational settings. While ref. 8 employed a 3 × 3 × 3 supercell, which is comparable to case (4), effectively modeling delocalized doping electrons, ref. 4 used a 2 × 2 × 2 supercell with a DFT+U functional, similar to case (1), thereby modeling localized doping electrons.
The present study also clarifies a fundamental distinction between proton and hydride migration by examining the influence of vibrational zero-point energy (ZPE) correction on Ea (see the SI for details on the calculation of the ZPE correction). For proton migration, ZPE corrections typically reduce Ea, as observed in various systems.39–41 In contrast, we find that ZPE corrections increase Ea by approximately 0.01 eV in BaTiO2.75H0.125□0.125. A comparison of the vibrational density of states between the initial/final states and the transition state reveals that one of the three H− normal modes becomes imaginary at the transition state, while the other two exhibit an upward energy shift, resulting in the observed positive ZPE correction. The stiffening of these vibrational modes is interpreted as evidence of enhanced binding of H− to the host lattice at the transition state and indicates that the H− does not migrate on a single, adiabatic potential energy surface (PES) as a proton would,42 but instead follows a non-adiabatic path involving breaking and forming bonds governed by transitions between intersecting PESs associated with the initial and final states. This finding suggests that the migration barrier is predominantly electronic in origin, governed by non-adiabatic coupling between the initial and final state PES, in contrast to proton migration, where the barrier is primarily determined by ion–ion interactions.
It is also interesting to compare our calculated Ea with the values obtained for localized H− diffusion from quasielastic neutron scattering (QENS) experiments. QENS studies on BaTiO2.3H0.04 reported an activation energy of Ea = 0.094 ± 0.002 eV,7 which aligns most closely with the value obtained for case (4) of full delocalization of the doping electrons. This consistency aligns with a previous combined DFT and INS study on BaTiO2.82H0.1□0.08, which demonstrated that the doping electrons form a band state.15 Similarly, QENS studies on the oxyhydrides SrVO2H and LaSrCoO3H0.7 – where H− ions are incorporated into a perovskite lattice, occupying sites that share the corners of oxygen octahedra – have shown that migration proceeds via the same nearest-neighbor hopping mechanism observed in BaTiO2.3H0.04. These studies reported activation energies of Ea = 0.20 ± 0.035 eV43 and Ea = 0.23 ± 0.045 eV,44 respectively. These values are in closest agreement with the activation energy obtained for case (4), corresponding to full delocalization of the doping electrons. Interestingly, these materials are Mott insulators,45–48 characterized by high electron localization on the V and Co ions, respectively. Our data thereby indicate that the doping electrons introduced in SrVO2H and LaSrCoO3H0.7 by H− and  do not form localized states within the VO but are instead localized near V and Co ions, or form band states. These observations suggest that H− migration is a local feature, independent of whether the oxyhydride material as a whole is a Mott insulator or a conductor, as evidenced by the fact that no electrons, except those forming the H− ion, are displaced during the migration in case (4) of full delocalization.
do not form localized states within the VO but are instead localized near V and Co ions, or form band states. These observations suggest that H− migration is a local feature, independent of whether the oxyhydride material as a whole is a Mott insulator or a conductor, as evidenced by the fact that no electrons, except those forming the H− ion, are displaced during the migration in case (4) of full delocalization.
Finally, we compare our results to other classes of mixed electronic–ionic conductors, such as transition metal oxide cathode materials, where the interplay between electron polarons and ionic conductivity has been extensively studied.18–20,49–52 In these materials, the ionic charge carrier is a lithium cation (Li+), which interacts attractively with negatively charged electron polarons, leading to different outcomes compared to our findings. For example, in LiV3O8, LiV2O5, and LiCoO2, it has been observed that the presence of electron polarons lowers Ea for Li+ diffusion, and their simultaneous migration with Li+ may be the driving force for Li+ diffusion.18,19,49 Notably, Ea in these materials is generally around 0.3 eV, which is comparable to our determined Ea = 0.29 eV for case (4) – full delocalization of doping electrons. This suggests that our results cannot be generalized to all mixed electronic–ionic conductors and that the diffusing ionic species drastically affects the effect of electron polarons.
4 Conclusions
To conclude, we have shown that the degree of electron localization has a considerable impact on the activation energy Ea for the inter-site migration of a H− in BaTiO3−2xHx□x. Specifically, we find that Ea exhibits a systematic decrease with less localization of the doping electrons. Since the degree of electron localization is an intrinsic material property, this may open up avenues for the rational design of oxyhydrides with tailored H− conductivity, e.g., by reducing the bandgap via cation substitutions to the Ti site53,54 to promote the delocalization of doping electrons into the conduction band. Additionally, we have highlighted the application of vibrational zero-point energy corrections as a conceptual tool to distinguish between proton and H− transport mechanisms in oxyhydride materials.Conflicts of interest
There are no conflicts of interest to declare.Data availability
Data from this study are openly available from the Swedish National Data Service (SND) at https://doi.org/10.71870/x76p-bp52.Supplementary information (SI): additional band structure, PDOS, and real-space electron density plots. See DOI: https://doi.org/10.1039/d5ma00521c.
Acknowledgements
M. K. acknowledges the support from the Swedish Research Council (Grant No. 2021-04807 and 2016-06958) and the Swedish Energy Agency (Grant No. 48712-1). M. K. and M. M. K. acknowledge the support from the ILL (Grant No. ILL-1880.1). The computational resources were provided by the Swedish National Infrastructure for Computing (SNIC) in the C3SE center, Gothenburg, and by the ILL.References
- Y. Kobayashi, O. J. Hernandez, T. Sakaguchi, T. Yajima, T. Roisnel, Y. Tsujimoto, M. Morita, Y. Noda, Y. Mogami and A. Kitada, et al., An oxyhydride of BaTiO3 exhibiting hydride exchange and electronic conductivity, Nat. Mater., 2012, 11, 507–511 CrossRef CAS PubMed.
- Y. Tang, Y. Kobayashi, N. Masuda, Y. Uchida, H. Okamoto, T. Kageyama, S. Hosokawa, F. Loyer, K. Mitsuhara, K. Yamanaka, Y. Tamenori, C. Tassel, T. Yamamoto, T. Tanaka and H. Kageyama, Metal-Dependent Support Effects of Oxyhydride-Supported Ru, Fe, Co Catalysts for Ammonia Synthesis, Adv. Energy Mater., 2018, 8, 1801772 CrossRef.
- Y. Kobayashi, Y. Tang, T. Kageyama, H. Yamashita, N. Masuda, S. Hosokawa and H. Kageyama, Titanium-Based Hydrides as Heterogeneous Catalysts for Ammonia Synthesis, J. Am. Chem. Soc., 2017, 139, 18240–18246 CrossRef CAS PubMed.
- Y. Tang, Y. Kobayashi, K. Shitara, A. Konishi, A. Kuwabara, T. Nakashima, C. Tassel, T. Yamamoto and H. Kageyama, On Hydride Diffusion in Transition Metal Perovskite Oxyhydrides Investigated via Deuterium Exchange, Chem. Mater., 2017, 29, 8187–8194 CrossRef CAS.
- T. Sakaguchi, et al., Oxyhydrides of (Ca,Sr,Ba)TiO3 Perovskite Solid Solutions, Inorg. Chem., 2012, 51, 11371–11376 CrossRef CAS PubMed.
- Y. Goto, M. Kikugawa, K. Kobayashi, Y. Manaka, T. Nanba, H. Matsumoto, M. Matsumoto, M. Aoki and H. Imagawa, Facile Formation of Barium Titanium Oxyhydride on a Titanium Hydride Surface as an Ammonia Synthesis Catalyst, RSC Adv., 2023, 13, 15410–15415 RSC.
- C. Eklöf-Österberg, R. Nedumkandathil, U. Häussermann, A. Jaworski, A. J. Pell, M. Tyagi, N. H. Jalarvo, B. Frick, A. Faraone and M. Karlsson, Dynamics of Hydride Ions in Metal Hydride-Reduced BaTiO3 Samples Investigated with Quasielastic Neutron Scattering, J. Phys. Chem. C, 2019, 123, 2019–2030 CrossRef.
- X. Liu, T. S. Bjørheim and R. Haugsrud, Formation and migration of hydride ions in BaTiO3−xHx oxyhydride, J. Mater. Chem. A, 2017, 5, 1050–1056 RSC.
- H. Guo, A. Jaworski, Z. Ma, A. Slabon, Z. Bacsik, R. Nedumkandathil and U. Häussermann, Trapping of different stages of BaTiO3 reduction with LiH, RSC Adv., 2020, 10, 35356–35365 RSC.
- R. Nedumkandathil, A. Jaworski, J. Grins, D. Bernin, M. Karlsson, C. Eklöf-Österberg, A. Neagu, C.-W. Tai, A. J. Pell and U. Häussermann, Hydride Reduction of BaTiO3 – Oxyhydride Versus O Vacancy Formation, ACS Omega, 2018, 3, 11426–11438 CrossRef CAS PubMed.
- G. Bouilly, T. Yajima, T. Terashima, W. Yoshimune, K. Nakano, C. Tassel, Y. Kususe, K. Fujita, K. Tanaka, T. Yamamoto, Y. Kobayashi and H. Kageyama, Electrical Properties of Epitaxial Thin Films of Oxyhydrides ATiO3−xHx (A = Ba and Sr), Chem. Mater., 2015, 27, 6354–6359 CrossRef CAS.
- M. Schrader, D. Mienert, T.-S. Oh, H.-I. Yoo and K. D. Becker, An optical, EPR and electrical conductivity study of blue barium titanate, BaTiO3−δ, Solid State Sci., 2008, 10, 768–775 CrossRef CAS.
- R. Scharfschwerdt, A. Mazur, O. F. Schirmer, H. Hesse and S. Mendricks, Oxygen vacancies in BaTiO3, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 15284–15290 CrossRef CAS PubMed.
- D. D. Cuong and J. Lee, Electronic structure of oxygen deficient BaTiO3, Integr. Ferroelectr., 2006, 84, 23–30 CrossRef CAS.
- E. J. Granhed, A. Lindman, C. Eklöf-Österberg, M. Karlsson, S. F. Parker and G. Wahnström, Band vs. polaron: vibrational motion and chemical expansion of hydride ions as signatures for the electronic character in oxyhydride barium titanate, J. Mater. Chem. A, 2019, 7, 16211–16221 RSC.
- R. Aleksis, R. Nedumkandathil, W. Papawassiliou, J. P. Carvalho, A. Jaworski, U. Häussermann and A. J. Pell, Probing the Electronic Structure and Hydride Occupancy in Barium Titanium Oxyhydride through DFT-assisted Solid-State NMR, Phys. Chem. Chem. Phys., 2022, 24, 28164–28173 RSC.
- T. Yajima, A. Kitada, Y. Kobayashi, T. Sakaguchi, G. Bouilly, S. Kasahara, T. Terashima, M. Takano and H. Kageyama, Epitaxial thin films of ATiO3−xHx (A = Ba, Sr, Ca) with metallic conductivity, J. Am. Chem. Soc., 2012, 134, 8782–8785 CrossRef CAS PubMed.
- M. Chotsawat, L. Ngamwongwan, P. Komen, P. Falun, S. Jungthawan, A. Junkaew and S. Suthirakun, Insight into the Effect of Oxygen Vacancies on Ion Intercalation and Polaron Conduction in LiV3O8 Cathodes of Li-Ion Batteries, J. Phys. Chem. C, 2022, 126, 18216–18228 CrossRef CAS.
- S. Suthirakun, A. Genest and N. Rösch, Modeling Polaron-Coupled Li Cation Diffusion in V2O5 Cathode Material, J. Phys. Chem. C, 2018, 122, 150–157 CrossRef CAS.
- T. Maxisch, F. Zhou and G. Ceder, Ab initio study of the migration of small polarons in olivine LixFePO4 and their association with lithium ions and vacancies, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 104301 CrossRef.
- J. Zhang, G. Gou and B. Pan, Study of Phase Stability and Hydride Diffusion Mechanism of BaTiO3 Oxyhydride from First-Principles, J. Phys. Chem. C, 2014, 118, 17254–17259 CrossRef CAS.
- Y. Iwazaki, T. Suzuki and S. Tsuneyuki, Negatively charged hydrogen at oxygen-vacancy sites in BaTiO3: Density-functional calculation, J. Appl. Phys., 2010, 108, 083705 CrossRef.
- W. Lyu, Y. Liu, D. Chen, F. Wang and Y. Li, Engineering the electron localization of metal sites on nanosheets assembled periodic macropores for CO2 photoreduction, Nat. Commun., 2024, 15, 10589 CrossRef CAS PubMed.
- Y. Zhou, et al., Dopant-induced electron localization drives CO2 reduction to C2 hydrocarbons, Nat. Chem., 2018, 10, 974–980 CrossRef CAS PubMed.
- S. Yan, C. Peng, C. Yang, Y. Chen, J. Zhang, A. Guan, X. Lv, H. Wang, Z. Wang, T.-K. Sham, Q. Han and G. Zheng, Electron Localization and Lattice Strain Induced by Surface Lithium Doping Enable Ampere-Level Electrosynthesis of Formate from CO2, Angew. Chem., 2021, 133, 25945–25949 CrossRef.
- G. Kresse and J. Hafner, Ab initio molecular dynamics for liquid metals, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, From ultrasoft pseudopotentials to the projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, A Climbing Image Nudged Elastic Band Method for Finding Saddle Points and Minimum Energy Paths, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- J. P. Perdew, A. Ruzsinszky, G. I. Csonka, O. A. Vydrov, G. E. Scuseria, L. A. Constantin, X. Zhou and K. Burke, Erratum: Restoring the Density-Gradient Expansion for Exchange in Solids and Surfaces [Phys. Rev. Lett. 100, 136406 (2008)], Phys. Rev. Lett., 2009, 102, 039902 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- V. I. Anisimov, J. Zaanen and O. K. Andersen, Band Theory and Mott Insulators: Hubbard U Instead of Stoner I, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 44, 943–954 CrossRef CAS PubMed.
- H. J. Kulik, Perspective: Treating Electron over-Delocalization with the DFT+U Method, J. Chem. Phys., 2015, 142, 240901 CrossRef PubMed.
- J. P. Perdew, R. G. Parr, M. Levy and J. L. Balduz, Density-Functional Theory for Fractional Particle Number: Derivative Discontinuities of the Energy, Phys. Rev. Lett., 1982, 49, 1691–1694 CrossRef CAS.
- G. Henkelman, A. Arnaldsson and H. Jónsson, A Fast and Robust Algorithm for Bader Decomposition of Charge Density, Comput. Mater. Sci., 2006, 36, 354–360 CrossRef.
- C. Eklöf-Österberg, L. Mazzei, E. J. Granhed, G. Wahnström, R. Nedumkandathil, U. Häussermann, A. Jaworski, A. J. Pell, S. F. Parker, N. H. Jalarvo, B. Lars and M. Karlsson, The role of oxygen vacancies on the vibrational motions of hydride ions in the oxyhydride of barium titanate, J. Mater. Chem. A, 2020, 8, 6360–6371 RSC.
- A. V. Krukau, O. A. Vydrov, A. F. Izmaylov and G. E. Scuseria, Influence of the Exchange Screening Parameter on the Performance of Screened Hybrid Functionals, J. Chem. Phys., 2006, 125, 224106 CrossRef PubMed.
- P. G. Sundell, M. E. Björketun and G. Wahnström, Density-functional calculations of prefactors and activation energies for H diffusion in BaZrO3, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 094301 CrossRef.
- M. G. Wardle, J. P. Goss and P. R. Briddon, First-Principles Study of the Diffusion of Hydrogen in ZnO, Phys. Rev. Lett., 2006, 96, 205504 CrossRef CAS PubMed.
- J. A. Ryder, A. K. Chakraborty and A. T. Bell, Density Functional Theory Study of Proton Mobility in Zeolites: Proton Migration and Hydrogen Exchange in ZSM-5, J. Phys. Chem. B, 2000, 104, 6998–7011 CrossRef CAS.
- M. Björketun, P. Sundell, G. Wahnström and D. Engberg, A Kinetic Monte Carlo Study of Proton Diffusion in Disordered Perovskite Structured Lattices Based on First-Principles Calculations, Solid State Ion, 2005, 176, 3035–3040 CrossRef.
- R. Lavén, U. Häussermann, A. Perrichon, M. S. Andersson, M. S. Targama, F. Demmel and M. Karlsson, Diffusional Dynamics of Hydride Ions in the Layered Oxyhydride SrVO2H, Chem. Mater., 2021, 33, 2967–2975 CrossRef PubMed.
- C. A. Bridges, F. Fernandez-Alonso, J. P. Goff and M. J. Rosseinsky, Observation of Hydride Mobility in the Transition-Metal Oxide Hydride LaSrCoO3H0.7, Adv. Mater., 2006, 18, 3304–3308 CrossRef CAS.
- T. Katayama, A. Chikamatsu, K. Yamada, K. Shigematsu, T. Onozuka, M. Minohara, H. Kumigashira, E. Ikenaga and T. Hasegawa, Epitaxial growth and electronic structure of oxyhydride SrVO2H thin films, J. Appl. Phys., 2016, 120, 085305 CrossRef.
- T. Yamamoto, D. Zeng, T. Kawakami, V. Arcisauskaite, K. Yata, M. A. Patino, N. Izumo, J. E. McGrady, H. Kageyama and M. A. Hayward, The role of π-blocking hydride ligands in a pressure-induced insulator-to-metal phase transition in SrVO2H, Nat. Commun., 2017, 8, 1217 CrossRef PubMed.
- C. A. Bridges, G. R. Darling, M. A. Hayward and M. J. Rosseinsky, Electronic Structure, Magnetic Ordering, and Formation Pathway of the Transition Metal Oxide Hydride LaSrCoO3H0.7, J. Am. Chem. Soc., 2005, 127, 5996–6011 CrossRef CAS PubMed.
- M. A. Hayward, E. J. Cussen, J. B. Claridge, M. Bieringer, M. J. Rosseinsky, C. J. Kiely, S. J. Blundell, I. M. Marshall and F. L. Pratt, The Hydride Anion in an Extended Transition Metal Oxide Array: LaSrCoO3H0.7, Science, 2002, 295, 1882–1884 CrossRef CAS.
- S. Ahn, J. Kim, B. Kim and S. Kim, First-principles study on small polaron and Li diffusion in layered LiCoO2, Phys. Chem. Chem. Phys., 2023, 25, 27848–27853 RSC.
- K. M. Bui, V. A. Dinh and T. Ohno, Diffusion Mechanism of Polaron–Li Vacancy Complex in Cathode Material Li2FeSiO4, Appl. Phys. Express, 2012, 5, 125802 CrossRef.
- F. Amano, M. Nakata, A. Yamamoto and T. Tanaka, Effect of Ti3+ Ions and Conduction Band Electrons on Photocatalytic and Photoelectrochemical Activity of Rutile Titania for Water Oxidation, J. Phys. Chem. C, 2016, 120, 6467–6474 CrossRef CAS.
- V. A. Dinh, J. Nara and T. Ohno, A New Insight into the Polaron–Li Complex Diffusion in Cathode Material LiFe1−yMnyPO4 for Li Ion Batteries, Appl. Phys. Express, 2012, 5, 045801 CrossRef.
- S. Mukherjee, D. Phuyal, C. U. Segre, S. Das, O. Karis, T. Edvinsson and H. Rensmo, Structure and Electronic Effects from Mn and Nb Co-doping for Low Band Gap BaTiO3 Ferroelectrics, J. Phys. Chem. C, 2021, 125, 14910–14923 CrossRef CAS.
- R. B. Comes, P. V. Sushko, S. M. Heald, R. J. Colby, M. E. Bowden and S. A. Chambers, Band-Gap Reduction and Dopant Interaction in Epitaxial La,Cr Co-doped SrTiO3 Thin Films, Chem. Mater., 2014, 26, 7073–7082 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |