
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unveiling the impact of the mpg-C3N4@Pa@Ni nanocomposite in the reduction of nitroaromatic derivatives by comparative solvent-free methods†
Fatemeh
Eshrati
,
Hossein
Ghafuri
 *,
Peyman
Hanifehnejad
and
Haniyeh
Dogari
*,
Peyman
Hanifehnejad
and
Haniyeh
Dogari
Catalysts and Organic Synthesis Research Laboratory, Department of Chemistry, Iran University of Science and Technology, 16846-13114 Tehran, Iran. E-mail: ghafuri@iust.ac.ir
First published on 4th December 2024
Abstract
In this research, the impact of the mesoporous graphitic carbon nitride–papain–nickel (mpg-C3N4@Pa@Ni) nanocomposite in the reduction of hazardous nitroaromatic derivatives was investigated under solvent-less and solvent-free conditions. The mpg-C3N4@Pa@Ni composite was synthesized in four steps; synthesizing bulk and mesoporous g-C3N4, and functionalization with 1,3-dibromopropane, papain, and Ni nanoparticles. Papain was found to be a suitable composite material due to its ability to form covalent and coordination bonds with the substrate and Ni. Several solvent-free and solvent-less methods, including using mortar and pestle, ball mill, microwave, and magnetic stirrer, were employed to investigate the reduction of nitroaromatic compounds due to their fast, simple, and economical green nature. The synthesized nanocomposite demonstrated high efficiency rates in reducing toxic nitroaromatic compounds ranging from 80–98.6%. Structural confirmation of the mpg-C3N4@Pa@Ni nanocomposite was carried out using various techniques such as Fourier-Transform Infrared spectroscopy (FT-IR), N2 adsorption analysis (BET), Field Emission Scanning Electron Microscopy (FE-SEM), Energy-dispersive X-ray spectroscopy (EDS), X-ray Diffraction spectroscopy (XRD), and Thermogravimetric Analysis (TGA). Furthermore, the mpg-C3N4@Pa@Ni nanocomposite showed promising recoverability without significant decreases in efficiency for up to eight cycles, indicating its potential as a sustainable and efficient catalyst. The synthesis of mpg-C3N4@Pa@Ni nanocomposite and its efficient performance in reducing hazardous nitroaromatic compounds pave the way for a sustainable and environmentally friendly alternative to traditional methods.
1. Introduction
Recently, carbon-based nanomaterials, including graphene, carbon nanotubes, and g-C3N4, have been applied in various scientific fields, including disinfection, supercapacitor, carbon dioxide reduction, degradation of pollutants, and catalysis.1,2Based on the reported literature, g-C3N4 has superior advantages in terms of tunable band gap, abundant and low-cost precursors, biocompatibility, and structural versatility.3–8 Pristine g-C3N4 has a low surface area and limited applications, while its other morphologies can broadly cover these limitations.9 mpg-C3N4 has advantages over pristine g-C3N4 in terms of surface area, tunable pore size, chemical stability, and catalytic selectivity.10,11 In addition, by compositing mpg-C3N4, its properties can be improved to a great extent.12 Enzymes are biocatalysts that offer high specificity and efficiency in catalyzing reactions, making them valuable components for composite material development.13 Integrating enzymes with substrates like g-C3N4 can enhance catalytic activity, stability, and functionality, opening up different application opportunities in various fields.3 Papain, a proteolytic enzyme derived from papaya, exhibits unique properties that make it an attractive candidate for composite formation with g-C3N4.14 So far, much research has been published on the applications of g-C3N4 composites. Li et al. utilized graphene oxide (GO) to enhance the interfacial conductivity of g-C3N4, leading to the construction of a 3D porous g-C3N4/GO (p-CNG) framework through a template-assisted thermal treatment process.
Moreover, as electron acceptors, precious metal (Au, Pd, Pt) co-catalysts were individually anchored onto the 3D p-CNG framework to enhance active site density and facilitate electron–hole separation. Then, they tested its ability in the hydrogen evolution reaction and reported its activity to be 2565.81 μmol g−1 h−1 at pH = 10.5.15 Gao et al. prepared a CoNiSx-g-C3N4 composite catalyst through a straightforward hydrothermal method. Then, they applied it in CO2 reduction with methane and carbon monoxide production rates of 0.904 and 11.77 μmol, respectively. g-C3N4 composites are being explored to address various challenges that still require significant improvements. One of the global challenges today is water pollution by chemical industries.16 Chemical industries often cause pollution by releasing harmful substances such as dyes, hazardous by-products, volatile organic compounds (VOCs), and air-suspended particles.17,18 Nitroaromatic compounds have been widely utilized in pesticides, producing explosives, dyes, perfumes, pigments, industrial fabrication of pharmaceuticals, and insecticides.19–22 Based on the information provided, the yearly output of nitroaromatic compounds exceeds 225000 metric tons, and approximately 9000 metric tons of this substance are released into water sources yearly.23 The United States Environmental Protection Agency has classified nitrobenzene (simplest nitroaromatic) as a priority contaminant and has set maximum allowable concentrations of 1 mg L−1 and 17 μg L−1 in wastewater and drinking water, respectively.24 The choice of the most effective method for removing nitroaromatic compounds, such as reduction, adsorption, advanced oxidation, or biodegradation, depends on various factors, including the specific properties of the contaminants, the desired treatment outcomes, and the efficiency and feasibility of the treatment process.25
While each method has advantages and limitations, reduction methods can offer fast reaction rates, potential for regeneration, and selective removal.26–28 When nitroaromatic compounds are selectively reduced, they become amino aromatic derivatives that are more biodegradable, less toxic, and can be used as crucial synthetic intermediates in commercial processes.29
Metal-catalyzed reduction reactions may benefit from the cooperative interactions between metals and g-C3N4, enhancing catalytic performance and selectivity.30 Pd, Ag, Cu, and Ni are the metals used with various substrates for nitroaromatic reduction reactions, including GO, MOFs, and mesoporous carbons.31 For instance, Amit Saha et al. have asserted that the chemoselective reduction of nitroaromatic compounds to the corresponding amino derivatives has been achieved by combining Cu nanoparticles and ammonium formate in ethylene glycol as a solvent.32 Moreover, the reaction yield was in the range of 75–90% at 120 °C in 8–12 h. Sean M. Kelly et al. have succeeded in reducing nitroaromatic compounds with zinc dust as a catalyst in the presence of water solvent at room temperature, with the best efficiency of 99% in 4 h.33 Despite the innumerable efforts to design the optimal conditions for removing and reducing nitroaromatic compounds, these methods have limitations, such as equal solvent amount, solvent toxicity, high temperature, and long reaction time.34 Thus, clean and efficient methods for reducing nitroaromatic compounds must be selected.35 Mechanochemical methods are a set of techniques that involve the use of mechanical force to induce chemical reactions.36–38 Unlike traditional chemical synthesis methods that rely on heat and solvents, mechanochemical methods utilize mechanical energy to drive reactions.39
In this research, we carried out a comprehensive study involving the synthesis and identification of the mpg-C3N4@Pa@Ni nanocomposite. Our primary objective was to investigate the catalytic performance of this nanocomposite in reducing nitroaromatic compounds using solvent-free and solvent-less methods, including using mortar and pestle, ball mill, microwave, and magnetic stirrer.40–42 Through experiments, we identified that the magnetic stirrer method had the lowest energy consumption due to its short reaction time, environmentally friendly nature, high yield, and minimal solvent usage. Moreover, we observed an extraordinary recovery of the nanocomposite after eight reaction cycles in the magnetic stirrer method, indicating its remarkable performance in this reaction. One of the key challenges in the field of catalyst design for the reduction of nitroaromatic derivatives is the identification and optimization of active sites that can efficiently drive the reduction process. In the case of the mpg-C3N4@Pa@Ni nanocomposite, the active site responsible for the reduction of nitroaromatic derivatives is primarily the Ni nanoparticles. These nanoparticles provide the necessary surface for the adsorption of nitroaromatic compounds and facilitate the electron transfer required for their reduction.
Compared to other reported catalysts, the mpg-C3N4@Pa@Ni nanocomposite offers significant advantages. It achieves high efficiency (up to 98.6%) in significantly shorter reaction times and under mild conditions. Additionally, this catalyst demonstrates excellent recyclability, maintaining its catalytic activity after multiple cycles without a significant loss in efficiency. Furthermore, the solvent-free nature of the reduction process adheres to green chemistry principles, making this nanocomposite a more sustainable and environmentally friendly alternative to conventional methods. Overall, our research provides valuable insights into the synthesis and properties of the mpg-C3N4@Pa@Ni nanocomposite, highlighting its potential applications in catalytic reactions. This study not only addresses the challenge of designing efficient catalysts for nitroaromatic reduction but also demonstrates the advantages of mpg-C3N4@Pa@Ni as a high-performance, recyclable, and green catalyst.
2. Experimental
2.1. Materials
Phosphoric acid (95%), sulfuric acid (96%), hydrazine hydrate (98%), and other solvents and materials were purchased from Sigma-Aldrich and Merk Companies. FT-IR, FE-SEM, EDS, XRD, TGA, BET, NMR spectroscopy, and melting point analyses were carried out utilizing a Tensor 27, TESCAN-MIRA III, Numerix DXP–X10P, Dron-8 diffractometer, STA504, micromeritics ASAP 2020, VARIAN Inova 500 MHz, and Electrothermal 9100, respectively. Moreover, an Elma ultrasonic bath (80 kHz) was utilized for sonication. In addition, a Retsch Ball-mill apparatus was used with a 20 mL iron cell and two 12 mm diameter iron balls, operating at a frequency of 20 Hz. Additionally, a national model NN-6653 microwave with a maximum power output of 900 W was also utilized.2.2. Preparation of mesoporous graphitic carbon nitride (mpg-C3N4)
10 g melamine was heated in a furnace at 550 °C for 4 h to obtain bulk g-C3N4. Then, 2 g obtained bulk g-C3N4 was mixed with 20 mL sulfuric acid and 20 mL phosphoric acid in a 600 mL beaker, for 5 h at 90 °C. Subsequently, 100 mL ethanol was added to the mixture and stirred at 25 °C. After 2 h, the beaker was placed overnight at 25 °C until the precipitate settles well. In the next step, solvent spilled over, and then the resulting mixture was sonicated for 7 h to obtain the desired morphology and improve the catalyst's efficiency. Eventually, the mpg-C3N4 powder was obtained by separation, washing with deionized water (DW) and ethanol (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) by centrifugation, and drying at 60 °C.43 A schematic for the preparation of mpg-C3N4 is shown in Fig. 1.
:1) by centrifugation, and drying at 60 °C.43 A schematic for the preparation of mpg-C3N4 is shown in Fig. 1.
 | ||
| Fig. 1 The schematic preparation of mpg-C3N4. | ||
2.3. Preparation of mpg-C3N4@Pr–Br
In a round-bottom flask (50 mL), 1 g mpg-C3N4 was sonicated with 40 mL dry toluene for 1 h. Next, 2 mL 1,3-dibromopropane and 0.163 g potassium iodide (KI) were added to the previous mixture and refluxed for 24 h under N2 atmosphere. Subsequently, the obtained precipitate was separated by centrifugation and washed several times with ethanol:ethyl acetate (2:1) to remove unreacted 1,3-dibromopropane. In the last step, the final product was dried at 60 °C to obtain mpg-C3N4@Pr–Br powder. A schematic of mpg-C3N4@Pr–Br is illustrated in Fig. 2.
 | ||
| Fig. 2 Schematic of mpg-C3N4@Pr–Br. | ||
2.4. Preparation of mpg-C3N4@papain
1 g mpg-C3N4@Pr–Br was dissolved in 3 mL dry toluene for 30 min. Thereafter, 1.6 g KI and 0.83 g papain were added to the mixture and refluxed for 12 h. Ultimately, the final product was separated, washed several times with DW:methanol (1:1), and dried at 60 °C. A schematic of mpg-C3N4@Pr@papain is depicted in Fig. 3.
 | ||
| Fig. 3 Schematic of mpg-C3N4@Pr@papain. | ||
2.5. Preparation of mpg-C3N4@Pa@Ni
0.5 g NiCl2 was poured into a round-bottom flask (25 mL) containing 20 mL DW and stirred until it was completely dissolved. Then, 0.2 g mpg-C3N4@papain was added to the mixture and sonicated for 3 h. Moreover, 5 mL hydrazine hydrate and 2 mL sodium hydroxide (1 M) were added to the mixture and vigorously stirred for 3 h at 60 °C. Finally, mpg-C3N4@Pa@Ni was obtained by centrifugation, washed with DW:ethanol (1:1), and dried at 60 °C. A schematic of mpg-C3N4@Pa@Ni is shown in Fig. 4.
 | ||
| Fig. 4 Schematic of mpg-C3N4@Pa@Ni. | ||
2.6. Reduction of nitroaromatic compounds catalyzed by mpg-C3N4@Pa@Ni nanocomposite
Nitroaromatic derivatives (0.125 mmol), sodium borohydride (NaBH4) (1 mmol), mpg-C3N4@Pa@Ni (10 mg), and water as a solvent (5 μL) were added to a 25 mL round-bottom flask at room temperature. After 2 min, upon completion of the reaction (monitored by thin layer chromatography (TLC)), the catalyst was separated using a centrifuge. Then, the final product was purified via recrystallization.2.7. Selected spectra data
3. Results and discussion
3.1. mpg-C3N4@Pa@Ni nanocomposite characterizations
To characterize the mpg-C3N4@Pa@Ni functional groups in each synthesis step, FT-IR analysis was taken from mpg-C3N4 (Fig. 5a), mpg-C3N4@Pr–Br (Fig. 5b), papain (Fig. 5c), mpg-C3N4@Pr@papain (Fig. 5d), and mpg-C3N4@Pa@Ni (Fig. 5e). Fig. 5a shows a wide peak around 3100–3400 cm−1 which may be related to the stretching vibration of –NH2, –NH, groups of carbon nitride, and OH groups of adsorbed solvent. In addition, the peaks appearing around 1620, 1323, and 795 cm−1 are due to the stretching vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N, C–N, and triazine groups in mpg-C3N4. Moreover, in Fig. 5b, the peak around 2776 cm−1 is related to the stretching vibration of the C–H groups associated with the presence of the 1,3-dibromopropane linker in the nanocomposite structure. Furthermore, in Fig. 5c, the broad peak around 3288–3540 cm−1 is related to the stretching vibration of –OH and N–H groups can be observed. Also, the peak around 2800–2967 cm−1 is related to the stretching vibration of the C–H groups. Furthermore, the peaks around 1651 and 1550 cm−1 are attributed to CO stretching vibrations of amides and acids, which are related to papain. Besides, in Fig. 5d, all the peaks related to the previous steps are observed, which confirms the nanocomposite synthesis up to this step. Finally, in Fig. 5e, it was observed that no specific peak was detected for nickel, which may be due to its low loading in the nanocomposite.
N, C–N, and triazine groups in mpg-C3N4. Moreover, in Fig. 5b, the peak around 2776 cm−1 is related to the stretching vibration of the C–H groups associated with the presence of the 1,3-dibromopropane linker in the nanocomposite structure. Furthermore, in Fig. 5c, the broad peak around 3288–3540 cm−1 is related to the stretching vibration of –OH and N–H groups can be observed. Also, the peak around 2800–2967 cm−1 is related to the stretching vibration of the C–H groups. Furthermore, the peaks around 1651 and 1550 cm−1 are attributed to CO stretching vibrations of amides and acids, which are related to papain. Besides, in Fig. 5d, all the peaks related to the previous steps are observed, which confirms the nanocomposite synthesis up to this step. Finally, in Fig. 5e, it was observed that no specific peak was detected for nickel, which may be due to its low loading in the nanocomposite.
 | ||
| Fig. 5 The FT-IR spectra of (a) mpg-C3N4, (b) mpg-C3N4@Pr–Br, (c) papain, (d) mpg-C3N4@papain and (e) mpg-C3N4@Pa@Ni. | ||
FT-IR spectroscopy is a commonly used analytical technique in chemistry that helps identify functional groups in a given sample. However, due to its inherent limitations, it was impossible to confirm the presence of nickel particles in the final nanocomposite. In order to confirm the presence of mpg-C3N4@Pa@Ni elements in each step of the synthesis process, EDS analysis was performed on the final nanocomposite (as shown in Fig. 6). The EDS analysis confirmed that the first step in the synthesis process had yielded mpg-C3N4, as evidenced by the presence of carbon and nitrogen elements. The second step, which involved the addition of papain, was also evident from the appearance of oxygen elements. Finally, the presence of nickel element indicated the successful composition of Ni nanoparticles in the final step of mpg-C3N4@Pr@papain nanocomposite synthesis. Thus, the results of the EDS analysis provide conclusive evidence that the desired nanocomposite has been synthesized accurately in a step-by-step fashion.
 | ||
| Fig. 6 The EDS spectrum of mpg-C3N4@Pa@Ni. | ||
The synthesized nanocomposite of mpg-C3N4@Pa@Ni has undergone crystallographic analysis using wide-angle XRD spectra ranging from 10° to 80° to determine its structure and composition. The results of this analysis are presented in Fig. 7. The XRD pattern of mpg-C3N4 was referenced to previous literature.44 Upon analyzing the diffractograms of the nanocomposite, it was observed that distinctive peaks were present at 2θ values of 13.05° and 26.87°. These peaks were attributed to the mpg-C3N4, which confirmed its presence in the composite material.45 Additionally, peaks at 2θ values of 13.05°, 19.98°, and 38.02° were observed, indicating papain's presence in the structure. According to the published literature, the presence of nickel nanoparticles in the final nanocomposite structure was confirmed by peaks at 2θ values of 41.56°, 49.91°, and 77.34°.46 The improved crystallinity of the Ni nanoparticles on the mpg-C3N4 substrate suggests that they have been effectively composited with the mpg-C3N4. Therefore, it can be concluded that the synthesis of Ni nanoparticles on the mpg-C3N4 substrate with modifications has resulted in a successful fabrication of mpg-C3N4@Pa@Ni nanocomposite with improved crystallinity.
 | ||
| Fig. 7 The XRD pattern of mpg-C3N4@Pa@Ni. | ||
The morphological images of mpg-C3N4@Pa@Ni nanocomposite were examined using FE-SEM to understand its structural characteristics (Fig. 8). The FE-SEM images in Fig. 8a and b display the synthetic porous carbon nitride substrate, which confirms the porosity of the structure. The porous substrate has numerous cavities, as evident from the images. As seen in Fig. 8c and d, the mpg-C3N4 surface becomes rough after being modified and through the composition with papain and nickel nanoparticles. The final nanocomposite displays an intricate network of cavities and materials, revealing the potential applications of mpg-C3N4@Pa@Ni nanocomposites in various fields.
 | ||
| Fig. 8 FE-SEM images of (a) and (b) mpg-C3N4 and (c) and (d) mpg-C3N4@Pa@Ni nanocomposite. | ||
The mesoporous nature of the mpg-C3N4@Pa@Ni nanocomposite is confirmed through both BET (Fig. 9) and pore volume distribution analysis (Fig. 10). The pore volume distribution reveals two prominent peaks in the ranges of 40–50 Å and 100–200 Å, indicating the predominance of mesopores within the material. These peaks suggest a highly concentrated distribution of mesoporous structures, contributing to a high surface area and optimal accessibility for surface reactions. The first peak, around 40–50 Å, represents smaller mesopores, while the second peak, in the range of 100–200 Å, indicates the presence of larger mesopores. The gradual decrease in pore volume beyond 200 Å suggests that the nanocomposite is primarily composed of mesoporous structures, with fewer larger pores.
 | ||
| Fig. 9 Isotherm linear plots of (a) mpg-C3N4 and (b) mpg-C3N4@Pr@papain@Ni. | ||
 | ||
| Fig. 10 Pore size distribution patterns of mpg-C3N4@Pa@Ni. | ||
Further confirmation of the mesoporous structure comes from the BET analysis, which determined the surface area and average pore size of the mpg-C3N4@Pa@Ni nanocomposite. As shown in Table 1, the BET surface area of the synthesized mpg-C3N4 is 85.9976 m2 g−1, with an average pore size of 24.8 nm. In comparison, literature values for g-C3N4 typically range from 24.1 to 59.4 m2 g−1, indicating that the synthesized version exhibits a significantly higher surface area, suggesting a more porous morphology. This increase in surface area, attributed to the augmentation of the mesoporous surface area, is critical for enhancing the material's functionality.
| Sample | Surface area (m2 g−1) | Pore volumea (cm3 g−1) | Average size (nm) | Ref. |
|---|---|---|---|---|
| a Determined at p/p0 = 0.97, where p is the equilibrium pressure and p0 is the saturation pressure of nitrogen at −196 °C. | ||||
| g-C3N4 nanosheet | 24.1 | — | — | 47 |
| g-C3N4 nanosheet | 59.4 | — | — | 48 |
| mpg-C3N4 | 85.9976 | 0.438832 | 24.8 | This work |
| mpg-C3N4@Pa@Ni | 98.9716 | 0.388148 | 15.5 | This work |
For the final mpg-C3N4@Pa@Ni nanocomposite, the BET surface area increased further to 98.9716 m2 g−1, though the average pore size decreased to 15.5 nm. This reduction in pore size is expected during the integration of additional components into the mesoporous substrate, a common occurrence in nanocomposite synthesis. However, this reduction also results in the creation of new surface structures that can improve the material's properties, particularly its reactivity. The higher surface area leads to greater exposure of active sites, enabling more efficient interactions with other substances. This characteristic is especially valuable in fields such as catalysis, energy storage, and environmental remediation, where enhanced surface interactions are crucial.
Moreover, the N2 adsorption/desorption isotherms for both mpg-C3N4 and mpg-C3N4@Pa@Ni exhibit a type (IV) adsorption isotherm, further confirming the mesoporous structure of the synthesized nanocomposite. This mesoporosity, combined with the increased surface area and new active surfaces, underscores the potential of mpg-C3N4@Pa@Ni for various applications.
The thermal stability of the mpg-C3N4@Pa@Ni nanocomposite has been thoroughly investigated using TGA analysis, as shown in Fig. 11. The TGA analysis revealed some interesting findings about the nanocomposite's thermal properties. Specifically, it was observed that the weight loss below 100 °C may be attributed to the volatilization of water or the solvent absorbed by the synthesized nanocomposite. Additionally, the weight loss observed between 225 °C and 285 °C would be credited to the thermal decomposition of papain. Moreover, the weight loss observed between 415 °C and 530 °C is related to the destruction of the structure of the mpg-C3N4 substrate. It is noteworthy that the remaining weight of the nanocomposite structure, which has not been destroyed up to 800 °C, is related to nickel nanoparticles occupying the mineral part of the structure. Furthermore, TGA analysis was used to calculate the mass percentage of different components of the mpg-C3N4@Pa@Ni nanocomposite (see the whole calculation in the ESI†). Based on Fig. 11, the solvent present in the structure contributes to approximately 8% of the total mass. Moreover, the TGA analysis of the mpg-C3N4@Pa@Ni nanocomposite revealed that papain and 1,3-dibromopropane constitute 50% of the total mass, while mpg-C3N4 contributes 19% and nickel nanoparticles contribute 23%. This information provides detailed insight into the composition of the unique mpg-C3N4@Pa@Ni nanocomposite, which has potential applications in various fields due to its remarkable thermal stability and composition.
 | ||
| Fig. 11 The TGA pattern of mpg-C3N4@Pa@Ni. | ||
3.2. Survey of different solvent-less methods toward nitroaromatics reduction
Reduction reactions, which involve the addition of electrons to the nitro group, are key to detoxifying nitroaromatic compounds. These reactions transform highly reactive and harmful nitro groups into less toxic amine or hydroxylamine derivatives. This conversion not only reduces the immediate toxicity of the compounds but also facilitates further degradation through microbial or abiotic processes. In this research, various parameters, such as solvent, reaction time, amount of catalyst, and reaction temperature, were investigated to optimize the reduction reaction conditions. To find the optimal reaction conditions, the reaction between 4-nitrophenol (0.25 mmol) and NaBH4 (1 mmol) along with mpg-C3N4@Pa@Ni as a catalyst (10 mg) and water as a solvent (5 μL) at 25 °C for 2 min was considered as the model reaction for reducing nitroaromatic compounds (Fig. 12). To achieve the highest possible product yield, the anti-solvent method (using a combination of ethyl acetate and n-hexane) was employed for product isolation. With these optimized conditions, the highest possible yield of the desired product was obtained. | ||
| Fig. 12 mpg-C3N4@Pa@Ni catalyzed reduction of nitroaromatic compounds. | ||
During the initial phase of optimizing the reaction conditions, it was observed that the reaction proceeded favorably at ambient temperature (25 °C) and in the presence of water as the solvent. To further optimize the reaction, these two parameters were kept constant. Afterward, the study aimed to explore the interaction of nitroaromatic derivatives (0.25 mmol) with sodium borohydride under various conditions and according to the principles of green chemistry. The study was performed in line with the principles of green chemistry, which emphasize the use of eco-friendly and sustainable chemical processes. These conditions included the presence or absence of catalyst and solvent. The reactions occurred at room temperature, which was set at 25 °C, using solvent-free methods such as using magnetic stirrer, microwave, ball mill, and mortar and pestle. During the study, the progress and completion of the reactions were monitored using TLC.
| Entry | Nitro compound (mmol) | NaBH4 (mmol) | Catalyst (mg) | Solvent (μL) | Time (min) | Yield (%) |
|---|---|---|---|---|---|---|
| 1 | 0.25 | 1 | 10 | — | 5 | Trace |
| 2 | 0.25 | 1 | 10 | 5 | 5 | 98.6 |
| 3 | 0.25 | 1 | 10 | 5 | 4 | 83 |
| 4 | 0.25 | 1 | 10 | 5 | 3 | 48 |
| Entry | Nitro compound (mmol) | NaBH4 (mmol) | Catalyst (mg) | Solvent (μL) | Ball mill (ball) | Time (min) | Yield (%) |
|---|---|---|---|---|---|---|---|
| 1 | 0.25 | 1 | 10 | — | 30 Hz (1) | 5 | 41 |
| 2 | 0.25 | 1 | 10 | — | 30 Hz (3) | 5 | 29 |
| 3 | 0.25 | 1 | 10 | 5 | 20 Hz (1) | 3 | 70 |
| 4 | 0.25 | 1 | 10 | 5 | 25 Hz (1) | 7 | 98.6 |
| 5 | 0.25 | 1 | 10 | 5 | 30 Hz (2) | 5 | 98.6 |
| 6 | 0.25 | 1.5 | 10 | 5 | 30 Hz (2) | 5 | 98.6 |
| 7 | 0.25 | 0.73 | 10 | 5 | 30 Hz (1) | 5 | 98.6 |
| 8 | 0.25 | 1 | 8 | 5 | 30 Hz (1) | 5 | 86 |
Firstly, the reaction was performed using 1 mmol NaBH4 and 10 mg catalyst, in the absence of water as a solvent with one ball, achieving an efficiency of 41% (entry 1). However, when the same reaction was performed using 3 balls, the efficiency dropped to 29% (entry 2). This difference can be attributed to the use of only one ball, which reduces the number of ineffective collisions and enhances the effective ones by providing the necessary energy to initiate the reaction. In the presence of 5 μL water and 3 min reaction time, using only one ball at a frequency of 20 Hz, the efficiency was significantly improved to 70% (entry 3). Further raising the frequency to 25 Hz resulted in achieving the desired product in 7 min with 98.6% efficiency, thanks to enhanced effective collision and the provision of necessary energy to initiate the reaction (entry 4). By increasing the number of balls to 2, adjusting the frequency to 30 Hz, utilizing 1 mmol NaBH4, and reducing the reaction time to 5 min, the reaction yield was attained to be 98.6% (entry 5). Comparing the results from entries 6 and 7, it is evident that while both achieved a high efficiency of 98.6%, several key reaction parameters were varied. In entry 6, two balls were used at a frequency of 30 Hz with 1.5 mmol NaBH4. In contrast, entry 7 involved the use of only one ball at a frequency of 30 Hz but with a reduced NaBH4 quantity of 0.73 mmol. Despite the reduction in the amount of NaBH4, the efficiency remained at 98.6%. This can be attributed to the fact that decreasing the number of balls from two to one reduces the number of ineffective collisions, allowing more efficient energy transfer and a higher proportion of effective collisions. Consequently, while the amount of reducing agent was lowered in entry 7, the optimized collision dynamics compensated for this change, maintaining the reaction efficiency. Therefore, both the number of balls and the amount of NaBH4 contribute significantly to the reaction's overall performance and must be considered together when optimizing conditions. However, diminishing the catalyst amount to 8 mg resulted in an 86% decrease in reaction efficiency within 5 min (entry 8). This decline can be ascribed to the reduction in catalyst quantity, consequently diminishing the available active sites for the reduction reaction.
Conclusively, the optimal reaction conditions involved 5 μL water, 0.73 mmol NaBH4, and 10 mg catalyst, using 1 ball at a frequency of 30 Hz for a duration of 5 min (entry 7). These conditions were found to be the most efficient in terms of reaction yield, time, and cost.
The optimal conditions were explored in the microwave method in the presence and absence of water as a solvent, various amounts of catalyst, and NaBH4 for different reaction times (see Table 4). Initially, the results showed that in the absence of water and using 1 and 1.5 mmol NaBH4 in the presence of 10 mg of catalyst, and within 5 min, no significant product was obtained (entries 1 and 2). In the trial, the addition of 5 μL water to the reaction within 2 min resulted in 98.6% efficiency (entry 3). Furthermore, by reducing the reaction time to 1 min, an efficiency of 42% was obtained (entry 4). These results indicate that the reaction is incomplete in less than 2 min, and adjusting the reaction time is markedly effective in efficiency. Furthermore, the influence of NaBH4 amount was examined, and it was discovered that reducing the NaBH4 amount to 0.73 mmol resulted in 91% efficiency (entry 5). Thus, a marginal decrease in NaBH4 quantity does not adversely impact reaction efficiency. Conversely, exploring the impact of the catalyst amount revealed that decreasing it to 8 mg led to a decline in reaction efficiency to 76% within 2 min (entry 6). This can be ascribed to the reduced catalyst amount, subsequently diminishing the active sites available for the reduction reaction. Ultimately, the best reaction conditions were identified using microwaves in the presence of water (5 μL), 1 mmol NaBH4, and 10 mg of catalyst in 2 min (entry 3). These findings provide valuable insights into the optimization of microwave-assisted reactions and can be useful for further development and improvement of chemical processes.
| Entry | Nitro compound (mmol) | NaBH4 (mmol) | Catalyst (mg) | Solvent (μL) | Time (min) | Yield (%) |
|---|---|---|---|---|---|---|
| 1 | 0.25 | 1 | 10 | — | 5 | Trace |
| 2 | 0.25 | 1.5 | 10 | — | 5 | Trace |
| 3 | 0.25 | 1 | 10 | 5 | 2 | 98.6 |
| 4 | 0.25 | 1 | 10 | 5 | 1 | 42 |
| 5 | 0.25 | 0.73 | 10 | 5 | 2 | 91 |
| 6 | 0.25 | 1 | 8 | 5 | 2 | 76 |
The mortar and pestle method required the least amount of energy, as it only relied on manual force and did not need any external energy input. However, the reaction time was longer, taking 5 min to reach 98.6% efficiency. Reducing the reaction time to 3 min led to a significant drop in yield (48%), showing that consistent manual mixing is challenging for shorter reaction times. This method is suitable for small-scale laboratory experiments, where simplicity and low cost are important, but its limited scalability and inconsistent mixing make it less ideal for larger, industrial applications.
The ball milling method provided more controlled and homogeneous mixing due to mechanical energy input. It reached 98.6% efficiency in 5 min at an optimal frequency of 30 Hz using 1 ball. While this method consumes more energy compared to using a mortar and pestle, it offers better precision and is scalable, making it suitable for both small- and large-scale applications. The mechanical agitation ensures uniform particle size reduction and consistent results, but its energy consumption is higher, which can be a limitation for energy-conscious processes.
The microwave method was the fastest, achieving 98.6% efficiency in just 2 min. Microwave heating provides selective energy transfer, accelerating the reaction kinetics. However, reducing the reaction time to 1 min drastically lowered the efficiency to 42%, which indicates that time control is critical. While microwaves are excellent for rapid reactions and energy-efficient in terms of speed, the overall energy usage can be higher compared to mechanical methods. Scaling up microwave reactors for industrial applications can also be challenging, as ensuring uniform heating across larger volumes requires specialized equipment.
The magnetic stirrer method stood out as the most energy-efficient and practical for scale-up. It achieved 98.6% efficiency in 2 min, with minimal energy consumption for stirring and very low solvent use. Additionally, the method demonstrated excellent catalyst recovery and reusability, maintaining high efficiency over eight cycles. Its simplicity, low energy requirements, and scalability make it the most suitable method for sustainable industrial-scale applications, where energy efficiency and ease of use are crucial.
In conclusion, although all methods reached similar catalytic efficiency, each method has specific advantages depending on the application. The magnetic stirrer method is ideal for large-scale, energy-efficient processes due to its low energy consumption and high performance. The ball milling method is suitable for applications requiring precise control and homogeneous mixing, though it is more energy-intensive. The microwave method excels in rapid reactions but requires careful time optimization and specialized equipment for scaling. Lastly, the mortar and pestle method, while simple and cost-effective, is best suited for small-scale, less demanding experiments where energy consumption is not a major concern.
| Entry | Nitro compound (mmol) | NaBH4 (mmol) | Catalyst (mg) | Solvent (μL) | Time (min) | Yield (%) |
|---|---|---|---|---|---|---|
| 1 | 0.25 | 2 | 10 | — | 9 | Trace |
| 2 | 0.25 | 2 | 10 | 5 | 9 | 98.6 |
| 3 | 0.25 | 1 | 10 | 5 | 2 | 98.6 |
| 4 | 0.25 | 1.5 | 7 | 5 | 5 | 84 |
| 5 | 0.25 | 1.5 | 10 | 5 | 1 | 48 |
| 6 | 0.25 | 1.6 | 5 | 5 | 2 | 40 |
| 7 | 0.25 | 0.73 | 15 | 5 | 2 | 95 |
In addition, the amount of catalyst and NaBH4 was varied to determine their effect on the reaction efficiency. By reducing the amount of catalyst to 7 mg, the active sites available for the reduction reaction were diminished, resulting in 84% efficiency (entry 4). Furthermore, by reducing the catalyst to 5 mg and increasing the amount of NaBH4 to 1.6 mmol, an efficiency of 40% was attained (entry 6), which can be attributed to the reduced amount of catalyst and fewer active sites for the reduction reaction. In the following, by reducing the amount of NaBH4 to 0.73 mmol and increasing the catalyst to 15 mg, a 95% efficiency was achieved within 2 min (entry 7). Thus, a negligible decrease in NaBH4 amount does not adversely impact reaction efficiency, and the reaction efficiency has been improved due to the increase in the amount of catalyst and, as a result, the increase in active sites. Finally, the optimal reaction conditions for the reduction of nitroaromatic compounds with NaBH4 were achieved in the presence of 5 μL water, at ambient temperature, and when stirred for 2 min (entry 3). These findings highlight the importance of optimizing reaction conditions to achieve maximum efficiency and can pave the way for future studies in this field.
In addition to optimizing the reaction conditions, the composition and quantity of materials in the nanocomposite also have a significant impact on catalytic performance. The nickel (Ni) content in the mpg-C3N4@Pa@Ni nanocomposite is pivotal in enhancing its catalytic performance by providing essential active sites. Therefore, the effect of increasing the nickel content in the nanocomposite was investigated. Doubling the Ni loading effectively increased the number of these active sites, leading to improved electron transfer and a boost in the reduction efficiency of nitroaromatic compounds to 99%. The well-dispersed Ni nanoparticles on the mesoporous g-C3N4 matrix facilitated synergistic interactions, enhancing catalytic activity without causing agglomeration. This optimized Ni loading maintained high selectivity and a balanced combination of activity and stability, as shown by TGA analysis indicating that Ni also contributes to the thermal stability of the catalyst. This stability enables effective reuse over at least eight cycles with minimal performance loss. However, while the increased Ni content did enhance reduction efficiency, the marginal improvement in catalytic performance may not justify the cost of doubling the Ni nanoparticle content on the composite. Therefore, careful consideration of Ni loading is crucial for achieving both maximum catalytic efficiency and cost-effectiveness, supporting the composite's long-term reusability in sustainable catalytic applications.
After examining various solvent-free and solvent-less methods to reduce nitroaromatic derivatives, even though 98.6% efficiency was obtained in all methods, the magnetic stirrer method was selected as the optimal method due to its ease of use and the lack of high energy consumption (Table 6). By using the magnetic stirrer method, the desired products were produced with remarkable performance. This was achieved by utilizing 1 mmol NaBH4, 10 mg catalyst, and 5 μL solvent, for 2 min at ambient temperature. Overall, the magnetic stirrer method proved to be a reliable and effective approach for regenerating derivatives.
| Entry | Method | NaBH4 (mmol) | Time (min) | Yield (%) |
|---|---|---|---|---|
| a Reaction conditions: nitroaromatic compounds (0.25 mmol), catalyst (10 mg), and solvent (5 μL), at 25 °C. | ||||
| 1 | Mortar and pestle | 1 | 5 | 98.6 |
| 2 | Ball milling (1 ball, 30 Hz) | 0.73 | 5 | 98.6 |
| 3 | Microwave | 1 | 2 | 98.6 |
| 4 | Magnetic stirrer | 1 | 2 | 98.6 |
Also, the reaction was performed under optimal conditions to evaluate the effect of each component on the reaction catalyst (Table 7). The first step involved using mpg-C3N4@Pr@Pa with NaBH4 as the reducing agent. However, the results showed a relatively low percentage of the intended product due to the limited reduction by NaBH4 as a reducing agent. On the other hand, the final nanocomposite (mpg-C3N4@Pa@Ni) without NaBH4 reduced the final product's percentage due to the lack of a proton-donating agent. Furthermore, using NaBH4 alone as the auxiliary reducing agent also did not yield significant results, with the product percentage being negligible. However, when Ni nanoparticles were added to the NaBH4, a more favorable percentage of 60% was observed due to the protonation of Ni by NaBH4, leading to an increase in reduction efficiency. Finally, using the mpg-C3N4@Pa@Ni nanocomposite with NaBH4 resulted in the best product percentage (98.6%) due to the favorable surface area and the presence of abundant active sites. In conclusion, the investigation results confirmed that the most effective catalyst for reducing nitroaromatic compounds is the mpg-C3N4@Pa@Ni nanocomposite with NaBH4. This study provides valuable insights into the development of efficient catalytic systems for reducing nitroaromatic compounds in the chemical industry.
In order to exhibit the catalytic merits of mpg-C3N4@Pa@Ni nanocomposite, the optimized conditions were applied to nitroaromatic compounds containing electron-donating or withdrawing groups, as shown in Table 8. The study aimed to determine the effects of different electron-donating and -withdrawing groups on the reaction process. The results showed that the reaction was not significantly influenced by the presence of either electron-donating or -withdrawing groups. In fact, all the nitroaromatic compounds were reduced in high yields, as demonstrated in Table 8. These findings suggest that using the mpg-C3N4@Pa@Ni nanocomposite could be a promising catalyst for reducing nitroaromatic compounds, regardless of their electronic properties (Table 8).
|
|
||||
|---|---|---|---|---|
| Entry | R1 | Yieldb (%) | M.P. (°C) | |
| Found | Reported (ref.) | |||
| a Reaction conditions: nitroaromatic compounds (0.25 mmol), NaBH4 (1 mmol), catalyst (10 mg), and solvent (5 μL), at 25 °C for 2 min. b The yields relate to the isolated product. | ||||
| 1 | H | 98.6 | Liquid sample | |
| 2 | 3-NO2 | 97.4 | 64–66 | 65–66 (49) |
| 3 | 2-NO2 | 97.2 | 99–103 | 100–102 (50) |
| 4 | 2-COPh | 98.4 | 106–107 | 105–107 (51) |
| 5 | 4-OH | 98.6 | 186–189 | 187–189 (52) |
| 6 | 4-CH2OH | 97.4 | 62–64 | 60–65 (50) |
| 7 | 3-CHO | 98 | 28–31 | 29–32 (53) |
| 8 | 4-COMe | 97.6 | 94–95 | 93–95 (54) |
| 9 | 3-COMe | 97.4 | 104–105 | 102–106 (55) |
| 10 | 2-CHO | 98.2 | 40–42 | 40–44 (56) |
| 11 | 4-NO2 | 97.6 | 143–146 | 144–147 (57) |
| 12 | 2-NO2-4-OMe | 97.4 | 46–48 | 45–46 (58) |
| 13 | 2-OH-5-Cl | 98.5 | 135–138 | 136.2–138.1 (59) |
| 14 | 4-COOH | 93 | 186–188 | 187–189 (60) |
| 15 | 2-Cl,5-NO2 | 97.1 | 64–68 | 65–68 (61) |
| 16 | 2-COOH | 84 | 145–146 | 145 (62) |
| 17 | 4-Br | 97.4 | 60–62 | 60–61 (63) |
| 18 | 4-COPh | 80 | 121–123 | 120.1–122.2 (64) |

3.3. Comparing the efficiency of mpg-C3N4@Pa@Ni nanocomposite with other published literature
For indicating the catalytic merits of mpg-C3N4@Pa@Ni nanocomposite the model reaction was compared with some other published literature, shown in Table 9. The results of the assessment, which are presented in Table 9, indicate that the synthesized nanocomposite has demonstrated high efficiency as a catalyst in the target reaction. These outcomes suggest that mpg-C3N4@Pa@Ni has the potential to be a promising catalyst for nitroaromatic compound reduction, aligning with green chemistry principles due to its shorter reaction time and reduced solvent usage in comparison to other methods.3.4. Catalytic mechanism
A schematic representation of the proposed mechanism for the reduction of nitroaromatic compounds using the mpg-C3N4@Pa@Ni nanocomposite is depicted in Fig. 13. The conversion of nitrobenzene to aniline by the mpg-C3N4@Pa@Ni nanocomposite involves several steps. Step (I): nitrobenzene (Ar-NO2) is adsorbed onto the Ni metal surfaces of the nanocomposite, which serves as the primary active site for the reduction process. The Ni nanoparticles play a crucial role by providing the surface necessary for the adsorption and activation of nitrobenzene. Step (II): NaBH4 reacts with water to produce hydride ions (H−), which are then adsorbed onto the surface of the Ni metal. Step (III): these hydride ions interact with the positively charged nitrogen atom of the nitro group in nitrobenzene, initiating the reduction process. Step (IV): a hydrogen shift occurs, wherein the hydrogen atom attached to the nitrogen transfers to one of the oxygens of the nitro group, resulting in resonance stabilization and forming a nitrogen-oxygen double bond. Step (V): another hydride ion attaches to the oxygen of the nitro group, converting it into a good leaving group (–H2O+) that subsequently departs from the molecule. Step (VI): the departure of the leaving group results in forming a nitroso intermediate. Step (VII): a hydride ion on the Ni surface subsequently interacts with the oxygen of the nitroso intermediate, facilitating its reduction. Step (VIII): another hydride ion then interacts with the nitroso intermediate, creating another suitable leaving group (–H2O+). Simultaneously, a hydride ion interacts with the nitrogen of the nitroso group. The elimination of the leaving group and the release of another water molecule leads to the formation of an amine group, resulting in the release of aniline from the catalyst surface. This mechanism underscores the efficiency of the mpg-C3N4@Pa@Ni nanocomposite as a catalyst in reducing nitrobenzene to aniline. Additionally, the role of mpg-C3N4 and papain in the nanocomposite should not be overlooked. The mesoporous structure of mpg-C3N4 enhances the dispersion of Ni nanoparticles, increasing the available surface area for catalytic reactions. Papain, on the other hand, contributes to the structural stability and coordination, further improving the catalytic performance of the nanocomposite. The combined effects of these components make the mpg-C3N4@Pa@Ni an efficient and robust catalyst for nitroaromatic reduction. Moreover, the nitroso intermediate (MP: 132–133 °C)69 was synthesized, and the reaction proceeded from step (VI) to validate the mechanism. | ||
| Fig. 13 The proposed mechanism for the reduction of nitroaromatic compounds. | ||
3.5. Reusability
The process of catalyst recycling is highly significant in green chemistry and environmental preservation, as it allows for the development of catalysts that are efficient, durable, and sustainable. This parameter is also essential in practical applications, as it aids researchers in identifying catalysts capable of enduring multiple reuse cycles. To ensure a thorough investigation, the quantities of all reaction components were doubled, which negligible losses during the separation of the catalyst after each cycle, as demonstrated in Fig. 14. This doubling of components was implemented to enhance experimental accuracy and to achieve the desired results with maximum efficiency. Initially, the catalyst's recyclability was tested up to five cycles and subsequently extended to eight cycles to assess its performance over a longer reuse period. During this extended evaluation, a slight reduction in efficiency was observed from 92% at cycle five to 87% at cycle eight indicating that the catalytic activity gradually decreases when the catalyst is used beyond five cycles. The study of the mpg-C3N4@Pa@Ni nanocomposite revealed that the content of the nanocomposite before washing was 20 mg, while after eight reuses, it decreased to 19.2 mg. This indicates a minimal reduction in reaction efficiency, which can be attributed to the occupation of the catalyst's active sites. The study revealed that the catalyst was effective and could be reused multiple times with minimal loss of efficiency. Upon conducting a comparative analysis of the FT-IR and EDS spectra of the recovered catalyst with the original catalysts depicted in Fig. S1 and S2 (ESI†), respectively, it can be inferred that the functional groups and elements have been retained. The analysis also indicates that the catalyst exhibits excellent performance even after undergoing eight cycles of use. These findings demonstrate that the catalyst maintains exceptional performance and structural integrity even after eight reuse cycles, underscoring its robustness and suitability for multiple applications with minimal efficiency loss.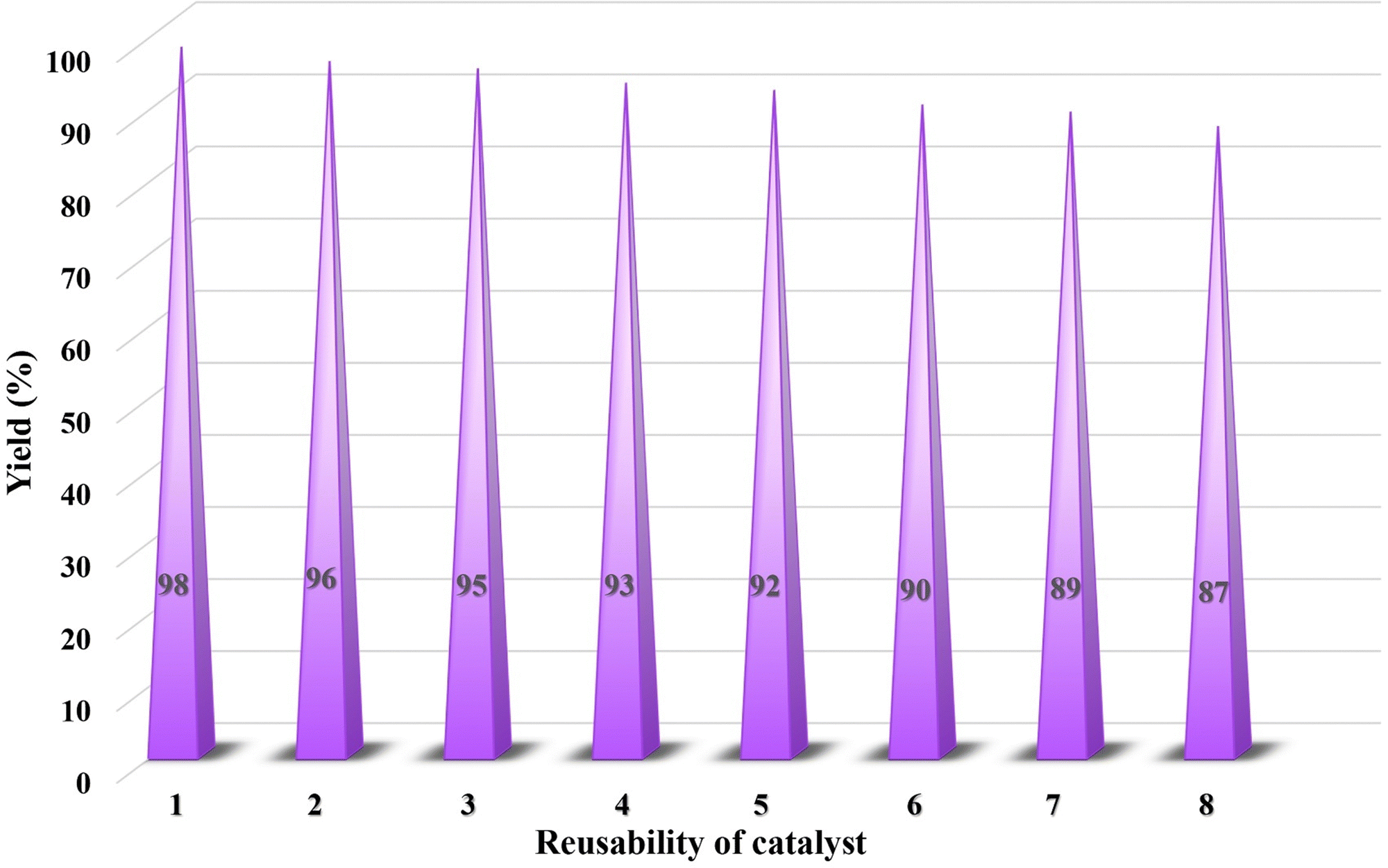 | ||
| Fig. 14 Recycling ability of the mpg-C3N4@Pa@Ni catalyst in the reduction of nitrobenzene. | ||
4. Conclusion
In this research, the mpg-C3N4@Pa@Ni nanocomposite, a highly effective catalyst for reducing harmful nitroaromatic compounds, was synthesized through a four-step process. Evidence-based reduction of harmful nitroaromatic compounds with several solvent-free methods such as using a mortar and pestle, ball mill, microwave, and magnetic stirrer were evaluated to identify the most effective way to perform this reaction. In the course of the investigation, it was discovered that all the methods under scrutiny achieved 98.6% efficiency. The results showed that the magnetic stirrer method was the most efficient, consumed the least amount of energy, and produced the desired outcome in the shortest time. Also, using the magnetic stirrer method, the recovery of the mpg-C3N4@Pa@Ni nanocomposite was investigated up to 8 cycles, and exhibited impressive performance. Moreover, even after eight recovery cycles, this nanocomposite demonstrated a significant yield for catalytic reactions. Using the solvent-less method as a green method and the absence of the need for high temperature, large amounts of solvent, and high energy made this method highly useful in reducing harmful nitroaromatic compounds. The results obtained from the study were deemed acceptable, indicating the potential of this nanocomposite to be applied in various industries to reduce harmful nitroaromatic compounds.Author contributions
Hossein Ghafuri was responsible for the conceptualization and supervision. Fatemeh Eshrati performed the experiments, writing of the original draft of the manuscript, and data analysis. Haniyeh Dogari analyzed data. Peyman Hanifehnejad assisted in data analysis, the original draft of the manuscript, and the methodology.Data availability
The data that support the findings of this study are available from the corresponding author, Hossein Ghafuri, upon reasonable request. The data include raw experimental data, analyzed data, and supporting information. Additionally, any supplementary information related to this study is included in the supplementary materials section of this manuscript. For any additional information or data requests, please contact ghafuri@iust.ac.ir.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge support from the Research Council of the Iran University of Science and Technology.References
- N. T. Dung, P. T. H. Hanh, V. D. Thao, N. T. Thuy, D. T. M. Thanh, N. T. Phuong, K.-Y. A. Lin and N. N. Huy, Environ. Sci.: Water Res. Technol., 2023, 9, 221–234 RSC.
- F. Besharat, F. Ahmadpoor, Z. Nezafat, M. Nasrollahzadeh, N. R. Manwar, P. Fornasiero and M. B. Gawande, ACS Catal., 2022, 12, 5605–5660 CrossRef CAS.
- A. Rashidizadeh, H. Ghafuri, N. Goodarzi and N. Azizi, Solid State Sci., 2020, 109, 106427 CrossRef CAS.
- H. Ghafuri, Z. Tajik, N. Ghanbari and P. Hanifehnejad, Sci. Rep., 2021, 11, 19792 CrossRef CAS PubMed.
- Y. Cao, G. Zhou, X. Chen, Q. Qiao, C. Zhao, X. Sun, X. Zhong, G. Zhuang, S. Deng and Z. Wei, J. Mater. Chem. A, 2020, 8, 124–137 RSC.
- S. Samanta and R. Srivastava, Mater. Adv., 2020, 1, 1506–1545 RSC.
- M. Singh, A. Kumar and V. Krishnan, Mater. Adv., 2020, 1, 1262–1272 RSC.
- V. SP, S. DS and A. B. Shbil, Adv. Mater. Process. Technol., 2024, 1–16 Search PubMed.
- H. Ghafuri, A. Rashidizadeh, M. G. Gorab and G. Jafari, Sci. Rep., 2022, 12, 2331 CrossRef CAS PubMed.
- A. Rashidizadeh, H. Ghafuri, H. R. Esmaili Zand and N. Goodarzi, ACS Omega, 2019, 4, 12544–12554 CrossRef CAS.
- H. Li, L. Wang, Y. Liu, J. Lei and J. Zhang, Res. Chem. Intermed., 2016, 42, 3979–3998 CrossRef CAS.
- C. Dai, Z. Feng, Q. Hu, J. Qiu, J. You, R. Guo and H. Zhang, Mater. Sci. Semicond. Process., 2023, 167, 107807 CrossRef CAS.
- B. A. Babalola, A. I. Akinwande, A. A. Otunba, G. E. Adebami, O. Babalola and C. Nwufo, Arabian J. Chem., 2024, 17, 105369 CrossRef.
- M. Akhgari, E. Mosaffa, H. Dogari, N. A. Ramsheh, H. Ghafuri and A. Banerjee, Environ. Sci.: Water Res. Technol., 2023, 9, 2112–2127 RSC.
- W. Li, X.-s Chu, F. Wang, Y.-y Dang, X.-y Liu, X.-c Wang and C.-y Wang, Appl. Catal., B, 2021, 288, 120034 CrossRef CAS.
- M. A. D. Fard, H. Ghafuri and A. Rashidizadeh, Microporous Mesoporous Mater., 2019, 274, 83–93 CrossRef.
- L. K. Mueller, M. Ågerstrand, T. Backhaus, M. Diamond, W. R. Erdelen, D. Evers, K. J. Groh, M. Scheringer, G. Sigmund and Z. Wang, Environ. Sci.: Adv., 2023, 2, 151–161 Search PubMed.
- J.-P. Zou, D.-D. Wu, J. Luo, Q.-J. Xing, X.-B. Luo, W.-H. Dong, S.-L. Luo, H.-M. Du and S. L. Suib, ACS Catal., 2016, 6, 6861–6867 CrossRef CAS.
- D. Zhang, Y. Li, A. Sun, S. Tong, G. Su, X. Jiang, J. Li, W. Han, X. Sun and L. Wang, Sci. Total Environ., 2019, 694, 133701 CrossRef CAS PubMed.
- H. Wang, H.-P. Zhao and L. Zhu, Environ. Sci. Technol., 2020, 54, 8760–8769 CrossRef CAS PubMed.
- S. Doherty, J. G. Knight, T. Backhouse, R. J. Summers, E. Abood, W. Simpson, W. Paget, R. A. Bourne, T. W. Chamberlain and R. Stones, ACS Catal., 2019, 9, 4777–4791 CrossRef CAS.
- Q. Zhang, J. Bu, J. Wang, C. Sun, D. Zhao, G. Sheng, X. Xie, M. Sun and L. Yu, ACS Catal., 2020, 10, 10350–10363 CrossRef CAS.
- V. A. R. Villegas, J. I. D. L. Ramírez, E. H. Guevara, S. P. Sicairos, L. A. H. Ayala and B. L. Sanchez, J. Saudi Chem. Soc., 2020, 24, 223–235 CrossRef.
- L. R. Shultz, B. McCullough, W. J. Newsome, H. Ali, T. E. Shaw, K. O. Davis, F. J. Uribe-Romo, M. Baudelet and T. Jurca, Molecules, 2019, 25, 89 CrossRef PubMed.
- A. Gawel, S. Sühnholz, A. Georgi, F.-D. Kopinke and K. Mackenzie, J. Hazard. Mater., 2023, 459, 132125 CrossRef CAS.
- S. Payamifar and A. Poursattar Marjani, Appl. Organomet. Chem., 2023, 37, e7287 CrossRef CAS.
- D. Cheng, Y. Tan, R. Ma, H. Ding, W. Liao, K. He, R. Sun, H. Ni and F. He, Environ. Sci. Technol., 2023, 57, 19827–19837 CrossRef CAS PubMed.
- S. Wang, J. Zhu, T. Li, F. Ge, Z. Zhang, R. Zhu, H. Xie and Y. Xu, Environ. Sci. Technol., 2022, 56, 7924–7934 CrossRef CAS PubMed.
- G. Pandey, N. Singh, N. Rajput, M. K. Saini, S. Kothari, J. Prasad, N. P. Lamba and M. S. Chauhan, Sci. Rep., 2024, 14, 2077 CrossRef CAS PubMed.
- M. Kashihara and Y. Nakao, Acc. Chem. Res., 2021, 54, 2928–2935 CrossRef CAS PubMed.
- F. Ferretti, D. R. Ramadan and F. Ragaini, ChemCatChem, 2019, 11, 4450–4488 CrossRef CAS.
- A. Saha and B. Ranu, J. Org. Chem., 2008, 73, 6867–6870 CrossRef CAS PubMed.
- S. M. Kelly and B. H. Lipshutz, Org. Lett., 2014, 16, 98–101 CrossRef CAS.
- J. Wen, J. Xie, X. Chen and X. Li, Appl. Surf. Sci., 2017, 391, 72–123 CrossRef CAS.
- H. Zhao and C.-H. Kong, Chem. Eng. J., 2018, 339, 424–431 CrossRef CAS.
- S. P. Swain, N. Prasad, L. Reddy, G. Subrahmanyam and S. Mohanty, ChemistrySelect, 2021, 6, 1088–1092 CrossRef CAS.
- A. Porcheddu, E. Colacino, L. De Luca and F. Delogu, ACS Catal., 2020, 10, 8344–8394 CrossRef CAS.
- J. D. Jiménez, L. E. Betancourt, M. Danielis, H. Zhang, F. Zhang, I. Orozco, W. Xu, J. Llorca, P. Liu and A. Trovarelli, ACS Catal., 2022, 12, 12809–12822 CrossRef.
- L. Gong, L. Qiu, X. Xing, J. Zhu, M. Lu, F. Dong, Y. Yu and W. Yu, Sci. Total Environ., 2024, 912, 169161 CrossRef CAS PubMed.
- H. Ahmad and M. K. Hossain, Mater. Adv., 2022, 3, 859–887 RSC.
- M. Cronau, M. Szabo and B. Roling, Mater. Adv., 2021, 2, 7842–7845 RSC.
- S. Mancillas-Salas, P. Hernández-Rodríguez, A. Reynosa-Martínez and E. López-Honorato, MRS Adv., 2020, 5, 3133–3140 CrossRef CAS.
- Z. Jahani, E. Mosaffa, M. Oroujzadeh and H. Ghafuri, Polym. Adv. Technol., 2023, 34, 3803–3817 CrossRef CAS.
- S. Elavarasan, B. Baskar, C. Senthil, P. Bhanja, A. Bhaumik, P. Selvam and M. Sasidharan, RSC Adv., 2016, 6, 49376–49386 RSC.
- M. Tasbihi, F. Fresno, I. Alvarez-Prada, A. Acharjya, A. Thomas, L. Escriche, N. Romero, X. Sala and J. García-Antón, J. CO2 Util., 2021, 50, 101574 CrossRef CAS.
- Y. Sahoo, Y. He, M. Swihart, S. Wang, H. Luo, E. Furlani and P. Prasad, J. Appl. Phys., 2005, 98, 5 CrossRef.
- W.-J. Niu, J.-Z. He, Y.-P. Wang, Q.-Q. Sun, W.-W. Liu, L.-Y. Zhang, M.-C. Liu, M.-J. Liu and Y.-L. Chueh, Nanoscale, 2020, 12, 19644–19654 RSC.
- Q. Lin, L. Li, S. Liang, M. Liu, J. Bi and L. Wu, Appl. Catal., B, 2015, 163, 135–142 CrossRef CAS.
- R. Taheri-Ledari, M. Saeidirad, F. S. Qazi, A. Fazeli, A. Maleki and A. E. Shalan, RSC Adv., 2021, 11, 25284–25295 RSC.
- A. Ghatak and S. Bhar, Synth. Commun., 2022, 52, 368–379 CrossRef CAS.
- S. B. Lee, S. Chun, S. H. Choi, J. Hong, D.-C. Oh and S. Hong, J. Org. Chem., 2023, 88, 1234–1245 Search PubMed.
- M. Gholinejad, M. Shojafar, J. M. Sansano, V. N. Mikhaylov, I. A. Balova and R. Khezri, J. Organomet. Chem., 2022, 970, 122359 CrossRef.
- V. Kandathil, T. S. Koley, K. Manjunatha, R. B. Dateer, R. S. Keri, B. Sasidhar, S. A. Patil and S. A. Patil, Inorg. Chim. Acta, 2018, 478, 195–210 CrossRef CAS.
- D. Peña-Solórzano, M. Scholler, G. N. Bernhardt, A. Buschauer, B. König and C. Ochoa-Puentes, ACS Med. Chem. Lett., 2018, 9, 854–859 CrossRef PubMed.
- M. Jang, T. Lim, B. Y. Park and M. S. Han, J. Org. Chem., 2022, 87, 910–919 CrossRef CAS PubMed.
- P. K. Lakshmi, S. V. Markandeya, C. Sridhar and N. Annapurna, Synth. Commun., 2022, 52, 1628–1634 CrossRef CAS.
- S. Fekri, Y. Mansoori and A. Bezaatpour, J. Organomet. Chem., 2023, 993, 122724 CrossRef CAS.
- S. S. Mahajan and R. G. Nandre, Mater. Adv., 2006, 37(46) DOI:10.1002/chin.200646019.
- Z. Hu, Y. Ai, L. Liu, J. Zhou, G. Zhang, H. Liu, X. Liu, Z. Liu, J. Hu and Hb Sun, Adv. Synth. Catal., 2019, 361, 3146–3154 Search PubMed.
- M. Guidetti, R. Hilfiker, M. Kuentz, A. Bauer-Brandl and F. Blatter, Cryst. Growth Des., 2022, 23, 842–852 CrossRef.
- M. Moradi, N. Rastakhiz, M. Ghaedi and R. Zhiani, Catal. Lett., 2021, 151, 1653–1662 CrossRef CAS.
- P. Karthikeyan, R. V. Jagadeesh, Y. Sree Sandhya, P. Puttaswamy, P. Nithya, S. Senthil Kumar and P. Bhagat, Appl. Organomet. Chem., 2011, 25, 34–46 CrossRef CAS.
- N. R. Lee, A. A. Bikovtseva, M. Cortes-Clerget, F. Gallou and B. H. Lipshutz, Org. Lett., 2017, 19, 6518–6521 CrossRef CAS.
- A. Sumita, Y. Otani and T. Ohwada, Chem. Commun., 2017, 53, 1482–1485 RSC.
- P. Ghamari Kargar, A. Ravanjamjah and G. Bagherzade, J. Chin. Chem. Soc., 2021, 68, 1916–1933 CrossRef CAS.
- M. Karthik and P. Suresh, ChemistrySelect, 2017, 2, 6916–6928 CrossRef CAS.
- D. Andreou, D. Iordanidou, I. Tamiolakis, G. S. Armatas and I. N. Lykakis, Nanomaterials, 2016, 6, 54 CrossRef PubMed.
- R. Rai and D. K. Chand, J. Chem. Sci., 2021, 133, 87 CrossRef CAS.
- A. Hashidoko, T. Kitanosono, Y. Yamashita and S. Kobayashi, Org. Lett., 2024, 26, 5517–5521 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ma00785a |
| This journal is © The Royal Society of Chemistry 2025 |