
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Simple size tuning of magnetic nanoparticles using a microwave solvothermal method and their application in facilitating the solid-phase synthesis of molecularly imprinted polymers†
Andrei N.
Stephen
a,
Tim
Mercer
 b,
William
Stockburn
a,
Sarah R.
Dennison
a,
Jennifer E.
Readman
a and
Subrayal M.
Reddy
*a
b,
William
Stockburn
a,
Sarah R.
Dennison
a,
Jennifer E.
Readman
a and
Subrayal M.
Reddy
*a
aDepartment of Chemistry, Institute of Materials and Investigative Sciences, UCLan Centre for Smart Materials, School of Pharmacy and Biomedical Sciences, University of Central Lancashire, Preston, PR1 2HE, UK. E-mail: smreddy@uclan.ac.uk
bMagnetic Materials Research Group, Jeremiah Horrocks Institute for Mathematics, Physics & Astronomy, University of Central Lancashire, Preston, PR1 2HE, UK
First published on 11th February 2025
Abstract
Herein, we have demonstrated a simple, economical, rapid and scalable microwave method to produce magnetite-based magnetic nanoparticles (MNPs) with desired sizes and their application in the facile synthesis of high-value polymer products. Solvothermal method is gaining traction in microwave synthesis as it offers a rapid and green method for MNP production. In this work, we report a novel, simple and reliable microwave synthesis method, where adjusting the temperature gradient from 20 °C to a dwell temperature of 200 °C enabled the size control of superparamagnetic aldehyde-functionalised nanoparticles (MNP@CHO). The size distribution of nanoparticles was measured using dynamic light scattering, which revealed values of 14 nm ± 8 nm at 90 °C min−1 (a 2-minute ramp time to dwell temperature) and 122 nm ± 49 nm at 18 °C min−1 (a 10-minute ramp time to dwell temperature), and these nanoparticles were produced within 20–30 minutes. Magnetic size analysis using the Chantrell method confirmed that the iron–oxide core size increased as a function of ramp time, with the median diameter in the range of 7.91 to 11.25 nm and lognormal σ values within 0.22 ≤ σ ≤ 0.33. The particle cluster size increased with an increase in the ramp time, which was measured using transmission electron microscopy, and it was found to be a function of particle agglomeration. Furthermore, we demonstrated that MNP@CHO functionalised with a protein of interest can be applied for the rational solid-phase synthesis of molecularly imprinted polymer nanoparticles (nanoMIPs) with high affinity for protein biomarkers. Thus, we demonstrated that an optimal MNP size is required for the highly efficient production of MNP-based nanoMIPs, which is the key to the mass production and commercialisation of low-cost and sustainable size-tuned MNPs and artificial antibodies.
1. Introduction
Magnetite (Fe3O4)-based magnetic nanoparticles (MNPs), also referred to as iron oxide nanoparticles (IONPs), are receiving a lot of attention in research and commercial applications.1–3 Fe3O4 is preferred over other nanomaterials owing to the relatively low toxicity of magnetite4–6 as well as the easy availability and low cost of its reaction precursors.6,7 Furthermore, the superparamagnetic property7,8 of superparamagnetic iron oxide nanoparticles (SPIONs) has enabled their use in a wide range of applications.For superparamagnetism (SPM) to occur, the size of magnetite particles should typically be smaller than about 20 nm;8–10 however, the SPM onset size is affected by several factors, including the shape effects on anisotropy and particle size distribution in any SPION assemblies, where it may be observed at sizes up to ∼50 nm.11,12 Unlike the ferrimagnetic behaviour of bulk materials, at these small sizes, the particles demonstrate superparamagnetic properties with no net magnetisation in a zero applied field.8 In this state, magnetic moments of the particles are randomly aligned by the agitation of thermal energy at room temperature and hence show no magnetic interaction with each other (similar to paramagnets). In contrast, in a magnetic field, superparamagnetic nanoparticles exhibit significantly increased magnetization due to the ready alignment of their magnetic moments with an applied field. Their ability to be easily moved and manipulated by an external magnetic field resulting from their superparamagnetic properties makes SPIONS useful in a range of applications in the biomedical field, including targeted anti-cancer drug delivery,13 as MRI contrast agents,14–16 for biological extraction/purification when functionalised with suitable receptors,17,18 for cancer treatment under magnetic hyperthermia conditions,19–22 and, more recently, in the molecular imprinting field.23
A range of approaches has been explored using low-cost reagents. The main methods are focused on producing Fe3O4 nanoparticle clusters via coprecipitation,24–27 solvothermal,28–31 and hydrothermal32,33 reactions. Generally, traditional co-precipitation methods are rapid but require the use of inert gases such as argon and nitrogen to prevent the creation of other, less useful iron oxides, and thus maintain the correct iron oxidation states.34–37 They also require an additional step to neutralize the resultant solution, requiring strong bases such as urea and sodium hydroxide, which increases the cost of the process. Furthermore, to achieve an adequate level of size control, additional equipment, such as magnetic arrays38 and ultrasonicators,39 are necessary. These requirements make scaling up the reaction very challenging.
Hydrothermal methods involve the reaction of iron precursors in sealed specialized vessels,40,41 which are autoclaved under high-temperature and high-pressure aqueous conditions over the lengthy course of 6–20 h,42 typically with the aid of stabilizing agents or surfactants. The hydrothermal environment promotes the nucleation and growth of iron oxide nanocrystals, leading to highly uniform and monodisperse particles, as shown by Mizutani et al.43 Among these methods, solvothermal reactions offer the best monodispersity, typically utilizing diethylene glycol (DEG) and ethylene glycol (EG) as the reducing solvent, sodium citrate tribasic as the ligand, and a basic salt such as sodium acetate (NaOAc). This method typically takes 8 h for the synthesis of MNPs and their subsequent functionalization, and a further 24 h to purify the resulting MNPs.31 The conventional heating provides a large temperature gradient, leading to variable nucleation rates, but the obtained particles can be produced with narrow size distributions, albeit over a much longer timescale compared with the microwave method.
The microwave synthesis of magnetic nanoparticles offers a simplified production process with reduced costs compared to traditional methods, while also presenting significant environmental advantages, making it a more sustainable and greener alternative.44–46 Traditional co-precipitation methods often necessitate an inert atmosphere such as argon or nitrogen to prevent oxidation during the synthesis, which increases the energy demand and environmental impact.47–49 Conversely, microwave synthesis can be performed under ambient conditions, eliminating the need for inert gases, and thereby reducing the overall carbon footprint of the process. Furthermore, co-precipitation and hydrothermal methods typically require extended heating durations, often lasting several hours, to synthesize magnetic nanoparticles, leading to considerable energy consumption.50 Alternatively, microwave synthesis is inherently more energy efficient due to its rapid and localized heating mechanism, which enables the formation of nanoparticles within an hour or less, drastically minimizing the energy input. Also, the one-pot nature of microwave synthesis reduces the need for additional reagents or multi-step processing due to the fact that the magnetic nanoparticles are formed with a coating through this process,46 further reducing the generation of waste. Moreover, unlike the co precipitation and hydrothermal methods, which rely on the application of a coating after the synthesis,51 adding extra cost and complexity, or the addition of additives to provide size control, microwave synthesis enables precise size control through simple adjustments in the ramping parameters, generating less chemical waste. Overall, the microwave synthesis of magnetic nanoparticles represents a greener and more sustainable alternative to the conventional techniques. By reducing energy consumption, avoiding the use of inert gases, and minimizing waste, this approach aligns with modern environmental sustainability goals, while efficiently delivering high-quality nanoparticles.
Microwave heating offers more controlled and homogeneous heating throughout the medium, resulting in the reproducible syntheses of colloidal materials. The microwave-based one-pot solvothermal synthesis of bare and functionalized superparamagnetic Fe3O4 MNPs in the <20 nm category is gaining traction given that it offers a low energy and rapid (<30 min) route to the product.46 Although small MNPs (<15 nm) can be useful, they are prone to drag fluctuations due to Brownian motion even under the influence of a magnetic field.52 Recently, the synthesis of larger MNP clusters composed of smaller superparamagnetic nanoparticles has been reported.53–55 Employing these methods, it is possible to obtain larger particles (25 nm to approximately 1 μm), and this increase in size scale offers advantageous applications compared with the smaller regime. An increase in MNP volume to surface area (i.e. production of a lower concentration of larger agglomerated particles) enables the chemical functionalization (conjugation) of more than one molecule or biomolecule to each MNP, allowing an increase in the capture of more than one complementary molecule per MNP from a sample of interest, while still retaining the superparamagnetic properties of the MNPs.56
Methods to predictably control the size of MNPs within a batch-type synthesis, while not altering their other properties, remain highly desirable. As the size of nanoparticles increases, they become less superparamagnetic, but their magnetic saturation is enhanced.57 Magnetic saturation is one of the most important properties when considering applications based on magnetic nanoparticles.58,59 A high magnetic saturation leads to a strong response at a low magnetic field, which can, for example, facilitate the rapid collection of analytes when they are used for biological extraction and biosensing. Moreover, for imaging, a strong response results in much more sharply defined images.60,61 Therefore, the use of a method that can tune the size of MNPs over a specific range can facilitate an increase in saturation magnetisation, while still maintaining their superparamagnetic properties.
There have been reports on tuning the size of nanoparticle clusters by adjusting the ratio of DEG/EG62,63 and adjusting the concentration of citrate.62 However, these methods do not offer fine control of the final particle size without affecting the monodispersity or other parameters, such as composition and yield, and offer only a limited tuning range. The use of polyol solvents in microwave-assisted techniques offers several advantages beyond their reducing capabilities. In the polyol method, diethylene glycol (DEG) and ethylene glycol (EG) function not only as solvents and reducing agents but also as surfactants, and are chosen for their relative high dielectric constants, which enable efficient microwave absorption and heating.64 Mascolo et al.65 demonstrated a size tuning in magnetite clusters through simple stoichiometric (chemical) control of the reaction solution basicity in the presence of a cationic surfactant and at room temperature. An excess concentration of OH− led to the stabilisation of smaller particles (<10 nm). The aggregate particle size (range of 40 to 100 nm) could be increased by decreasing the hydroxide concentration. Other researchers66–68 used microfluidics and flow chemistry to control the rate at which the reaction solution transits a microwave reactor to control the size of synthesised iron oxide nanoparticles and associated clusters. However, this method required significant engineering to control the size and volume of the employed micro/milli-fluidic reactor, minimise laminar flow and the need for scaling up the synthesis at high speed. Alternatively, the microwave synthesis method is inherently scalable and well-suited to industrial applications, given the availability of industrial-scale microwave reactors. Also, unlike the conventional co-precipitation or hydrothermal methods, the microwave method requires no additional specialized equipment, thereby eliminating the need for complex fabrication and testing processes.
Herein, we focused on tuning the physical conditions and parameters used in microwave synthesis as a means to control the final MNP nanoparticle size. We report an approach to control and tune the size of aldehyde-functionalised iron oxide magnetic nanoparticles and their clustering by simply changing the microwave temperature gradient during MNP synthesis. We investigated the sizing using dynamic light scattering, transmission electron microscopy and magnetometry. The produced magnetic materials possessed a hydrodynamic diameter in the range of 36 nm to 122 nm, which was measured using dynamic light scattering. Also, we propose a mechanism where with a change in the temperature ramp time, there is an accompanying change in the rate of decomposition of an iron acetate intermediate in the reaction as the route to tune the MNP entity size. We also propose that oligomerisation and integration of glutaraldehyde during the MNP growth phase contribute to the formation of uniform MNP cluster sizes. The proposed method not only tunes the particle size but also facilitates uniformity and surface functionalization in a single step.
Recently, aldehyde-functionalised MNPs have been applied in the synthesis of artificial antibody receptors, namely nanoscale molecularly imprinted polymers (NanoMIPs).23,69 MIPs are produced in a facile self-assembly and polymerisation process in the presence of a target template molecule. When the template is removed, polymeric materials with high affinity for the target are produced. Suitably functionalised MNPs have been used as the nucleation site for the production of nanoMIPs. MNPs have also been modified with esoteric chemistry via the silanisation of their surface70,71 or use of borane chemistry72,73 and subsequent bioconjugation with a template molecule to enable nanoMIP synthesis at the MNP surface. However, although these methodologies have resulted in the production of high-affinity nanoMIPs, they are laborious, time-consuming (up to 3 days) and require large amounts of reagent for production, ultimately resulting in low (mg) yields. Recently, we published a solid-phase synthesis method using microwaves to produce aldehyde MNPs as the core for protein (template) attachment and subsequent production of nanoMIPs.23 Herein, we present the application of size-tuned nanoMIPs and demonstrate that the MNP size is critical to optimising the yield of high-affinity nanoMIPs.
2. Experimental
2.1 Materials
N-(Hydroxymethyl)acrylamide (NHMA, 48% w/v), N,N′-methylenebisacrylamide (MBAm; 99% pure), ethylene glycol ((CH2OH)2; 99% pure), iron chloride (FeCl3·6H2O; 96% pure), methylhydroquinone (MHQ; 99% pure), sodium acetate (NaOAc; ≥99% pure), phosphate buffered saline tablets (PBS, 10 mM, pH 7.4 ± 0.2), potassium ferricyanide (K3Fe(CN)6; 99% pure), potassium chloride (KCl; 99% pure), sodium nitrate (NaNO3; ≥99% pure), ammonium persulphate (APS; 98% pure), N,N,N′,N′-tetramethylethylenediamine (TEMED; 99% pure), potassium peroxydisulfate (KPS; ≥99% pure (RT)), haemoglobin from bovine blood (BHb), bovine serum albumin (BSA), sodium dodecyl sulphate (SDS; ≥98.5% pure) and glutaraldehyde (25% v/v)) were used as received from Merck. Buffers were prepared in MilliQ water (resistivity 18.2 ± 0.2 MΩ cm). DropSens disposable screen-printed electrodes (Au-BT) comprised of a gold working electrode (0.4 cm diameter), a platinum counter electrode and silver reference electrode were purchased from Metrohm (Runcorn, Cheshire, UK).2.2 Instrumentation
A BioDrop μLITE UV/visible spectrometer was purchased from Biochrom Ltd Cambridge, UK. A Nicolet AVATAR 330 FTIR spectrophotometer with Pike MIRacle accessory and FEI Tecnai 12 TEM at 100 kV with a Tietz F214 2k × 2k CCD camera were purchased from Thermo Fisher Scientific, Loughborough, UK. An Anton Paar Monowave 200 microwave oven was purchased from Anton Paar Ltd Hertfordshire, UK. An SLS Lab Basics centrifuge was purchased from Scientific Laboratory Supplies, Nottingham, UK. All electrochemical experiments were performed using a Metrohm Autolab PGSTAT204 potentiostat and NOVA2.1.4 software. Magnetisation curves were obtained using a 6 kOe Vibrating Sample Magnetometer (VSM) built in-house at UCLan.2.3 MNP production using microwave synthesis
Bare and aldehyde-functionalised magnetic particles were produced following our previously published solvothermal microwave method.20 Briefly, 0.5 g of FeCl3·6H2O and 1.8 g of NaOAc were dissolved in 15 mL of ethylene glycol in a 30 mL Anton Parr G30 microwave reaction vial (MRV). Then, glutaraldehyde (3.5 mL) was added to the resulting solution with stirring for a further 5 min. Subsequently, the stirrer bar was removed and the MRV was placed into an Anton Paar Monowave 200 microwave oven and the reaction was heated up to a dwell temperature of 200 °C. We investigated various ramp times to the dwell temperature from a slow ramp time (10 min; 18 °C min−1) to a fast ramp time (2 min; 90 °C min−1). The reaction was held at the dwell temperature for 20 min under pressure (9 bar). An aliquot (10 mL) of the MNP suspension was oven dried (110 °C for 2 days) for use in the TEM analysis. The MNP production method was repeated, but in the absence of glutaraldehyde to obtain bare MNPs.2.4 X-ray diffraction analysis
X-ray powder diffraction data were collected using a Bruker D2 Phaser diffractometer in the θ–θ geometry, using Cu Kα radiation (λ = 1.5418 Å) and operating at 30 keV and 30 mA. A nickel filter was used to remove the Kβ radiation and the detector was a LynxEye. Data were collected in the 2θ range of 5–80°, with a step size of 0.020194° and a total scan time of 1 h per sample. The energy discrimination of the detector was modified to suppress the fluorescence from the iron-containing samples. The sample holder was rotated at 30 rpm to maximise the powder averaging. The crystallite size analysis was performed using the Bruker EVA software. The peak width of the peak at the 2θ value of approximately 35.5° was measured at FWHM and used in the Scherrer calculation.2.5 DLS characterization of MNPs
The size distribution of the nanoparticles was characterized using a Zetasizer Nano ZS. The produced MNPs/nanoMIPs/NanoNIPs were suspended in 1 mL of PBS. The sample was loaded into a disposable cuvette with the refractive index set to 1.32. The solution was equilibrated for 60 s before the measurement was performed. Measurements were performed in triplicate.2.6 Magnetic measurements
Magnetic measurements on dried powder samples were carried out at room temperature using a 6 kOe vibrating sample magnetometer (VSM). Given that large agglomerates were formed after drying, a pestle and mortar were required to break them up for packing into cuboid glass slides (Camlab) of a given internal thickness and width of (0.40 ± 0.04) mm and (4.0 ± 0.4) mm, respectively. The slides were cut at ∼10 mm in length within the range of (9.75 ≥ length ≥ 11.60) mm, resulting in errors in the order 10−2 mm from a minimum of 5 measurements along the length at different points across the width. Based on these dimensions, the magnetometric demagnetisation factor, Nd, was found to be low and in the range of (0.037 ≥ Nd ≥ 0.044).742.7 Transmission electron microscopy of MNPs
The aldehyde-functionalized MNPs were suspended in ultra-pure water (0.1 g in 50 μL water) and a 5 μL droplet was deposited onto a Formvar/carbon-coated 200 mesh copper TEM grid (Agar Scientific, UK). After 1 min the grid was blotted, washed for 30 s in ultra-pure water, blotted again, and allowed to dry. Images were collected using a FEI Tecnai 12 TEM at 100 kV with a Tietz F214 2k × 2k CCD camera.2.8 Protein functionalization of MNPs
A suspension (1 mL) equivalent to 0.010 g of the produced aldehyde (–CHO)-functionalised magnetic nanoparticles (MNP@CHO; 10 mg mL−1) was placed in an Eppendorf centrifuge tube. A neodymium magnet was placed on the side of the tube to rapidly pull the magnetic nanoparticles from the solution (10 minutes). The supernatant was removed and replaced with 1 mL of a 1 mg mL−1 PBS solution of bovine haemoglobin (BHb). The Eppendorf tube was then sonicated for 2 min, followed by vigorous shaking and vortexing to ensure that the nanoparticles were fully dispersed. The reaction mixture was left undisturbed at room temperature (22 °C) for 30 min, allowing the protein to conjugate with MNP@CHO. Conjugation occurs due to the free –NH2 groups in the protein undergoing a nucleophilic addition–elimination reaction with –CHO on MNP, resulting in an imine bond between the protein and MNP. After 30 min, the particles were once again separated from the solution and the supernatant exchanged with fresh buffer in triplicate to remove any non-conjugated protein. The amount of protein conjugated with the MNPs (functionalized and bare) was calculated by comparing the initial and final concentrations of protein remaining in the supernatant. The concentration of the non-adsorbed protein was measured by spectrophotometry (405 nm for haemoglobin) using a BioDrop μLITE UV/visible spectrometer. The resulting MNP@CHO@BHb particles were stored wet at 4 °C until further use.2.9 NanoMIP production using MNPs
The MNP@CHO@BHb magnetic nanoparticles (0.023 g) were resuspended in 906 μL of PBS (pH 7.4) and transferred to a 15 mL Falcon tube. The contents in the tube were then mixed at 400 rpm at room temperature. The sample was then degassed using nitrogen for 15 min with stirring. After, the nitrogen line was removed and 37 mg of NHMA monomer (77 μL of 48% v/v solution) and MBAm (6 mg) together with SDS (0.4 mg) were immediately added to the reaction mixture, followed by 20 μL of a solution containing 10% (v/v) TEMED and 5% (w/v) APS. Then, a nitrogen headspace was created, and the Falcon tube sealed with a cap, and then wrapped in parafilm. The solution was left to mix at 400 rpm for 15 min to allow nanoMIP particles to be produced at the surface of the MNP@CHO@BHb particles.At 15 min, the reaction was rapidly quenched with 1 mL of 10 mM methylhydroquinone (MHQ). The reaction solution was exchanged three times with fresh PBS to remove any unreacted monomers and quencher. Then, the solution was then resealed, and the tube placed on its side on a neodymium magnet (2 min). After, the supernatant was removed. The MNP@CHO@BHb∼nanoMIP particles were then dispersed in 600 μL of e-pure water and placed in a sonicator (using a VWR ultrasonicator (600 W, 45 kHz) for 5 min at 37 °C. The Falcon tube was then once again placed on a neodymium magnet and the supernatant now containing the released nanoMIPs was placed in a 1.5 mL volume Eppendorf and stored at 4 °C until further use. The preparation was repeated by using either bare MNP and MNP@CHO instead of MNP@CHO@BHb to produce non-imprinted control polymer (nanoNIP).
2.10 Electrochemical deposition and analysis of nanoMIP
NanoMIPs were eluted using sonication, and then entrapped within an electropolymerized layer (E-layer). E-Layers were fabricated directly onto BT-Au screen-printed electrodes (SPEs; Metrohm) using cyclic voltammetry (CV) according to the procedure in ref. 75. Briefly, a 50 μL solution in PBS comprising 0.1 mg of nanoMIP, 641 mM of NHMA as the functional monomer, 41.5 mM MBAm as the cross-linker, 0.29 M NaNO3, and 48.15 mM KPS was deposited onto the SPE. Then, the potential was cycled between −0.2 V and −1.4 V for 7 cycles at 50 mV s−1 (10 min, RT, 22 °C ± 2 °C) to produce the E-layer with entrapped nanoMIP. E-Layers in the absence of nanoMIP were also produced as a control.The E-layer comprised of entrapped nanoMIP islands (E-NMI) or control E-layer (in the absence of nanoMIP) was exposed to varying concentrations of target protein (haemoglobin) template solutions over a wide concentration range (1 fM to 100 μM) for a period of 5 min at each concentration and analysed using electrochemical impedance spectroscopy (EIS) post-rebinding and subsequent rinsing to determine the degree of target rebound to the nanoMIP islands.
Selective protein binding was tracked using electrochemical impedance spectroscopy (EIS) of an external 5 mM potassium ferricyanide solution in PBS containing 0.5 M KCl as the supporting electrolyte. Electrochemical impedance spectroscopy (EIS) measurements were conducted at a standard potential of 0.1 V (±0.01 V) with 10 scans, and a sinusoidal potential peak-to-peak with amplitude 0.01 V in the frequency range of 0.1–100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Hz. A Randles equivalent circuit was fitted for all EIS experiments using the FRA32 module (see Fig. S1, ESI†).
000 Hz. A Randles equivalent circuit was fitted for all EIS experiments using the FRA32 module (see Fig. S1, ESI†).
3. Results and discussion
3.1 Characterisation of MNPs produced using the microwave technique
We previously reported46 a microwave synthesis method for the rapid production of magnetic nanoparticles, where the temperature gradient from 20 °C to 200 °C was fixed at 90 °C min−1 (representing a 2 min ramp time), resulting in MNPs with an average size of 7 ± 2 nm, which was measured using transmission electron microscopy.Herein, we varied the time taken to reach the dwell temperature (200 °C). We investigated ramp times of 2, 4, 6, 8, 10 and 15 min, corresponding to temperature gradients of 90 °C min−1, 45 °C min−1, 30 °C min−1, 22.5 °C min−1, 18 °C min−1 and 12 °C min−1, respectively. This resulted in the production of aldehyde-functionalised magnetic nanoparticles (MNP@CHO). The size of the particles in the dispersion was in the range of 14 nm to 120 nm, which was measured using dynamic light scattering spectroscopy, as summarised in Fig. 1 (see Fig. S1(a)–(e), ESI†). The particles produced at a ramp time of 15 min had the consistency of an oily slurry and could not be easily dispersed in aqueous solution. The DLS analysis indicated that the average particle size was in the range of 1–2 μm. Additionally, these particles produced at a ramp time of 15 min were no longer susceptible to an external magnetic field using a neodymium magnet.
 | ||
| Fig. 1 Effect of microwave temperature ramp time from room temperature to dwell temperature (200 °C) on the size of final MNP@CHO nanoparticles. Hydrodynamic diameter of particles measured using dynamic light spectroscopy. (Data represents mean ± S.E.M., n = 3). | ||
We propose that the difference in particle size is related to the rate at which the reactants are consumed as a function of ramp time (Fig. 2).
 | ||
| Fig. 2 Schematic of the reaction mixture during microwave synthesis, demonstrating the different states present depending on the reaction temperature transition. The time lapse in any temperature range depending on ramp rate will impact the nature and predominance of the species present. | ||
Ethylene glycol is the primary solvent, but can act as a mild reducing agent, resulting in the production of Fe2+ ions en route to producing Fe3O4 according to the following equations:76
| 2HOCH2 − CH2OH → 2CH3CHO + 2H2O | (1) |
| 2CH3CHO + 2Fe3+ → CH3CO − COCH3 + 2Fe2+ + 2H+ | (2) |
| 2Fe2+ + 4OH− ⇌ 2Fe(OH)2 | (3) |
| 4Fe(OH)3 + 2Fe(OH)2 → 2Fe3O4 + 8H2O | (4) |
| Fe(CH3COO)3 + 3H2O → Fe(OH)3 + 3CH3COOH | (5) |
| Fe(OH)3 → Fe2O3 + Fe3O4 | (6) |
| 12Fe(CH3COO)3 → 4 Fe3O4 + 18 CH3COCH3 + 18 CO2 + O2 |
We propose that the time taken to reach the microwave dwell temperature of 200 °C influences the composition of the reaction mixture, and importantly that the level of acetate present influences the final particle and aggregate sizes. Acetate acts as a weak buffer to produce hydroxide ions in situ, supporting the production of [Fe(OH)3] and resulting in iron oxide precipitation and subsequent aggregation. Therefore, by altering the ramp time, we controlled the degree of FeOAc conversion to Fe(OH)3 in the early stages of MNP production, consequently controlling the size of the initial particles produced. At a fast (2 min) temperature ramp (i.e. 90 °C min−1) to the dwell temperature, less iron hydroxide was produced during the ramping period. At a slow (10 min) temperature ramp (i.e. 18 °C min−1) to the dwell temperature, there was more time for iron acetate to be converted to iron hydroxide during the ramping period, resulting in more maghemite and magnetite production during the ramping phase. Slowing down the time at which acetate decomposition takes place led to further precipitation and aggregation, and the controlled production of larger magnetic nanoparticles.
Fig. 3 shows the FTIR spectrum obtained for MNP@CHO. The absorption band at 520 cm−1 is attributed to the octahedral Fe–O vibrational stretching of the iron–oxygen bond The slight non-symmetry of this peak suggests that most of the iron present is in the form of magnetite, with only a small amount of maghemite.79 The peak at 1724 cm−1 corresponds to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration of the carbonyl bond. The peak at 2820 cm−1 is associated with the asymmetric stretching of the C–H bonds. These peaks indicate the presence of a magnetic core surrounded by aldehyde groups, as synthesised via the described one-pot microwave method.
O stretching vibration of the carbonyl bond. The peak at 2820 cm−1 is associated with the asymmetric stretching of the C–H bonds. These peaks indicate the presence of a magnetic core surrounded by aldehyde groups, as synthesised via the described one-pot microwave method.
 | ||
| Fig. 3 Infrared spectrum of MNP@CHO produced at 10 min ramp time, followed by 20 min dwell time. The particles were oven dried at 110 °C for 2 days prior to the measurement at room temperature. | ||
Our IR analysis (Fig. 3) did not indicate a covalent link between Fe and aldehyde,46 but we cannot rule it out. Further, due to the ability of glutaraldehyde to polymerise when aged or heated,80,81 we believe that we achieved the coating of growing superparamagnetic iron oxide crystal structures with glutaraldehyde oligomers, which still retained aldehyde groups. We believe that the glutaraldehyde polymer chains became entrapped as the nanoparticle was forming, allowing the glutaraldehyde groups to cover the MNP in a core–shell fashion. Although we do not fully understand the mechanism of agglomeration (clustering), we propose that it is associated with the glutaraldehyde oligomerising (growing in chain length) and partly acting as a binding agent (glue) between individual growing particles. Our assertion is consistent with the work by others who have shown that structures and assemblies of single cores can be stabilised into clusters of multi-core magnetic systems in the presence of hydrophilic and polymeric molecules82,83 such as heparin and carbohydrates such as dextran.
The X-ray diffraction patterns of the samples are shown in Fig. S2 (ESI†). All the samples contain predominately Fe3O4 (space group Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m, a = 8.400 Å) with α-Fe2O3 (space group RcH, a = 5.0324 Å and c = 13.7643 Å), both appearing as broad peaks in the diffraction patterns. Sharp peaks attributed to NaCl (marked with *) are also present. The broad peak widths observed for the MNP mean together with the close proximity of the expected peak positions of Fe3O4 and α-Fe2O3 resulted in some uncertainty in the exact ratios of Fe3O4 and α-Fe2O3. For example, the most intense MNP peak in the diffraction patterns was observed at the 2θ value of 35.5° and the (1 3 1) peak of Fe3O4 is located at 35.4°, while the (1 −2 0) peak of α-Fe2O3 appears at 35.7°. Any variation in the amount of α-Fe2O3 will cause asymmetry in the peaks and uncertainty in the Scherrer calculation.
m, a = 8.400 Å) with α-Fe2O3 (space group RcH, a = 5.0324 Å and c = 13.7643 Å), both appearing as broad peaks in the diffraction patterns. Sharp peaks attributed to NaCl (marked with *) are also present. The broad peak widths observed for the MNP mean together with the close proximity of the expected peak positions of Fe3O4 and α-Fe2O3 resulted in some uncertainty in the exact ratios of Fe3O4 and α-Fe2O3. For example, the most intense MNP peak in the diffraction patterns was observed at the 2θ value of 35.5° and the (1 3 1) peak of Fe3O4 is located at 35.4°, while the (1 −2 0) peak of α-Fe2O3 appears at 35.7°. Any variation in the amount of α-Fe2O3 will cause asymmetry in the peaks and uncertainty in the Scherrer calculation.
The particles after oven drying were imaged using transmission electron microscopy. Fig. 4(a)–(d) show the TEM images of the MNP@CHO particles produced at ramp times of 2, 6, 8 and 10 min, respectively. Increasing the ramp time between 2 min (Fig. 4a) and 10 min (Fig. 4d) resulted in a corresponding increase in the MNP@CHO core particle size between 7 ± 2 nm and 12.6 ± 3.2 nm, respectively, and cluster size of 91 nm ± 15 nm at 10 min (Fig. 4e). We could not identify any clustering at the ramp time of 2 min.
 | ||
| Fig. 4 TEM images of magnetic nanoparticles produced at ramp time of (a) 2 min (90 °C min−1), (b) 6 min (30 °C min−1), (c) 8 min (22.5 °C min−1) and (d) 10 min (12 °C min−1). (e) 10 min particles clustering at lower magnification. The cluster size of the particles increased with an increase in the ramp time. The average particle size in (d) for individual magnetic nanoparticles was calculated to be 12.7 nm ± 3.7 nm (data represents mean ± S.E.M., n = 100). | ||
The TEM sizing of the MNP@CHO formulations is on average smaller than the corresponding DLS sizes. The DLS sizing is conducted in aquo, and therefore represents the hydrodynamic diameter, whereas the TEM measurements are conducted in vacuo and in a dried state. This has also been observed by Dingchen-Wen et al.84 in their study on the chemical synthesis of MNPs.
3.2 Magnetic measurements and sizing of MNP@CHO
The magnetisation curves as a function of applied field are shown in Fig. 5 from the series of samples with microwave ramp times in the range of 2 to 10 min. Only the first quadrants of the full M–H loops are shown for clarity, with the near closed curves of the loops having negligible coercivity and remanence, indicating the superparamagnetic state. | ||
| Fig. 5 Magnetisation curves of samples with increasing microwave ramp time. The increasing mass saturation magnetisation is consistent with the increasing particle size as expected for magnetite particles on the nanoscale. The full loops are near-closed and therefore have very small coercivity and remanence, as shown for the sample with the highest values in the inset. | ||
It is well known (e.g. Ref. 85) that the saturation magnetisation, Ms, for magnetite decreases from the bulk value of 92 emu g−1 when in a multi-domain ferrimagnetic state to lower values as a function of decreasing particle size when in the single-domain superparamagnetic state of size-order tens of nm. It is widely accepted that there are effectively ‘magnetically dead’ layers at, or near, the particle surface,86 leaving only the core that is magnetically responsive, and thereby diluting the magnetic content within the volume (or mass) of the particle and subsequent reduction in Ms values. In the bare particle case, this is assigned to surface oxidation and/or crystallographic disorder. Further dilution occurs when the nanoparticles are coated with surfactants, lipids and other functional agents, such as aldehyde, in the magnetic measurements. Given that the surface effects become more dominant with a decrease in particle size, and subsequent increase in surface area, the reduction in Ms observed here is also consistent with the decrease in particle size because of the decreasing ramp time.
The results in Fig. 5 were further investigated using the magnetic sizing method reported by Chantrell.87 Briefly, the median particle diameter, Dm, and standard deviation, σ, of a lognormal distribution of particle sizes are calculated using the following equations:
 | (7) |
 | (8) |
 is the particle volume fraction, k is the Boltzmann constant and T is the absolute temperature. The Langevin function provides a good theoretical description of superparamagnetic curves and is used in the Chantrell method to derive eqn (7) and (8). At a large field, H, it is reduced to a linear expression such that the plot of M as a function of 1/H will result in a linear fit that crosses the abscissa when M = zero at the point 1/H0. Subsequently, experimental measurements of χi, 1/H0 and
is the particle volume fraction, k is the Boltzmann constant and T is the absolute temperature. The Langevin function provides a good theoretical description of superparamagnetic curves and is used in the Chantrell method to derive eqn (7) and (8). At a large field, H, it is reduced to a linear expression such that the plot of M as a function of 1/H will result in a linear fit that crosses the abscissa when M = zero at the point 1/H0. Subsequently, experimental measurements of χi, 1/H0 and  may be used to determine Dm and σ using eqn (7) and (8).
may be used to determine Dm and σ using eqn (7) and (8).
The outcome is shown in Fig. 6, which shows a clear trend of an overall increase in particle size with an increase in ramp time, as found in the in the DLS and TEM results. This confirms that the increase in saturation magnetisation is a result of the increase in particle size due to the increase in ramp times, as can be seen in the inset of Fig. 6, where the Ms values are those extrapolated from the data from Fig. 5 using M verses 1/H at high applied fields, to the crossing point of the ordinate i.e. when the applied field is tending to infinity.
 | ||
| Fig. 6 Median particle diameter and lognormal σ values as a function of ramp time following the Chantrell method.87 The decrease in particle size with a decrease in ramp time confirms that this is the cause of the drop in saturation magnetisation in the inset obtained by extrapolation of the data from Fig. 5. There is no overall trend in the σ values that indicate the 4-minute sample has the widest range of particle sizes in its distribution and the 10-minute sample has the narrowest. | ||
There is no obvious trend in the values of σ shown on the right-hand axis of Fig. 6. The largest value of 0.33 is associated with the 4-min sample and suggests that it has the widest particle size distribution range. Careful observation of the magnetisation curve of the same sample in Fig. 5 also shows this is further away from saturation than the other samples, with a steeper gradient on the approach to 6 kOe. The assumption inherent in the sizing method is of a lognormal distribution and any deviation from this along with its largest σ value may explain, in part at least, the noticeable difference in the magnetisation curve towards the maximum applied field.
Table 1 compares the median particle size determined using magnetic measurements with the agglomerate size results determined using TEM and DLS at selected ramp times. The magnetic core size measurement and calculations refer to the size of individual magnetic cores i.e. single particle core size, not agglomerates. The recorded TEM images suggest that we achieved clustering or agglomeration with an increase in ramp time. It was difficult to discern individual particles at all ramp times using TEM but where we could, for example, at a ramp time of 10 min (Fig. 4d), the average individual particle size determined by TEM (12.6 nm ± 3.2 nm) is in good agreement with the magnetic core size determination of 11.25 nm. Given that the TEM measurements include all the particles, including the magnetically dead outer layers, they are expected to be larger than that of the magnetic measurements. DLS gives the hydrodynamic diameter of particles in an aqueous suspension. We believe that the DLS size is the summation of the MNP magnetic core size plus a glutaraldehyde shell layer plus some agglomeration of the MNPs. Therefore, although all the measurement methods used show a correlation with the ramp time, the size increases in the order of Mag core < TEM < DLS. The crystallite size was determined based on the XRD measurements using the Scherrer calculation using the 2 and 10 min MNP@CHO particles, giving crystallite sizes of 7.7 nm and 9.3 nm, respectively. Although the 2 min particles are in good agreement with the magnetic and TEM sizing, there is some significant deviation in the 10 min crystallite size calculation.
| Ramp time (min) | Measurement techniques | |||
|---|---|---|---|---|
| Magnetic core size | TEM size clusters (nm) | DLS size (nm) | ||
| D median (nm) | σ | |||
| 2 | 7.91 | 0.27 | 8.5 ± 2 | 14.9 ± 8 |
| 6 | 9.77 | 0.32 | 23 ± 6 | 60 ± 7 |
| 10 | 11.25 | 0.22 | 91 ± 15 | 122 ± 49 |
Our results demonstrate a correlation (across measurement techniques) between an increase in particle/agglomerate size and increase in ramp time.
3.3 Impact of MNP size on solid-phase synthesis of molecularly imprinted polymers
Recently, there has been growing interest in the synthesis of polymers with biorecognition capability and their application in diagnostics, biological extraction and therapeutics. Molecularly imprinted polymers (MIPs) are a class of artificial receptors. They can be synthetically grown around a biological target,46,75,88,89 imparting complementary recognition sites within the crosslinked polymer. Recently, we reported that MNPs modified with a protein can be used as a solid substrate to facilitate the manufacture of nanoscale MIPs.23 Subsequently, we showed that the nanoMIPs could be harvested and the MNP@protein could be recycled and re-used to scale up the yield of nanoMIPs. Here, we show that the MNP size is critical to the effective functioning of the material for the solid-phase synthesis of nanoMIPs (see Fig. 7 for a schematic of the process).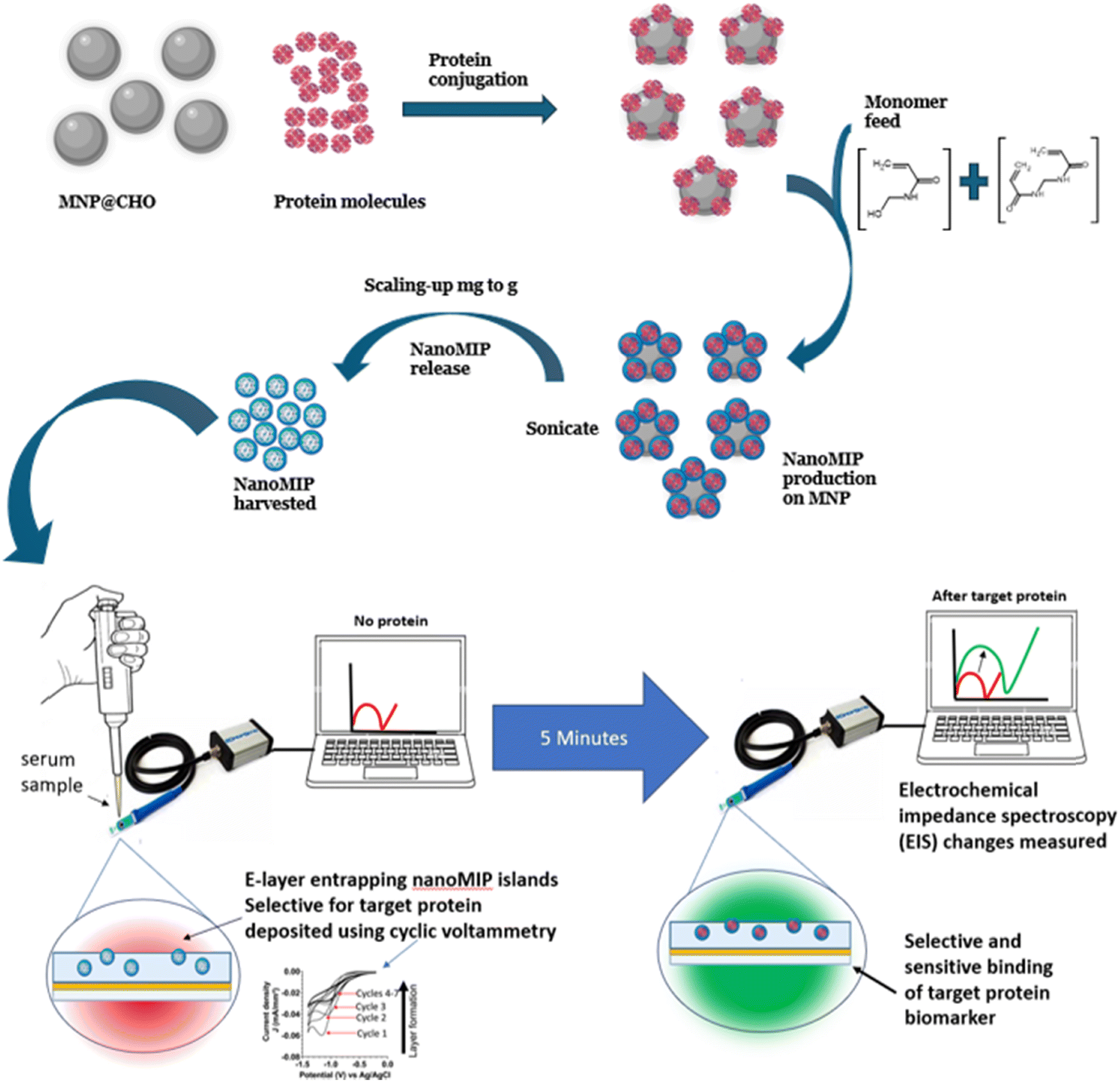 | ||
| Fig. 7 Schematic of nanoMIP polymer synthesis on MNP solid phase. MNP@CHO is first conjugated with the target protein to give MNP@protein. In the presence of a monomer and crosslinker feed, the nanoMIPs grow specifically around MNP@protein. Once released and harvested, the nanoMIP is integrated into a disposable screen-printed electrode for biosensor determination of protein biomarker. | ||
We used mass equivalents of the as-produced MNP@CHO particles at 2–10 min ramp times for the synthesis of nanoMIPs. MNP@CHO was first conjugated with bovine haemoglobin (BHb) as the target (template). The resulting MNP@CHO@BHb particles were used as the solid phase to produce nanoMIPs selective for BHb. Subsequently, the nanoMIPs produced were released from the MNP and size characterised using DLS. The isolated nanoMIPs were integrated into a disposable screen-printed gold electrode for electrochemical determination of protein and non-target protein rebinding from test solutions. Electrochemical impedance spectroscopy (EIS) was used to interrogate and quantify protein binding. EIS is a suitable sensitive technique to measure nanomolar to picomolar levels of target binding to a synthetic receptor.90 It relies on interrogating the electrochemical properties of the nanoMIP/electrode interface in the presence of a suitable redox marker (ferrocyanide was used here) at a standard potential of 0.1 V (±0.01 V) at multiple frequencies, and a sinusoidal potential peak-to-peak with an amplitude of 0.01 V in the frequency range of 0.1–100000 Hz. The interface is modelled on the Randles circuit. We measured the change in charge transfer resistance (ΔRCT) when the electrode was modified with nanoMIP, which was a function of resistance of ferrocyanide redox marker diffusion to the working electrode.23 When the target protein was added, it selectively bound to the nanoMIP at the nanoMIP/electrode interface, creating an additional barrier to ferrocyanide redox marker diffusion. There was a corresponding increase in ΔRCT with an increase in the target protein binding. Fig. S1–S5 (ESI†) compare the plots of [BHb] versus ΔRCT for the nanoMIPs synthesised on BHb functionalised MNP@CHO magnetic nanoparticles produced at ramp times of 2 min (Fig. S3, ESI†), 4 min (Fig. S4, ESI†), 6 min (Fig. S5, ESI†), 8 min (Fig. S6, ESI†) and 10 min (Fig. S7, ESI†). Table 2 summarises the impact of MNP size (measured using DLS) on the subsequent nanoMIP synthesis parameters including nanoMIP particle size, yield and affinity factors such as the equilibrium dissociation constant (KD) and the relative response of the biosensor to target protein (BHb) and non-target protein (bovine serum albumin; BSA). The equilibrium dissociation constant KD for each nanoMIP batch was determined using the Hill–Langmuir method using data from Fig. S2–S6 (ESI†).
| Microwave ramp time (min) | DLS size of MNP@CHO (nm) | DLS size of nanoMIP (nm) | Yield of nanoMIP (mg mL−1) | K D (mol L−1) | Selectivity factor (target:non-target signal ratio @1 nM) |
|---|---|---|---|---|---|
| 2 | 14 ± 8 | 80 ± 14 | 0.13 ± 0.06 | 1.40 × 10−10 ± 2.79 × 10−12 | 49:1 |
| 4 | 46 ± 12 | 123 ± 41 | 1.6 ± 0.3 | 2.01 × 10−11 ± 5.05 × 10−12 | 75:1 |
| 6 | 60 ± 7 | 119 ± 51 | 3.7 ± 0.3 | 1.75 × 10−11 ± 2.61 × 10−12 | 166:1 |
| 8 | 84 ± 11 | 120 ± 57 | 6.5 ± 0.3 | 2.40 × 10−11 ± 9.21 × 10−12 | 100:1 |
| 10 | 122 ± 49 | 125 ± 43 | 12.3 ± 2.5 | 3.47 × 10−11 ± 2.35 × 10−12 | 188:1 |
Although a low KD in the range of 10−9 to 10−11 mol L−1 gives an indication of the tendency of the nanoMIP to tightly bind with the target with affinities akin to a monoclonal antibody, the selectivity factor is an effective measure of how more effective the MIP is at picking out its target protein (complement) compared with a non-target (non-complementary) protein. We demonstrated a direct correlation between the MNP@CHO size (and subsequently MNP@protein size) with nanoMIP yield. Although all the particles resulted in the production of nanoMIPs with high affinity and nanoMIP selectivity, the nanoMIP yield increased with an increase in ramp time, with the 10 min ramp time returning the best yield of nanoMIPs. The least effective nanoMIPs were produced using the 2-min ramp time particles. Interestingly, the DLS size of nanoMIP is approximately 120 nm and independent of the ramp time between 4 and 10 min. We did not study the 15-min ramp time particles given that their clumping sludge-like characteristics did not make them ideal candidates for the manufacture of nanoMIPs.
We have demonstrated a simple, economical, rapid and scalable microwave method to produce magnetite-based magnetic nanoparticles (MNPs) at the desired size and their application in the facile synthesis of high-value polymer products such as nanoMIPs. Our size-tuned MNPs have many potential applications in biological extraction (when conjugated with antibodies or nanoMIPs), which we are currently investigating, as well as applications in medical imaging and therapeutics.
4. Conclusions
Aldehyde-functionalised magnetic nanoparticles (MNP@CHO) with a tuneable size could be produced within 20–30 min. The initial temperature ramp used prior to the 20 min dwell time for the MNP synthesis was crucial in influencing both the MNP particle and clustering size, as determined using transmission electron microscopy. We presented a mechanism based on the rate of acetate decomposition during the MNP particle and cluster formation. Altering the ramp time between 2 and 10 min resulted in the corresponding increase in MNP@CHO particle size between 7 nm and 91 nm, as measured using TEM, and cluster (stable agglomerate) sizes of between 36 nm and 122 nm, which were measured using DLS.We also demonstrated their application in the development of nanoscale molecularly imprinted polymer (NanoMIP) receptor-based electrochemical sensors. We demonstrated that there is an optimal MNP size for highly efficient MNP-based nanoMIP production, which is the key to the mass production and commercialisation of low-cost and sustainable bespoke size-tuned MNPs and antibody replacement technologies.
Author contributions
SMR conceived, designed and directed the study and wrote the manuscript. ANS and WJS prepared the MNPs. ANS performed nanoMIP synthesis, DLS and TEM characterization and electrochemical studies. JER performed XRD. TM performed magnetic measurements and data analysis. SMR, ANS, JER and TM performed the analysis. All authors contributed to manuscript revision, read, and approved the submitted version.Data availability
All data are available within the article and its ESI,† and from the authors upon request.Conflicts of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.Acknowledgements
The authors are grateful to the University of Central Lancashire, the Royal Society of Chemistry COVID-19 Action fund (H20-188), the Daiwa Anglo-Japanese Foundation (13094/13916) and The Royal Society (IES\R3\193093) for funding this work. We wish to thank Dr Jennifer Simpson (The Pirbright Institute) for the providing the service in obtaining transmission electron microscope images of the MNPs.References
- H. M. Williams, Biosci. Horiz., 2017, 10, 1–10 CrossRef CAS.
- D. A. Alromi, S. Y. Madani and A. Seifalian, Polymers, 2021, 13, 4146 CrossRef CAS PubMed.
- A. Akbarzadeh, M. Samiei and S. Davaran, Nanoscale Res. Lett., 2012, 7, 144 Search PubMed.
- H. Markides, M. Rotherham and A. J. El Haj, J. Nanomater., 2012, 2012, 614094 CrossRef.
- N. Malhotra, J. S. Lee, R. A. D. Liman, J. M. S. Ruallo, O. B. Villaflores, T. R. Ger and C. D. Hsiao, Molecules, 2020, 25, 3159 CrossRef CAS PubMed.
- Y. Chen and S. Hou, Cell Death Discovery, 2023, 9, 195 CrossRef CAS PubMed.
- C. Das, N. N. Ghosh, V. Pulhani, G. Biswas and P. Singhal, RSC Adv., 2023, 13, 15015–15023 RSC.
- J. Dulińska-Litewka, A. Łazarczyk, P. Hałubiec, O. Szafrański, K. Karnas and A. Karewicz, Materials, 2019, 12, 617 CrossRef PubMed.
- C. T. Yavuz, J. T. Mayo, W. W. Yu, A. Prakash, J. C. Falkner, S. Yean, L. Cong, H. J. Shipley, A. Kan, M. Tomson, D. Natelson and V. L. Colvin, Science, 2006, 314, 964–967 CrossRef PubMed.
- Y. W. Jun, Y. M. Huh, J. S. Choi, J. H. Lee, H. T. Song, S. Kim, S. Yoon, K. S. Kim, J. S. Shin, J. S. Suh and J. Cheon, J. Am. Chem. Soc., 2005, 127, 5732–5733 CrossRef CAS PubMed.
- N. Lamichhane, M. E. Sharifabad, B. Hodgson, T. Mercer and T. Sen, in Nanoparticle Therapeutics, ed. P. Kesharwani and K. K. Singh, Academic Press, 2022, pp. 455–497 DOI:10.1016/B978-0-12-820757-4.00003-X.
- D. J. Dunlop, J. Geophys. Res., 1973,(78), 1780–1793 CrossRef.
- M. Z. Iqbal, G. I. Dar, I. Ali and A. Wu, in Nanomedicine in Brain Diseases: Principles and Application, ed. X. Xue, Springer Singapore, Singapore, 2019, pp. 269–313 DOI:10.1007/978-981-13-8731-9_10.
- Z. R. Stephen, F. M. Kievit and M. Zhang, Mater. Today, 2011, 14, 330–338 Search PubMed.
- T. Tegafaw, S. Liu, M. Y. Ahmad, A. Saidi, D. Zhao, Y. Liu, S. W. Nam, Y. Chang and G. H. Lee, Pharmaceutics, 2023, 15, 1745 CrossRef PubMed.
- Y. Patil-Sen, E. Torino, F. De Sarno, A. M. Ponsiglione, V. Chhabria, W. Ahmed and T. Mercer, Nanotechnology, 2020, 31, 375102 CrossRef CAS PubMed.
- M. Wierucka and M. Biziuk, TrAC, Trends Anal. Chem., 2014, 59, 50–58 Search PubMed.
- M. Khajeh and A. Khajeh, Int. J. Green Nanotechnol., 2009, 1, P51–P56 CrossRef.
- A. J. Giustini, A. A. Petryk, S. M. Cassim, J. A. Tate, I. Baker and P. J. Hoopes, Nano LIFE, 2010, 1, 17–32 CrossRef CAS PubMed.
- X. Liu, Y. Zhang, Y. Wang, W. Zhu, G. Li, X. Ma, Y. Zhang, S. Chen, S. Tiwari, K. Shi, S. Zhang, H. M. Fan, Y. X. Zhao and X. J. Liang, Theranostics, 2020, 10, 3793–3815 CrossRef CAS PubMed.
- L. Lartigue, P. Hugounenq, D. Alloyeau, S. P. Clarke, M. Lévy, J.-C. Bacri, R. Bazzi, D. F. Brougham, C. Wilhelm and F. Gazeau, ACS Nano, 2012, 6, 10935–10949 CrossRef CAS PubMed.
- S. Dutz, M. Kettering, I. Hilger, R. Müller and M. Zeisberger, Nanotechnology, 2011, 22, 265102 CrossRef PubMed.
- S. M. Reddy, A. N. Stephen, M. A. Holden, W. J. Stockburn and S. R. Dennison, Biomater. Sci., 2024, 12, 5845–5855 Search PubMed.
- D. Hu, Y. Wang and Q. Song, Particuology, 2009, 7, 363–367 Search PubMed.
- Y. Mizukoshi, T. Shuto, N. Masahashi and S. Tanabe, Ultrason. Sonochem., 2009, 16, 525–531 Search PubMed.
- I. Nedkov, T. Merodiiska, L. Slavov, R. E. Vandenberghe, Y. Kusano and J. Takada, J. Magn. Magn. Mater., 2006, 300, 358–367 CrossRef CAS.
- H. Pardoe, W. Chua-anusorn, T. G. St. Pierre and J. Dobson, J. Magn. Magn. Mater., 2001, 225, 41–46 Search PubMed.
- S. Fakurpur Shirejini, S. M. Dehnavi and M. Jahanfar, Chem. Eng. Res. Des., 2023, 190, 580–589 Search PubMed.
- J. Liu, L. Wang, J. Wang and L. Zhang, Mater. Res. Bull., 2013, 48, 416–421 CrossRef CAS.
- Y. Chen, J.-G. Zhang, Z. Wang and Z. Zhou, Appl. Sci., 2019, 9, 5157 CrossRef CAS.
- Z. Huang, K. Wu, Q.-H. Yu, Y.-Y. Wang, J. Xing and T.-L. Xia, Chem. Phys. Lett., 2016, 664, 219–225 CrossRef CAS.
- X. Yang, W. Jiang, L. Liu, B. Chen, S. Wu, D. Sun and F. Li, J. Magn. Magn. Mater., 2012, 324, 2249–2257 Search PubMed.
- M. Tadic, M. Panjan, V. Damnjanovic and I. Milosevic, Appl. Surf. Sci., 2014, 320, 183–187 CrossRef CAS.
- N. A. Yazid and Y. C. Joon, AIP Conf.: Proc., 2019, 2124, 1–10 Search PubMed.
- H. Köçkar and O. Karaagac, J. Mater. Sci.: Mater. Electron., 2021, 32, 13673–13684 Search PubMed.
- O. u Rahman, S. C. Mohapatra and S. Ahmad, Mater. Chem. Phys., 2012, 132, 196–202 CrossRef CAS.
- K. Petcharoen and A. Sirivat, Mater. Sci. Eng., B, 2012, 177, 421–427 Search PubMed.
- D. Lokhat, S. Brijlal, D. E. Naidoo, C. Premraj and E. Kadwa, Ind. Eng. Chem. Res., 2022, 16980–16991 Search PubMed.
- M. Jafari Eskandari and I. Hasanzadeh, Mater. Sci. Eng., B, 2021, 266, 115050 CrossRef CAS.
- M. Zahid, N. Nadeem, M. A. Hanif, I. A. Bhatti, H. N. Bhatti and G. Mustafa, in Magnetic Nanostructures: Environmental and Agricultural Applications, ed. K. A. Abd-Elsalam, M. A. Mohamed and R. Prasad, Springer International Publishing, Cham, 2019, pp. 181–212 DOI:10.1007/978-3-030-16439-3_10.
- G. F. Stiufiuc and R. I. Stiufiuc, Appl. Sci., 2024, 14, 1623 Search PubMed.
- A. Ali, H. Zafar, M. Zia, I. Ul Haq, A. R. Phull, J. S. Ali and A. Hussain, Nanotechnol., Sci. Appl., 2016, 9, 49–67 Search PubMed.
- N. Mizutani, T. Iwasaki, S. Watano, T. Yanagida and T. Kawai, Curr. Appl. Phys., 2010, 10, 801–806 Search PubMed.
- E. Aivazoglou, E. Metaxa and E. Hristoforou, AIP Adv., 2017, 8, 1–14 Search PubMed.
- L. M. Kustov, E. M. Kostyukhin, E. Y. Korneeva and A. L. Kustov, Russ. Chem. Bull., 2023, 72, 583–601 CrossRef CAS.
- M. Sullivan, W. Stockburn, P. Hawes, T. Mercer and S. Reddy, Nanotechnology, 2020, 32, 095502 CrossRef PubMed.
- Z. Klencsár, A. Ábrahám, L. Szabó, E. G. Szabó, S. Stichleutner, E. Kuzmann, Z. Homonnay and G. Tolnai, Mater. Chem. Phys., 2019, 223, 122–132 CrossRef.
- L. M. Cursaru, R. M. Piticescu, D. V. Dragut, R. Morel, C. Thébault, M. Carrière, H. Joisten and B. Dieny, Nanomaterials, 2020, 10, 1500 CrossRef CAS PubMed.
- H. Al-Madhagi, V. Yazbik, W. Abdelwahed and L. Alchab, Bionanoscience, 2023, 13, 853–859 CrossRef.
- M. S. Islam, Y. Kusumoto, J. Kurawaki, M. D. Abdulla-Al-Mamun and H. Manaka, Bull. Mater. Sci., 2012, 35, 1047–1053 CrossRef CAS.
- A. R. Trifoi, E. Matei, M. Râpă, A.-C. Berbecaru, C. Panaitescu, I. Banu and R. Doukeh, React. Kinet., Mech. Catal., 2023, 136, 2835–2874 CrossRef CAS.
- X. Wu, H. Choe, J. Strayer, J. Gómez-Pastora, M. Zborowski, B. Wyslouzil and J. Chalmers, Nanoscale, 2024, 16, 7041–7057 RSC.
- A. J. Willis, S. P. Pernal, Z. A. Gaertner, S. S. Lakka, M. E. Sabo, F. M. Creighton and H. H. Engelhard, Int. J. Nanomed., 2020, 15, 4105–4123 CrossRef CAS PubMed.
- F. L. Durhuus, L. H. Wandall, M. H. Boisen, M. Kure, M. Beleggia and C. Frandsen, Nanoscale, 2021, 13, 1970–1981 Search PubMed.
- Z. Cai, C. Wu, L. Yang, D. Wang and H. Ai, ACS Biomater. Sci. Eng., 2020, 6, 2533–2542 CrossRef CAS PubMed.
- S. Sharma, H. Sharma and R. Sharma, Chem. Inorg. Mater., 2024, 2, 100035 Search PubMed.
- A. G. Kolhatkar, A. C. Jamison, D. Litvinov, R. C. Willson and T. R. Lee, Int. J. Mol. Sci., 2013, 14, 15977–16009 CrossRef PubMed.
- Y. Hadadian, H. Masoomi, A. Dinari, C. Ryu, S. Hwang, S. Kim, B. K. Cho, J. Y. Lee and J. Yoon, ACS Omega, 2022, 7, 15996–16012 Search PubMed.
- L. Gloag, M. Mehdipour, D. Chen, R. D. Tilley and J. J. Gooding, Adv. Mater., 2019, 31, 1904385 CrossRef CAS PubMed.
- L. C. Wu, Y. Zhang, G. Steinberg, H. Qu, S. Huang, M. Cheng, T. Bliss, F. Du, J. Rao, G. Song, L. Pisani, T. Doyle, S. Conolly, K. Krishnan, G. Grant and M. Wintermark, Am. J. Neuroradiol., 2019, 40, 206 CrossRef CAS PubMed.
- S. M. Dadfar, K. Roemhild, N. I. Drude, S. von Stillfried, R. Knüchel, F. Kiessling and T. Lammers, Adv. Drug Delivery Rev., 2019, 138, 302–325 CrossRef CAS PubMed.
- J. Medinger, M. Nedyalkova and M. Lattuada, Nanomaterials, 2021, 11, 360 CrossRef CAS PubMed.
- Y. Chen, J. Zhang, Z. Wang and Z. Zhou, Appl. Sci., 2019, 9, 5157 CrossRef CAS.
- F. Fiévet, S. Ammar-Merah, R. Brayner, F. Chau, M. Giraud, F. Mammeri, J. Peron, J. Y. Piquemal, L. Sicard and G. Viau, Chem. Soc. Rev., 2018, 47, 5187–5233 Search PubMed.
- M. C. Mascolo, Y. Pei and T. A. Ring, Materials, 2013, 6, 5549–5567 Search PubMed.
- L. Panariello, M. O. Besenhard, S. Damilos, A. Sergides, V. Sebastian, S. Irusta, J. Tang, N. T. K. Thanh and A. Gavriilidis, Chem. Eng. Process., 2022, 182, 109198 Search PubMed.
- M. O. Besenhard, L. Panariello, C. Kiefer, A. P. LaGrow, L. Storozhuk, F. Perton, S. Begin, D. Mertz, N. T. K. Thanh and A. Gavriilidis, Nanoscale, 2021, 13, 8795–8805 RSC.
- M. O. Besenhard, A. P. LaGrow, A. Hodzic, M. Kriechbaum, L. Panariello, G. Bais, K. Loizou, S. Damilos, M. Margarida Cruz, N. T. K. Thanh and A. Gavriilidis, Chem. Eng. J., 2020, 399, 125740 CrossRef CAS.
- F. Canfarotta, A. Poma, A. Guerreiro and S. Piletsky, Nat. Protoc., 2016, 11, 443–455 CrossRef CAS PubMed.
- T. Hix-Janssens, J. R. Davies, N. W. Turner, B. Sellergren and M. V. Sullivan, Anal. Bioanal. Chem., 2024, 416, 7305–7316 Search PubMed.
- R. Mahajan, M. Rouhi, S. Shinde, T. Bedwell, A. Incel, L. Mavliutova, S. Piletsky, I. A. Nicholls and B. Sellergren, Angew. Chem., Int. Ed., 2019, 58, 727–730 Search PubMed.
- S. Lyons, P. Baile Pomares, L. Vidal, K. McGarry, A. Morrin and D. F. Brougham, Langmuir, 2023, 39, 8100–8108 CrossRef CAS PubMed.
- C. Comanescu, Coatings, 2023, 13, 1772 CrossRef CAS.
- A. Aharoni, J. Appl. Phys., 1998, 83, 3432–3434 Search PubMed.
- A. Stephen, S. Dennison, M. Holden and S. Reddy, The Analyst, 2023, 148, 5476–5485 RSC.
- Z. Kozakova, I. Kuritka, N. E. Kazantseva, V. Babayan, M. Pastorek, M. Machovsky, P. Bazant and P. Saha, Dalton Trans., 2015, 44, 21099–21108 RSC.
- W. Ma, P. M. Gehret, R. E. Hoff, L. P. Kelly and W. H. Suh, Nanomaterials, 2019, 9, 453 CrossRef CAS PubMed.
- A. Laurikėnas, J. Barkauskas, J. Reklaitis, G. Niaura, D. Baltrūnas and A. Kareiva, Lith. J. Phys., 2016, 56, 35–41 Search PubMed.
- M. Stoia, R. Istratie and C. Păcurariu, J. Therm. Anal. Calorim., 2016, 125, 1185–1198 CrossRef CAS.
- K. E. Rasmussen and J. Albrechtsen, Histochemistry, 1974, 38, 19–26 CAS.
- I. Migneault, C. Dartiguenave, M. J. Bertrand and K. C. Waldron, Biotechniques, 2004, 37, 790–802 CrossRef CAS PubMed.
- D. Sarkar and P. Somasundaran, Langmuir, 2004, 20, 4657–4664 CrossRef CAS PubMed.
- E. P. K. Currie, W. Norde and M. A. Cohen Stuart, Adv. Colloid Interface Sci., 2003, 100–102, 205–265 CrossRef CAS PubMed.
- D. Wen, T. Ralph, J. Han, S. Bradley, M. J. Giansiracusa, V. Mitchell, C. Boskovic and N. Kirkwood, J. Phys. Chem. C, 2023, 127, 9164–9172 CrossRef CAS.
- M. C. Mascolo, Y. Pei and T. A. Ring, Materials, 2013, 6, 5549–5567 CrossRef CAS PubMed.
- K. O'Grady, G. V. Fernandez and A. Hirohata, Particulate and Granular Magnetism: Nanoparticles and Thin Films, Oxford University Press, 2023 Search PubMed.
- R. Chantrell, J. Popplewell and S. Charles, IEEE Trans. Magn., 1978, 14, 975–977 CrossRef.
- D. Hawkins, D. Stevenson and S. Reddy, Anal. Chim. Acta, 2005, 542, 61–65 Search PubMed.
- E. Saridakis, S. Khurshid, L. Govada, Q. Phan, D. Hawkins, G. Crichlow, E. Lolis, S. Reddy and N. Chayen, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 11081–11086 CrossRef CAS PubMed.
- A. N. Stephen, S. R. Dennison, M. A. Holden and S. M. Reddy, Analyst, 2023, 148, 5476–5485 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ma01115e |
| This journal is © The Royal Society of Chemistry 2025 |