
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The conversion of ethanol over 3d-metal saponite-like smectites†
Marc
Greuel
 ac,
Clara Maria
Watermann
a,
Heiko
Lohmann
a,
Stefan
Kaluza
b,
Ulf-Peter
Apfel
*ac and
Barbara
Zeidler-Fandrich
*a
ac,
Clara Maria
Watermann
a,
Heiko
Lohmann
a,
Stefan
Kaluza
b,
Ulf-Peter
Apfel
*ac and
Barbara
Zeidler-Fandrich
*a
aFraunhofer UMSICHT, Osterfelder Straße 3, 46047 Oberhausen, Germany. E-mail: ulf.apfel@rub.de; barbara.zeidler-fandrich@umsicht.fraunhofer.de
bUniversity of Applied Science Düsseldorf, Münsterstraße 156, 40476 Düsseldorf, Germany
cRuhr University Bochum, Universitätsstraße 150, 44801 Bochum, Germany
First published on 21st March 2025
Abstract
The replacement of fossil fuel processes with renewable pathways is critical to circumvent climate change and environmental risks. A process of interest is the production of 1,3-butadiene (BD), which is primarily derived as a by-product of naphtha steam cracking. A sustainable alternative path involves converting renewable ethanol to BD via the Lebedev process, which requires a catalyst with balanced redox, acidic, and basic sites. Due to the necessity of a multifunctional catalyst, existing materials for this reaction are either compirsed of catalyst mixtures or supported catalysts. In this study, we introduce a bulk material, saponite, containing M–O–Si sites, which combine required catalytic sites for the ethanol to BD (ETB) reaction in one material. The product composition in ethanol conversion were strongly dependent on the type of 3d-metal used, while no conclusive correlation between surface properties, conversion, and product composition was observed. Herein, using V-Sap*, we achieved an ethene productivity of 448 gethene kgcat−1 h−1 (74%) at 573.15 K. High acetaldehyde productivity was maintained with Cu-Sap* (466 gAcA kgcat−1 h−1, 49%) at 573.15 K and Zn-Sap (528 gAcA kgcat−1 h−1, 55%) at 723.15 K. Mg-Sap primarily produced ethene but also yielded 10 gBD kgcat−1 h−1 BD at 723.15 K. Higher BD outputs were observed with Ni-Sap (31 gBD kgcat−1 h−1 at 523.15 K) and Mn-Sap* (51 gBD kgcat−1 h−1 at 723.15 K). This underscores the potential of saponite-based materials for flexible product outputs in ethanol conversion, influenced by the choice of integrated 3d-metal.
Introduction
To reduce carbon dioxide (CO2) emissions, it is essential to shift from fossil-based to renewable chemical processes. A notable reaction is the conversion of ethanol to 1,3-butadiene (BD), known as the ethanol-to-1,3-butadiene (ETB) process.1–3 It can be performed via a one-step (Lebedev, ethanol → BD) or two-step (Ostromisslensky, ethanol → acetaldehyde → BD) route.4,5 Recently, interest in BD production from ethanol has increased, mainly because 95% of BD is currently produced as a by-product from the steam cracking of fossil naphtha.4–6 Furthermore, the application of shale gas in the cracking process leads to lower BD yields and production rates, as well as an increasing BD price.7–9 Moreover, BD is crucial for producing polymers like polybutadiene rubber (PBR) and styrene–butadiene rubber (SBR), used in tires, adhesives, and automotive parts.10–12 As a result, the BD market is closely linked to the automotive sector and is expected to grow at a compound annual growth rate of 3–5.6% from 2022 to 2035.13–15Different life-cycle assessments (LCA) and techno-economic analyses (TEA) showed that the production of BD via the Lebedev process can achieve lower CO2 emissions compared to the fossil-based process.16–21
Due to a complex reaction path (Fig. 1), this process requires multifunctional catalysts with a balanced ratio of redox, Lewis acid, Brønsted acid, Lewis basic and Brønsted basic sites to obtain industrial relevant BD productivity (>150 gBD kgcat−1 h−1) at a weight hourly space velocity (WHSV) of around 0.5 h−1 and to avoid by-products such as ethylene, diethyl ether.22
 | ||
| Fig. 1 Reaction scheme for the reaction of ethanol to 1,3-butadiene via the Toussaint–Kagan mechanism23,24 (green arrows) and the most commonly detected by-products via the respective side reactions (orange arrows). Main reactions (green): (i) dehydrogenation, (ii) aldol addition, (iii) dehydration, (iv) Meerwein–Ponndorf–Verley (MPV)25–27 reduction, (v) dehydration and side reactions (orange): (a) condensation, (b) dehydration, (c) di- or polymerization, (d) cracking, (e) Tishchenko reaction,28,29 (f) decarboxylative ketonization mechanism, (g) consecutive aldol condensations, (h) dehydrogenation and dehydration, (j) hydrogenation.10,24,30–32 | ||
The commonly accepted mechanism for the ETB reaction is the Toussaint–Kagan pathway (Fig. 1, green arrows) which consists of five consecutive reaction steps.23,33 Due to necessity of various catalytic sites several side reactions can occur (Fig. 1, orange arrows).10,24,30–32
Various multifunctional catalytic systems for the Lebedev process have been investigated, focusing on combinations of metals and supports.10,24,34–36 The state-of-the-art catalysts are mostly based on ZnO/Al2O3, ZrO2/SiO2 and MgO/SiO2. Notably, Ag/ZrO2/SiO2 systems were extensively studied by Dagle et al.37,38 and Sushkevich et al.,39–42 achieving conversion rates (X) up to 99% with BD selectivities (S) over 70% at 598 K and WHSVs of 0.2–0.5 h−1. Comparable results were reported by Dai et al.43 and De Beardemaeker et al.,21 using CuZnY/zeolite and CuZnHf/SiO2 as catalysts, reaching BD yields of 73% and 72%, respectively. Additionally, extensive research on the MgO/SiO2 catalyst, an industrial version of the original ZnO/SiO2 used by Lebedev,34 was conducted by Kvisle et al.,44 who highlighted the significance of Mg–O–Si sites for the catalytic selectivity towards BD. Modifications of the MgO/SiO2 system like support changes, incorporation of transition metals, and adjustments in acid–base properties were also investigated.10,24,34–36
Overall, bulk materials containing M–O–Si sites seem promising to produce BD from ethanol in industrial relevant amounts. Along this line, synthetic clays are promising materials for this purpose and can be tailored with specific acid–base ratios, elemental compositions, or pore size distributions.45–47 However, only one clay mineral, sepiolite (Mg8Si12O30(OH)4(H2O)4·nH2O), has been reported by Kitayama et al.48–50 and Gruver et al.51,52 as a catalyst for the Lebedev process. Although natural sepiolite was initially ineffective in producing BD, its modification through the substitution of Si with Al, Mg with Cu, cation exchange with Ag, or impregnation with 3d metals (Mn, Ni, Co, V, Zn, Cu) significantly enhanced BD yield, achieving up to 67% BD with Zn impregnation.
We recently reported on the use of saponites for the conversion of ethanol, which are mesoporous minerals of the smectite group.53,54 Saponites consist of two tetrahedral –O–Si–O– layers (T) surrounding a central octahedral –O–Mg–O– layer (–O–) and providing Mg–O–Si sites. The typical molecular formula for saponites is Az+x/z[Mg6][Si8−xAlx]O20(OH)4·nH2O (with x = 0.4 to 1.2; Az+x/z = counterion). The substitution of Si4+ ions with Al3+ leads to negative charges in the tetrahedral layer, which are balanced by exchangeable cations Az+x/z (such as Na+, K+, Li+, NH4+, Mg2+) located in the intermediate layer of the T–O–T sandwich structure. Additionally, natural saponites show substitution of octahedral cations (Mg2+) by other metal cations (e.g., Fe2+, Fe3+, Al3+, Li+, Mn2+, Ni2+, and Ti4+), enhancing their adaptability. This exchangeability provides a multifunctional bulk material with various possibilities to tailor its properties for the Lebedev process. Moreover, the advantages of using saponites are their high specific surface areas, surface acidity and cation exchangeability.55–58 Our previous work introduced saponites as promising catalysts in the conversion of ethanol with different heating methods (resistive and inductive), but the tailoring of the saponite composition for the Lebedev process is currently an uninvestigated point of research.53,54
Therefore, the aim of this work is to improve BD yields by developing and tailoring a saponite composition combining MOx and SiO2 sites in one material. Moreover, we examine possible correlations between the catalytic performance and material properties, focusing on structure and surface characteristics like area, pore structure, acidity, and basicity. Finally, we discuss the influence of the used 3d-metals on these trends and investigate the impact of temperature on the catalytic behavior of the synthesized materials.
Experimental
Materials
If not otherwise stated, all chemicals were obtained from commercial suppliers and used without further purification. Water used for synthesis and washing was desalinated via a reverse osmosis system. The following chemicals were utilized during synthesis. Sodium silicate (Na2SiO3) solution (VWR, 25.5–28.5 wt% SiO2 in H2O), aluminum(III) nitrate nonahydrate (Al(NO3)3·9H2O, Carl Roth, >98%), sodium hydroxide (NaOH, Carl Roth, ≥99%), urea (CH4N2O, Sigma Aldrich, ≥99%), magnesium(II) nitrate hexahydrate (Mg(NO3)2·6H2O, Carl Roth, ≥99%), vanadium(II) chloride (VCl2, Sigma Aldrich, 85%), chromium(II) chloride (CrCl2, Alfa Aesar, >96%), manganese(II) nitrate tetrahydrate (Mn(NO3)2·4H2O, Carl Roth, ≥98%), iron(II) sulfate heptahydrate (FeSO4·7H2O, Alfa Aesar, ≥98%), cobalt(II) nitrate hexahydrate (Co(NO3)2·6H2O, Carl Roth, ≥98%), nickel(II) nitrate hexahydrate (Ni(NO3)2·6H2O, Carl Roth, ≥98%), copper(II) nitrate trihydrate (Cu(NO3)2·3H2O, Carl Roth, ≥98%), zinc(II) nitrate hexahydrate (Zn(NO3)2·6H2O, Carl Roth, ≥98%), synthetic air (20% O2, 80% N2, Linde, 5.0), carbon dioxide (CO2, Westfalen AG, 4.8) and helium (He, Linde, 5.0). The actual SiO2 content in the sodium silicate solution determined via ICP-OES amounted to 26.1%. For the catalytic experiments ethanol (C2H5OH, Carl Roth, ≥99.5%) and nitrogen gas (N2, Linde GmbH, 5.0) were used.Preparation of Az+x/z[M6][Si8−xAlx]O20(OH)4·nH2O catalysts
The catalyst synthesis was performed based on the non-hydrothermal saponite synthesis reported by Vogels et al.55 for a Si to Al molar ratio of 5.67. In the first step of the synthesis, a Na2SiO3 solution (5.075 g, 22.0 mmol Si) was diluted with 25 mL H2O (Solution A). While vigorously stirring Solution A, a solution of aluminum(III) nitrate nonahydrate (1.459 g, 3.9 mmol) in 2 M NaOH (10 mL) (Solution B) was added within 30 seconds. After formation of an aluminosilicate gel, it was left to age for 1 hour. Then, the gel was diluted with H2O (250 mL), stirred and the resulting suspension was heated to 363.15 K. In the second step of the synthesis, a solution (Solution C) consisting of urea (4.44 g, 73.9 mmol), H2O (50 mL) and the respective metal(II) salt(s) (19.5 mmol of M2+, with M2+ = Mg2+, V2+, Cr2+, Mn2+, Fe2+, Co2+, Ni2+, Cu2+, and Zn2+) was added dropwise to the suspension. Subsequently, the resulting suspension was stirred for 20 h at 363.15 K. After cooling, filtration and washing of the resulting product, the solid was dried over night at 403.15 K before grinding the sample with a ball mill to obtain a fine powder (particle size <500 μm). Finally, the powder was heated with a rate of 10 K min−1 to 773.15 K (673.15 K in case of Zn containing samples) and calcined in synthetic air at this temperature for 3 h. Additionally, one sample was prepared without the addition of a metal(II) salt (M2+ = 0) in Solution C. The resulting materials were denoted as M-Sap for pure saponite phases or M-Sap* for impure samples. For example, for pure saponite with M2+ = Mg2+ the resulting catalyst was denoted as Mg-Sap and for impure material M2+ = Cu2+ the resulting catalyst was denoted as Cu-Sap*.Catalysts characterization
X-ray diffraction (XRD) measurements were performed with an X’pert Powder (PANalytical) equipment with a copper anode (Cu Kα λ = 1.54184 Å) operated at 45 kV and 40 mA and a PIXcel1D detector with 256 channels. Angles from 10°–85° 2θ were measured with a step size of 0.05° and a scan speed of 0.026° s−1.X-ray photoelectron spectroscopy (XPS) was performed using a polychromatic Al Kα X-ray source (hν = 1486.6 eV) in a vacuum chamber at a pressure of 10−9 mbar. The anode current was 13 mA at a voltage of 14 kV. As detector, a VG-CLAM2 (Thermo Fischer Scientific) was used. The results were calibrated based on the binding energy of carbon (C 1s = 285 eV) and measured from 0 to 1100 eV with a step size of 0.500 eV. A pass energy of 200.0 eV for the survey scan and 50.0 eV for the elemental scans was used. CasaXPS (Casa Software Ltd) was used for data evaluation.
Inductive coupled plasma with optical emission spectroscopy (ICP-OES) was performed using a SpectroAcros (SPECTRO Analytical Instruments GmbH). For analysis, the samples were first dissolved using a melt digestion with lithium metaborate (Alfa Aesar, Spectroflux 110A) and di-lithium tetraborate (Merck, Spectromelt A 100) before dissolving the sample in diluted hydrochloric acid (1HCl![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6H2O). Yttrium was added as an internal standard.
:6H2O). Yttrium was added as an internal standard.
Scanning electron microscopy (SEM) with energy-dispersive X-ray spectroscopy (EDX) was performed on a Merlin Gemini 2 HR-FESEM (ZEISS) equipped with an N-Max silicon drift detector (Oxford Instruments). Acceleration voltages of 5 kV and 20 kV at a WD of 10 mm were used to obtain SEM images and EDX analysis, respectively. Prior to analysis the samples were coated with carbon to improve the conductivity of the sample.
For N2 physisorption measurements a 3Flex (Micrometrics) instrument was used to determine the surface area of the materials. After degassing the sample in vacuum, an N2 physisorption isotherm was determined at 77 K. Brunauer, Emmett and Teller (BET) method was used to determine the surface area, while Barett, Joyner and Halenda (BJH) method was used to determine the pore volume and the pore size distribution.
To determine the acidic and basic properties of the catalyst surface temperature programmed desorption (TPD) measurements were preformed using ammonia (NH3) and carbon dioxide (CO2) as probe gas molecules, respectively. A BELCAT A v2.6.0 (BEL Japan) equipped with a thermal conductivity detector (TCD) for the desorbed exhaust gas and a quartz glass sample holder (Microtrac) was used for both measurements. The sample (0.050 g) was fixed in the glass tube between two glass wool plugs. For NH3 desorption the sample was first heated with a heating rate of 10 K min−1 to 673.15 K and kept at this temperature for 30 minutes under inert gas atmosphere (He, 50 mL min−1). Subsequently, the sample was cooled to 373.15 K and then the gas feed was changed to NH3 (5% NH3 in He, 50 mL min−1) for 30 minutes. Afterwards, the reactor was flushed again with inert gas (He, 50 mL min−1) for 15 minutes. Finally, the reactor was heated from 373.15 K to 773.15 K with a linear heating ramp of 10 K min−1 under helium flow (30 mL min−1). For CO2 desorption the sample was first heated to 673.15 K and kept at this temperature for 30 minutes under inert gas atmosphere (He, 50 mL min−1). Subsequently, the sample was cooled to 303.15 K and then gas feed was changed to CO2 gas (CO2, 50 mL min−1) for 40 minutes. Afterwards, the reactor was flushed again with inert gas (He, 50 mL min−1) for 15 minutes. Finally, the reactor was heated from 303.15 K to 773.15 K with a linear heating ramp of 10 K min−1 under helium flow (30 mL min−1). For data analysis the software ChemMaster from BEL Japan was used.
Particle-size analysis using laser diffraction was performed in a batch cell of a Shimadzu SALD-2300 instrument. A spatula point of sample was pre-dispersed in isopropanol for 3 minutes using an ultrasonic bath before adding it dropwise into the batch cuvette. The refractive index using 1.60–1.00 for saponites was used to calculate the particle size distribution from the diffractogram. Particle number-based distributions were calculated using the Shimadzu WingSALD II software.
Catalytic conversion of ethanol
Catalytic tests for the conversion of ethanol were conducted in a continuous flow, fixed-bed stainless steel tube reactor with a 10 mm inner diameter in a tube furnace. A mesh was fixed at 330 mm from the top of the tube to place the catalytic bed on top. 0.5 g of the catalyst (particle size <100 μm, Fig. S1, ESI†) and 2.5 g of an inert material (α-Al2O3, 355–500 μm, 99.7%, Herfeldt GmbH) were mixed, fixed between two glass wool plugs, and placed on top of the mesh. All reactions were performed at atmospheric pressure and with the feed flow running vertically through the catalyst bed from top to bottom. The catalysts were in situ dried at 473.15 K under 100 mL min−1 N2 flow for 30 min. Subsequently, two by-pass measurements were preformed prior to testing to determine the initial ethanol concentration (cEtOH,i). Ethanol (C2H5OH, 0.5 g h−1) was fed to the carrier gas (N2, 94 mL min−1) with a controlled evaporator mixer (CEM) (W-101A-111-K, Bronkhorst GmbH) at 373.15 K to obtain a reactant flow of 100 mL min−1 at standard temperature and pressure (STP) containing 4.6 mol% of ethanol (WHSV = 1 gEtOH gcat h−1). After every measurement at a specific reaction temperature (starting at 473.15 K) the temperature was increased by 50 K with a heating rate of 5 K min−1 and kept at that temperature for 45 minutes time-on-stream (TOS) before taking the sample at the respective temperature. These steps were repeated for all temperatures up to 723.15 K. Sample analysis of the product stream was performed with an inline GC 2014 gas chromatograph (Shimadzu Corporation) equipped with a flame ionization detector (FID) to measure hydrocarbons and oxygenates. For the separation of the sample an SH-Rxi-624Sil MS column (30 m × 0.25 mm × 1.4 μm) was used. Carbon-based conversions XC were calculated using eqn (1). In the temperature range from 473.15 to 623.15 K and low conversions, the carbon-based selectivity for component a SP,a was related to all formed products and calculated according to eqn (2), while at temperatures above 673.15 K, the carbon-based selectivity SC,a was conversion-related and calculated according to eqn (3). | (1) |
 | (2) |
 | (3) |
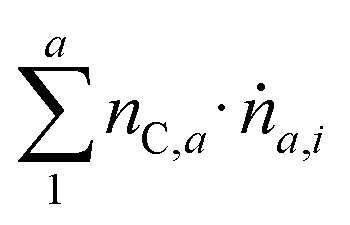 with nC,a equal to the number of carbon atoms in component a and ṅa,i equal to the molar flow of component a in the outgoing gas flow at the respective reaction temperature i. For eqn (3)ṅEtOH,0 and ṅEtOH,i represent the ingoing and outgoing molar flows of ethanol, respectively. While nC,EtOH represents the number carbon atoms per molecule of ethanol. Gaps in the carbon balance at higher temperatures (above 673.15 K) occurred due to unknown products of aldol condensation with higher carbon numbers. The standard error on the conversion was determined using Gaussian error propagation.
with nC,a equal to the number of carbon atoms in component a and ṅa,i equal to the molar flow of component a in the outgoing gas flow at the respective reaction temperature i. For eqn (3)ṅEtOH,0 and ṅEtOH,i represent the ingoing and outgoing molar flows of ethanol, respectively. While nC,EtOH represents the number carbon atoms per molecule of ethanol. Gaps in the carbon balance at higher temperatures (above 673.15 K) occurred due to unknown products of aldol condensation with higher carbon numbers. The standard error on the conversion was determined using Gaussian error propagation.
Productivity of a product a was calculated with eqn (4), with ṅa,i in mol min−1 and the mass of the catalyst mcatalyst in grams.
 | (4) |
000 converts the units from minute to hour (60) and gram to kilogram (1000).
Finally, the yield of BD YBD was calculated using eqn (5).
 | (5) |
Results and discussion
Different materials were synthesized via the non-hydrothermal synthesis route of Vogels et al.55 and substitution of Mg with 3d-metals (V, Cr, Mn, Fe, Co, Ni, Cu, Zn). Various colored fine powders (Fig. S2, ESI†) were obtained by variation of the octahedral metal centers.Characterization of the obtained 3d-materials
The obtained X-ray diffraction (XRD) patterns confirmed saponite-like structure for Mg, Fe, Co, Ni, and Zn samples by the specific broad reflections (dashed lines, Fig. 2) at diffraction angles (2θ) of around 20°, 27°, 35° and 60°, corresponding to lattice planes (020)/(110), (113), (202), and (060), respectively. Here, the (060) reflection indicates a dominant trioctahedral structure of the materials.59 Variations in the reflection positions are attributed to atom sizes, while the asymmetric nature of the (020)/(110), (113) and (060) reflections are attributed to different orientations of –T–O–T– layers (turbostratic disorders) in the stacking of the saponite sheets.60,61 Zn-Sap showed the sharpest reflections and a distinct (001) reflection at 2θ of about 5°, suggesting an increase in crystallite size and higher degree of layer stacking. The other samples lacked this specific reflection, possibly due to a higher degree of turbostratic disorders as shown by Lutteroti et al.,61 and is therefore not shown in the XRD results. Exact determination of the crystallite sizes was not possible with the Scherrer equation due to asymmetry and different full width at half maximum (FWHM) of the reflexes. Notably, Fe-Sap* exhibited faint saponite reflections and additional peaks attributed to FeO(OH)62 suggesting oxidation of Fe(II) to Fe(III) and incomplete ferrous saponite synthesis. Thus, in addition to an iron (Fe(II) or Fe(III)) containing saponite, FeO(OH) is formed as a secondary phase. Moreover, the XRD patterns of Cu-, Mn-, and Cr-Sap* displayed sharp reflexes characteristic for CuO,63 Mn2O364,65 and Cr2O366 respectively, without typical saponite peaks. Chrysocolla [Cu2Si2O5(OH)2] formation as described by Vogels et al.55 in Cu-Sap* was not observed. V-Sap* and 0-Sap* showed a broad signal from 2θ of 15° to 40°, attributed to amorphous aluminosilicates,67 indicating little vanadium uptake for V-Sap*. | ||
| Fig. 2 XRD patterns of the resulting materials with exchange of octahedral cations, 0-Sap* represents the resulting aluminosilicate by omission of a M2+ salt in the second reaction step and dashed lines denote the saponite reflections,59 References: □ Cr2O3,66 ● Mn2O3,64,65 ■ FeO(OH),62 ○ CuO63. | ||
The XRD results are supported by XPS measurements with determined binding energies (BE) shown in Tables 1 and 2 (Fig. S3, S4 and Table S1, ESI†). Considering the measured binding energies (BE) for O 1s shown in Table 1 the additional signals at 530.3, 531.3, 530.8, and 529.9 eV for Cr-,68 Mn-,69 Fe-,70 and Cu-Sap*,71 respectively, are attributed to lattice oxygen and hydroxides expected for the various oxides identified via XRD. Moreover, the metal-specific 2p orbitals (Table 2) showed distinct BEs that are in good agreement with the presence of these oxides. For Cr-Sap*, the Cr 2p3/2 BE at 577.1 eV was deconvoluted to reveal peaks at 576.8 and 578.9 eV, indicating a Cr2O3 surface being partially oxidized from Cr3+ to Cr6+.68 The Mn 2p3/2 peak at 642.8 eV matched the Mn3+ state associated with Mn2O3 on silica.72
| Sample | Al 2p tetrahedral (eV) (area) | Al 2p octahedral (eV) (area) | Si 2p (eV) | O 1s (eV) | O 1s oxides(eV) |
|---|---|---|---|---|---|
| a Overlay with Ni 3p. b Overlay with Cu 3p leads to higher BEs, raw data are shown in Fig. S3 (ESI). | |||||
| Ma-Sap | 74.5 (66.7%) | 75.5 (33.3%) | 103.1 | 532.4 | — |
| V-Sap* | 74.8 (87.6%) | 75.8 (12.4%) | 103.0 | 532.5 | 531.0 |
| Cr-Sap* | 74.8 (80.1%) | 75.8 (19.9%) | 103.3 | 532.7 | 530.3 |
| Mn-Sap* | 74.6 (79.2%) | 75.6 (20.8%) | 103.1 | 532.5 | 531.3 |
| Fe-Sap* | 74.6 (97.5%) | 75.6 (2.6%) | 103.0 | 532.3 | 530.8 |
| Co-Sap | 74.5 (100%) | — | 103.0 | 532.2 | — |
| Ni-Sap | 74.2 (100%)a | — | 103.0 | 532.3 | — |
| Cu-Sap* | 75.2 (38.4%)b | 78.0 (61.6%)b | 102.9 | 532.2 | 529.9 |
| Zn-Sap | 74.7 (100%) | — | 102.9 | 532.2 | — |
| Sample | M 2p3/2-1 [eV] | M 2p1/2-1 [eV] | ΔBE [eV] | M 2p3/2-2 [eV] | M 2p1/2-2 [eV] | ΔBE [eV] |
|---|---|---|---|---|---|---|
| Ma-Sap | 1304.5 (Mg 1s) | — | — | |||
| V-Sap* | 518.3 | 525.5 | 7.2 | |||
| Cr-Sap* | 577.1 | 586.6 | 9.5 | |||
| Mn-Sap* | 642.8 | 654.6 | 11.8 | |||
| Fe-Sap* | 712.3 | 725.8 | 13.5 | |||
| Co-Sap | 781.5 | 797.8 | 16.3 | |||
| Ni-Sap | 857.0 | 874.6 | 17.6 | |||
| Cu-Sap* | 933.6 | 953.6 | 20.0 | 935.4 | 955.4 | 20.0 |
| Zn-Sap | 1022.8 | 1045.9 | 23.1 |
Fe-Sap* exhibited a Fe 2p3/2 BE of 712.3 eV, deconvoluted into signals at 711.7 and 713.7 eV for Fe2+ (69%) and Fe3+ (31%), respectively, suggesting the presence of both FeO(OH)73 and ferrous saponite.74 Cu-Sap* revealed a Cu 2p3/2 BE at 933.4 eV with a shake-up satellite at 944.0 eV (Table S1, ESI†), characteristic for CuO,71 and a second species at 935.6 eV, attributed to a copper-containing phyllosilicate.75 Furthermore, a O 1s signal at 531.0 eV in the V-Sap* shows the presence of an additional oxide which was attributed to V2O5 in good agreement with the V 2p3/2 BE at 518.3 eV for V5+, despite its absence in the XRD pattern.76,77 The absence of the V2O5 reflections in the XRD diffractogram could be a result of a small quantity or low crystallinity of V2O5 particles.
The other metal-specific BEs of Mg-Sap, Co-Sap, Ni-Sap and Zn-Sap are also shown in Table 2 and correspond to the saponite structure observed with XRD. For Mg-Sap, the Mg 1s BE at 1304.5 eV was deconvoluted into signals at 1304.4 and 1305.5 eV, reflecting Mg2+ in cis- and trans-sites within the octahedral layer of saponite.78,79
Co-Sap showed a Co 2p3/2 BE at 781.9 eV, consistent with previous reports on Co-Saponite, which included a shake-up satellite about 6 eV above the main Co 2p signal.80 The Ni 2p3/2 BE of 857.0 eV, along with a satellite peak at around 6 eV higher BEs, confirmed its identity as Ni-Saponite, aligning with reported values.81,82 Using the difference of the Ni 2p and Si 2p ΔBENi–Si for Ni-phyllosilicates as reported by Coenen et al. are also in good agreement with the here reported ΔBENi–Si = 754 eV for Ni-Sap.81–83 Lastly, the Zn 2p BE for Zn-Saponite (sauconite) was reported at 1022.8 eV, differentiating it from typical ZnO values (1020.7–1021.2 eV).84,85
In line with the XRD results, the silicon coordination was confirmed by Si 2p BEs of 102.9 to 103.1 eV, corresponding to tetrahedrally coordinated silicon in saponites, phyllosilicates and similar clay minerals.79 Moreover, in terms of the clay mineral structure Al 2p peaks were observed at 74.2 to 75.2 eV which were deconvoluted into a duplet, with the lower BEs (74.2 to 74.8 eV) corresponding to tetrahedrally coordinated aluminum and the higher BEs (75.5 to 75.8 eV) to octahedrally coordinated aluminum. The octahedral coordination was attributed to partial occupation of the octahedral layer in smectites.86–88
The XRD and XPS results were further supported via scanning electron microscopy (SEM) (Fig. 3) with energy-dispersive X-ray spectroscopy (EDX) measurements (Table S2, ESI†). The Cu-Sap* (a) shows rod-shaped monoclinic crystals (encircled in orange), typical for CuO, on a porous surface.63 In contrast to the overall composition of the Cu-Sap* showing an Si/Al ratio of 7.07 and a Cu/Si ratio of 0.80, areas with surface crystals showed a Cu/O ratio of 1 to 1.2, aligning with the stoichiometry of CuO. Thus, the EDX analysis of this crystals confirmed the presence of CuO crystals on the surface of a Cu-phyllosilicate. The uneven and porous surface observed for Cr-Sap* (e) is covered with small hexagonal crystals. This is likely due to Cr2O3 crystallizing in the eskolaite structure with space group R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c.66,89 In combination with the observed characteristic dark green color of the material (Fig. S2, ESI†), this supports the thesis of the formation of Cr2O3 crystals on the surface. For Mn-Sap*, in addition to porous and irregular particles, cubic crystals (f) are observed. Mn2O3 crystallizes in space group Ia3 in large crystals.64,65 For Mn-Sap*, in addition to porous and irregular particles, cubic crystals (f) are observed. Mn2O3 crystallizes in space group Ia3 in large crystals.64,65
c.66,89 In combination with the observed characteristic dark green color of the material (Fig. S2, ESI†), this supports the thesis of the formation of Cr2O3 crystals on the surface. For Mn-Sap*, in addition to porous and irregular particles, cubic crystals (f) are observed. Mn2O3 crystallizes in space group Ia3 in large crystals.64,65 For Mn-Sap*, in addition to porous and irregular particles, cubic crystals (f) are observed. Mn2O3 crystallizes in space group Ia3 in large crystals.64,65
 | ||
| Fig. 3 SEM images of selected products, (a) Cu-Sap* with CuO crystals (orange), (b) Mg-Sap, (c) Zn-Sap, (d) Ni-Sap, (e) Cr-Sap* with hexagonal Cr2O3 crystals, and (f) Mn-Sap* with cubic Mn2O3 crystals; the magnification for (a)–(d) is 10000× and for (e) and (f) 6480× and 8200×, respectively. | ||
While the Mg-Sap (Fig. 3b) and Ni-Sap (d) display uneven, porous surfaces, Zn-Sap (c) and Co-Sap (Fig. S5, ESI†) exhibited more uniform and crystalline surfaces. These observations are in good agreement with N2-physisorption measurements (Table 3) showing the highest surface areas for Mg- (458 m2 g−1) and Ni-Sap (498 m2 g−1), while the lowest surface area was obtained for the Zn-Sap (107 m2 g−1) and the smallest pore size of 2 nm for the Co-Sap (Fig. S6, ESI†). Moreover, the surface acid/base ratio (Ra/b) (Table S3, ESI†) was determined via NH3- and CO2-TPD, showing no trend regarding the order in the periodic table.
| Sample | BET surface area [m2 g−1] | BJH pore volume [cm3 g−1] | Isotherme/hysteresis [type] |
|---|---|---|---|
| Ma-Sap | 458 | 0.88 | Type IV/H3 hysteresis |
| V-Sap* | 198 | 0.56 | Type IV/H1 hysteresis |
| Cr-Sap* | 247 | 0.39 | Type IV/H1 hysteresis |
| Mn-Sap* | 293 | 0.55 | Type IV/H3 hysteresis |
| Fe-Sap* | 343 | 0.65 | Type IV/H3 hysteresis |
| Co-Sap | 308 | 0.23 | Type IV/H2 hysteresis |
| Ni-Sap | 498 | 1.03 | Type IV/H3 hysteresis |
| Cu-Sap* | 324 | 0.41 | Type IV/H4 hysteresis |
| Zn-Sap | 107 | 0.13 | Type IV/H2 hysteresis |
Despite the differences in the material structure, the composition of the materials was determined via inductively coupled plasma with optical emission spectroscopy (ICP-OES), and the metal contents were normalized to 6.8 moles of silicon (Si) based on the molecular formula of saponites (Table S4, ESI†). Protons (H+) compensated for the absence of sodium (Na+). The elemental analysis of V-Sap* revealed a stoichiometric coefficient of only 0.6 for vanadium, against the expected 6, supporting the XRD and XPS findings that only a small amount of V2O5 has been incorporated into the aluminosilicate structure presumably due to oxidation with oxygen and the formation of soluble vanadium anionic complexes such as HVO42−, HV2O73−, and VO43− in alkaline solutions, which show repulsive interactions with the negatively charged aluminosilicate gel.90 Moreover, the Mg-Sap, showed 30% less magnesium incorporation as desired. This value is in good agreement with the results of Vogels et al. and can be attributed to the final pH during the synthesis at which Mg2+ partially stays in solution.55 For all other materials, the expected stoichiometric ratios of the individual metals (Si = 6.8, Al = 1.2 and M = 6) were obtained. The analysis of the interlayer cations across all samples revealed a stoichiometric coefficient for sodium (Na+) ranging from 0.0 to 0.5, which is significantly lower than the expected value of 1.2. This observation suggests that sodium cations were exchanged with protons (H+), introduced during the washing process due to the autoprotolysis of water.
Synthesis and characterization results are summarized in Fig. 4. Saponites were formed with Mg, Co, Ni, and Zn. For Fe, a mix of a saponite-like material and FeO(OH) was observed. The oxides Cr2O3, Mn2O3, and CuO were found on an aluminosilicate support for Cr, Mn, and Cu, respectively. Additionally, Cu-Sap* showed the formation of a Cu-phyllosilicate. Only low amounts of V2O5 were detected in the V-Sap* sample, primarily resulting in an aluminosilicate. Besides the different structures and compounds of the materials, the expected elemental compositions were obtained except for V. Moreover, high mesoporous surfaces (demonstrated by type IV hysteresis (Table 3 and Fig. S7, ESI†) could be obtained for all samples showing their potential for conversion of ethanol using heterogeneous catalysis.
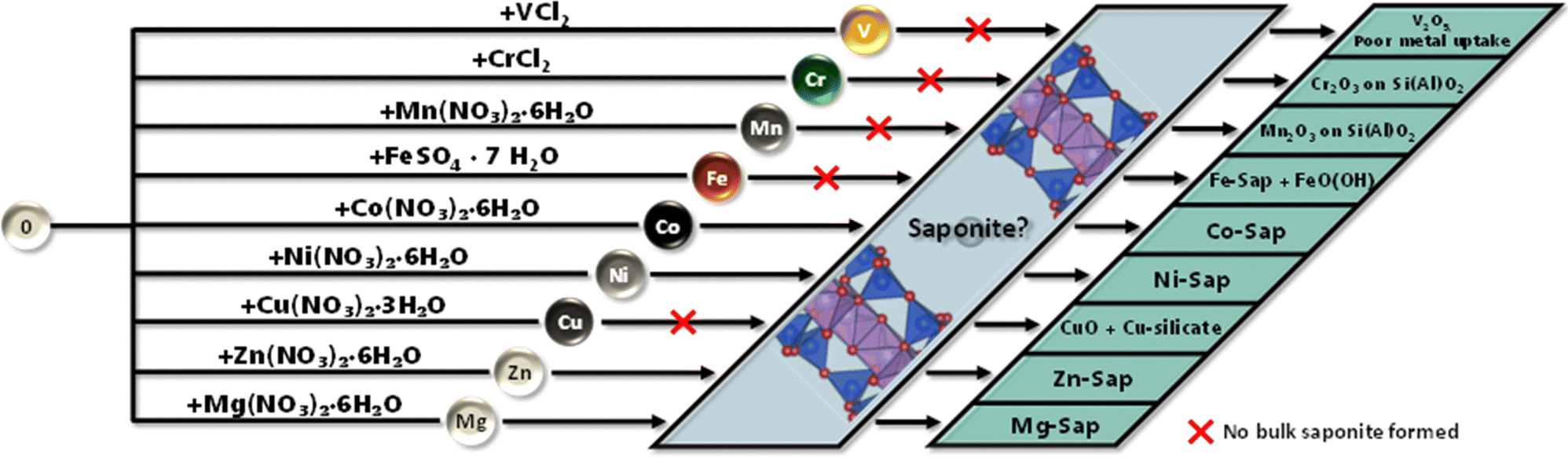 | ||
| Fig. 4 Graphical summary of the 3d-material synthesis, from left to right: in a first step an aluminosilicate is formed, then a 3d-metal salt is added, forming the M-Sap, which were extensively characterized to obtain their final composition. | ||
Influence of 3d-metal catalyst and temperature on the conversion of ethanol
To test the catalytic activity of the synthesized materials a continuous plug-flow reactor was used with 100 mL min−1 EtOH (5 mol%) in N2 at a WHSV of 1 h−1 and temperatures ranging from 473.15 K to 723.15 K. Notable products, intermediates and by-products are 1,3-butadiene (BD), acetaldehyde (AcA) and ethylene (ethene), respectively. The following chapter discusses the temperature influence on the catalytic conversion of the synthesized materials (Fig. 5). Moreover, since the surface area (Table 3) and surface acidity (Table S3, ESI†) are important in the ETB reaction, we discuss the influence of these two factors on the catalytic behavior of the materials.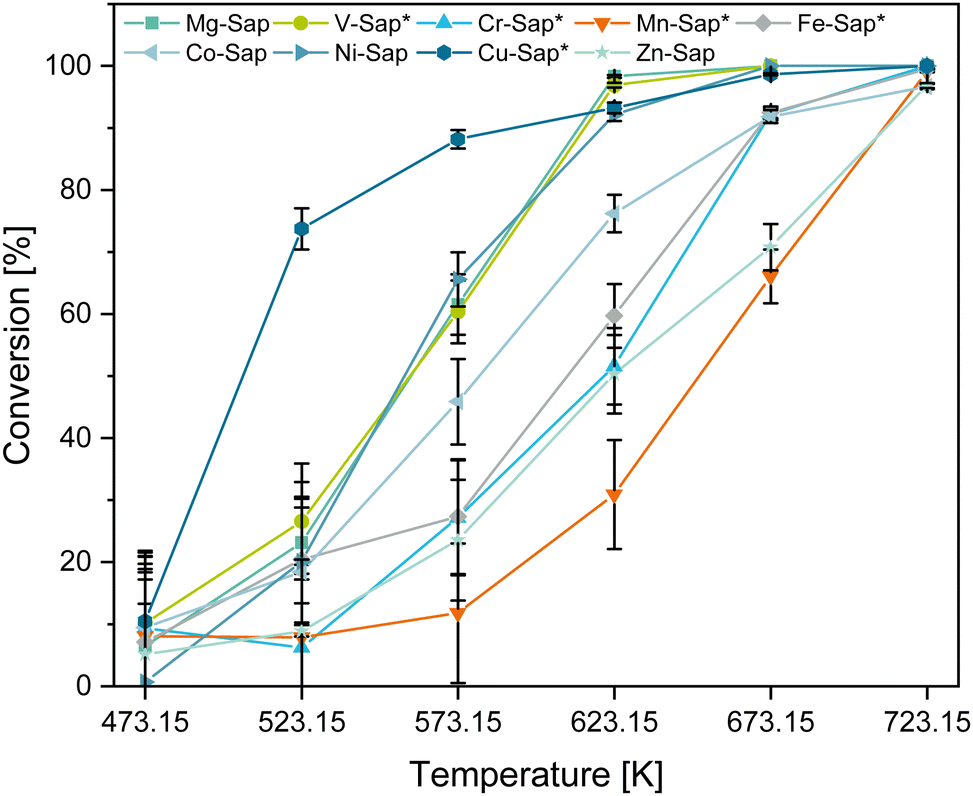 | ||
| Fig. 5 Ethanol conversion with the synthesized materials at 473.15 to 723.15 K. | ||
To better understand whether the catalytic performance is an effect of the saponite structure or of specific metal centers characteristics, both phase pure and non-phase pure saponite materials were tested.
The Cu-Sap* shows the highest conversion at lower temperatures with 88 ± 2% at 573.15 K, which can be attributed to the overall highest concentration of surface acid (4.6 mmol g−1) and basic (1.5 mmol g−1) sites (Table S3, ESI†). Consistent with this observation, the Mn-Sap* and Zn-Sap samples show the overall lowest concentrations of surface acid (0.9 and 0.7 mmol g−1) and basic (0.6 and 0.2 mmol g−1) sites, hence they exhibit inferior ethanol conversions of 12 ± 11% and 23 ± 10% at 573.15 K, respectively. Regarding the surface area, even though the Zn-Sap has the lowest surface area (107 m2 g−1) and surface-active site concentration (0.7 mmol g−1 acidic and 0.2 mmol g−1 basic sites), it shows a globally higher ethanol conversion than the Mn-Sap* sample. Additionally, although Ni-Sap features the largest surface area with 498 m2 g−1, the Cu-Sap*, with a 35% lower surface area (324 m2 g−1) but an 88% higher total concentration of active sites (3.2 to 6.1 mmol g−1), exhibits a 22% greater ethanol conversion at 573.15 K. These findings indicate that the concentration of catalytic active sites is a more crucial factor for high ethanol conversion activity than surface area.
In general, the main products for all materials, as displayed in Tables S5–S10 (ESI†), are diethyl ether (DEE), ethene, AcA, and BD. Besides these products propene, 1-butene, methane, methanol, ethyl acetate, butyraldehyde (butanal), and crotonaldehyde were detected ranging from 0.3 g kgcat−1 h−1 propene with the Zn-Sap at 623.15 K to 37.2 g kgcat−1 h−1 ethyl acetate with the Co-Sap at 723.15 K. Crotyl alcohol was not detected, which can be attributed to the high dehydration activity of the materials leading to an instant dehydration to BD. The observation of these products is in good agreement with the reaction scheme displayed above (Fig. 1). Thus, it can be assumed that the proposed mechanism and side reactions apply to the investigated catalytic systems. Moreover, at temperatures above 623.15 K higher C.
A key performance indicator for the catalytic activity and industrial relevance of a material is the productivity for a desired product. The product composition varies with the choice of catalyst, leading to a classification of the investigated catalysts into three categories, namely ethene-producing, acetaldehyde-producing, and 1,3-butadiene-producing, which will be discussed in detail below.
Ethene-producing catalysts
Fig. 6 displays the productivity for DEE, ethene, AcA and BD for the Mg-Sap, V-Sap*, and Cr-Sap*. These catalysts show the highest ethene productivity of the investigated materials. Notably, especially for these catalysts, and for the others as well, the productivity of DEE first increases and then decreases with rising temperature. The decrease in DEE productivity is accompanied by a significant increase in ethene productivity. This observation suggests a consecutive reaction from DEE to ethene, following reactions (a) and (b) in Fig. 1. Alternatively, the reaction way changes to a direct dehydration ethanol at higher temperatures. For further clarification, kinetic studies need to be conducted. | ||
| Fig. 6 Conversion of ethanol and productivity of DEE, ethene, AcA and BD for the ethylene-producing catalysts; (a) Mg-Sap, (b) V-Sap*, and (c) Cr-Sap*. | ||
Using the Mg-Sap and V-Sap* catalysts the highest DEE and ethene productivities of 166 to 194 gDEE kgcat−1 h−1 and 404 to 448 gethene kgcat−1 h−1 were reached, demonstrating excellent condensation and dehydration capabilities of these catalysts. While both materials reach similar ethene productivities of around 400 gethene kgcat−1 h−1 at 673.15 K, at higher temperatures, they achieve full conversion yet display distinct catalytic behaviors. While the dehydration capability of V-Sap* increases, resulting in a higher ethene productivity and lower AcA productivity, the activity of the Mg-Sap in the Toussaint–Kagan pathway (Fig. 1, green arrows) intensifies, leading to elevated formation of AcA and BD. As the V- and Cr-Sap* exhibit AcA productivities of 82 to 96 gAcA kgcat−1 h−1 but show almost no BD (4 to 7 gBD kgcat−1 h−1) formation, it can be concluded that the necessary combinations of catalytic sites for the aldol condensation of AcA are not present on these catalysts. For the Mg-Sap, on the other hand, small amounts of BD (10 gBD kgcat−1 h−1) and AcA (24 gAcA kgcat−1 h−1) were observed, while ethene represents the main product. These results suggest that the availability of Mg–O–Si sites is not the only requirement for a bulk material to selectively form BD from ethanol. Other catalytically active sites, such as Brønsted acids (H+) involved in ethene formation as shown by Lewandowski et al.,91 can also compete in the reaction. However, the V-Sap* was identified as the best catalyst out of the investigated materials for the dehydration of ethanol as the observed ethene productivity (448 gethene kgcat−1 h−1) corresponds to a yield of 74% with the used ethanol feed (eqn (4)). Since the V-Sap* primarily comprises an aluminosilicate with a minimal amount of incorporated V, comparable Al2O3/SiO2 catalysts reaches high ethene yields of >97%,92 showing the potential of the obtained aluminosilicate for the dehydration of ethanol. To understand the catalytic behavior, the acidic and basic properties of the materials were investigated by NH3- and CO2-TPD, respectively (Table S3, ESI†). From the three catalysts, the V-Sap* exhibited the highest acid/base ratio (Ra/b) (Ra/b = 5.6), with a distinct M and Al to Si ratio compared to the two other materials. With the Cr-Sap* and Mg-Sap showing intermediate acidities (Ra/b = 2.3 and 2.4), no correlation between the acid/base ratio and the catalytic selectivity of the samples was observed. Instead, variations in material composition, metal site characteristics, and structure were attributed to more significantly influence the activity in converting ethanol to ethene.
Acetaldehyde-producing catalysts
Since AcA is one of the major intermediates in the reaction of ethanol to 1,3-butadiene (BD), materials with a high AcA productivity could be beneficial to produce BD in the Ostromisslensky or in the Lebedev process, in combination with other metals or by adjusting the acid–base properties of these material.10 In our study, the Cu-Sap* sample (Fig. 7a) showed the highest AcA productivity at 573.15 K of 466 gAcA kgcat−1 h−1, while the Zn-Sap (Fig. 7b) shows the second highest AcA productivity of 62 gAcA kgcat−1 h−1 at this temperature due to a high selectivity towards AcA (48.8%). Additionally, the AcA productivity of Zn-Sap increased with rising temperature to reach the overall highest AcA productivity of 528 gAcA kgcat−1 h−1 at 723.15 K. Thus, these two catalysts were identified as good dehydrogenation catalyst for ethanol at the respective temperatures.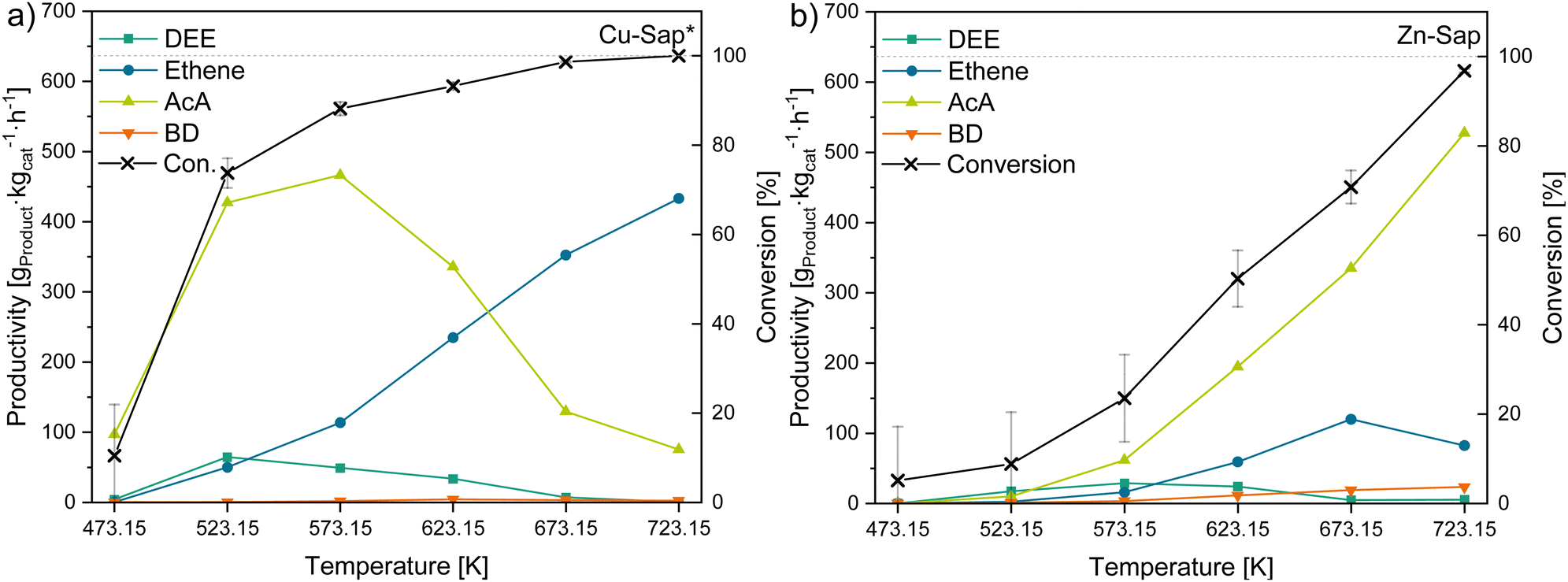 | ||
| Fig. 7 Conversion of ethanol and productivity of DEE, ethene, AcA and BD for the AcA-producing catalysts; (a) Cu-Sap* and (b) Zn-Sap*. | ||
While for the Cu-Sap* the AcA productivity declines above 573.15 K, the ethene productivity increases linearly from 623.15 K to 723.15 K reaching 433 gethene kgcat−1 h−1. This observation implies an alteration in the reaction mechanism of the catalyst, transitioning from functioning as a dehydrogenation agent to acting as a dehydration agent of ethanol with higher temperatures. As the V-Sap* and Cr-Sap* showed mostly ethene formation above 573.15 K, this change in catalytic behavior can presumably be attributed to the phyllosilicate obtained in the Cu-Sap* sample, which can act as dehydration catalyst.92 As both catalysts show a similar Ra/b value of 3.2, the difference in the catalytic behavior is attributed to their different composition, leading to different catalytically active sites for the dehydrogenation of ethanol to AcA. Furthermore, an increase in ethyl acetate selectivity with rising AcA productivity (Tables S5–S10, ESI†) was observed for these catalysts as well as for the 1,3-butadiene producing catalysts discussed below. This increase could be attributed to the higher partial pressure of AcA at elevated levels of conversions, which enhances the thermodynamic favorability of the dehydrogenation–condensation reaction between AcA and ethanol (reaction e, Fig. 1). Additionally, the Zn-Sap started to produce small quantities of BD, butanal and crotonaldehyde at temperatures above 623.15 K, which can be also attributed to increased AcA concentrations leading to the formation of these products via the Toussaint–Kagan pathway and the hydrogenation of crotonaldehyde to butanal (reaction j, Fig. 1). These results suggests that the aldol condensation is the rate determining step with the Zn-Sap, which could require additional Lewis acid-weak base pairs as shown by Sushkevich et al.93 Overall, the two dehydrogenation catalysts obtained high AcA yields of 49% with the Cu-Sap* and 55% with the Zn-Sap at 573.15 K and 723.15 K, respectively. For the Zn-Sap the obtained yields are comparable to Zn containing zeolites (YAcA = 62%) investigated by Gao et al.94 Whereas, Zhang et al. obtained AcA yields of 88% with Cu phyllosilicates synthesized via an ammonia evaporation method, showing the potential of the Cu-Sap*.95,96
1,3-Butadiene-producing catalysts
Although the predominant products for the Mn-, Fe-, Co-, and Ni-Sap materials were DEE, ethene, and AcA, they showed higher BD productivity compared to the ethene and AcA-specific catalysts. Consequently, the focus with these catalysts will be on the products generated via the Toussaint–Kagan pathway. Fig. 9 displays the productivities of DEE, ethene, AcA, and BD for the Mn-Sap*, Fe-Sap*, Co-, and Ni-Sap.It was observed that the lowest temperature necessary for BD production increased in the reverse order of the 3d-metals in the periodic table, ranging from 523.15 K to 673.15 K, with the order Ni < Co < Fe < Mn. A possible explanation can be concluded from the mechanism proposed by Ordomsky et al.93 (Fig. 8) who showed that the abstraction of the β-H is an important step in aldol condensation, with the activation of the second acetaldehyde for the aldol coupling reaction occurring on a Brønsted acid site of a neighboring silanol group.
 | ||
| Fig. 8 Mechanism for the aldol condensation on metal oxide centers adapted from Ordomsky et al.93 | ||
 | ||
| Fig. 9 Conversion of ethanol and productivity of DEE, ethene, AcA and BD for the BD-producing catalysts; (a) Mn-Sap*, (b) Fe-Sap*, (c) Co-Sap, and (d) Ni-Sap. | ||
From this we expect an interaction of an AcA carbonylic oxygen with the electron accepting metal center. Thus, the observed trend of the decreasing temperature for BD formation could be attributed to the increase in electronegativity (EN) from Mn towards Ni in the periodic table. We assume that the higher EN difference in the M–O bound enhances the positive inductive effect (+I) of the metal center leading to a stronger fractional positive charge (δ+) on the carbonylic C atom as well as higher basicity of the lattice oxygen weakening the C–H-bond on the β-methyl group, reducing the activation energy for an aldol coupling reaction.
This trend regarding the EN is also represented in the conversions, showing higher conversion over the whole temperature range for Ni > Co > Fe > Mn, which can be attributed to the lower activation energy required with different catalysts. Additionally, the Ni-Sap showed the highest BD productivities at temperatures below 623.15 K with an increasing BD productivity up to 38 gBD kgcat−1 h−1. Above this temperature the BD and AcA productivity rapidly decreases until at 723.15 K only methane and ethene were detected. Moreover, a clogging of the reactor occurred, which was attributed to decomposition of ethanol leading to the formation of coke similar to ethanol reforming over nickel containing catalysts at 573 to 1073 K as described by Ni et al.97
The Mn-, Fe-, and Co-Sap exhibited a different behavior reaching their highest BD productivities at 723.15 K with 51, 18, and 38 gBD kgcat−1 h−1, respectively. While these samples showed an increase in AcA productivity with rising temperature, the behavior of the BD productivity was different. Besides the varying initial temperatures for BD formation, the Fe-and Co-Sap showed stagnation of BD productivity at 673.15 K, while the Mn-Sap* displayed a linear increase up to 723.15 K. While for the Fe-Sap* and Co-Sap crotonaldehyde and ethyl acetate were detected at 673.15 K (Table S9, ESI†) and butanal at 723.15 K (Table S10, ESI†), these products were observed for the Mn-Sap* only at 723.15 K. The occurrence of ethyl acetate was attributed to the increase in acetaldehyde concentration following reaction e) (Fig. 1) as described for the AcA producing catalysts. The observation of butanal and crotonaldehyde can be attributed to low concentrations of ethanol at high conversion rates. Due to the absence of ethanol, the Meerwein–Ponndorf–Verley (MPV) reduction (reaction iv) can no longer proceed, leading to an accumulation of crotonaldehyde, which can then be reduced to butanal by the hydrogen produced in reaction i. Thus, a stagnation of BD productivity occurred.
Although Mn-Sap* and Ni-Sap, as well as Fe-Sap* and Co-Sap exhibit similar Ra/b values respectively, the initial temperature of BD production varied between all samples. Moreover, the Fe-Sap*, Co-Sap, Cu-Sap*, and Zn-Sap catalysts exhibit similar Ra/b values (3.2 to 3.3), yet the product composition shows significant differences. For instance, DEE and ethene are the primary products for Fe-Sap* and Co-Sap, following reactions (a) and (b) as shown in Fig. 1. In contrast, the main product for Cu-Sap* (at 573.15 K) and Zn-Sap (at 723.15 K) was AcA, following reaction (i) of the Toussaint–Kagan pathway. Thus, no correlation between the Ra/b values and the product selectivity was found.
Conclusion
We herein present the capability for customizing the octahedral layers of saponite-based bulk catalysts to produce BD and other valuable chemicals, such as ethene and acetaldehyde. Saponite structures were successfully obtained for Mg, Co, Ni, and Zn. However, V, Cr, Mn, Fe, and Cu showed the formation of various kinds of oxides on the surface of aluminosilicate supports.Regarding the conversion, it was found that the concentration of catalytically active sites plays a more important role for high ethanol conversion than surface area and acid to base ratio. Additionally, it was proposed, that a higher electronegativity of the used metals reduces the activation energy for the aldol coupling and the ethanol dehydrogenation using the BD producing (Mn-Sap*, Fe-Sap*, Co-Sap, and Ni-Sap) and AcA producing catalysts (Cu-Sap* and Zn-Sap), respectively. Thus, the more pronounced factors influencing the conversion and BD productivity were found to be the amount of catalytically active sites and composition of the catalysts.
At 573.15 K, Cu-Sap* and Ni-Sap exhibited the highest productivity for acetaldehyde (466 gAcA kgcat−1 h−1) and BD (31 gBD kgcat−1 h−1), respectively. At an increased temperature of 735.15 K, Zn- and Mn-Sap* demonstrated the highest productivity for acetaldehyde (466 gAcA kgcat−1 h−1) and BD (51 gBD kgcat−1 h−1), correspondingly. Thus, combining each of the two respective metals in a saponite-like bulk material seems promising to increase BD productivity towards industrial relevant production rates, as AcA is an important intermediate in the Toussaint–Kagan mechanism. Overall, the investigated catalysts require further optimization to achieve competitive BD yields compared to other studies on multifunctional catalysts reporting BD yields above 70%.21,38,39
Saponite structure was only obtained for Mg-, Ni-, Co-, and Zn-Sap, suggesting that M–O–Si sites are not necessary for BD formation if different catalytic sites are in close spatial proximity. This assumption is supported by the catalytic results obtained with the base saponite (Mg-Sap), which showed ethene (353 gethene kgcat−1 h−1) as the main product with small amounts of BD (10 gBD kgcat−1 h−1) and AcA (24 gAcA kgcat−1 h−1). Thus, the availability of Mg–O–Si sites is not the only requirement for a bulk MgO/SiO2 material to selectively produce BD from ethanol. However, M–O–Si sites could reduce deactivation mechanisms such as sintering and reduction by hydrogen produced in the first step of the Toussaint–Kagan mechanism. To understand the full potential of the cheap multifunctional and tailorable bulk saponite-based catalysts to reach industrial relevant BD productivity (300 gBD kgcat−1 h−1) further modification of the composition of the octahedral layer and the interlayer cations is necessary. The adjustment of the saponite composition by introducing AcA producing metals in the magnesium saponite structure seems promising to obtain higher BD productivity. Moreover, modification of interlayer cations could potentially influence the electronic properties of the T–O–T saponite layer to obtain more active saponite materials for the Lebedev process. This work highlights the importance of the electronic effects in multifunctional metal oxide catalysts for the ETB reaction besides their acid properties showing the need for further investigations focusing on both electronic and acidic properties of ETB catalysts.
Author contributions
Marc Greuel: conceptualization, methodology, investigation, writing – original draft, writing – review & editing, visualization. Stefan Kaluza: methodology, writing – reviewing & editing. Ulf-Peter Apfel: methodology, writing – reviewing & editing. Clara Watermann: methodology. Heiko Lohmann: methodology. Barbara Zeidler-Fandrich: funding acquisition, project administration.Data availability
All data supporting the findings can be found in the article and ESI,† or is available from the authors upon request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial support of the European Regional Development Fund (EFRE-0400156), and the measurements provided by the Department of Inorganic Chemistry I at the Ruhr University Bochum.Notes and references
- W. Ipatiew, J. Prakt. Chem., 1903, 421 Search PubMed.
- A. Requardt, Chem. Unserer Zeit, 2020, 54, 106–115, DOI:10.1002/ciuz.201900867.
- J. U. Nef, Justus Liebigs Ann. Chem., 1901, 318, 137–230, DOI:10.1002/jlac.19013180202.
- Great Britain Pat., GB331482A, 1929.
- I. I. Ostromisslensky, J. Russ. Phys.-Chem. Soc., 1915, 1472–1506 Search PubMed.
- D. Hradsky, P. Machac, D. Skoda, L. Leonova, P. Sazama, J. Pastvova, D. Kaucky, D. Vsiansky, Z. Moravec and A. Styskalik, Appl. Catal., A, 2023, 652, 119037, DOI:10.1016/j.apcata.2023.119037.
- P. C. A. Bruijnincx and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2013, 52, 11980–11987, DOI:10.1002/anie.201305058.
- S. Kim, S. Jeong and E. Heo, Energy Sources, Part B, 2019, 14, 49–66, DOI:10.1080/15567249.2019.1607631.
- K. Wagemann, Chem. Ing. Tech., 2014, 86, 2115–2134, DOI:10.1002/cite.201400108.
- I. Bin Samsudin, H. Zhang, S. Jaenicke and G.-K. Chuah, Chem. – Asian J., 2020, 15, 4199–4214, DOI:10.1002/asia.202001023.
- D. C. Blackley, in Synthetic Rubbers: Their Chemistry and Technology, ed. D. C. Blackley, Springer Netherlands, Dordrecht, 1983, pp. 93–163 Search PubMed.
- E4tech, RE-CORD, WUR, From the Sugar Platform to biofuels and biochemicals, Final report for the European Commission contract No. ENER/C2/423-2012/SI2.673791, https://biobasedeconomy.nl/wp-content/uploads/2015/10/EC-Sugar-Platform-final-report.pdf, (accessed 14 February 2023).
- ChemAnalyst, Butadiene Market Analysis: Plant Capacity, Production, Operating Efficiency, Demand & Supply, End-User Industries, Sales Channel, Regional Demand, Company Share, Foreing Trade, 2015–2035; Decode the Future of Butadiene, https://www.chemanalyst.com/industry-report/butadiene-market-623, (accessed 14 February 2023).
- Mordor Intelligence, Butadiene Market – Growth, Trends, COVID-19 Impact, and Forecasts (2023–2028), https://www.mordorintelligence.com/industry-reports/butadiene-market, (accessed 14 February 2023).
- Polaris Market Research Analysis, 1,3 Butadiene (BD) Market Share, Size, Trends, Industry Analysis Report, By Type (Extractive Distillation, and Oxidative Dehydrogenation); By Application; By Region; Segment Forecast, 2022–2030 PM1251, https://www.polarismarketresearch.com/industry-analysis/1-3-butadiene-market, (accessed 14 February 2023).
- C. E. Cabrera Camacho, B. Alonso-Fariñas, A. L. Villanueva Perales, F. Vidal-Barrero and P. Ollero, ACS Sustainable Chem. Eng., 2020, 8, 10201–10211, DOI:10.1021/acssuschemeng.0c02678.
- D. Cespi, F. Passarini, I. Vassura and F. Cavani, Green Chem., 2016, 18, 1625–1638, 10.1039/c5gc02148k.
- S. Farzad, M. A. Mandegari and J. F. Görgens, Bioresour. Technol., 2017, 239, 37–48, DOI:10.1016/j.biortech.2017.04.130.
- A. D. Patel, K. Meesters, H. den Uil, E. de Jong, K. Blok and M. K. Patel, Energy Environ. Sci., 2012, 5, 8430, 10.1039/C2EE21581K.
- S. Shylesh, A. A. Gokhale, C. D. Scown, D. Kim, C. R. Ho and A. T. Bell, ChemSusChem, 2016, 9, 1462–1472, DOI:10.1002/cssc.201600195.
- T. de Baerdemaeker, M. Feyen, U. Müller, B. Yilmaz, F.-S. Xiao, W. Zhang, T. Yokoi, X. Bao, H. Gies and D. E. de Vos, ACS Catal., 2015, 5, 3393–3397, DOI:10.1021/acscatal.5b00376.
- E. V. Makshina, W. Janssens, B. F. Sels and P. A. Jacobs, Catal. Today, 2012, 198, 338–344, DOI:10.1016/j.cattod.2012.05.031.
- M. Y. Kagan, G. D. Lyubarskii and O. M. Podurovskaya, Izv. Akad. Nauk SSSR, Ser. Khim., 1947, 173 CAS.
- G. Pomalaza, P. Arango Ponton, M. Capron and F. Dumeignil, Catal. Sci. Technol., 2020, 10, 4860–4911, 10.1039/d0cy00784f.
- H. Meerwein and R. Schmidt, Justus Liebigs Ann. Chem., 1925, 444, 221–238, DOI:10.1002/jlac.19254440112.
- W. Ponndorf, Z. Angew. Chem., 1926, 39, 138–143, DOI:10.1002/ange.19260390504.
- A. Verley, Bull. Soc. Chim. Fr., 1925, 37, 537–542 CAS.
- W. Tishchenko, Chem. Zentralbl., 1906, 38, 482–540 Search PubMed.
- W. Tishchenko, Chem. Zentralbl., 1906, 38, 355–418 Search PubMed.
- M. Iwamoto, Catal. Today, 2015, 242, 243–248, DOI:10.1016/j.cattod.2014.06.031.
- C. R. V. Matheus, L. H. Chagas, G. G. Gonzalez, E. Falabella, S. Aguiar and L. G. Appel, ACS Catal., 2018, 8, 7667–7678, DOI:10.1021/acscatal.8b01727.
- P. Gallezot and D. Richard, Catal. Rev., 1998, 40, 81–126, DOI:10.1080/01614949808007106.
- W. J. Toussaint, J. T. Dunn and D. R. Jachson, Ind. Eng. Chem., 1947, 39, 120–125, DOI:10.1021/ie50446a010.
- E. V. Makshina, M. Dusselier, W. Janssens, J. Degrève, P. A. Jacobs and B. F. Sels, Chem. Soc. Rev., 2014, 43, 7917–7953, 10.1039/c4cs00105b.
- P. I. Kyriienko, O. V. Larina, S. O. Soloviev and S. M. Orlyk, Theor. Exp. Chem., 2020, 56, 213–242, DOI:10.1007/s11237-020-09654-2.
- B. B. Corson, E. E. Stahly, H. E. Jones and H. D. Bishop, Ind. Eng. Chem., 1949, 41, 1012–1017, DOI:10.1021/ie50473a028.
- A. D. Winkelman, V. L. Dagle, T. L. Lemmon, L. Kovarik, Y. Wang and R. A. Dagle, Catal. Sci. Technol., 2023, 13, 975–983, 10.1039/d2cy01722a.
- V. L. Dagle, M. D. Flake, T. L. Lemmon, J. S. Lopez, L. Kovarik and R. A. Dagle, Appl. Catal., B, 2018, 236, 576–587, DOI:10.1016/j.apcatb.2018.05.055.
- V. L. Sushkevich and I. I. Ivanova, ChemSusChem, 2016, 9, 2216–2225, DOI:10.1002/cssc.201600572.
- V. L. Sushkevich and I. I. Ivanova, Appl. Catal., B, 2017, 215, 36–49, DOI:10.1016/j.apcatb.2017.05.060.
- V. L. Sushkevich, I. I. Ivanova and E. Taarning, Green Chem., 2015, 17, 2552–2559, 10.1039/c4gc02202e.
- V. L. Sushkevich, D. Palagin and I. I. Ivanova, ACS Catal., 2015, 5, 4833–4836, DOI:10.1021/acscatal.5b01024.
- H. Dai, T. Ye, K. Wang, M. Zhang, L.-M. Wu and G. Ouyang, Catalysts, 2022, 12, 1147, DOI:10.3390/catal12101147.
- S. Kvisle, A. Aguero and R. Sneeden, Appl. Catal., 1988, 43, 117–131, DOI:10.1016/S0166-9834(00)80905-7.
- P. Akisanmi, in Mineralogy, ed. M. René, IntechOpen, 2022 Search PubMed.
- K. Emmerich, Handbook of Clay Science, Elsevier, 2013, 381–404 Search PubMed.
- S. M. Auerbach, K. A. Carrado and P. K. Dutta, Handbook of Layered Materials, CRC Press, 2004 Search PubMed.
- Y. Kitayama, K. Shimizu, T. Kodama, S. Murai, T. Mizusima, M. Hayakawa and M. Muraoka, Impact of Zeolites and other Porous Materials on the new Technologies at the Beginning of the New Millennium, Proceedings of the 2nd International FEZA (Federation of the European Zeolite Associations) Conference, Elsevier, 2002, pp. 675–682 Search PubMed.
- Y. Kitayama and A. Abe, Nippon Kagaku Kaishi, 1989, 1824–1829, DOI:10.1246/nikkashi.1989.1824.
- Y. Kitayama and A. Michishita, J. Chem. Soc., Chem. Commun., 1981, 401, 10.1039/C39810000401.
- V. Gruver, A. Sun and J. J. Fripiat, Catal. Lett., 1995, 34, 359–364, DOI:10.1007/BF00806885.
- J. Delacaillerie, V. Gruver and J. J. Fripiat, J. Catal., 1995, 151, 420–430, DOI:10.1006/jcat.1995.1044.
- M. Greuel, C. M. Watermann, K. K. Matschuk, M. M. Krisam, A. Menne and B. Zeidler-Fandrich, Chem. Ing. Tech., 2024, 96, 671–678, DOI:10.1002/cite.202300207.
- J. Maier, M. Greuel, M. Hausruckinger, M. Oppmann, C. Waterman and B. Schug, Chem. Ing. Tech., 2024, 96, 679–687, DOI:10.1002/cite.202300209.
- R. Vogels, Am. Mineral., 2005, 90, 931–944, DOI:10.2138/am.2005.1616.
- C. H. Zhou, Q. Zhou, Q. Q. Wu, S. Petit, X. C. Jiang, S. T. Xia, C. S. Li and W. H. Yu, Appl. Clay Sci., 2019, 168, 136–154, DOI:10.1016/j.clay.2018.11.002.
- C. P. Ponce and J. T. Kloprogge, Life, 2020, 10, 168–200, DOI:10.3390/life10090168.
- F. Carniato, G. Gatti and C. Bisio, New J. Chem., 2020, 44, 9969–9980, 10.1039/d0nj00253d.
- S. Marchesi, F. Carniato, M. Guidotti, M. Botta, L. Marchese and C. Bisio, New J. Chem., 2020, 44, 10033–10041, 10.1039/c9nj05983k.
- R. Besselink, T. M. Stawski, H. M. Freeman, J. Hövelmann, D. J. Tobler and L. G. Benning, Cryst. Growth Des., 2020, 20, 3365–3373, DOI:10.1021/acs.cgd.0c00151.
- L. Lutterotti, M. Voltolini, H.-R. Wenk, K. Bandyopadhyay and T. Vanorio, Am. Mineral., 2010, 95, 98–103, DOI:10.2138/am.2010.3238.
- N. Yoshinaga and N. Kanasaki, Clay Sci., 1993, 9, 43–51, DOI:10.11362/jcssjclayscience1960.9.43.
- D. Zhu, L. Wang, W. Yu and H. Xie, Sci. Rep., 2018, 8, 5282, DOI:10.1038/s41598-018-23174-z.
- H. Klein and J. David, Acta Crystallogr., Sect. A: Found. Crystallogr., 2011, 67, 297–302, DOI:10.1107/S0108767311006581.
- N. F. M. Yusoff, N. H. Idris, M. F. M. Din, S. R. Majid, N. A. Harun and M. M. Rahman, Sci. Rep., 2020, 10, 9207, DOI:10.1038/s41598-020-66148-w.
- Z. Cao and C. Zuo, RSC Adv., 2017, 7, 40243–40248, 10.1039/c7ra06188a.
- I. Buljan, C. Kosanović, B. Subotić, N. Novak Tušar, A. Ristić, R. Gabrovšek, V. Kau
![[c with combining breve]](https://www.rsc.org/images/entities/char_0063_0306.gif) i and T. Mišić Radić, Cryst. Growth Des., 2010, 10, 838–844, DOI:10.1021/cg901194k.
i and T. Mišić Radić, Cryst. Growth Des., 2010, 10, 838–844, DOI:10.1021/cg901194k. - A. Bumajdad, S. Al-Ghareeb, M. Madkour and F. A. Sagheer, Sci. Rep., 2017, 7, 14788, DOI:10.1038/s41598-017-14779-x.
- A. Ramírez, P. Hillebrand, D. Stellmach, M. M. May, P. Bogdanoff and S. Fiechter, J. Phys. Chem. C, 2014, 118, 14073–14081, DOI:10.1021/jp500939d.
- L. Li, C. Guo, J. Ning, Y. Zhong, D. Chen and Y. Hu, Appl. Catal., B, 2021, 293, 120203, DOI:10.1016/j.apcatb.2021.120203.
- M. A. Dar, S. H. Nam, Y. S. Kim and W. B. Kim, J. Solid State Electrochem., 2010, 14, 1719–1726, DOI:10.1007/s10008-010-1022-z.
- Y.-F. Han, F. Chen, Z. Zhong, K. Ramesh, L. Chen and E. Widjaja, J. Phys. Chem. B, 2006, 110, 24450–24456, DOI:10.1021/jp064941v.
- N. S. McIntyre and D. G. Zetaruk, Anal. Chem., 1977, 49, 1521–1529, DOI:10.1021/ac50019a016.
- J. Zhang, Q. Yin, Y. Wu, S. Zhang, K.-J. Wang and J. Han, J. Energy Chem., 2022, 67, 92–100, DOI:10.1016/j.jechem.2021.10.007.
- C. Mosser, A. Mosser, M. Romeo, S. Petit and A. Decarreau, Clays Clay Miner., 1992, 40, 593–599, DOI:10.1346/CCMN.1992.0400514.
- G. Silversmit, D. Depla, H. Poelman, G. B. Marin and R. de Gryse, J. Electron Spectrosc. Relat. Phenom., 2004, 135, 167–175, DOI:10.1016/j.elspec.2004.03.004.
- Z.-B. Wang, X. Xu, S.-L. Bai, Y.-J. Guan and S.-T. Jiang, Appl. Phys. A: Mater. Sci. Process., 2017, 123, 1–8, DOI:10.1007/s00339-017-0775-y.
- J. T. Kloprogge and C. P. Ponce, Minerals, 2021, 11, 112, DOI:10.3390/min11020112.
- J. Theo Kloprogge and B. J. Wood, Clay Sci., 2018, 22, 85–94, DOI:10.11362/jcssjclayscience.22.4_85.
- V. S. Kshirsagar, A. C. Garade, K. R. Patil, R. K. Jha and C. V. Rode, Ind. Eng. Chem. Res., 2009, 48, 9423–9427, DOI:10.1021/ie801941e.
- Y. Wei, J. Horlyck, M. Song, J. Scott, R. Amal and Q. Cao, Appl. Catal., A, 2020, 592, 117418, DOI:10.1016/j.apcata.2020.117418.
- T. Lehmann, T. Wolff, C. Hamel, P. Veit, B. Garke and A. Seidel-Morgenstern, Microporous Mesoporous Mater., 2012, 151, 113–125, DOI:10.1016/j.micromeso.2011.11.006.
- J. W. Coenen, Appl. Catal., 1991, 75, 193–223, DOI:10.1016/S0166-9834(00)83132-2.
- R. Al-Gaashani, S. Radiman, A. R. Daud, N. Tabet and Y. Al-Douri, Ceram. Int., 2013, 39, 2283–2292, DOI:10.1016/j.ceramint.2012.08.075.
- S. Yokoyama, K. Tamura, T. Hatta, S. Nemoto, Y. Watanabe and H. Yamada, Clay Sci., 2006, 13, 75–80, DOI:10.11362/jcssjclayscience1960.13.75.
- A. R. Gonzalez-Elipe, J. P. Espinos, G. Munuera, J. Sanz and J. M. Serratosa, J. Phys. Chem., 1988, 92, 3471–3476, DOI:10.1021/j100323a031.
- T. Ebina, T. Iwasaki, A. Chatterjee, M. Katagiri and G. D. Stucky, J. Phys. Chem. B, 1997, 101, 1125–1129, DOI:10.1021/jp9622647.
- T. L. Barr, S. Seal, K. Wozniak and J. Klinowski, J. Chem. Soc., Faraday Trans., 1997, 93, 181–186, 10.1039/A604061F.
- T. Ogneva, K. Emurlaev, Y. Malyutina, E. Domarov, I. Chakin, A. Ruktuev, P. Riabinkina, A. Yurgin and I. Bataev, Metals, 2023, 13, 1689, DOI:10.3390/met13101689.
- P. Sun, K. Huang and H. Liu, Hydrometallurgy, 2016, 165, 370–380, DOI:10.1016/j.hydromet.2015.10.026.
- M. Lewandowski, G. S. Babu, M. Vezzoli, M. D. Jones, R. E. Owen, D. Mattia, P. Plucinski, E. Mikolajska, A. Ochenduszko and D. C. Apperley, Catal. Commun., 2014, 49, 25–28, DOI:10.1016/j.catcom.2014.02.003.
- M. Zhang and Y. Yu, Ind. Eng. Chem. Res., 2013, 52, 9505–9514, DOI:10.1021/ie401157c.
- V. V. Ordomsky, V. L. Sushkevich and I. I. Ivanova, J. Mol. Catal. A: Chem., 2010, 333, 85–93, DOI:10.1016/j.molcata.2010.10.001.
- K. Gao, J. Mielby and S. Kegnæs, Catal. Today, 2022, 405–406, 144–151, DOI:10.1016/j.cattod.2022.06.023.
- J. Pang, M. Yin, P. Wu, X. Li, H. Li, M. Zheng and T. Zhang, Green Chem., 2021, 23, 7902–7916, 10.1039/D1GC02799A.
- H. Zhang, H.-R. Tan, S. Jaenicke and G.-K. Chuah, J. Catal., 2020, 389, 19–28, DOI:10.1016/j.jcat.2020.05.018.
- M. Ni, D. Y. Leung and M. K. Leung, Int. J. Hydrogen Energy, 2007, 32, 3238–3247, DOI:10.1016/j.ijhydene.2007.04.038.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ma01036a |
| This journal is © The Royal Society of Chemistry 2025 |