
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChitin nanowhiskers: a review of manufacturing, processing, and the influence of content on composite reinforcement and property enhancement†
Reshma
Panackal Shibu‡
a,
Mona
Jafari‡
a,
Sophia L.
Sagala‡
 a and
Julia L.
Shamshina
*ab
a and
Julia L.
Shamshina
*ab
aFiber and Biopolymer Research Institute, Department of Plant and Soil Science, Texas Tech University, TX 79409, USA. E-mail: jshamshi@ttu.edu
bDepartment of Chemistry and Biochemistry, Texas Tech University, TX 79403, USA
First published on 19th June 2025
Abstract
This review critically examines the potential of chitin nanowhiskers (ChNWs) as high-performance reinforcement materials for the plastics industry, with a specific emphasis on their impact on composite properties. It provides a structured overview of established ChNW preparation techniques—acid hydrolysis, oxidation, mechanical disintegration, and green solvent processing—and discusses advanced fabrication strategies for producing ChNW-reinforced composites of both natural (e.g., cellulose, chitosan, starch, hyaluronan) and synthetic (e.g., polyvinyl alcohol, poly(α-cyanoacrylate), polyaniline, polyethylene terephthalate, polypropylene) types. Key performance enhancements include increased mechanical strength, tensile strength, Young's modulus, thermal stability, and water resistance. These enhancements make ChNW-based composites suitable for real-world applications in aerospace, biomedical devices, packaging, and construction. Unlike previous reviews that emphasize only processing methods, this work identifies and highlights structure–property relationships as a central theme, bridging nanoscale morphology with macroscopic functionality.
 Reshma Panackal Shibu | Reshma Panackal Shibu is a Ph.D. student and Graduate Research Assistant at Texas Tech University in Dr Shamshina's research group. She specializes in the processing, isolation, and application of biopolymers—primarily nanochitin—for advanced material synthesis. Her current research aims to create high-performance materials by harnessing the unique properties of nanochitin. |
 Sophia L. Sagala | Dr Sophia L. Sagala is a Postdoctoral Research Associate (PDRA) at the Fiber and Biopolymer Research Institute, Texas Tech University. She earned her Ph.D. in Chemistry in 2024, with a specialization in the synthesis, physicochemical properties, and dynamics of ionic liquids for diverse applications. Currently, she is conducting postdoctoral research in Dr Julia Shamshina's group, where her work focuses on the development of advanced biopolymer-based materials enabled by ionic liquid technologies. |
 Julia L. Shamshina | Dr Julia L. Shamshina is an Assistant Professor at the Fiber and Biopolymer Research Institute, Texas Tech University, where she leads research on the development of advanced functional materials from biopolymers and biomass. She earned her Ph.D. in Chemistry with a focus on organic synthesis, and later completed postdoctoral training at The University of Alabama, specializing in ionic liquids. Before returning to academia in 2021, Dr Shamshina spent nearly eight years in industry, where she led the development of patentable technologies involving chitin polymer. Her current research spans both the fundamental science and industrial applications of sustainable biopolymer processing. |
1. Introduction
1.1. Overview of chitin and its potential as a reinforcement material
The field of nanotechnology has witnessed significant growth, with increasing focus on nanocomposites due to their improved mechanical strength, electrical conductivity, and thermal stability. A substantial portion of research centers on polysaccharide nanomaterials derived from renewable semi-crystalline biopolymers, such as chitin, valued for their high strength, large surface area, abundance, and renewability. Chitin is a linear polysaccharide found in the exoskeletons of crustaceans, and the protective tissues of invertebrates such as insects. As a waste product, rather than a nonrenewable commodity based on fossil fuels, chitin is expected to have a low environmental impact.2 Structurally, chitin is similar to cellulose, but features an acetamide group on the C-2 carbon atom replacing the hydroxyl group found in cellulose (Fig. 1).3 | ||
| Fig. 1 Chitin structure. | ||
Chitin exists in nanosized microfibril form within the structural scaffold of crustaceans, insects, or fungi.4,5 Among these, crustaceans represent the largest source of chitin, with global aquaculture production of crustaceans exceeding 11.88 million metric tons (MT) per annum in 2021.6 Studies on the lobster cuticle (Homarus americanus) have demonstrated that the crustacean shells exhibit a highly organized hierarchical structure7 primarily composed of chitin, proteins, calcium carbonate, and smaller quantities of lipids and astaxanthin. Sugar monomers of N-glucosamine form the chitin chains which assemble into semicrystalline structures featuring alternating amorphous and nanocrystalline domains stabilized by hydrogen bonds and van der Waals forces.8–10 These nanocrystalline units typically have diameters of ∼2–5 nm.11 The semicrystalline structures form chitin nanofibrils, which are further bundled into microfibrils with diameters of ∼20–100 nm, oriented along the longitudinal axis. The microfibrils are encased in a sheath of proteins and assembled into elongated fibrils. The chitin–protein fibrils are organized into planar layers that stack to create a Bouligand structure, a higher-order 3D helicoidal arrangement that enables the exoskeleton to resist fractures.12 To achieve this helicoidal structure, adjacent planar layers of chitin–protein fibrils rotate at a constant angle, completing a full 180° rotation. This Bouligand structure is also mineralized allowing it to withstand high-velocity impacts. The anisotropy ratio of the elastic modulus varies throughout the structure, decreasing progressively from 4.25 for single-crystalline chitin to 1.75 for mineralized chitin–protein nanofibrils and 1.4 in the fully developed helicoidal Bouligand structure.11
To utilize chitin in nanoform from mineralized chitin–protein nanofibrils for the development of promising biomaterials, the amorphous domains must first be removed. This process, detailed in the next section, can be achieved through methods such as mechanical shearing, high-pressure homogenization, chemical or enzymatic hydrolysis, or oxidation. Once the amorphous domains are eliminated, the remaining crystalline domains can be isolated on a nanoscale. These nanosized crystalline particles can be categorized as chitin nanocrystals (ChNCs), nanowhiskers (ChNWs), or nanofibers (ChNFs), collectively referred to as nanofillers, which differ in size and morphology. ChNCs or ChNWs are rod-like, whisker-shaped particles with a crystalline structure. Their dimensions typically range from 50–500 nm in length and 5–20 nm in width, depending on the source biomass.13–15 ChNFs exist as long, flexible fiber networks with a fibril diameter of 2–5 nm and lengths extending from several hundred nanometers to several microns, making them significantly larger than ChNWs.14,16,17 The distinct size and shape of these nanofillers contribute to their versatility and applicability in advanced biomaterial systems.
These nanosized crystalline particles have emerged as novel nanomaterials offering unique size-dependent properties.18 Particle size influences key characteristics such as diffusivity,19,20 viscosity, chirality, phase transitions from isotropic to anisotropic states, immunogenicity,21 nanocomposite transparency,3 and light transmission,22 all of which are important for material manufacturing. These nanomaterials are lightweight, with an average density of 1.45 g m−1,3,19 and feature a high aspect ratio ranging from 20–100.19,23–25 They also exhibit exceptional mechanical properties, including an elastic modulus of ∼120–150 GPa, a transverse modulus of ∼15 GPa,26 Young's modulus of ∼40 GPa,27 and tensile strength of ∼170 MPa.16,28 From a materials engineering perspective, chitin nanofillers are particularly appealing as renewable and sustainable materials, suitable for a diverse range of applications. These include adhesives29 and coatings,30,31 rheology32 and barrier property modifiers,33,34 food/cosmetic additives,35 iridescent sensors,36 liquid crystals (LC),37 polymer reinforcements, cement38 and concrete strength and durability enhancements,39 and more. Furthermore, chemical modifications such as oxidation, esterification, and grafting have expanded the potential of nanochitin-based products endowing them with fascinating new properties.2,40 Due to their high longitudinal and transverse modulus, these particles are particularly valuable as reinforcing materials in thermoplastic and thermoset plastics.41 The incorporation of nanochitin particles into various plastics enables the production of advanced materials with superior properties. These enhanced plastics find applications across various industries, from aerospace to medical devices.
The currently used industrial fillers – fiberglass,42 carbon nanotubes,43 graphene nanoplatelets,44 nanoclays,45,46 and mineral fillers,23,47,48 – are plagued by several significant issues, including: (1) heterogeneity and lack of proper bonding with polymers of organic origin;24,49–52 (2) high density, which increases the weight of the composites,13 impacting applications requiring lightweight materials; (3) high abrasiveness, which causes wear and tear on processing equipment, shortening device lifespan;53,54 (4) high hardness, which complicates polishing and coating processes;55,56 (5) reduced toughness and ductility which leads to brittleness in composites;13 (6) high cost which limits widespread adoption in cost-sensitive applications;25,51 and (7) non-compostability which restricts use to non-biodegradable plastics, contributing to environmental concerns.24,49,52,57
More specific problems with “traditional” filler materials are as follows. Fiberglass fiber-reinforced polymer composites (so-called GFRP composites) common in electronics, aerospace, and automotive industries58 suffer from poor rigidity, low fatigue resistance, and weak strength perpendicular to fiber alignment (as low as 5%).59 Manufacturing and testing are complex and specialized, and the material's density adds weight.60 Carbon nanotube (CNT)-based nanomaterials and graphene nanoplatelets exhibit remarkable mechanical, electrical, and electron transport properties expanding their applications in areas ranging from energy harvesting to electronic skin to reinforced plastics.61,62 However, their high costs limit their usage. Clay and mineral filler materials (e.g., silica, talc, mica, kaolin) are widely used in the plastics industry, e.g., for automotive parts, multilayer films, high-barrier bottles and containers, packaging applications, furniture, and household appliances, due to their excellent mechanical properties (tensile, stress, and elongation), high thermal stability, reduction of gas and liquid permeability, improvement of dynamic mechanical performance, and flame retardancy.63 However, their use is necessitated by thermoplastics’ inherent limitations, including shrinkage during molding, stiffness, and they contribute to weight and brittleness.
Optimizing the benefits of fillers while minimizing their drawbacks is an essential industrial goal that significantly influences product properties, including cost, processing efficiency, shrinkage, stiffness, heat resistance, flammability, appearance, aging, and toughness.64 Chitin nanofillers offer a sustainable solution as they are organic, bind effectively with organic polymers, and exhibit low density, hardness, and abrasiveness, all while enhancing material stability. Due to their nanoscale dimensions and exceptionally high surface-to-volume ratios, they provide a significantly larger interface area between the matrix and filler. The addition of a small amount of ChNWs, typically 0.1–0.5 wt%, leads to marked improvements in composite properties.2
This review focuses on the potential of ChNWs as reinforcement materials for the plastic industry, emphasizing their role in improving composite properties. It will cover the methods for manufacturing ChNWs (summarized briefly due to extensive existing literature), technologies for preparing biocompatible and biodegradable chitin nanofiller composites, and factors affecting property enhancements, such as the amount and dispersion of chitin, matrix–filler interactions, and processing methods (e.g., in situ polymerization, casting, nonaqueous solvent dispersion, and post-solution blending). Critical processing conditions, including reaction time and temperature, will also be analyzed, along with an evaluation of various plastics reinforced with nanostructured chitin and the impact of ChNWs on mechanical, thermal, and optical properties. This review excludes the use of biopolymers other than chitin (e.g., chitosan, cellulose) and reinforced plastics that are mentioned in the literature but lack supporting characterization data. The novelty of this review lies in its focused analysis of structure–property relationships of nanochitin in polymer matrices—a crucial aspect largely overlooked in the current literature. Unlike previous reviews that emphasize only processing methods, this work identifies and highlights structure–property relationships as a central theme, bridging nanoscale morphology with macroscopic functionality.
Despite the extensive literature on ChNW extraction and processing, few reviews address how the intrinsic structure and surface chemistry of nanochitin directly influence composite performance. This review fills that gap by analyzing structure–property relationships, thereby offering a deeper understanding of how nanochitin's nanoscale characteristics affect dispersion, interfacial bonding, and bulk mechanical properties.
The incorporation of nanochitin into synthetic and natural polymers has yielded reinforced plastics with significant enhancements in tensile strength, thermal stability, and water vapor barrier properties—critical for applications in biomedical devices, sustainable packaging, and lightweight construction materials. These real-world applications underscore the value of ChNWs as multifunctional, sustainable additives capable of replacing mineral and petroleum-derived fillers in high-performance plastic systems.
1.2. Summary of preparation methods for nanochitin
The preparation of ChNWs primarily relies on hydrolysis, a method first developed in 195965 and extensively utilized since (Table 1). This process involves treating chitin polymer with hydrochloric acid (HCl) which removes disordered regions of chitin, leaving acid-resistant crystallized sections (Table 1).10,66–68 Boco Technology, Inc. (Toronto, Canada) has commercialized the preparation of chitin nanowhisker-based polymer composites using hydrogen chloride (HCl) as a hydrolyzing agent and holds the exclusive patents for the preparation and use of ChNWs as a reinforcing material in plastics.69,70 In addition to HCl, sulfuric acid, H2SO4, has also been employed for hydrolysis of chitin and ChNWs synthesis (Table 1).71,72 An alternative to using harsh chemicals such as strong acids and alkalis is a pressure-assisted technique called steam explosion that utilizes high-pressure steam under neutral conditions accompanied by rapid decompression to precisely break down the chitin structure, fragmenting it into individualized nanowhiskers (Table 1).73,74| Chitin source | Extraction method | Structural parameters of nanowhiskers | Ref. | ||
|---|---|---|---|---|---|
| Length (nm) | Width (nm) | Aspect ratio (L/d) | |||
| a While the manuscript claims to use chitin powder, the additional deproteinization was employed; b no concentration was provided in the manuscript; c demineralization step is erroneously claimed to be achieved with KOH treatment; ND = not determined. | |||||
| Hydrochloric acid hydrolysis | |||||
| Chitin powdera | Purification through the removal of residual proteins with 5% NaOH, followed by hydrolysis with 3 N HCl | 200–400 | 10–25 | 8–40 | 40 |
| Chitin flakes | Chitin flakes hydrolyzed with concentrated HClb | ND | 13.5 | ND | 75 |
| Chitin | Hydrolysis with 3 N HCl | 200–600 | 20–60 | 3.3–30 | 76 |
| Crab α-chitin | Hydrolysis with 3 M HCl | ND | ND | ND | 77 |
| Shrimp α-chitin | Hydrolysis with 3 N HCl | 206 ± 72 | 18 ± 5 | 5.8–21.4 | 78 |
| Crab α-chitin | Hydrolysis with 3 N HCl | 114 ± 28 | 15 ± 2 | 5.05–10.9 | 79 |
| Chitin (practical grade) | Purification through the removal of residual proteins with 5% KOH, bleaching with NaClO2. Then hydrolysis with 3 N HCl | ND | ND | ND | 80 |
| Chitin | Hydrolysis with 3 N HCl | 100–900 | 20 | 5–45 | 81 |
| Shrimp α-chitin | Hydrolysis with 3 N HCl | 383 | 33.0 | 11.6 | 82 |
| Chitin | Hydrolysis with 3 N HCl | 20 | 40 | 0.5 | 83 |
| Crab α-chitin | Purification through the removal of residual proteins with 3% KOH and bleaching with 1.7% NaClO2. Then hydrolysis with 3 N HCl | 100–350 (214 ± 49) | 5–30 (16 ± 5.2) | 13 (15 ± 5.4) | 84 |
| 85 | |||||
| Chitin | Purification through the removal of residual proteins with 5% KOH. Hydrolysis with 3 N HCl | 200–400 | 10–25 | 8–40 | 86 |
| Squid pen β-chitin | Hydrolysis with 3 N HCl | 30–250 | 4–22 | 1.36–62.5 | 87 |
| Crab α-chitin | Extraction of α-chitin from the speckled swimming crabs: deproteinization with 5% NaOH, a demineralization with 7% HCl solution. Then hydrolysis with 3 M HCl | 410 | 19 | 21.6 | 88 |
| Shrimp α-chitin | Hydrolysis with 3 N HCl | 180.9 ± 86.0 | 30.8 ± 9.0 | 2.4–12.2 | 89 |
| Shrimp shells | Extraction of α-chitin from shrimp shells: demineralization with 2 N oxalic acid, bleaching with 15% H2O2, and deproteinization with 10% NaOH. Then hydrolysis with 3 N HCl | 400 | 25–32 | 12–16 | 18 |
| Chitin powder | Hydrolysis with 3 N HCl | 100–200 | 10–40 | 2.5–20 | 90 |
| Crab α-chitin | Hydrolysis with 3 N HCl | 253 ± 100 | 23 ± 5 | 11 ± 3 | 1 |
| Chitin | 2× hydrolysis with 3 N HCl | 209.63 ± 23.13 | 17.50 ± 3.75 | 12 | 91 |
| Crab α-chitin | 2× hydrolysis with 3 N HCl | 590 ± 180 | 58 ± 14 | 10 | 92 |
| Lobster shells | Extraction of α-chitin from lobster wastes: deproteinization with 2 M NaOH followed by demineralization with 2 N HCl and depigmentation with acetone and ethanol. Then hydrolysis with 3 N HCl | 300 | 60 | 5 | 20 |
| Crab α-chitin | Hydrolysis with 3 N HCl | 100–200 | 6–8 | 12.5–33 | 24 |
| Chitin from crab shellsa | Purification through the removal of residual proteins with 5% KOH, bleaching with NaClO2. Then hydrolysis with 3 N HCl | 100–600 (∼240) | 4–40 (∼15) | ∼16 | 25 and 51 |
| α-Chitin flakes prepared from crab shells | Hydrolysis with 3 N HCl | 114–320 | 8–17 | 6.5–40 | 93 |
| α-Chitin from shells of Penaeus merguiensis shrimps | Hydrolysis with 3 N HCl | 231–969 (∼549) | 12–65 (∼31) | ∼17.7 | 49 |
| α-Chitin flakes from crab shells | Hydrolysis with 3 N HCl | 100–500 | 10–50 | 15 | 94 |
| Chitin (practical grade) | Hydrolysis with 3 N HCl | 200–1000 | ∼50 | 4–20 | 13 |
| Shells of Penaeus merguiensis shrimps | Extraction of α-chitin from Penaeus merguiensis shrimp: deproteinization with NaOH followed by demineralization with HCl. Then hydrolysis with 3 N HCl | 150–800 (∼417) | 5–70 (∼33) | 17 | 95 |
| Chitin powder from shrimp shells | Purification through the removal of residual proteins with 1 N NaOH and bleaching with NaClO2. Then hydrolysis with 3 N HCl | 156–426 (∼226) | 16–47 (∼27) | ∼8.3 | 96 |
| Chitin powder | Hydrolysis with 3 N HCl | 200–500 | 15 | 13–33 | 41 |
| Riftia tubes | Extraction of β-chitin from Riftia tubes: deproteinization with 5% KOH followed by bleaching with NaClO2 (no demineralization required). Then hydrolysis with 3 N HCl | >500 (∼2200 μm) | 18 | 120 | 97 |
| α-Chitin from crab | Purification through the removal of residual proteins with 5% KOH and bleaching with NaClO2. Then hydrolysis with 4 M HCl | 200–500 | 5–20 | 15–20 | 68 |
| Lobster wastes powder | Extraction of α-chitin from the yellow lobster wastes: deproteinization with 2 M NaOH, a demineralization with 2 M HCl solution. Then hydrolysis with 3 N HCl | 300 | 60 | 5 | 98 |
| Chitin powder from crab shells | Hydrolysis with 3 M HCl | 150–260 | 8–40 | ∼8.8 | 86 |
| Fresh speckled swimming crabs | Extraction of α-chitin from the speckled swimming crabs: deproteinization with 5% NaOH, a demineralization with 7% HCl solution. Then hydrolysis with 3 M HCl | 100–700 | 3–25 | 25.6 | 99 |
| Coarse flakes of α-chitin from shrimp shells | Purification through the removal of residual proteins with 5% KOH and bleaching with NaClO2. Then hydrolysis with 3 M HCl | 229–258 | 9–10 | 23–29 | 100 |
| Dried shrimp flakes (shells) | Extraction of α-chitin from dried shrimp flakes: demineralization with 0.25 M HCl, deproteinization with 1 M NaOH, and chlorine bleaching. Then hydrolysis with 3 M HCl | 262 ± 122 | 8.7 ± 3.8 | ∼30 | 101 |
| Shrimp chitin powder | 3× hydrolysis with 3 M HCl | 50–150 | 30–50 | 1–5 | 102 |
| Native crab shell powder | Extraction of α-chitin from native crab shell powder: demineralization with 3 N HCl, deproteinization with 5% KOH, and bleaching with NaClO2 and C2H3NaO2. Then hydrolysis with 3 M HCl | 100–500 | 5–10 | 10–100 | 103 |
| Sulfuric acid hydrolysis | |||||
| Fresh lobster (Homarus americanus) | Extraction of α-chitin from lobster (Homarus americanus): demineralization with 2 M HCl, depigmentation in ethanol, deproteinization with NaOHb. Then hydrolysis with 64 wt% H2SO4 | >500 | 10–40 | 12.5–50 | 72 |
| Chitin powder | Hydrolysis with 15–40 wt% H2SO4 | 446 | ∼43 | ∼10 | 104 |
| Shrimp shell-derived α-chitin | Hydrolysis with H2SO4 | 162 ± 56 | 22 ± 8 | 7.4 | 105 |
| Bleached shrimp shell flakes | Extraction of α-chitin from bleached shrimp shell flakes: deproteinization with 5% KOH, bleaching with NaClO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) c. Then hydrolysis with H2SO4b c. Then hydrolysis with H2SO4b |
20–100 | ND | ND | 15 |
| Chitin powder from shrimp shells | Hydrolysis with H2SO4 | 150–250 | 15 | 10–16.67 | 106 |
| Steam explosion technique | |||||
| Shrimp shell flakes | 40 g of raw chitin flakes were treated with 3 N NaOH for deproteinization followed by demineralization with 2 N HCl | 100–400 | ND | ND | 73 |
| Chitin from prawn shell | Prawn shells waste was treated with 3 N NaOH, after that chitin was taken from the autoclave and washed with water, the suspension was then treated with 2 N HCl, treated with DI water to neutral Ph | 100–400 | 10–50 | 2–40 | 74 |
Beyond acid hydrolysis, alternative methods for ChNW preparation include oxidation mediated by 2,2,6,6-tetramethylpiperidine-1-oxyl radical (TEMPO),107–109 periodate-anions,110–112 and ammonium persulfate (APS)113 (Table 2). In these processes, the amorphous regions of chitin are oxidized, releasing ChNWs. For instance, in TEMPO/NaBr/NaClO oxidation, the primary hydroxyl groups of chitin are selectively converted to carboxyl groups. Sodium hypochlorite (NaClO) acts as the primary oxidant, oxidizing the TEMPO radical to its N-oxoammonium form (TEMPO+). This cation reacts with dissociated primary hydroxy groups forming a covalent bond producing an intermediate structure. Subsequent cleavage of this covalent bond generates aldehyde groups and N-hydroxy-TEMPO molecules. Further hydration of aldehydes and dissociation of hydroxy groups under alkaline conditions result in additional covalent bond formation with TEMPO+, ultimately yielding carboxyl groups and N-hydroxy-TEMPO.114
| Chitin source | Extraction method | Structural parameters of nanowhiskers | Ref. | ||
|---|---|---|---|---|---|
| Length (nm) | Width (nm) | Aspect ratio (L/d) | |||
| 2,2,6,6-Tetramethylpiperidine-1-oxy-radical (TEMPO)-mediated oxidation | |||||
| Chitin powder from crab shell | TEMPO-mediated oxidation | 50–500 | 8 | 6–62 | 107 |
| Chitin powder from crab shell | TEMPO-mediated oxidation | 242 ± 75 | 12.67 ± 3.43 | ∼18 | 115 |
| Chitin | TEMPO-mediated oxidation | 200–400 | 10–15 | 13–40 | 116 |
| Chitin | TEMPO-mediated oxidation | 347.63 ± 113 | 5 ± 1.9 | ∼70 | 117 |
| α-Chitin powder | Purification through the removal of residual proteins with 1 M NaOH and 1 M HCl, respectively, followed by bleaching with NaClO2 and NaClO. Subsequent partial deacetylation (%DA 77.2%) with 30% NaOH. Then TEMPO-mediated oxidation | ∼544 | ∼10 | ∼54 | 118 |
| Chitin particles from crab shells | O2/laccase/TEMPO oxidation | 480 ± 200 | 24 ± 17 | 11–20 | 119 |
| Chitin | TEMPO oxidation followed by high-pressure homogenization | 100–500 | ND | ND | 120 |
| Chitin powder | Purification through the removal of residual proteins with 5% NaOH, residual minerals with 5% HCl, and bleaching with 0.3% NaClO. TEMPO oxidation followed | 100–500 | ∼5–10 | 10–100 | 121 |
| Ammonium persulfate (APS)-mediated oxidation | |||||
| Chitin from crab shell | APS-mediated oxidation | 400–500 | 15 | 26–33 | 113 |
| Periodate-anions mediated oxidation | |||||
| Chitin from shrimp shells | Periodate oxidation | 250–400 | 10–30 | 8–40 | 110 |
| Chitin from crab shells | Periodate oxidation | 227 ± 76 | 11.7 ± 5 | ∼19.5 | 111 |
| Chitin from shrimp shells | Periodate oxidation | 241.8 ± 80 | 12.2 ± 5 | ∼21 | 111 |
Deep eutectic solvents (DESs), composed of hydrogen bond acceptors (HBAs) and hydrogen bond donors (HBDs) linked by intermolecular hydrogen bonding, have also been used for the formation of ChNWs from raw materials. The acidic HBDs facilitate demineralization while strongly electronegative HBAs enable deproteinization. DESs dissolve chitin by forming new hydrogen bonds with chitin molecules, disrupring their intra- and inter-molecular hydrogen bonds. During this process, the surface chemistry of chitin is simultaneously modified, producing acylated or deacetylated chitin derivatives. Subsequent mechanical disintegration results in ChNCs and ChNFs with distinctive yields, morphologies, and surface chemistries.122 Additionally, ChNWs can be formed by dissolving commercially available chitin powder in 1-allyl-3-methylimidazolium bromide ([Amim]Br) ionic liquid, followed by the mechanical breaking of the formed gelated polymeric chains.108,123 The dimensional characteristics of ChNWs extracted through various DES- and IL-type of treatments from diverse sources are summarized in Table 3.
| Chitin source | Extraction method | Structural parameters of nanowhiskers | Ref. | ||
|---|---|---|---|---|---|
| Length (nm) | Width (nm) | Aspect ratio (L/d) | |||
| a Betaine hydrochloride/ferric chloride hexahydrate DES; ND = not determined. | |||||
| Deep eutectic solvents (DESs) | |||||
| Chitin from shrimp shells | Pretreatment with deep eutectic solvent (DESs) ([HBet]Cl/FeCl3·6H2O),a followed by ultrasonication | 201–268 | 10 | 20–27 | 124 |
| Ionic liquids | |||||
| Chitin powder from crab shells | Dissolution in [Amim]Br, gelation, regeneration with methanol, and subsequent ultrasonication | Several hundred | 20–60 | ND | 123 |
| Crustacean biomass | No prior chitin isolation. Hydrolysis with 1-butyl-3-methylimidazolium hydrogen sulfate, and subsequent ultrasonication | 400–600 | 5 | 80–120 | 14 |
| Chitin | Hydrolysis with 1-butyl-3-methylimidazolium hydrogen sulfate, and subsequent ultrasonication | 200 | 20 | 10 | 14 |
Mechanical disintegration, as the term suggests, involves applying physical forces such as ultrasonication,125 grinding,126 high-pressure homogenization,126 or magnetic stirring127 under controlled conditions to break down chitin fibrils into individual nanoscale fibrils or whiskers (Table 4). It is typically part of a multistep process, preceded by deproteinization, demineralization, and depigmentation, which serve to weaken the H-bonding between fibrils.126,128 It also promotes protonation of amino groups on the fibril surface under acidic conditions, enhancing electrostatic repulsion and facilitating fibril individualization. The nanofibrils isolated through this approach retain their native properties without significant chemical modification, thus preserving their structural integrity. The goal of gentle mechanical processing is crucial to avoid structural degradation while still allowing for efficient isolation. Furthermore, this method can yield nanofibrils with high aspect ratios, making them structurally intact and effective as reinforcing agents in composite materials.
| Chitin source | Extraction method | Structural parameters of nanowhiskers | Ref. | ||
|---|---|---|---|---|---|
| Length (nm) | Width (nm) | Aspect ratio (L/d) | |||
| Mechanical disintegration | |||||
| Chitin powder from crab shell | Partial deacetylation (%DA 70–77%) with 33% NaOH. Then mechanical disintegration via a magnetic stirring at 1200 rpm for 5 days, and ultrasonication for 1 min | 250 ± 140 | 6.2 ± 1.1 | 18–40 | 127 |
| Speckled swimming crab shells | Extraction of α-chitin from swimming crab shells: deproteinization with 5% NaOH, demineralization with 7% HCl, depigmentation in ethanol. Then mechanical disintegration via wet grinding and high-pressure homogenization | 500–1000 | 10–80 | 6–100 | 126 |
| α-Chitin from crab shell | Mechanical disintegration via ultrasonic homogenization | ND | ND | ND | 125 |
| Crab tendon (Chionoecetes opilio) | Extraction of β-chitin from crab tendon (Chionoecetes opilio): deproteinization with 0.5 M NaOH, demineralization with 0.5 M HCl. Then mechanical disintegration via ultrasonic homogenization | ND | ND | ND | 125 |
| Tubeworm (Lamellibrachia satsuma) | Extraction of β-chitin from tubeworm (Lamellibrachia satsuma): deproteinization with 0.5 M NaOH, demineralization with 0.5 M HCl. Then mechanical disintegration via ultrasonic homogenization | ND | ND | ND | 125 |
| Squid pen | Extraction of β-chitin from squid pen: deproteinization with 0.5 M NaOH, demineralization with 0.5 M HCl. Then mechanical disintegration via ultrasonic homogenization at pH 3–4 | A few microns | 3–4 | >250 | 125 |
| Lobster shells (Homarus americanus) | Extraction of α-chitin from lobster (Homarus americanus): demineralization with 2 M HCl, depigmentation in ethanol, deproteinization with 8–20% NaOH. Then mechanical disintegration via blending and micro-fluidization | 200–2000 | 4–5.6 | 36–500 | 128 |
The preparation of partially deacetylated chitin nanowhiskers (ChNWs) involves the removal of residual proteins and minerals through a series of sequential chemical pretreatments, including deproteinization and demineralization, followed by an additional treatment with concentrated NaOH or KOH solutions (commonly 10–50 wt%)121,127,129–131 (Table 5). This final alkaline treatment selectively removes a controlled fraction of acetyl groups at the C2 position, introducing protonated amino groups that enhance the surface charge of the chitin fibrils. The degree of deacetylation plays a critical role in tailoring the functional properties of ChNWs, including improved dispersibility in aqueous media, increased surface reactivity, tunable morphology, enhanced swelling and antimicrobial activity, as well as the mechanical and crystalline properties, which could ensure compatibility with various polymer matrices.
| Chitin source | Extraction method | Structural parameters of nanowhiskers | Ref. | ||
|---|---|---|---|---|---|
| Length (nm) | Width (nm) | Aspect ratio (L/d) | |||
| a In chitin, the acetylated units prevail (the degree of acetylation (%DA) is >50%), while in chitosan the deacetylated units prevail (%DA is <50%). Therefore, this material is classified as chitin by definition; ND = not determined. | |||||
| Nanowhiskers from partially deacetylated chitin (DA > 0.50) | |||||
| α-Chitin (source not shown) | Purification through the removal of residual proteins with 5% KOH, and bleaching with 0.17 wt% NaClO2 followed by hydrolysis with 3 N HCl to obtain ChNWs. CNWs then partially deacetylated to %DA = 60%a |
100–500 | 6–15 | 6.7–83.3 | 132 |
| Chitin flakes | Flakes were deacetylated with 40 wt% NaOH (%DA = 62%a) and homogenized under high pressure |
120 ± 46 | 6.2 ± 1.5 | 9.6–35.3 | 129 |
| Crab shells | Shells were deproteinized with 5 wt% KOH and demineralized with 7 wt% HCl. The purified chitin was deacetylated with 40 wt% NaOH (%DA = 76%a) and homogenized under high-pressure |
114 | 3.4 | 33.5 | 130 |
| Crab shells | Shells were deproteinized with 5 wt% and demineralized with 7 wt% HCl. The purified chitin was deacetylated with 30 wt% NaOH (%DA = 85%a) and homogenized under high pressure |
230 | 3.8 | 60.5 | 130 |
| Chitin powder | Purification through the removal of residual proteins with 5% NaOH, residual minerals with 5% HCl, and bleaching with 0.3% NaClO. The powder was suspended in 36% NaOH solution, and 0.15 wt% NaBH4 was added. Then the partially deacetylated chitin (%DA = 66%a) was washed to pH 7 and homogenized under high pressure |
100–500 | ∼5–10 | 10–100 | 121 |
| Crab shells (Portunus trituberculatus) | Extraction of α-chitin from the speckled swimming crabs: deproteinization with 1 M NaOH, a demineralization with 1 M HCl solution, and bleaching with NaClO2. Partial deacetylation in 33% NaOH (%DA = 72%a), then mechanical disintegration |
ND | ND | ND | 131 |
| α-Chitin | Partial deacetylation in 33% NaOH (DA 0.60–0.90), then mechanical disintegration | 250 ± 140 | 6.2 ± 1.1 | 15.1–76.5 | 127 |
| Crab and shrimp shells | Extraction of α-chitin from the speckled swimming crabs: deproteinization with 1 M NaOH, a demineralization with 1 M HCl solution, and bleaching with 0.3% v/w NaClO2. The powder was suspended in 33% NaOH solution, and NaBH4 was added. Then the partially deacetylated (DA not shown) chitin was washed to pH > 4 and homogenized under high pressure | 800 | 10–50 | 16–80 | 133 |
| α-Chitin nanocrystals from yellow lobster | α-Chitin nanocrystals from yellow lobster was extracted using microwave-assisted extraction (MAE) technique with optimal isolation condition of 1 M HCl, 10 min, and 124.75 W | 314.74 ± 62.50 | 41.16 ± 10.92 | 7 | 134 |
| α-Chitin nanocrystals from shrimp | α-Chitin nanocrystals from shrimp was extracted using MAE technique with optimal isolation condition of 1 M HCl, 14.34 min, and 50.21 W | 386.12 ± 47.49 | 42.16 ± 4.62 | 9 | 134 |
1.3. Reinforced plastics with chitin
ChNWs are highly crystalline, rigid, whisker-like particles with exceptional mechanical properties.40,135 They are non-toxic and exhibit liquid crystalline behavior, making them ideal candidates for the preparation and reinforcement of nanocomposites with unique properties across different polymer matrices.82,94,135,136 A list of polymers used as a matrix reinforced with ChNWs can be found in Table 6, which provides a concise summary. Detailed explanations and discussions are presented in the subsequent sections. These polymers include various fossil fuel-based polymers, such as poly(vinyl) alcohol (PVA) and polypropylene (PP); thermoset polymers, such as epoxy resin and bio-based polyurethane; rubber, such as natural rubber (NR), styrene–butadiene rubber latex (SBR), carboxylated styrene butadiene rubber latex (XSBR), and nitrile butadiene rubber (NBR); biodegradable polyesters, such as polycaprolactone (PCL) and polylactic acid (PLA); and natural polymers, such as corn starch, gelatin, potato starch, and chitosan.14,18,40,108,137–139| Polymer name | Abbreviation (if any) | Polymer type | Ref. |
|---|---|---|---|
| Acrylonitrile-butadiene rubber | NBR | Synthetic rubber/elastomer | 18 |
| Bio-based polyurethane | Bio-PUR | Renewable thermoset | 139 |
| Carboxymethyl chitosan | CMCS | Derivative of natural polysaccharide | 106 |
| Cellulose | — | Natural polysaccharide | 121 |
| (Microcrystalline) cellulose | MCC powder | Natural biopolymer, biodegradable | 79 |
| Cellulose acetate | CA | Derivative of natural polysaccharide | 140 |
| Cellulose acetate | CA | Derivative of natural polysaccharide | 141 |
| Cellulose acetate | CA | Derivative of natural polysaccharide | 142 |
| Chitosan | Cs | Natural polysaccharide | 24 |
| Corn starch | — | Natural polysaccharide | 20 |
| Epoxy resin | — | Thermoset | 23 |
| Epoxy resin | EPON828 | Biodegradable synthetic resin | 23 |
| Gelatin | — | Protein | 143 |
| Gelatin | Gel | Protein | 120 |
| Gelatin | — | Protein | 78 |
| Hyaluronan | HA | Natural polysaccharide | 88 |
| Methacrylate polymer | MA | Non-biodegradable synthetic polymer | 77 |
| Natural rubber | NR | Natural occurring rubber/elastomer | 25 |
| Poly(butylene adipate-co-furanoate) | PBAF | Synthetic polyester, biodegradable | 105 |
| Poly(ethylene terephthalate) | PET | Synthetic polyester, non-biodegradable | 129 |
| Poly(ethylene terephthalate) | PET | Synthetic polyester, non-biodegradable | 130 |
| Poly(ethylene terephthalate) | PET | Synthetic polyester, non-biodegradable | 89 |
| Poly(vinyl alcohol) | PVA | Synthetic polymer, biodegradable | 80 |
| Poly(vinyl alcohol) | PVA | Synthetic polymer, biodegradable | 40 |
| Poly(vinyl alcohol) | PVA | Synthetic polymer, biodegradable | 133 |
| Poly(vinyl alcohol) | PVA/PVOH | Synthetic polymer, biodegradable | 144 |
| Poly(vinyl alcohol)/modified boron nitride | PVA | Synthetic polymer, biodegradable, inorganic | 81 |
| Poly(vinyl alcohol)/montmorillonite | PVA | Synthetic polymer, biodegradable, clay mineral | 83 |
| Poly(3-hydroxybutyrate-co-3-hydroxyvalerate) | PHBV | Natural polymer, microbially produced, biodegradable | 91 |
| Poly-(ε-caprolactone) | PCL | Synthetic polyester, biodegradable | 145 |
| Polyaniline | PANI | Synthetic polymer, non-biodegradable | 87 |
| Polycarbonate | PC | Synthetic polyester, non-biodegradable | 76 |
| Polyethylene glycol di-methacrylate | PEGDA | Synthetic polyester, non-biodegradable | 90 |
| Polylactic acid | PLA | Synthetic polyester, biodegradable | 91 |
| Polypropylene | PP | Synthetic polymer, non-biodegradable | 19 |
| Polypropylene | PP | Synthetic polymer, non-biodegradable | 82 |
| Potato starch | — | Natural polysaccharide | 48 |
| Silk | — | Sericin and fibroin proteins | 131 |
| Sodium alginate | SA | Natural polysaccharide | 75 |
| Sodium alginate | ALG | Natural polysaccharide | 132 |
| Soy protein isolate | SPI | Natural biopolymer, biodegradable | 73 |
| Soy protein isolate | SPI | Protein | 74 |
| Styrene-butadiene rubber/carboxylated styrene butadiene rubber latex (XSBR latex) | SBR/XSBR | Synthetic rubber/elastomer, non-biodegradable | 146 |
2. Preparation methods for chitin-reinforced plastics
The methods to prepare polymer nanocomposites reinforced with nanostructured chitin can be broadly categorized into ex situ processing and in situ polymerization (Fig. 2). In ex situ preparation, nanomaterials are dispersed into a pre-existing polymer matrix, either in solid form or in solution, to create the composite. This method is particularly suited for large-scale applications. However, a significant challenge is achieving a uniform dispersion of nanomaterials within the polymer matrix, along with maintaining long-term stability against aggregation. Sonication is commonly employed to enhance the dispersion of nanomaterials in polymer solutions.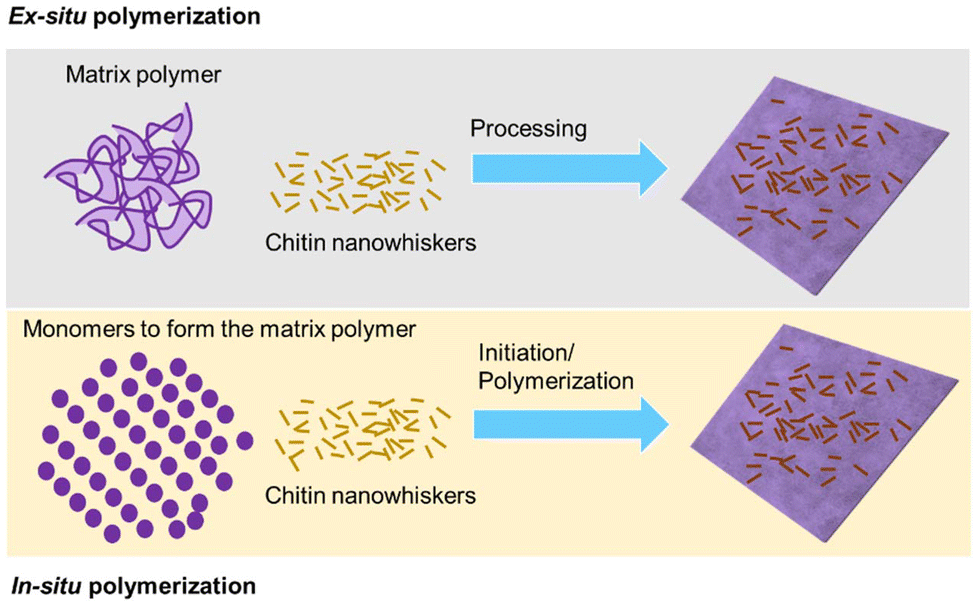 | ||
| Fig. 2 Schematics of ex situ and in situ polymerization. | ||
In contrast, in situ preparation involves synthesizing the polymer matrix in the presence of nanomaterials, allowing nanoparticles to be trapped within the polymer as it forms. This approach prevents particle agglomeration and ensures a uniform spatial distribution of nanoparticles within the polymer matrix. Additionally, in situ polymerization is a simple and effective method for producing nanocomposites with improved dispersion stability.
2.1. Casting
Casting is an ex situ method for preparing nanocomposites, where a suspension containing base polymer(s) and fillers—such as chitin nanowhiskers (ChNWs) or chitin nanofibers (ChNFs)—is poured into molds. These molds are typically made from or coated with materials such as polytetrafluoroethylene (PTFE),13,25,29,41,51,73,74,83,93,147 polyethylene (PE),40,80 or silicone rubber;3 Petri dishes are also used.81,148 After casting, the solvent is removed through evaporation (referred to in this review as casting-evaporation), freeze-drying, or hot pressing under controlled conditions (temperature, pressure, relative humidity) tailored to the specific properties of the polymers.In casting-evaporation method (Fig. 3), ChNWs or CNFs and the base polymer are suspended or dissolved either in the same solvent, or two miscible solvents and mixed (Table 7, entries 1–27). The resulting mixture is then cast into films, and the solvent is evaporated. Water,40,48,49,88,146 alcohols,3 or weak organic acids (e.g., formic acid149,150) are typically used as solvents for hydrophilic polymers such as PVA, rubber latex, carboxylated styrene butadiene, polycarbonate, biopolymers – starch and hyaluronic acid, while organic solvents (e.g., toluene,147 1-methylpyrrolidinone,76 methylene chloride93) are employed for hydrophobic polymers such as polysulfone, acrylics, polylactic acid, epoxy-polymers, polyethylene, polystyrene, and polyvinyl chloride.
 | ||
| Fig. 3 Schematics of commonly used fabrication methods. | ||
| Polymers | Method | Ratio of components in final composite, g g−1 | Protocol | Notes | Ref. | |
|---|---|---|---|---|---|---|
| a ChNWs are the essential component rather than a reinforcement filler. b Chitin is not usually soluble in acids, so it was likely a misleading title, and the paper might have used chitosan. | ||||||
| Casting | ||||||
| Casting followed by solvent evaporation | ||||||
| 1 | PVA (Mw = 75000 Da)/α-ChNWs |
Casting | ChNWs/PVA = 2.55/97.45 | The suspension of ChNWs in water was mixed with PVAaq solution, then the resulting suspension was cast into film | Solvent removal by evaporation at room temperature | 49 |
| ChNWs/PVA = 5.11/94.89 | ||||||
| ChNWs/PVA = 7.66/92.34 | ||||||
| ChNWs/PVA = 10.11/89.89 | ||||||
| ChNWs/PVA = 12.76/87.24 | ||||||
| ChNWs/PVA = 15.17/84.83 | ||||||
| ChNWs/PVA = 17.74/82.26 | ||||||
| ChNWs/PVA = 25.38/74.62 | ||||||
| 2 | Potato starch powder/chitin nanoparticles (ChNPs) | Casting | ChNPs/potato starch = 1/99 | ChNPs were dispersed in a solution of distilled water and glycerol plasticizer and ultrasonicated before adding starch. The mixture was heated at 90 °C to plasticize the starch, and then cast into a dish | Dried in an air-circulating oven at 50 °C (∼6 h) | 48 |
| ChNPs/potato starch = 2/98 | ||||||
| ChNPs/potato starch = 3/97 | ||||||
| ChNPs/potato starch = 4/96 | ||||||
| ChNPs/potato starch = 5/95 | ||||||
| 3 | XSBR latex/ChNWs | Casting | ChNWs/XSBR latex = 4.1/95.9 | Chitin and latex were suspended in water, mixed at certain volume ratio, and cast | Solvent (water) removal by evaporation, in 40 °C oven | 146 |
| ChNWs/XSBR latex = 6.6/93.4 | ||||||
| ChNWs/XSBR latex = 9.0/91.0 | ||||||
| 4 | Unvulcanized natural rubber latex (unvulc. NR)/ChNCs | Casting | ChNCs/unvulc. NR = 1.0/99.0 | ChNCs were added into water, and then ultrasonicated. Meanwhile, NR latex was diluted using water. Afterward, ChNCs dispersions were added to the uniform NR latex with stirring, to obtain a homogeneous mixture. The NR/ChNCs composite films were cast in a square PTFE mold | Solvent removal by oven drying at 60 °C for 16 h | 13 |
| ChNCs/unvulc. NR = 2.4/97.6 | ||||||
| ChNCs/unvulc. NR = 4.8/95.2 | ||||||
| ChNCs/unvulc. NR = 7.0/93.0 | ||||||
| ChNCs/unvulc. NR = 9.0/91.0 | ||||||
| 5 | XSBR latex/ChNWs | Casting | ChNWs/XSBR = 1/99 | The dispersion of chitin nanowhiskers was mixed with XSBR latex with the addition of distilled water and the mixture was cast on a PTFE mold | Solvent removal by evaporation in an oven at 40 °C for 12 h and then at 60 °C under vacuum for 2 h | 41 |
| ChNWs/XSBR = 2/98 | ||||||
| ChNWs/XSBR = 3/97 | ||||||
| ChNWs/XSBR = 4/96 | ||||||
| ChNWs/XSBR = 5/95 | ||||||
| 6 | SPI/magnesium oxide (MgO)/ChNWs | Casting | ChNWs/SPI = 6/94 | The solution of SPI in distilled water and glycerol was mixed with required amounts of chitin nanowhiskers and MgO followed by casting the mixture on a Teflon-coated pan | Solvent removal by drying in a vacuum oven overnight and storage in a desiccator at 50% RH | 73 |
| ChNWs/SPI/MgO = 6/93/1 | ||||||
| ChNWs/SPI/MgO = 6/89/5 | ||||||
| 7 | Gel/dialdehyde microfibrillated cellulose (DAMFC)/ChNWs | Casting | DAMFC/gel/ChNWs = 14.90/84.30/0.80 | The aqueous solution of gel was mixed with the aqueous suspensions of DAMFC and chitin nanowhiskers and the mixture was cast to form hydrogels | Incubating for 48 h, at 25 °C | 120 |
| DAMFC/gel/ChNWs = 14.74/83.55/1.71 | ||||||
| DAMFC/gel/ChNWs = 14.62/82.82/2.56 | ||||||
| DAMFC/gel/ChNWs = 14.36/81.36/4.28 | ||||||
| DAMFC/gel/ChNWs = 13.71/77.66/8.63 | ||||||
| 8 | Gel/ChNWs | Casting | ChNWs/gel = 1/100 | The aqueous solution containing gelatin, glucose cross-linker, and glycerol plasticizer was mixed with the suspension of chitin nanowhiskers in deionized water and the mixture was cast | Solvent removal by evaporation in air-circulating fume hood at 20 °C and 56% RH for 24 h | 106 |
| ChNWs/gel = 2/98 | ||||||
| ChNWs/gel = 4/96 | ||||||
| 9 | Soy Protein Isolate (SPI)/ChNWs | Casting | ChNWs/SPI = 61.54/38.46 | The aqueous dispersion of SPI was mixed with glycerol, Clitoria ternatea flower extract (CTE extract), suspension of chitin nanowhiskers in water and the mixture was cast on Teflon plate | Solvent removal by evaporation in an air oven at 55 °C | 74 |
| ChNWs/SPI/CTE = 38.10/23.81/38.10 | ||||||
| 10 | Montmorillonite (MMT)/PVA/ChNWs | Casting | MMT/ChNWs = 50/50 | The dispersions of PVA in distilled water and MMT in deionized water were mixed with chitin nanowhiskers were dispersed in deionized water and were cast on Teflon plate | Solvent removal by evaporation in air at room temperature | 83 |
| PVA/ChNWs = 50/50 | ||||||
| ChNWs/MMT/PVA = 33.33/33.33/33.33 | ||||||
| 11 | Polyaniline (PANI)/ZnO nanoparticles (ZnO NPs)/β-chitin ChNWs | Casting | β ChNWs/ZnO = 97.09/2.91 | The suspension of chitin nanowhiskers in distilled water was mixed with aqueous glycerol suspension of: 1: ZnO NPs; 2. PANI; 3. ZnO NPs + PANI and the suspensions were cast | Solvent removal by drying in an oven at 40 °C for 18 h and conditioning at 25 °C for at least 24 h | 87 |
| β ChNWs/PANI = 99.01/0.99 | ||||||
| β ChNWs/ZnO/PANI = 96.15/2.88/0.96 | ||||||
| 12 | Carboxymethyl chitosan (CMCS) cross-linked with dextran dialdehyde (DDA)/ChNWs/ | Casting | ChNWs/CMCS = 0.5/99.5 | The dispersion of ChNWs in deionized water was mixed with CMCS and hydrogels were prepared by coextruding DDA solution and CMCS solution | Gelation time was monitored by a vial inversion test at 37 °C | 106 |
| ChNWs/CMCS = 1.0/99.0 | ||||||
| 13 | Hyaluronic Acid (HA)/partially deacetylated chitin nanowhiskers (ChNWs, DA 73%) | Casting | ChNWs/HA = 50.00/50.00 | The solution of HA in deionized water was mixed with partially deacetylated ChNWs and were cast | Solvent removal by drying in an oven at 125 °C for 10 min | 88 |
| ChNWs/HA = 90.91/9.09 | ||||||
| ChNWs/HA = 93.75/6.25 | ||||||
| ChNWs/HA = 97.22/2.78 | ||||||
| ChNWs/HA = 98.04/1.96 | ||||||
| 14 | PVA/ChNWs | Casting | ChNWs/PVA = 1.0/99.0 | The suspension of chitin nanowhiskers was mixed with PVA aqueous solution and NaCl and the mixture was cast on a polyethylene plate. The film was then soaked in NaOH to reconstruct the hydrogen bonds | Dried under vacuum | 80 |
| ChNWs/PVA = 1.5/98.5 | ||||||
| ChNWs/PVA = 2.0/98.0 | ||||||
| ChNWs/PVA = 2.5/97.5 | ||||||
| ChNWs/PVA = 3.0/97.0 | ||||||
| 15 | PVA/ionic shielded chitin nanowhiskers (IsChNWs) | Casting | IsChNWs/PVA = 80/20 | The dispersion of chitin nanowhiskers in HCl was mixed with aqueous solution of PVA. NaCl aqueous was then added in the mixture as an ion-shielding agent, centrifuged and cast into a film on a polyethylene (PE) plate | Solvent removal by drying at room temperature | 40 |
| IsChNWs/PVA = 70/30 | ||||||
| IsChNWs/PVA = 60/40 | ||||||
| IsChNWs/PVA = 50/50 | ||||||
| 16 | PVA (DP = 2500) and β-ChNWs | Casting | β-ChNWs/PVA = 90/10 | β-Chitina was dissolved in formic acid and mixed with PVA-formic acid solution, at room temperature, then the resulting solution was cast into film | Solvent removal by drying in an oven at 40 °C for 24 h | 149 |
| β-ChNWs/PVA = 80/20 | ||||||
| β-ChNWs/PVA = 70/30 | ||||||
| β-ChNWs/PVA = 60/40 | ||||||
| β-ChNWs/PVA = 50/50a | ||||||
| β-ChNWs/PVA = 40/60 | ||||||
| β-ChNWs/PVA = 30/70 | ||||||
| 17 | PVA/ChNWs | Casting | β-ChNWs/PVA = 80/20 | β-Chitinb was dissolved in formic acid and mixed with PVA–formic acid solution, at room temperature, then the resulting solution was cast into film | Solvent removal by drying in a drying oven at 60 °C for 12 h | 150 |
| β-ChNWs/PVA = 60/40 | ||||||
| β-ChNWs/PVA = 50/50 | ||||||
| β-ChNWs/PVA = 40/60 | ||||||
| β-ChNWs/PVA = 20/80 | ||||||
| 18 | Chitosan (CS)/ChNWs | Casting | ChNWs/CS = 2.97/97.03 | Chitosan solution in acetic acid was mixed with ChNWs aqueous suspension. To each of these suspensions, isocyanate cross-linker HDS was added and mixed. The mixtures were transferred to a mold, and gelation was allowed to occur by standing at 60 °C for 48 h. The resulting chitosan/chitin nanowhisker gels were washed in water | Obtained as gels, not dried | 24 |
| ChNWs/CS = 5.77/94.23 | ||||||
| ChNWs/CS = 10.90/89.10 | ||||||
| ChNWs/CS = 13.3/86.70 | ||||||
| 19 | Sorbitol polyglycidyl ether (SPE)/ChNFs/CS | Casting | ChNFs/SPE/CS = 3/66.3/30.7 | The slurry of ChNFs in aqueous acetic acid was mixed with SPE and an aqueous acetic acid solution of CS, then the resulting solution was poured into Petri dish | Dried at 80 °C for 12 h followed by post-curing at 120 °C for 6 h | 148 |
| ChNFs/SPE/CS = 5/64.9/30.1 | ||||||
| ChNFs/SPE/CS = 10/61.5/28.5 (footnote: calculated w/o inclusion of hardener) | ||||||
| 20 | NR latex/ChNWs grafted with alkenyl succinic anhydride (ASA), phenyl isocyanate (PI), and isopropenyl-α,α′-dimethylbenzyl isocyanate (TMI) | Casting | ChNWs-ASA/NR = 90/10 | The ChNWs (grafted with alkenyl succinic anhydride (ASA) and phenyl isocyanate (PI)) isopropenyl-α,α′-dimethylbenzyl isocyanate (TMI)and lyophilized NR latex (neat and treated with) were separately dispersed in toluene. The two dispersions were then mixed, after which toluene was removed in a rotavapor. The thick slurry was then cast in a Teflon mold | Drying in an oven at 60 °C for several days | 147 |
| ChNWs-PI/NR = 90/10 | ||||||
| ChNWs-TMI/NR = 90/10 | ||||||
| 21 | PLA/acetylated chitin nanocrystals (Ac-ChNCs) | Casting | Ac-ChNCs/PLA = 1/99 | Ac-ChNCs and PLA were added to CH2Cl2, and then stirred to produce a mixture which was conditioned overnight to eliminate bubbles before being cast into a Teflon mold | Solvent removal by evaporation at ambient temperature for 24 h | 93 |
| Ac-ChNCs/PLA = 2/98 | ||||||
| Ac-ChNCs/PLA = 4/96 | ||||||
| Ac-ChNCs/PLA = 6/94 | ||||||
| Ac-ChNCs/PLA = 8/92 | ||||||
| Ac-ChNCs/PLA = 10/90 | ||||||
| 22 | Recycled polycarbonate (rPC, MW 45000)/polysulfone (PSf, MW 75000–81000)/ChNWs |
Casting | ChNWs/rPC/PSf = 2/49/49 | PSf was added to a solution of rPC in 1-methyl-2-pyrrolidinone solvent (NMP). Pluronic F127 porogen and ChNWs followed by casting the mixture on a glass plate using casting knife. The fabricated membrane was then washed with water, to remove NMP | Solvent removal by washing with water at 70 °C for 1 h, and then at room temperature for 24 h. Not dried | 76 |
| ChNWs/rPC/PSf = 4//48/48 | ||||||
| ChNWs/rPC/PSf = 6/47/47 | ||||||
| ChNWs/rPC/PSf = 8/46/46 | ||||||
| ChNWs/rPC/PSf = 10/45/45 | ||||||
| 23 | Modified boron nitride modified with silane (BNSi)/ChNWs | Compounding | ChNWs/BNSi = 16.7/83.3 | Pre-made BNSi and ChNWs were mixed in ethanol at room temperature with magnetic stirring for 5 h. The hybrid material was collected, cast in Petri dishes, and dried | Dried at room temperature | 81 |
| 24 | BNSi/ChNWs/PVA | Compounding followed by casting | ChNWs/BNSi/PVA = 0.79/3.97/95.24 | Pre-made BNSi and ChNWs were mixed in ethanol at room temperature with magnetic stirring for 5 h. The hybrid material was collected. PVA was dissolved in deionized water, then hybrid materials were mixed into the PVA solution. After mixing evenly, mixtures were poured cast in Petri dishes, and dried | Dried in ventilate oven at 60 °C for 24 h | 81 |
| ChNWs/BNSi/PVA = 2.17/10.87/86.96 | ||||||
| ChNWs/BNSi/PVA = 3.33/16.67/80.00 | ||||||
| ChNWs/BNSi/PVA = 4.35/21.74/73.91 | ||||||
| 25 | Modified boron nitride modified with polyvinylpyrrolidone (BNPVP)/ChNWs | Compounding followed by casting | ChNWs/BNPVP = 16.7/83.3 | Pre-made BNPVP and ChNWs were mixed in ethanol at room temperature with magnetic stirring for 5 h. The hybrid material was collected, cast in Petri dishes, and dried | Dried at room temperature | 81 |
| 26 | BNPVP/ChNWs/PVA | Compounding followed by casting | ChNWs/BNPVP/PVA = 0.79/3.97/95.24 | Pre-made BNPVP and ChNWs were mixed in ethanol at room temperature with magnetic stirring for 5 h. The hybrid material was collected and dried | Dried in ventilate oven at 60 °C for 24 h | 81 |
| ChNWs/BNPVP/PVA = 2.17/10.87/86.96 | ||||||
| ChNWs/BNPVP/PVA = 3.33/16.67/80.00 | ||||||
| ChNWs/BNPVP/PVA = 4.35/21.74/73.91 | ||||||
| 27 | Epoxy resin bisphenol A diglycidyl ether (DGEBA)/ChNWs | Slurry compounding followed by casting | ChNWs/DGEBA = 0.25/99.75 | The suspension of chitin nanowhiskers was mixed with the solution of DGEBA in ethanol through slurry compounding followed by the addition of triethylenetetramine (TETA) curing agent, and the mixture was cast on silicone rubber molds | Post curing at 100 °C for 24 hours | 3 |
| ChNWs/DGEBA = 0.50/99.50 | ||||||
| ChNWs/DGEBA = 0.75/99.25 | ||||||
| Casting followed by freeze drying | ||||||
| 28 | ALG/α-ChNWs | n Casting | ChNWs/ALG = 33.3/66.7 | The ChNWs suspension in deionized water was mixed with sodium alginate and glycerol and were cast | Solvent removal by overnight freezing at −40 °C and lyophilization at 0.11 mbar for 8 h | 75 |
| 29 | ALG/partially deacetylated α-ChNWs (DDA 0.40 ± 0.03) | Casting | ChNWs/ALG = 2.5/97.5 | The partially deacetylated ChNWs aqueous suspension was mixed with concentrated solutions of ALG and were cast. To isolate the ALG-ChNW microgels, the ALG-ChNW hydrogel was diluted with water, centrifuged, washed with water, and freeze-dried | Solvent removal by drying by refrigerator at 4 °C for 2 days, and freeze-dried | 132 |
| ChNWs/ALG = 7.5/92.5 | ||||||
| ChNWs/ALG = 14.5/85.5 | ||||||
| 30 | PVA/ChNWs | Casting | ChNWs/PVA = 25/75 | The chitin nanowhisker suspension was mixed with PVA solution and added with water to reach the final volume of 100 mL. The mixture was gently stirred at room temperature for 4 h, then transferred into a glass vial, and frozen by two methods | Two methods of solvent removal: | 153 |
| ChNWs/PVA = 40/60 | 1. Plunging in liquids N2 for 5 min (−196 °C) | |||||
| ChNWs/PVA = 50/50 | 2. Directional freezing in which only the bottom vial containing samples was in contact with liquid N2, followed by freeze-dried for 24 h | |||||
| 31 | PVA/partially deacetylated α-ChNWs | Casting | ChNWs/PVA = 20/80 | The partially deacetylated ChNWs suspension in deionized water and acetic acid was mixed with the suspension of PVA in deionized water followed by adding glutaraldehyde as the cross linker and finally the well mixed suspensions were molded | Solvent removal by freezing at −20 °C for about 24 h, followed by thawing for 12 h for three continuous cycles (repeating freeze–thawing cycle) | 133 |
| ChNWs/PVA = 30/70 | ||||||
| ChNWs/PVA = 40/60 | ||||||
| ChNWs/PVA = 50/50 | ||||||
| ChNWs/PVA = 60/40 | ||||||
| Casting followed by hot pressing | ||||||
| 32 | Poly-(ε-caprolactone) (PCL)/ChNWs | Casting | ChNWs/PCL = 0.5/99.5 | The colloidal microcrystalline dispersion of chitin nanowhiskers in water was mixed with the suspension of PCL and were molded | Freeze drying and hot pressing at 21 bar (2.1 MPa) and 100 °C | 97 |
| ChNWs/PCL = 1.0/99.0 | ||||||
| ChNWs/PCL = 2.5/97.5 | ||||||
| ChNWs/PCL = 5.0/95.0 | ||||||
| ChNWs/PCL = 10.0/90.0 | ||||||
| 33 | NR latex/ChNWs | Casting | ChNWs/NR = 2/98 | The aqueous suspensions of chitin whiskers and NR latex were mixed and cast in a Teflon mold | Solvent removal by either one: 1. evaporation (unvulcanized NR) at 40 °C for 10–15 days; 2. hot pressing (pre-vulcanized NR) at 138 bar (13.8 MPa) for 2 min at 100 °C; and 3. (pre-vulcanized NR) freeze drying | 25 and 51 |
| ChNWs/NR = 5/95 | ||||||
| ChNWs/NR = 10/90 | ||||||
| ChNWs/NR = 15/85 | ||||||
| ChNWs/NR = 20/80 | ||||||
| 34 | SPI plastics/ChNWs | Casting | ChNWs/SPI = 5/95 | The homogenous dispersions of SPI and various content of ChNWs were freeze-dried and 30% glycerol was added. The resulting mixture were hot pressed, and slowly cooled to room temperature | Hot pressing at 20 MPa, for 10 min at 140 °C | 155 |
| ChNWs/SPI = 10/90 | ||||||
| ChNWs/SPI = 15/85 | ||||||
| ChNWs/SPI = 20/80 | ||||||
| ChNWs/SPI = 25/75 | ||||||
| ChNWs/SPI = 30/70 | ||||||
| 35 | Polylactic acid (PLA)/chitin nanoparticle (ChNP) | ChNP/PLA = 0.5/99.5 | Nano-chitin was mixed with PLA containing 20 wt% glycerol triacetate and the resulting mixture was hot-pressed into films | Hot pressing at 15 kPa and 190 °C | 156 | |
| ChNP/PLA = 1/99 | ||||||
| ChNP/PLA = 2/98 | ||||||
| ChNP/PLA = 3/97 | ||||||
| 36 | Cellulose nanofibers/ChNC | ChNC/cellulose = 1/99 | The self-bonded composite films were obtained by mixing cellulose nanofibers and chitin nanocrystals at 15000 rpm, 10 min to ensure well-dispersed slurry. The wet mats were dried at 50 °C and molded |
Water removal by vacuum filtration. Hot pressing at 18 MPa, for 15 min, at 110°C | 154 | |
| ChNC/cellulose = 2.5/97.5 | ||||||
| ChNC/cellulose = 5/95 | ||||||
| ChNC/cellulose = 7.5 /92.5 | ||||||
| ChNC/cellulose = 9/91 | ||||||
| ChNC/cellulose = 10/90 | ||||||
| Casting followed by the combination of freeze-drying and hot pressing | ||||||
| 37 | Unvulc.NR/ChNWs | Casting | ChNWs/unvulc. NR = 5/95 | The aqueous suspension of ChWs and unvulc. NR were mixed, stirred and then cast to obtain solid composite films | Freeze-drying and subsequent hot pressing | 51 |
| ChNWs/unvulc. NR = 10/90 | ||||||
| ChNWs/unvulc. NR = 15/85 | ||||||
| ChNWs/unvulc. NR = 20/80 | ||||||
| 38 | PCL/ChNWs | Casting | ChNWs/PCL = 0.5/99.5 | The colloidal chitin dispersion was mixed with the suspension of latex and cast to obtain composite films | Freeze-drying and hot pressing at 21 bar, 100 °C | 97 |
| ChNWs/PCL = 1/99 | ||||||
| ChNWs/PCL = 1.5/98.5 | ||||||
| ChNWs/PCL = 5/95 | ||||||
| ChNWs/PCL = 10/90 | ||||||
| Casting followed by (autoclave) heat treatment | ||||||
| 39 | PVA/α-ChNWs | Casting | ChNWs/PVA = 0.74/99.26 | The dispersion of ChNWs in HCl were mixed with a solution of PVA in distilled water and were cast onto a plastic mold. After solvent removal, ChNWs-reinforced PVA films were heat treated in an autoclave at 110 °C for 5 min | Solvent removal by drying at 40 °C for 24 h | 95 |
| ChNWs/PVA = 1.48/98.52 | ||||||
| ChNWs/PVA = 2.96/97.34 | ||||||
| ChNWs/PVA = 7.40/92.60 | ||||||
| ChNWs/PVA = 14.8/85.20 | ||||||
| ChNWs/PVA = 22.2/77.80 | ||||||
| ChNWs/PVA = 29.6/70.40 | ||||||
| Casting using ionic liquids as dissolving or stabilizing media | ||||||
| 40 | Epoxy resin bisphenol A diglycidyl ether (DGEBA)/ChNWs | Casting | ChNWs/DGEBA = 0.8/98.2 | The CNWs ethanol suspension was mixed with DGEBA epoxy, ethanol was evaporated, and small amount of [C2mim][OAc] IL was added as a dispersion aid/plasticizer. Then the solution was degassed in a vacuum chamber and cast in customized two-part rectangular glass molds to a sheet | Dried in an oven at 100 °C for 24 h to complete the cure | 22 |
| ChNWs/DGEBA = 1.0/99.0 | ||||||
| ChNWs/DGEBA = 1.5/98.5 | ||||||
| ChNWs/DGEBA = 2.0/98.0 | ||||||
| ChNWs/DGEBA = 3.0/97.0 | ||||||
| 41 | PVA (DP 4300)/ChNWs | Gel casting | ChNWs/PVA = 76.92/23.08 | The dispersion of chitin in [Amim]Br ionic liquid (IL) was heated at 100 °C for 48 hours to dissolve chitin and form a gel. A solution of PVA in hot water was then mixed to the gel. The resulting mixture was then soaked in methanol was added to the mixture, followed by sonication, filtration and finally Soxhlet extraction with methanol | Dried under reduced pressure at 60 °C for 12 h | 123 |
| ChNWs/PVA = 83.33/16.66 | ||||||
| ChNWs/PVA = 90.91/9.09 | ||||||
| ChNWs/PVA = 95.24/4.76 | ||||||
| ChNWs/PVA = 99.01/0.99 | ||||||
| Compression molding/curing | ||||||
| 42 | SPE epoxy/ChNFs/chitosan (CS) | Compression molding then curing | ChNFs/SPE/CS = 3/78.7/18.3 | The slurry of SPE and CNFs in aqueous acetic acid were mixed with a solution of CS in acetic acid, poured on a Petri dish made of stainless steel, and stirred using a spatula on a hot plate of 100–140 °C while distilling most of the solvent. Polyamidoamine- (PAA- or PEA-) hardeners were added to the reaction mixture, and stirred at 100 °C for 15 min. The mixture was compression-molded at 6 MPa for 6 h to form a cured biocomposite | The mixture was dried at 80 °C for 12 h and then post-cured at 120 °C for 6 h in an electric oven | 148 |
| ChNFs/SPE/CS = 5/77.1/17.9 | ||||||
| ChNFs/SPE/CS = 10/73.1/16.9 (footnote: calculated without inclusion of hardener) | ||||||
| 43 | SPE epoxy resin/ChNFs/lysine (Lys) | Compression molding/casting | ChNFs/SPE/Lys = 3.00/82.94/14.07 | The dispersion of ChNFs in aqueous acetic acid were mixed with a solution of SPE and lysine (acting as a curing agent) in ethanol and aqueous acetic acid. The suspension was poured on a Petri dish and stirred using a spatula on a hot plate of 100–110 °C while distilling most of the solvent. The mixture was compression-molded at 3 MPa for 3 h to form SPE/Lys/ChNFs | Cured at 60 °C for 24 h and subsequently at 140 °C for 6 h in a vacuum oven | 161 |
| ChNFs/SPE/Lys = 5.00/81.23/13.78 | ||||||
| ChNFs/SPE/Lys = 10.00/76.95/13.05 | ||||||
| 44 | SPE epoxy resin/ChNFs/arginine (Arg) | Compression molding/casting | ChNFs/SPE/Arg = 3.00/84.79/12.21 | The dispersion of ChNFs in aqueous acetic acid were mixed with a solution of SPE and arginine (acting as a curing agent) in ethanol and aqueous acetic acid. The suspension was poured on a Petri dish and stirred using a spatula on a hot plate of 100–110 °C while distilling most of the solvent. The mixture was compression-molded at 3 MPa for 3 h to form SPE/Arg/ChNFs. | Cured at 60 °C for 24 h and subsequently at 140 °C for 6 h in a vacuum oven | 161 |
| ChNFs/SPE/Arg = 5.00/83.05/11.95 | ||||||
| ChNFs/SPE/Arg = 10.00/78.68/11.32 | ||||||
| 45 | SPE epoxy resin/ChNFs/cysteine (Cys) | Compression molding/casting | ChNFs/SPE/Cys = 3.00/84.79/12.21 | The dispersion of ChNFs in aqueous acetic acid were mixed with a solution of SPE and cysteine (acting as a curing agent) in ethanol and aqueous acetic acid. The suspension was poured on a Petri dish and stirred using a spatula on a hot plate of 100–110 °C while distilling most of the solvent. The mixture was compression-molded at 3 MPa for 3 h to form SPE/Cys/ChNFs | Cured at 60 °C for 24 h and subsequently at 140 °C for 6 h in a vacuum oven | 161 |
| ChNFs/SPE/Cys = 5.00/83.05/11.95 | ||||||
| ChNFs/SPE/Cys = 10.00/78.68/11.32 | ||||||
| Compounding | ||||||
| 46 | Acrylonitrile butadiene rubber (NBR)/ChNWs | Masterbatch preparation followed by compounding | ChNWs/NBR = 3.84/96.16 | The ChNWs were dispersed in NBR latex and were coagulated using CaCl2 solution to obtain sheets. The masterbatch was then compounded with solid NBR and other vulcanizing agents to obtain NBR/ChNWs composites | Solvent removal by drying in vacuum at 70 °C for 48 h and conditioning for 24 h | 18 |
| ChNWs/NBR = 7.41/92.59 | ||||||
| ChNWs/NBR = 10.71/89.29 | ||||||
| 47 | Eucalyptus pulp/partially deacetylated chitin nanowhiskers (DeChNWs)/TEMPO-oxidized chitin nanowhiskers (TOChNWs) | Compounding | ChNWs/dry pulp = 0.2/99.8 | The bleached eucalyptus pulp soaked in water was dispersed with a standard pulp defibrilizer and chitin nanowhiskers were added. The mizture was compounded and made into handsheets by drying | Equilibrating at 23 °C and 50% RH for at least 24 h | 121 |
| ChNWs/dry pulp = 0.4/99.6 | ||||||
| ChNWs/dry pulp = 0.6/99.4 | ||||||
| ChNWs/dry pulp = 0.8/99.2 | ||||||
| ChNWs/dry pulp = 1.0/99.0 | ||||||
| ChNWs/dry pulp = 2.0/98.0 | ||||||
| ChNWs/dry pulp = 5.0/95.0 | ||||||
| 48 | Epoxy resin bisphenol A diglycidyl ether (DGEBA)/ChNWs | Slurry compounding then curing | ChNWs/DGEBA = 0.5/99.5 | Suspension of ChNWs in ethanol was added into DGEBA and the mixture was heated at 60 °C. Then amine hardener was added into the ChNWs/DGEBA mixture and homogenized. The mixture was degassed under vacuum at 80 °C for 1 h and cured | Cured at 100 °C for 2 h and at 200 °C for 5 h | 27 |
| ChNWs/DGEBA = 1.0/99.0 | ||||||
| ChNWs/DGEBA = 2.5/97.5 | ||||||
| Melt extrusion | ||||||
| 49 | Polypropylene (PP)/ChNWs | Twin screw micro compounding | ChNWs/PP = 1/99 | The chitin nanowhiskers was added into the mixture of PP and maleated PP (MAPP) and compounded using co-rotating twin screw micro compounder followed by compression molding to obtain composite films | Compression molding at 180 °C and 1000 psi | 19 |
| ChNWs/PP = 2/98 | ||||||
| ChNWs/PP = 5/95 | ||||||
| ChNWs/PP = 10/90 | ||||||
| 50 | PLA/polyhydroxybutyrate-co-valerate (PHBV)/ChNWs | Twin screw micro compounding | ChNWs/PLA = 0.5/99.5 | The dispersions of ChNWs in N,N-dimethyl acetamide (DMAc) solution and oven-dried PLA were blended in co-rotating twin screw micro compounder and molded into a dog-bone shaped tensile specimen | Sealing in high pressure chambers at 4.14 MPa for 48 h followed by soaking at 95 °C for 10 s | 91 |
| ChNWs/PLA = 1.0/99.0 | ||||||
| ChNWs /PLA = 2.0/98.0 | ||||||
| ChNWs /PLA = 5.0/95.0 | ||||||
| 51 | ChNWs/PLA/PHBV | Twin screw micro compounding | ChNWs/PLA/PHBV = 0.5/84.6/14.9 | The dispersions of ChNWs in N,N-dimethyl acetamide (DMAc) solution and oven-dried PLA and PHBV pellets were blended in co-rotating twin screw micro compounder at 180 °C with the screw speed of 200 rpm for 10 min, and shaped into a dog-bone shaped tensile specimen | Compositions were air cooled, cut into pellet-sized pieces, and oven dried at 45 °C for 24 h | 91 |
| ChNWs/PLA/PHBV = 1.0/84.5/14.8 | ||||||
| ChNWs/PLA/PHBV = 2.0/84.4/14.7 | ||||||
| ChNWs/PLA/PHBV = 5.0/80.7/14.3 | ||||||
| 52 | PLA/α-ChNWs | Melt blending/batch-foaming | ChNWs/PLA = 2/98 | PLA was compounded with chitin nanocomposite using benchtop twin screw compounder to yield compatibilized chitin nanocomposite. Then the foams of PLA–chitin composites were prepared by two-step batch foaming technique using CO2 | Heated in hot water at 95 °C for a period of 20 s and then quenched in cold water | 92 |
| ChNWs/PLA = 5/95 | ||||||
| ChNWs/PLA = 1/99 | ||||||
| 53 | PLA grafted with maleic anhydride (PLA-(grafted)-MA)/α-ChNWs | Melt blending/batch-foaming | ChNWs/(PLA-(grafted)-MA) = 2/98 | PLA-(grafted)-MA with peroxide-based free radical initiator were compounded with chitin nanocomposite and PLA using benchtop twin screw compounder to yield compatibilized chitin nanocomposite. Then the foams of PLA–chitin composites were prepared by two-step batch foaming technique using CO2 | Heated in hot water at 95 °C for a period of 20 s and then quenched in cold water | 92 |
| ChNWs/(PLA-(grafted)-MA) = 5/95 | ||||||
| ChNWs/(PLA-(grafted)-MA) = 1/99 | ||||||
| 54 | PLA (Mw 170000)/chitin nanofibrils (ChNFs)/polyethylene glycol (PEG, Mw 400, and 8000) |
Extrusion | PEG400/ChNFs/PLA = 10/2/88 | The dispersions of chitin nanofibrils (ChNFs) in water were mixed with poly(ethylene glycol) (PEG) and dried by water evaporation to prepare a solid pre-composite. The pre-composite is then added to PLA in the injection molding extruder to obtain transparent nanocomposites | Injection molding at 180 °C, 650 bars holding time of 15 s, mold temperature of 35 °C | 165 |
| PEG8000/ChNFs/PLA = 10/2/88 | ||||||
| PEG400/ChNFs/PLA = 1/2/97 | ||||||
| PEG400/ChNFs/PLA = 5/2/93 | ||||||
| PEG400/ChNFs/PLA = 10/5/85 | ||||||
| PEG400/ChNFs/PLA = 10/12/78 | ||||||
| 55 | Corn starch granules/α-ChNFs/α-ChNCs | Melt-mixing in micro compounder, then injection molding | ChNFs or ChNCs/corn starch = 5/95 | The dispersions of ChNWs or ChNFs in distilled water with glycerol plasticizer were mixed with starch using micro compounder, then molded using an injection molding | Conditioning at 50% relative humidity (RH) at 25 °C for 7 days | 20 |
| ChNFs or ChNCs s/corn starch = 10/90 | ||||||
| ChNFs or ChNCs/corn starch = 15/85 | ||||||
| ChNFs or ChNCs/corn starch = 20/80 | ||||||
| 56 | PLA/α-ChNFs | Extrusion with Minilab (small twin-screw extruder) | ChNFs/PLA = 10/90 | The ChNWs (pre-treated in water, polyethylene glycol (PEG), or polyethylene oxide (PEO) followed by freeze-drying), were mixed with PLA powder followed by blending, compounding, and ultimately extrusion | Not conducted | 26 |
| ChNFs/PLA = 20/80 | ||||||
| ChNFs/PLA = 30/70 | ||||||
| ChNFs PLA = 40/60 | ||||||
| ChNFs/PLA/PEG or PEO = 10/80/10 | ||||||
| ChNFs/PLA/PEG or PEO = 20/70/10 | ||||||
| ChNFs/PLA/PEG or PEO = 30/60/10 | ||||||
| ChNFs/PLA/PEGorPEO = 40/50/10 | ||||||
| In situ polymerization | ||||||
| 57 | Poly(butylene adipate-co-furanoate) (PBAF) made in situ from adipic acid (AA) and dimethyl 2,5-furandicarboxylate (DMFD)/sulfated α-chitin nanowhiskers (S-ChNWs) | In situ polymerization, then hot-pressing | S-ChNWs/(AA + DMFD) = 0.1/99.9 | Respective amount of S-ChNW in butanediol were added to a liquid mixture of AA and DMFD monomers, and titanium(IV) butoxide (TBT, 97%) catalyst by heating at 180 °C for 2 h for transesterification, then increased to 210 °C for 2 h, and to 240 °C for complete polymerization, finally polycondensation for in situ formation of PBAF | Films were prepared by pressing the in situ composites at 150 °C at 10 Pa for 5 min and then quenching in cold water | 105 |
| S-ChNWs/(AA + DMFD) = 0.3/99.7 | ||||||
| S-ChNWs/(AA + DMFD) = 0.7/99.3 | ||||||
| S-ChNWs/(AA + DMFD) = 1.0/99.0 | ||||||
| 58 | Polyethylene glycol dimethacrylate (PEGDA, Mn = 6000 Da)/CS (Mw = 100000 Da)/ChNWs |
In situ free radical polymerization using UV light (365 nm, 50 mW cm−2) | PEGDA/CS = 88.24/11.76 | In situ free radical polymerization of PEGDA/chitosan/ChNWs aqueous suspension initiated by UV light | Not conducted | 90 |
| PEGDA/CS/ChNWs = 68.18/9.09/22.73 | ||||||
| 59 | Poly(butylene adipate-co-terephtalate) (PBAT) made in situ from adipic acid (AA) and dimethylterephtalate/sulfated α-chitin nanowhiskers (S-ChNWs) | In situ polymerization, then hot-pressing | S-ChNWs/(AA + DMT) = 0.1/99.9 | Respective amounts of S-ChNW in butanediol were added to a mixture of AA and DMT monomers, and titanium(IV) butoxide (TBT, 97%) catalyst by heating at 180 °C for 2 h for transesterification, then increased to 210 °C for 2 h, and to 240 °C for complete polymerization, finally polycondensation for in situ formation of PBAT | Films were prepared by pressing the in situ composites at 150 °C at 10 Pa for 5 min and then quenching in cold water | 105 |
| S-ChNWs/(AA + DMT) = 0.3/99.7 | ||||||
| S-ChNWs/(AA + DMT) = 0.7/99.3 | ||||||
| S-ChNWs/(AA + DMT) = 1.0/99.0 | ||||||
| 60 | PANI-grafted (g)/chitin nanocrystals (ChNCs) | In situ oxidative polymerization | ChNC/PANI = 14.3/85.7 | PANI-g-ChNCs were prepared via in situ oxidative polymerization of aniline monomers with ammonium persulfate serving as the oxidant and HCl as an acid dopant for PANI. The ChNC suspension was sonicated for 30 s in acidic HCl solution, after which the corresponding amount of aniline was added. The suspension was centrifuged, washed several times with 0.5 M HCl to remove byproducts and remaining agents, and the pH was adjusted to 3. For the preparation of PANI-g-ChNC/latex nanocomposites the desired amounts of PANI-g-ChNCs suspension were sonicated for 20 s then added to 1.5 g of latex under stirring, and cast into Teflon mold | Dried at room temperature for 24 h | 29 |
| ChNC/PANI = 50/50 | ||||||
| 3D printing | ||||||
| 61 | Methacrylate (MA)/α-chitin photocurable nanowhiskers (pChNWs, DDA ∼6.4%, Mw ∼150 kDa) | Stereolithography (SLA) printing via radical photo-polymerization | pChNWs/MA = 0.2/99.8 | The ChNWs of varying amounts were mixed with the MA resin and underwent SLA printing via radical photopolymerization to form 3D-printed nanocomposites | Solvent was removed by drying in vacuum oven at 60 °C for 15 min | 81 |
| pChNWs/MA = 0.5/99.5 | ||||||
| pChNWs/MA = 1.0/99.0 | ||||||
| 62 | MA resin/ChNWs | Modified slurry compounding | ChNWs/MA = 0.5/99.5 | The dispersions of ChNWs in ethanol was mixed with MA-ethanol solution and the nanocomposite resin obtained were 3D printed | The solvent was removed by drying in vacuum at 60 °C | 1 |
| ChNWs/MA = 1.0/99.0 | ||||||
| ChNWs/MA = 1.5/98.5 | ||||||
| Gel spinning and electrospinning | ||||||
| 63 | PVA/α-ChNWs | Gel spinning | ChNWs/PVA = 3/97 | The dispersion of ChNWs and water was mixed with a solution of PVA in distilled water in the rotary mixer and fibers were fabricated using a laboratory-scale gel-spinning apparatus | Cooling in a methanol bath for 24 h followed by solvent removal by evaporation for 48 h | 94 |
| ChNWs/PVA = 5/95 | ||||||
| ChNWs/PVA = 10/90 | ||||||
| ChNWs/PVA = 15/85 | ||||||
| ChNWs/PVA = 20/80 | ||||||
| ChNWs/PVA = 30/70 | ||||||
| 64 | PVA (Mw = 78000)/α-ChNWs |
Electro-spinning | ChNWs/PVA = 2.5/97.5 | The dispersions of ChNWs was mixed with a solution of PVA in distilled water and electrospun using a single-needle electrospinning to form nanofibrous membranes | Solvent removal by drying at 155 °C for 15 min | 96 |
| ChNWs/PVA = 5.0/95.0 | ||||||
| ChNWs/PVA = 7.5/92.5 | ||||||
| 65 | Cellulose acetate (CA Mn ≈ 30000 Da)/ChNWs |
Electro-spinning | ChNWs/CA = 0.5/99.5 | The solution of CA in acetone and DMAc was mixed with ChNWs and electrospun using a single-needle electrospinning to form nanofiber mats | Solvent removal by vacuum drying at ∼25 °C and conditioned at ∼53% RH | 142 |
| ChNWs/CA = 2.5/97.5 | ||||||
| 66 | PVA (Mw = 75000 Da)/α-ChNWs |
Electro-spinning | ChNWs/PVA = 2.55/97.45 | The suspension of ChNWs in water was mixed with PVAaq solution, then the resulting suspension was electrospun | Solvent removal by evaporation at room temperature for 3 days | 49 |
| ChNWs/PVA = 5.11/94.89 | ||||||
| ChNWs/PVA = 7.66/92.34 | ||||||
| ChNWs/PVA = 10.11/89.89 | ||||||
| ChNWs/PVA = 12.76/87.24 | ||||||
| ChNWs/PVA = 15.17/84.83 | ||||||
| ChNWs/PVA = 17.74/82.26 | ||||||
| ChNWs/PVA = 25.38/74.62 | ||||||
| Coating | ||||||
| 67 | Polyethylene terephthalate (PET)/ChNWs/cellulose nanocrystals (CNCs) | Spray-coating | ChNWs/CNCs = 50/50 coated in sequence on PET film | Two separate dispersions of ChNWs and CNCs in water were spray-coated on PET film in sequence, to form a bilayer-coated PET film | Solvent removal by drying at room temperature for 24 h | 129 |
| 68 | Cellulose acetate (CA)/α-ChNWs/CNCs | Spray-coating | ChNWs/CNCs = 50/50 coated in sequence on CA film | Two separate dispersions of ChNWs and CNCs in water were coated on the CA substrate to form a bilayer-coated CA film | Solvent removal by drying at room temperature for 96 h | 140 |
| 69 | CA/ChNWs/cellulose nanocrystals (CNCs) | Spray coating followed by thermal treatment | ChNWs/CNC = 50/50 coated in sequence on CA film | Two separate dispersions of ChNWs and CNCs in water were spray-coated on CA-film, to form a bilayer-coated CA film | Solvent removal by drying at room temperature for 24 h and conditioned at ∼53% RH | 141 |
| 70 | α-ChNWs/CS/PET | Blade-coating | ChNWs/CS = 66.66/33.33 and ChNWs/CS = 50.0/50.0 and ChNWs/CS = 33.33/66.66 spread over PET substrate | The dispersion of ChNWs and CS (mixed together) in water was spread over PET substrate | Solvent removal by drying at 60 °C for 30 min | 130 |
| 71 | α-ChNWs/PET | Blade-coating | ChNWs spread over PET substrate | The dispersion of ChNWs in water was spread over PET substrate | Solvent removal by drying at 60 °C for 30 min | 130 |
| 72 | α-Short chitin nanowhiskers (SChNWs 114 nm) or α-long chitin nanowhiskers (LChNWs, 230 nm)/chitosan (CS)/polyethylene terephthalate (PET) | Blade-coating | SChNWs/CS = 66.66/33.33 and SChNWs/CS = 50.0/50.0 and SChNWs/CS = 33.33/66.66 | The dispersion of (ChNWs + CS) mixed together spread over PET substrate pre-coated with CNC in layer-by-layer (LBL) fashion | Solvent removal by drying at 60 °C for 30 min | 130 |
| LChNWs/CS = 66.67/33.33 | ||||||
| LChNWs/CS = 50/50 | ||||||
| LChNWs/CS = 33.3366.67 spread over PET substrate pre-coated with CNC | ||||||
| 73 | PP/α-ChNWs/TEMPO cellulose nanofibers (TCNFs) | Layer-by-Layer (LBL) coating | ChNWs and TCNFs coated in sequence on PP film, ratio not shown | The ultraviolet/ozone treated PP substrates were alternately immersed into two separate dispersions: ChNWs and TCNF in water forming bi-layered coated films | Solvent removal by drying at 80 °C for 24 h | 82 |
| 74 | PET/α-ChNWs/cellulose nanofibers (CNFs) | Layer-by-Layer (LBL) coating | ChNWs and CNFs coated in sequence on PET film, ratio not shown | The dispersion of ChNWs in aqueous acetic acid and CNFs in water were sprayed on the upper surface of the PET substrate in a zigzag pattern completing a single bilayer structure | Solvent removal by hot-pressing at 80 °C at 32 MPa for 10 min | 89 |
| Impregnation | ||||||
| 75 | PVA (99% hydrolyzed)/ChNFs | Impregnation | ChNFs/PVA = 60/40 | ChNFs dispersed in water were vacuum filtered to make a wet sheet. After drying, ChNFs pre-made film was impregnated into the pre-prepared PVAaq solution for 12 h as a sandwich-like structure | Solvent removal by drying in an oven at 55 °C for 12 h | 16 |
| 76 | Gelatin (gel)/ChNFs | Immersion | ChNFs/gel = 73.0/27.0 | ChNF suspensions were vacuum filtered to prepare a wet film. The gelatin powder was dissolved in distilled water at different concentrations. Afterwards the wet ChNF films were immersed into gelatin solutions and kept at 60 °C for 12 h, respectively | The ChNF/gelatin films were dried at room temperature for 2 days | 143 |
| ChNFs/gel = 62.5/37.5 | ||||||
| ChNFs/gel = 50.2/49.8 | ||||||
| 77 | PVA (Mw = 26000 Da)/α-ChNWs |
Solution impregnation | Sandwich-like structure, ratio not shown | ChNWs aqueous suspension was cast, dried and ChNWs film was obtained. After that, ChNWs pre-made film was impregnated in the pre-prepared PVAaq solution for 12 h as a sandwich-like structure | Solvent removal by evaporation at room temperature overnight, followed by drying at 40 °C for 12 h | 52 |
| 78 | Microcrystalline cellulose (MCC, Mw 1 × 105)/α-ChNWs | Impregnation | ChNWs/MCC = 1.0/99.0 | The dispersion of ChNWs was introduced into cellulose solutions in phosphoric acid at the stage of cellulose regeneration | The obtained material was a gel, that was freeze-dried and vacuum dried for the characterization | 79 |
| ChNWs/MCC = 2.5/97.5 | ||||||
| ChNWs/MCC = 5.0/95.0 | ||||||
| ChNWs/MCC = 10.0/90.0 | ||||||
The solvents are often evaporated at room temperature, elevated temperatures, under vacuum, through incubation, or using a combination of these methods. For instance, various studies by different authors on the reinforcement of PVA have reported solvent removal being conducted at room temperature, elevated temperatures, or under vacuum. The specific conditions varied depending on factors such as the solution stability and uniformity of dispersion,78,137 the use of additional reagents (e.g. NaOH, NaCl),40,80 or the types of solvents employed.40,49,80,83,149,150 In the casting-evaporation process, this treatment provides sufficient time for ChNWs to form a percolated network through hydrogen bonding interaction, resulting in composites with enhanced mechanical properties.
Lyophilization, or freeze-drying (Table 7, entries 28–31), is another technique used for the solvent removal to obtain solid nanocomposite films. This process involves freezing the material, followed by sublimation under reduced pressure,151 creating a porous network within the matrix while preserving the material's structure. Freeze-drying begins with the formation of ice crystals, which is achieved through different freezing methods. Slow freezing occurs at approximately −20 °C, while fast freezing uses dry ice and organic solvents at −78 °C. Ultra-fast freezing is typically done with liquid nitrogen at around −196 °C.152 The size and structure of the ice crystals formed during freezing significantly influence the pore size and morphology of the composite matrix. Fast freezing generally results in smaller ice crystals, which cause minimal disruption to the material's original morphology. In contrast, slow freezing forms larger ice crystals that can penetrate the structure, potentially creating fractures or cavities, which may compromise the material's integrity. For example, under freezing conditions of −40 °C followed by lyophilization, alginate dressings with ChNWs (Table 7, entry 28) exhibited a soft, porous morphology, and reduced surface roughness (9.17 ± 4.06 μm).75 In another study, sodium alginate (ALG)–ChNW hydrogels formed under slow freezing (−20 °C) followed by lyophilization (Table 7, entry 29) developed a honeycomb structure.132 A directional freeze-drying which involves freezing the material in a controlled direction, often achieved by applying a temperature gradient during freezing, is also used. This method promotes the formation of aligned ice crystals, which, upon sublimation during lyophilization, result in a structured matrix with oriented channels. Thus, a directional freeze-casting method using fast freezing with liquid nitrogen (−196 °C) followed by lyophilization (Table 7, entry 30) produced structured ChNW/PVA biofoams with an oriented and continuous channel architecture.153
2.2. Hot pressing
The hot-pressing technique is another widely used for drying chitin nanoparticle–polymer suspensions or solutions. This process involves placing the material into a mold, typically made of high heat- and pressure-resistance materials, and subjecting it to elevated temperatures and pressures simultaneously. The technique shapes and reinforces the polymer composites into a more compact and structurally sound form. In general polymer applications, heat is often provided by induction or resistance heating, with temperatures reaching up to 2400 °C and pressures up to 50 MPa. However, for chitin nanocomposite fabrication, much lower temperatures and pressures are employed, to prevent chitin nanomaterials from degradation.154 Examples include 100 °C and 2.1–13.8 MPa (Table 7, entries 32–33),25,51,97 140 °C and 20 MPa (Table 7, entry 34),155 190 °C and 15 kPa (Table 7, entry 35),156 and 110 °C and 18 MPa (Table 7, entry 36).154 This technique improves mechanical performance by densification of materials and eliminating porosity. The combination of high temperature and pressure compacts the material, reducing voids and improving interfacial bonding between components in the ChNWs composites.The combination of two drying techniques can also be applied in the fabrication of ChNW-reinfored composites. For instance, freeze-drying is often combined with hot pressing to produce ChNW-reinforced NR composite films (Table 7, entry 37).51 However, the obtained composite films were up to 20 times mechanically weaker than those prepared by casting-evaporation (10.2 vs. 229 MPA, respectively). This disparity is likely due to the insufficient formation of a robust 3-D rigid network through hydrogen bond interaction as a result of fast solvent removal process as opposed to relatively slow solvent removal in casting-evaporation. The same freeze-drying/hot pressing combination in the case of ChNW/poly-(caprolactone) composites97 (Table 7, entry 38) also produced materials with great mechanical attributes.51,97
Autoclave heat treatment has been reported as another solvent removal method to prepare ChNW-reinforced PVA composite films (Table 7, entry 39).95 This treatment reduces the water affinity of the ChNW-reinforced composites. Here, heat treatment has been shown to improve the mechanical properties and dimensional stability of PVA-based materials. While the mechanical properties of heat-treated ChNW/PVA composites were not explicitly reported, it can be presumed that they exhibit similar improvements due to the enhanced structural integrity imparted by heat treatment.95
2.3. Methods that utilize ionic liquids
The use of ionic liquids (ILs) has also been explored as processing aids in chitin nanocomposite fabrication. For example ILs such as [Amim]Br,123 and [C2mim][OAc]23 (Table 7, entries 40 and 41) have demonstrated utility in various roles. Thus, [C2mim][OAc] has been employed as both a dispersion aid and a plasticizer, effectively disrupting the hydrogen bonds in chitin (Table 7, entry 40). This disruption facilitates a more uniform ChNW distribution in the epoxy–polymer matrix.23 In another example [Amim]Br,123 was used not merely as a dispersant but as a solvent for the dissolution of ChNWs (Table 7, entry 41). This approach differs from traditional ChNWs dispersion, allowing for improved homogeneity of ChNWs within the matrix. Specifically, the use of ILs such as [Amim]Br enables a more uniform distribution of ChNWs within the PVA matrix, contributing to enhanced material properties.2.4. Compression molding and curing
Compression molding and curing are integral processes in the fabrication of ChNWs-reinforced composite materials. Compression molding is a widely used manufacturing process where a preheated polymer or composite is placed into a preheated mold cavity (120–200 °C) using a two-segment mold system. Preheating ensures that the thermoplastic polymer melts uniformly, preventing inconsistent flow.157 The mold is then tightly closed with a plug and subjected to high pressure ensuring the resin remains in contact with the entire mold surface and conforms to its shape. One segment of the compression molder (lower plate) is static, while the other (upper plate) is movable.158 The vacuum generated by the compression lower plate increases pressure on the material, creating uniform Preheating ensures thermoplastic polymer melts pressure and temperature across the mold cavity. This uniformity allows the material to flow, take the shape of the cavity, and undergo chemical or physical changes necessary for achieving the desired properties.159 Biocomposites composed of sorbitol polyglycidyl ether (SPE) as a bio-based epoxy resin and ChNFs were reportedly reinfored by this method (Table 7, entries 42–45). Additionally, this expoxy-based composites often require curing after the applied compression molding to achieve the final hardened state and optimal properties.Curing, in the context of ChNW-reinforced polymer processing combined with compression molding, refers to the process of hardening or setting the material by applying heat and/or pressure on the molds, inducing crosslinking of the polymer chains. It can occur during the compression molding process or as a separate step, particularly with thermosetting polymers, such as epoxy resins which form permanent bonds by strong cross-linking (Table 7, entries 43–45).160,161 This method is frequently favored for nanofiber-based composites (specifically chitin nanofiber biocomposites), where the ChNFs are dispersed in solvents, often organic acids such as acetic acid,148,161 formic acid,149,150etc., and then combined with polymer solutions to form a paste-like mixture. The paste is then molded and can be used ‘as is’ or treated with a cross-linking curing agent, such as amino acids or chitosan.148,161 After molding, curing is typically completed by drying in oven with or without vacuum. Examples include temperatures of 120–140 °C during 6 h treatment (Table 7, entries 42–45). Compared to injection molding, compression molding takes longer in the shaping process and may have less consistent results, but it is cost-effective for producing fabrics, fiber mats, strands, and tapes.
2.5. Compounding and extrusion
Compounding refers to the process of integrating reinforcement additives (ChNWs or ChNFs) into a polymer matrix to achieve specific material properties. This process is distinct from simple blending or mixing, as it typically requires the melting of the matrix polymer to ensure uniform dispersion and effective integration of the additives. Specifically, in this process, materials are mixed in certain ratios, the mixture is melted and homogenized in a compounding machine, and the compounded material is cooled and pelletized for further use. It is possible to be processed and performed using extruders, high-shear mixers, or pelletizers (Table 7, entries 46–48).Before the advent of continuous mixing using extruders, blends were commonly prepared using batch mixing. This was typically conducted in open mills equipped with two-roll systems featuring contra-rotating cylindrical rolls. The base polymers and fillers were repeatedly passed between these rolls to ensure consistent and uniform dispersion of the fillers throughout the matrix and to prevent particle aggregation.162 Batch mixing using roll mills is particularly suited for elastomers (rubbers) due to their high elasticity and resistance to mechanical stress. Both thermoplastics, such as PP and PLA, and thermosetting polymers (such as epoxy resin) can be processed using this method, because of their chemical and thermal stability. For instance, the preparation of NBR/ChNW composites utilized a two-roll mill,18 starting with the formulation of a mixture of polymers and ChNWs, which was then compounded with vulcanizing agents (Table 7, entry 46). In a separate study involving ChNWs/epoxy nanocomposites, a solvent-mediated slurry compounding was employed to create a homogeneous dispersion of chitin nanofillers and hardeners within the epoxy matrix27 (Table 7, entry 48). The solvent-mediated approach demonstrated advantages in cost efficiency, time savings, reduced solvent consumption, and improved sustainability. Additionally compounding has been applied to the preparation of nanocomposite ChNWs/dry pulp paper handsheets. This process involves the formation of a homogeneous pulp suspension prior to the application of the standard handsheet formation method121 (Table 7, entry 47).
Extrusion offers a continuous mixing process for composite fabrication. In this method, the ChNWs–polymer blends pass through extruders where they are uniformly blended, devolatilized, and then transferred to the molds via injection or compression molding163 (Table 7, entries 49–56). The screw speed can be adjusted for either more intense or moderate mixing, depending on the final material. Two common types of extruders are Single-Screw Extruders (SSE) and Twin-Screw Extruders (TSE). SSEs feature simple designs, low operational costs, and high reliability for small to moderate-scale production. However, their mixing capabilities are limited, often requiring additional devices or screw designs for improved efficiency. The fixed screw design may lead to prolonged residence times, causing inconsistent processing and potential material degradation.164
Conversely, TSEs feature two intermeshing screws that can be customized for better mixing, melting, and conveying. This makes the twin-screw compounder a preferred choice, particularly for its ability to efficiently blend the polymer dispersions prior to molding. Its compatibility with thermoplastic polymers has led to multiple studies exploring the blending of ChNWs with polymers such as PP,19 PLA,26,91,92,165 and PHBV.91 In this process, the thermoplastics fed into the extruders are melted by heat generated from both the rotating screws and external heaters. The melted samples are then mixed both dispersively and distributively by the action of the screws, ensuring uniform distribution of the components throughout the blend.164 The molten mixtures are then directed toward the die, where they are shaped into desired forms such as pellets, films, discs, or hand sheets. Typical operating parameters for a micro-compounder include temperatures of 150–200 °C, screw speeds of 200 rpm, and processing time 1–10 minutes. Extruded materials are often pelletized for further processing or molded into different shapes. Depending on the target material, they may also undergo batch foaming techniques to produce foams (Table 7, entries 52–53). After molding or extrusion, materials are stabilized under specific conditions such as vacuum, hot/cold water, high pressure, or oven-drying. These treatments ensure the final properties of the materials are preserved and optimized for their intended applications.
2.6. In situ polymerization
In situ polymerization involves the polymerization of monomers directly in the presence of a reinforcing material such as nanochitin. During in situ polymerization, ChNWs or ChNFs are dispersed in a liquid monomer or a liquid mixture of monomers, leading to the formation of a polymer matrix that encapsulates nanochitin particles (Table 7, entries 57–60).29,90,105 Polymerization process can be initiated by heat (Table 7, entry 57),105 radical initiator or UV-light (Table 7, entry 58),90 a catalyst (Table 7, entry 59),105 or an oxidizing agent (Table 7, entry 60).29 Here, the nanochitin acts as a template through the surface amino groups of its nanocrystals,29 and serves as a reinforcement phase,90,105 providing composite materials with remarkable strength and durability. When used as a structural skeleton, nanochitin provides a hierarchical framework that guides the formation and alignment of the polymer matrix around it, during the in situ polymerization process. For instance, the PANI-grafted chitin nanocrystals were synthesized by polymerizing the aniline monomer onto ChNCs in an acidic HCl solution.29 the process began with the protonation of amino groups on the ChNC followed by their oxidation by ammonium persulfate into the ammonium cation. Polymerization was then initiated by the addition of the aniline cation radicals, leading to chain propagation driven by interactions between the growing oligomer radical cations and aniline radical cations. This continued until the aniline was fully consumed, resulting in the formation of PANI-grafted ChNC polymers.29In situ polymerization incorporates nanofillers directly into the polymer matrix at the monomeric stage, achieving superior dispersion compared to ex situ methods.2.7. 3D-printing
3D printing, or additive manufacturing, is a process used to create three-dimensional objects. This technology facilitates the development of printable inks or filaments by mixing ChNWs with polymers. This integration can be achieved with various printing methods such as fused deposition modeling (FDM),166 stereolithography (SLA, see Table 7, entries 61 and 62),1,77 and inkjet printing. SLA is particularly favored for the fabrication of ChNWs-reinforced composites due to its precision, speed, and ability to create highly customized and complex structures.77 For instance, SLA 3D printing with photocurable methacrylate-based resin reinforced with 0.5 wt% ChNWs improved the tensile strength and stiffness of material up to 78 and 71%, respectively without compromising the toughness and ductility77 (Table 7, entry 61). This method enables the creation of complex, customized shapes and structures that are difficult to achieve with traditional manufacturing methods such as molding or extrusion. Additionally, 3D printing can produce multi-functional features such as regions with varying stiffness or porosity tailored to particular uses.2.8. Gel spinning and electrospinning
Gel spinning94 and electrospinning,49,96,142 are often combined with solution mixing to produce nanocomposite fibers or nanofibrous mats and enhance the alignment of nanochitin within the polymer matrix. Due to polymer chain alignment, both processes result in fibers with superior strength compared to those made using other techniques. In these processes, a uniform polymer–nanochitin suspension is first prepared via adding ChNWs to a polymeric solution and the resulting mixture is then subjected to conditions that promote gelation, forming a semi-solid gel. The gel is extruded through a spinneret to form continuous fibers which are then solidified by drying in air or coagulation in a liquid bath, such as methanol or water, to remove the solvent. Examples include mixing the dispersion of ChNWs and water with an aqueous solution of PVA fabricating fibers using a laboratory-scale gel-spinning apparatus (Table 7, entry 63).167 Post-treatment processes, such as stretching or drawing, are often applied to further align the polymer chains and enhance mechanical properties, while additional treatments like heat-setting or annealing stabilize the fibers. Since the jets are only stretched to a limited extent, the resulting fibers typically have diameters ranging from 10 to 100 μm.167 A combination of solution mixing and gel spinning leverages the benefits of both methods, yielding nanocomposite fibers with smooth surfaces, uniform diameters, and enhanced mechanical properties.94 Despite its advantages, including the ability to produce ultra-strong, lightweight fibers, gel spinning requires precise control of processing conditions and may involve the use of hazardous solvents, making it more complex and expensive than other spinning methods.Unlike conventional fiber spinning techniques (wet spinning, dry spinning, melt spinning, gel spinning), electrostatic spinning (or electrospinning) is capable of fabricating ultrafine fibers with diameters in the nm to sub-μm range.168 In this process, a high-voltage electrostatic field stretches a polymer suspension containing ChNWs into fine fibers, which are collected on a grounded screen (Table 7, entries 64–66).168 The polymer solution is ejected from a single-needle syringe or a multi-needle spinneret, with the applied voltage (typically 14–21 kV)49,96,142 controlling the alignment of the nanochitin within the fibers. As fibers form, solvent evaporation leads to the production of solid nanocomposite fibers or mats. ChNWs-reinforced electrospun fibers have been successfully created using water soluble polymers, such as PVA or CA, with solvent evaporation occurring under various conditions such as at room temperature,49 under vacuum drying, or at elevated temperatures,96 with or without additional conditioning142 (Table 7, entries 64–66). This technique consistently produces fibers in the nanometer range, offering significant potential for advanced nanocomposite applications, with ChNWs assisting in reducing fiber diameter. Thus, in the fabrication of electrospun CA–ChNWs composites, the addition of as little as 0.5 wt% ChNWs reduced the fiber diameters more than twice (223 ± 76 nm for the CA–ChNWs nanofibers vs. 563 ± 222 nm for the CA nanofibers).142 This reduction was attributed to the increased electrical conductivity of the ChNW-containing solution, which accelerated and stretched the polymer jet during electrospinning. Electrospinning is a highly adaptable method for creating ChNW–polymer forms such as mats, membranes, or individual fibers tailored to specific applications.
2.9. Coating methods
Another commonly used method for fabricating reinforced nanochitin composites is coating. This method involves applying a thin layer of nanochitin (or a nanochitin-containing mixture) onto a substrate surface, typically through methods such as spraying,129,140,141 blade-coating,130 or layer-by-layer (LbL) assembly82,89 (Table 7, entries 67–74). In this process, nanochitin is dispersed in a solvent and applied to a surface of polymeric material, where it subsequently dried or cured to form a thin film. Coating primarily modifies the surface of composites, with the addition of nanochitin concentrated in thin surface layers, typically on the micrometer scale, to improve attributes such as water resistance and barrier performance.For oxygen barrier coatings in the manufacture of biomaterials, nanochitin is often combined with nanocellulose to form bilayers or multiple-layer films (Table 7, entries 73 and 74).82,89 This technique utilizes surface-modified, oppositely charged cellulose nanofibers (CNFs) and chitin nanowhiskers (ChNWs) to create a stable polymer coating through sequential layer-by-layer (LbL) assembly driven by electrostatic interactions. First, surface modifications were applied to both cellulose nanofibers (CNFs) and ChNWs to enhance their compatibility and functionality. TEMPO-mediated oxidation was applied to CNFs, introducing carboxylate groups and producing negatively charged CNFs. In turn, ChNWs underwent deacetylation to expose amino groups, resulting in positively charged ChNWs. These modified nanomaterials were subsequently coated onto the polymer surface using a layer-by-layer (LbL) assembly technique. This process involved the sequential deposition of the oppositely charged CNFs and ChNWs, driven by strong electrostatic interactions, to create a stable and uniform coating. Hydrogen bonding and electrostatic interactions between chitin and cellulose nanomaterials promotes densification at the interface, improving barrier properties. Since this method focuses on surface enhancement and does not significantly reinforce the bulk material, it is particularly well-suited for improving surface properties such as biocompatibility, and barrier performance particularly in food packaging applications.
2.10. Impregnation techniques
In the fabrication of nanochitin reinforced composites, impregnation refers to the process of incorporating nanochitin into polymeric matrix. The impregnation process typically begins with the preparation of a stable suspension of nanochitin in a liquid medium, often water or an organic solvent. This suspension is then processed into a pre-made film of nanochitin using methods like vacuum filtration or casting. The resulting film serves as a scaffold that will later interact with the polymeric matrix. To achieve impregnation, the nanochitin film is usually immersed in a solution of a second polymer, allowing the polymer molecules to infiltrate the porous structure of the nanochitin film and form a cohesive composite upon solidification. Reduced air pressure or applied force can be utilized to enhance the polymer's penetration into the nanochitin framework. These techniques improve the polymer's ability to uniformly infiltrate and fill the nanochitin's internal network, ensuring thorough embedding of the polymer and maximizing the interfacial bonding between the nanochitin and the polymer matrix. Ultimately, the choice of impregnation method depends on the desired composite properties, the compatibility between the polymer and nanochitin, and the manufacturing setup. Immersion-based impregnation remains the most commonly used approach due to its simplicity and effectiveness in producing uniform composites16,52,143 (Table 7, entries 75–77). Following impregnation, the composite is dried (at room temperature,143 or elevated temperatures16,52) to solidify the material. This method emphasizes using nanochitin films as scaffolds for polymer infiltration and is suitable for a wide variety of polymers and matrices.Contrarily it is possible to directly integrate chitin nanowhiskers into a polymeric gel. Thus, in another study,79 the impregnation of ChNWs into cellulose cryogels was achieved by dispersing ChNWs into the cellulose matrix during the regeneration of cellulose from a phosphoric acid solution (Table 7, entry 78), followed by freeze-drying. In this process, ChNWs served a dual role as both nanofillers and natural crosslinking agents, which significantly contributed to the formation of denser structures with reduced shrinkage and smaller composite cryogel volumes. This tighter packing resulted in a more robust cryogel structure, with the incorporation of ChNWs leading to a threefold increase in the strength of the cellulose cryogels.79 This impregnation method is relatively simple, and applicable to a wide range of matrices, including porous, fibrous, or layered materials. Its versatility makes it effective for reinforcing bulk materials by infusing nanochitin into a matrix. However, challenges such as achieving uniform dispersion and ensuring solvent compatibility may limit its scalability and effectiveness in certain applications.
Fig. 4 presents examples of surface micrographs illustrating how different processing methods affect the performance of ChNW-based composites. As previously discussed, the casting-evaporation method generally yields composites with superior mechanical properties compared to hot pressing (thermal processing) as demonstrated in the rubber/ChNW system25 (Fig. 4a1 and a2) because solvent evaporation occurs gradually, allowing sufficient time for the nanofiller to form percolation network. In contrast, the shear stress during hot pressing tends to cause ChNW agglomeration. In the case of the PVA reinforcement with nanochitin, the casting method123 and gel spinning136 produced films and fibers with low and high molecular orientation, respectively (Fig. 4b1 and b2). Electrospinning, however, is employed to fabricate highly porous nanofibers with improved mechanical and functional properties49 (Fig. 4b3). From a thermodynamic perspective, in situ polymerization incorporates nanochitin into the polymer matrix from the monomeric stage, achieving better dispersion than ex situ mixing. This approach has been widely used to develop various hybrid and green biocomposites such as the nylon/ChNW system169 shown in Fig. 4c.
 | ||
| Fig. 4 Nanochitin as reinforcing fillers in various polymer systems. (a) Cryofractured surfaces of natural rubber/ChNW composites at 20 wt% ChNW load. (a1) Prepared by casting-evaporation method. SEM micrographs are obtained under 7 kV, scalebar – 5 μm, magnification not provided. Reproduced with permission.25 (a2) Prepared by hot pressing method. SEM micrographs were obtained under 7 kV, scalebar – 5 μm, magnification not provided. Reproduced with permission.25 (b) PVA/ChNW composites fabricated by different methods. (b1) Fabricated by casting, at 76.92 wt% nanowhiskers load. SEM micrographs are obtained under 5 kV, scalebar – 500 nm, magnification 70000×. Adapted with permission.123 (b2) Fabricated by gel spinning at 5 wt% nanowhiskers load. SEM micrographs are obtained under 25 kV, scalebar – 100 μm, magnification 300×. Reproduced with permission.136 (b3) Fabricated by electrospinning at 5.11 wt% nanowhiskers load. SEM micrographs are obtained under 15 kV, scalebar – 5 μm, magnification 5000×. Reproduced with permission.49 (c) Composites of nylon 6,6/ChNW at 0.4 wt% nanowhiskers load. (c1) Pictures of cast film (top) and dumbbell-shaped film specimens (bottom). Reproduced with permission.169 (c2) Tensile-fractured surface of in situ polymerized film. SEM micrographs are obtained under 15 mA current, scalebar – 5 μm, magnification not provided. Reproduced with permission.169 (c3) SEM image of ex situ blended film. SEM micrographs are obtained under 15 mA current, scalebar – 5 μm, magnification not provided. Reproduced with permission.169 | ||
3. Reinforcing mechanism of chitin in nanocomposites
The strength of polymer composites depends on multiple factors, including the inherent properties of the polymer, the characteristics of the reinforcement agent, the compatibility and interaction between the two, and the dispersion of the reinforcement filler within the polymer matrix. Among these, the dispersion of the reinforcement filler and the interfacial interactions between the reinforcement and the polymer are critical for determining composite strength. Enhancing these interactions often involves functionalizing the surface of nanoparticles with suitable groups. Additionally, the spatial arrangement of fillers and their mutual interactions—both among themselves and with the polymer matrix—are key considerations for a comprehensive understanding of composite material performance.170The reinforcement of nanocomposites with ChNWs could be explained by several interconnected mechanisms such as mechanical reinforcement (load transfer and stress distribution),144 and interfacial interaction and bonding.2,170 As a high-strength material, nanochitin acts as a load-bearing element when incorporated into a polymer matrix. Its large surface area facilitates efficient mechanical load transfer from the matrix to the nanofiller,144 significantly enhancing the tensile strength, stiffness, and modulus of the composites. Additionally, the high aspect ratio and alignment of ChNFs within the matrix contribute to uniform stress distribution improving mechanical performance under tensile, bending, and impact forces.77,97
Interfacial interaction and bonding in the ChNW-reinforced composites are primarily governed by long-range electrostatic interactions and hydrogen bonding.2,25,97,170 Here the amine and acetamide groups on the chitin surface, influenced by its degree of acetylation (%DA), play a crucial role. Below a pH of 6, positively charged ammonium ions promote long-range electrostatic interactions that dominate over short-range hydrogen bonding. At a pH above 6, the hydroxyl, ether, acetyl, and amine groups form an intricate network of hydrogen bonds within the crystal structure and at the interface. The number and distribution of amine groups, affected by processing conditions and source material, impacts the surface charge characteristics. Moreover, the less polar ether and acetyl groups may contribute to material amphiphilicity, leading to varying charge fractions within chitin samples.2
Strong hydrogen bonding between chitin and polymer matrix, combined with the large surface area of ChNWs, facilitates the formation of a rigid network of fillers within the host matrix, often referred to as a “percolation network” (Fig. 5). This network significantly enhances the mechanical properties of the chitin-based polymer composites. Factors influencing the formation of this network include interparticle interactions, particles’ orientation, aspect ratio, concentration, as well as the interfacial phenomena arising from the combination of the high surface area and responsive surface of the chitin.97,170
 | ||
| Fig. 5 Formation of a percolation network. | ||
The presence of amine/amide groups further facilitates modification compared to cellulose, offering a variety of functionalization routes. Amine groups, in particular, offer increased reactivity and are often used for surface modifications such as hydrophobic group attachment to reduce hydrophilicity144 or to improve its compatibility with different surfaces.2 Surface modification method is essential for enhancing the compatibility and interaction at the interface between nanoparticles and the polymer matrix. However, such modification may also reduce hydrogen bonding interaction, which in turn can affect the formation of a rigid percolation network.170
4. Change in properties of chitin-reinforced plastics
4.1. Reinforcement of biopolymers with nanochitin
Biopolymers are macromolecules derived from natural sources such as plants, animals, or microorganisms. These polymers are biodegradable, environmentally friendly, and attractive for various applications. Common natural polymers include polysaccharides (e.g., chitosan, cellulose, alginate, starch, and hyaluronic acid), proteins (e.g., gelatin, collagen, silk), and bacterial polyesters (e.g., polyhydroxyalkanoates). Their biocompatibility, biodegradability, and renewability make them suitable for applications in drug delivery, tissue engineering, stem cell morphogenesis, wound healing, regenerative medicine, food packaging, etc.171 Despite their advantages, natural polymers face limitations such as poor mechanical strength, moisture sensitivity, and processing challenges. For example, cellulose is strong but brittle while starch and gelatin are highly moisture-sensitive, which compromises their stability in humid environments. Additionally, complex processing techniques are often required to improve their mechanical properties or enable large-scale production.172 Reinforcing these polymers with nanochitin has been extensively studied to improve their properties addressing these limitations and expanding their potential applications.Recent studies have extensively investigated the reinforcement of natural polymers with chitin nanomaterials demonstrating significant improvements in their mechanical, thermal, and functional (i.e., water-vapor transmission) properties (summarized in Table 8). These enhancements arise from the unique structural and interactive characteristics of ChNWs and ChNFs, which integrate effectively into polymer matrices, addressing inherent limitations like weak mechanical strength and high moisture sensitivity.
| No. | Polymers | Affected properties | Composition | Value | Ref. |
|---|---|---|---|---|---|
| a The values were estimated from the graphs. b The amounts of tannic acid as the crosslinking agent are given in mg of tannic acid per 1 g of chitosan. c ChNWs suspensions with different degrees of deacetylation (%DA) were formed under different environmental conditions by optimizing three variables in the deacetylation step: concentration of NaOH (% NaOH), temperature of the oil bath, and reaction time. d While coating, the CA film was affixed to a hot plate regulated at a consistent temperature (spray coating temperature, Tspray). e Once the films were formed, they were placed in an oven at a specific temperature (thermal coating temperature, Tthermal) for one hour and then conditioned. f For the optimization of the film properties, the Tspray and Tthermal were tuned. g T n indicates films that were not thermally treated. | |||||
| Chitosan | |||||
| 1 | Chitosan (CS)/cross-linked with isocyanate hexamethylene-1,6-di-(aminocarboxysulfonate) (HDS)/ChNWs | Young's modulus, MPa | CS | 2.5 | 24 |
| ChNWs/CS = 2.97/97.03 | 17.8 | ||||
| ChNWs/CS = 5.77/94.23 | 59.1 | ||||
| ChNWs/CS = 10.90/89.10 | 130 | ||||
| ChNWs/CS = 13.30/86.70 | 169 | ||||
| Stress at break, kPa | CS | 3.2 | |||
| ChNWs/CS = 2.97/97.03 | 17.7 | ||||
| ChNWs/CS = 5.77/94.23 | 42.9 | ||||
| ChNWs/CS = 10.90/89.10 | 117 | ||||
| ChNWs/CS = 13.30/86.70 | 135 | ||||
| Swelling ratio, Q | CS | 357 | |||
| ChNWs/CS = 2.97/97.03 | 211 | ||||
| ChNWs/CS = 5.77/94.23 | 53.3 | ||||
| ChNWs/CS = 10.90/89.10 | 40.7 | ||||
| ChNWs/CS = 13.30/86.70 | 33 | ||||
| 2 | CS/cross-linked with tannic acid (TA)/ChNWs | Tensile strengtha, MPa | CS | 22.02 | 173 |
| TAb/CS = 20/100 | 39.75 | ||||
| TA/CS = 40/100 | 41.88 | ||||
| ChNWs/CS = 10/90 | 42 | ||||
| ChNWs/CS = 20/80 | 49.9 | ||||
| ChNWs/CS = 30/70 | 52.23 | ||||
| ChNWs/TA/CS = 10/20/90 | 37.8 | ||||
| ChNWs/TA/CS = 20/20/80 | 38.5 | ||||
| ChNWs/TA/CS = 30/20/70 | 48.9 | ||||
| ChNWs/TA/CS = 10/40/90 | 34.5 | ||||
| ChNWs/TA/CS = 20/40/80 | 34.8 | ||||
| ChNWs/TA/CS = 30/40/70 | 48.9 | ||||
| Elongation at breaka, % | CS | 50.3 | |||
| TAb/CS = 20/100 | 40 | ||||
| TA/CS = 40/100 | 33 | ||||
| ChNWs/CS = 10/90 | 26.8 | ||||
| ChNWs/CS = 20/80 | 23.5 | ||||
| ChNWs/CS = 30/70 | 21.3 | ||||
| ChNWs/TA/CS = 10/20/90 | 25 | ||||
| ChNWs/TA/CS = 20/20/80 | 23.2 | ||||
| ChNWs/TA/CS = 30/20/70 | 24.8 | ||||
| ChNWs/TA/CS = 10/40/90 | 23 | ||||
| ChNWs/TA/CS = 20/40/80 | 21.5 | ||||
| ChNWs/TA/CS = 30/40/70 | 22.5 | ||||
| Moisture contenta, % | CS | 555 | |||
| TAb/CS = 20/100 | 340 | ||||
| TA/CS = 40/100 | 320 | ||||
| ChNWs/CS = 10/90 | 530 | ||||
| ChNWs/CS = 20/80 | 415 | ||||
| ChNWs/CS = 30/70 | 400 | ||||
| ChNWs/TA/CS = 10/20/90 | 330 | ||||
| ChNWs/TA/CS = 20/20/80 | 275 | ||||
| ChNWs/TA/CS = 30/20/70 | 273 | ||||
| ChNWs/TA/CS = 10/40/90 | 275 | ||||
| ChNWs/TA/CS = 20/40/80 | 270 | ||||
| ChNWs/TA/CS = 30/40/70 | 260 | ||||
| Water solubilitya, % | CS | 21.8 | |||
| TAb/CS = 20/100 | 15.5 | ||||
| TA/CS = 40/100 | 13.2 | ||||
| ChNWs/CS = 10/90 | 18.8 | ||||
| ChNWs/CS = 20/80 | 16.7 | ||||
| ChNWs/CS = 30/70 | 13.5 | ||||
| ChNWs/TA/CS = 10/20/90 | 14.2 | ||||
| ChNWs/TA/CS = 20/20/80 | 13.4 | ||||
| ChNWs/TA/CS = 30/20/70 | 12.6 | ||||
| ChNWs/TA/CS = 10/40/90 | 12.2 | ||||
| ChNWs/TA/CS = 20/40/80 | 9.2 | ||||
| ChNWs/TA/CS = 30/40/70 | 8.4 | ||||
| Carboxymethyl chitosan (CMCS) | |||||
| 3 | Carboxymethyl chitosan (CMCS) cross-linked with dextran dialdehyde (DDA)/ChNWs | Young's modulus, kPa | CMCS/DDA | 43.2 ± 4.6 | 106 |
| ChNWs/CMCS = 0.5/99.5 | 47.2 ± 5.6 | ||||
| ChNWs/CMCS = 1.0/99.0 | 67.0 ± 6.2 | ||||
| Swelling ratio at 36 h, % | CMCS/DDA | 52.62 ± 8.59 | |||
| ChNWs/CMCS = 0.5/99.5 | 61.62 ± 6.10 | ||||
| ChNWs/CMCS = 1.0/99.0 | 64.20 ± 4.93 | ||||
| Bonding strength, kPa | CMCS/DDA | 21.93 | |||
| ChNWs/CMCS = 0.5/99.5 | 25.2 | ||||
| ChNWs/CMCS = 1.0/99.0 | 33.21 | ||||
| Alginate | |||||
| 4 | Sodium alginate (ALG)/α-ChNWs | Tensile strength, N mm−2 | ALG | 0.0256 ± 0.0031 | 75 |
| ChNWs/ALG = 33.3/66.7 | 0.0618 ± 0.0054 | ||||
| Elongation, % | ALG | 51.52 ± 2.61 | |||
| ChNWs/ALG = 33.3/66.7 | 37.67 ± 2.31 | ||||
| 5 | ALG/partially deacetylated α-chitin nanowhiskers (ChNWs, DDA 0.40 ± 0.03) | Yield stress, Pa | ALG | 1120 | 132 |
| ChNWs/ALG = 2.5/97.5 | 1560 | ||||
| ChNWs/ALG = 7.5/92.5 | 18000–44000 |
||||
| ChNWs/ALG = 14.5/85.5 | 17000 |
||||
| 6 | Pectin-sodium alginate (pectin-ALG)/surface functionalized chitin nanocrystals (i.e., acid-hydrolyzed ChNCs (AH-ChNCs, DDA = 10%), and carboxylated ChNCs (OX-ChNCs, DDA = 12%)) | Tensile strength, MPa | Pectin-ALG | 25.9 ± 1.4 | 175 |
| AH-ChNCs/pectin-ALG = 20/80 | 33.5 ± 1.7 | ||||
| OX-ChNCs/pectin-ALG = 20/80 | 23 ± 1 | ||||
| Elongation at break, mm | Pectin-ALG | 2.1 ± 0.7 | |||
| AH-ChNCs/pectin-ALG = 20/80 | 2.6 ± 0.5 | ||||
| OX-ChNCs/pectin-ALG = 20/80 | 1.4 ± 0.3 | ||||
| Young's modulus, MPa | Pectin-ALG | 1132 ± 49 | |||
| AH-ChNCs/pectin-ALG = 20/80 | 1810 ± 341 | ||||
| OX-ChNCs/pectin-ALG = 20/80 | 1458 ± 90 | ||||
| 7 | ALG/ChNWs | Compressive modulus, MPa | ALG | 5.08 | 176 |
| ChNWs/ALG = 0.5/99.5 | 9.56 | ||||
| ChNWs/ALG = 1/99 | 12.12 | ||||
| ChNWs/ALG = 2/98 | 13.72 | ||||
| ChNWs/ALG = 2.5/97.5 | 13.81 | ||||
| Compressive strength, MPa | ALG | 1.39 | |||
| ChNWs/ALG = 0.5/99.5 | 2.01 | ||||
| ChNWs/ALG = 1/99 | 2.52 | ||||
| ChNWs/ALG = 2/98 | 3.18 | ||||
| ChNWs/ALG = 2.5/97.5 | 1.97 | ||||
| Strain, % | ALG | 76.05 | |||
| ChNWs/ALG = 0.5/99.5 | 78.57 | ||||
| ChNWs/ALG = 1/99 | 78.78 | ||||
| ChNWs/ALG = 2/98 | 78.15 | ||||
| ChNWs/ALG = 2.5/97.5 | 74.8 | ||||
| Swelling ratio, g g−1 | ALG | 34.6 | |||
| ChNWs/ALG = 0.5/99.5 | 24.6 | ||||
| ChNWs/ALG = 1/99 | 20.7 | ||||
| ChNWs/ALG = 2/98 | 17 | ||||
| ChNWs/ALG = 2.5/97.5 | 14 | ||||
| Eucalyptus pulp | |||||
| 8 | Eucalyptus pulp/partially deacetylated chitin nanowhiskers (DeChNWs)/TEMPO-oxidized chitin nanowhiskers (TOChNWs) | Tensile index, N m g−1 | Dry pulp (paper) | 18.4 | 121 |
| DeChNWs/dry pulp = 0.2/99.8 | 19 | ||||
| DeChNWs/dry pulp = 0.4/99.6 | 21 | ||||
| DeChNWs/dry pulp = 0.6/99.4 | 22.5 | ||||
| DeChNWs/dry pulp = 0.8/99.2 | 24.2 | ||||
| DeChNWs/dry pulp = 1.0/99.0 | 23 | ||||
| DeChNWs/dry pulp = 2.0/98.0 | 22.5 | ||||
| DeChNWs/dry pulp = 5.0/95.0 | 22 | ||||
| Tensile index, N m g−1 | Dry pulp (paper) | 17.5 | |||
| TOChNWs/dry pulp = 0.2/99.8 | 18 | ||||
| TOChNWs/dry pulp = 0.4/99.6 | 19.5 | ||||
| TOChNWs/dry pulp = 0.6/99.4 | 19.52 | ||||
| TOChNWs/dry pulp = 0.8/99.2 | 20 | ||||
| TOChNWs/dry pulp = 1.0/99.0 | 20.5 | ||||
| TOChNWs/dry pulp = 2.0/98.0 | 21 | ||||
| TOChNWs/dry pulp = 5.0/95.0 | 18 | ||||
| Tear index, mN m2 g−1 | Dry pulp (paper) | 2.4 | |||
| DeChNWs/dry pulp = 0.2/99.8 | 2.6 | ||||
| DeChNWs/dry pulp = 0.4/99.6 | 3 | ||||
| DeChNWs/dry pulp = 0.6/99.4 | 3.1 | ||||
| DeChNWs/dry pulp = 0.8/99.2 | 3.5 | ||||
| DeChNWs/dry pulp = 1.0/99.0 | 3.6 | ||||
| DeChNWs/dry pulp = 2.0/98.0 | 3.7 | ||||
| DeChNWs/dry pulp = 5.0/95.0 | 3.4 | ||||
| Tear index, mN m2 g−1 | Dry pulp (paper) | 2 | |||
| TOChNWs/dry pulp = 0.2/99.8 | 2.2 | ||||
| TOChNWs/dry pulp = 0.4/99.6 | 2.2 | ||||
| TOChNWs/dry pulp = 0.6/99.4 | 2.4 | ||||
| TOChNWs/dry pulp = 0.8/99.2 | 2.4 | ||||
| TOChNWs/dry pulp = 1.0/99.0 | 2.3 | ||||
| TOChNWs/dry pulp = 2.0/98.0 | 2.1 | ||||
| TOChNWs/dry pulp = 5.0/95.0 | 2.4 | ||||
| Microcrystalline cellulose (MCC) | |||||
| 9 | Microcrystalline cellulose (MCC, Mw 1 × 105)/α-ChNWs | Compressive modulus, kPa (w.r.t freezing days) | MCC (3 days) | 1231 ± 132 | 79 |
| ChNWs/MCC = 1.0/99.0 (3 days) | 1238 | ||||
| ChNWs/MCC = 2.5/97.5 (3 days) | 1570 | ||||
| ChNWs/MCC = 5.0/95.0 (3 days) | 273 | ||||
| ChNWs/MCC = 10.0/90.0 (3 days) | 383 ± 150 | ||||
| ChNWs/MCC = 1.0/99.0 (10 days) | 3120 ± 120 | ||||
| ChNWs/MCC = 2.5/97.5 (10 days) | 3050 ± 160 | ||||
| Cellulose acetate | |||||
| 10 | Cellulose acetate (CA)/ChNCs | Young's modulusa, MPa | CA | 183 | 178 |
| ChNCs/CA = 0.5/99.5 | 219 | ||||
| ChNCs/CA = 1.0/99.0 | 220 | ||||
| ChNCs/CA = 2.0/98.0 | 255 | ||||
| Tensile strengtha, MPa | CA | 2.95 | |||
| ChNCs/CA = 0.5/99.5 | 2.41 | ||||
| ChNCs/CA = 1.0/99.0 | 2.5 | ||||
| ChNCs/CA = 2.0/98.0 | 2.99 | ||||
| Elongation at breaka, % | CA | 1.95 | |||
| ChNCs/CA = 0.5/99.5 | 1.53 | ||||
| ChNCs/CA = 1.0/99.0 | 1.63 | ||||
| ChNCs/CA = 2.0/98.0 | 1.78 | ||||
| Contact angle measurementsa, O | CA | 48 | |||
| ChNCs/CA = 0.5/99.5 | 45.4 | ||||
| ChNCs/CA = 1.0/99.0 | 43.9 | ||||
| ChNCs/CA = 2.0/98.0 | 44.3 | ||||
| Anti-fungal activities, expressed as fungal growth inhibition (FGI)a, % | CA | 7.5 | |||
| ChNCs/CA = 0.5/99.5 | 33.5 | ||||
| ChNCs/CA = 1.0/99.0 | 15 | ||||
| ChNCs/CA = 2.0/98.0 | 35.2 | ||||
| Optical transmitance (at 400 nm)a, % | CA | 20.6 | |||
| ChNCs/CA = 0.5/99.5 | 16.1 | ||||
| ChNCs/CA = 1.0/99.0 | 13.9 | ||||
| ChNCs/CA = 2.0/98.0 | 12.8 | ||||
| 11 | Cellulose acetate (CA)/α-ChNWs/cellulose nanocrystals (CNCs) | Ultimate tensile strength, MPa | ChNWs/CNCs = 50/50 coated in sequence on CA film | 140 | |
| CA | 36.0 ± 0.9 | ||||
| ChNWs suspensionc (%DA = 88.8 ± 1.4) | 33.3 ± 1.2 | ||||
| ChNWs suspension (%DA = 88.8 ± 3.1) | 31.9 ± 0.9 | ||||
| ChNWs suspension (%DA = 93.1 ± 0.8) | 32.3 ± 1.4 | ||||
| ChNWs suspension (%DA = 93.7 ± 1.9) | 33.6 ± 1.1 | ||||
| ChNWs suspension (%DA = 85.7 ± 1.5) | 31.1 ± 0.9 | ||||
| ChNWs suspension (%DA = 93.4 ± 1.0) | 30.6 ± 1.8 | ||||
| ChNWs suspension (%DA = 86.5 ± 1.1) | 30.9 ± 1.7 | ||||
| ChNWs suspension (%DA = 89.0 ± 0.8) | 31.9 ± 1.0 | ||||
| ChNWs suspension (%DA = 84.1 ± 1.7) | 33.5 ± 1.0 | ||||
| ChNWs suspension (%DA = 75.1 ± 1.5) | 33.3 ± 1.2 | ||||
| ChNWs suspension (%DA = 6.8 ± 2.8) | 32.5 ± 0.9 | ||||
| Failure strain, % | CA | 22.7 ± 2.7 | |||
| ChNWs suspensionc (%DA = 88.8 ± 1.4) | 21.6 ± 1.1 | ||||
| ChNWs suspension (%DA = 88.8 ± 3.1) | 19.0 ± 2.7 | ||||
| ChNWs suspension (%DA = 93.1 ± 0.8) | 20.5 ± 1.4 | ||||
| ChNWs suspension (%DA = 93.7 ± 1.9) | 23.6 ± 1.8 | ||||
| ChNWs suspension (%DA = 85.7 ± 1.5) | 22.7 ± 1.4 | ||||
| ChNWs suspension (%DA = 93.4 ± 1.0) | 22.1 ± 3.4 | ||||
| ChNWs suspension (%DA = 86.5 ± 1.1) | 23.4 ± 3.8 | ||||
| ChNWs suspension (%DA = 89.0 ± 0.8) | 21.6 ± 1.0 | ||||
| ChNWs suspension (%DA = 84.1 ± 1.7) | 23.5 ± 2.6 | ||||
| ChNWs suspension (%DA = 75.1 ± 1.5) | 22.1 ± 1.5 | ||||
| ChNWs suspension (%DA = 6.8 ± 2.8) | 20.9 ± 1.5 | ||||
| Water vapor transmission rate, g mm m−2 day−1 kPa−1 | CA | 12.5 | |||
| ChNWs suspensionc (%DA = 88.8 ± 1.4) | 10.2 | ||||
| ChNWs suspension (%DA = 88.8 ± 3.1) | 10.5 | ||||
| ChNWs suspension (%DA = 93.1 ± 0.8) | 10 | ||||
| ChNWs suspension (%DA = 93.7 ± 1.9) | 10 | ||||
| ChNWs suspension (%DA = 85.7 ± 1.5) | 10.1 | ||||
| ChNWs suspension (%DA = 93.4 ± 1.0) | 10 | ||||
| ChNWs suspension (%DA = 86.5 ± 1.1) | 10.1 | ||||
| ChNWs suspension (%DA = 89.0 ± 0.8) | 9 | ||||
| ChNWs suspension (%DA = 84.1 ± 1.7) | 8 | ||||
| ChNWs suspension (%DA = 75.1 ± 1.5) | 9.8 | ||||
| ChNWs suspension (%DA = 6.8 ± 2.8) | 9 | ||||
| Oxygen permeability, cm3 μm m−2 day−1 kPa−1 | CA | 1553 | |||
| ChNWs suspensionc (%DA = 88.8 ± 1.4) | 132 | ||||
| ChNWs suspension (%DA = 88.8 ± 3.1) | 90 | ||||
| ChNWs suspension (%DA = 93.1 ± 0.8) | 41 | ||||
| ChNWs suspension (%DA = 93.7 ± 1.9) | 70 | ||||
| ChNWs suspension (%DA = 85.7 ± 1.5) | 50 | ||||
| ChNWs suspension (%DA = 93.4 ± 1.0) | 45 | ||||
| ChNWs suspension (%DA = 86.5 ± 1.1) | 75 | ||||
| ChNWs suspension (%DA = 89.0 ± 0.8) | 50 | ||||
| ChNWs suspension (%DA = 84.1 ± 1.7) | 45 | ||||
| ChNWs suspension (%DA = 75.1 ± 1.5) | 30 | ||||
| ChNWs suspension (%DA = 76.8 ± 2.8) | 16.7 | ||||
| 12 | Cellulose acetate (CA)/ChNWs/cellulose nanocrystals (CNCs) | Oxygen permeability, μm m−2 day−1 kPa−1 (with different Tsprayd at a controlled Tthermale of 40 °C) |
ChNWs/CNC = 50/50 coated in sequence on CA film | 141 | |
| CA | 535 | ||||
| 40 °Cf |
44.4 | ||||
| 60 °C | 27.9 | ||||
| 80 °C | 28.6 | ||||
| 100 °C | 23.3 | ||||
| Oxygen permeability, μm m−2 day−1 kPa−1 (with different Tthermal at a controlled Tspray of 80 °C) | CA | 535 | |||
| 40 °C | 28.6 | ||||
| 60 °C | 22.1 | ||||
| 80 °C | 24.1 | ||||
| 100 °C | 20.1 | ||||
| 120 °C | 15.9 | ||||
| 140 °C | 11.5 | ||||
|
T
ng |
20.2 | ||||
| Water vapor transmission rate, g mm m−2 day−1 (with different Tspray at a controlled Tthermal of 40 °C) | CA | 8.3 | |||
| 40 °C | 7.5 | ||||
| 60 °C | 7.7 | ||||
| 80 °C | 7.7 | ||||
| 100 °C | 7.8 | ||||
| Water vapor transmission rate, g mm m−2 day−1 (with different Tthermal at a controlled Tspray of 80 °C) | CA | 8.3 | |||
| 40 °C | 7.7 | ||||
| 60 °C | 7.6 | ||||
| 80 °C | 7.4 | ||||
| 100 °C | 7.4 | ||||
| 120 °C | 7.3 | ||||
| 140 °C | 6.6 | ||||
| T n | 6.8 | ||||
| Breaking force, N | CA | 38.5 ± 0.8 | |||
|
T
spray40 Tthermal40f |
47.8 ± 2.0 | ||||
| T spray60 Tthermal40 | 45.6 ± 2.5 | ||||
| T spray80 Tthermal40 | 47.6 ± 3.4 | ||||
| T spray100 Tthermal40 | 46.1 ± 4.3 | ||||
| T spray80 Tn | 47.7 ± 2.3 | ||||
| T spray80 Tthermal60 | 41.6 ± 2.3 | ||||
| T spray80 Tthermal80 | 44.0 ± 1.3 | ||||
| T spray80 Tthermal100 | 45.2 ± 1.3 | ||||
| T spray80 Tthermal120 | 47.9 ± 1.6 | ||||
| T spray80 Tthermal140 | 45.9 ± 2.1 | ||||
| Modulus, GPa | CA | 2.39 ± 0.25 | |||
| T spray40 Tthermal40 | 2.44 ± 0.26 | ||||
| T spray60 Tthermal40 | 2.58 ± 0.13 | ||||
| T spray80 Tthermal40 | 2.62 ± 0.22 | ||||
| T spray100 Tthermal40 | 2.45 ± 0.12 | ||||
| T spray80 Tn | 2.87 ± 0.10 | ||||
| T spray80 Tthermal60 | 2.81 ± 0.05 | ||||
| T spray80 Tthermal80 | 2.82 ± 0.09 | ||||
| T spray80 Tthermal100 | 2.78 ± 0.07 | ||||
| T spray80 Tthermal120 | 2.71 ± 0.14 | ||||
| T spray80 Tthermal140 | 2.73 ± 0.22 | ||||
| Yield stress, MPa | CA | 42.9 ± 5.9 | |||
| T spray40 Tthermal40 | 49.2 ± 3.9 | ||||
| T spray60 Tthermal40 | 47.8 ± 1.7 | ||||
| T spray80 Tthermal40 | 49.9 ± 4.3 | ||||
| T spray100 Tthermal40 | 50.5 ± 1.5 | ||||
| T spray80 Tn | 57.2 ± 5.0 | ||||
| T spray80 Tthermal60 | 53.3 ± 1.4 | ||||
| T spray80 Tthermal80 | 53.8 ± 2.0 | ||||
| T spray80 Tthermal100 | 53.6 ± 2.4 | ||||
| T spray80 Tthermal120 | 53.2 ± 2.2 | ||||
| T spray80 Tthermal140 | 52.3 ± 7.4 | ||||
| Strain at break, % | CA | 3.3 ± 1.0 | |||
| T spray40 Tthermal40 | 8.9 ± 6.1 | ||||
| T spray60 Tthermal40 | 8.7 ± 6.1 | ||||
| T spray80 Tthermal40 | 5.5 ± 3.3 | ||||
| T spray100 Tthermal40 | 14.5 ± 5.3 | ||||
| T spray80 Tn | 3.4 ± 0.9 | ||||
| T spray80 Tthermal60 | 3.5 ± 0.4 | ||||
| T spray80 Tthermal80 | 10.4 ± 6.4 | ||||
| T spray80 Tthermal100 | 6.6 ± 3.0 | ||||
| T spray80 Tthermal120 | 3.9 ± 0.4 | ||||
| T spray80 Tthermal140 | 11.3 ± 7.9 | ||||
| Transmittance, % (with different Tspray at a controlled Tthermal of 40 °C) | CA | 90 | |||
| 40 °C | 75 | ||||
| 60 °C | 78 | ||||
| 80 °C | 80 | ||||
| 100 °C | 81 | ||||
| Transmittance, % (with different Tthermal at a controlled Tspray of 80 °C) | CA | 90 | |||
| 40 °C | 81 | ||||
| 60 °C | 85 | ||||
| 80 °C | 86 | ||||
| 100 °C | 84 | ||||
| 120 °C | 82 | ||||
| 140 °C | 80 | ||||
| Haze, % (with different Tspray at a controlled Tthermal of 40 °C) | CA | 0.5 | |||
| 40 °C | 13 | ||||
| 60 °C | 8 | ||||
| 80 °C | 5 | ||||
| 100 °C | 2 | ||||
| Haze, % (with different Tthermal at a controlled Tspray of 80 °C) | CA | 0.5 | |||
| 40 °C | 8 | ||||
| 60 °C | 2 | ||||
| 80 °C | 4 | ||||
| 100 °C | 1 | ||||
| 120 °C | 0.9 | ||||
| 140 °C | 4 | ||||
| Potato Starch | |||||
| 13 | Potato starch powder/chitin nanoparticles (ChNPs) | Tensile strength, MPa | Potato starch | 2.84 | 48 |
| ChNPs/potato starch = 1/99 | 3.5 | ||||
| ChNPs/potato starch = 2/98 | 5.2 | ||||
| ChNPs/potato starch = 3/97 | 6.25 | ||||
| ChNPs/potato starch = 4/96 | 7.25 | ||||
| ChNPs/potato starch = 5/95 | 7.79 | ||||
| Elongation at break, % | Potato starch | 59.3 | |||
| ChNPs/potato starch = 1/99 | 45 | ||||
| ChNPs/potato starch = 2/98 | 34.25 | ||||
| ChNPs/potato starch = 3/97 | 26 | ||||
| ChNPs/potato starch = 4/96 | 22 | ||||
| ChNPs/potato starch = 5/95 | 19.3 | ||||
| Water vapor permeability, g m−1 s−1 Pa−1 | Potato starch | 5.62 × 10−10 | |||
| ChNPs/potato starch = 1/99 | 4.45 × 10−10 | ||||
| ChNPs/potato starch = 2/98 | 4.15 × 10−10 | ||||
| ChNPs/potato starch = 3/97 | 3.45 × 10−10 | ||||
| ChNPs/potato starch = 4/96 | 3.42 × 10−10 | ||||
| ChNPs/potato starch = 5/95 | 3.41 × 10−10 | ||||
| Corn starch | |||||
| 14 | Corn starch granules/α-ChNFs/α-chitin nanocrystals (ChNCs) | Young's modulus, MPa | Corn starch | 80 | 20 |
| ChNCs/corn starch = 5/95 | 222 | ||||
| ChNCs/corn starch = 10/90 | 280 | ||||
| ChNCs/corn starch = 15/85 | 367 | ||||
| ChNCs/corn starch = 20/80 | 390 | ||||
| ChNFs/corn starch = 5/95 | 330 | ||||
| ChNFs/corn starch = 10/90 | 390 | ||||
| ChNFs/corn starch = 15/85 | 410 | ||||
| ChNFs/corn starch = 20/80 | 520 | ||||
| Tensile strength, MPa | Corn starch | 4.5 | |||
| ChNCs/corn starch = 5/95 | 7.1 | ||||
| ChNCs/corn starch = 10/90 | 7.5 | ||||
| ChNCs/corn starch = 15/85 | 9 | ||||
| ChNCs/corn starch = 20/80 | 10.8 | ||||
| ChNFs/corn starch = 5/95 | 9.9 | ||||
| ChNFs/corn starch = 10/90 | 12 | ||||
| ChNFs/corn starch = 15/85 | 12 | ||||
| ChNFs/corn starch = 20/80 | 15 | ||||
| Elongation at break, % | Corn starch | 87 | |||
| ChNCs/corn starch = 5/95 | 38 | ||||
| ChNCs/corn starch = 10/90 | 26.5 | ||||
| ChNCs/corn starch = 15/85 | 17 | ||||
| ChNCs/corn starch = 20/80 | 15 | ||||
| ChNFs/corn starch = 5/95 | 26 | ||||
| ChNFs/corn starch = 10/90 | 19 | ||||
| ChNFs/corn starch = 15/85 | 11 | ||||
| ChNFs/corn starch = 20/80 | 9 | ||||
| Gelatin | |||||
| 15 | Gel/ChNWs | Young's modulus, MPa | Gelatin | 2455 ± 74 | 78 |
| ChNWs/gel = 1/99 | 2435 ± 50 | ||||
| ChNWs/gel = 2/98 | 2496 ± 56 | ||||
| ChNWs/gel = 4/96 | 2921 ± 76 | ||||
| Ultimate tensile strength, MPa | Gelatin | 71.0 ± 1.8 | |||
| ChNWs/gel = 1/99 | 69.5 ± 2.0 | ||||
| ChNWs/gel = 2/98 | 68.9 ± 3.3 | ||||
| ChNWs/gel = 4/96 | 77.4 ± 3.1 | ||||
| Elongation at break, % | Gelatin | 7.7 ± 0.5 | |||
| ChNWs/gel = 1/99 | 7.8 ± 0.6 | ||||
| ChNWs/gel = 2/98 | 9.1 ± 1.2 | ||||
| ChNWs/gel = 4/96 | 6.8 ± 1.1 | ||||
| Transparency | Gelatin | 1.39 ± 0.07 | |||
| ChNWs/gel = 1/99 | 1.77 ± 0.14 | ||||
| ChNWs/gel = 2/98 | 1.90 ± 0.23 | ||||
| ChNWs/gel = 4/96 | 2.03 ± 0.08 | ||||
| Absorbance (289 nm) | Gelatin | 0.42 ± 0.01 | |||
| ChNWs/gel = 1/99 | 0.45 ± 0.03 | ||||
| ChNWs/gel = 2/98 | 0.44 ± 0.03 | ||||
| ChNWs/gel = 4/96 | 0.41 ± 0.03 | ||||
| Solubility | Gelatin | 13.1 ± 1.2 | |||
| ChNWs/gel = 1/99 | 12.0 ± 0.3 | ||||
| ChNWs/gel = 2/98 | 11.0 ± 0.3 | ||||
| ChNWs/gel = 4/96 | 10.2 ± 0.2 | ||||
| 16 | Gel/ChNFs | Stress, MPa | Gelatin | 87.2 ± 3.2 | 143 |
| ChNFs | 65.8 ± 5.3 | ||||
| ChNFs/gel = 73.0/27.0 | 74.0 ± 3.1 | ||||
| ChNFs/gel = 62.5/37.5 | 84.7 ± 4.5 | ||||
| ChNFs/gel = 50.2/49.8 | 101.0 ± 5.3 | ||||
| Strain, % | Gelatin | 8.0 ± 0.4 | |||
| ChNFs | 1.4 ± 0.1 | ||||
| ChNFs/gel = 73.0/27.0 | 1.4 ± 0.2 | ||||
| ChNFs/gel = 62.5/37.5 | 1.8 ± 0.1 | ||||
| ChNFs/gel = 50.2/49.8 | 3.1 ± 0.2 | ||||
| Young's modulus, MPa | Gelatin | 2576 ± 264 | |||
| ChNFs | 6705 ± 342 | ||||
| ChNFs/gel = 73.0/27.0 | 7039 ± 277 | ||||
| ChNFs/gel = 62.5/37.5 | 6372 ± 313 | ||||
| ChNFs/gel = 50.2/49.8 | 5192 ± 194 | ||||
| Transmittance, % | Gelatin | 90.6 | |||
| ChNFs | 65 | ||||
| ChNFs/gel = 73.0/27.0 | 86.9 | ||||
| ChNFs/gel = 62.5/37.5 | 87.6 | ||||
| ChNFs/gel = 50.2/49.8 | 88.7 | ||||
| 17 | Gel/surface-deacetylated ChNFs (S-ChNFs, DA = 63 ± 2%) | Stress, MPa | Gelatin | 89.2 ± 2.83 | 181 |
| S-ChNFs | 100.4 ± 5.26 | ||||
| S-ChNFs/gel = 45.5/54.5 | 126.3 ± 3.07 | ||||
| S-ChNFs/gel = 62.5/37.5 | 119.4 ± 3.54 | ||||
| S-ChNFs/gel = 22.1/77.9 | 103.2 ± 4.72 | ||||
| Strain, % | Gelatin | 8.07 ± 0.33 | |||
| S-ChNFs | 2.87 ± 0.19 | ||||
| S-ChNFs/gel = 45.5/54.5 | 3.67 ± 0.23 | ||||
| S-ChNFs/gel = 62.5/37.5 | 6.96 ± 0.27 | ||||
| S-ChNFs/gel = 22.1/77.9 | 7.58 ± 0.34 | ||||
| Young's modulus, MPa | Gelatin | 2598 ± 214 | |||
| S-ChNFs | 7075 ± 297 | ||||
| S-ChNFs/gel = 45.5/54.5 | 6356 ± 261 | ||||
| S-ChNFs/gel = 62.5/37.5 | 5569 ± 413 | ||||
| S-ChNFs/gel = 22.1/77.9 | 4627 ± 306 | ||||
| 18 | Gel/dialdehyde microfibrillated cellulose (DAMFC)/ChNWs | Compressive stress, MPa | Gelatin | 0.522 | 120 |
| DAMFC/gel = 15.0/85.0 | 1.949 | ||||
| DAMFC/gel/ChNWs = 14.90/84.30/0.80 | 2.419 | ||||
| DAMFC/gel/ChNWs = 14.74/83.55/1.71 | 2.753 | ||||
| DAMFC/gel/ChNWs = 14.62/82.82/2.56 | 2.729 | ||||
| DAMFC/gel/ChNWs = 14.36/81.36/4.28 | 1.944 | ||||
| DAMFC/gel/ChNWs = 13.71/77.66/8.63 | 1.504 | ||||
| Silk | |||||
| 19 | Silk nanowhiskers (SNWs)/partially deacetylated α-chitin nanofibers (DeChNFs) | Tensile strength, MPa | DeChNFs | 102.00 ± 1.09 | 131 |
| DeChNFs/SNWs = 98/2 | 184.05 ± 13.86 | ||||
| DeChNFs/SNWs = 95/5 | 171.25 ± 16.16 | ||||
| DeChNFs/SNWs = 90/10 | 156.97 ± 9.09 | ||||
| DeChNFs/SNWs = 50/50 | 66.38 ± 8.77 | ||||
| Elongation at break, % | DeChNFs | 9.06 ± 2.80 | |||
| DeChNFs/SNWs = 98/2 | 6.35 ± 0.76 | ||||
| DeChNFs/SNWs = 95/5 | 6.00 ± 0.84 | ||||
| DeChNFs/SNWs = 90/10 | 4.93 ± 0.59 | ||||
| DeChNFs/SNWs = 50/50 | 4.74 ± 2.06 | ||||
| Young's modulus, MPa | DeChNFs | 3802.912 ± 563.04 | |||
| DeChNFs/SNWs = 98/2 | 7987.962 ± 403.88 | ||||
| DeChNFs/SNWs = 95/5 | 6448.212 ± 1121.18 | ||||
| DeChNFs/SNWs = 90/10 | 6422.663 ± 1023.48 | ||||
| DeChNFs/SNWs = 50/50 | 2381.890 ± 450.36 | ||||
| Soy protein isolate (SPI) | |||||
| 20 | SPI/ChNWs/Clitoria ternatea flower extract (CTE) | Tensile strength, MPa | SPI | 4.67 ± 0.79 | 74 |
| SPI/CTE = 61.54/38.46 | 3.61 ± 0.92 | ||||
| ChNWs/SPI = 61.54/38.46 | 15.45 ± 0.95 | ||||
| ChNWs/SPI/CTE = 38.10/23.81/38.10 | 13.25 ± 0.47 | ||||
| Elongation at break, % | SPI | 59.02 ± 1.12 | |||
| SPI/CTE = 61.54/38.46 | 90.1 ± 5.52 | ||||
| ChNWs/SPI = 61.54/38.46 | 7.05 ± 1.75 | ||||
| ChNWs/SPI/CTE = 38.10/23.81/38.10 | 38.46 ± 3.2 | ||||
| 21 | SPI/MgO/ChNWs | Antibacterial activity against Gram-positive Bacillus subtilis (inhibition zones, mm) | ChNWs/SPI = 6/94 | 5.2 ± 0.25 | 73 |
| SPI/MgO = 95/5 | 6 ± 0.18 | ||||
| ChNWs/SPI/MgO = 6/93/1 | 5 ± 0.28 | ||||
| ChNWs/SPI/MgO = 6/89/5 | 6.5 ± 0.25 | ||||
| Thermal stability (residual mass, %) | SPI | 12.23 | |||
| ChNWs/SPI/MgO = 6/93/1 | 18.62 | ||||
| ChNWs/SPI/MgO = 6/89/5 | 17.95 | ||||
| 22 | Acid-induced SPI gel/ChNCs | Storage modulus (G′), Pa | SPI gel | 75.6 ± 3.2 | 183 |
| ChNCs/SPI = 0.25/99.75 | 265.7 ± 9.1 | ||||
| ChNCs/SPI = 0.50/99.50 | 573.2 ± 9.5 | ||||
| ChNCs/SPI = 0.75/99.25 | 897.3 ± 76.3 1024.3 ± 81.6 | ||||
| ChNCs/SPI = 1.00/99.00 | |||||
| Recovery rate in creep-recovery test, % | SPI gel | 80.27 ± 1.11 | |||
| ChNCs/SPI = 0.25/99.75 | 83.53 ± 1.69 | ||||
| ChNCs/SPI = 0.50/99.50 | 84.47 ± 0.81 | ||||
| ChNCs/SPI = 0.75/99.25 | 84.73 ± 1.21 | ||||
| ChNCs/SPI = 1.00/99.00 | 85.47 ± 1.16 | ||||
| Thermal transition temperature, °C | SPI gel | 121.43 | |||
| ChNCs/SPI = 0.25/99.75 | 128.35 | ||||
| ChNCs/SPI = 0.50/99.50 | 129.87 | ||||
| ChNCs/SPI = 0.75/99.25 | 131.72 | ||||
| ChNCs/SPI = 1.00/99.00 | 132.43 | ||||
| Hyaluronic acid (HA) | |||||
| 23 | HA/partially deacetylated chitin nanowhiskers (ChNWs, DA 73%) | Crossover modulus, Pa | HA | 82 | 88 |
| ChNWs/HA = 90.91/9.09 | 82 | ||||
| ChNWs/HA = 97.22/2.78 | 120 | ||||
| ChNWs/HA = 98.04/1.96 | 83 | ||||
| Tensile modulus, GPa | HA | 2.58 | |||
| ChNWs/HA = 50.00/50.00 | 2.6 | ||||
| ChNWs/HA = 90.91/9.09 | 3.2 | ||||
| ChNWs/HA = 93.75/6.25 | 3.5 | ||||
| ChNWs/HA = 97.22/2.78 | 3.95 | ||||
| ChNWs/HA = 98.04/1.96 | 4.54 | ||||
| Yield stress, MPa | HA | 69.64 | |||
| ChNWs/HA = 50.00/50.00 | 75 | ||||
| ChNWs/HA = 90.91/9.09 | 77 | ||||
| ChNWs/HA = 93.75/6.25 | 79.00 | ||||
| ChNWs/HA = 97.22/2.78 | 84 | ||||
| ChNWs/HA = 98.04/1.96 | 80.54 | ||||
Another study demonstrated that the addition of up to 1.0 wt% ChNWs into carboxymethyl chitosan (CMCS) crosslinked with dextran dialdehyde (DDA) (Table 8, entry 3) increased compressive stress (up to 1.87 times) and Young's modulus (up to 1.56 times) while preserving the swelling ratio.106 The improvements are likely attributed to ChNWs ability to strengthen interactions among the polymer coils across different layers, significantly restricting segment movement under applied stretching force.174 These mechanisms also help explain the enhanced mechanical performance observed in ChNWs-incorporated CMCS/DDA hydrogels.106
Similar findings were reported in another study, where alginate hydrogels reinforced with ChNWs showed improved tensile properties and reduced elongation at break (Table 8, entry 5).132 Additionally, the incorporation of ChNWs into the hydrogels increased yield stress, enhanced viscosity, modulated rheological properties, and prolonged drug release.132 A recent study by Santos et al.175 highlighted surface functionalization of ChNCs can improve pectin-alginate films. Two types of surface-functionalized ChNCs – acid hydrolyzed ChNCs (AH-ChNCs) and carboxylated ChNCs (OX-ChNCs) – were studied to reinforce an edible pectin alginate film. The results showed that despite AH-ChNCs having lower rheological characteristics than OX-ChNCs, the films reinforced with AH-ChNCs exhibited higher tensile properties (Table 8, entry 6). This underscores the crucial role of amines in effectively strengthening the films.175
Huang et al.176 successfully fabricated alginate-based nanocomposite hydrogels reinforced with chitin whiskers utilizing the pH-induced charge shifting behavior of ChNWs. The addition of chitin whiskers to the hydrogels not only greatly improved the mechanical properties, but also inhibited swelling (Table 8, entry 7), attributed to the strong electrostatic interaction between ChNWs and alginate. Furthermore, the introduction of ChNWs significantly promoted osteoblast cell adhesion and proliferation, making the resulting materials suitable candidates for bone tissue engineering.176
Reinforcement with DeChNWs and TOChNWs also enhanced the tear index of paper handsheets, again with DeChNWs being more effective. The maximum tear index for DeChNWs (3.7 mN m2 g−1) was observed at a 2 wt% loading, while TOChNWs achieved a peak tear index of 2.4 mN m2 g−1 at 0.8–1 wt%. Similar to the tensile index, the tear index reaches a threshold value of 3.7 mN m2 g−1 at a 2 wt% DeChNW load, with both lower and higher concentrations resulting in a decreased tear index. The superior performance of DeChNWs is attributed to their ability to form hydrogen bonds with fiber surfaces and neutralize charges between the positively charged DeChNWs and negatively charged fibers. This improves binding and retention at low concentrations. However, excessive DeChNWs result in uneven dispersion and reduced strength. In contrast, TOChNWs only improve paper strength through hydrogen bonding, lacking the charge-neutralization benefit, making DeChNWs overall more effective.121
Similarly, microcrystalline cellulose composite cryogels incorporating low concentrations of ChNWs (1 and 2.5 wt%) showed enhanced mechanical properties, as indicated by increased compressive modulus (Table 8, entry 9). However, higher concentrations (5 and 10 wt% ChNWs) reduced the compressive modulus, likely due to ChNW aggregation, which disrupted their uniform distribution, stress redistribution, and reduced cellulose molecule mobility.79 The study also evaluated the effect of freezing time on the properties of the MCC cryogels. Extending freezing time to 10 days resulted in a threefold increase in compressive modulus,79 highlighting the importance of optimizing both nanofiller concentration and processing conditions to achieve maximum mechanical performance.
Cellulose acetate (CA), an ester derivative of cellulose, is formed by partially or completely O-alkylating the hydroxyl groups in the cellulosic glucose units with acetyl groups. While CA has been used in the biocomposites and bioplastics formation for different industries, it suffers from limitations, such as low stability, mechanical resistance, and reduced adsorption levels. To address these challenges, CA has been reinforced with plant- or animal-derived materials such as chitin, chitosan, etc.177 Incorporating ChNCs into CA films enhanced their hydrophilicity, antifungal activity, and slightly improved (up to 1.2 times) the Young's modulus at a 0.5 wt% concentration (Table 8, entry 10).178 However, ChNCs negatively affected tensile strength and elongation at break and generally decreased light transmittance in the composite films.178
Other studies explored the combined effects of chitin nanowhiskers (ChNWs) and cellulose nanocrystals (CNCs) as coatings for CA-based films, particularly for food packaging applications.140,141 One of the objectives of the studies was to optimize deacetylation conditions for crab shell chitin to tune the charge and size of the resulting ChNWs and investigate their impact on oxygen permeability in layered film structures (Table 8, entry 11). It was observed that more aggressive deacetylation conditions (higher NaOH concentration, temperature, and longer time) produced shorter ChNWs, but significant surface charge alterations only occurred beyond a specific threshold condition (35% NaoH, 140 °C, 140 min).140 Despite a ∼10% decrease in ultimate tensile strength and no significant change in failure strain, the ChNW–CNC coatings reduced the water vapor transmission rate by ∼20% compared to uncoated CA films. Optimized coatings achieved a 91–99% reduction in oxygen permeability (132–16.7 cm3 μm m−2 day−1 kPa−1 compared to 1553 cm3 μm m−2 day−1 kPa−1 for uncoated CA films).140 The follow-up study further evaluated spray-coated ChNWs and CNCs on CA films and tuned the film properties by adjusting the spray coating temperature (Tspray) and the post thermal treatment temperature (Tthermal).141 By increasing Tspray (from 40 to 100 °C) and Tthermal (from 40 to 140 °C), the oxygen permeability (OP) decreased by 48% and 62%, respectively. The optimal OP of 11.5 cm3 μm m−2 day−1 kPa−1 was achieved at Tthermal of 140 °C with a controlled Tspray of 80 °C (Table 8, entry 12), which is comparable to that of commercial oriented poly(ethylene terephthalate). Higher temperatures also enhanced transmittance and reduced haze, approaching the clarity of bare CA films. The mechanical and water vapor barrier properties of the coated films were improved by an average of 20% and 11%, respectively, compared to pure CA, with minimal impact from temperature variations.141 These findings highlight the potential of chitin nanomaterials as effective reinforcing materials for cellulose acetate-based composites.
In thermoplastic corn starch-based biocomposites, chitin nanofillers notably enhanced mechanical properties (Table 8, entry 14).20 To understand the role of the chitin filler's morphology and aspect ratio in determining the final properties of the composite material, the study individually evaluated two types of chitin nanofillers – nanocrystals (ChNCs) and nanofibers (ChNFs) – by incorporating them one at a time into a corn-starch matrix via melt-mixing.20 The incorporation of both ChNC and ChNF improved mechanical performance, including increased Young's modulus and tensile strength. For instance, increase of ChNCs loading from 5 to 20 wt% enhanced Young's modulus from 222 to 390 MPa and tensile strength from 7.1 to 10.8 MPa. The addition of ChNFs produced significantly better results, with Young's modulus increasing from 330 MPa to 520 MPa and tensile strength rising from 9.9 MPa to 15.0 MPa at 5% and 20% ChNF content, respectively (Table 8, entry 14). These improvements resulted in a notable reduction in elongation at break, decreasing from 38% to 15% for 5 wt% and 20 wt% ChNC loadings, respectively, and from 26% to 9% for 5 wt% and 20 wt% ChNF loadings, respectively. The enhanced mechanical performance of the ChNCs/corn-starch and ChNFs/corn-starch composites is primarily attributed to: (a) the formation of a rigid network of ChNCs or ChNFs, driven by intra- and intermolecular hydrogen bonds; (b) the interlocking or mutual entanglement of the chitin nanofillers with the starch matrix; and (c) the effective stress transfer from the matrix to the chitin nanofillers.48,180
It also appears from the study that the mechanical behavior of the ChNCs/corn-starch composites may be enhanced by increased crystallinity attributed to the nucleating effect of ChNCs. In contrast, the superior mechanical properties of ChNFs/corn-starch composites are primarily due to the percolation effect. ChNFs, due to their high aspect ratio (L/d = 60) and extended fibrillar structure, form an interconnected network within the starch matrix. The entanglement of long chitin fibrils with the starch molecules also reinforces the matrix by creating multiple points of interaction, further increasing the composite's resistance to mechanical stress. This percolation effect is particularly significant because it allows for the formation of a continuous and rigid phase even at relatively low filler concentrations. As a result, the ChNFs/corn-starch composites exhibited better mechanical performance, such as higher tensile strength and Young's modulus, compared to the ChNC/corn-starch counterparts.20 Furthermore, incorporating chitin nanofillers into corn-starch matrix improved both thermal and storage modulus compared to the starch matrix, with the nanofiber-based materials outperforming those with nanoparticles.20
For example, Etxabide et al. developed glucose-crosslinked gelatin films reinforced with ChNWs and observed a significant increase in Young's modulus and tensile strength, reaching 2921 MPa, and 77 MPa, respectively at the 4 wt% ChNW content (Table 8, entry 15).78 These property alterations were reportedly attributed to the following factors. First, the reaction enhances the organized structure of the matrix (i.e., increases crystallinity), contributing to greater stiffness and thermal stability. Second, the Maillard reaction, which involves the interaction of glucose (used as a crosslinking agent) with amino groups in gelatin, forms covalent bonds that improve mechanical properties. Finally, to a lesser extent, the formation of noncovalent interactions between ChNWs and gelatin also plays a role.78
Additionally, the addition of ChNWs in the films notably improve UV–Vis light absorption capacity and thermal stability of the films. However, the reinforced films became less flexible and less transparent likely due to increased density of crosslinks which reduces molecular mobility and changes the films’ light-scattering characteristics.78
Another study used the immersion method for composites preparation, where the gelatin films have been soaked in a solution containing the chitin nanofibers, followed by drying at room temperature to form the composite. The improved mechanical performance along with high transparency were achieved (Table 8, entry 16).143 The Young's modulus of the 50.2%-ChNFs/gelatin (with a ChNF content of 50.2%) was approximately 5192 MPa – nearly double that of pure gelatin. Furthermore, this parameter increased as the ChNF content in the gelatin increased attributed to the reinforcing effect of the ChNF nano-networks and the hydrogen bond formation between gelatin and chitin molecules.
This group also investigated the effect of surface-deacetylated chitin nanofibers (S-ChNFs, DA = 63 ± 2%) on gelatin nanocomposites and observed a significant improvement in composites’ tensile properties.181 Furthermore, when comparing the performance of the S-ChNFs/gelatin to ChNWs/gelatin composites at the same component ratio, the S-ChNFs/gelatin composite exhibited higher stress and Young's modulus (Table 8, entry 17). This enhancement is attributed to surface deacetylation, which reduces fiber diameter and increases the aspect ratio of nanofibers, facilitating the formation of densely interconnected nanonetworks within the gelatin matrix.182 The strong interactions between chitin and gelatin molecules further contributed to the improved mechanical properties of S-ChNFs/gelatin composites.181
A recent study provided a promising approach for developing biocompatible and conductive hydrogels, leveraging the synergistic effects of gelatin, cellulose-based nanofibers, and chitin-derived nanowhiskers. It successfully fabricated conductive gelatin-based hydrogels by integrating a gelatin solution with suspensions of dialdehyde microfibrillated cellulose (DAMFC) and carboxylated chitin nanowhiskers (ChNWs). The fabrication process involved mixing these components, casting the resulting mixture, and incubating it at room temperature. The improved mechanical strength along with good electrical conductivity and freezing tolerance were achieved (Table 8, entry 18).120 When DAMFC was incorporated without ChNWs, the resulting hydrogel exhibited an increased compressive stress reaching 1.949 MPa, which is ∼4 times higher than that of pure gelatin. This enhancement was attributed to the formation of a robust dynamic 3D network, arising from the Schiff base reaction between the aldehyde groups on the surface of the DAMFC fibrils and the amino groups of the gelatin.120 When both DAMFC and ChNWs were introduced, the compressive stress of the composite hydrogels increased further, reaching a the highest level at 2 wt% of ChNWs (Table 8, entry 18). At higher ChNWs loadings (>2 wt%), aggregation of the excessive ChNWs led to the stress concentration in the hydrogel ultimately impairing its mechanical performance.120
This group also investigated the conductivity and freezing-tolerance of 2 wt% ChNW-reinforced DAMFC/Gel hydrogels through ion immersion. The researchers found that soaking the hydrogels in NaCl solution significantly enhanced their ionic conductivity, making it approximately four times higher than that of hydrogels soaked in pure water. Additionally, the hydrogels maintained their flexibility even at −20 °C, demonstrating excellent freezing resistance.120 Overall, the reinforcing effects of chitin nanomaterials on gelatin composites are attributed to good compatibility and interactions between chitin and gelatin. These properties make ChNW-reinforced DAMFC/Gel hydrogels promising materials for applications in biomedical, food packaging, and active packaging industries where mechanical strength, conductivity, and temperature resilience are critical.
Another recent study successfully developed strong acid-induced SPI gels by incorporating ChNCs and proposing a gelatinization mechanism (Table 8, entry 22).183 The process involved mixing SPI powder with a ChNC suspension at pH 3 to form a SPI–ChNC solution, adding gluconolactone (GDL), which hydrolyzed into gluconic acid, gradually reducing the solution's pH, and lowering the net surface charge of protein aggregates, leading to acid-induced gel formation. The addition of ChNCs into SPI gels improved the storage modulus, recovery rate, and thermal stability of SPI gels. These improvements are attributed to strong interactions between ChNC and SPI matrix, including electrostatic interactions and hydrogen bonding.183
Raman spectroscopy revealed that ChNCs altered the secondary protein structure of SPI gels. With the addition of 1.00% ChNCs, the α-helix content decreased from 19.7% to 12.1% whereas β-sheet increased from 46.5% to 52.6%. This transformation indicates that ChNCs promote protein unfolding and orderly aggregation, forming a densely cross-linked gel network. Additionally, the incorporation of ChNCs strengthened the SPI gel by enhancing hydrophobic interactions, electrostatic interactions, and hydrogen bonding further stabilizing the gel matrix.183
The crossover modulus of ChNWs/HA composites increased nearly linearly from 82 Pa to 120 Pa as the ChNW content rose to 35 vol%. However, at 50 vol% of ChNWs, the modulus dropped to 83 Pa, suggesting excessive nanofiller aggregation compromised the mechanical network. Tensile modulus and stress was also considerably enhanced, with the highest improvements observed at 35–50 vol% of ChNWs in the HA matrix. Furthermore, the nanocomposite biofilms demonstrated strong antibacterial properties while maintaining cytocompatibility, which improved as the volume fraction of ChNWs in the HA matrix increased.88
4.2. Reinforcement of elastomers with chitin nanomaterials
Several studies have explored how nanochitin disperses in NR matrix with different modifications to evaluate the level of reinforcement as summarized in Table 9, entries 1–3. Comprehensive studies by Nair and coworkers25,51,147 explored the reinforcement effects of colloidal ChNW in NR latex. Their preliminary study exploited the role of ChNWs in reinforcing both unvulcanized and prevulcanized NR latex, with composites prepared using the casting-evaporation method (Table 9, entry 1).25,51 Both unvulcanized and prevulcanized NR latex exhibited similar tensile moduli, measuring 1.7 MPa and 1.6 MPa, respectively. The addition of ChNWs at all loadings significantly improved the tensile modulus, with the reinforced unvulcanized NR composites (229 MPa at 20 wt% load) outperforming vulcanized ones (111 MPa at the same load). A similar trend was also observed in the conventional modulus. It is suggested that the interference of chemical cross-linking reactions during vulcanization hinders the formation of chitin network, resulting in a lower reinforcing effect and consequently, lower tensile and conventional moduli in vulcanized samples.
| No. | Polymers | Property affected | Composition | Value | Ref. |
|---|---|---|---|---|---|
| a The values of the mechanical properties were estimated from the graphs. b The composition ratio is reported in wt.%, calculated from the ChNF or ChNW content expressed in phr. | |||||
| Natural rubber | |||||
| 1 | Unvulcanized natural rubber latex (unvulc. NR) and prevulcanized natural rubber latex (prevulc. NR)/ChNWs (casting-evaporation method) | Tensile modulus, MPa | Unvulc. NR | 1.7 | 25 and 51 |
| ChNWs/unvulc. NR = 2/98 | 5.6 | ||||
| ChNWs/unvulc. NR = 5/95 | 17.8 | ||||
| ChNWs/unvulc. NR = 10/90 | — | ||||
| ChNWs/unvulc. NR = 15/85 | 127 | ||||
| ChNWs/unvulc. NR = 20/80 | 229 | ||||
| Prevulc. NR | 1.6 | ||||
| ChNWs/prevulc. NR = 2/98 | 2.5 | ||||
| ChNWs/prevulc. NR = 5/95 | — | ||||
| ChNWs/prevulc. NR = 10/90 | 25.9 | ||||
| ChNWs/prevulc. NR = 15/85 | 52.8 | ||||
| ChNWs/prevulc. NR = 20/80 | 111 | ||||
| Conventional modulus, % | Unvulc. NR | 1.8 | |||
| ChNWs/unvulc. NR = 2/98 | 2.5 | ||||
| ChNWs/unvulc. NR = 5/95 | 5 | ||||
| ChNWs/unvulc. NR = 10/90 | — | ||||
| ChNWs/unvulc. NR = 15/85 | 17 | ||||
| ChNWs/unvulc. NR = 20/80 | 23.9 | ||||
| Prevulc. NR | 1.6 | ||||
| ChNWs/prevulc. NR = 2/98 | 2.1 | ||||
| ChNWs/prevulc. NR = 5/95 | — | ||||
| ChNWs/prevulc. NR = 10/90 | 6.8 | ||||
| ChNWs/prevulc. NR = 15/85 | 11.4 | ||||
| ChNWs/prevulc. NR = 20/80 | 15.3 | ||||
| Stress at break, MPa | Unvulc. NR | 25.5 | |||
| ChNWs/unvulc. NR = 2/98 | 21.7 | ||||
| ChNWs/unvulc. NR = 5/95 | 25 | ||||
| ChNWs/unvulc. NR = 10/90 | — | ||||
| ChNWs/unvulc. NR = 15/85 | 29.7 | ||||
| ChNWs/unvulc. NR = 20/80 | 9.8 | ||||
| Prevulc. NR | 395 | ||||
| ChNWs/prevulc. NR = 2/98 | 388 | ||||
| ChNWs/prevulc. NR = 5/95 | — | ||||
| ChNWs/prevulc. NR = 10/90 | 178 | ||||
| ChNWs/prevulc. NR = 15/85 | 128 | ||||
| ChNWs/prevulc. NR = 20/80 | 20.5 | ||||
| Elongation at break, % | Unvulc. NR | 248 | |||
| ChNWs/unvulc. NR = 2/98 | 213 | ||||
| ChNWs/unvulc. NR = 5/95 | 192 | ||||
| ChNWs/unvulc. NR = 10/90 | — | ||||
| ChNWs/unvulc. NR = 15/85 | 133 | ||||
| ChNWs/unvulc. NR = 20/80 | 126 | ||||
| Prevulc. NR | 11.8 | ||||
| ChNWs/prevulc. NR = 2/98 | 13.7 | ||||
| ChNWs/prevulc. NR = 5/95 | — | ||||
| ChNWs/prevulc. NR = 10/90 | 10.5 | ||||
| ChNWs/prevulc. NR = 15/85 | 9.4 | ||||
| ChNWs/prevulc. NR = 20/80 | 5.6 | ||||
| 2 | Unvulcanized natural rubber (unvulc. NR) latex/ChNWs (freeze-drying and hot pressing method) | Tensile modulus, MPa | Unvulc. NR | 1.1 | 25 and 51 |
| ChNWs/unvulc. NR = 2/98 | 1.4 | ||||
| ChNWs/unvulc. NR = 5/95 | 2.1 | ||||
| ChNWs/unvulc. NR = 10/90 | 4.6 | ||||
| ChNWs/unvulc. NR = 15/85 | 8.7 | ||||
| ChNWs/unvulc. NR = 20/80 | 10.2 | ||||
| Conventional modulus, % | Unvulc. NR | 0.71 | |||
| ChNWs/unvulc. NR = 2/98 | 0.99 | ||||
| ChNWs/unvulc. NR = 5/95 | 1.7 | ||||
| ChNWs/unvulc. NR = 10/90 | 3.4 | ||||
| ChNWs/unvulc. NR = 15/85 | 7.2 | ||||
| ChNWs/unvulc. NR = 20/80 | 9.4 | ||||
| Stress at break, MPa | Unvulc. NR | 41.6 | |||
| ChNWs/unvulc. NR = 2/98 | 5.9 | ||||
| ChNWs/unvulc. NR = 5/95 | 115.5 | ||||
| ChNWs/unvulc. NR = 10/90 | 21.7 | ||||
| ChNWs/unvulc. NR = 15/85 | 62.7 | ||||
| ChNWs/unvulc. NR = 20/80 | 83.9 | ||||
| Elongation at break, % | Unvulc. NR | 252 | |||
| ChNWs/unvulc. NR = 2/98 | 176 | ||||
| ChNWs/unvulc. NR = 5/95 | 175 | ||||
| ChNWs/unvulc. NR = 10/90 | 160 | ||||
| ChNWs/unvulc. NR = 15/85 | 179 | ||||
| ChNWs/unvulc. NR = 20/80 | 176 | ||||
| 3 | Natural rubber (NR) latex/chitin nanowhiskers grafted with alkenyl succinic anhydride (ASA), phenyl isocyanate (PI), and isopropenyl-α,α′-dimethylbenzylisocyanate (TMI) (casting-evaporation method) | Tensile modulus, MPa | NR | 0.96 | 147 |
| ChNWs-ASA/NR = 10/90 | 5.5 | ||||
| ChNWs-TMI/NR = 10/90 | 2.5 | ||||
| ChNWs-PI/NR = 10/90 | 1.7 | ||||
| Stress at break, MPa | NR | 2.4 | |||
| ChNWs-ASA/NR = 10/90 | 10.3 | ||||
| ChNWs-TMI/NR = 10/90 | 5.5 | ||||
| ChNWs-PI/NR = 10/90 | 5.5 | ||||
| Elongation at break, % | NR | 195 | |||
| ChNWs-ASA/NR = 10/90 | 150 | ||||
| ChNWs-TMI/NR = 10/90 | 128 | ||||
| ChNWs-PI/NR = 10/90 | 184 | ||||
| 4 | Natural rubber (NR) latex/chitin nanofibers (ChNFs) | Young's modulus, MPa | NR | 0.4 | 199 |
| ChNFs/NR = 0.99/99.01 | 0.5 | ||||
| ChNFs/NR = 1.96/98.04 | 0.8 | ||||
| ChNFs/NR = 9.09/90.91 | 8.8 | ||||
| ChNFs/NR = 16.67/83.33 | 10.7 | ||||
| Tensile strength, MPa | NR | 0.5 | |||
| ChNFs/NR = 0.99/99.01 | 0.5 | ||||
| ChNFs/NR = 1.96/98.04 | 0.6 | ||||
| ChNFs/NR = 9.09/90.91 | 1.7 | ||||
| ChNFs/NR = 16.67/83.33 | 2.3 | ||||
| Elongation at break, % | NR | 1125 | |||
| ChNFs/NR = 0.99/99.01 | 1002 | ||||
| ChNFs/NR = 1.96/98.04 | 945 | ||||
| ChNFs/NR = 9.09/90.91 | 602 | ||||
| ChNFs/NR = 16.67/83.33 | 247 | ||||
| 5 | Unvulcanized natural rubber latex (unvulc. NR)/chitin nanocrystals (ChNCs) | Tensile strength, MPa | Unvulc. NR | 0.92 ± 0.12 | 13 |
| ChNCs/unvulc. NR = 1.0/99.0 | 2.26 ± 0.18 | ||||
| ChNCs/unvulc. NR = 2.5/97.5 | 2.82 ± 0.14 | ||||
| ChNCs/unvulc. NR = 5.0/95.0 | 3.30 ± 0.21 | ||||
| ChNCs/unvulc. NR = 7.5/92.5 | 4.90 ± 0.15 | ||||
| ChNCs/unvulc. NR = 10.0/90.0 | 5.75 ± 0.14 | ||||
| Tensile modulus at 100% strain, MPa | Unvulc. NR | 0.40 ± 0.07 | |||
| ChNCs/unvulc. NR = 1.0/99.0 | 0.40 ± 0.09 | ||||
| ChNCs/unvulc. NR = 2.5/97.5 | 0.57 ± 0.03 | ||||
| ChNCs/unvulc. NR = 5.0/95.0 | 0.59 ± 0.10 | ||||
| ChNCs/unvulc. NR = 7.5/92.5 | 1.53 ± 0.04 | ||||
| ChNCs/unvulc. NR = 10.0/90.0 | 1.83 ± 0.06 | ||||
| Tensile modulus at 300% strain, MPa | Unvulc. NR | 0.49 ± 0.03 | |||
| ChNCs/unvulc. NR = 1.0/99.0 | 0.47 ± 0.07 | ||||
| ChNCs/unvulc. NR = 2.5/97.5 | 0.81 ± 0.05 | ||||
| ChNCs/unvulc. NR = 5.0/95.0 | 0.96 ± 0.14 | ||||
| ChNCs/unvulc. NR = 7.5/92.5 | 3.14 ± 0.07 | ||||
| ChNCs/unvulc. NR = 10.0/90.0 | 4.20 ± 0.10 | ||||
| Tensile modulus at 500% strain, MPa | Unvulc. NR | 0.56 ± 0.04 | |||
| ChNCs/unvulc. NR = 1.0/99.0 | 0.68 ± 0.10 | ||||
| ChNCs/unvulc. NR = 2.5/97.5 | 1.47 ± 0.08 | ||||
| ChNCs/unvulc. NR = 5.0/95.0 | 2.17 ± 0.11 | ||||
| ChNCs/unvulc. NR = 7.5/92.5 | — | ||||
| ChNCs/unvulc. NR = 10.0/90.0 | — | ||||
| Elongation at break, % | Unvulc. NR | 685.1 ± 28.0 | |||
| ChNCs/unvulc. NR = 1.0/99.0 | 825.8 ± 36.5 | ||||
| ChNCs/unvulc. NR = 2.5/97.5 | 699.2 ± 20.8 | ||||
| ChNCs/unvulc. NR = 5.0/95.0 | 606.7 ± 30.0 | ||||
| ChNCs/unvulc. NR = 7.5/92.5 | 416.2 ± 22.0 | ||||
| ChNCs/unvulc. NR = 10.0/90.0 | 382.5 ± 24.6 | ||||
| Tear strength, kN m−1 | Unvulc. NR | 2.27 | |||
| ChNCs/unvulc. NR = 1.0/99.0 | 4.93 | ||||
| ChNCs/unvulc. NR = 2.5/97.5 | 6.05 | ||||
| ChNCs/unvulc. NR = 5.0/95.0 | 6.91 | ||||
| ChNCs/unvulc. NR = 7.5/92.5 | 9.89 | ||||
| ChNCs/unvulc. NR = 10.0/90.0 | 12.33 | ||||
| Storage modulus, MPa (−100 °C) | Unvulc. NR | 279.4 | |||
| ChNCs/unvulc. NR = 1.0/99.0 | 206.36 | ||||
| ChNCs/unvulc. NR = 2.5/97.5 | 340.54 | ||||
| ChNCs/unvulc. NR = 5.0/95.0 | 602.68 | ||||
| ChNCs/unvulc. NR = 7.5/92.5 | 946.44 | ||||
| ChNCs/unvulc. NR = 10.0/90.0 | 2992.68 | ||||
| Storage modulus, MPa (−40 °C) | Unvulc. NR | 1.2 | |||
| ChNCs/unvulc. NR = 1.0/99.0 | 4.79 | ||||
| ChNCs/unvulc. NR = 2.5/97.5 | 7.69 | ||||
| ChNCs/unvulc. NR = 5.0/95.0 | 12.13 | ||||
| ChNCs/unvulc. NR = 7.5/92.5 | 22.71 | ||||
| ChNCs/unvulc. NR = 10.0/90.0 | 218.01 | ||||
| Storage modulus, MPa (30 °C) | Unvulc. NR | 0.13 | |||
| ChNCs/unvulc. NR = 1.0/99.0 | 0.42 | ||||
| ChNCs/unvulc. NR = 2.5/97.5 | 0.64 | ||||
| ChNCs/unvulc. NR = 5.0/95.0 | 1.65 | ||||
| ChNCs/unvulc. NR = 7.5/92.5 | 2.44 | ||||
| ChNCs/unvulc. NR = 10.0/90.0 | 12.48 | ||||
| 6 | Prevulcanized NR latex/ChNFs | Tensile strength,a MPa | Prevulc. NR | 6.37 | 201 |
| ChNFs/prevulc. NR = 0.3/99.7 | 13.42 | ||||
| ChNFs/prevulc. NR = 0.5/99.5 | 13.62 | ||||
| ChNFs/prevulc. NR = 1.0/99.0 | 15.71 | ||||
| ChNFs/prevulc. NR = 2.0/98.0 | 13.92 | ||||
| Elongation at breaka, % | Prevulc. NR | 843.9 | |||
| ChNFs/prevulc. NR = 0.3/99.7 | 913.2 | ||||
| ChNFs/prevulc. NR = 0.5/99.5 | 877.3 | ||||
| ChNFs/prevulc. NR = 1.0/99.0 | 842.5 | ||||
| ChNFs/prevulc. NR = 2.0/98.0 | 707.9 | ||||
| Toughnessa, MJ m−3 | Prevulc. NR | 17.87 | |||
| ChNFs/prevulc. NR = 0.3/99.7 | 35.18 | ||||
| ChNFs/prevulc. NR = 0.5/99.5 | 38.79 | ||||
| ChNFs/prevulc. NR = 1.0/99.0 | 41.58 | ||||
| ChNFs/prevulc. NR = 2.0/98.0 | 54.58 | ||||
| 7 | Prevulcanized NR latex/zwitterionic ChNFs (Z-ChNFs) and carboxylated ChNFs (C-ChNFs) | Young's modulusa, MPa | Prevulc. NR | 0.47 | 202 |
| C-ChNFs/prevulc. NRb ≈ 0.50/99.50 | 0.67 | ||||
| C-ChNFs/prevulc. NR ≈ 0.99/99.01 | 0.8 | ||||
| C-ChNFs/prevulc. NR ≈ 1.96/98.04 | 1.39 | ||||
| C-ChNFs/prevulc. NR ≈ 3.38/96.62 | 2.71 | ||||
| Z-ChNFs/prevulc. NRb ≈ 0.50/99.50 | 0.47 | ||||
| Z-ChNFs/prevulc. NR ≈ 0.99/99.01 | 0.52 | ||||
| Z-ChNFs/prevulc. NR ≈ 1.96/98.04 | 0.61 | ||||
| Z-ChNFs/prevulc. NR ≈ 3.38/96.62 | 1.98 | ||||
| Stress at 100% straina, MPa | Prevulc. NR | 0.58 | |||
| C-ChNFs/prevulc. NRb ≈ 0.50/99.50 | 0.76 | ||||
| C-ChNFs/prevulc. NR ≈ 0.99/99.01 | 1.01 | ||||
| C-ChNFs/prevulc. NR ≈ 1.96/98.04 | 0.76 | ||||
| C-ChNFs/prevulc. NR ≈ 3.38/96.62 | 2.17 | ||||
| Z-ChNFs/prevulc. NRb ≈ 0.50/99.50 | 0.55 | ||||
| Z-ChNFs/prevulc. NR ≈ 0.99/99.01 | 0.63 | ||||
| Z-ChNFs/prevulc. NR ≈ 1.96/98.04 | 0.7 | ||||
| Z-ChNFs/prevulc. NR ≈ 3.38/96.62 | 0.99 | ||||
| Stress at 300% straina, MPa | Prevulc. NR | 0.94 | |||
| C-ChNFs/prevulc. NRb ≈ 0.50/99.50 | 1.76 | ||||
| C-ChNFs/prevulc. NR ≈ 0.99/99.01 | 3.14 | ||||
| C-ChNFs/prevulc. NR ≈ 1.96/98.04 | 3.01 | ||||
| C-ChNFs/prevulc. NR ≈ 3.38/96.62 | 4.26 | ||||
| Z-ChNFs/prevulc. NRb ≈ 0.50/99.50 | 1.07 | ||||
| Z-ChNFs/prevulc. NR ≈ 0.99/99.01 | 1.07 | ||||
| Z-ChNFs/prevulc. NR ≈ 1.96/98.04 | 1.31 | ||||
| Z-ChNFs/prevulc. NR ≈ 3.38/96.62 | 1.82 | ||||
| Synthetic rubbers | |||||
| 8 | Acrylonitrile butadiene rubber (NBR)/ChNWs | Tensile strength, MPa | NBR | 2.75 ± 0.25 | 18 |
| ChNWs/NBRb = 3.84/96.16 | 5.95 ± 0.67 | ||||
| ChNWs/NBR = 7.41/92.59 | 5.02 ± 0.89 | ||||
| ChNWs/NBR = 10.71/89.29 | 4.69 ± 0.37 | ||||
| Tensile modulus at 300% strain, MPa | NBR | 1.95 ± 0.20 | |||
| ChNWs/NBRb = 3.84/96.16 | 4.27 ± 0.49 | ||||
| ChNWs/NBR = 7.41/92.59 | 4.71 ± 0.38 | ||||
| ChNWs/NBR = 10.71/89.29 | 5.87 ± 0.25 | ||||
| Elongation at break, % | NBR | 395 ± 15 | |||
| ChNWs/NBRb = 3.84/96.16 | 354 ± 11 | ||||
| ChNWs/NBR = 7.41/92.59 | 363 ± 12 | ||||
| ChNWs/NBR = 10.71/89.29 | 371 ± 14 | ||||
| Tear strength, N mm−1 | NBR | 19.76 ± 0.75 | |||
| ChNWs/NBRb = 3.84/96.16 | 24.85 ± 0.61 | ||||
| ChNWs/NBR = 7.41/92.59 | 28.98 ± 0.69 | ||||
| ChNWs/NBR = 10.71/89.29 | 31.25 ± 0.67 | ||||
| Hardness, shore A | NBR | 45 ± 1 | |||
| ChNWs/NBRb = 3.84/96.16 | 48 ± 1 | ||||
| ChNWs/NBR = 7.41/92.59 | 51 ± 1 | ||||
| ChNWs/NBR = 10.71/89.29 | 53 ± 1 | ||||
| Compression set, % | NBR | 10.23 ± 0.25 | |||
| ChNWs/NBRb = 3.84/96.16 | 18.43 ± 0.25 | ||||
| ChNWs/NBR = 7.41/92.59 | 14.38 ± 0.29 | ||||
| ChNWs/NBR = 10.71/89.29 | 15.75 ± 0.25 | ||||
| Abrasion resistance index, ARI | NBR | 180 ± 5 | |||
| ChNWs/NBRb = 3.84/96.16 | 192 ± 3 | ||||
| ChNWs/NBR = 7.41/92.59 | 186 ± 4 | ||||
| ChNWs/NBR = 10.71/89.29 | 183 ± 5 | ||||
| Swelling index, % | NBR | 207 ± 2 | |||
| ChNWs/NBRb = 3.84/96.16 | 204 ± 1 | ||||
| ChNWs/NBR = 7.41/92.59 | 205 ± 1 | ||||
| ChNWs/NBR = 10.71/89.29 | 209 ± 2 | ||||
| Cross-link density, mol g−1 × 10−4 | NBR | 1.31 ± 0.05 | |||
| ChNWs/NBRb = 3.84/96.16 | 1.38 ± 0.03 | ||||
| ChNWs/NBR = 7.41/92.59 | 1.34 ± 0.05 | ||||
| ChNWs/NBR = 10.71/89.29 | 1.29 ± 0.06 | ||||
| Storage modulus, MPa (25 °C) | NBR | 3.651 | |||
| ChNWs/NBRb = 3.84/96.16 | 6.157 | ||||
| ChNWs/NBR = 10.71/89.29 | 5.968 | ||||
| Storage modulus, MPa (−40 °C) | NBR | 1189 | |||
| ChNWs/NBRb = 3.84/96.16 | 1473 | ||||
| ChNWs/NBR = 10.71/89.29 | 1384 | ||||
| T g, °C | NBR | −4.09 | |||
| ChNWs/NBRb = 3.84/96.16 | 2.78 | ||||
| ChNWs/NBR = 10.71/89.29 | 1.07 | ||||
| tanδ maximum |
NBR | 1.535 | |||
| ChNWs/NBRb = 3.84/96.16 | 0.985 | ||||
| ChNWs/NBR = 10.71/89.29 | 1.119 | ||||
| 9 | Carboxylated styrene butadiene rubber (XSBR) latex/ChNWs | Storage modulus at 25 °C, MPa (non-annealed) | XSBR | 1.39 | 146 |
| ChNWs/XSBR = 4.1/95.9 | 3.95 | ||||
| ChNWs/XSBR = 6.6/93.4 | 8.9 | ||||
| ChNWs/XSBR = 9.0/91.0 | 17.7 | ||||
| Storage modulus at 25 °C, MPa (annealed at 100 °C) | XSBR | 1.44 | |||
| ChNWs/XSBR = 4.1/95.9 | 3.95 | ||||
| ChNWs/XSBR = 6.6/93.4 | 6.25 | ||||
| ChNWs/XSBR = 9.0/91.0 | 25.25 | ||||
| Loss modulus at 25 °C, MPa (non-annealed) | XSBR | 0.18 | |||
| ChNWs/XSBR = 4.1/95.9 | 1.03 | ||||
| ChNWs/XSBR = 6.6/93.4 | 3.08 | ||||
| ChNWs/XSBR = 9.0/91.0 | 1.7 | ||||
| Loss modulus at 25 °C, MPa (annealed at 100 °C) | XSBR | 0.15 | |||
| ChNWs/XSBR = 4.1/95.9 | 0.7 | ||||
| ChNWs/XSBR = 6.6/93.4 | 1.13 | ||||
| ChNWs/XSBR = 9.0/91.0 | 3.99 | ||||
| T g from DSC analysis, °C (non-annealed) | XSBR | 10 | |||
| ChNWs/XSBR = 4.1/95.9 | 14.2 | ||||
| ChNWs/XSBR = 6.6/93.4 | 12.2 | ||||
| ChNWs/XSBR = 9.0/91.0 | 16 | ||||
| T g from DSC analysis, °C (annealed at 100 °C) | XSBR | 13.8 | |||
| ChNWs/XSBR = 4.1/95.9 | 13.3 | ||||
| ChNWs/XSBR = 6.6/93.4 | 14.6 | ||||
| ChNWs/XSBR = 9.0/91.0 | 16.7 | ||||
| 10 | XSBR latex/chitin nanocrystals (ChNCs) | Young's modulus, MPa | XSBR | 14.1 | 41 |
| ChNCs/XSBR = 1/99 | 14.3 | ||||
| ChNCs/XSBR = 2/98 | 24.3 | ||||
| ChNCs/XSBR = 3/97 | 31.1 | ||||
| ChNCs/XSBR = 4/96 | 36.2 | ||||
| ChNCs/XSBR = 5/95 | 39.2 | ||||
| Tensile modulus at 100% strain, MPa | XSBR | 1.85 | |||
| ChNCs/XSBR = 1/99 | 2.07 | ||||
| ChNCs/XSBR = 2/98 | 3.17 | ||||
| ChNCs/XSBR = 3/97 | 4.12 | ||||
| ChNCs/XSBR = 4/96 | 4.63 | ||||
| ChNCs/XSBR = 5/95 | 5.54 | ||||
| Tensile modulus at 300% strain, MPa | XSBR | 3.7 | |||
| ChNCs/XSBR = 1/99 | 4.07 | ||||
| ChNCs/XSBR = 2/98 | 5.88 | ||||
| ChNCs/XSBR = 3/97 | 7.76 | ||||
| ChNCs/XSBR = 4/96 | 9.37 | ||||
| ChNCs/XSBR = 5/95 | 10.22 | ||||
| Tensile strength, MPa | XSBR | 4.29 | |||
| ChNCs/XSBR = 1/99 | 6.26 | ||||
| ChNCs/XSBR = 2/98 | 8.68 | ||||
| ChNCs/XSBR = 3/97 | 9.42 | ||||
| ChNCs/XSBR = 4/96 | 10.51 | ||||
| ChNCs/XSBR = 5/95 | 10.4 | ||||
| Elongation at break, % | XSBR | 374.5 | |||
| ChNCs/XSBR = 1/99 | 423.5 | ||||
| ChNCs/XSBR = 2/98 | 419.7 | ||||
| ChNCs/XSBR = 3/97 | 389.1 | ||||
| ChNCs/XSBR = 4/96 | 296.7 | ||||
| ChNCs/XSBR = 5/95 | 240.8 | ||||
| Storage modulus, MPa (−100 °C) | XSBR | 1768.4 | |||
| ChNCs/XSBR = 1/99 | 1718.9 | ||||
| ChNCs/XSBR = 2/98 | 1854 | ||||
| ChNCs/XSBR = 3/97 | 1823.5 | ||||
| ChNCs/XSBR = 4/96 | 1924.3 | ||||
| ChNCs/XSBR = 5/95 | 2184.3 | ||||
| Storage modulus, MPa (30 °C) | XSBR | 38.2 | |||
| ChNCs/XSBR = 1/99 | 31.5 | ||||
| ChNCs/XSBR = 2/98 | 41.8 | ||||
| ChNCs/XSBR = 3/97 | 43.7 | ||||
| ChNCs/XSBR = 4/96 | 51.7 | ||||
| ChNCs/XSBR = 5/95 | 80.4 | ||||
| Storage modulus, MPa (80 °C) | XSBR | 1.6 | |||
| ChNCs/XSBR = 1/99 | 1.1 | ||||
| ChNCs/XSBR = 2/98 | 2.4 | ||||
| ChNCs/XSBR = 3/97 | 3 | ||||
| ChNCs/XSBR = 4/96 | 4.3 | ||||
| ChNCs/XSBR = 5/95 | 7.5 | ||||
The study further investigated the impact of the processing techniques on the properties of the unvulcanized-based composites.51 Two techniques, i.e., water evaporation and freeze-drying followed by a hot-pressing step, were evaluated and compared. The results that the composite films produced by the later method were up to 20 times mechanically weaker than those prepared by casting-evaporation (10.2 vs. 229 MPA, respectively, see Table 9, entries 1 and 2). This discrepancy is likely due to the insufficient formation of a robust 3-D rigid network through hydrogen bond interaction, caused by the rapid solvent removal in freeze-drying compared to the slower solvent removal in casting-evaporation.
Surface modification is often necessary for effectively dispersing nanochitin in NR latex due to the negatively charged phospholipid layers and protein in NR latex, which remain stable only in basic medium,199 whereas amino groups in nanochitin are stable in mildly acidic medium.198 To address this, Nair et al.147 conducted a follow-up study where they modified ChNWs using three chemical coupling agents: phenyl isocyanate (PI), alkenyl succinic anhydride (ASA) (Accosize 18 from American Cyanamid), and 3-isopropenyl-R,R′dimethylbenzyl isocyanate (TMI). The study focused solely on 10 wt% ChNW reinforcement and used the casting-evaporation method to prepare NR composite films. SEM and swelling studies confirmed that the chemical modification improved the adhesion between NR and ChNWs. Regardless of the coupling agent used, the incorporation of surface-modified ChNWs moderately enhanced the tensile modulus (1.5 to 5.5-fold) and stress at break (2.3- to 4.3-fold) of the NR films, with ASA-modified ChNW films showing superior performance compared to TMI- and PI-modified ChNW films (Table 9, entry 3). The improved tensile properties of ASA-modified ChNW composite were attributed to the partial existence of a chitin–chitin network within the NR matrix. However, these enhance mechanical properties were significantly lower compared to the unmodified ChNWs/NR films in their previous study. For instance, the tensile modulus of the modified ChNW-ASA/NR films (Table 9, entry 3) was nearly half that of the unmodified ChNW/prevulcanized NR film (Table 9, entry 1). This significant reduction following chemical modification was likely due to the partial or total degration of 3-D chitin network present in the unmodified films, with a more pronounced reduction observed in the case of isocyanate modifications.
Another study reported the successful fabrication of self-assembled ChNF-NR composite sheets using re-dispersed ChNFs and NR latex.199 In this study, self-assembled ChNFs were redispersed in aqueous ammonia solution after being isolated as a film and then mixed with NR latex at various ChNF loadings (∼1 to 17 wt%). SEM and XRD results confirmed that ChNFs were well re-disperser in ammonia and evenly dispersed in the NR latex with increased crystallinity as the ChNF loading increased. With the addition of ChNFs up to 17 wt%, both tensile strength and Young's modulus of the NR latex increased by ∼4.6-fold and 26–8, respectively (Table 9, entry 4), demonstrating the reinforcing effect of ChNFs. Meanwhile, elongation at break consistently dropped from 1125% for neat NR sheet to 247% at ∼17 wt% loading, implying increased brittleness with higher ChNF content.
Rod-like structure ChNCs were used as a reinforcing agent for unvulcanized NR latex.13 When incorporated at different concentrations into the NR latex via the solution-casting method to form composite films, ChNCs formed liquid crystalline phases at concentrations above 5 wt%, which lead to enhanced mechanical properties (Table 9, entry 5). The tensile strength of ChNC/NR film containing 10 wt% ChNC is 5.75 MPa, which is 6.25 multiples of pure NR. Additionally, the tensile modulus at various strains and tear strength of the films increased with increasing ChNC loadings. For instance, the tear strength of the composites with 10 wt% ChNC content is 12.33 kN m−1, 5.43 times that of neat NR. Similarly, the modulus at 500% strain for the composite with 5 wt% ChNC content is 2.17 MPa, 3.88 times that of the pure NR.
The study further explored the impact of ChNCs on the dynamic properties of NR as a function of temperature.13 The results revealed that all composites displayed a glassy-rubbery state transition within the temperature range of −100 °C to 50 °C. The storage modulus (E′) of the composites increased with ChNC loading, except for the composite with 1 wt% ChNCs in the glassy state at −100 °C, when compared to neat NR. For example, at 10 wt% loading, the storage modulus at −40 °C and 30 °C were 218.01 and 12.48 MPa, respectively −181.7 and 96 times greater than that of neat NR. These findings demonstrate that ChNC networks effectively transfer load from the rubber matrix to the rigid nanoparticles. The slight decrease in E′ for the rubber composite with 1 wt% ChNCs in the glassy state likely stems from changes in the rubber's conformation and free volume due to the addition of a small amount of ChNCs.13,200 Additionally, the incorporation of ChNCs improved the thermal stability of ChNCs/NR composites, which is attributed to the restricted mobility of the rubber chains resulting from interfacial interactions.
Another study reinforced a prevulcanized NR matrix with rigid ChNFs.201 In this study, ChNFs were self-assembled from a dilute α-chitin/potassium hydroxide (KOH)/urea aqueous solution. The highly stable ChNFs demonstrated better compatibility with NR latex and dispersed at nanoscale within the NR matrix. The mechanical properties of the ChNFs/NR nanocomposites showed a remarkable improvement with small loadings (0.3–2 wt%, see Table 9, entry 6). At just 0.3 wt% ChNFs, both the tensile strength and toughness of the composites were more than doubled. These improvements were attributed to the large aspect ratio of ChNFs and their homogeneous dispersion in the NRL matrix.201
Another study by Yin et al.202 fabricated prevulcanized NR (pervulc.NR)/ChNFs nanocomposites by incorporating zwitterionic ChNFs (Z-ChNFs) and carboxylated ChNFs (C-ChNFs) via dip molding technique. The oxidation of ChNFs with 2,2,6,6-tetramethylpiperidinyloxy (TEMPO) produced C-ChNFs with negatively charged COO groups, which was stable in basic medium, but unstable in acidic medium due to the possibility of hydrogen bonding tempted by COOH groups. In contrast, zwitterionic ChNFs, formed through partial deacetylation and subsequent TEMPO oxidation, contain both carboxyl and amino groups, making them stable across various pH conditions. The study found that C-ChNFs exhibited better dispersion in the pervulcanized NR matrix than Z-ChNFs. As shown in Table 9, entry 7, incorporating both C-ChNFs and Z-ChNFs into NR improved stresses at 100% and 300% strain. When C-ChNFs reached approximately 3.4 wt%, the stress at 100% strain and 300% strain in C-ChNFs/pervulc.NR composites was about four times greater than that of neat NR. In contrast, at the same loading, Z-ChNFs increased the stress at both 100% strain and 300% strain by 2-fold compared to neat NR. However, for ChNF content below 3.4 wt%, the stress at 100% strain and 300% strain in Z-ChNFs/pervulc.NR increased more gradually with higher nanofiber content compared to C-ChNFs/pervulc.NR. The Young's modulus followed a similar trend, with C-ChNFs enhancing the modulus by nearly 1.37 times more than Z-ChNFs reinforcement.
A previous study18 successfully fabricated ChNWs/acrylonitrile-butadiene rubber (NBR) composites utilized a two-roll mill, starting with the formulation of a mixture of polymers and ChNWs, which was then compounded with vulcanizing agents. Subsequently, the reinforcing effects of ChNWs on processing and performance of NBR based nanocomposites were evaluated. The tensile strength of NBR increased from 2.75 MPa to 5.95 MPa at 3.84 wt% ChNWs, but decreased at higher ChNW concentrations (Table 9, entry 8). This improvement was attributed to several factors, including hydrogen bonding interactions between the –OH groups of ChNWs and the nitrile group of NBR rubber at the interface, the 3-D intra- and inter-hydrogen bonding networks within the ChNWs, their uniform dispersion, and ChNW high aspect ratio. However, at higher ChNW loadings, ChNWs was prone to agglomerating into bundles, with filler–filler interactions dominating over matrix–filler interactions. This caused debonding at the interface and created voids that may act as stress concentration sites, reducing the tensile strength.18 Meanwhile, the modulus at 300% strain and tear strength increased with the increasing ChNW loadings, attributed to improved interfacial compatibility and the reinforcing efficiency of ChNWs, as well as the obstruction they created in the tear path. Although the tensile strength peaked at 3.84 wt% ChNW loading, the tear strength was highest at 10.71 wt%, showing a 58% increase. This trend aligned with their previous study, where cellulosic nanofibers were used as a filler in NR.207 A similar trend in tear strength was also observed in the hardness property of the ChNW/NBR composites, which was ascribed to the improved rigidity.
Incorporating ChNWs also increased cross-link density, peaking at 3.84 wt% ChNW loading due to hydrogen bond interaction between ChNWs and NBR. However, at 10.71 wt% ChNW loading, cross-link density decreased as increased intramolecular hydrogen bonding within ChNWs reduced matrix–filler interactions. Dynamical mechanical analysis (DMA) studies revealed that the glass transition temperature of the composite at 3.84 wt% ChNW loading was approximately 6 °C higher than that of neat NBR. Additionally, the addition of ChNWs lowered the loss tangent peak height and shifted the maximum tan δ peak to a higher temperature region (Table 9, entry 8), indicating effective interaction of ChNW with the NBR matrix.
ChNWs derived from crab shell waste have also effectively reinforced the thermo-mechanical properties of carboxylated styrene–butadiene rubber (XSBR) composites.146 DMA results revealed that the storage modulus (G′) of the composites increased across the entire studied temperature range with higher ChNW content. At 25 °C, the addition of up to 9 wt% ChNWs increased the storage modulus of the composite nearly 13 times compared to neat XSBR, while the loss modulus doubled (Table 9, entry 9). Differential scanning calorimetry results indicated a slight increase in glass transition temperature (Tg) with increasing ChNW content.
The study also assessed the effect of annealing the XSBR-based composites at 100 °C to improve their durability for specific applications (back coating for textiles and carpets).146 Compared to non-annealed composites, the annealing treatment had a limited effect on the loss modulus profile, with only a small increase of the G′ values at 25 °C, likely due to stiffening. A slight increase in Tg was also observed following the annealing process. However, SEM results highlighted an exfoliation of the rubber matrix upon toluene absorption, indicating the weak interface between the XSBR and ChNW aggregates.
On the other hand, a study by Liu et al.41 reported that when added to XSBR via casting-evaporation method, with loadings below 4 wt%, improved both the tensile strength and elongation at break of the composites (Table 9, entry 10). For instance, the composite with 3 wt% ChNCs exhibited a tensile strength of 9.16 MPa and elongation at break of 396%, which were 2.23 times and 11.0% higher, respectively, compared to neat XSBR. The strength continued to increase with higher ChNC loading, reaching a maximum value of 10.4 MPa; however, the elongation at break decreased compared to neat XSBR. A similar trend was also observed in the Young's modulus and tensile moduli at 100% and 300% strain, indicating that the small amount of ChNCs acted as the physical crosslinker of the XSBR, enhancing the mobility of the polymer chains. On the other hand, excessive ChNCs loading resulted in a stiffer network, reducing the flexibility of the rubber chains and decreasing elongation at break.
The reinforcement effect of ChNCs was also evident in the DMA properties of the composites under dynamic stress, facilitated by a 3-D network formed by the interconnection of polysaccharide nanocrystals, stabilized by hydrogen bonding.41 As the ChNC concentration increased, both the rubbery and glassy regions showed a significant increase in E′, except for the composite with 1 wt% ChNCs. The reinforcement effect became more pronounced with rising temperature. For example, the E′ values for the composite with 5 wt% CNCs at −100, 30, and 80 °C were 2184.3, 80.4, and 7.5 MPa, respectively −23.5%, 110.5% and 368.8% higher than those of neat XSBR (Table 9, entry 10). This trend aligned with the results from the static tensile tests.
4.3. Reinforcement of poly(lactic) acid with nanochitin
Polylactic acid (PLA) is a widely recognized thermoplastic bio-based polymer, almost exclusively sourced from renewable resources like potato, corn, cassava, rice, cane sugar, or other carbohydrate-rich crops through a fermentation process into lactic acid followed by polymerization.208,209 It has captured broad interest because it offers high mechanical strength and stiffness, UV stability, clarity, and easy processability – it can be molded, extruded, or 3D-printed.210 The properties of PLA are affected by various factors, such as the component isomers (L-, D-, or DL-), the molecular weights, and processing routes. Thus, the tensile strength of poly(L-lactide) (PLLA) is 15.5–150 MPa, whereas the tensile strength of poly(DL-lactide) (PDLLA) is 0.04–0.05 MPa.211PLA could replace many petroleum-based plastics to fabricate biomedical materials such as sutures, drug delivery systems, and tissue engineering devices, due to its biocompatibility. In packaging and consumer goods, PLA is a popular material for biodegradable food containers, disposable cutlery, and sustainable textiles. The limitations of PLA are associated with its intrinsic brittleness, low impact resistance, slow crystallization, and insufficient thermal stability.212 Several researchers have reported on the use of polysaccharide nanofillers such as cellulose, starch, clay, wood fibers etc., to reinforce PLA by forming a hydrogen-bonded or chemically crosslinked network,213via blending them with a non-polar thermoplastic matrix.214 A few of the studies have probed into the dispersion of chitin nanoparticles in PLA based matrix using several techniques to assess the degree of reinforcement (Table 10, entries 1–10). Zhang et al.93 reported a significant improvement in tensile strength and Young's modulus of PLA composites reinforced with acetylated chitin nanocrystals (Ac-ChNCs). The enhancements surpassed those of neat PLA (25 MPa and 1148 MPa, respectively) across all loading levels (1 to 10 wt%), with the highest improvement observed at 4 wt% Ac-ChNCs, where tensile strength and Young's modulus peaked at 38 MPa and 1580 MPa, respectively (Table 10, entry 1). However, exceeding this threshold resulted in nanocrystal agglomeration, leading to the formation of a percolation network that disrupted the homogeneity and mechanical integrity of the PLA matrix. Additionally, elongation at break decreased at all concentrations, indicating increased stiffness and reduced flexibility of the composite.
| No. | Polymers | Affected properties | Composition | Value | Ref. |
|---|---|---|---|---|---|
| Polylactic acid (PLA) | |||||
| a Four methods were conducted to evaluate the dispersion effect of nanofibers for the composite preparation. Two of these methods utilized either PEG or PEO as a surfactant and dispersing agent to optimize the dispersion of nanofibers within the PLA matrix. b The storage modulus of chitin nanofibers/PLA composites remains stable below 60 °C, while it gradually decreases with rising temperature. Additionally, compared to neat PLA, the storage modulus values of the nanocomposites show a notable increase. The loss modulus rises with temperature until it reaches the glass transition (Tg) of 55 °C, after which it decreases for all composites. In comparison to pure PLA, the loss modulus values of the nanocomposites are notably lower, indicating that adding fibers reduces the loss modulus. | |||||
| 1 | Polylactic acid (PLA)/acetylated chitin nanocrystals (Ac-ChNCs) | Young's modulus, MPa | PLA | 1148 | 93 |
| Ac-ChNCs/PLA = 1/99 | 1200 | ||||
| Ac-ChNCs/PLA = 2/98 | 1400 | ||||
| Ac-ChNCs/PLA = 4/96 | 1580 | ||||
| Ac-ChNCs/PLA = 6/94 | 1500 | ||||
| Ac-ChNCs/PLA = 8/92 | 1250 | ||||
| Ac-ChNCs/PLA = 10/90 | 1100 | ||||
| Tensile strength, MPa | PLA | 25 | |||
| Ac-ChNCs/PLA = 1/99 | 33 | ||||
| Ac-ChNCs/PLA = 2/98 | 35 | ||||
| Ac-ChNCs/PLA = 4/96 | 38 | ||||
| Ac-ChNCs/PLA = 6/94 | 28 | ||||
| Ac-ChNCs/PLA = 8/92 | 23 | ||||
| Ac-ChNCs/PLA = 10/90 | 21 | ||||
| Elongation at break, % | PLA | 33 | |||
| Ac-ChNCs/PLA = 1/99 | 10 | ||||
| Ac-ChNCs/PLA = 2/98 | 10 | ||||
| Ac-ChNCs/PLA = 4/96 | 7 | ||||
| Ac-ChNCs/PLA = 6/94 | 6 | ||||
| Ac-ChNCs/PLA = 8/92 | 4 | ||||
| Ac-ChNCs/PLA = 10/90 | 4 | ||||
| 2 | PLA-grafted maleic anhydride (PLA-g-MA)/α-ChNWs | Elongation at break, % | PLA-g-MA | 0.6 | 91 |
| ChNWs/PLA-g-MA = 0.5/99.5 | 0.175 | ||||
| ChNWs/PLA-g-MA = 1.0/99.0 | 0.175 | ||||
| ChNWs/PLA-g-MA = 2.0/98.0 | 0.16 | ||||
| ChNWs/PLA-g-MA = 5.0/95.0 | 0.15 | ||||
| Ultimate tensile strength, MPa | PLA-g-MA | 45 | |||
| ChNWs/PLA-g-MA = 0.5/99.5 | 53 | ||||
| ChNWs/PLA-g-MA = 1.0/99.0 | 50 | ||||
| ChNWs/PLA-g-MA = 2.0/98.0 | 48.5 | ||||
| ChNWs/PLA-g-MA = 5.0/95.0 | 35 | ||||
| Young's modulus, MPa | PLA-g-MA | 550 | |||
| ChNWs/PLA-g-MA = 0.5/99.5 | 650 | ||||
| ChNWs/PLA-g-MA = 1.0/99.0 | 675 | ||||
| ChNWs/PLA-g-MA = 2.0/98.0 | 650 | ||||
| ChNWs/PLA-g-MA = 5.0/95.0 | 600 | ||||
| 3 | PLA-(grafted)-MA/ChNWspoly(3-hydroxybutyrate-co-3-hydroxyvalerate) (PHBV)/ChNWs | Elongation at break, % | PLA-g-MA/PHBV | 0.56 | 91 |
| ChNWs/PLA-g-MA/PHBV = 0.5/84.6/14.9 | 0.25 | ||||
| ChNWs/PLA-g-MA/PHBV = 1.0/84.5/14.8 | 0.25 | ||||
| ChNWs/PLA-g-MA/PHBV = 2.0/84.4/14.7 | 0.38 | ||||
| ChNWs/PLA-g-MA/PHBV = 5.0/80.7/14.3 | 0.07 | ||||
| Ultimate tensile strength, MPa | PLA-g-MA/PHBV | 39 | |||
| ChNWs/PLA-g-MA/PHBV = 0.5/84.6/14.9 | 44 | ||||
| ChNWs/PLA-g-MA/PHBV = 1.0/84.5/14.8 | 42 | ||||
| ChNWs/PLA-g-MA/PHBV = 2.0/84.4/14.7 | 37 | ||||
| ChNWs/PLA-g-MA/PHBV = 5.0/80.7/14.3 | 27 | ||||
| Young's modulus, MPa | PLA-g-MA/PHBV | 550 | |||
| ChNWs/PLA-g-MA/PHBV = 0.5/84.6/14.9 | 545 | ||||
| ChNWs/PLA-g-MA/PHBV = 1.0/84.5/14.8 | 575 | ||||
| ChNWs/PLA-g-MA/PHBV = 2.0/84.4/14.7 | 500 | ||||
| ChNWs/PLA-g-MA/PHBV = 5.0/80.7/14.3 | 495 | ||||
| 4 | Polylactic acid (PLA)/α-ChNWs | Young's modulus, MPa | PLA | 350 | 92 |
| ChNWs/PLA = 1/99 | 460 | ||||
| ChNWs/PLA = 2/98 | 525 | ||||
| ChNWs/PLA = 5/95 | 480 | ||||
| Ultimate tensile strength, MPa | PLA | 56 | |||
| ChNWs/PLA = 1/99 | 52.25 | ||||
| ChNWs/PLA = 2/98 | 51.25 | ||||
| ChNWs/PLA = 5/95 | 48 | ||||
| 5 | Polylactic acid (PLA) grafted with maleic anhydride (PLA-g-MA)/α-ChNWs | Young's modulus, MPa | PLA-g-MA | 350 | 92 |
| ChNWs/(PLA-g-MA) = 2/98 | 475 | ||||
| ChNWs/(PLA-g-MA) = 5/95 | 480 | ||||
| ChNWs/(PLA-g-MA) = 1/99 | 490 | ||||
| Ultimate tensile strength, MPa | PLA-g-MA | 56 | |||
| ChNWs/(PLA-g-MA) = 2/98 | 52.5 | ||||
| ChNWs/(PLA-g-MA) = 5/95 | 51.5 | ||||
| ChNWs/(PLA-g-MA) = 1/99 | 48.5 | ||||
| 6 | Polylactic acid (PLA, Mw 170000)/chitin nanofibrils (ChNFs)/polyethylene glycol (PEG, Mw 400, and 8000) |
Young's modulus, GPa | PLA | 3.5 ± 0.1 | 165 |
| PEG400/PLA = 10/90 | 2.3 ± 0.3 | ||||
| PEG400/ChNFs/PLA = 10/2/88 | 1.8 ± 0.3 | ||||
| PEG8000/ChNFs/PLA = 10/2/88 | 2.5 ± 0.1 | ||||
| PEG400/ChNFs/PLA = 1/2/97 | 3.2 ± 0.8 | ||||
| PEG400/ChNFs/PLA = 5/2/93 | 2.8 ± 0.8 | ||||
| PEG400/ChNFs/PLA = 10/5/85 | 1.8 ± 0.3 | ||||
| PEG400/ChNFs/PLA = 10/12/78 | 1.7 ± 0.3 | ||||
| Stress at yield, MPa | PLA | 60.4 ± 0.3 | |||
| PEG400/PLA = 10/90 | 26 ± 0.3 | ||||
| PEG400/ChNFs/PLA = 10/2/88 | 23 ± 5.0 | ||||
| PEG8000/ChNFs/PLA = 10/2/88 | 45 ± 5.0 | ||||
| PEG400/ChNFs/PLA = 1/2/97 | 52 ± 6.0 | ||||
| PEG400/ChNFs/PLA = 5/2/93 | 47 ± 3.0 | ||||
| PEG400/ChNFs/PLA = 10/5/85 | 34 ± 2.0 | ||||
| PEG400/ChNFs/PLA = 10/12/78 | 23 ± 5.0 | ||||
| Stress at break, MPa | PLA | 57 ± 1 | |||
| PEG400/PLA = 10/90 | 33 ± 2 | ||||
| PEG400/ChNFs/PLA = 10/2/88 | 32 ± 2 | ||||
| PEG8000/ChNFs/PLA = 10/2/88 | 34 ± 2 | ||||
| PEG400/ChNFs/PLA = 1/2/97 | 40 ± 7 | ||||
| PEG400/ChNFs/PLA = 5/2/93 | 32 ± 2 | ||||
| PEG400/ChNFs/PLA = 10/5/85 | 23 ± 2 | ||||
| PEG400/ChNFs/PLA = 10/12/78 | 22 ± 6 | ||||
| Elongation at break, % | PLA | 4.1 ± 0.5 | |||
| PEG400/PLA = 10/90 | 180 ± 10.0 | ||||
| PEG400/ChNFs/PLA = 10/2/88 | 160 ± 10.0 | ||||
| PEG8000/ChNFs/PLA = 10/2/88 | 160 ± 10.0 | ||||
| PEG400/ChNFs/PLA = 1/2/97 | 10 ± 2.0 | ||||
| PEG400/ChNFs/PLA = 5/2/93 | 11.4 ± 0.9 | ||||
| PEG400/ChNFs/PLA = 10/5/85 | 160 ± 10.0 | ||||
| PEG400/ChNFs/PLA = 10/12/78 | 181 ± 6.0 | ||||
| 7 | Polylactic acid (PLA)/α-ChNFs (water pretreatment method)a | Bending strength, MPa | PLA | 14.6 | 26 |
| ChNFs/PLA = 10/90 | 22.5 | ||||
| ChNFs/PLA = 20/80 | 27.5 | ||||
| ChNFs/PLA = 30/70 | 37.5 | ||||
| ChNFs/PLA = 40/60 | 41.2 | ||||
| Bending modulus, MPa | PLA | 1697 | |||
| ChNFs/PLA = 10/90 | 1750 | ||||
| ChNFs/PLA = 20/80 | 2000 | ||||
| ChNFs/PLA = 30/70 | 3250 | ||||
| ChNFs/PLA = 40/60 | 4153 | ||||
| Impact toughness, J m−1 | PLA | 8.4 | |||
| ChNFs/PLA = 10/90 | 10 | ||||
| ChNFs/PLA = 20/80 | 15 | ||||
| ChNFs/PLA = 30/70 | 28 | ||||
| ChNFs/PLA = 40/60 | 35 | ||||
| Storage modulus (temperature range −30 to 60 °C)b, GPa | PLA | Significantly lower | |||
| ChNFs/PLA = 10/90 | 7.8 | ||||
| ChNFs/PLA = 20/80 | 7.8 | ||||
| ChNFs/PLA = 30/70 | 7.8 | ||||
| ChNFs/PLA = 40/60 | 7.8 | ||||
| 8 | Polylactic acid (PLA)/α-ChNFs/polyethylene glycol (PEG) PEG pretreatment method | Bending strength, MPa | PLA | 14.6 | 26 |
| ChNFs/PLA/PEG = 10/80/10 | 22.5 | ||||
| ChNFs/PLA/PEG = 20/70/10 | 25 | ||||
| ChNFs/PLA/PEG = 30/60/10 | 35 | ||||
| ChNFs/PLA/PEG = 40/50/10 | 37.5 | ||||
| Bending modulus, MPa | PLA | 1697 | |||
| ChNFs/PLA/PEG = 10/80/10 | 2250 | ||||
| ChNFs/PLA/PEG = 20/70/10 | 2500 | ||||
| ChNFs/PLA/PEG = 30/60/10 | 3250 | ||||
| ChNFs/PLA/PEG = 40/50/10 | 3550 | ||||
| Impact strength, J m−1 | PLA | 8.4 | |||
| ChNFs/PLA/PEG = 10/80/10 | 10 | ||||
| ChNFs/PLA/PEG = 20/70/10 | 15 | ||||
| ChNFs/PLA/PEG = 30/60/10 | 26 | ||||
| ChNFs/PLA/PEG = 40/50/10 | 30 | ||||
| Storage modulus (temperature range −30 to 60 °C)b, GPa | PLA | Significantly lower | |||
| ChNFs/PLA/PEG = 10/80/10 | 6.5 | ||||
| ChNFs/PLA/PEG = 20/70/10 | 6.5 | ||||
| ChNFs/PLA/PEG = 30/60/10 | 6.5 | ||||
| ChNFs/PLA/PEG = 40/50/10 | 6.5 | ||||
| 9 | Polylactic acid (PLA)/α-ChNFs/polyethylene oxide (PEO) PEO pretreatment method | Bending strength, MPa | PLA | 14.6 | 26 |
| ChNFs/PLA/PEO = 10/80/10 | 24 | ||||
| ChNFs/PLA/PEO = 20/70/10 | 26 | ||||
| ChNFs/PLA/PEO = 30/60/10 | 30 | ||||
| ChNFs/PLA/PEO = 40/50/10 | 34 | ||||
| Bending modulus, MPa | PLA | 1697 | |||
| ChNFs/PLA/PEO = 10/80/10 | 1550 | ||||
| ChNFs/PLA/PEO = 20/70/10 | 2250 | ||||
| ChNFs/PLA/PEO = 30/60/10 | 2500 | ||||
| ChNFs/PLA/PEO = 40/50/10 | 2900 | ||||
| Impact strength, J m−1 | PLA | 8.4 | |||
| ChNFs/PLA/PEO = 10/80/10 | 9.5 | ||||
| ChNFs/PLA/PEO = 20/70/10 | 14 | ||||
| ChNFs/PLA/PEO = 30/60/10 | 22.5 | ||||
| ChNFs/PLA/PEO = 40/50/10 | 28 | ||||
| Storage modulus (temperature range −30 to 60 °C)b, GPa | PLA | Significantly lower | |||
| ChNFs/PLA/PEO = 10/80/10 | 6.1 | ||||
| ChNFs/PLA/PEO = 20/70/10 | 6.1 | ||||
| ChNFs/PLA/PEO = 30/60/10 | 6.1 | ||||
| ChNFs/PLA/PEO = 40/50/10 | 6.1 | ||||
| 10 | Polylactic acid (PLA)/α-ChNFs (freeze-dried, compounding method) | Bending strength, MPa | PLA | 14.6 | 26 |
| ChNFs/PLA = 10/90 | 24 | ||||
| ChNFs/PLA = 20/80 | 26 | ||||
| ChNFs/PLA = 30/70 | 27.5 | ||||
| ChNFs/PLA = 40/60 | 27.5 | ||||
| Bending modulus, MPa | PLA | 1697 | |||
| ChNFs/PLA = 10/90 | 1550 | ||||
| ChNFs/PLA = 20/80 | 1555 | ||||
| ChNFs/PLA = 30/70 | 1600 | ||||
| ChNFs/PLA = 40/60 | 1655 | ||||
| Impact strength, J m−1 | PLA | 8.4 | |||
| ChNFs/PLA = 10/90 | 9 | ||||
| ChNFs/PLA = 20/80 | 12 | ||||
| ChNFs/PLA = 30/70 | 12.5 | ||||
| ChNFs/PLA = 40/60 | 15 | ||||
| Storage modulus (temperature range −30 to 60 °C)b, GPa | PLA | Markedly lower | |||
| ChNFs/PLA = 10/90 | 5 | ||||
| ChNFs/PLA = 20/80 | 5 | ||||
| ChNFs/PLA = 30/70 | 5 | ||||
| ChNFs/PLA = 40/60 | 5 | ||||
Guan and Naguib91 investigated PLA-based and PLA/PHBV-based nanocomposites, incorporating maleic anhydride (MA) as a compatibilizing agent via grafting (Table 10, entries 2 and 3). It is well known that the hydrophobic nature of PLA results in poor interfacial adhesion when compounded with hydrophilic natural reinforcements. A strategic approach to address this challenge is compatibilizing one of the phases to improve interfacial interactions. Grafted MA, a widely studied compatibilizer, was employed in this study to promote interfacial wetting between PLA and chitin. For PLA-grafted maleic anhydride (PLA-g-MA)/α-ChNWs, nanochitin loadings ranged from 0.5 to 5 wt%, with tensile strength increasing from 45 MPa for neat PLA-g-MA to a peak of 53 MPa at 0.5 wt%, before declining at higher loadings. Young's modulus improved from 550 MPa to a maximum of 675 MPa at 1 wt%, then gradually decreased. Elongation at break consistently dropped from 0.600 for neat PLA-g-MA to 0.150 at 5 wt%, indicating increased brittleness with higher nanochitin content. At elevated ChNW loadings (∼5 wt%), mechanical property deterioration was attributed to solvolytic degradation caused by the dispersing agent (DMAc), which likely disrupted the polymer matrix. Similarly, for PLA-g-MA/PHBV-ChNWs, nanochitin loadings from 0.5 to 5 wt% exhibited a comparable trend. Tensile strength, which was 39 MPa for PLA-g-MA/PHBV, peaked at 44 MPa at 0.5 wt% but declined at higher loadings. Young's modulus increased from 550 MPa to a maximum of 575 MPa at 1 wt%, before gradually decreasing. Meanwhile, elongation at break consistently declined from 0.56 for PLA/PHBV to 0.07 at 5 wt%, further demonstrating the increased stiffness and brittleness with rising nanochitin content.
Rizvi et al.92 investigated the mechanical behavior of PLA-grafted maleic anhydride (PLA-g-MA)/α-ChNWs composites, where MA was again used as a compatibilizer to enhance interfacial wetting. Here, however, to assess the influence of MA addition on mechanical properties, the study compared composites with and without MA (Table 10, entries 4 and 5). For neat PLA, Young's modulus was 350 MPa, which peaked at 525 MPa with 2 wt% ChNWs before declining at higher loadings. Similarly, for PLA-g-MA, Young's modulus increased from 350 MPa to 490 MPa at 1 wt% ChNWs, followed by a decrease at higher concentrations. However, tensile strength, which was 56 MPa for neat PLA, consistently decreased with the addition of ChNWs, regardless of loading percentage. A similar trend was observed in PLA-g-MA/ChNWs composites, indicating that while stiffness improved with nanochitin reinforcement, overall strength diminished which could be due to the reduction in the molecular weight on account of the hydrolysis of PLA throughout the fabrication process, as explained by the authors.
Concerning the need for better dispersing agents for ChNWs to inhibit particle agglomeration, Coltelli et al.165 employed PEGs with different molecular weights in combination with PLA (Table 10, entry 6). Similar to how MA was used in the study by Rizvi et al.92 as a compatibilizer to improve interfacial wetting between PLA and chitin, PEGs were utilized here as dispersing agents to enhance the uniform distribution of ChNWs within the PLA matrix. The incorporation of PEGs aimed to improve the dispersion and interaction of ChNWs within PLA, addressing the common challenge of nanoparticle agglomeration, which can negatively impact the mechanical properties of the composite.
The initial study on PEG/PLA composites without nanowhiskers revealed that adding PEG to PLA at 10/90 w/w significantly reduced mechanical properties, decreasing Young's modulus by 1.5-fold (from 3.5 to 2.3 GPa), stress at yield by 2.3-fold (from 60 to 26 MPa), and stress at break by 1.7-fold (from 57 to 33 MPa). However, elongation at break improved nearly 45-fold due to PEG's plasticizing effect. When nanofibers were introduced, the PEG400/ChNFs/PLA formulation with the same PEG content (10 wt%) exhibited similar or worse mechanical properties compared to PEG400/PLA alone. Specifically, Young's modulus decreased further to 1.7–1.8 GPa, regardless of nanofiber content (2, 5, or 12 wt%). Similarly, stress at break remained at 33 MPa for 2% ChNFs but dropped to 22–23 MPa at higher ChNFs loadings (5 and 12 wt%). Stress at yield showed mixed behavior, staying at 23 MPa for 2% and 12% ChNFs but increasing to 34 MPa at 5% ChNFs. These findings indicate that ChNFs failed to provide reinforcement, likely due to PEG's strong affinity for chitin, which inhibited proper interfacial bonding between ChNFs and PLA, preventing effective stress transfer. Instead, PEG's dominant plasticizing effect continued to weaken the composite. Regarding elongation at break, the PEG400/ChNFs/PLA formulations exhibited an elongation identical to that of PEG/PLA, indicating that the presence of nanowhiskers regardless of amount did not further alter the ductility introduced by PEG.
The inability of PEG to facilitate reinforcement in the previous study165 contrasts with findings from another study (Table 10, entries 7–9),26 where chitin nanofibers (ChNFs) extracted from crab shells were successfully used to reinforce PLA through extrusion molding. In this case, the dispersion issue of nanofibers within the PLA matrix was addressed using three different pretreatment methods: water pretreatment, polyethylene glycol (PEG) pretreatment, and polyethylene oxide (PEO) pretreatment.
In the first water-pretreatment method (Table 10, entry 7),26 PLA powder was added to a 1 wt% ChNF water slurry, stirred, filtered, and freeze-dried to remove water while preserving nanofiber dispersion. The dried ChNF/PLA mixture was mechanically broken down before compounding and extrusion. Increasing ChNFs content from 10 to 40 wt% led to improvements, reaching 41.2 MPa, a 180% increase compared to neat PLA. Similarly, the bending modulus increased to 4153 MPa, a 144% improvement, and impact toughness rose from 8.4 J m−1 in neat PLA to 35 J m−1. Storage modulus was significantly improved, reaching 7.8 GPa, but remained independent of ChNF content, suggesting effective load distribution across the polymer matrix.
In the PEG pretreatment method (Table 10, entry 8),26 PEG powder was dissolved in distilled water at 2 wt% and heated to 80 °C before adding a 1 wt% ChNF aqueous slurry. The thickened mixture was freeze-dried, broken down, and compounded with PLA. The results were comparable to the water pretreatment method, with bending strength increasing to 37.5 MPa, bending modulus to 3550 MPa, and impact toughness to 30 J m−1 at 40 wt% ChNFs. Storage modulus improved to 6.5 GPa, again independent of ChNF content. While PEG facilitated dispersion, the absolute mechanical improvements were slightly lower than those from water pretreatment, suggesting that residual PEG at the interface might have reduced stiffness and interfacial bonding between ChNFs and PLA.
The PEO pretreatment method (Table 10, entry 9) conducted in the same manner as PEG-pretreatment26 yielded similar results to PEG, though mechanical properties were slightly lower: bending strength reached 34 MPa, bending modulus 2900 MPa, and impact toughness 28 J m−1, while storage modulus stabilized at 6.1 GPa. Unlike the previous study, where direct PEG addition hindered reinforcement by coating ChNFs and preventing stress transfer, these pretreatment strategies improved dispersion and integration within PLA. The use of pre-dissolved PEG or PEO as a dispersing medium before compounding likely reduced aggregation and enhanced interfacial adhesion.
The freeze-dried chitin nanofibers were also directly extruded with PLA granules as a control (Table 10, entry 10). In the control sample, where freeze-dried ChNFs were directly extruded with PLA, bending strength improved from 14.6 MPa to 27.5 MPa with 40 wt% ChNFs. However, bending modulus slightly decreased to 1550–1655 MPa compared to the original 1697 MPa. Impact strength increased from 8.4 J m−1 to 15 J m−1, while storage modulus reached 5 GPa.
These results indicate that although the direct extrusion method provided some improvement in strength and impact resistance, it did not achieve the same level of reinforcement as the water, PEG, or PEO pretreatment methods. The lower bending modulus suggests that without proper dispersion, the chitin nanofibers might not have been fully integrated into the PLA matrix, leading to suboptimal load transfer. The comparative study underscores the importance of pretreatment strategies in enhancing the dispersion and interfacial adhesion of ChNFs, ultimately leading to better mechanical performance in PLA composites.
4.4. Reinforcement of poly(vinyl) alcohol with nanochitin
Polyvinyl alcohol (PVA) is a linear, synthetic, nontoxic, biodegradable polymer, with a semicrystalline structure.215,216 Its physical and chemical properties vary depending on the molecular weight.217 Owing to its excellent optical properties, high storage capacity, and superior dielectric strength, PVA is widely utilized in electronics and bioengineering.218 However, its poor mechanical and thermal stability, along with limited compatibility with other polymers often necessitate doping with nanofillers to enhance its physicochemical properties for demanding applications. This includes customizing shape, size, and interactions with other polymers.219,220 In recent decades, nano-dimensional fillers – such as quantum dots, metal oxides, metal sulfides, silicates, nanotubes – have been broadly explored as candidates for the reinforcement of PVA.221 Among these, cellulose nanofillers have been extensively examined as sustainable, high-performing biofillers compatible with PVA.222 More recently, chitin nanofillers have gained increasing research interest, particularly for fabricating PVA-based nanocomposite materials such as hydrogels, beads, fibers, films, membranes, scaffolds and many more.Numerous studies have reported the reinforcement of PVA with chitin nanomaterials, particularly highlighting improvements in tensile properties as summarized in Table 11, entries 1–18. However, direct comparisons between these studies are challenging due to the use of PVA with different MWs (DP 550–2700) and alcoholysis degree, which significantly influence the material's mechanical properties. Despite this variability, the results consistently show that nanochitin incorporation enhances mechanical performance, attributed to the strong interfacial interactions between chitin nanofillers and the PVA matrix.
| Polyvinyl alcohol (PVA) | |||||
|---|---|---|---|---|---|
| No. | Polymers | Property affected | Composition | Value | Ref. |
| a The dried ChNWs/PVA composite films were swollen and then stretched at various draw ratios ranging from 1 to 3, using a stretching speed of 10 mm min−1. Here, “x” in x-ChNWs/PVA denotes the specific stretch ratio applied to the films, while maintaining a constant ChNWs/PVA composition. b Four distinct films were created: MMT/PVA, MMT/ChNWs, PVA/ChNWs, and ChNWs/MMT/PVA and the properties of each film were then compared. | |||||
| 1 | Polyvinyl alcohol (PVA, 99% hydrolyzed, DP = 1700)/ChNFs | Ultimate tensile strength, MPa | ChNFs | 163 | 16 |
| PVA | 65 | ||||
| ChNFs/PVA = 60/40 | 127 | ||||
| Young's modulus, MPa | ChNFs | 7160 | |||
| PVA | 2500 | ||||
| ChNFs/PVA = 60/40 | 5700 | ||||
| Elongation at break, % | ChNFs | 4.0 | |||
| PVA | 22.0 | ||||
| ChNFs/PVA = 60/40 | 4.4 | ||||
| 2 | Polyvinyl alcohol (PVA, DP = 2500) and β-ChNWs | Tensile strength, MPa (dry state) | β-ChNWs | 5.00 ± 0.12 | 149 |
| PVA | 2.17 ± 0.04 | ||||
| β-ChNWs/PVA = 90/10 | 4.46 ± 0.08 | ||||
| β-ChNWs/PVA = 80/20 | 3.04 ± 0.09 | ||||
| β-ChNWs/PVA = 70/30 | 5.17 ± 0.16 | ||||
| β-ChNWs/PVA = 60/40 | 4.09 ± 0.09 | ||||
| β-ChNWs/PVA = 50/50 | 3.18 ± 0.05 | ||||
| β-ChNWs/PVA = 40/60 | 2.53 ± 0.02 | ||||
| β-ChNWs/PVA = 30/70 | 1.75 ± 0.02 | ||||
| Elongation at break, % (dry state) | β-ChNWs | 5.8 ± 0.15 | |||
| PVA | 487 ± 11.50 | ||||
| β-ChNWs/PVA = 90/10 | 13.1 ± 0.31 | ||||
| β-ChNWs/PVA = 80/20 | 13.6 ± 0.42 | ||||
| β-ChNWs/PVA = 70/30 | 26.1 ± 0.94 | ||||
| β-ChNWs/PVA = 60/40 | 41.1 ± 0.62 | ||||
| β-ChNWs/PVA = 50/50 | 87.5 ± 1.97 | ||||
| β-ChNWs/PVA = 40/60 | 77.4 ± 2.05 | ||||
| β-ChNWs/PVA = 30/70 | 129.4 ± 5.97 | ||||
| Tensile strength, MPa (wet state) | β-ChNWs | 0.4 ± 0.01 | |||
| PVA | 3.10 ± 0.01 | ||||
| β-ChNWs/PVA = 90/10 | 0.9 ± 0.02 | ||||
| β-ChNWs/PVA = 80/20 | 0.35 ± 0.01 | ||||
| β-ChNWs/PVA = 70/30 | 0.38 ± 0.01 | ||||
| β-ChNWs/PVA = 60/40 | 0.37 ± 0.01 | ||||
| β-ChNWs/PVA = 50/50 | 0.25 ± 0.01 | ||||
| β-ChNWs/PVA = 40/60 | 0.12 ± 0.00 | ||||
| β-ChNWs/PVA = 30/70 | 0.24 ± 0.01 | ||||
| Elongation at break, % (wet state) | β-ChNWs | 20 ± 0.65 | |||
| PVA | 423 ± 15.64 | ||||
| β-ChNWs/PVA = 90/10 | 15.6 ± 0.42 | ||||
| β-ChNWs/PVA = 80/20 | 21.3 ± 0.12 | ||||
| β-ChNWs/PVA = 70/30 | 29.2 ± 0.12 | ||||
| β-ChNWs/PVA = 60/40 | 45.4 ± 1.24 | ||||
| β-ChNWs/PVA = 50/50 | 47.2 ± 1.35 | ||||
| β-ChNWs/PVA = 40/60 | 44.3 ± 1.02 | ||||
| β-ChNWs/PVA = 30/70 | 77.1 ± 2.34 | ||||
| 3 | Polyvinyl alcohol (PVA, DP = 4300)/ChNWs | Stress, MPa | Chitin | 4.0 | 123 |
| ChNWs/PVA = 76.92/23.08 | 9.0 | ||||
| ChNWs/PVA = 83.33/16.66 | 7.0 | ||||
| ChNWs/PVA = 90.91/9.09 | 6.5 | ||||
| ChNWs/PVA = 95.24/4.76 | 5.5 | ||||
| ChNWs/PVA = 99.01/0.99 | 5.0 | ||||
| Strain, % | Chitin | 0.20 | |||
| ChNWs/PVA = 76.92/23.08 | 0.34 | ||||
| ChNWs/PVA = 83.33/16.66 | 0.30 | ||||
| ChNWs/PVA = 90.91/9.09 | 0.25 | ||||
| ChNWs/PVA = 95.24/4.76 | 0.20 | ||||
| ChNWs/PVA = 99.01/0.99 | 0.15 | ||||
| 4 | Polyvinyl alcohol (PVA)/β-ChNWs | Tensile strength, MPa | Pure β-ChNWs | 5.1 | 150 |
| PVA | 0.7 | ||||
| β-ChNWs/PVA = 20/80 | 2.0 | ||||
| β-ChNWs/PVA = 40/60 | 2.2 | ||||
| β-ChNWs/PVA = 50/50 | 2.5 | ||||
| β-ChNWs/PVA = 60/40 | 2.7 | ||||
| β-ChNWs/PVA = 80/20 | 5.1 | ||||
| Elongation at break, % | Pure β-ChNWs | 2.9 | |||
| PVA | 165.2 | ||||
| β-ChNWs/PVA = 20/80 | 15.0 | ||||
| β-ChNWs/PVA = 40/60 | 5.0 | ||||
| β-ChNWs/PVA = 50/50 | 4.0 | ||||
| β-ChNWs/PVA = 60/40 | 3.0 | ||||
| β-ChNWs/PVA = 80/20 | 2.9 | ||||
| 5 | Polyvinylalcohol (PVA)/partially deacetylated α-ChNWs | Compressive fracture stress, MPa | PVA | 1.55 | 133 |
| ChNWs/PVA = 20/80 | 3.70 | ||||
| ChNWs/PVA = 30/70 | 6.22 | ||||
| ChNWs/PVA = 40/60 | 26.59 | ||||
| ChNWs/PVA = 50/50 | <20.22 | ||||
| ChNWs/PVA = 60/40 | <20.22 | ||||
| 6 | Poly(vinyl alcohol) (PVA, DP = 1799)/ionic shielded chitin nanowhiskers (IsChNWs), followed by alkali treatment (at) | Elongation at break, % | PVA | 9.6 ± 2.2 | 40 |
| IsChNWs/PVA = 80/20 | 1.0 ± 0.2 | ||||
| IsChNWs/PVA = 70/30 | 3.7 ± 0.5 | ||||
| IsChNWs/PVA = 60/40 | 13.2 ± 0.7 | ||||
| IsChNWs/PVA = 50/50 | 78.0 ± 2.5 | ||||
| ChNWs/PVA = 70/30 | 4.7 ± 0.9 | ||||
| At-ChNWs/PVA = 70/30 | 4.1 ± 1.3 | ||||
| At-IsChNWs/PVA = 70/30 | 4.1 ± 0.5 | ||||
| Tensile strength, MPa | PVA | 52.1 ± 1.4 | |||
| IsChNWs/PVA = 80/20 | 30.3 ± 1.6 | ||||
| IsChNWs/PVA = 70/30 | 39.4 ± 1.2 | ||||
| IsChNWs/PVA = 60/40 | 34.3 ± 1.1 | ||||
| IsChNWs/PVA = 50/50 | 32.9 ± 1.2 | ||||
| ChNWs/PVA = 70/30 | 33.4 ± 1.7 | ||||
| At-ChNWs/PVA = 70/30 | 50.4 ± 0.6 | ||||
| At-IsChNWs/PVA = 70/30 | 67.0 ± 1.2 | ||||
| Young's modulus, MPa | PVA | 2464.9 ± 391.7 | |||
| IsChNWs/PVA = 80/20 | 4334.0 ± 153.5 | ||||
| IsChNWs/PVA = 70/30 | 3619.6 ± 351.7 | ||||
| IsChNWs/PVA = 60/40 | 1092.3 ± 61.3 | ||||
| IsChNWs/PVA = 50/50 | 870.9 ± 68.4 | ||||
| ChNWs/PVA = 70/30 | 1895 ± 187.0 | ||||
| At-ChNWs/PVA = 70/30 | 3588.8 ± 169.5 | ||||
| At-IsChNWs/PVA = 70/30 | 5307.1 ± 106.2 | ||||
| 7 | Polyvinyl alcohol (PVA, DP = 1799)/ionic shielded chitin nanowhiskers (IsChNWs), followed by alkali treatment (at) | Elongation at break, % | At-IsCHNWs/PVA | 9.3 ± 0.7 | 80 |
| 1.0a-at-IsChNWs/PVA = 60/40 | 7.2 ± 2.2 | ||||
| 1.5-at-IsChNWs/PVA = 60/40 | 20.3 ± 2.3 | ||||
| 2.0-at-IsChNWs/PVA = 60/40 | 31.6 ± 4.1 | ||||
| 2.5-at-IsChNWs/PVA = 60/40 | 28.0 ± 5.6 | ||||
| 3.0-at-IsChNWs/PVA = 60/40 | 40.7 ± 5.6 | ||||
| Young's modulus, MPa | At-IsChNWs/PVA | 1133.4 ± 31.7 | |||
| 1.0-at-IsChNWs/PVA = 60/40 | 2830.1 ± 102.5 | ||||
| 1.5-at-IsChNWs/PVA = 60/40 | 983.7 ± 220.1 | ||||
| 2.0-at-IsChNWs/PVA = 60/40 | 1003.5 ± 108.9 | ||||
| 2.5-at-IsChNWs/PVA = 60/40 | 1246.2 ± 154.9 | ||||
| 3.0-at-IsChNWs/PVA = 60/40 | 1155.2 ± 89.7 | ||||
| Tensile strength, MPa | At-IsChNWs/PVA | 38.6 ± 1.0 | |||
| 1.0-at-IsChNWs/PVA = 60/40 | 46.3 ± 1.8 | ||||
| 1.5-at-IsChNWs/PVA = 60/40 | 65.9 ± 2.7 | ||||
| 2.0-at-IsChNWs/PVA = 60/40 | 76.2 ± 2.6 | ||||
| 2.5-at-IsChNWs/PVA = 60/40 | 94.4 ± 3.0 | ||||
| 3.0-at-IsChNWs/PVA = 60/40 | 115.2 ± 9.3 | ||||
| 8 | Polyvinyl alcohol (PVA)/α-ChNWs | Tensile strength, MPa | PVA | 55.5 | 95 |
| ChNWs/PVA = 0.74/99.26 | 65.0 | ||||
| ChNWs/PVA = 1.48/98.52 | 70.0 | ||||
| ChNWs/PVA = 2.96/97.34 | 83.3 | ||||
| ChNWs/PVA = 7.40/92.60 | 82.0 | ||||
| ChNWs/PVA = 14.80/85.20 | 81.0 | ||||
| ChNWs/PVA = 22.20/77.80 | 81.0 | ||||
| ChNWs/PVA = 29.60/70.40 | 80.0 | ||||
| Elongation at break, % | PVA | 15.0 | |||
| ChNWs/PVA = 0.74/99.26 | 12.0 | ||||
| ChNWs/PVA = 1.48/98.52 | 9.8 | ||||
| ChNWs/PVA = 2.96/97.34 | 7.8 | ||||
| ChNWs/PVA = 7.40/92.60 | 7.8 | ||||
| ChNWs/PVA = 14.8/85.20 | 6.0 | ||||
| ChNWs/PVA = 22.2/77.80 | 6.5 | ||||
| ChNWs/PVA = 29.6/70.40 | 5.0 | ||||
| 9 | Polyvinyl alcohol (PVA, DP = 1773)/α-ChNWs | Tensile strength, MPa | PVA | 3.94 | 96 |
| ChNWs/PVA = 2.5/97.5 | 5.92 | ||||
| ChNWs/PVA = 5.0/95.0 | 4.50 | ||||
| ChNWs/PVA = 7.5/92.5 | 4.00 | ||||
| 10 | Polyvinyl alcohol (PVA, DP = ∼1705)/α-ChNWs (electrospun mats) | Tensile strength, MPa | PVA | 4.3 | 49 |
| ChNWs/PVA = 2.55/97.45 | 4.9 | ||||
| ChNWs/PVA = 5.11/94.89 | 5.7 | ||||
| ChNWs/PVA = 7.66/92.34 | 5.0 | ||||
| ChNWs/PVA = 10.11/89.89 | 5.1 | ||||
| ChNWs/PVA = 12.76/87.24 | 4.2 | ||||
| ChNWs/PVA = 15.17/84.83 | 4.0 | ||||
| ChNWs/PVA = 17.74/82.26 | 3.8 | ||||
| ChNWs/PVA = 25.38/74.62 | 3.4 | ||||
| Young's modulus, MPa | PVA | 35 | |||
| ChNWs/PVA = 2.55/97.45 | 170 | ||||
| ChNWs/PVA = 5.11/94.89 | 140 | ||||
| ChNWs/PVA = 7.66/92.34 | 230 | ||||
| ChNWs/PVA = 10.11/89.89 | 255 | ||||
| ChNWs/PVA = 12.76/87.24 | 270 | ||||
| ChNWs/PVA = 15.17/84.83 | 280 | ||||
| ChNWs/PVA = 17.74/82.26 | 275 | ||||
| ChNWs/PVA = 25.38/74.62 | 220 | ||||
| Elongation at break, % | PVA | 60 | |||
| ChNWs/PVA = 2.55/97.45 | 32 | ||||
| ChNWs/PVA = 5.11/94.89 | 53 | ||||
| ChNWs/PVA = 7.66/92.34 | 20 | ||||
| ChNWs/PVA = 10.11/89.89 | 11 | ||||
| ChNWs/PVA = 12.76/87.24 | 24 | ||||
| ChNWs/PVA = 15.17/84.83 | 5 | ||||
| ChNWs/PVA = 17.74/82.26 | 4 | ||||
| ChNWs/PVA = 25.38/74.62 | 6 | ||||
| 11 | Polyvinyl alcohol (PVA, DP = ∼1705)/α-ChNWs (solution-casted films) | Tensile strength, MPa | PVA | 20 | 49 |
| ChNWs/PVA = 2.55/97.45 | 32 | ||||
| ChNWs/PVA = 5.11/94.89 | 36 | ||||
| ChNWs/PVA = 7.66/92.34 | 30 | ||||
| ChNWs/PVA = 10.11/89.89 | 35 | ||||
| ChNWs/PVA = 12.76/87.24 | 37 | ||||
| ChNWs/PVA = 15.17/84.83 | 34 | ||||
| ChNWs/PVA = 17.74/82.26 | 45 | ||||
| ChNWs/PVA = 25.38/74.62 | 34 | ||||
| Young's modulus, MPa | PVA | 50 | |||
| ChNWs/PVA = 2.55/97.45 | 150 | ||||
| ChNWs/PVA = 5.11/94.89 | 300 | ||||
| ChNWs/PVA = 7.66/92.34 | 350 | ||||
| ChNWs/PVA = 10.11/89.89 | 650 | ||||
| ChNWs/PVA = 12.76/87.24 | 725 | ||||
| ChNWs/PVA = 15.17/84.83 | 850 | ||||
| ChNWs/PVA = 17.74/82.26 | 1300 | ||||
| ChNWs/PVA = 25.38/74.62 | 1500 | ||||
| Elongation at break, % | PVA | 300 | |||
| ChNWs/PVA = 2.55/97.45 | 265 | ||||
| ChNWs/PVA = 5.11/94.89 | 290 | ||||
| ChNWs/PVA = 7.66/92.34 | 275 | ||||
| ChNWs/PVA = 10.11/89.89 | 235 | ||||
| ChNWs/PVA = 12.76/87.24 | 230 | ||||
| ChNWs/PVA = 15.17/84.83 | 225 | ||||
| ChNWs/PVA = 17.74/82.26 | 220 | ||||
| ChNWs/PVA = 25.38/74.62 | 105 | ||||
| 12 | Polyvinyl alcohol (PVA)/α-ChNWs | Storage modulus, GPa (25 °C) | PVA | 1.66 | 94 |
| ChNWs/PVA = 3/97 | 3.35 | ||||
| ChNWs/PVA = 5/95 | 3.77 | ||||
| ChNWs/PVA = 10/90 | 4.48 | ||||
| ChNWs/PVA = 15/85 | 4.90 | ||||
| ChNWs/PVA = 20/80 | 6.16 | ||||
| ChNWs/PVA = 30/70 | 6.18 | ||||
| Storage modulus, GPa (150 °C) | PVA | 0.14 | |||
| ChNWs/PVA = 3/97 | 0.22 | ||||
| ChNWs/PVA = 5/95 | 0.36 | ||||
| ChNWs/PVA = 10/90 | 0.47 | ||||
| ChNWs/PVA = 15/85 | 0.57 | ||||
| ChNWs/PVA = 20/80 | 0.86 | ||||
| ChNWs/PVA = 30/70 | 1.18 | ||||
| Retention ratio, % | PVA | 8 | |||
| ChNWs/PVA = 3/97 | 7 | ||||
| ChNWs/PVA = 5/95 | 10 | ||||
| ChNWs/PVA = 10/90 | 11 | ||||
| ChNWs/PVA = 15/85 | 12 | ||||
| ChNWs/PVA = 20/80 | 14 | ||||
| ChNWs/PVA = 30/70 | 19 | ||||
| 13 | Polyvinyl alcohol (PVA, DP = 591)/α-ChNWs | Young modulus, MPa | Sandwich-like structure, ratio not shown | 52 | |
| PVA | 207.41 | ||||
| Heat treated PVA/ChNWs | 311.23 | ||||
| Elongation at break, % | PVA | 85.4 | |||
| PVA/ChNWs | 51.0 | ||||
| Heat treated PVA/ChNWs | 46.0 | ||||
| Tensile strength, MPa | PVA | 42.5 | |||
| PVA/ChNWs | 46.5 | ||||
| Heat treated PVA/ChNWs | 48.0 | ||||
| Water-resistant pressure, mm | PVA | 375 | |||
| PVA/ChNWs | 385 | ||||
| Heat treated PVA/ChNWs | 768.72 | ||||
| Oxygen transmission rate, cc m−2 day−1 | PVA | 6.32 | |||
| PVA/ChNWs | 2.75 | ||||
| Heat treated PVA/ChNWs | 0.20 | ||||
| 14 | Boron nitride modified with silane (BNSi)/ChNWs/PVA | Electrical resistance, Ω | PVA | 5.7 × 1010 | 81 |
| BNSi–ChNWs/PVA = 4.35/21.74/73.91 | 5.5 × 109 | ||||
| Thermal conductivity, W m−1 K−1 | PVA | 0.353 | |||
| BNSi–ChNWs/PVA = 4.35/21.74/73.91 | 1.034 | ||||
| 15 | Boron nitride modified with poly-vinylpyrrolidone (BNPVP)/ChNWs/PVA | Electrical resistance, Ω | PVA | 5.70 × 1010 | |
| BNPVP–ChNWs/PVA = 4.35/21.74/73.91 | 1.57 × 109 | ||||
| Thermal conductivity, W m−1 K−1 | PVA | 0.353 | |||
| BNPVP–ChNWs/PVA = 4.35/21.74/73.91 | 1.049 | ||||
| 16 | Montmorillonite/PVA/ChNWsb | Tensile strength, MPa | MMT/PVA = 50/50 | 68.99 ± 5.26 | 83 |
| MMT/ChNWs = 50/50 | 10.89 ± 2.10 | ||||
| PVA/ChNWs = 50/50 | 45.37 ± 6.14 | ||||
| ChNWs/MMT/PVA = 33.33/33.33/33.33 | 263.50 ± 4.01 | ||||
| Elongation at break, % | MMT/PVA = 50/50 | 1.54 ± 0.34 | |||
| MMT/ChNWs = 50/50 | 0.41 ± 0.12 | ||||
| PVA/ChNWs = 50/50 | 0.26 ± 0.03 | ||||
| ChNWs/MMT/PVA = 33.33/33.33/33.33 | 0.60 ± 0.03 | ||||
| Young's modulus, GPa | MMT/PVA = 50/50 | 4.48 ± 1.12 | |||
| MMT/ChNWs = 50/50 | 2.66 ± 0.97 | ||||
| PVA/ChNWs = 50/50 | 17.45 ± 1.04 | ||||
| ChNWs/MMT/PVA = 33.33/33.33/33.33 | 43.92 ± 1.28 | ||||
| Toughness, J m−3 | MMT/PVA = 50/50 | 61.34 | |||
| MMT/ChNWs = 50/50 | 2.33 | ||||
| PVA/ChNWs = 50/50 | 5.88 | ||||
| ChNWs/MMT/PVA = 33.33/33.33/33.33 | 102.15 | ||||
| Transmittance, % | MMT/PVA = 50/50 | 20 | |||
| MMT/ChNWs = 50/50 | 10 | ||||
| PVA/ChNWs = 50/50 | 80 | ||||
| ChNWs/MMT/PVA = 33.33/33.33/33.33 | 40 | ||||
| 17 | PVA (DP = ∼2727) crosslinked with borax (PB)/acid hydrolyzed ChNCs (AChNCs)/TEMPO-oxidized ChNCs (TChNCs) | Tensile strength, kPa | Borax crosslinked PVA (PB) | 1.3 | 223 |
| AChNCs/PB = 0.5/99.5 | 13.8 | ||||
| AChNCs/PB = 1.0/99.0 | 18.3 | ||||
| AChNCs/PB = 1.5/98.5 | 31.5 | ||||
| AChNCs/PB = 2.0/98.0 | 54.3 | ||||
| TChNCs/PB = 0.5/99.5 | 3.3 | ||||
| TChNCs/PB = 1.0/99.0 | 5.0 | ||||
| TChNCs/PB = 1.5/98.5 | 6.8 | ||||
| TChNCs/PB = 2.0/98.0 | 9.3 | ||||
| Elongation at break, % | PB | 1426 | |||
| AChNCs/PB = 0.5/99.5 | 746 | ||||
| AChNCs/PB = 1.0/99.0 | 658 | ||||
| AChNCs/PB = 1.5/98.5 | 582 | ||||
| AChNCs/PB = 2.0/98.0 | 424 | ||||
| TChNCs/PB = 0.5/99.5 | 1132 | ||||
| TChNCs/PB = 1.0/99.0 | 787 | ||||
| TChNCs/PB = 1.5/98.5 | 745 | ||||
| TChNCs/PB = 2.0/98.0 | 542 | ||||
| Work of fracture, kJ m−3 | PB | 1074 | |||
| AChNCs/PB = 0.5/99.5 | 5918 | ||||
| AChNCs/PB = 1.0/99.0 | 7786 | ||||
| AChNCs/PB = 1.5/98.5 | 8989 | ||||
| AChNCs/PB = 2.0/98.0 | 10995 |
||||
| TChNCs/PB = 0.5/99.5 | 2721 | ||||
| TChNCs/PB = 1.0/99.0 | 3184 | ||||
| TChNCs/PB = 1.5/98.5 | 3732 | ||||
| TChNCs/PB = 2.0/98.0 | 4541 | ||||
| Self-healing efficiency, % | PB | 99.4 | |||
| AChNCs/PB = 1.0/99.0 | 90.4 | ||||
| AChNCs/PB = 1.5/98.5 | 87.8 | ||||
| TChNCs/PB = 1.0/99.0 | 96.0 | ||||
| TChNCs/PB = 1.5/98.5 | 89.5 | ||||
| Conductivity, S m−1 | PB | 2.02 × 10−5 | |||
| AChNCs/PB = 1.0/99.0 | 2.08 × 10−6 | ||||
| AChNCs/PB = 1.5/98.5 | 1.51 × 10−6 | ||||
| TChNCs/PB = 1.0/99.0 | 6.94 × 10−6 | ||||
| TChNCs/PB = 1.5/98.5 | 1.25 × 10−5 | ||||
It has also been reported that nanochitin can function both as a primary component in PVA-based composites (Table 11, entries 1–7), and as a reinforcement filler (Table 11, entries 8–17), depending on the desired material properties. Generally, when added at lower concentrations, nanochitin primarily enhances tensile strength and mechanical properties while still allowing PVA to retain flexibility and processability, whereas at higher concentrations, nanochitin contributes significantly to the composite's rigidity, strength, and structural integrity, making it the dominant phase rather than just a reinforcing agent.80,149 This dual functionality allows nanochitin–PVA composites to be tailored for various applications, ranging from flexible, high-strength materials to rigid, high-performance biopolymers, depending on the composition.
Previous studies indicated that when chitin function as the primary component in PVA-based composites, higher concentrations of nanochitin improve tensile strength, whereas the addition of PVA reduced material rigidity.150 This implies PVA, rather than acting as the matrix material, served as an adhesive to combine the ChNWs.80 Optimal tensile strength was observed at nanochitin concentrations between 60–80% in all cases, with lower or higher concentrations yielding less favorable results (Table 11, entries 1–7). When incorporating PVA at higher concentrations, tensile strength of the composites decreased to levels comparable to or even lower than pure PVA (Table 11, entry 2). It is also important to note that the reinforcement of PVA composites with ChNFs (Table 11, entry 1) is substantially amplified compared to that with ChNWs (Table 11, entries 2–7). While tensile strength and Young's modulus increased significantly compared to neat PVA, elongation showed only a modest increase at a 60 wt% nanochitin concentration.
Other studies developed novel strategies to prepare ChNWs/PVA films with improved strength and durability, where ChNWs acted as building blocks and essential components and PVA acted as adhesive.40,80 One of the strategies of the studies was introducing a two-step treatment process using salt and alkali, respectively into the mixture of ChNWs and PVA.40 It was observed that among all ionic shielded ChNWs (IsChNWs) composite films, the film with 70 wt% ionic shielded ChNWs exhibited the optimal overall performance (Table 11, entry 6). At the same ChNW load, the Young's modulus of the IsChNWs/PVA film is nearly twice that of the ChNWs/PVA film, while maintaining comparable elongation at break, suggesting that the ionic shield effect of salt greatly weakened the electrostatic repulsion on the ChNW surface, resulting in aggregation of the ChNWs in a certain extent. Furthermore, integrating the ionic shielding effect of salt and hydrogen bond remolding effect of alkali resulted in significantly improved performance, with Young's modulus and tensile strength increasing to 5307.1 ± 106.2 MPa and 67.0 ± 1.2 MPa, respectively −1.7 and 1.5 times higher those of the IsChNW/PVA film. Alkali treatment of IsChNWs/PVA films disrupted interparticle hydrogen bonds among ChNWs, allowing PVA to penetrate ChNW aggregates and fill the interspace, thereby enhancing the hydrogen bond interaction between ChNWs and PVA.40
The follow-up study further evaluated the alkali-treated IsChNWs/PVA films and tuned the film properties by developing a tensile-induced orientation and hydrogen bond reconstruction strategy80 to initiate inter-layer sliding (Table 11, entry 7). The alkali-treated IsChNWs/PVA films were first swollen in deionized water and stretched at a certain draw ratio, then soaked in alkali to reconstruct the hydrogen bonds and stabilize the orientational structure. The films, which underwent stretching and hydrogen-bond reconstruction, exhibited enhanced tensile strength and elongation from 38.6 to 115.2 MPa and 9.37% to 40.7%, respectively. Additionally, morphological analysis of these films before and after tensile failure revealed that the mechanisms responsible for strengthening and toughening were linked to intra-layer ordering and inter-layer sliding, respectively. The stacked multilayer structure and organized arrangement of the ChNWs allowed external stress to be efficiently transferred through the aligned ChNWs within the layers. This stress was then dissipated through the layering effect and inter-layer sliding, leading to a combination of reinforcing and toughening effects.80
When nanochitin was utilized specifically as a reinforcement filler, the tensile strength peaked at 3–5 wt% nanochitin concentrations of (Table 11, entries 8–10), depending on the nature of the material, the preparation method, and the material's intended brittleness and strength. Consistently, elongation at break decreased with increasing chitin content, but increased with higher PVA content when chitin was the primary substrate. Other mechanical properties, such as Young's modulus, storage modulus, and compressive fracture stress, also improved with higher nanoparticles content.
Junkasem et al. fabricated electrospun nanocomposite fibers and solvent-casted films with PVA serving as the matrix.49 This group evaluated the ChNW reinforcement effect, and the compatibility of the composite components prepared from different methods on the composite mechanical integrity (Table 11, entries 10 and 11). Although the tensile strengths of both as-spun PVA/ChNWs fiber mats and as-cast PVA/ChNWs films increased with the addition of ChNWs, the critical thresholds were distinct. The tensile strength of the as-spun fiber mats reached its threshold (5.7 ± 0.6 MPa) at approximately 5.1 wt% and decreased with further increases in ChNW content (Table 11, entry 10). In contrast, the tensile strength of the as-cast films reached its threshold (45.2 ± 3.6 MPa) at 17.7 wt% (Table 11, entry 11). A similar disparity was observed in the case of Young's modulus for both composites. The strength initially increased for the as-spun fibers, reaching a threshold at 15.2%, whereas for the as-cast films, it continued to increase with each addition of ChNWs. The author reported that the discrepancy was possibly attributed to the difference in the dimensionality of the two sample types. The observed decrease in Young's modulus of the as-spun fibers above 15.2% ChNW load was likely due to the ChNW aggregation arising from the confinement effect, which was not observed in the as-cast films, unless the thickness of the films approached the sizes of the nanosized reinforcing fillers.49
Another study focused on the preparation of PVA blends with hybrid materials, that combined ChNWs and modified boron nitride, to enhance the thermal and electrical conductivity of the final composites (Table 11, entries 14 and 15).81 Leveraging the high thermal conductivity of boron nitride to create an efficient thermal conductivity network within the polymer matrix, two modified boron nitrides – silane coupling agent-treated boron nitride (BNSi) and poly-vinylpyrrolidone-treated boron nitride (BNPVP) – were combined with ChNWs before being compounded with PVA to produce BNSi-ChNWs/PVA and BNPVP-ChNWs/PVA composites. The incorporation of the hybrid materials into the PVA matrix resulted in enhanced thermal conductivity (192.9–197.2% higher than neat PVA), with the BNPVP-ChNWs-reinforced composite outperforming the BNSi-ChNWs version (1.049 W m−1 K−1vs. 1.034 W m−1 K−1, respectively). The improvement in both reinforced composites was attributed to the enhanced interface compatibility of the hybrid materials, which allowed for more even dispersion in PVA and the formation of effective thermal conductivity networks.81 However, no significant improvements were observed in the electrical insulation performance in the composites with these hybrid materials. Instead, the electrical resistance of the reinforced composites is an order of magnitude lower than that of the neat PVA, with BNPVP-ChNW/PVA showing the lowest resistance (1.57 × 109 Ω), which was attributed to the proton-conductive nature of PVP. Meanwhile, the slightly lower electrical resistance of BNSi-ChNW/PVA indicated that silanization primarily improved the dispersion the boron nitride in the matrix and enhanced interfacial bonding between boron nitride particles, ChNWs, and PVA. Despite their relatively low electrical resistance, both PVA composites still exhibited stable electrical insulation performance for practical applications.
Wu et al.83 evaluated the degree of ChNW reinforcement on the properties of montmorillonite (MMT)/PVA films (Table 11, entry 16). Four types of films, each with the same weight percent ratio – MMT/PVA, MMT/ChNWs, PVA/ChNWs, and ChNWs/MMT/PVA – were prepared using the casting-evaporation approach, and the properties of each film were then compared. Among all the films, the ChNWs/MMT/PVA composites demonstrated superior tensile strength and Young's modulus, although toughness decreased (Table 11, entry 16). As highlighted by the authors, the enhancement was attributed to presence of a new crystalline structure arising from the intercalation of PVA and ChNW in the layer of MMT sheet through hydrogen bond and van der Waals forcing.83
Another recent study by Jiao et al.223 highlighted how the surface charge state of ChNCs significantly affects the structure and properties of borax-crosslinked PVA (PB) hydrogels (Table 11, entry 17). The study examined two types of ChNCs with opposite surface charges—positively charged ChNCs (AChNCs) prepared by acid hydrolysis and negatively charged ChNCs (TChNCs) prepared by TEMPO oxidation—as reinforcements for PB hydrogels. The results showed that among hydrogels containing from 0.5 to 2 wt% AChNCs, those with 2 wt% exhibited the highest tensile strength, a 17-fold increase compared to unreinforced PB hydrogels, while maintaining high elongation at break. A similar trend was observed with TChNCs, but their reinforcement was less effective. Additionally, both AChNCs and TChNCs improved the fracture work of the hydrogels, with AChNCs showing greater effectiveness. Maximum fracture work was achieved at a 2 wt% load for both reinforcements. The superior performance of AChNCs is associated with electrostatic attraction between the hydroxyl groups of AChNCs and borax ions, forming a second network in addition to the borax-crosslinked PVA network, which enhances mechanical performance. In contrast, the TChNCs created charge repulsion with the borax crosslinker and attracted a lesser amount of borax ions, leading to a relatively loose network structure in the composite hydrogels.223
The study also evaluated the impact of charged ChNCs on the self-healing efficiency and conductivity of the composite hydrogels. It was found that TChNCs enhance the self-healing ability and ionic conductivity more effectively than AChNCs, with values of 89.5% and 1.25 × 10−5 S m−1 compared to 87.8% and 1.51 × 10−6, respectively, at a 1 wt% load. The authors suggest that this difference is due to the energy required during healing and the mobility of borax ions in the composite hydrogels. Specifically, the strong electrostatic attraction in the AChNCs/PB hydrogel leads to more dynamic borate bonds, which increases the fracture work and requires more energy for healing, resulting in lower self-healing efficiency. Furthermore, this attraction restricts the movement of borax ions in the AChNCs/PB hydrogel, reducing conductivity. In contrast, the repulsive interaction between TChNCs and borax ions facilitates ion movement, improving both conductivity and self-healing performance.223
4.5. Reinforcement of epoxy resins with nanochitin
Epoxy resins are synthetic thermosetting polymers formed from two or more active epoxide groups. Known for their strong adhesion and effective crosslinking with various curing agents,224 epoxy resins typically cure between 0 and 180 °C without producing by-products, transforming into an insoluble 3-D network polymer.225 Despite their advantageous properties, epoxy resins face limitations, including brittleness, weak impact resistance, and poor resilience.226 To address these issues, they are often reinforced with nanofillers such as graphene oxide, carbon nanotubes, nano silica, and more.227–229 Recently, research has focused on reinforcing epoxy resins with renewable, and sustainable nanofillers like nanochitin to improve their performance.230 These nanocomposite materials have wide-ranging applications in biomedical fields, aerospace, adhesives, packaging, and coatings.231Several studies have explored the reinforcement of epoxy resin with chitin nanoparticles under various conditions (Table 12, entries 1–10). (DGEBA) based chitin nanocomposites substantially brought about an improvement in much of the mechanical properties (Table 12, entries 1–3). Wang et al.23 synthesized high-strength ChNWs/epoxy resin bisphenol A diglycidyl ether (DGEBA) nanocomposites using 1-ethyl-3-methylimidazolium acetate ([Emim][OAc]) ionic liquid for effective dispersion of ChNWs (Table 12, entry 1). Results showed a significant improvement in mechanical properties with the ChNWs incorporation from 0.75 to 2 wt%. The addition of 2 wt% led to a maximum increase of 8.3% in Young's modulus, 34.5% in tensile strength, and 80% in strain compared to the neat polymer. Energy to failure and impact strength also exhibited substantial increases of 174.7% and 91.4%, respectively, at the same critical concentration. However, upon comparing the percentage increase in all the defined properties, a minimal improvement in Young's modulus was observed, likely due to the plasticizing effect of the ionic liquid that might have counteracted the reinforcing impact of ChNWs.
| No. | Polymers | Affected properties | Composition | Value | Ref. |
|---|---|---|---|---|---|
| a The abbreviation of CA11 represents CS/PAA active hydrogen ratio 1/1. b The abbreviation of CA21 represents CS/PAA active hydrogen ratio 2/1. c The abbreviation of CE21 represents CS/PEA active hydrogen ratio 2/1. | |||||
| Epoxy resins | |||||
| Epoxy resin: bisphenol A diglycidyl ether (DGEBA) | |||||
| 1 | Epoxy resin bisphenol A diglycidyl ether (DGEBA)/ChNWs | Young's modulus, MPa | DGEBA | 948.83 ± 8.70 | 23 |
| ChNWs/DGEBA = 0.75/99.25 | 978.92 ± 20.89 | ||||
| ChNWs/DGEBA = 1.0/99.0 | 956.33 ± 15.28 | ||||
| ChNWs/DGEBA = 1.5/98.5 | 967.62 ± 7.93 | ||||
| ChNWs/DGEBA = 2.0/98.0 | 1028.06 ± 37.31 | ||||
| ChNWs/DGEBA = 3.0/97.0 | 946.70 ± 13.34 | ||||
| Tensile strength, MPa | DGEBA | 69.76 ± 2.93 | |||
| ChNWs/DGEBA = 0.75/99.25 | 84.33 ± 0.17 | ||||
| ChNWs/DGEBA = 1.0/99.0 | 89.59 ± 2.47 | ||||
| ChNWs/DGEBA = 1.5/98.5 | 91.08 ± 3.09 | ||||
| ChNWs/DGEBA = 2.0/98.0 | 93.80 ± 2.06 | ||||
| ChNWs/DGEBA = 3.0/97.0 | 83.92 ± 2.00 | ||||
| Tensile strain, mm mm−1 | DGEBA | 0.10 ± 0.0048 | |||
| ChNWs/DGEBA = 0.75/99.25 | 0.12 ± 0.0067 | ||||
| ChNWs/DGEBA = 1.0/99.0 | 0.15 ± 0.012 | ||||
| ChNWs/DGEBA = 1.5/98.5 | 0.16 ± 0.017 | ||||
| ChNWs/DGEBA = 2.0/98.0 | 0.18 ± 0.0098 | ||||
| ChNWs/DGEBA = 3.0/97.0 | 0.13 ± 0.011 | ||||
| Energy to failure, J cm−3 | DGEBA | 4.07 ± 0.36 | |||
| ChNWs/DGEBA = 0.75/99.25 | 5.90 ± 0.43 | ||||
| ChNWs/DGEBA = 1.0/99.0 | 8.08 ± 1.08 | ||||
| ChNWs/DGEBA = 1.5/98.5 | 9.14 ± 0.55 | ||||
| ChNWs/DGEBA = 2.0/98.0 | 11.18 ± 1.08 | ||||
| ChNWs/DGEBA = 3.0/97.0 | 6.43 ± 0.85 | ||||
| Impact strength, J m−2 | DGEBA | 68.17 ± 3.83 | |||
| ChNWs/DGEBA = 0.75/99.25 | 90.70 ± 6.07 | ||||
| ChNWs/DGEBA = 1.0/99.0 | 120.44 ± 1.93 | ||||
| ChNWs/DGEBA = 1.5/98.5 | 124.03 ± 2.84 | ||||
| ChNWs/DGEBA = 2.0/98.0 | 130.48 ± 1.21 | ||||
| ChNWs/DGEBA = 3.0/97.0 | 115.48 ± 3.08 | ||||
| 2 | Epoxy resin bisphenol A diglycidyl ether (DGEBA)/ChNWs | Tensile strength, MPa | DGEBA | 51.6 | 27 |
| ChNWs/DGEBA = 0.5/99.5 | 67.4 | ||||
| ChNWs/DGEBA = 1.0/99.0 | 72.8 | ||||
| ChNWs/DGEBA = 2.5/97.5 | 77.1 | ||||
| Tensile modulus, MPa | DGEBA | 2410 | |||
| ChNWs/DGEBA = 0.5/99.5 | 2733 | ||||
| ChNWs/DGEBA = 1.0/99.0 | 2760 | ||||
| ChNWs/DGEBA = 2.5/97.5 | 2812 | ||||
| Elongation at break, % | DGEBA | 3.31 | |||
| ChNWs/DGEBA = 0.5/99.5 | 5.02 | ||||
| ChNWs/DGEBA = 1.0/99.0 | 6.35 | ||||
| ChNWs/DGEBA = 2.5/97.5 | 8.27 | ||||
| Toughness J m−3 104 | DGEBA | 99.7 | |||
| ChNWs/DGEBA = 0.5/99.5 | 213.0 | ||||
| ChNWs/DGEBA = 1.0/99.0 | 309.0 | ||||
| ChNWs/DGEBA = 2.5/97.5 | 456.0 | ||||
| 3 | Epoxy resin bisphenol A diglycidyl ether (DGEBA)/ChNWs | Tensile modulus, MPa | DGEBA | 1000 | 3 |
| ChNWs/DGEBA = 0.25/99.75 | 1250 | ||||
| ChNWs/DGEBA = 0.50/99.50 | 1200 | ||||
| ChNWs/DGEBA = 0.75/99.25 | 1050 | ||||
| Tensile strength, MPa | DGEBA | 70 | |||
| ChNWs/DGEBA = 0.25/99.75 | 78 | ||||
| ChNWs/DGEBA = 0.50/99.50 | 76 | ||||
| ChNWs/DGEBA = 0.75/99.25 | 75 | ||||
| Tensile strain at failure, mm/mm | DGEBA | 0.120 | |||
| ChNWs/DGEBA = 0.25/99.75 | 0.100 | ||||
| ChNWs/DGEBA = 0.50/99.50 | 0.110 | ||||
| ChNWs/DGEBA = 0.75/99.25 | 0.124 | ||||
| Storage modulus, MPa | DGEBA | 2240 | |||
| ChNWs/DGEBA = 0.25/99.75 | 2450 | ||||
| ChNWs/DGEBA = 0.50/99.50 | 2420 | ||||
| ChNWs/DGEBA = 0.75/99.25 | 2400 | ||||
| Loss modulus, MPa | DGEBA | 50 | |||
| ChNWs/DGEBA = 0.25/99.75 | 53 | ||||
| ChNWs/DGEBA = 0.50/99.50 | 54 | ||||
| ChNWs/DGEBA = 0.75/99.25 | 63 | ||||
| Energy to tensile failure, J cm−3 | DGEBA | 5.2 | |||
| ChNWs/DGEBA = 0.25/99.75 | 5.0 | ||||
| ChNWs/DGEBA = 0.50/99.50 | 5.2 | ||||
| ChNWs/DGEBA = 0.75/99.25 | 6.5 | ||||
| Epoxy resin: sorbitol polyglycidyl ether (SPE) | |||||
| 4 | Epoxy resin sorbitol polyglycidyl ether (SPE)/ChNFs/chitosan nanofibers (CSNFs)/chitosan (CS) | Tensile strength, MPa | SPE/CS | 46.9 | 148 |
| ChNFs/SPE/CS = 33/66.25/30.75 | 49.6 | ||||
| ChNFs/SPE/CS = 5/64.88/30.11 | 37 | ||||
| ChNFs/SPE/CS = 10/61.47/28.53 | 59.3 | ||||
| Tensile modulus, MPa | SPE/CS | 2078 | |||
| ChNFs/SPE/CS = 33/66.25/30.75 | 2390 | ||||
| ChNFs/SPE/CS = 5/64.88/30.11 | 1723 | ||||
| ChNFs/SPE/CS = 10/61.47/28.53 | 1736 | ||||
| Elongation at break, % | SPE/CS | 2.78 | |||
| ChNFs/SPE/CS = 33/66.25/30.75 | 3.96 | ||||
| ChNFs/SPE/CS = 5/64.88/30.11 | 3.58 | ||||
| ChNFs/SPE/CS = 10/61.47/28.53 | 5.16 | ||||
| 5 | Epoxy resin sorbitol polyglycidyl ether (SPE)/ChNFs/chitosan (CS)/polyamidoamine (PAA) | Tensile strength, MPa | SPE/CA11 | 11.1 | 148 |
| ChNFs/SPE/CA11 = 3/53.54/43.46 | 16.1 | ||||
| ChNFs/SPE/CA11 = 5/52.44/42.56 | 15.6 | ||||
| ChNFs/SPE/CA11 = 10/49.68/40.32 | 11.7 | ||||
| Tensile modulus, MPa | SPE/CA11 | 46.2 | |||
| ChNFs/SPE/CA11 = 3/53.54/43.46 | 622.0 | ||||
| ChNFs/SPE/CA11 = 5/52.44/42.56 | 498.0 | ||||
| ChNFs/SPE/CA11 = 10/49.68/40.32 | 416.0 | ||||
| Elongation at break, % | SPE/CA11 | 11.8 | |||
| ChNFs/SPE/CA11 = 3/53.54/43.46 | 7.51 | ||||
| ChNFs/SPE/CA11 = 5/52.44/42.56 | 7.42 | ||||
| ChNFs/SPE/CA11 = 10/49.68/40.32 | 4.50 | ||||
| 6 | Epoxy resin sorbitol polyglycidyl ether (SPE)/ChNFs/chitosan (CS)/polyamidoamine (PAA) | Tensile strength, MPa | SPE/CA21 | 11.8 | 148 |
| ChNFs/SPE/CA21 = 3/53.54/43.46 | 25.5 | ||||
| ChNFs/SPE/CA21 = 5/52.44/42.56 | 17.9 | ||||
| ChNFs/SPE/CA21 = 10/49.68/40.32 | 17.1 | ||||
| Tensile modulus, MPa | SPE/CA21 | 37.3 | |||
| ChNFs/SPE/CA21 = 3/53.54/43.46 | 1051.0 | ||||
| ChNFs/SPE/CA21 = 5/52.44/42.56 | 394.0 | ||||
| ChNFs/SPE/CA21 = 10/49.68/40.32 | 753.0 | ||||
| Elongation at break, % | SPE/CA21 | 8.94 | |||
| ChNFs/SPE/CA21 = 3/53.54/43.46 | 5.12 | ||||
| ChNFs/SPE/CA21 = 5/52.44/42.56 | 5.15 | ||||
| ChNFs/SPE/CA21 = 10/49.68/40.32 | 3.39 | ||||
| 7 | Epoxy resin sorbitol polyglycidyl ether (SPE)/ChNFs/chitosan (CS)/polyetheramine (PEA) | Tensile strength, MPa | SPE/CE21 | 9.07 | 148 |
| ChNFs/SPE/CE21 = 3/53.54/43.46 | 14.10 | ||||
| ChNFs/SPE/CE21 = 5/52.44/42.56 | 18.30 | ||||
| ChNFs/SPE/CE21 = 10/49.68/40.32 | 11.50 | ||||
| Tensile modulus, MPa | SPE/CE21 | 36.8 | |||
| ChNFs/SPE/CE21 = 3/53.54/43.46 | 453.0 | ||||
| ChNFs/SPE/CE21 = 5/52.44/42.56 | 676.0 | ||||
| ChNFs/SPE/CE21 = 10/49.68/40.32 | 147.0 | ||||
| Elongation at break, % | SPE/CE21 | 7.03 | |||
| ChNFs/SPE/CE21 = 3/53.54/43.46 | 4.77 | ||||
| ChNFs/SPE/CE21 = 5/52.44/42.56 | 4.20 | ||||
| ChNFs/SPE/CE21 = 10/49.68/40.32 | 3.92 | ||||
| 8 | Epoxy resin (sorbitol poly glycidyl ether (SPE))/ChNFs/lysine (Lys) | Tensile modulus, MPa | SPE/Lys | 855 | 161 |
| ChNFs/SPE/Lys = 3.00/82.94/14.07 | 1257 | ||||
| ChNFs/SPE/Lys = 5.00/81.23/13.78 | 537 | ||||
| ChNFs/SPE/Lys = 10.00/76.95/13.05 | 220 | ||||
| Tensile strength, MPa | SPE/Lys | 26.7 | |||
| ChNFs/SPE/Lys = 3.00/82.94/14.07 | 45.6 | ||||
| ChNFs/SPE/Lys = 5.00/81.23/13.78 | 33.5 | ||||
| ChNFs/SPE/Lys = 10.00/76.95/13.05 | 28.7 | ||||
| Elongation at break, % | SPE/Lys | 2.1 | |||
| ChNFs/SPE/Lys = 3.00/82.94/14.07 | 3.9 | ||||
| ChNFs/SPE/Lys = 5.00/81.23/13.78 | 7.9 | ||||
| ChNFs/SPE/Lys = 10.00/76.95/13.05 | 5.3 | ||||
| 9 | Epoxy resin (sorbitol poly glycidyl ether (SPE))/ChNFs/arginine (Arg) | Tensile modulus, MPa | SPE/Arg | 422 | 161 |
| ChNFs/SPE/Arg = 3.00/84.79/12.21 | 263 | ||||
| ChNFs/SPE/Arg = 5.00/83.05/11.95 | 64 | ||||
| ChNFs/SPE/Arg = 10.00/78.68/11.32 | 505 | ||||
| Tensile strength, MPa | SPE/Arg | 8.5 | |||
| ChNFs/SPE/Arg = 3.00/84.79/12.21 | 4 | ||||
| ChNFs/SPE/Arg = 5.00/83.05/11.95 | 2.2 | ||||
| ChNFs/SPE/Arg = 10.00/78.68/11.32 | 9.1 | ||||
| Elongation at break, % | SPE/Arg | 3.3 | |||
| ChNFs/SPE/Arg = 3.00/84.79/12.21 | 3.6 | ||||
| ChNFs/SPE/Arg = 5.00/83.05/11.95 | 10.6 | ||||
| ChNFs/SPE/Arg = 10.00/78.68/11.32 | 11.4 | ||||
| 10 | Epoxy resin (sorbitol poly glycidyl ether (SPE))/ChNFs/cysteine (Cys) | Tensile modulus, MPa | SPE/Cys | 584 | 161 |
| ChNFs/SPE/Cys = 3.00/84.79/12.21 | 30 | ||||
| ChNFs/SPE/Cys = 5.00/83.05/11.95 | 128 | ||||
| ChNFs/SPE/Cys = 10.00/78.68/11.32 | 236.8 | ||||
| Tensile strength, MPa | SPE/Cys | 10 | |||
| ChNFs/SPE/Cys = 3.00/84.79/12.21 | 4.8 | ||||
| ChNFs/SPE/Cys = 5.00/83.05/11.95 | 8.45 | ||||
| ChNFs/SPE/Cys = 10.00/78.68/11.32 | 7.99 | ||||
| Elongation at break, % | SPE/Cys | 19.5 | |||
| ChNFs/SPE/Cys = 3.00/84.79/12.21 | 30.7 | ||||
| ChNFs/SPE/Cys = 5.00/83.05/11.95 | 27.74 | ||||
| ChNFs/SPE/Cys = 10.00/78.68/11.32 | 11.8 | ||||
A similar trend was observed by Wang et al.27 where ChNWs were loaded at concentrations ranging from 0.5 to 2.5 wt%. The properties such as tensile strength, modulus, elongation at break, and toughness showed remarkable improvements (49%, 16%, 150%, and 357%, respectively) up to 2.5 wt% of ChNWs (Table 12, entry 2). Comparing these studies,23,27 it can be inferred that beyond 3 wt%, all properties began to decline,23 highlighting the optimal ChNW concentration at around 2 to 2.5 wt%.
On the other hand, Anwer et al.3 studied the effects of ChNWs in epoxy resins cured with triethylenetetramine (TETA), with ChNW loadings ranging from 0.25 to 0.75 wt% (Table 12, entry 3). This group observed significant increases in tensile modulus, tensile strength, and storage modulus, with 0.25 wt% ChNWs achieving the highest improvements. The addition of ChNWs resulted in a stable covalent network among the epoxy, fillers, and hardeners (e.g., TETA), providing high strength without immediate failure.27
In studies involving bio-based sorbitol polyglycidyl ether (SPE) epoxy resins, tensile properties such as tensile strength, modulus, and elongation at break were investigated before and after the addition of ChNFs, under the influence of several curing agents at different conditions (Table 12, entries 4–10).148,161 Shibata et al.148 fabricated bio-nanocomposites composed of SPE, ChNFs, and chitosan (CS) and/or a commercial water-soluble polyamidoamine (PAA) or polyetheramine (PEA) as amine-type hardeners via the casting or compression molding method (Table 12, entries 4–7). They synthesized the nanocomposite resins by curing ChNFs/SPE with CS alone, or with CS-PAA (CA) and CS-PEA (CE) under different active hydrogen ratios. The CS/PAA was evaluated at two ratios, i.e., 1:1 (CA11) and 2:1 (CA21), while CS/PEA was assessed only at 2:1 ratio (CE21). For the cured resins without ChNFs, SPE-CS exhibited higher tensile strength and modulus (46.9 MPa and 2078 MPa, respectively) compared to the other three resins (11.1 MPa and 46.2 MPa for SPE-CA11, 11.8 MPa and 37.3 MPa for SPE-CA21, and 9.07 MPa and 36.8 MPa for SPE-CE21). However, SPE-CS showed a much lower elongation at break, indicating that the CS-cured SPE has higher rigidity, while the PAA- or PEA-cured SPE shows greater flexibility. Adding ChNFs into all the bio-nanocomposite resins significantly improved their strength and rigidity with tensile strength and modulus peaking at 3 wt% ChNFs for most SPE-cured resins, except for the CS-PEA system, which reached its threshold at 5 wt% ChNFs. Among all the ChNWs/SPE composites with CA and CE curing agents, the ChNFs/SPE/CA21 nanocomposites showed the highest values for tensile strength (25.5 MPa) and modulus (1051 MPa) at 3 wt% ChNFs. Meanwhile, the ChNFs/SPE/CS composites showed even higher values of strength (49.6 MPa) and modulus (2390 MPa). However, elongation at break decreased consistently across all ChNFs/SPE-CA and ChNFs/SPE-CE composites. In contrast, the elongation at break increased in the ChNFs/SPE/CS resins with all ChNFs additions (up to 10 wt%). Upon comparing all the properties, the addition of ChNFs acted as an efficient reinforcing agent, while CS served as a more effective hardener, providing high rigidity, and PAA enhanced brittleness.148
Further studies by the same authors investigated the use amino acids including lysine (Lys), arginine (Arg), and cysteine (Cys), as curing agents for SPE based composites (Table 12, entries 8–10).161 The results indicated that, except for ChNFs/SPE/Lys bionanocomposites, no reinforcement effect was observed in ChNFs/SPE-Arg and ChNFs/SPE-Cys systems. Specifically, the tensile strength (26.7 MPa) and modulus (855 MPa) of neat SPE/Lys composites improved with the addition of ChNFs, reaching 45.6 MPa and 1257 MPa, respectively, at a concentration of 3 wt% (Table 12, entry 8). Nevertheless, the addition of ChNFs significantly enhanced the elongation at break in all nanocomposites, reaching peak improvement at 5 wt% for ChNFs/SPE/Lys and ChNFs/SPE-Cys (Table 12, entries 8 and 10), and at 10 wt% for ChNFs/SPE-Arg (Table 12, entry 9), implying the reduction in crosslink density.
4.6. Reinforcement of other plastics with nanochitin
The incorporation of nanochitin into PCL resulted in only a marginal improvement in tensile properties, regardless of whether the casting and evaporation method or hot pressing was used for composite fabrication (Table 13, entry 1).97 However, a slight increase in tensile strength was observed with higher nanochitin concentrations, with tensile values increasing from 8.50 Pa for neat PCL to 8.86 Pa and 8.98 Pa for composites containing 5 wt% and 10 wt% chitin nanowhiskers (ChNW), respectively. Similarly, the incorporation of ChNWs into poly(S-co-BuA) led to only a minor improvement in tensile modulus, independent of the fabrication method used (Table 13, entry 2).97 For cast-evaporated poly(S-co-BuA), the tensile modulus increased from 5.75 Pa to 6.75 Pa at 5 wt% ChNWs. In hot-pressed poly(S-co-BuA), the tensile modulus increased from 8.5 Pa to 8.9 Pa at 5 wt% ChNWs. These findings suggest that while nanochitin incorporation provides some enhancement in mechanical properties, the improvements remain limited.97 The likely reason is the poor interaction between ChNWs and the polymers due to differences in surface chemistry, which leads to ineffective stress transfer from the polymer to the nanowhiskers, resulting in minimal reinforcement.
| No. | Polymers | Affected properties | Composition | Value | Ref. |
|---|---|---|---|---|---|
|
a Two methods were utilized to produce nanocomposite films: water evaporation and hot-pressing. The mechanical tests were conducted on both the evaporated and hot-pressed materials to evaluate their properties.
b The PP substrates were dipped alternatively in a suspension of ChNWs (forming the first coating layer), then rinsed with deionized water, followed by immersion in a TCNF suspension and rinsed again with deionized water. This process constituted one complete dip coating cycle, resulting in one bilayer of ChNWs/TCNFs. The cycle was repeated until the desired number of (ChNWs/TCNF) bilayers was achieved. The concentrations of the suspensions for TCNF and ChNWs were adjusted in order to evaluate the barrier performance of the layers.
c The oxygen transmission rates were measured for films coated with 0.4 wt% TCNF suspension and ChNWs suspensions under different concentrations.
d Data were estimated from the graphs.
e PET substrates were coated with layers of ChNW/CS suspension and CNCs suspension. A single layer was created using either SChNW/CS or LChNW/CS coatings on PET while a bilayer was formed with (SChNW/CS)-CNC or (LChNW/CS)-CNC coatings on PET, where the mass ratio of ChNWs to CS for bilayers was 1:1.
f Gas barrier coatings were generated using multilayers of (ChNWs/CNFs)n (in which n indicates the number of bilayers, that tend to vary from 10 to 50 bilayers). For example, the sequential assembly of ChNWs and CNFs afforded a bilayer of (ChNWs/CNFs)1.
|
|||||
| Poly-(ε-caprolactone) | |||||
| 1 | Poly-(ε-caprolactone) (PCL)/ChNWs | Tensile modulus, Pa at 330 K (water evaporation)a | PCL | 8.52 | 97 |
| ChNWs/PCL = 0.5/99.5 | 8.36 | ||||
| ChNWs/PCL = 1.0/99.0 | 8.70 | ||||
| ChNWs/PCL = 2.5/97.5 | 8.78 | ||||
| Tensile modulus, Pa at 298 K (hot pressed)a | PCL | 8.50 | |||
| ChNWs/PCL = 0.5/99.5 | 8.58 | ||||
| ChNWs/PCL = 1.0/99.0 | 8.64 | ||||
| ChNWs/PCL = 2.5/97.5 | 8.72 | ||||
| ChNWs/PCL = 5/95.0 | 8.86 | ||||
| ChNWs/PCL = 10/90.0 | 8.98 | ||||
| 2 | Poly(styrene-co-butyl acrylate) (poly(S-co-BuA))/ChNWs | Tensile modulus, Pa at 298 K (water evaporation)a | Poly(S-co-BuA) | 5.75 | 97 |
| ChNWs/poly(S-co-BuA) = 0.5/99.5 | 5.85 | ||||
| ChNWs/poly(S-co-BuA) = 1.0/99.0 | 6.05 | ||||
| ChNWs/poly(S-co-BuA) = 2.5/97.5 | 6.50 | ||||
| ChNWs/poly(S-co-BuA) = 5.0/95.0 | 6.75 | ||||
| Tensile modulus, Pa (hot pressed)a | Poly(S-co-BuA) | 8.50 | |||
| ChNWs/poly(S-co-BuA) = 0.5/99.5 | 8.55 | ||||
| ChNWs/poly(S-co-BuA) = 1.0/99.0 | 8.70 | ||||
| ChNWs/poly(S-co-BuA) = 2.5/97.5 | 8.80 | ||||
| ChNWs/poly(S-co-BuA) = 5.0/95.0 | 8.90 | ||||
| Recycled polycarbonate/polysulfone | |||||
| 3 | Recycled polycarbonate (rPC, MW 45000)/polysulfone (PSf, MW 75000–81000)/ChNWs |
Tensile strength, MPa | rPC/PSf = 50/50 | 2.0 | 76 |
| ChNWs/rPC/PSf = 2/49/49 | 1.6 | ||||
| ChNWs/rPC/PSf = 4/48/48 | 1.6 | ||||
| ChNWs/rPC/PSf = 6/47/47 | 1.8 | ||||
| ChNWs/rPC/PSf = 8/46/46 | 2.2 | ||||
| ChNWs/rPC/PSf = 10/45/45 | 2.2 | ||||
| Elongation at break, % | rPC/PSf = 50/50 | 12.0 | |||
| ChNWs/rPC/PSf = 2/49/49 | 8.5 | ||||
| ChNWs/rPC/PSf = 4/48/48 | 8.0 | ||||
| ChNWs/rPC/PSf = 6/47/47 | 10.0 | ||||
| ChNWs/rPC/PSf = 8/46/46 | 15.0 | ||||
| ChNWs/rPC/PSf = 10/45/45 | 11.0 | ||||
| Pure water flux (PWF) rejection, L m−2 h−1 | rPC/PSf = 50/50 | 225 | |||
| ChNWs/rPC/PSf = 2/49/49 | 275 | ||||
| ChNWs/rPC/PSf = 4/48/48 | 350 | ||||
| ChNWs/rPC/PSf = 6/47/47 | 250 | ||||
| ChNWs/rPC/PSf = 8/46/46 | 255 | ||||
| Bovine serum albumin (BSA) rejection, % | rPC/PSf = 50/50 | 58 | |||
| ChNWs/rPC/PSf = 2/49/49 | 70 | ||||
| ChNWs/rPC/PSf = 4/48/48 | 90 | ||||
| ChNWs/rPC/PSf = 6/47/47 | 60 | ||||
| ChNWs/rPC/PSf = 8/46/46 | 62 | ||||
| Flux recovery ratio, % | rPC/PSf = 50/50 | 60 | |||
| ChNWs/rPC/PSf = 2/49/49 | 80 | ||||
| ChNWs/rPC/PSf = 4/48/48 | 78 | ||||
| ChNWs/rPC/PSf = 6/47/47 | 98 | ||||
| ChNWs/rPC/PSf = 8/46/46 | 98 | ||||
| Polyaniline | |||||
| 4 | Polyaniline (PANI)/ZnO nanoparticles (ZnO NPs)/β-chitin chitin nanowhiskers (β ChNWs) | Elastic modulus, MPa | ChNWs | 27.0 | 87 |
| βChNWs/ZnO = 97.09/2.91 | 32.5 | ||||
| β ChNWs/PANI = 99.01/0.99 | 4.0 | ||||
| βChNWs/ZnO/PANI = 96.15/2.88/0.96 | 2.5 | ||||
| Elongation at break, % | ChNWs | 3.0 | |||
| ChNWs/ZnO = 97.09/2.91 | 2.0 | ||||
| ChNWs/PANI = 99.01/0.99 | 2.5 | ||||
| ChNWs/ZnO/PANI = 96.15/2.88/0.96 | 2.2 | ||||
| Tensile strength, MPa | ChNWs | 13.8 | |||
| ChNWs/ZnO = 97.09/2.91 | 11.8 | ||||
| ChNWs/PANI = 99.01/0.99 | 10.0 | ||||
| ChNWs/ZnO/PANI = 96.15/2.88/0.96 | 8.0 | ||||
| Polyolefins | |||||
| 5 | Polypropylene (PP)b/α-ChNWs/TEMPO cellulose nanofibers (TCNFs) | Oxygen transmission rate,c cc m−2 day−1 | ChNWs and TCNFs coated in sequence on PP film, ratio not shown | 82 | |
| PP | 1118.00 | ||||
| ChNWs suspension = 0.8 wt% | 108.30 | ||||
| ChNWs suspension = 1.6 wt% | 13.10 | ||||
| ChNWs suspension = 2.4 wt% | 21.67 | ||||
| ChNWs suspension = 3.2 wt% | 49.71 | ||||
| Water vapor transmission rate, g m−2 day−1 | PP | 2.43 | |||
| PP/20 (ChNWs/TCNFs) bilayers | 2.13 | ||||
| Young's modulus, MPa | PP | 1290 | |||
| PP/10(ChNWs/TCNFs) bilayers | 1215 | ||||
| PP/20(ChNWs/TCNFs) bilayers | 1371 | ||||
| PP/30(ChNWs/TCNFs) bilayers | 1356 | ||||
| Tensile strength, MPa | PP | 45.96 | |||
| PP/10(ChNWs/TCNFs) bilayers | 36.83 | ||||
| PP/20(ChNWs/TCNFs) bilayers | 29.01 | ||||
| PP/30(ChNWs/TCNFs) bilayers | 36.01 | ||||
| Elongation at break, % | PP | 636 | |||
| PP/10(ChNWs/TCNFs) bilayers | 495 | ||||
| PP/20(ChNWs/TCNFs) bilayers | 367 | ||||
| PP/30(ChNWs/TCNFs) bilayers | 461 | ||||
| 6 | PP/ChNWs | Elastic modulus, MPa | PP | 1120 | 19 |
| ChNWs/PP = 1/99 | 1330 | ||||
| ChNWs/PP = 2/98 | 1370 | ||||
| ChNWs/PP = 5/95 | 1280 | ||||
| ChNWs/PP = 10/90 | 1180 | ||||
| Ultimate tensile strength, MPa | PP | 34.0 | |||
| ChNWs/PP = 1/99 | 37.0 | ||||
| ChNWs/PP = 2/98 | 39.0 | ||||
| ChNWs/PP = 5/95 | 37.5 | ||||
| ChNWs/PP = 10/90 | 34.0 | ||||
| Elongation at break, % | PP | 2.00 | |||
| ChNWs/PP = 1/99 | 0.65 | ||||
| ChNWs/PP = 2/98 | 0.10 | ||||
| ChNWs/PP = 5/95 | 0.15 | ||||
| ChNWs/PP = 10/90 | 0.05 | ||||
| 7 | Maleated polypropylene (MAPP)/ChNWs | Elastic modulus, MPa | PP | 1120 | 19 |
| ChNWs/MAPP = 1/99 | 1280 | ||||
| ChNWs/MAPP = 2/98 | 1300 | ||||
| ChNWs/MAPP = 5/95 | 1320 | ||||
| ChNWs/MAPP = 10/90 | 1080 | ||||
| Ultimate tensile strength, MPa | PP | 34.0 | |||
| ChNWs/MAPP = 1/99 | 36.5 | ||||
| ChNWs/MAPP = 2/98 | 38.0 | ||||
| ChNWs/MAPP = 5/95 | 40.0 | ||||
| ChNWs/MAPP = 10/90 | 34.0 | ||||
| Elongation at break, % | PP | 2.00 | |||
| ChNWs/MAPP = 1/99 | 0.60 | ||||
| ChNWs/MAPP = 2/98 | 0.20 | ||||
| ChNWs/MAPP = 5/95 | 0.15 | ||||
| ChNWs/MAPP = 10/90 | 0.40 | ||||
| Biodegradable polyesters | |||||
| 8 | PBAT/ChNFs | Tensile strengthd, MPa | PBAT | 12.5 | 240 |
| ChNFs/PBAT = 0.5/99.5 | 22.8 | ||||
| ChNFs/PBAT = 1.0/99.0 | 14.7 | ||||
| ChNFs/PBAT = 2.0/98.0 | 16.3 | ||||
| ChNFs/PBAT = 4.0/96/0 | 9.7 | ||||
| ChNFs/PBAT = 8.0/92.0 | 8.1 | ||||
| Elongation at breakd, % | PBAT | 536 | |||
| ChNFs/PBAT = 0.5/99.5 | 882 | ||||
| ChNFs/PBAT = 1.0/99.0 | 673 | ||||
| ChNFs/PBAT = 2.0/98.0 | 645 | ||||
| ChNFs/PBAT = 4.0/96/0 | 428 | ||||
| ChNFs/PBAT = 8.0/92.0 | 231 | ||||
| Glass transition temperature (Tg), °C | PBAT | −22.1 | |||
| ChNFs/PBAT = 0.5/99.5 | −21.5 | ||||
| ChNFs/PBAT = 1.0/99.0 | −20.9 | ||||
| ChNFs/PBAT = 2.0/98.0 | −21.0 | ||||
| ChNFs/PBAT = 4.0/96/0 | −20.9 | ||||
| ChNFs/PBAT = 8.0/92.0 | −20.0 | ||||
| Degree of crystallinity (χc), % | PBAT | 8.6 | |||
| ChNFs/PBAT = 0.5/99.5 | 11.4 | ||||
| ChNFs/PBAT = 1.0/99.0 | 10.8 | ||||
| ChNFs/PBAT = 2.0/98.0 | 10.9 | ||||
| ChNFs/PBAT = 4.0/96/0 | 9.7 | ||||
| ChNFs/PBAT = 8.0/92.0 | 8.7 | ||||
| The melting enthalphy (ΔHm), J g−1 | PBAT | 9.9 | |||
| ChNFs/PBAT = 0.5/99.5 | 12.9 | ||||
| ChNFs/PBAT = 1.0/99.0 | 12.2 | ||||
| ChNFs/PBAT = 2.0/98.0 | 12.2 | ||||
| ChNFs/PBAT = 4.0/96/0 | 10.6 | ||||
| ChNFs/PBAT = 8.0/92.0 | 9.2 | ||||
| The enthalphy of crystallization (ΔHc), J g−1 | PBAT | 14.7 | |||
| ChNFs/PBAT = 0.5/99.5 | 15.2 | ||||
| ChNFs/PBAT = 1.0/99.0 | 14.2 | ||||
| ChNFs/PBAT = 2.0/98.0 | 16.3 | ||||
| ChNFs/PBAT = 4.0/96/0 | 15.3 | ||||
| ChNFs/PBAT = 8.0/92.0 | 13.3 | ||||
| 9 | Poly(butylene adipate-co-terephtalate) (PBAT) made in situ from adipic acid (AA) and dimethylterephtalate/sulfated α-chitin nanowhiskers (S-ChNWs) | Young's modulus, MPa | PBAT | 170 ± 4 | 105 |
| S-ChNWs/PBAT = 0.1/99.9 | 136 ± 0.4 | ||||
| S-ChNWs/PBAT = 0.3/99.7 | 146 ± 3 | ||||
| S-ChNWs/PBAT = 0.7/99.3 | 150 ± 5 | ||||
| S-ChNWs/PBAT = 1.0/99.0 | 149 ± 3 | ||||
| Tensile strength, MPa | PBAT | 57 ± 4 | |||
| S-ChNWs/PBAT = 0.1/99.9 | 62 ± 2 | ||||
| S-ChNWs/PBAT = 0.3/99.7 | 70 ± 0.5 | ||||
| S-ChNWs/PBAT = 0.7/99.3 | 64 ± 2 | ||||
| S-ChNWs/PBAT = 1.0/99.0 | 63 ± 2 | ||||
| Elongation at break, % | PBAT | 980 ± 39 | |||
| S-ChNWs/PBAT = 0.1/99.9 | 926 ± 7 | ||||
| S-ChNWs/PBAT = 0.3/99.7 | 919 ± 11 | ||||
| S-ChNWs/PBAT = 0.7/99.3 | 962 ± 21 | ||||
| S-ChNWs/PBAT = 1.0/99.0 | 988 ± 14.7 | ||||
| 10 | Poly(butylene adipate-co-furanoate) (PBAF) made in situ from adipic acid (AA) and dimethyl 2,5-furandicarboxylate (DMFD)/sulfated α-chitin nanowhiskers (S-ChNWs) | Young's modulus, MPa | PBAF | 60 ± 0.5 | 105 |
| S-ChNWs/PBAF = 0.1/99.9 | 63 ± 2.0 | ||||
| S-ChNWs/PBAF = 0.3/99.7 | 65 ± 1.0 | ||||
| S-ChNWs/PBAF = 0.7/99.3 | 55 ± 0.2 | ||||
| S-ChNWs/PBAF = 1.0/99.0 | 53 ± 0.9 | ||||
| Tensile strength, MPa | PBAF | 53 ± 0.6 | |||
| S-ChNWs/PBAF = 0.1/99.9 | 83 ± 3.0 | ||||
| S-ChNWs/PBAF = 0.3/99.7 | 73 ± 3.0 | ||||
| S-ChNWs/PBAF = 0.7/99.3 | 47 ± 0.4 | ||||
| S-ChNWs/PBAF = 1.0/99.0 | 47 ± 3.0 | ||||
| Elongation at break, % | PBAF | 1007 ± 11 | |||
| S-ChNWs/PBAF = 0.1/99.9 | 1107 ± 19 | ||||
| S-ChNWs/PBAF = 0.3/99.7 | 933 ± 14 | ||||
| S-ChNWs/PBAF = 0.7/99.3 | 1116 ± 13 | ||||
| S-ChNWs/PBAF = 1.0/99.0 | 1128 ± 51 | ||||
| Ultimate tear strength, N cm−1 | PBAF | 730 ± 20 | |||
| S-ChNWs/PBAF = 0.1/99.9 | 1029 ± 39 | ||||
| S-ChNWs/PBAF = 0.3/99.7 | 814 ± 8 | ||||
| S-ChNWs/PBAF = 0.7/99.3 | 827 ± 20 | ||||
| S-ChNWs/PBAF = 1.0/99.0 | 761 ± 45 | ||||
| Extension at break, mm | PBAF | 94 ± 9 | |||
| S-ChNWs/PBAF = 0.1/99.9 | 93 ± 11 | ||||
| S-ChNWs/PBAF = 0.3/99.7 | 91 ± 4 | ||||
| S-ChNWs/PBAF = 0.7/99.3 | 82 ± 6 | ||||
| S-ChNWs/PBAF = 1.0/99.0 | 96 ± 6 | ||||
| Tear toughness, J cm−1 | PBAF | 52 ± 4 | |||
| S-ChNWs/PBAF = 0.1/99.9 | 73 ± 2 | ||||
| S-ChNWs/PBAF = 0.3/99.7 | 58 ± 2 | ||||
| S-ChNWs/PBAF = 0.7/99.3 | 52 ± 6 | ||||
| S-ChNWs/PBAF = 1.0/99.0 | 47 ± 7 | ||||
| Methacrylate | |||||
| 11 | Methacrylate (MA) resin/ChNWs | Tensile strength, MPa | MA | 32 ± 1.5 | 1 |
| ChNWs/MA = 0.5/99.5 | 37 | ||||
| ChNWs/MA = 1.0/99.0 | 46 ± 1.2 | ||||
| ChNWs/MA = 1.5/98.5 | 36 ± 1.6 | ||||
| Elongation at break, % | MA | 6.4 ± 0.5 | |||
| ChNWs/MA = 0.5/99.5 | 7.8 | ||||
| ChNWs/MA = 1.0/99.0 | 8.4 ± 0.5 | ||||
| ChNWs/MA = 1.5/98.5 | 3.6 ± 0.4 | ||||
| Tensile modulus, GPa | MA | 0.8 ± 0.1 | |||
| ChNWs/ MA = 0.5/99.5 | 0.9 | ||||
| ChNWs/ MA = 1.0/99.0 | 1.1 | ||||
| ChNWs/ MA = 1.5/98.5 | 1.4 ± 0.1 | ||||
| 12 | Methacrylate (MA)/α-chitin photocurable nanowhiskers (pChNWs, DDA ∼ 6.4%, Mw ∼ 150 kDa) | Storage modulus, MPa (−50 °C) | MA | 3165.4 | 77 |
| pChNWs/MA = 0.2/99.8 | 3585.4 | ||||
| pChNWs/MA = 0.5/99.5 | 4179.5 | ||||
| pChNWs/MA = 1.0/99.0 | 4688.9 | ||||
| Storage modulus, MPa (85 °C) | MA | 59.1 | |||
| pChNWs/MA = 0.2/99.8 | 71.6 | ||||
| pChNWs/MA = 0.5/99.5 | 89.3 | ||||
| pChNWs/MA = 1.0/99.0 | 125.2 | ||||
| Storage modulus, MPa (200 °C) | MA | 8.1 | |||
| pChNWs/MA = 0.2/99.8 | 11.4 | ||||
| pChNWs/MA = 0.5/99.5 | 17.7 | ||||
| pChNWs/MA = 1.0/99.0 | 15.5 | ||||
| Tensile strength, MPa | MA | 36.8 ± 2.6 | |||
| pChNWs/MA = 0.2/99.8 | 50 | ||||
| pChNWs/MA = 0.5/99.5 | 65.6 ± 3.7 | ||||
| pChNWs/MA = 1.0/99.5 | 30 | ||||
| Elongation at break, % | MA | 6 | |||
| pChNWs/MA = 0.2/99.8 | 8.9 | ||||
| pChNWs/MA = 0.5/99.5 | 6.7 | ||||
| pChNWs/MA = 1.0/99.0 | ∼2.0 | ||||
| Tensile modulus, GPa | MA | 0.85 ± 0.13 | |||
| pChNWs/MA = 0.2/99.8 | 1.1 ± 0.13 | ||||
| pChNWs/MA = 0.5/99.5 | 1.45 ± 0.13 | ||||
| pChNWs/MA = 1.0/99.5 | 1.7 ± 0.13 | ||||
| Polyethylene glycol dimethacrylate | |||||
| 13 | Polyethylene glycol dimethacrylate (PEGDMA, Mn = 6000 Da)/chitosan (CS, Mw = 100000 Da)/ChNWs |
Adhesion, kPa | PEGDMA | 2.1 | 90 |
| PEGDMA/CS = 88.24/11.76 | 3.1 | ||||
| PEGDMA/CS/ChNWs = 68.18/9.09/22.73 | 4.2 | ||||
| Elongation at break, % | PEGDMA | 100 | |||
| PEGDMA/CS = 88.24/11.76 | 125 | ||||
| PEGDMA/CS/ChNWs = 68.18/9.09/22.73 | 145 | ||||
| Tensile strength, kPa | PEGDMA | 5.0 | |||
| PEGDMA/CS | 9.0 | ||||
| PEGDMA/CS/ChNWs = 68.18/9.09/22.73 | 11.2 | ||||
| Swelling, % | PEGDMA | 277 | |||
| PEGDMA/CS | 300 | ||||
| PEGDMA/CS/ChNWs = 68.18/9.09/22.73 | 289 | ||||
| Polyesters | |||||
| 14 | Polyethylene terephthalate (PET)/ChNWs/cellulose nanocrystals (CNCs) | Oxygen permeability, cm3 μm m−2 day−1 kPa−1 | ChNWs/CNCs = 50/50 coated in sequence on PET film | 129 | |
| PET | 17.3 ± 0.0 | ||||
| PET-ChNWs | 12.6 ± 0.9 | ||||
| PET-CNCs | 5.9 ± 0.7 | ||||
| PET-ChNWs–CNCs | 5.2 ± 0.4 | ||||
| PET-blend | 10.2 ± 1.2 | ||||
| Water vapor transmission rate, g μm m−2 day−1 | PET | 148 | |||
| PET-ChNWs | 141 | ||||
| PET-CNCs | 148 | ||||
| PET-ChNWs–CNCs | 155 | ||||
| PET-blend | 140 | ||||
| Transmittance, % | PET | 87.1 | |||
| PET-ChNWs | 87.1 | ||||
| PET-CNCs | 87.7 | ||||
| PET-ChNWs–CNCs | 86.9 | ||||
| PET-blend | 86.9 | ||||
| Haze, % | PET | 1.8 | |||
| PET-ChNWs | 1.3 | ||||
| PET-CNCs | 2.4 | ||||
| PET-ChNWs–CNCs | 1.2 | ||||
| PET-blend | 3.3 | ||||
| 15 | Polyethylene terephthalate (PET)/α-short chitin nanowhiskers (SChNWs 114 nm) or α-long chitin nanowhiskers (LChNWs, 230 nm) | Oxygen permeability, cm3 μm m−2 day−1 kPa−1 at RH 50% | PET | 15.9 ± 0.4 | 130 |
| PET/SChNWs | 11.1 ± 0.3 | ||||
| PET/LChNWs | 16.5 ± 0.4 | ||||
| Transmittance, % | PET | 85 | |||
| PET/SChNWs | 85 | ||||
| PET/LChNWs | 87.5 | ||||
| Tensile strength, MPa | PET | 90 | |||
| PET/SChNWs | 78 | ||||
| PET/LChNWs | 80 | ||||
| Failure strain, % | PET | 42 | |||
| PET/SChNWs | 40 | ||||
| PET/LChNWs | 46 | ||||
| 16 | Polyethylene terephthalate (PET)/α-ChNWs/chitosan (CS); α-short chitin nanowhiskers (SChNWs 114 nm) or α-long chitin nanowhiskers (LChNWs, 230 nm)/chitosan (CS)/polyethylene terephthalate (PET) | Oxygen permeability, cm3 μm m−2 day−1 kPa−1 at RH 50% | PET | 15.9 ± 0.4 | 130 |
| CS | 11.0 ± 0.3 | ||||
| SChNWs/CS = 66.67/33.33 | 7.0 ± 0.3 | ||||
| SChNWs/CS = 50/50 | 5.1 ± 0.1 | ||||
| SChNWs/CS = 33.33/66.67 | 6.9 ± 0.2 | ||||
| LChNWs/CS = 66.67/33.33 | 13.1 ± 0.3 | ||||
| LChNWs/CS = 50/50 | 13.2 ± 0.3 | ||||
| LChNWs/CS = 33.33/66.66 | 8.2 ± 0.3 | ||||
| Transmittance, % | PET | 85 | |||
| CS | 85 | ||||
| SChNWs/CS = 66.67/33.33 | 85 | ||||
| LChNWs/CS = 66.67/33.33 | 85 | ||||
| SChNWs/CS = 50/50 | 87.5 | ||||
| LChNWs/CS = 50/50 | 85 | ||||
| SChNWs/CS = 33.3366.67 | 87.5 | ||||
| LChNWs/CS = 33.3366.67 | 85 | ||||
| Tensile strength, MPa | PET | 90 | |||
| CS | 70 | ||||
| SChNWs/CS = 66.67/33.33 | 78 | ||||
| LChNWs/CS = 66.67/33.33 | 76 | ||||
| SChNWs/CS = 50/50 | 77 | ||||
| LChNWs/CS = 50/50 | 78 | ||||
| SChNWs/CS = 33.3366.67 | 80 | ||||
| LChNWs/CS = 33.3366.67 | 77 | ||||
| Failure strain, % | PET | 42 | |||
| CS | 42 | ||||
| SChNWs/CS = 66.67/33.33 | 42 | ||||
| LChNWs/CS = 66.67/33.33 | 41 | ||||
| SChNWs/CS = 50/50 | 38 | ||||
| LChNWs/CS = 50/50 | 46 | ||||
| SChNWs/CS = 33.3366.67 | 46 | ||||
| LChNWs/CS = 33.3366.67 | 42 | ||||
| Water vapor transmission rate, g μm m−2 day−1 (under RH 45%, without thermal treatment) | PET | 148 | |||
| SChNWs/CS = 50/50 | 200 | ||||
| LChNWs/CS = 50/50 | 200 | ||||
| Water vapor transmission rate, g μm m−2 day−1 (under RH 75%, with thermal treatment) | PET | 175 | |||
| SChNWs/CS = 50/50 | 180 | ||||
| LChNWs/CS = 50/50 | 180 | ||||
| 17 | Polyethylene terephthalate (PET)/α-short chitin nanowhiskers (SChNWs 114 nm) or α-long chitin nanowhiskers (LChNWs, 230 nm)/chitosan (CS)/cellulose nanocrystals (CNCs) | Oxygen permeability, cm3 μm m−2 day−1 kPa−1 at RH 50% | ChNWs spread over PET substratee | 130 | |
| PET | 15.9 ± 0.4 | ||||
| (SChNW/CS)-CNC | 3.6 ± 0.1 | ||||
| (LChNW/CS)-CNC | 4.0 ± 0.1 | ||||
| CNCs | 8.0 ± 0.2 | ||||
| Transmittance, % | PET | 85 | |||
| SChNWs/CS-CNCs | 85 | ||||
| LChNWs/CS-CNC | 85 | ||||
| CNCs | 85.5 | ||||
| Tensile strength, MPa | PET | 90 | |||
| SChNWs/CS-CNCs | 77 | ||||
| LChNWs/CS-CNCs | 74 | ||||
| CNCs | 76 | ||||
| Failure strain, % | PET | 42 | |||
| SChNWs/CS-CNCs | 40 | ||||
| LChNWs/CS-CNCs | 40 | ||||
| CNCs | 38 | ||||
| Water vapor transmission rate, g μm m−2 day−1 (under RH 45%, without thermal treatment) | PET | 148 | |||
| SChNWs/CS-CNCs | 150 | ||||
| LChNWs/CS-CNCs | 149 | ||||
| CNCs | 175 | ||||
| Water vapor transmission rate, g μm m−2 day−1 (under RH 75%, with thermal treatment) | PET | 175 | |||
| SChNWs/CS-CNCs | 175 | ||||
| LChNWs/CS-CNCs | 155 | ||||
| CNCs | 175 | ||||
| 18 | Polyethylene terephthalate (PET)/α-ChNWs/cellulose nanofibers (CNFs)f | Oxygen transmission rate, mL m−2 day−1 | ChNWs and CNFs coated in sequence on PET film, ratio not shown | 89 | |
| PET | 38.28 | ||||
| (ChNWs/CNFs)10 | 0.97 | ||||
| (ChNWs/CNFs)20 | 0.48 | ||||
| (ChNWs/CNFs)30 | 0.57 | ||||
| (ChNWs/CNFs)40 | 0.59 | ||||
| (ChNWs/CNFs)50 | 0.51 | ||||
| Transmittance, % | PET | 92 | |||
| (ChNWs/CNFs)10 | 89 | ||||
| (ChNWs/CNFs)20 | 89 | ||||
| (ChNWs/CNFs)30 | 88.5 | ||||
| (ChNWs/CNFs)40 | 88 | ||||
| (ChNWs/CNFs)50 | 87.5 | ||||
| Haze, % | PET | 2.9 | |||
| (ChNWs/CNFs)10 | 2.5 | ||||
| (ChNWs/CNFs)20 | 2.0 | ||||
| (ChNWs/CNFs)30 | 1.8 | ||||
| (ChNWs/CNFs)40 | 1.7 | ||||
| (ChNWs/CNFs)50 | 1.6 | ||||
When composite membranes were fabricated using a phase inversion process, incorporating polycarbonate (PC) recovered from waste compact discs, polysulfone (PSf), and chitin nanowhiskers (ChNWs) to enhance their mechanical properties and performance,76 the improvements observed were also minor (Table 13, entry 3). The tensile strength of the composite membranes exhibited only minor improvements with the incorporation of chitin nanowhiskers (ChNWs). As the ChNW content increased from 2% to 10% in 2% increments, the corresponding tensile strength (MPa) values increased from 2.0 MPa for rPC/PSf = 1/1 to 2.2 MPa for the same composite with 10% ChNWs incorporated. These results suggest that the mechanical enhancement achieved with increasing ChNW concentration remains limited.
The elongation at break of the composite membranes varied with the incorporation of chitin nanowhiskers (ChNWs), see Table 13, entry 3. For rPC/PSf = 50/50, the elongation at break was 12.0%. As the ChNW content increased from 2% to 10% in 2% increments, the corresponding values fluctuated, decreasing to 8.5% at 2% ChNW and further to 8.0% at 4% ChNW. A slight recovery was observed at 6% ChNW, reaching 10.0%, followed by a peak of 15.0% at 8% ChNW before declining to 11.0% at 10% ChNW. These results indicate a non-linear trend in elongation at break, with the highest value observed at 8% ChNW.
The pure water flux (PWF), which represents the volume of pure water passing through a membrane per unit area and time (L m−2 h−1), is a key parameter in evaluating membrane permeability and efficiency.235,236 It is crucial in membrane applications such as water treatment and filtration, where maintaining high permeability while ensuring effective separation is essential. In this study, the PWF of the composite membranes varied with the incorporation of chitin nanowhiskers (ChNWs), see Table 13, entry 3. For rPC/PSf = 50/50, the PWF was 225 L m−2 h−1. As the ChNW content increased from 2% to 10% in 2% increments, the PWF initially increased, peaking at 350 L m−2 h−1 at 4% ChNW. However, at higher ChNW concentrations, a decline was observed, suggesting that excessive ChNW content may lead to structural modifications that reduce water permeability. These results indicate that while lower ChNW concentrations enhance water flux, an optimal balance is required to prevent a decline in efficiency due to excessive filler incorporation.
The bovine serum albumin (BSA) rejection, which refers to the membrane's ability to prevent BSA molecules from passing through, is a critical measure of separation efficiency in filtration applications, particularly in biomedical processes.76,237 A higher BSA rejection indicates improved selectivity, reducing protein contamination. In this study, the addition of 4% ChNW resulted in the highest BSA rejection of 90%, compared to 58% in membranes without ChNWs, demonstrating a significant enhancement in filtration performance (Table 13, entry 3). However, the flux recovery ratio (FRR), which indicates the membrane's ability to regain permeability after cleaning, showed an unclear trend, with the highest values observed at 6% and 8% ChNW. This suggests that while moderate ChNW incorporation enhances protein rejection efficiency, its impact on membrane fouling resistance and cleaning efficiency varies with concentration, highlighting the need for an optimal balance between permeability, selectivity, and antifouling properties.
The study conducted by Andre et al.87 reported the development of a disposable ethanol sensor based on nanochitin composite films (Table 13, entry 4), designed as a nature-friendly and cost-effective electronic device without the need for metallic electrodes. Unlike traditional reinforcement fillers, ChNWs served as a primary electrode component. The sensor was fabricated using a casting method, incorporating ZnO nanoparticles and polyaniline (PANI) into chitin-based films. Three different suspensions were prepared with varying compositions of ZnO and PANI, followed by ultrasound treatment and homogenization. The films were cast into acrylic molds, dried, and conditioned before characterization and ethanol vapor detection tests. The resulting films were classified as ChNWs–ZnO, ChNWs–PANI, and ChNWs–ZnO/PANI, along with a pure ChNW control film. While ChNWs were not used as a filler in this study, we considered it relevant to include them in this review due to their significant role as a primary electrode component.
The incorporation of ChNWs was first attempted by coating. Nguyen et al.82 fabricated polypropylene (PP) films through a sequential dip-coating process, alternating between chitin nanowhiskers (ChNWs) and TEMPO-oxidized cellulose nanofibers (TCNFs) (Table 13, entry 5). The PP substrates were first immersed in a ChNW suspension to form the initial coating layer, rinsed with deionized water, then dipped into a TCNF suspension, followed by another rinse. This sequence constituted a complete dip-coating cycle, resulting in a single bilayer of ChNWs/TCNFs, with the process repeated until 10 to 30 bilayers were achieved. The concentrations of ChNWs and TCNF suspensions were systematically adjusted to evaluate the barrier performance of the multilayer coatings.
The incorporation of chitin nanowhiskers (ChNWs) and TEMPO-oxidized cellulose nanofibers (TCNFs) as sequential coatings on polypropylene (PP) films significantly influenced the oxygen transmission rate (OTR), water vapor transmission rate (WVTR), and mechanical properties. The OTR of uncoated PP was 1118.00 cc m−2 day−1, but after coating with ChNWs at different suspension concentrations, a drastic reduction was observed, reaching 13.10 cc m−2 day−1 at 1.6 wt% ChNWs. However, a slight increase was seen at higher concentrations, with values of 21.67 cc m−2 day−1 at 2.4 wt% and 49.71 cc m−2 day−1 at 3.2 wt%, indicating that an optimal filler concentration enhances barrier properties before diminishing effects arise. Similarly, the WVTR showed a minor reduction, from 2.43 g m−2 day−1 for uncoated PP to 2.13 g m−2 day−1 for PP with 20 bilayers of ChNWs/TCNFs, reflecting improved moisture resistance.
Regarding mechanical properties, the Young's modulus increased with the addition of bilayers, rising from 1290 MPa in pure PP to 1371 MPa in PP with 20 bilayers, with a slight decrease at 30 bilayers (1356 MPa), suggesting increased stiffness due to nanocoating. However, tensile strength declined, from 45.96 MPa in pure PP to 29.01 MPa at 20 bilayers, before partially recovering at 30 bilayers (36.01 MPa). Likewise, elongation at break followed a decreasing trend, from 636% in pure PP to 367% in the 20-bilayer coated film, but slightly increased to 461% at 30 bilayers. These results indicate that while multilayer coatings enhance stiffness and barrier performance, they also reduce flexibility and tensile strength, likely due to stress retention at the interface and increased rigidity of the coatings.
Another study evaluated the mechanical properties of polypropylene (PP) and maleated polypropylene (MAPP) composites reinforced with chitin nanowhiskers (ChNWs), with a focus on elastic modulus, ultimate tensile strength, and elongation at break across different ChNW loadings (Table 13, entries 6 and 7).19 For PP/ChNW composites (Table 13, entry 6), the elastic modulus increased with ChNW content up to 5 wt% (1370 MPa) but decreased at 10 wt% (1180 MPa), suggesting that while ChNWs enhance stiffness at lower concentrations, excessive filler content may lead to agglomeration, reducing reinforcement efficiency. The ultimate tensile strength followed a similar trend, increasing from 34 MPa in pure PP to 39 MPa at 5 wt% ChNWs, before slightly decreasing at higher concentrations. However, elongation at break significantly declined, from 2.00% in pure PP to just 0.05% at 10 wt% ChNWs, indicating a drastic loss in ductility due to the rigid nanofiller.
In MAPP/ChNW composites (Table 13, entry 7), a more gradual increase in elastic modulus was observed, peaking at 1320 MPa at 10 wt% ChNWs, which suggests better interfacial adhesion between MAPP and ChNWs compared to unmodified PP. Similarly, ultimate tensile strength improved consistently with ChNW content, reaching 40 MPa at 10 wt% ChNWs, showing a stronger reinforcing effect. However, elongation at break followed a decreasing trend, from 2.00% in pure MAPP to 0.15% at 10 wt% ChNWs, with slightly better retention than in the unmodified PP composites. Overall, the results indicate that ChNWs improve stiffness and strength in both PP and MAPP, with MAPP composites showing better mechanical performance due to enhanced interfacial adhesion. However, higher ChNW concentrations reduce ductility, which could limit applications where flexibility is required.
The incorporation of chitin nanofibers (ChNFs) into biodegradable PBAT was successfully achieved through melt bending and compression molding methods. SEM revealed that the nanochitin was uniformly dispersed throughout the PBAT matrix at low contents (<2 wt%). The physical, thermal and mechanical properties of the composites were found to be highly influenced by both the concentration of ChNFs and their dispersion within the matrix (Table 13, entry 8). A significant 82.5% increase in the tensile strength was observed when 0.5 wt% ChNFs was melt blended with PBAT, but this increase sharply declined at higher ChNF concentrations. This result suggests that the addition of a small amount of ChNFs improved ChNF dispersion within the PBAT matrix, promoting effective heterogeneous nucleation of PBAT crystallites and providing excellent interfacial adhesion between ChNFs and PBAT chains. These factors likely contributed to enhanced rigidity and tensile strength of the composite. A closely similar trend was observed in the elongation at break, with 64.5% enhancement at the 0.5 wt% ChNF loading. This indicates that the small amount of ChNFs not only improved the tensile strength but also enhanced the composite's flexibility. However, as with the tensile strength, the elongation at break decreased at higher ChNF concentrations, suggesting that excessive ChNFs may hinder the polymer's ability to stretch, likely due to agglomeration or poor dispersion.240
Dynamic mechanical analysis results revealed an increase in the glass transition temperature (Tg) as the nanochitin content in the composites increased. This suggests that the movement of PBAT polymer chains was more restricted due to the interfacial bonding between ChNFs and PBAT matrix. The differential scanning calorimetry results indicated that the addition of ChNFs increased the enthalpy of melting, the enthalphy of crystallinization, and the degree of crystallinity, however, the optimal enhancements were still observed at 0.5 wt%, further supporting the idea that a small amount of ChNFs indeed created a heterogeneous nucleation effect, promoting the formation of crystallites in the PBAT matrix during cooling from the melt.240
Another study first investigated the use of sulfated ChNWs (S-ChNWs) to enhance the mechanical properties of PBAT and PBAF through an in situ polycondensation method. S-ChNWs were produced by sulfuric acid hydrolysis of bulk α-ChNWs, which removed the amorphous regions and simultaneously converted some primary hydroxy (C6-OH) and acetamide groups on the surface into sulfate-half esters (OSO3−) and protonated amino (NH3+) groups. This surface sulfation allowed for better dispersion of S-ChNWs in the monomer solution, maximizing their reinforcing effect in the final polymer matrix (Table 13, entries 9 and 10). For PBAT's mechanical properties, the tensile strength increased with S-ChNW addition, rising from 57 MPa in neat PBAT to 70 MPa at 0.3 wt% loading, though it slightly decrease at 0.7 and 1.0 wt% (Table 13, entry 9). This result alings with the previous study by Meng et al.,240 which found significant improvements in PBAT tensile strength with low nanochitin addition. However, the Young's modulus decreased from 170 MPa in neat PBAT to 146 MPa at 0.3 wt% S-ChNW loading, before partially recovering to 150 MPa and 149 MPa at 0.7 wt% and 1 wt% loading, respectively. Elongation at break followed a similar trend, decreasing from 980% in neat PBAT to 919% at 0.3 wt% S-ChNW loading, but increasing to 988% at 1.0 wt% loading. This result indicate that S-ChNWs were efficiently dispersed in the PBAT matrix during in situ polymerization, facilitating beneficial interactions between the NH3+ groups of S-ChNWs and the polymer's carbonyl groups, which led to improved mechanical attributes.105
The mechanically reinforcing effects of the S-ChNWs on PBAF are more pronounced than on PBAT. Adding just 0.1 wt% S-ChNWs increased the tensile strength by 1.6 times (53 MPa in neat PBAF vs. 83 MPa in the S-ChNWs/PBAF), and enhanced tear toughness by 1.4 times (52 J cm−1 in neat PBAF vs. 73 J cm−1 in the S-ChNWs/PBAF; see Table 13, entry 10). The in situ PBAF composite with 0.1 wt% S-ChNWs exhibited 1.2 times higher tensile strength and slightly lower elongation at break compared to the in situ PBAT composite with 0.3 wt% S-ChNWs. The superior performance of the PBAF composite with 0.1 wt% S-ChNWs was attributed to the higher polarity of the furan ring compared to the nonpolar benzene ring, promoting better dispersion of the hydrophilic nanofiller.105,243 Additionally, the angular structure of the furan ring in PBAF allowed S-ChNWs to fit between polymer chains, and their high aspect ratio created large filler/matrix interfaces.105,244
The optimal reinforcement for all properties, except elongation at break, was observed at 0.1 wt% for PBAF and 0.3 wt% for PBAT. Beyond these points, the properties of both composites declined, indicating unfavorable reinforcement effects. This decline is attributed to particle aggregation due to excessive hydrogen bonding from the excess nanoparticles in the matrix. Additionally, the increasing elongation at break is likely due to the less mobile chitin nanoparticles, which may act as a plasticizer, enhancing elongation at break.105
Tensile tests revealed that neat MA polymer had a tensile strength of 32 ± 1.5 MPa, which increased by 44% with the addition of 1.0 wt% ChNWs (46 ± 1.2 MPa) as shown in Table 13, entry 11. The maximum strain at break also improved with ChNW inclusion at the same loading (8.4 ± 0.5%), marking 31% increase compared to neat MA (6.4 ± 0.5%). Consequently, the combined increase in the tensile strength and strain at break enhanced the tensile toughness. These improvements were attributed to the enhanced interfacial adhesion between ChNWs and MA, as confirmed by their TEM results1 and previous FTIR analysis.3,41 However, further increasing ChNWs loading to 1.5 wt% led to a reduction in both tensile strength and strain at break by 22% and 57%, respectively compared to the 1.0 wt% loading. This decline was ascribed to the aggregation of excess ChNWs in the MA matrix.
The study also observed an improvement in the tensile modulus with increasing ChNW loading. Incorporating ChNWs at 1.5 wt% loading increased the tensile modulus by 75% (1.4 ± 0.1 GPa) compared to neat MA (0.8 ± 0.1 GPa), demonstrating the direct impact of ChNW intrinsic stiffness on the composite modulus. Furthermore, the overall property improvements were primarily influenced by matrix–filler interactions, driven by hydrogen-bonded functional groups between MA and ChNWs.1,41,247 Upon application in SLA 3D printing, the resulting 3D-printed nanocomposites, even at higher ChNW loadings, demonstrated high resolution and accuracy without requiring adjustments to the settings of a standard SLA 3D printer, confirming the enhanced applicability of MA for 3D printing.
A subsequent study by this group77 evaluated the use of surface modified ChNWs to reinforce SLA MA-based resin (Table 13, entry 12). In this study, ChNWs were functionalized with reactive acrylate groups to produce surface-modified, photocurable ChNWs (pChNWs). The DMA temperature studies showed that incorporation pChNWs into the MA resin improved the polymer chain mobility and energy dissipation. For instance, at −50 °C, the inclusion of 0.2, 0.5, and 1.0 wt% pCNW increased storage modulus of neat MA from 3165.4 to 3585.4 MPa (13.3%), 4179.5 MPa (32.0%), as shown in Table 13, entry 12. This improvement was attributed to the stiffer nature of ChNWs compared to the polymer matrix at this temperature. As temperature increased, the storage modulus decreased, with a rapid drop observed in the transition zone at 85 °C. At this temperature, the storage modulus of neat MA increased from 59.1 to 71.6 MPa (21.2%), 89.3 MPa (51.1%), and 125.2 MPa (111.8%) MPa, with the addition of 0.2, 0.5, and 1.0 wt% pCNW, respectively, due to the reinforcing ability of pCNW in the transition zone where the chain mobility begins.77 The modulus improvement for pChNW-filled composites was still evident at 200 °C, a temperature corresponding to second rubbery plateu region. At this temperature, the storage modulus of neat MA increased from 8.1 MPa to 17.7 MPa (118.5%) with the addition of 0.5 wt% pChNWs and slightly decreased to 15.5 MPa at 1.0 wt% pChNW loading. Likewise, the tensile modulus also increased with increasing pChNW content.
The incorporation of pChNWs also increased the composite tensile strength, reaching a maximum strength of 65.6 MPa at 0.5 wt% pChNW loading. The elongation at break of the composites increased with addition of 0.2 and 0.5 wt% (from 6.0 for neat MA to 8.9 and 6.7%, respectively). However, the addition of 1.0 wt% decreased significantly to ∼2.0%, indicating brittle behavior. These improvements were attributed to the strong interfacial adhesion induced by the physical and chemical crosslinking between pChNW filler and MA matrix.77
Another study examined the reinforcement effects of ChNWs on PEGDMA and PEGDMA/Chitosan (CS) hydrogels by assessing their adhesion, tensile properties, and swelling behavior (Table 13, entry 13).90 The reinforced hydrogels were synthesize via free radical polymerization under the UV light, enabling the crosslinking of PEGDMA and CS to form interpenetrating network hydrogels. The addition of CS and ChNWs improved adhesion strength, increasing it from 2.1 kPa in neat PEGDMA to 3.1 kPa in PEGDMA/CS and 4.2 kPa in PEGDMA/CS/ChNWs, suggesting that CS enhanced interface adhesion and ChNWs further strengthened it. The hydrogels’ tensile properties also improved with the incorporation of CS and ChNWs. Elongation at break increased from 100% in neat PGEDMA to 125% with CS, and further to 145% with both CS and ChNWs. Breaking strength also rose from 5.0 kPa to 9.0 kPa, and further to 11.2 kPa, demonstrating excellent toughness. Swelling tests showed that all hydrogels – PEGDA, PEGDA/CS, and PEGDA/CS/ChNWs – had high swelling rates, reaching 277%, 300%, and 289% respectively. This suggested that the hydrogels expand to a larger volume, which supports faster wound healing and provides an ideal mechanical microenvironment for tissue regeneration.90
Several studies have highlighted the use of nanochitin as reinforcing agent in the coating layer of PET films.89,129,130 Ji et al. investigated multilayer films with PET as the model substrate, using ChNWs and cellulose nanocrystals (CNCs) as bilayer barrier coatings (Table 13, entry 14). ChNWs and CNCs were applied in equal ratios as sequential coatings, with ChNW as the bottom layer and CNCs as the top layer on PET films. The properties – including oxygen permeability, water vapor transmission rate, transmittance, and haze – were then evaluated. To further understand the reinforcement effects of ChNWs compared to CNCs, additional PET films were coated with single layers of ChNW, CNC, or a blend of both (1:1 mass ratio, PET-blend), and their properties were also assessed. The bilayer ChNWs–CNCs–PET film exhibited an oxygen permeability (OP) 5.2 cm3 μm m−2 day−1 kPa−1, representing a 70% reduction compared to uncoated PET (17.3 cm3 μm m−2 day−1 kPa−1). However, the OP value of this bilayer film was not significantly different from the single-layer CNCs–PET film. In contrast, the OP value of PET-blend (10.2 cm3 μm m−2 day−1 kPa−1) was lower than that of ChNWs–PET but higher than that of CNCs–PET, confirming that applying ChNWs and CNCs as bilayers provides a better oxygen barrier performance than blending. While the water vapor barrier properties of the coated films were similar to those of the uncoated PET films, the bilayer ChNWs–CNCs–PET film exhibited the lowest transmittance and haze.
Yu et al.130 reported the use of blade coating to apply single-layer coatings of ChNWs and blended ChNW/CS, and bilayer coatings of these materials layered with CNCs, on PET substrates (Table 13, entries 15–17). Two ChNWs with different sizes and degrees of acetylation were selected as the basic coating materials. Short ChNWs (114 nm, SChNWs), with lower degree of acetylation (76.4% DA) than long ChNW (230 nm, 85.1% DA, LChNWs), showed better barrier performance with 30% reduction in OP (11.1 cm3 μm m−2 day−1 kPa−1 under 50% RH) compared to uncoated PET (15.9 cm3 μm m−2 day−1 kPa−1 under 50% RH, see Table 13, entry 15). SChNWs were found to be efficient in forming a dense packed structure over LChNWs. Further, CS was added to fill and close the pores formed in the SChNW-based coating layer. With an optimal mass ratio of 1:1 between these two components, the OP dropped to 5.1 ± 0.1 cm3 μm m−2 day−1 kPa−1 under 50% RH (Table 13, entry 16). This ChNW/CS-based coating led to a less than 2% and 15% decrease in light transmittance and tensile strength compared to uncoated PET films, respectively. The O2 barrier properties were further improved by adding a CNC layer to the ChNW/CS-coated PET. Under 50% RH, the OP of (SChNW/CS)-CNC-coated PET film was as low as 3.6 ± 0.1 cm3 μm m−2 day−1 kPa−1 (Table 13, entry 17). Moreover, after thermal treatment (120 °C, 2.5 h), the OP dropped to 2.2 cm3 μm m−2 day−1 kPa−1 (50% RH) and 4.8 cm3 μm m−2 day−1 kPa−1 (80% RH). This improvement was attributed to the formation of a densely compacted interface between SChNW/CS and CNC.130 The tensile properties of the coated PET substrates, in all cases, decreased compared to the uncoated PET due to substantial residual stress at the interface between the coatings and the base polymer.
A study by Kim et al.89 reported the design of a highly flexible coating on commercial PET film that is transparent to visible light and radio frequency (Table 13, entry 18). Nanoscale blending was achieved through spray-assisted layer-by-layer assembly, where negatively charged cellulose nanofibers (CNFs) and positively charged ChNWs were combined. The degree of reinforcement of ChNWs in combination with CNFs was evaluated across multilayers (ranging from 10 to 50 numbers of bilayers) on commercial PET film. The oxygen transmission rate (OTR) of PET film decreased sharply to below 1.0 mL m2 day−1 upon multilayer coating of ChNWs and CNCs (compared to 38.28 mL m2 day−1 for pristine PET film). This enhanced oxygen barrier performance plateaued at 0.48 ± 0.02 mL m2 day−1 at 20 bilayers. This indicates that the synergistic interplay between these highly crystalline nanomaterials results in a flexible film with superior barrier properties. This synergy arised from electrostatic interactions or ionic cross-linking between the respective amine and carboxylate groups, which reduced the free volume in the film. The coating also notably reduces the haziness of PET (2.9% for neat PET vs. 87.5 for 50 bilayers of ChNWs and CNFs coated on PET), with a negligible loss of transparency, and provides effective inhibition of antibacterial growth. The reduced light scattering of the coated PET is due to the reduction in surface roughness, as confirmed by AFM results.
5. Applications
The improvements observed across various property categories with the addition of chitin nanomaterials, as demonstrated in different studies, mark a significant advancement in materials science. The inclusion of nanochitin as fillers has enhanced the strength of several polymers, enabling them to meet the desired specifications for applications in diverse industries, including biomedicine, packaging, automotive, electronics, and the paper industry, among others.5.1. In biomedical sciences
Nanochitin is highly regarded for its beneficial properties, including its biodegradability, nontoxicity, and antimicrobial activity, making it an excellent substrate and filler for creating various biomedical frameworks.248 These characteristics make nanochitin particularly valuable in the development of advanced drug delivery systems, as biobased matrices commonly used in these applications often face significant challenges.249,250 This is why fillers, especially in the form of chitin nanomaterials have been extensively studied in conjunction with various biopolymers, particularly in materials like hydrogels, which are essential for drug delivery.A growing body of research has focused on designing advanced, biocompatible wound healing scaffolds, hydrogels, films, and nano-based drug delivery systems using chitin nanomaterials. Conventional wound dressings, for instance, often suffers from issues such as low mechanical strength, poor flexibility, reduced antimicrobial activity, and inadequate adhesion and absorption capacity.4,90 Furthermore, scaffolds made from polymers like polycaprolactone and gelatin, which are typically used to support cell growth, tissue regeneration, and tissue/organ replacement, also face challenges such as improper pore size, incorrect shape, and structural instability and fragility.251,252 However, studies reviewed in this article confirm that the integration of nanochitin significantly improves the morphology, mechanical strength, swelling behavior, compression strength, cytotoxicity, thermal stability, and antibacterial activity of nanocomposite hydrogels made from biobased polymers such as PEGDA,90 alginate,132 PVA,133 chitosan,106 and gelatin.120 These enhancements make hydrogels more stable, flexible, and stronger, positioning them as improved drug carriers for advancing medical therapies.
5.2. In packaging
Another significant application of chitin nanomaterials is their use as fillers in packaging materials. As the shift towards replacing petroleum-based products gain momentum, the adoption of bio-based polymers in packaging is becoming a key concern. While a wide range of biomaterials is available to create food packaging materials, their potential is often limited by poor mechanical, barrier, and thermal properties, making it difficult for them to compete with conventional polymers. However, the crosslinking of polymers with nanochitin has proven to substantially enhance the inherent characteristics of these base polymers.Barrier properties are especially critical in packaging applications, and reports indicate that the use of nanochitin as a filler has significantly improved the performance of polymers such as soy protein isolate,73 gelatin,78 polypropylene,82 and PVA.52 Specifically, nanochitin integration has enhanced properties such as water vapor transmission rate, oxygen permeability, UV-blocking capacity, and thermal tolerance, often surpassing the required standards for food storage. These enhancements make bio-based packaging materials more competitive with traditional petroleum-based polymers. Furthermore, the addition of nanochitin improves antimicrobial activity, which extends the longevity and preservation of sealed materials without compromising food quality.73 The improvements in mechanical properties, biodegradability, and overall enhancement support multifunctionality, biocompatibility, and sustainability, aligning with the growing demand for eco-friendly packaging solutions.
5.3. In other applications
Chitin nanomaterials also have significant applications in various industries, including rubber, paper, electronics, adhesives, hardeners, and more. For example, the inclusion of nanochitin as fillers in cellulose papers has enhanced their antibacterial properties (∼99% resistance), crystallinity, tensile strength, and color detection properties.72 These improved properties papers are crucial for a wide range of applications, whether in packaging, medical and healthcare supplies, stationery, construction, or industrial uses. The enhanced antimicrobial properties are particularly valuable for applications like filters, wound dressings, food packaging, and personal care products. The improved tensile strength benefits materials such as laminates, insulation components, wall coverings, and surgical materials. Additionally, the enhanced color detection properties are useful for environmental monitoring, including water and air quality testing, diagnostic substrates, and indicators.In the rubbery industry, nanochitin has proven effective in reinforcing elastomeric materials, offering advantages such as improved color, lightweight characteristics, increased thermal stability, and superior mechanical properties (tensile, tear, and swelling potential) compared to traditional natural rubber.13,18,25,51,53,147 Since natural rubber is not ideal for engineering applications in its raw form, its vulcanized version is more commonly used. The incorporation of nanochitin into these materials shows promising results for advanced rubber-based elastomeric applications, such as in the automotive industry (tires, seals, gaskets, belts), flooring mats, aerospace components (vibration dampers, conveyor belts, rollers), electronics, and medical uses. Furthermore, ChNWs contribute to producing mechanically enhanced materials and are also believed to exhibit strong bio-adhesive properties. Their higher adhesive performance, which is comparable to cellulose, offers significant advantages in structuring various material frameworks for a wide range of interdisciplinary applications.90,101,106
In summary, chitin nanomaterials have emerged as promising green nanofillers and base polymers in material science. While their advantages over conventional polymers are still being explored, their exceptional properties, which surpass those of many natural and synthetic polymers, are gradually becoming more evident. However, studies clearly indicate that achieving optimal material performance requires careful optimization of the amount of chitin nanowhiskers used. It is also important to note that the potential use of chitin in its nanoform as has not yet been fully explored across a wide range of polymers. Moreover, there is still an exciting scope that remains unexplored in utilizing nanochitin as natural polymers in sustainable chemistry. For instance, they can be explored as a catalyst support for carbon monoxide oxidation reaction253 and as activated chitin-based carbon sheet for oxygen reduction and evolution reactions.254 Therefore, it is essential to conduct further research to enhance the characteristics of nanochitin. Overcoming challenges related to its production, processing, and compatibility with various polymers is crucial for advancing its use in diverse applications.
6. Conclusion and outlook
In this review, we summarized various strategies for preparing nanochitin-reinforced composite plastics and examined how nanochitin influences polymer properties for diverse applications. Nanochitin, with its varying size and morphology, can be synthesized through several methods discussed in this review. We analyzed these strategies from the perspective of identifying simpler, more efficient methods compared to more complex ones. For example, researchers have explored casting methods and ways to optimize these processes to enhance both efficiency and environmental sustainability. While composites reinforced with nanochitin can be fabricated using various techniques and conditions, the effectiveness of chitin reinforcement and its interactions with polymer matrix can vary even when the same method is used. The nature, size, shape, loading and interaction of chitin nanomaterials are crucial factors that determine their role in reinforcing polymers.Additionally, the large amount of free reactive hydroxyl and acetyl amine groups on the nanochitin surface enables a range of chemical modifications and functionalization through chemical derivatization or post-functionalization. These modifications have distinct effects on different polymers, showcasing the tunability of nanochitin to meet specific application needs. For instance, the incorporation of carboxylated nanochitin in the form of ChNWs notably improves the mechanical properties of paper handsheets at low loading (1 wt%),121 whereas for natural rubber reinforcements, a higher loading (3.4 wt%) resulted in a more significant improvement in mechanical properties.202
Several challenges remain in fully realizing the potential of nanochitin as a reinforcing agent in polymer matrices and as a natural polymer. As highlighted in this review, many studies have reported the agglomeration of nanochitin particles at higher loading levels, with the extent of agglomeration varying across different polymers. This agglomeration negatively impacts the reinforcement efficiency of nanochitin, as the agglomerates hinder uniform dispersion and disrupt interactions within the polymer matrix, ultimately influencing the material's overall properties. The degree of agglomeration is influenced by factors such as the polymer type, nanochitin loading, and processing conditions during composite preparation. In certain cases, pretreatment methods like PEG or PEO to improve nanochitin dispersion in PLA have shown some positive effects, but this has also led to a decrease in the tensile indices of the composites. On the other hand, curing agents like chitosan and lysine contributed to both improved nanochitin dispersion in the epoxy resin matrix and enhanced mechanical properties of the composites. However, agents like arginine and cysteine improved nanochitin dispersion but had adverse effects on the epoxy mechanical properties. The scalability of nanochitin-based composites is another crucial concern. While the production of nanochitin may be scalable, its success depends on the techniques used to produce chitin, create nanochitin/polymer composites, and ensure their efficiency. Another major concern is the cost of nanochitin and nanochitin-reinforced composites, as high production costs could limit their widespread use in various applications.
Finally, in most cases nanochitin has been used primarily as nanofillers or supportive materials, although its potential for other applications remains to be explored. Therefore, further investigation into its full potential is essential to realize a circular economy with sustainable practices. Overall, challenges remain in the areas of large-scale manufacturing, consistent dispersion in hydrophobic matrices, and comprehensive biodegradation profiling under both laboratory and real-world conditions. Advancing understanding in these areas—particularly regarding long-term environmental fate and degradation behavior—will be critical for unlocking the full commercial and ecological potential of ChNW-based composites across diverse industries.
Conflicts of interest
The authors declare the following competing financial interests: J. L. Shamshina is an inventor of the related patents and applications and has partial ownership of Chitalyst, LLC and Mirus Boletus, LLC.Data availability
The data supporting this article have been included as part of the article and the ESI.†Acknowledgements
The authors gratefully acknowledge support from the Cotton Incorporated project titled “Chitin Nanowhisker-Encapsulated Herbicides for Sustainable Release, Superior Leaf Adhesion, and Enhanced Safety in Cotton Protection” (Project Number: 25-123, Project Year: 2025).References
- R. D. Maalihan, B. B. Pajarito and R. C. Advincula, 3D-printing methacrylate/chitin nanowhiskers composites via stereolithography: Mechanical and thermal properties, Mater. Today: Proc., 2020, 33, 1819–1824, DOI:10.1016/j.matpr.2020.05.063.
- L. Bai, L. Liu, M. Esquivel, B. L. Tardy, S. Huan, X. Niu, S. Liu, G. Yang, Y. Fan and O. J. Rojas, Nanochitin: Chemistry, structure, assembly, and applications, Chem. Rev., 2022, 122(13), 11604–11674, DOI:10.1021/acs.chemrev.2c00125.
- M. A. S. Anwer, J. Wang, A. Guan and H. E. Naguib, Chitin nano-whiskers (CNWs) as a bio-based bio-degradable reinforcement for epoxy: Evaluation of the impact of CNWs on the morphological, fracture, mechanical, dynamic mechanical, and thermal characteristics of dgeba epoxy resin, RSC Adv., 2019, 9(20), 11063–11076, 10.1039/C9RA00769E.
- M. Jones, M. Kujundzic, S. John and A. Bismarck, Crab vs. Mushroom: A review of crustacean and fungal chitin in wound treatment, Mar. Drugs, 2020, 18(1), 64, DOI:10.3390/md18010064.
- N. A. Z. Abidin, F. Kormin, N. A. Z. Abidin, N. A. F. M. Anuar and M. F. A. Bakar, The potential of insects as alternative sources of chitin: An overview on the chemical method of extraction from various sources, Int. J. Mol. Sci., 2020, 21(14), 1–25, DOI:10.3390/ijms21144978.
- A. G. J. Tacon, Word aquaculture trends and future prospects, in World Aquaculture, 2023. https://www.was.org/Magazine/2023/04/22/#zoom=true Search PubMed.
- P. Romano, H. Fabritius and D. Raabe, The exoskeleton of the lobster homarus americanus as an example of a smart anisotropic biological material, Acta Biomater., 2007, 3(3 SPEC. ISS.), 301–309, DOI:10.1016/j.actbio.2006.10.003.
- L. Barandiaran, B. Alonso, A. Reifs, I. Larraza, R. Olmos-juste, A. F. Calvo, A. Eceiza and R. Perez-jimenez, Enzymatic upgrading of nanochitin using an ancient lytic polysaccharide monooxygenase, Commun. Mater., 2022, 3(55), 1–23, DOI:10.1038/s43246-022-00277-9.
- N. E. Mushi, S. Utsel and L. A. Berglund, Nanostructured biocomposite films of high toughness based on native chitin nanofibers and chitosan, Front. Chem., 2014, 2, 1–11, DOI:10.3389/fchem.2014.00099.
- B. Joseph, R. Mavelil Sam, P. Balakrishnan, H. J. Maria, S. Gopi, T. Volova, S. C. M. Fernandes and S. Thomas, Extraction of nanochitin from marine resources and fabrication of polymer nanocomposites: Recent advances, Polymers, 2020, 12(8), 1664, DOI:10.3390/polym12081664.
- S. Ling, D. L. Kaplan and M. J. Buehler, Nanofibrils in nature and materials engineering, Nat. Rev. Mater., 2018, 3, 1–15, DOI:10.1038/natrevmats.2018.16.
- S. N. Patek and R. L. Caldwell, Extreme impact and cavitation forces of a biological hammer: Strike forces of the peacock mantis shrimp odontodactylus scyllarus, J. Exp. Biol., 2005, 208(19), 3655–3664, DOI:10.1242/jeb.01831.
- Y. Liu, M. Liu, S. Yang, B. Luo and C. Zhou, Liquid crystalline behaviors of chitin nanocrystals and their reinforcing effect on natural rubber, ACS Sustainable Chem. Eng., 2018, 6(1), 325–336, DOI:10.1021/acssuschemeng.7b02586.
- J. L. Shamshina and N. Abidi, Isolation of chitin nano-whiskers directly from crustacean biomass waste in a single step with acidic ionic liquids, ACS Sustainable Chem. Eng., 2022, 10(36), 11846–11855, DOI:10.1021/acssuschemeng.2c02461.
- S. Gopi, P. Balakrishnan, C. Divya, S. Valic, E. Govorcin Bajsic, A. Pius and S. Thomas, Facile synthesis of chitin nanocrystals decorated on 3D cellulose aerogels as a new multi-functional material for waste water treatment with enhanced anti-bacterial and anti-oxidant properties, New J. Chem., 2017, 41(21), 12746–12755, 10.1039/c7nj02392h.
- Q. Deng, J. Li, J. Yang and D. Li, Optical and flexible α-chitin nanofibers reinforced poly(vinyl alcohol) (PVA) composite film: Fabrication and property, Composites, Part A, 2014, 67, 55–60, DOI:10.1016/j.compositesa.2014.08.013.
- Y. Fan, H. Fukuzumi, T. Saito and A. Isogai, Comparative characterization of aqueous dispersions and cast films of different chitin nanowhiskers/nanofibers, Int. J. Biol. Macromol., 2012, 50(1), 69–76, DOI:10.1016/j.ijbiomac.2011.09.026.
- C. D. M. Dominic, R. Joseph, P. M. Sabura Begum, A. Raghunandanan, N. T. Vackkachan, D. Padmanabhan and K. Formela, Chitin nanowhiskers from shrimp shell waste as green filler in acrylonitrile-butadiene rubber: Processing and performance properties, Carbohydr. Polym., 2020, 245, 116505, DOI:10.1016/j.carbpol.2020.116505.
- S. C.-Y. Li, Y.-C. Sun, Q. Guan and H. Naguib, Effects of chitin nanowhiskers on the thermal, barrier, mechanical, and rheological properties of polypropylene nanocomposites, RSC Adv., 2016, 6(76), 72086–72095, 10.1039/C6RA11623J.
- A. M. Salaberria, J. Labidi and S. C. M. Fernandes, Chitin nanocrystals and nanofibers as nano-sized fillers into thermoplastic starch-based biocomposites processed by melt-mixing, Chem. Eng. J., 2014, 256, 356–364, DOI:10.1016/j.cej.2014.07.009.
- F. J. Alvarez, The effect of chitin size, shape, source and purification method on immune recognition, Molecules, 2014, 19(4), 4433–4451, DOI:10.3390/molecules19044433.
- Y. Feng, J. Li, B. Wang, X. Tian, K. Chen, J. Zeng, J. Xu and W. Gao, Novel nanofibrillated cellulose/chitin whisker hybrid nanocomposites and their use for mechanical performance enhancements, BioResources, 2018, 13(2), 3030–3044 CAS.
- J. Wang, Z. Chen, A. Guan, N. R. Demarquette and H. E. Naguib, Ionic liquids facilitated dispersion of chitin nanowhiskers for reinforced epoxy composites, Carbohydr. Polym., 2020, 247, 116746, DOI:10.1016/j.carbpol.2020.116746.
- J. Araki, Y. Yamanaka and K. Ohkawa, Chitin-chitosan nanocomposite gels: Reinforcement of chitosan hydrogels with rod-like chitin nanowhiskers, Polym. J., 2012, 44(7), 713–717, DOI:10.1038/pj.2012.11.
- K. Gopalan Nair and A. Dufresne, Crab shell chitin whisker reinforced natural rubber nanocomposites. 1. Processing and swelling behavior, Biomacromolecules, 2003, 4(3), 657–665, DOI:10.1021/bm020127b.
- J. Li, Y. Gao, J. Zhao, J. Sun and D. Li, Homogeneous dispersion of chitin nanofibers in polylactic acid with different pretreatment methods, Cellulose, 2017, 24(4), 1705–1715, DOI:10.1007/s10570-017-1216-y.
- M. Wang, H. Xue, Z. Feng, B. Cheng and H. Yang, Increase of tensile strength and toughness of bio-based diglycidyl ether of bisphenol a with chitin nanowhiskers, PLoS One, 2017, 12(6), 1–12, DOI:10.1371/journal.pone.0177673.
- S. Ifuku, S. Morooka, N. Nakagaito and H. Saimoto, Preparation and characterization of optically transparent chitin nanofiber/(meth)acrylic resin composites, Green Chem., 2011, 1708–1711, 10.1039/c1gc15321h.
- E. Ben Ayed, N. Ghorbel, A. Kallel, J. L. Putaux and S. Boufi, Polyaniline-grafted chitin nanocrystals as conductive reinforcing nanofillers for waterborne polymer dispersions, Biomacromolecules, 2022, 23(10), 4167–4178, DOI:10.1021/acs.biomac.2c00635.
- M. Esmailzadeh, E. Tammari, T. Safarpour, S. M. Razavian and L. Pezzato, Anti-corrosion effect of chitin and chitosan nanoparticles in epoxy coatings, Mater. Chem. Phys., 2024, 317, 129097–129097, DOI:10.1016/j.matchemphys.2024.129097.
- M. Zhou, Z. Liu, T. Liu, Y. Zhu and N. Lin, Tradeoff between amino group and crystallinity of chitin nanocrystals as a functional component in fluorescent nail coatings, ACS Sustainable Chem. Eng., 2022, 10(31), 10327–10338, DOI:10.1021/acssuschemeng.2c03024.
- S. Y. Baraki, Y. Zhang, X. Li, L. Ding, D. K. Debeli, D. K. Macharia, B. Wang, X. Feng, Z. Mao and X. Sui, Regenerated chitin reinforced polyhydroxybutyrate composites via pickering emulsion template with improved rheological, thermal, and mechanical properties, Compos. Commun., 2021, 25, 100655, DOI:10.1016/j.coco.2021.100655.
- J. Liao, Y. Zhou, B. Hou, J. Zhang and H. Huang, Nano-chitin: Preparation strategies and food biopolymer film reinforcement and applications, Carbohydr. Polym., 2023, 305, 120553–120553, DOI:10.1016/j.carbpol.2023.120553.
- C. Muñoz-Núñez, M. Fernández-García and A. Muñoz-Bonilla, Chitin nanocrystals: Environmentally friendly materials for the development of bioactive films, Coatings, 2022, 12(2), 1–17, DOI:10.3390/coatings12020144.
- P. Morganti, M. Palombo, G. Tishchenko, V. E. Yudin, F. Guarneri, M. Cardillo, P. Del Ciotto, F. Carezzi, G. Morganti and G. Fabrizi, Chitin-hyaluronan nanoparticles: A multifunctional carrier to deliver anti-aging active ingredients through the skin, Cosmetics, 2014, 1(3), 140–158, DOI:10.3390/cosmetics1030140.
- P. Heidarian and A. Z. Kouzani, Starch-g-acrylic acid/magnetic nanochitin self-healing ferrogels as flexible soft strain sensors, Sensors, 2023, 23(3), 1138, DOI:10.3390/s23031138.
- D. Liu, Y. Chang, D. Tian, W. Wu, A. Lu, N. Prempeh, M. Tan and Y. Huang, Lyotropic liquid crystal self-assembly of h2o2-hydrolyzed chitin nanocrystals, Carbohydr. Polym., 2018, 196, 66–72, DOI:10.1016/j.carbpol.2018.04.098.
- M. M. Haider, G. Jian, T. Zhong, H. Li, C. A. Fernandez, L. S. Fifield, M. Wolcott and S. Nassiri, Insights into setting time, rheological and mechanical properties of chitin nanocrystals- and chitin nanofibers-cement paste, Cem. Concr. Compos., 2022, 132, 104623–104623, DOI:10.1016/j.cemconcomp.2022.104623.
- T. Zhong, G. Jian, Z. Chen, M. Wolcott, S. Nassiri and C. A. Fernandez, Interfacial interactions and reinforcing mechanisms of cellulose and chitin nanomaterials and starch derivatives for cement and concrete strength and durability enhancement: A review, Nanotechnol. Rev., 2022, 11(1), 2673–2713, DOI:10.1515/ntrev-2022-0149.
- Y. Shi, G. Wu, S.-C. Chen, F. Song and Y.-Z. Wang, Green fabrication of high-performance chitin nanowhiskers/PVA composite films with a “brick-and-mortar” structure, ACS Sustainable Chem. Eng., 2020, 8(48), 17807–17815, DOI:10.1021/acssuschemeng.0c06736.
- M. Liu, Q. Peng, B. Luo and C. Zhou, The improvement of mechanical performance and water-response of carboxylated sbr by chitin nanocrystals, Eur. Polym. J., 2015, 68, 190–206, DOI:10.1016/j.eurpolymj.2015.04.035.
- M. Etcheverry and S. E. Barbosa, Glass fiber reinforced polypropylene mechanical properties enhancement by adhesion improvement, Materials, 2012, 5(6), 1084–1113, DOI:10.3390/ma5061084.
- S. Imani Yengejeh, S. A. Kazemi and A. Öchsner, Carbon nanotubes as reinforcement in composites: A review of the analytical, numerical and experimental approaches, Comput. Mater. Sci., 2017, 136, 85–101, DOI:10.1016/j.commatsci.2017.04.023.
- A. Kiziltas, W. Liu, S. Tamrakar and D. Mielewski, Graphene nanoplatelet reinforcement for thermal and mechanical properties enhancement of bio-based polyamide 6, 10 nanocomposites for automotive applications, Compos., Part C: Open Access, 2021, 6, 100177–100177, DOI:10.1016/j.jcomc.2021.100177.
- P. C. LeBaron and T. J. Pinnavaia, Clay nanolayer reinforcement of a silicone elastomer, Chem. Mater., 2001, 13(10), 3760–3765, DOI:10.1021/cm010982m.
- A. Barzegar and T. Ghaffari, Nanoclay-reinforced polymethylmethacrylate and its mechanical properties, Dent. Res. J., 2018, 15(4), 295–301, DOI:10.4103/1735-3327.237246.
- A. Y. Goldman, J. A. Montes, A. Barajas, G. Beall and D. D. Eisenhour, Effect of aging on mineral-filled nanocomposites, in Annual Technical Conference - ANTEC, Conference Proceedings, 1999, vol. 2, pp. 195–209. DOI: DOI:10.1016/B978-188420775-4.50022-2.
- P. R. Chang, R. Jian, J. Yu and X. Ma, Starch-based composites reinforced with novel chitin nanoparticles, Carbohydr. Polym., 2010, 80(2), 420–425, DOI:10.1016/j.carbpol.2009.11.041.
- J. Junkasem, R. Rujiravanit and P. Supaphol, Fabrication of α-chitin whisker-reinforced poly(vinyl alcohol) nanocomposite nanofibres by electrospinning, Nanotechnology, 2006, 17(17), 4519–4528, DOI:10.1088/0957-4484/17/17/039.
- G. Schmidt and M. M. Malwitz, Properties of polymer – nanoparticle composites, Curr. Opin. Colloid Interface Sci., 2003, 8(1), 103–108, DOI:10.1016/S1359-0294(03)00008-6.
- K. G. Nair and A. Dufresne, Crab shell chitin whisker reinforced natural rubber nanocomposites. 2. Mechanical behavior, Biomacromolecules, 2003, 666–674, DOI:10.1021/bm0201284.
- C. Peng and G. Chen, Preparation and assessment of heat-treated α-chitin nanowhiskers reinforced poly(viny alcohol) film for packaging application, Materials, 2018, 11(10), 1883, DOI:10.3390/ma11101883.
- W. O. Egbujuo, P. I. Anyanwu and H. C. Obasi, Utilization of chitin powder as a filler in natural rubber vulcanizates: In comparison with carbon black filler, Int. Rev. Appl. Sci. Eng., 2020, 11(1), 43–51, DOI:10.1556/1848.2020.00006.
- S. H. Kamarudin, M. S. Mohd Basri, M. Rayung, F. Abu, S. b. Ahmad, M. N. Norizan, S. Osman, N. Sarifuddin, M. S. Z. M. Desa and U. H. Abdullah, et al., A Review on natural fiber reinforced polymer composites (nfrpc) for sustainable industrial applications, Polymers, 2022, 14(17), 1–36, DOI:10.3390/polym14173698.
- A. Murawski, R. Diaz, S. Inglesby, K. Delabar and R. L. Quirino, Synthesis of bio-based polymer composites: Fabrication, fillers, properties, and challenges, in Lecture Notes in Bioengineering, 2019, pp. 29–55. DOI:10.1007/978-3-030-04741-2_2.
- E. Habib, R. Wang, Y. Wang, M. Zhu and X. X. Zhu, Inorganic fillers for dental resin composites: Present and future, ACS Biomater. Sci. Eng., 2016, 2(1), 1–11, DOI:10.1021/acsbiomaterials.5b00401.
- H. P. S. A. Khalil, E. W. N. Chong, F. A. T. Owolabi, M. Asniza, Y. Y. Tye, M. R. N. Fazita, M. K. M. Haa, Z. Nurmiati and M. T. Paridah, Enhancement of basic properties of polysaccharide-based composites with organic and inorganic fi llers: A review, J. Appl. Polym. Sci., 2018, 136(12), 47251, DOI:10.1002/app.47251.
- T. P. Sathishkumar, S. Satheeshkumar and J. Naveen, Glass fiber-reinforced polymer composites - a review, J. Reinf. Plast. Compos., 2014, 33(13), 1258–1275, DOI:10.1177/0731684414530790.
- I. S. Abbood, S. A. Odaa, K. F. Hasan and M. A. Jasim, Properties evaluation of fiber reinforced polymers and their constituent materials used in structures - a review, Mater. Today: Proc., 2021, 43, 1003–1008, DOI:10.1016/j.matpr.2020.07.636.
- B. S. Okan, Y. Menceloǧlu, B. G. Ozunlu and Y. E. Yagci, Graphene from waste tire by recycling technique for cost-effective and light-weight automotive plastic part production, AIP Conf. Proc., 2020, 2205(1), 020046, DOI:10.1063/1.5142961.
- P. Cataldi, A. Athanassiou and I. S. Bayer, Graphene nanoplatelets-based advanced materials and recent progress in sustainable applications, Appl. Sci., 2018, 8(9), 1438, DOI:10.3390/app8091438.
- N. M. Nurazzi, M. R. M. Asyraf, A. Khalina, N. Abdullah, F. A. Sabaruddin, S. H. Kamarudin, S. b. Ahmad, A. M. Mahat, C. L. Lee and H. A. Aisyah, et al., Fabrication, functionalization, and application of carbon nanotube-reinforced polymer composite: An overview, Polymers, 2021, 13(7), 1047, DOI:10.3390/polym13071047.
- M. Jawaid, A. e. k. Qaiss and R. Bouhfid, Nanoclay reinforced polymer composites, 2016 Search PubMed.
- R. N. Rothon, Mineral fillers in thermoplastics i, 1999 Search PubMed.
- R. H. Marchessault, F. F. Morehead and N. M. Walter, Liquid crystal systems from fibrillar polysaccharides, Nature, 1959, 184(4686), 632–633, DOI:10.1038/184632a0.
- S. P. Druzian, N. P. Zanatta, L. N. Côrtes, A. Fátima and M. Streit, Preparation of chitin nanowhiskers and its application for crystal violet dye removal from wastewaters, Environ. Sci. Pollut. Res. Int., 2019, 26, 28548–28557, DOI:10.1007/s11356-018-3547-0.
- J. Revel and R. H. Marchessaultf, In vitro chiral nematic ordering of chitin crystallites, Int. J. Biol. Macromol., 1993, 15, 329–335, DOI:10.1016/0141-8130(93)90049-r.
- Y. Yamamoto, T. Nishimura, T. Saito and T. Kato, CaCO3/chitin-whisker hybrids: Formation of CaCO3 crystals in chitin-based liquid-crystalline suspension, Polym. J., 2010, 42(7), 583–586, DOI:10.1038/pj.2010.32.
- Q. Guan, WIPO Patent, WO2015/000055A1, 2015 Search PubMed.
- Q. Guan, US2017/0183459A1, 2017.
- T. Jin, T. Liu, E. Lam and A. Moores, Chitin and chitosan on the nanoscale, Nanoscale Horiz., 2021, 6(7), 505–542, 10.1039/d0nh00696c.
- Z. Li, M. Zhang, D. Cheng and R. Yang, Preparation of silver nano-particles immobilized onto chitin nano-crystals and their application to cellulose paper for imparting antimicrobial activity, Carbohydr. Polym., 2016, 151, 834–840, DOI:10.1016/j.carbpol.2016.06.012.
- R. R. Koshy, A. Reghunadhan, S. K. Mary, L. A. Pothen and S. Thomas, Mgonp/chitin nanowhisker-based hybrid filler bound soy protein thin films for bioactive packaging applications, Int. J. Food Sci. Technol., 2022, 57(9), 5650–5662, DOI:10.1111/ijfs.15805.
- R. R. Koshy, A. Reghunadhan, S. K. Mary, P. S. Pillai, S. Joseph and L. A. Pothen, pH indicator films fabricated from soy protein isolate modified with chitin nanowhisker and clitoria ternatea flower extract, Curr. Res. Food Sci., 2022, 5, 743–751, DOI:10.1016/j.crfs.2022.03.015.
- S. C. Furtado, B. Srinivasan, J. Anbu and S. Abraham, Lyophilized biocomposite chitin-alginate matrices for wound healing application, Mater. Today: Proc., 2023 DOI:10.1016/j.matpr.2023.03.803.
- D. Saijun, P. Boonsuk and W. Chinpa, Conversion of polycarbonate from waste compact discs into antifouling ultrafiltration membrane via phase inversion, J. Polym. Res., 2022, 29(6), 220, DOI:10.1007/s10965-022-03073-8.
- R. D. Maalihan, Q. Chen, H. Tamura, J. R. H. Sta. Agueda, B. B. Pajarito, E. B. Caldona and R. C. Advincula, Mechanically and thermally enhanced 3D-printed photocurable polymer nanocomposites containing functionalized chitin nanowhiskers by stereolithography, ACS Appl. Polym. Mater., 2022, 4(4), 2513–2526, DOI:10.1021/acsapm.1c01816.
- A. Etxabide, P. A. Kilmartin, J. I. Maté and J. Gómez-Estaca, Characterization of glucose-crosslinked gelatin films reinforced with chitin nanowhiskers for active packaging development, LWT–Food Sci. Technol., 2022, 154, 112833, DOI:10.1016/j.lwt.2021.112833.
- I. V. Tyshkunova, I. V. Gofman, D. G. Chukhchin, A. V. Malkov, A. I. Mishanin, A. S. Golovkin, E. N. Pavlova, D. N. Poshina and Y. A. Skorik, Biophysical characterization and cytocompatibility of cellulose cryogels reinforced with chitin nanowhiskers, Polymers, 2022, 14(13), 1–18, DOI:10.3390/polym14132694.
- Y. Shi, S. C. Chen, W. T. Xiong and Y. Z. Wang, Simultaneous toughening and strengthening of chitin-based composites via tensile-induced orientation and hydrogen bond reconstruction, Carbohydr. Polym., 2022, 275, 118713–118713, DOI:10.1016/j.carbpol.2021.118713.
- Y. Zhang, H. He, B. Huang, S. Wang and X. He, Enhanced thermal conductivity of polyvinyl alcohol insulation composites with m-bn@cnw hybrid materials, Compos. Sci. Technol., 2021, 208, 108766–108766, DOI:10.1016/j.compscitech.2021.108766.
- H. L. Nguyen, T. H. Tran, L. T. Hao, H. Jeon, J. M. Koo, G. Shin, D. S. Hwang, S. Y. Hwang, J. Park and D. X. Oh, Biorenewable, transparent, and oxygen/moisture barrier nanocellulose/nanochitin-based coating on polypropylene for food packaging applications, Carbohydr. Polym., 2021, 271, 118421–118421, DOI:10.1016/j.carbpol.2021.118421.
- Y. Wu, Y. Guan, H. Gao, L. Zhou and F. Peng, Novel high-strength montmorillonite/polyvinyl alcohol composite film enhanced by chitin nanowhiskers, J. Appl. Polym. Sci., 2021, 138(18), e50344, DOI:10.1002/app.50344.
- A. G. B. Pereira, E. C. Muniz and Y.-L. Hsieh, Chitosan-sheath and chitin-core nanowhiskers, Carbohydr. Polym., 2014, 107, 158–166, DOI:10.1016/j.carbpol.2014.02.046.
- A. G. B. Pereira, E. C. Muniz and Y.-L. Hsieh, 1 h nmr and 1h–13c hsqc surface characterization of chitosan–chitin sheath-core nanowhiskers, Carbohydr. Polym., 2015, 123, 46–52, DOI:10.1016/j.carbpol.2015.01.017.
- A. A. Oun and J. W. Rhim, Preparation of multifunctional chitin nanowhiskers/zno-ag nps and their effect on the properties of carboxymethyl cellulose-based nanocomposite film, Carbohydr. Polym., 2017, 169, 467–479, DOI:10.1016/j.carbpol.2017.04.042.
- R. S. Andre, D. M. Santos, L. A. Mercante, M. H. M. Facure, S. P. Campana-filho, L. H. C. Mattoso and D. S. Correa, Nanochitin-based composite films as a disposable ethanol sensor, J. Environ. Chem. Eng., 2020, 8(5), 104163–104163, DOI:10.1016/j.jece.2020.104163.
- R. M. Abdelrahman, A. M. Abdel-Mohsen, M. Zboncak, J. Frankova, P. Lepcio, L. Kobera, M. Steinhart, D. Pavlinak, Z. Spotaz and R. Sklenářévá, et al., Hyaluronan biofilms reinforced with partially deacetylated chitin nanowhiskers: Extraction, fabrication, in vitro and antibacterial properties of advanced nanocomposites, Carbohydr. Polym., 2020, 235, 115951, DOI:10.1016/j.carbpol.2020.115951.
- T. Kim, T. H. Tran, S. Y. Hwang, J. Park, D. X. Oh and B. S. Kim, Crab-on-a-tree: All biorenewable, optical and radio frequency transparent barrier nanocoating for food packaging, ACS Nano, 2019, 13(4), 3796–3805, DOI:10.1021/acsnano.8b08522.
- X. Liu, S. Qin, L. Xu, G. Fu, Y. Huang, C. Yu, G. Cheng, Y. Li, Y. He and Y. Qi, et al., A tough and mechanically stable adhesive hydrogel for non-invasive wound repair, Front. Bioeng. Biotechnol., 2023, 11, 1–10, DOI:10.3389/fbioe.2023.1173247.
- Q. Guan and H. E. Naguib, Fabrication and characterization of PLA/phbv-chitin nanocomposites and their foams, J. Polym. Environ., 2014, 22(1), 119–130, DOI:10.1007/s10924-013-0625-8.
- R. Rizvi, B. Cochrane, H. Naguib and P. C. Lee, Fabrication and characterization of melt-blended polylactide-chitin composites and their foams, J. Cell. Plast., 2011, 47(3), 283–300, DOI:10.1177/0021955X11402549.
- Q. Zhang, S. Wei, J. Huang, J. Feng and P. R. Chang, Effect of surface acetylated-chitin nanocrystals on structure and mechanical properties of poly(lactic acid), J. Appl. Polym. Sci., 2014, 131(2), 2–9, DOI:10.1002/app.39809.
- A. J. Uddin, J. Araki, M. Fujie, S. Sembo and Y. Gotoh, Interfacial interaction and mechanical properties of chitin whisker-poly(vinyl alcohol) gel-spun nanocomposite fibers, Polym. Int., 2012, 61(6), 1010–1015, DOI:10.1002/pi.4174.
- J. Sriupayo, P. Supaphol, J. Blackwell and R. Rujiravanit, Preparation and characterization of α-chitin whisker-reinforced poly(vinyl alcohol) nanocomposite films with or without heat treatment, Polymer, 2005, 46(15), 5637–5644, DOI:10.1016/j.polymer.2005.04.069.
- S. J. Kim, B. M. Hong and W. H. Park, The effects of chitin/chitosan nanowhiskers on the thermal, mechanical and dye adsorption properties of electrospun PVA nanofibrous membranes, Cellulose, 2020, 27(10), 5771–5783, DOI:10.1007/s10570-020-03191-w.
- A. Morin and A. Dufresne, Nanocomposites of chitin whiskers from riftia tubes and poly(caprolactone), Macromolecules, 2002, 35(6), 2190–2199, DOI:10.1021/ma011493a.
- A. M. Salaberria, R. H. Diaz, J. Labidi and S. C. M. Fernandes, Preparing valuable renewable nanocomposite films based exclusively on oceanic biomass - chitin nanofillers and chitosan, React. Funct. Polym., 2015, 89, 31–39, DOI:10.1016/j.reactfunctpolym.2015.03.003.
- M. C. Li, Q. Wu, K. Song, A. D. French, C. Mei and T. Lei, Ph-responsive water-based drilling fluids containing bentonite and chitin nanocrystals, ACS Sustainable Chem. Eng., 2018, 6(3), 3783–3795, DOI:10.1021/acssuschemeng.7b04156.
- F. Larbi, A. García, L. J. Valle, A. Hamou, J. Puiggali, N. Belgacem and J. Bras, Comparison of nanocrystals and nanofibers produced from shrimp shell α-chitin: From energy production to material cytotoxicity and pickering emulsion, Carbohydr. Polym., 2018, 196, 385–397, DOI:10.1016/j.carbpol.2018.04.094.
- L. G. Greca, K. J. De France, J. Majoinen, N. Kummer, O. I. V. Luotonen, S. Campioni, O. J. Rojas, G. Nyström and B. L. Tardy, Chitin-amyloid synergism and their use as sustainable structural adhesives, J. Mater. Chem. A, 2021, 9(35), 19741–19753, 10.1039/d1ta03215a.
- Z. Li, H. Wang, S. An and X. Yin, Nanochitin whisker enhances insecticidal activity of chemical pesticide for pest insect control and toxicity, J. Nanobiotechnol., 2021, 19(1), 1–13, DOI:10.1186/s12951-021-00792-w.
- K. P. S. Kiliona, M. Zhou, Y. Zhu, P. Lan and N. Lin, Preparation and surface modification of crab nanochitin for organogels based on thiol-ene click cross-linking, Int. J. Biol. Macromol., 2020, 150, 756–764, DOI:10.1016/j.ijbiomac.2020.02.125.
- Y. Wang, L. Zhu, J. You, F. Chen, L. Zong, X. Yan and C. Li, Catecholic coating and silver hybridization of chitin nanocrystals for ultrafiltration membrane with continuous flow catalysis and gold recovery, ACS Sustainable Chem. Eng., 2017, 5(11), 10673–10681, DOI:10.1021/acssuschemeng.7b02633.
- T. V. Thanh, L. T. Hao, H. Y. Cho, H. Kim, S. A. Park, M. Lee, H. J. Kim, H. Jeon, S. Y. Hwang and J. Park, et al., Sustainable poly(butylene adipate- co-furanoate) composites with sulfated chitin nanowhiskers: Synergy leading to superior robustness and improved biodegradation, ACS Sustainable Chem. Eng., 2022, 10(26), 8411–8422, DOI:10.1021/acssuschemeng.2c01395.
- J. Pang, S. Bi, T. Kong, X. Luo, Z. Zhou, K. Qiu, L. Huang, X. Chen and M. Kong, Mechanically and functionally strengthened tissue adhesive of chitin whisker complexed chitosan/dextran derivatives based hydrogel, Carbohydr. Polym., 2020, 237, 116138–116138, DOI:10.1016/j.carbpol.2020.116138.
- Y. Fan, T. Saito and A. Isogai, Chitin nanocrystals prepared by tempo-mediated oxidation of α-chitin, Biomacromolecules, 2008, 9(1), 192–198, DOI:10.1021/bm700966g.
- A. M. Salaberria, J. Labidi and S. C. M. Fernandes, Different routes to turn chitin into stunning nano-objects, Eur. Polym. J., 2015, 68, 503–515, DOI:10.1016/j.eurpolymj.2015.03.005.
- C. Hou, J. Zhang, X. Zhang, Y. Wang, T. Li, B. Xia, J. Jiang and W. Dong, High strength chitin nanocrystal/alginate filament prepared by wet-spinning in “green” coagulating bath, Cellulose, 2022, 29(16), 8611–8621, DOI:10.1007/s10570-022-04814-0.
- K. Zhang and P. Liu, US Pat., US11718689B2, 2023 Search PubMed.
- P. Liu, H. Liu, T. Schäfer, T. Gutmann, H. Gibhardt, H. Qi, L. Tian, X. C. Zhang, G. Buntkowsky and K. Zhang, Unexpected selective alkaline periodate oxidation of chitin for the isolation of chitin nanocrystals, Green Chem., 2021, 23(2), 745–751, 10.1039/D0GC04054A.
- L. Zhang, B. Zhan and L. Yan, Preparation of nanochitin using deep eutectic solvents, iScience, 2024, 27(3), 109312, DOI:10.1016/j.isci.2024.109312.
- A. A. Oun and J. W. Rhim, Effect of oxidized chitin nanocrystals isolated by ammonium persulfate method on the properties of carboxymethyl cellulose-based films, Carbohydr. Polym., 2017, 175, 712–720, DOI:10.1016/j.carbpol.2017.08.052.
- A. Isogai, T. Hänninen, S. Fujisawa and T. Saito, Review: Catalytic oxidation of cellulose with nitroxyl radicals under aqueous conditions, Prog. Polym. Sci., 2018, 122–148, DOI:10.1016/j.progpolymsci.2018.07.007.
- R. Chen, W. C. Huang, W. Wang and X. Mao, Characterization of tempo-oxidized chitin nanofibers with various oxidation times and its application as an enzyme immobilization support, Mar. Life Sci. Technol., 2021, 3(1), 85–93, DOI:10.1007/s42995-020-00054-y.
- J. Sun, Y. Du, J. Ma, Y. Li, L. Wang, Y. Lu, J. Zou, J. Pang and C. Wu, Transparent bionanocomposite films based on konjac glucomannan, chitosan, and tempo-oxidized chitin nanocrystals with enhanced mechanical and barrier properties, Int. J. Biol. Macromol., 2019, 138, 866–873, DOI:10.1016/j.ijbiomac.2019.07.170.
- Y. Yue, X. Wang, Q. Wu, J. Han and J. Jiang, Highly recyclable and super-tough hydrogel mediated by dual-functional tio2 nanoparticles toward efficient photodegradation of organic water pollutants, J. Colloid Interface Sci., 2020, 564, 99–112, DOI:10.1016/j.jcis.2019.12.069.
- J. Jiang, J. Yu, L. Liu, Z. Wang, Y. Fan, T. Satio and A. Isogai, Preparation and hydrogel properties of pH-sensitive amphoteric chitin nanocrystals, J. Agric. Food Chem., 2018, 66(43), 11372–11379, DOI:10.1021/acs.jafc.8b02899.
- J. Jiang, W. Ye, J. Yu, Y. Fan, Y. Ono, T. Saito and A. Isogai, Chitin nanocrystals prepared by oxidation of α-chitin using the o2/laccase/tempo system, Carbohydr. Polym., 2018, 189, 178–183, DOI:10.1016/j.carbpol.2018.01.096.
- Y. Yue, J. Liu, S. Gao, Y. Pei, Y. Jiang, K. Tang, J. Zhang, J. Liu and X. Zheng, Ionically conductive gelatin-based hybrid composite hydrogels with high mechanical strength, self-healing, and freezing-tolerant properties, Eur. Polym. J., 2022, 172, 111230–111230, DOI:10.1016/j.eurpolymj.2022.111230.
- H. Tang, J. Wu, D. Li, C. Shi, G. Chen, M. He and J. Tian, High-strength paper enhanced by chitin nanowhiskers and its potential bioassay applications, Int. J. Biol. Macromol., 2020, 150, 885–893, DOI:10.1016/j.ijbiomac.2020.02.154.
- Z. Li, C. Liu, S. Hong, H. Lian, C. Mei, J. Lee, Q. Wu, M. A. Hubbe and M. C. Li, Recent advances in extraction and processing of chitin using deep eutectic solvents, Chem. Eng. J., 2022, 446(P2), 136953–136953, DOI:10.1016/j.cej.2022.136953.
- J.-i. Kadokawa, A. Takegawa, S. Mine and K. Prasad, Preparation of chitin nanowhiskers using an ionic liquid and their composite materials with poly (vinyl alcohol), Carbohydr. Polym., 2011, 84(4), 1408–1412, DOI:10.1016/j.carbpol.2011.01.049.
- S. Hong, Y. Yuan, K. Zhang, H. Lian and H. Liimatainen, Efficient hydrolysis of chitin in a deep eutectic solvent synergism for production of chitin nanocrystals, Nanomaterials, 2020, 10(5), 869, DOI:10.3390/nano10050869.
- Y. Fan, T. Saito and A. Isogai, Preparation of chitin nanofibers from squid pen β-chitin by simple mechanical treatment under acid conditions, Biomacromolecules, 2008, 9(7), 1919–1923, DOI:10.1021/bm800178b.
- M. C. Li, Q. Wu, K. Song, H. N. Cheng, S. Suzuki and T. Lei, Chitin nanofibers as reinforcing and antimicrobial agents in carboxymethyl cellulose films: Influence of partial deacetylation, ACS Sustainable Chem. Eng., 2016, 4(8), 4385–4395, DOI:10.1021/acssuschemeng.6b00981.
- Y. Fan, T. Saito and A. Isogai, Individual chitin nano-whiskers prepared from partially deacetylated α-chitin by fibril surface cationization, Carbohydr. Polym., 2010, 79(4), 1046–1051, DOI:10.1016/j.carbpol.2009.10.044.
- N. E. Mushi, N. Butchosa, M. Salajkova, Q. Zhou and L. A. Berglund, Nanostructured membranes based on native chitin nanofibers prepared by mild process, Carbohydr. Polym., 2014, 112, 255–263, DOI:10.1016/j.carbpol.2014.05.038.
- Y. Ji, D. E. Shen, Y. Lu, G. T. Schueneman, M. L. Shofner and J. C. Meredith, Aqueous-based recycling of cellulose nanocrystal/chitin nanowhisker barrier coatings, ACS Sustainable Chem. Eng., 2023, 11(29), 10874–10883, DOI:10.1021/acssuschemeng.3c02457.
- Z. Yu, Y. Ji and J. C. Meredith, Multilayer chitin–chitosan–cellulose barrier coatings on poly(ethylene terephthalate), ACS Appl. Polym. Mater., 2022, 4(10), 7182–7190, DOI:10.1021/acsapm.2c01059.
- Y. Hu, L. Liu, J. Yu, Z. Wang and Y. Fan, Preparation of silk nanowhisker-composited amphoteric cellulose/chitin nano fiber membranes, Biomacromolecules, 2020, 21(4), 1625–1635, DOI:10.1021/acs.biomac.0c00223.
- V. A. Petrova, V. Y. Elokhovskiy, S. V. Raik, D. N. Poshina, D. P. Romanov and Y. A. Skorik, Alginate gel reinforcement with chitin nanowhiskers modulates rheological properties and drug release profile, Biomolecules, 2019, 9(7), 291, DOI:10.3390/biom9070291.
- C. Peng, J. Xu, G. Chen, J. Tian and M. He, The preparation of α-chitin nanowhiskers-poly (vinyl alcohol) hydrogels for drug release, Int. J. Biol. Macromol., 2019, 131, 336–342, DOI:10.1016/j.ijbiomac.2019.03.015.
- R. Fernández-Marín, F. Hernández-Ramos, A. M. Salaberria, M.Á Andrés, J. Labidi and S. C. M. Fernandes, Eco-friendly isolation and characterization of nanochitin from different origins by microwave irradiation: Optimization using response surface methodology, Int. J. Biol. Macromol., 2021, 186, 218–226, DOI:10.1016/j.ijbiomac.2021.07.048.
- H. V. D. Nguyen, R. de Vries and S. D. Stoyanov, Chitin nanowhiskers with improved properties obtained using natural deep eutectic solvent and mild mechanical processing, Green Chem., 2022, 24(9), 3834–3844, 10.1039/D2GC00305H.
- A. J. Uddin, M. Fujie, S. Sembo and Y. Gotoh, Outstanding reinforcing effect of highly oriented chitin whiskers in PVA nanocomposites, Carbohydr. Polym., 2012, 87(1), 799–805, DOI:10.1016/j.carbpol.2011.08.071.
- M. Yanat and K. Schroën, Advances in chitin-based nanoparticle use in biodegradable polymers: A review, Carbohydr. Polym., 2023, 312, 120789, DOI:10.1016/j.carbpol.2023.120789.
- R. Fernández Marín, PhD Thesis, University of the Basque Country (UPV/EHU), 2021.
- N. Lin, S. Wei, T. Xia, F. Hu, J. Huang and A. Dufresne, Green bionanocomposites from high-elasticity “soft” polyurethane and high-crystallinity “rigid” chitin nanocrystals with controlled surface acetylation, RSC Adv., 2014, 4(90), 49098–49107, 10.1039/C4RA07899C.
- Y. Ji, S. Waters, E. Lim, A. W. Lang, P. N. Ciesielski, M. L. Shofner, J. R. Reynolds and J. C. Meredith, Minimizing oxygen permeability in chitin/cellulose nanomaterial coatings by tuning chitin deacetylation, ACS Sustainable Chem. Eng., 2022, 10(1), 124–133, DOI:10.1021/acssuschemeng.1c05051.
- Y. Ji, D. E. Shen, E. K. Young, C. L. Goins, J. R. Reynolds, M. L. Shofner and J. C. Meredith, Optimization of spray-coated nanochitin/nanocellulose films as renewable oxygen barrier layers via thermal treatment, Mater. Adv., 2022, 3(22), 8351–8360, 10.1039/d2ma00832g.
- A. G. B. Pereira, A. R. Fajardo, A. P. Gerola, J. H. S. Rodrigues, C. V. Nakamura, E. C. Muniz and Y. L. Hsieh, First report of electrospun cellulose acetate nanofibers mats with chitin and chitosan nanowhiskers: Fabrication, characterization, and antibacterial activity, Carbohydr. Polym., 2020, 250, 116954, DOI:10.1016/j.carbpol.2020.116954.
- C. Chen, S. Deng, Y. Yang, D. Yang, T. Ye and D. Li, Highly transparent chitin nanofiber/gelatin nanocomposite with enhanced mechanical properties, Cellulose, 2018, 25(9), 5063–5070, DOI:10.1007/s10570-018-1915-z.
- H. Lisboa, Reinforcement of poly (vinyl alcohol) films with alpha-chitin nanowhiskers, Polimeros, 2018, 28(1), 69–75, DOI:10.1590/0104-1428.07916.
- H. Jafari, M. Shahrousvand and B. Kaffashi, Preparation and characterization of reinforced poly (ε-caprolactone) nanocomposites by cellulose nanowhiskers, Polym. Compos., 2020, 41(2), 624–632, DOI:10.1002/pc.25393.
- P. M. Visakh, M. Monti, D. Puglia, M. Rallini, C. Santulli, F. Sarasini, S. Thomas and J. M. Kenny, Mechanical and thermal properties of crab chitin reinforced carboxylated sbr composites, eXPRESS Polym. Lett., 2012, 6(5), 396–409, DOI:10.3144/expresspolymlett.2012.42.
- K. G. Nair, A. Dufresne, A. Gandini and M. N. Belgacem, Crab shell chitin whiskers reinforced natural rubber nanocomposites. 3. Effect of chemical modification of chitin whiskers, Biomacromolecules, 2003, 4(6), 1835–1842, DOI:10.1021/bm030058g.
- M. Shibata, M. Enjoji, K. Sakazume and S. Ifuku, Bio-based epoxy/chitin nanofiber composites cured with amine-type hardeners containing chitosan, Carbohydr. Polym., 2016, 144, 89–97, DOI:10.1016/j.carbpol.2016.02.033.
- Y. M. Lee, S. H. Kim and S. J. Kim, Preparation and characteristics of β-chitin and poly (vinyl alcohol) blend, Polymer, 1996, 37(26), 5897–5905 CrossRef CAS.
- M. Peesan, R. Rujiravanit and P. Supaphol, Characterisation of beta-chitin/poly(vinyl alcohol) blend films, Polym. Test., 2003, 22(4), 381–387, DOI:10.1016/S0142-9418(02)00118-6.
- A. Amalraj, S. Jude and S. Gopi, Chapter 1 - polymer blends, composites and nanocomposites from chitin and chitosan; manufacturing, characterization and applications, in Handbook of chitin and chitosan, ed. S. Gopi, S. Thomas and A. Pius, Elsevier, 2020 Search PubMed.
- T. Basak and J. L. Shamshina, Design of chitin cell culture matrices for 3D tissue engineering: The importance of chitin types, solvents, cross-linkers, and fabrication techniques, Pharmaceutics, 2024, 16(6), 777, DOI:10.3390/pharmaceutics16060777.
- Y. Zhou, S. Fu, Y. Pu, S. Pan and A. J. Ragauskas, Preparation of aligned porous chitin nanowhisker foams by directional freeze–casting technique, Carbohydr. Polym., 2014, 112, 277–283, DOI:10.1016/j.carbpol.2014.05.062.
- E. Robles, A. M. Salaberria, R. Herrera, S. C. M. Fernandes and J. Labidi, Self-bonded composite films based on cellulose nanofibers and chitin nanocrystals as antifungal materials, Carbohydr. Polym., 2016, 144, 41–49, DOI:10.1016/j.carbpol.2016.02.024.
- Y. Lu, L. Weng and L. Zhang, Morphology and properties of soy protein isolate thermoplastics reinforced with chitin whiskers, Biomacromolecules, 2004, 5(3), 1046–1051, DOI:10.1021/bm034516x.
- L. Broers, S. van Dongen, V. de Goederen, M. Ton, J. Spaen, J. Boeriu and K. Schroën, Addition of chitin nanoparticles improves polylactic acid film properties, Nanotechnol. Adv. Mater. Sci., 2018, 1(2), 1–8, DOI:10.31038/NAMS.2018121.
- M. N. M. Azlin, S. M. Sapuan, E. S. Zainudin, M. Y. M. Zuhri and R. A. Ilyas, Chapter 3 - natural polylactic acid-based fiber composites: A Review, in Advanced processing, properties, and applications of starch and other bio-based polymers, ed. F. M. Al-Oqla and S. M. Sapuan, Elsevier, 2020 Search PubMed.
- M. Rajabi, A. Ali, M. McConnell and J. Cabral, Keratinous materials: Structures and functions in biomedical applications, Mater. Sci. Eng., C, 2020, 110, 110612, DOI:10.1016/j.msec.2019.110612.
- D. Dixit, R. Pal, G. Kapoor and M. Stabenau, 6 - lightweight composite materials processing, in Lightweight ballistic composites, ed. A. Bhatnagar, Woodhead Publishing, 2nd edn, 2016 Search PubMed.
- A. Hannibal and B. Weir, 11 - ballistic material processing, in Lightweight ballistic composites, ed. A. Bhatnagar, Woodhead Publishing, 2006 Search PubMed.
- M. Shibata, J. Fujigasaki, M. Enjoji, A. Shibita, N. Teramoto and S. Ifuku, Amino acid-cured bio-based epoxy resins and their biocomposites with chitin- and chitosan-nanofibers, Eur. Polym. J., 2018, 98, 216–225, DOI:10.1016/j.eurpolymj.2017.11.024.
- A. Wood, Practical methods of mixing additives with polymers, in Plastics additives: An a-z reference, ed. G. Pritchard, Springer Netherlands, Dordrecht, 1998 Search PubMed.
- L. A. Utracki, G. Z.-H. Shi, D. Rodrigue and R. Gonzalez-Núñez, Compounding polymer blends, in Polymer blends handbook, ed. L. A. Utracki and C. A. Wilkie, Springer Netherlands, Dordrecht, 2014 Search PubMed.
- P. Hold, Mixing of polymers— an overview part i, Adv. Polym. Technol., 1982, 2(2), 141–151, DOI:10.1002/adv.1993.060020205.
- M. B. Coltelli, P. Cinelli, V. Gigante, L. Aliotta, P. Morganti, L. Panariello and A. Lazzeri, Chitin nanofibrils in poly(lactic acid) (PLA) nanocomposites: Dispersion and thermo-mechanical properties, Int. J. Mol. Sci., 2019, 20(3), 504, DOI:10.3390/ijms20030504.
- N. Fijoł, A. Aguilar Sanchez, A. Mautner and A. P. Mathew, 3D-printed polylactic acid-based biocomposite filters reinforced with tempo-oxidized cellulose nanofibers and chitin nanofibers for removal of metal ions and microplastics from water, SSRN Electron. J., 2022, 4156467, DOI:10.2139/ssrn.4156467.
- J. Xue, T. Wu, Y. Dai and Y. Xia, Electrospinning and electrospun nanofibers: Methods, materials, and applications, Chem. Rev., 2019, 119(8), 5298–5415, DOI:10.1021/acs.chemrev.8b00593.
- A. Frenot and I. S. Chronakis, Polymer nanofibers assembled by electrospinning, Curr. Opin. Colloid Interface Sci., 2003, 8(1), 64–75, DOI:10.1016/S1359-0294(03)00004-9.
- L. T. Hao, Y. Eom, T. H. Tran, J. M. Koo, J. Jegal, S. Y. Hwang, D. X. Oh and J. Park, Rediscovery of nylon upgraded by interactive biorenewable nano-fillers, Nanoscale, 2020, 12(4), 2393–2405, 10.1039/C9NR08091K.
- Y. Chen, L. Gan, J. Huang and A. Dufresne, Reinforcing mechanism of cellulose nanocrystals in nanocomposites, in Nanocellulose: From Fundamentals to Advanced Materials, 2019, pp. 201–250. DOI:10.1002/9783527807437.ch7.
- G. Satchanska, S. Davidova and P. D. Petrov, Natural and synthetic polymers for biomedical and environmental applications, Polymers, 2024, 16(8), 1159, DOI:10.3390/polym16081159.
- F. A. Yihun, Nanochitin preparation and its application in polymer nanocomposites: A review, Emergent Mater., 2022, 5(6), 2031–2060, DOI:10.1007/s42247-022-00427-2.
- V. Rubentheren, T. A. Ward, C. Y. Chee and C. K. Tang, Processing and analysis of chitosan nanocomposites reinforced with chitin whiskers and tannic acid as a crosslinker, Carbohydr. Polym., 2015, 115, 379–387, DOI:10.1016/j.carbpol.2014.09.007.
- F. Zhu, C. Wang, S. Yang, Q. Wang, F. Liang, C. Liu, D. Qiu, X. Qu, Z. Hu and Z. Yang, Injectable tissue adhesive composite hydrogel with fibroblasts for treating skin defects, J. Mater. Chem. B, 2017, 5(13), 2416–2424, 10.1039/c7tb00384f.
- M. Santos, O. Del Carlo, J. M. Hong, Z. Liu, S. Jiang, S. Hrapovic, E. Lam, T. Jin and A. Moores, Effect of surface functionality on the rheological and self-assembly properties of chitin and chitosan nanocrystals and use in biopolymer films, Biomacromolecules, 2023, 24(9), 4180–4189, DOI:10.1021/acs.biomac.3c00513.
- Y. Huang, M. Yao, X. Zheng, X. Liang, X. Su, Y. Zhang, A. Lu and L. Zhang, Effects of chitin whiskers on physical properties and osteoblast culture of alginate based nanocomposite hydrogels, Biomacromolecules, 2015, 16(11), 3499–3507, DOI:10.1021/acs.biomac.5b00928.
- K. Kurita, Chitin and chitosan: Functional biopolymers from marine crustaceans, Mar. Biotechnol., 2006, 8(3), 203–226, DOI:10.1007/s10126-005-0097-5.
- P. Ravikumar and S. Sagadevan, Influence of the addition of chitin nanocrystals on the characteristics of cellulose acetate films, Polimery, 2021, 66(2), 98–104, DOI:10.14314/polimery.2021.2.2.
- J. Yu, J. Yang, B. Liu and X. Ma, Preparation and characterization of glycerol plasticized-pea starch/zno–carboxymethylcellulose sodium nanocomposites, Bioresour. Technol., 2009, 100(11), 2832–2841, DOI:10.1016/j.biortech.2008.12.045.
- F. Xie, E. Pollet, P. J. Halley and L. Avérous, Starch-based nano-biocomposites, Prog. Polym. Sci., 2013, 38(10), 1590–1628, DOI:10.1016/j.progpolymsci.2013.05.002.
- C. Chen, Y. Wang, Y. Yang, M. Pan, T. Ye and D. Li, High strength gelatin-based nanocomposites reinforced by surface-deacetylated chitin nanofiber networks, Carbohydr. Polym., 2018, 195, 387–392, DOI:10.1016/j.carbpol.2018.04.095.
- L. Liu, R. Wang, J. Yu, J. Jiang, K. Zheng, L. Hu, Z. Wang and Y. Fan, Robust self-standing chitin nanofiber/nanowhisker hydrogels with designed surface charges and ultralow mass content via gas phase coagulation, Biomacromolecules, 2016, 17(11), 3773–3781, DOI:10.1021/acs.biomac.6b01278.
- Q. Zhang, P. Sun, Z. Xu, W. Qu, Y. Zhang and X. Sui, Chitin nanocrystals as natural gel modifier for yielding stronger acid-induced soy protein isolate gel, Carbohydr. Polym., 2024, 323, 121446, DOI:10.1016/j.carbpol.2023.121446.
- Q. Zhang, L. Jiang and X. Sui, Incorporating chitin nanocrystal yields stronger soy protein gel: Insights into linear and nonlinear rheological behaviors by oscillatory shear tests, Food Hydrocolloids, 2023, 135, 108177, DOI:10.1016/j.foodhyd.2022.108177.
- A. M. Abdel-Mohsen, J. Jancar, R. M. Abdel-Rahman, L. Vojtek, P. Hyršl, M. Dušková and H. Nejezchlebová, A novel in situ silver/hyaluronan bio-nanocomposite fabrics for wound and chronic ulcer dressing: In vitro and in vivo evaluations, Int. J. Pharm., 2017, 520(1), 241–253, DOI:10.1016/j.ijpharm.2017.02.003.
- L. David, V. Dulong, D. Le Cerf, L. Cazin, M. Lamacz and J.-P. Vannier, Hyaluronan hydrogel: An appropriate three-dimensional model for evaluation of anticancer drug sensitivity, Acta Biomater., 2008, 4(2), 256–263, DOI:10.1016/j.actbio.2007.08.012.
- A. M. Abdel-Mohsen, D. Pavliňák, M. Čileková, P. Lepcio, R. M. Abdel-Rahman and J. Jančář, Electrospinning of hyaluronan/polyvinyl alcohol in presence of in situ silver nanoparticles: Preparation and characterization, Int. J. Biol. Macromol., 2019, 139, 730–739, DOI:10.1016/j.ijbiomac.2019.07.205.
- H. Tan, J. Wu, L. Lao and C. Gao, Gelatin/chitosan/hyaluronan scaffold integrated with plga microspheres for cartilage tissue engineering, Acta Biomater., 2009, 5(1), 328–337, DOI:10.1016/j.actbio.2008.07.030.
- J. Lou, F. Liu, C. D. Lindsay, O. Chaudhuri, S. C. Heilshorn and Y. Xia, Dynamic hyaluronan hydrogels with temporally modulated high injectability and stability using a biocompatible catalyst, Adv. Mater., 2018, 30(22), 1705215, DOI:10.1002/adma.201705215.
- V. C. Chandrasekaran, Chapter 11 - storage and service life of rubber seals, in Rubber seals for fluid and hydraulic systems, ed. V. C. Chandrasekaran, William Andrew Publishing, Oxford, 2010 Search PubMed.
- R. Amna, H. Mittal and S. M. Alhassan, Sulfur copolymers used for the removal of heavy metals from wastewater, in Reference module in materials science and materials engineering, Elsevier, 2024 Search PubMed.
- A. P. Mathew, Elastomeric nanocomposites reinforced with nanocellulose and nanochitin, in Handbook of nanocellulose and cellulose nanocomposites, ed. H. Kargarzadeh, I. Ahmad, S. Thomas and A. Dufresne, Wiley-VCH Verlag GmbH & Co. KGaA, Estolcomo, 2017 Search PubMed.
- R. A. Chowdhury, A. M. Sadri and M. E. Hoque, 18 - industrial implementations of biocomposites, in Green biocomposites for biomedical engineering, ed. M. E. Hoque, A. Sharif and M. Jawaid, Woodhead Publishing, 2021 Search PubMed.
- V. K. Abitha, S. Thomas, A. V. Rane and K. Kanny, 3 – raw materials—rubber dam, in Hydraulic rubber dam, ed. S. Thomas, A. V. Rane, V. K. Abitha, K. Kanny and A. Dutta, William Andrew Publishing, 2019 Search PubMed.
- A. Y. Coran, Chapter 7 - vulcanization, in The science and technology of rubber, ed. J. E. Mark, B. Erman and C. M. Roland, Academic Press, Boston, 4th edn, 2013 Search PubMed.
- A. bin Samsuri, 3.33 - degradation of natural rubber and synthetic elastomers, in Shreir's corrosion, ed. B. Cottis, M. Graham, R. Lindsay, S. Lyon, T. Richardson, D. Scantlebury and H. Stott, Elsevier, Oxford, 2010 Search PubMed.
- A. Dorigato, D. Rigotti and G. Fredi, Recent advances in the devulcanization technologies of industrially relevant sulfur-vulcanized elastomers, Adv. Ind. Eng. Polym. Res., 2023, 6(3), 288–309, DOI:10.1016/j.aiepr.2022.11.003.
- A. S. Sethulekshmi, A. Saritha and K. Joseph, A comprehensive review on the recent advancements in natural rubber nanocomposites, Int. J. Biol. Macromol., 2022, 194, 819–842, DOI:10.1016/j.ijbiomac.2021.11.134.
- A. Kawano, K. Yamamoto and J.-i. Kadokawa, Preparation of self-assembled chitin nanofiber-natural rubber composite sheets and porous materials, Biomolecules, 2017, 7(3), 47, DOI:10.3390/biom7030047.
- M. E. Mackay, T. T. Dao, A. Tuteja, D. L. Ho, B. Van Horn, H.-C. Kim and C. J. Hawker, Nanoscale effects leading to non-einstein-like decrease in viscosity, Nat. Mater., 2003, 2(11), 762–766, DOI:10.1038/nmat999.
- B. Ding, S. Huang, K. Shen, J. Hou, H. Gao, Y. Duan and J. Zhang, Natural rubber bio-nanocomposites reinforced with self-assembled chitin nanofibers from aqueous koh/urea solution, Carbohydr. Polym., 2019, 225, 115230, DOI:10.1016/j.carbpol.2019.115230.
- J. Yin, J. Hou, S. Huang, N. Li, M. Zhong, Z. Zhang, Y. Geng, B. Ding, Y. Chen and Y. Duan, et al., Effect of surface chemistry on the dispersion and pH-responsiveness of chitin nanofibers/natural rubber latex nanocomposites, Carbohydr. Polym., 2019, 207, 555–562, DOI:10.1016/j.carbpol.2018.12.025.
- H. Angellier, S. Molina-Boisseau, L. Lebrun and A. Dufresne, Processing and structural properties of waxy maize starch nanocrystals reinforced natural rubber, Macromolecules, 2005, 38(9), 3783–3792, DOI:10.1021/ma050054z.
- Y.-P. Wu, G.-H. Liang and L.-Q. Zhang, Influence of starch on the properties of carbon-black-filled styrene–butadiene rubber composites, J. Appl. Polym. Sci., 2009, 114(4), 2254–2260, DOI:10.1002/app.30649.
- H. M. Eyssa, D. E. Abulyazied, M. Abdulrahman and H. A. Youssef, Mechanical and physical properties of nanosilica/nitrile butadiene rubber composites cured by gamma irradiation, Egypt. J. Pet., 2018, 27(3), 383–392, DOI:10.1016/j.ejpe.2017.06.004.
- E. S. Abdul Rashid, N. B. Muhd Julkapli and W. Abdul Hadi Yehya, Reinforcement effect of nanocellulose on thermal stability of nitrile butadiene rubber (NBR) composites, J. Appl. Polym. Sci., 2018, 135(32), 46594, DOI:10.1002/app.46594.
- M. C. D. Dominic, R. Joseph, P. M. S. Begum, M. Joseph, D. Padmanabhan, L. A. Morris, A. S. Kumar and K. Formela, Cellulose nanofibers isolated from the cuscuta reflexa plant as a green reinforcement of natural rubber, Polymers, 2020, 12(4), 814, DOI:10.3390/POLYM12040814.
- A. Paul, K. Sreedevi, S. S. Sharma and V. N. Anjana, Polylactic acid (PLA), in Handbook of biopolymers, ed. S. Thomas, A. Ar, C. Jose Chirayil and B. Thomas, Springer Nature Singapore, Singapore, 2022 Search PubMed.
- D. Garlotta, A literature review of poly(lactic acid), J. Polym. Environ., 2001, 9(2), 63–84, DOI:10.1023/A:1020200822435.
- M. F. Ashby, Chapter 15 - material profiles, in Materials and the environment, ed. M. F. Ashby, Butterworth-Heinemann, Boston, 2nd edn, 2013 Search PubMed.
- T. C. Mokhena, J. S. Sefadi, E. R. Sadiku, M. J. John, M. J. Mochane and A. Mtibe, Thermoplastic processing of PLA/cellulose nanomaterials composites, Polymers, 2018, 10(12), 1363, DOI:10.3390/polym10121363.
- R. Scaffaro, L. Botta, F. Lopresti, A. Maio and F. Sutera, Polysaccharide nanocrystals as fillers for PLA based nanocomposites, Cellulose, 2017, 24(2), 447–478, DOI:10.1007/s10570-016-1143-3.
- S. Farah, D. G. Anderson and R. Langer, Physical and mechanical properties of PLA, and their functions in widespread applications—a comprehensive review, Adv. Drug Delivery Rev., 2016, 107, 367–392, DOI:10.1016/j.addr.2016.06.012.
- . Mayilswamy and B. Kandasubramanian, Green composites prepared from soy protein, polylactic acid (PLA), starch, cellulose, chitin: A review, Emergent Mater., 2022, 5(3), 727–753, DOI:10.1007/s42247-022-00354-2.
- Z. W. Abdullah, Y. Dong, I. J. Davies and S. Barbhuiya, PVA, PVA blends, and their nanocomposites for biodegradable packaging application, Polym.-Plast. Technol. Eng., 2017, 56(12), 1307–1344, DOI:10.1080/03602559.2016.1275684.
- I. Saini, A. Sharma, R. Dhiman, S. Aggarwal, S. Ram and P. K. Sharma, Grafted sic nanocrystals: For enhanced optical, electrical and mechanical properties of polyvinyl alcohol, J. Alloys Compd., 2017, 714, 172–180, DOI:10.1016/j.jallcom.2017.04.183.
- M. Aslam, M. A. Kalyar and Z. A. Raza, Fabrication of reduced graphene oxide nanosheets doped PVA composite films for tailoring their opto-mechanical properties, Appl. Phys. A: Mater. Sci. Process., 2017, 123(6), 424, DOI:10.1007/s00339-017-1035-x.
- M. Aslam, M. A. Kalyar and Z. A. Raza, Polyvinyl alcohol: A review of research status and use of polyvinyl alcohol based nanocomposites, Polym.-Plast. Technol. Eng., 2018, 58(12), 2119–2132, DOI:10.1002/pen.24855.
- G. S. A. Suleiman, X. Zeng, R. Chakma, I. Y. Wakai and Y. Feng, Recent advances and challenges in thermal stability of PVA-based film: A review, Polym. Adv. Technol., 2024, 35(2), e6327, DOI:10.1002/pat.6327.
- X. Zhou, X. Zhou, L. Zhou, M. Jia and Y. Xiong, Nanofillers in novel food packaging systems and their toxicity issues, Foods, 2024, 13(13), 2014, DOI:10.3390/foods13132014.
- S. Moulay, Review: Poly(vinyl alcohol) functionalizations and applications, Polym.-Plast. Technol. Eng., 2015, 54(12), 1289–1319, DOI:10.1080/03602559.2015.1021487.
- N. L. Bhandari, G. Bhandari, K. Bist, D. Adhikari, K. N. Dhakal, R. Adhikari, R. Lach, A. A. Kim, D. J. Yoo and M. B. Poudel, Comparative investigation of fillers loading effect on morphological, micromechanical, and thermal properties of polyvinyl alcohol/cellulosicfillers-based composites, Int. J. Biol. Macromol., 2024, 280, 136192, DOI:10.1016/j.ijbiomac.2024.136192.
- S. Jiao, X. Yang, X. Zheng, Y. Pei, J. Liu and K. Tang, Effects of charge state of nano-chitin on the properties of polyvinyl alcohol composite hydrogel, Carbohydr. Polym., 2024, 330, 121776, DOI:10.1016/j.carbpol.2024.121776.
- J. L. Massingill and R. S. Bauer, Epoxy resins, in Applied polymer science: 21st century, ed. C. D. Craver and C. E. Carraher, Pergamon, Oxford, 2000 Search PubMed.
- S. Bello, J. Agunsoye, B. Hassan, M. Zebaze Kana and I. Raheem, Epoxy resin based composites, mechanical and tribological properties: A Review, Tribol. Ind., 2015, 37, 500–524 Search PubMed.
- M. A. Rashid, M. A. Islam, M. N. Hasan, M. N. N. Anu and M. H. Ikbal, A critical review of dynamic bonds containing curing agents for epoxy resin: Synthesis, challenges, and emerging applications, Polym. Degrad. Stab., 2024, 229, 110980, DOI:10.1016/j.polymdegradstab.2024.110980.
- M. Wang, L. Ma, L. Shi, P. Feng, X. Wang, Y. Zhu, G. Wu and G. Song, Chemical grafting of nano-sio2 onto graphene oxide via thiol-ene click chemistry and its effect on the interfacial and mechanical properties of go/epoxy composites, Compos. Sci. Technol., 2019, 182, 107751, DOI:10.1016/j.compscitech.2019.107751.
- Q. Hao, S. Liu, X. Wang, P. Zhang, Z. Mao and X. Zhang, Progression from graphene and graphene oxide to high-performance epoxy resin-based composite, Polym. Degrad. Stab., 2024, 223, 110731, DOI:10.1016/j.polymdegradstab.2024.110731.
- H. Dong, Y. Qiao, K. Yang, Y. Zhen, W. Tan, Q. Cheng and Y. Wang, Epoxy resin coatings doped with layered double hydroxide for enhanced anti-permeation performance, Appl. Clay Sci., 2024, 258, 107505, DOI:10.1016/j.clay.2024.107505.
- A. J. Chandran, S. M. Rangappa, I. Suyambulingam and S. Siengchin, Micro/nano fillers for value-added polymer composites: A comprehensive review, J. Vinyl Addit. Technol., 2024, 30(5), 1083–1123, DOI:10.1002/vnl.22106.
- T. Aziz, F. Haq, A. Farid, L. Cheng, L. F. Chuah, A. Bokhari, M. Mubashir, D. Y. Y. Tang and P. L. Show, The epoxy resin system: Function and role of curing agents, Carbon Lett., 2024, 34(1), 477–494, DOI:10.1007/s42823-023-00547-7.
- B. Azimi, P. Nourpanah, M. Rabiee and S. Arbab, Poly (ε-caprolactone) fiber: An overview, J. Eng. Fibers Fabr., 2014, 9(3), 155892501400900320, DOI:10.1177/155892501400900309.
- F. Taghizadeh, M. Heidari, S. Mostafavi, S. M. Mortazavi and A. Haeri, A review of preparation methods and biomedical applications of poly(ε-caprolactone)-based novel formulations, J. Mater. Sci., 2024, 59(24), 10587–10622, DOI:10.1007/s10853-024-09774-3.
- E. V. Antonakou and D. S. Achilias, Recent advances in polycarbonate recycling: A Review of degradation methods and their mechanisms, Waste Biomass Valorization, 2013, 4(1), 9–21, DOI:10.1007/s12649-012-9159-x.
- Y. H. Teow, A. W. Mohammad, S. Ramli, M. S. Sajab and N. Mohamad Mazuki, Potential, of membrane technology for treatment and reuse of water from old mining lakes, Sains Malays., 2018, 47, 2887–2897, DOI:10.17576/jsm-2018-4711-32.
- A. Tiraferri, M. Malaguti, M. Mohamed, M. Giagnorio and F. J. Aschmoneit, Standardizing practices and flux predictions in membrane science via simplified equations and membrane characterization, npj Clean Water, 2023, 6(1), 58, DOI:10.1038/s41545-023-00270-w.
- N. Alele and M. Ulbricht, Membrane-based purification of proteins from nanoparticle dispersions: Influences of membrane type and ultrafiltration conditions, Sep. Purif. Technol., 2016, 158, 171–182, DOI:10.1016/j.seppur.2015.11.031.
- F. Christakopoulos, P. M. H. van Heugten and T. A. Tervoort, Additive manufacturing of polyolefins, Polymers, 2022, 14(23), 5147, DOI:10.3390/polym14235147.
- M. Zakaria, M. A. R. Bhuiyan, M. S. Hossain, N. M. M. U. Khan, M. A. Salam and K. Nakane, Advances of polyolefins from fiber to nanofiber: Fabrication and recent applications, Discover Nano, 2024, 19(1), 24, DOI:10.1186/s11671-023-03945-y.
- D. Meng, J. Xie, G. I. N. Waterhouse, K. Zhang, Q. Zhao, S. Wang, S. Qiu, K. Chen, J. Li and C. Ma, et al., Biodegradable poly(butylene adipate-co-terephthalate) composites reinforced with bio-based nanochitin: Preparation, enhanced mechanical and thermal properties, J. Appl. Polym. Sci., 2020, 137(12), 48485, DOI:10.1002/app.48485.
- C. Zhang, Biodegradable polyesters: Synthesis, properties, applications, in Biodegradable polyesters, 2015 Search PubMed.
- H. Kim, T. Kim, S. Choi, H. Jeon, D. X. Oh, J. Park, Y. Eom, S. Y. Hwang and J. M. Koo, Remarkable elasticity and enzymatic degradation of bio-based poly(butylene adipate-co-furanoate): Replacing terephthalate, Green Chem., 2020, 22(22), 7778–7787, DOI:10.103s9/D0GC01688H.
- U. M. Shrestha, L. Han, T. Saito, K. S. Schweizer and M. D. Dadmun, Mechanism of soft nanoparticle diffusion in entangled polymer melts, Macromolecules, 2020, 53(17), 7580–7589, DOI:10.1021/acs.macromol.0c00870.
- Y. Liu, J. Zhao, Y. Peng, J. Luo, L. Cao and X. Liu, Comparative study on the properties of epoxy derived from aromatic and heteroaromatic compounds: The role of hydrogen bonding, Ind. Eng. Chem. Res., 2020, 59(5), 1914–1924, DOI:10.1021/acs.iecr.9b05904.
- J. Z. Manapat, Q. Chen, P. Ye and R. C. Advincula, 3D printing of polymer nanocomposites via stereolithography, Macromol. Mater. Eng., 2017, 302(9), 1600553, DOI:10.1002/mame.201600553.
- H. Liu, X. Hu, W. Li, M. Zhu, J. Tian, L. Li, B. Luo, C. Zhou and L. Lu, A highly-stretchable and adhesive hydrogel for noninvasive joint wound closure driven by hydrogen bonds, Chem. Eng. J., 2023, 452, 139368, DOI:10.1016/j.cej.2022.139368.
- N. B. Palaganas, J. D. Mangadlao, A. C. C. de Leon, J. O. Palaganas, K. D. Pangilinan, Y. J. Lee and R. C. Advincula, 3D printing of photocurable cellulose nanocrystal composite for fabrication of complex architectures via stereolithography, ACS Appl. Mater. Interfaces, 2017, 9(39), 34314–34324, DOI:10.1021/acsami.7b09223.
- J. Zhang, F. Mohd Said, N. F. S. Daud and Z. Jing, Present status and application prospects of green chitin nanowhiskers: A comprehensive review, Int. J. Biol. Macromol., 2024, 278, 134235, DOI:10.1016/j.ijbiomac.2024.134235.
- K. Elumalai, S. Srinivasan and A. Shanmugam, Review of the efficacy of nanoparticle-based drug delivery systems for cancer treatment, Biomed. Technol., 2024, 5, 109–122, DOI:10.1016/j.bmt.2023.09.001.
- A. Haider, S. Khan, D. N. Iqbal, M. Shrahili, S. Haider, K. Mohammad, A. Mohammad, M. Rizwan, Q. Kanwal and G. Mustafa, Advances in chitosan-based drug delivery systems: A comprehensive review for therapeutic applications, Eur. Polym. J., 2024, 210, 112983, DOI:10.1016/j.eurpolymj.2024.112983.
- M. E. Hoque, Y. L. Chuan and I. Pashby, Extrusion based rapid prototyping technique: An advanced platform for tissue engineering scaffold fabrication, Biopolymers, 2012, 97(2), 83–93, DOI:10.1002/bip.21701.
- D. Gogoi, M. Kumar and J. Singh, A comprehensive review on hydrogel-based bio-ink development for tissue engineering scaffolds using 3D printing, Ann. 3D Print. Med., 2024, 15, 100159, DOI:10.1016/j.stlm.2024.100159.
- X. Pei, Y. Li, Y. Deng, L. Lu, W. Li, R. Shi, A. Lei and L. Zhang, Chitin microsphere supported Pd nanoparticles as an efficient and recoverable catalyst for CO oxidation and heck coupling reaction, Carbohydr. Polym., 2021, 251, 117020, DOI:10.1016/j.carbpol.2020.117020.
- H. Yuan, L. Deng, X. Cai, S. Zhou, Y. Chen and Y. Yuan, Nitrogen-doped carbon sheets derived from chitin as non-metal bifunctional electrocatalysts for oxygen reduction and evolution, RSC Adv., 2015, 5, 56121–56129, 10.1039/C5RA05461C.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5lp00104h |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |