
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStimuli-responsive thiocarbamate-based polymeric particles for hydrogen sulfide generation†
Daniel A.
Paterson
,
Aggie
Lawer
 ,
Jared
Davidson
,
Sarah
Hook
and
Allan B.
Gamble
*
,
Jared
Davidson
,
Sarah
Hook
and
Allan B.
Gamble
*
School of Pharmacy, University of Otago, 18 Frederick St, Dunedin, New Zealand 9054. E-mail: allan.gamble@otago.ac.nz
First published on 28th May 2025
Abstract
Hydrogen sulfide (H2S) imbalance has been implicated in pathologies, and reinstating H2S homeostasis could be a useful therapeutic strategy. However, delivery of H2S to the disease site remains a challenge. Functionalised nanoformulations could be used as a strategy to deliver high concentrations of H2S in a targeted manner. Use of a disease-associated trigger that activates and releases H2S would provide therapeutic selectivity. As proof-of-concept, synthesis and formulation of block co-polymers bearing a thiocarbamate bond, a carbonyl sulfide (COS) precursor, is described. Activation by hydrogen peroxide (H2O2), and a subsequent 1,6-self-immolation process leads to release of COS, which in the presence of carbonic anhydrase is hydrolysed to H2S. H2S generation was exemplified by reduction of an azido-pro-fluorophore. Formulation of the polymer resulted in compound vesicles that were able to encapsulate a model drug and could be useful in future biological studies exploring delivery of H2S as a therapeutic, or to activate azido-masked prodrug/pro-fluorophore in areas of high reactive oxygen species (ROS).
Introduction
Hydrogen sulfide (H2S) is a gaseous cell signalling molecule1 that is essential in maintaining homeostasis.2–4 Low levels of H2S are associated with, but not limited to, neurodegenerative disease (e.g., Alzheimer's disease),5 cardiovascular disease (e.g., myocardial infarction),6,7 pain2 and liver disease (e.g., non-alcoholic Steatohepatitis).8 Abnormal or unregulated levels of H2S have been linked to cancer.9,10 The loss of anti-oxidant effects when H2S homeostasis is not maintained is thought to play a key role in disease progression,11 hence therapeutics that can deliver H2S (H2S-donors) are of great interest.12,13 H2S, being a noxious gas, is delivered as a precursor, generally in the form of a salt (NaSH or Na2S) or small molecule (e.g., AP39 [1,2-dithiolethione]14 or GYY4137 [modified Lawesson's reagent]15). There are limitations in using salt-based H2S-donors, including rapid hydrolysis in water and an uncontrolled burst release.16 Non-specific, slow-releasing H2S-donors and rapid triggerable H2S-donors that can be activated by a non-specific stimuli (e.g., AP39 hydrolysis14), or specific endogenous stimuli (e.g., cysteine-sensitive dithioesters17 and self-amplifying H2S azide capped thiocarbamates17,18) have been documented. However, the concentration of H2S that can be generated in situ is limited for small molecules,19 and careful consideration of the pharmacokinetics and distribution profiles in designing small molecule H2S-donors is required.13H2S delivered in particulate systems, such as liposomes and polymeric particles (e.g., micelles, polymersomes and bicontinuous nanospheres), could improve delivery. Particles could enable prolonged release and increased local levels of H2S as well as improving stability and aqueous solubility of the H2S-donor.20 In addition to the delivery of a H2S payload, particles could co-deliver a second payload, for example a drug or diagnostic cargo,21,22 to gain synergistic benefits. Due to polymers versatility, polymeric particles are of great interest in stimuli-responsive drug delivery and more recently H2S generation.18,23,24 The amphiphilic block co-polymers (BCPs) that form particles in solution are synthetically adaptable and enable a precursory H2S-donor to be built-in to the hydrophobic polymer backbone.17,20,23 Upon formulation, any cargo is protected from the environment, and in the case of H2S donors,20,24,25 the stimuli-responsive/triggerable H2S precursor functional group is protected in the hydrophobic part of the particle.
Much like small molecule prodrugs26,27 and H2S-donors,14,20 the stimulus for particle activation can be endogenous to the disease (e.g., reactive oxygen species [ROS]) or exogenous (e.g., light-activation). Recent examples of stimuli-responsive H2S-donor particles include those that have used the S-aroylthiooxime (SATO) group and perthiols.24,28 However, there are no reports of the p-aminobenzyloxycarbonyl (PABC) and p-hydroxybenzyloxycarbonyl (PHBC) self-immolative linkers, commonly used in prodrug strategies, in H2S-donor particles. Due to the ease in which the stimuli-responsive trigger group on the PABC/PHBC linker can be modified to suit the disease-associated stimulus, we designed a BCP that contains a thiocarbamate group as part of an arylboronate self-immolative linker (p-boronate-benzyloxythiocarbonyl; BBOT), using existing aryl boronate trigger moieties linked with methacrylates to form novel BCPs. Combined with a hydrophilic PEG unit, the synthesised BCP can be readily formulated into particles that are responsive to hydrogen peroxide (H2O2),29–31 a ROS produced by the immune system in response to various pathological conditions (Fig. 1).
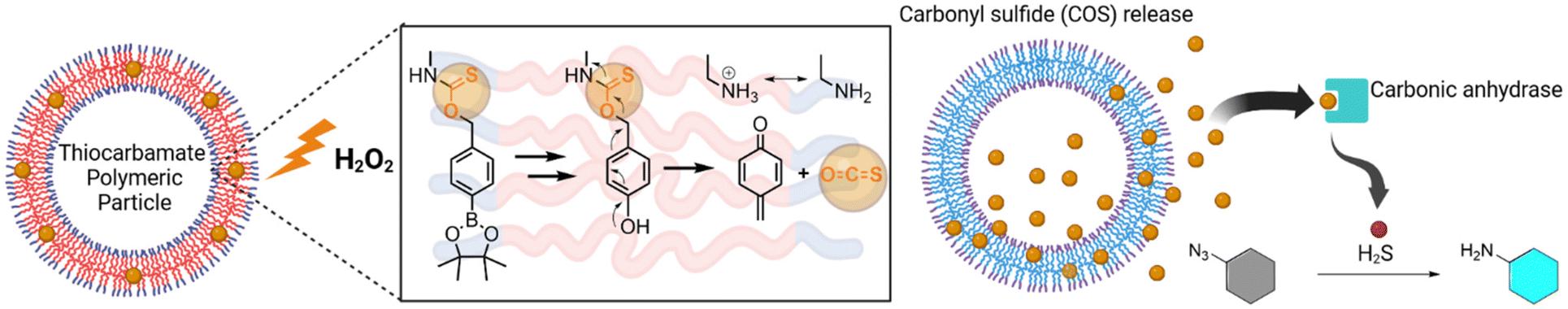 | ||
| Fig. 1 Proposed activation and release of carbonyl sulfide (COS) from thiocarbamate-modified polymeric nanoparticles via a hydrogen peroxide (H2O2)-triggered arylboronate oxidation and self-immolation. COS is catalytically and rapidly hydrolysed to hydrogen sulfide (H2S) by carbonic anhydrase. The H2S generated by the particles has potential as a direct H2S therapeutic or in the reductive activation of diagnostic or therapeutic aryl azides (i.e., pro-fluorophores and prodrugs). Created in BioRender. Gamble, A. (2025) https://BioRender.com/a62d458. | ||
Herein, we report on the synthesis of stimuli-responsive PEG-BBOT polymers and explore their H2S-producing capabilities in polymer and particle form (compound vesicles). The production of H2S via carbonic anhydrase-catalysed hydrolysis of carbonyl sulfide (COS) is exemplified by activation of an azido-functionalised pro-fluorescent dye. The particles show potential to generate high local concentrations of H2S for activation of azido-functionalised prodrugs or probes (Fig. 1).
Results and discussion
Synthesis of thiocarbamate and carbamate methacrylates
Small molecule and macromolecular H2S-donors normally lead to the direct generation of H2S,9 with mechanisms that fall into the following classes: (i) hydrolysis-mediated, (ii) thiol-promoted, (iii) photoactivation-mediated or enzyme-mediated.32 More recently, the generation of H2S via carbonic anhydrase-mediated hydrolysis of carbonyl sulfide (COS)33 has been gaining traction in H2S-donor research, with the ubiquitous carbonic anhydrase facilitating COS hydrolysis at the site of COS release. The precursor to COS is a thiocarbamate bond which is easily incorporated into PABC/PHBC-type self-immolative linkers.34 The use of a self-immolative linker as a COS donor provides an immediate advantage over other H2S generation methods as simple modification of the linker can enable the user to select the stimulus required for activation of COS/H2S. Pluth and co-workers have reported on the activation of self-immolative linkers that generate COS/H2S,34 using stimuli such as H2O2.35–37 The synthetic flexibility of the self-immolative linkers and responsive capping groups facilitates their incorporation into macromolecules as hydrophobic segments of BCPs.Herein, COS-releasing p-boronate-benzyloxythiocarbamate (BBOT, 1) was prepared by reacting 4-(hydroxymethyl)phenylboronic acid pinacol ester 4 with 2-isothiocyanatoethyl methacrylate 6 (Scheme 1A). As a control, a non-ROS-responsive p-fluoro-benzyloxythiocarbamate (FBOT, 2) was prepared from the reaction of benzyl alcohol 5 with isothiocyanate 6 (Scheme 1A) and a CO2-releasing carbamate methacrylate p-boronate-benzyloxycarbamate (BBOC, 3) was synthesised by reacting benzyl alcohol 4 with isocyanate 7 (Scheme 1B).38,39
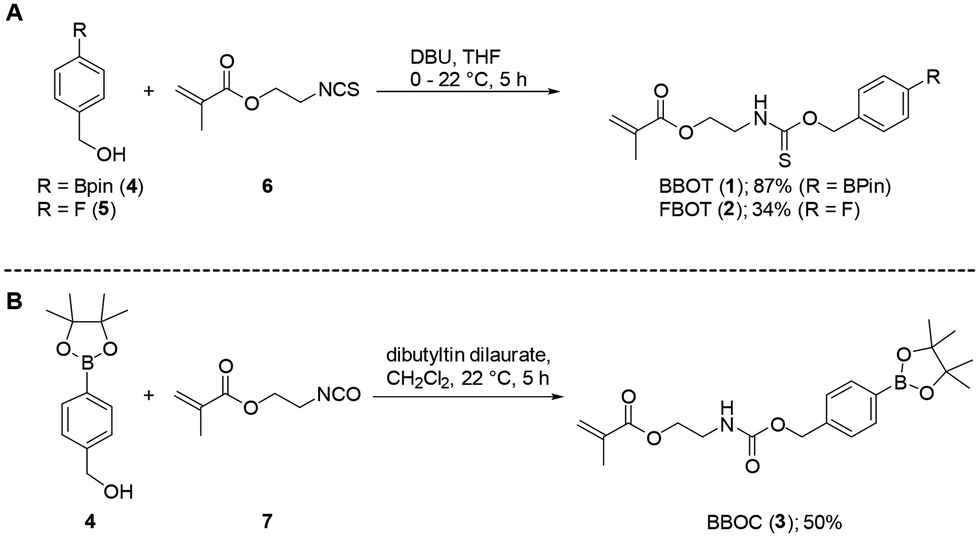 | ||
| Scheme 1 Synthesis of self-immolative (A) thiocarbamate and (B) carbamate methacrylate monomers. | ||
Prior to polymerisation with mPEG, the stimuli-responsiveness of methacrylates 1–3 to an oxidant (H2O2) (Scheme 2) was examined. Previous thiocarbamate-based COS-donors have used anilines as the leaving group,18,37 but in this instance an alkyl amine is released from the BBOT monomers after oxidatively driven self-immolation. Excess H2O2 was added to BBOT (1), FBOT (2) and BBOC (3), with the oxidation and self-immolation progress monitored by 1H NMR spectroscopy (Scheme 2, Fig. 2 [BBOT], Fig. S1 [FBOT] and S2/S3 [BBOC]†). Exposure of the FBOT control polymer to H2O2 resulted in no change to the 1H NMR (Fig. S1†). This suggests that the other functional groups in the monomers are stable towards H2O2, and the response observed in BBOT (1) can be attributed to boronic ester oxidation.
 | ||
| Scheme 2 Oxidation of arylboronate thiocarbamate monomer (BBOT; 1) with H2O2 and proposed trapping of methide observed using 1H NMR spectroscopy. | ||
 | ||
| Fig. 2 1H NMR of BBOT monomer (1) at 0, 0.25, 2 and 6 hours post-exposure to excess H2O2 in DMSO-d6:D2O-PBS solution (25 °C). The key changes in chemical shift that indicate rapid oxidation to the phenol and subsequent self-immolation are annotated as A to A1, B to B1, C to C1 (refer to Scheme 2) and D to D1 (pinacol protons). In BBOT (1) (top spectrum) the thiocarbamate bond gives rise to rotamers.40 | ||
The initial oxidation of the arylboronate BBOT (1) (Scheme 2) to the corresponding phenol and free pinacol boronic ester was confirmed by an upfield shift in the aromatic proton resonances (Fig. 2, peaks A to A1). Small amounts of arylboronic ester hydrolysis to the arylboronic acid was observed in the aromatic region (minor peaks at δ 7.25 to 7.75 ppm), and the upfield shift of the pinacol protons (peak D to D1) indicated hydrolysis of the released pinacol boronate ester (after phenol formation) to orthoboric acid and 2,3-dimethylbutane-2,3-diol. The oxidation step for 1 was comparable to that of the control BBOC monomer 3, which was transformed to the phenol within 2 hours (Fig. S2,† peak A to A1).
Substantial self-immolation of BBOT (1) occurred within 2–6 hours, supported by the shift of the benzylic peak from 5.43 to 3.91 (Fig. 2, B to B1). For BBOC (3), the benzylic proton peak had a relatively small upfield shift (δ 4.99 to 4.85 ppm, Scheme S2†), possibly due to a slower self-immolation of the resultant carbamate-phenol intermediate under the organic solvent conditions (Scheme S2, peak B†). Both reactions were conducted in a mixture of organic![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :aqueous solvent (DMSO-d6/D2O-PBS, 4:1), which was expected to slow the rate of self-immolation compared to pure aqueous conditions.41,42
:aqueous solvent (DMSO-d6/D2O-PBS, 4:1), which was expected to slow the rate of self-immolation compared to pure aqueous conditions.41,42
Based on our previous aryl azide self-immolative methacrylates activated by H2S,39 the quinone methide formed was expected to quench with phosphate or water;39,42 however this was not observed for the arylboronate methacrylate BBOT (1). Instead, the NMR evidence pointed towards attack of the released alkyl amine on the electrophilic methide to afford amine-linked methacylate analogue 8 (Scheme 2 and Fig. 2). From the 1H NMR study (Fig. 2), evidence suggesting the structure of 8 was provided by the upfield shift in the benzylic protons (B to B1; δ 5.44 to δ 3.92) and alkyl chain protons (C to C1; δ 4.20/3.69 to 4.06/3.38). If water or phosphate had reacted with the methide in place of the amine group, the benzylic protons at B/B1 would be expected to shift to δ ∼4.35 ppm (4-hydroxybenzyl alcohol) and δ ∼5 ppm (4-hydroxybenzyl phosphate);39,42 absent peaks in the NMR spectrum for 1 (Fig. 2). Alternatively, H2S generated from the released COS could itself act as a nucleophile and form a benzyl thiol compound (aromatic–CH2–SH). However, there was no observed benzylic proton (CH2) shift at δ ∼3.92 ppm in our previous report,39 even though the methide was generated in a large excess of H2S. This further supports the likely product with benzylic protons at 3.92 ppm as compound 8 and not a benzyl thiol.
The generation of COS from BBOT (1) via carbonic anhydrase (enzymatic) catalysed hydrolysis of COS to H2S was detected by measuring the activation of pro-fluorophore 7-azido-4-methylcoumarin (AzMC)43 (Fig. 3). BBOT (1), FBOT (2) and BBOC (3) were solubilized in PBS:DMSO (9:1) and incubated at 37 °C in the presence of H2O2 (10-fold excess) at pH 7.4. Activation of the pro-fluorophore AzMC was fast, with a rapid increase in fluorescence being observed when COS/H2S-releasing BBOT (1) was incubated with H2O2 and CA (Fig. 3, red). The control polymers, non-triggerable FBOT (2) and triggerable but CO2-releasing BBOC (3), produced no fluorescent signal (Fig. 3, green and blue). Further support for H2S generation from BBOT (1) was provided by comparison to an equimolar amount of peroxyTCM-2, the control H2O2-responsive small molecule COS/H2S donor37 (Fig. 3, black). Compared to BBOT (1), peroxyTCM-2 (synthesis described in ESI, section 1.5†) produced a slightly faster increase in fluorescence output than BBOT (1). However, the fluorescent output was the same after approximately 6 hours incubation.
 | ||
| Fig. 3 Activation of pro-fluorophore 7-azido-4-methylcoumarin (AzMC) after exposure to H2O2 in the presence of carbonic anhydrase in PBS (pH 7.4) at 37 °C from peroxyTCM-2 (black), BBOT (1) (red), FBOT (2) (green) and BBOC (3) (blue), as measured by fluorescence ex/em: 355/460 nm. | ||
Block co-polymer (BCP) synthesis from methacrylates
The methacrylate monomers 1, 2 and 3 were polymerised using Activators ReGenerated by Electron Transfer (ARGET) Atom Transfer Radical Polymerisation (ATRP).39 ARGET-ATRP was selected for its relatively mild conditions so that the boronic ester and thiocarbamate bond would remain intact during polymerisation.39 mPEG2000 macroinitiator (calculated at 49 repeating units via NMR spectroscopic analysis) was used to initiate the reaction with sodium ascorbate as the reducing agent.39,44 Targeted polymers of 10 kDa were synthesised to achieve a hydrophobic/hydrophilic balance of 0.2,45 with consistent conversion and narrow polydispersity (Đ) (Table 1). BCPs containing the BBOT monomer (1) were isolated as mPEG49-BBOT24 (10, ∼10 kDa, 24 × BBOT units, Đ = 1.19). The control polymers, mPEG49-FBOT38 (11) and mPEG49-BBOC23 (12) were synthesized with similar molecular weights and Đ (Table 1). The relative integration of the benzylic protons to the PEG protons (Fig. S16, S18 and S21†) indicated that connection of methacrylate to the PEG unit had occurred without any undesired degradation of the capping group (including the thiocarbamate bond).|
|
||||||
|---|---|---|---|---|---|---|
| Methacrylate | Polymer | DPnb |
Conversionc (%) |
M
nd |
Đ | n |
| a Number of moles monomer/methacrylate used (1, 2, or 3) described in the ESI.† b Degree of polymerisation. c Conversion of monomers into polymer, determined from molar input and DPn. d Number average molecular weight, determined via1H NMR analysis. e Determined via GPC analysis (Mw/Mn, see ESI†). n = replicate polymerisations. TPMA = tris(2-pyridylmethyl)amine. | ||||||
| 1 | mPEG49-BBOT24 (10) | 24 | 93 ± 3 | 11859 ± 906 |
1.19 ± 0.03 | 8 |
| 2 | mPEG49-FBOT38 (11) | 38 | 94 | 13643 |
1.17 | 1 |
| 3 | mPEG49-BBOC23 (12) | 23 | 79 ± 16 | 11439 ± 627 |
1.30 ± 0.08 | 4 |

Formulation of polymers and H2O2 triggered activation of particles producing H2S
Polymers were self-assembled via nanoprecipitation, using THF and the rapid addition of PBS. Compound vesicle-like particles39 were generated (Fig. S4†) but these were unstable at 37 °C, with aggregation occurring and average measured particle size increasing over 8–24 hours (Fig. S5†). To increase steric stabilization, 9% w/w Pluronic F127 was added.46,47 This prevented aggregation of the mPEG49-BBOT24 (10) particles (Fig. S5†) and resulted in particles of 130 ± 4 nm and narrow distribution (PDI = 0.106). Control polymers formed similar sized particles (Table S1†). The addition of Pluronic F127 altered particle morphology, giving a mixed morphology of polymersomes (vesicles) and large compound vesicles (Fig. 4A) with a unimodal particle size distribution of 130 ± 4 nm (Fig. 4C). The formation of these types of structures was in line with the increased time to kinetic entrapment.39 | ||
| Fig. 4 Cryo-TEM of mPEG49-pBBOT24 particles (10) formulated with Pluronic F127 9% w/w prior to (A) and after (B) exposure to 1 mM H2O2. Scale bar is 200 nm. (C) DLS histogram of particles after exposure to PBS (control, black) or 1 mM H2O2 (red). (D) Remaining DLS derived count rate of particles after exposure to PBS control (blue), 0.1 mM (orange) or 1 mM (red) H2O2. (E) Activation of pro-fluorophore AzMC via H2O2-triggering (∼10-fold excess) of mPEG49-BBOT24 (10, red), mPEG49-BBOC23 (12, green) or mPEG49-FBOT38 (11, blue) particles (all formulated with 9% w/w Pluronic and incubated with carbonic anhydrase), as measured by fluorescence ex/em: 355/460 nm. (F) GPC analysis of recovered polymers after exposure to PBS (blue) or 1 mM H2O2 (red) for 24 hours. | ||
Oxidation of the arylboronate within the mPEG49-pBBOT24 (10) particles was achieved by exposure to 1 mM H2O2. Triggered activation of the mPEG49-BBOT24 particles (10) was explored using NMR, DLS, GPC, electron microscopy and fluorescence assays (Fig. 4).
For oxidation of the arylboronate to occur in the particles, H2O2 must partition into the hydrophobic bilayer. The time for diffusion is expected to influence the kinetics of COS production20 which is expected to be slower than that of the free monomer.31,42 As predicted, after incubating mPEG49-BBOT24 (10) with H2O2 (∼10-fold excess), the generation of H2S via conversion of COS was observed to increase slowly over 8 hours, with a maximal fluorescent output at 24 hours (Fig. 4E). This was slower than the triggering of BBOT (1) monomer and peroxyTCM-2 (seen in Fig. 3) and represented an approx. 3-fold increase in fluorescent signal output compared to the control particles 11 and 12 (Fig. 4E) which did not produce any COS/H2S in the presence of H2O2 and carbonic anhydrase (background fluorescence only observed). While release was relatively slow for the mPEG49-BBOT10 particles, a challenge in H2S delivery is the selective and controlled release of the gas.12,13,20 The slower, steady H2S profile shown in Fig. 4E could prove beneficial over more rapidly generating small molecule donors if the future application of the COS/H2S donor particles is reinstating physiologically relevant concentrations of H2S to mimic endogenous levels (instead of rapid bolus release).
1H NMR spectroscopy was performed on recovered mPEG49-BBOT24 (10) particles (Fig. S6†). Exposure to control conditions (PBS only) resulted in no change to the 1H NMR spectrum of mPEG49-BBOT24 (10) particles. Exposure to H2O2 reduced the relative intensity of aromatic and benzylic protons (derived from methacrylate backbone) to the PEG backbone, suggesting that self-immolation had occurred with diffusion of these groups out of the bilayers and removal during dialysis. Exposure of the control mPEG49-FBOT38 (11) particles to H2O2 showed no changes in the 1H NMR spectrum (Fig. S7†).
Qualitative GPC was performed on control (PBS only) and triggered (H2O2) particles after lyophilization (Fig. 4F). The polymers had an apparent increase in MW and polydispersity, suggesting extensive cross-linking had occurred within the bilayers, although no quantification was performed. The cross-linking resulted in stable particles that retained their initial morphology. DLS showed an unchanged particle size distribution (Fig. 4C) and particle counts for the triggered and non-triggered particles (Fig. 4D). This suggests particles do not lyse upon H2O2 exposure, similar to the non-triggerable control particles, mPEG49-FBOT38 (11) (Fig. S8†). Triggerable control mPEG49-BBOC23 (12) particles exhibited a rapid reduction in DLS count rates when exposed to H2O2 (Fig. S9†). Cryo-TEM (Fig. 4B) on H2O2 exposed mPEG49-BBOT24 (10) particles showed well-defined vesicles, but with a potential loss of the internal particle structures seen prior to H2O2 exposure (Fig. 4A). This suggests polymer cross-linking may occur when the primary amines within the bilayer are liberated48,49 in the more non-polar bilayer of the thiocarbamate in mPEG49-BBOT24 (10), as compared to the carbamate mPEG49-BBOC23 (12) particles which underwent lysis.39 The crosslinking via an amide bond-forming reaction was further supported by FTIR analysis (Fig. S10†), with a new amide carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) bond stretch at 1654 cm−1.39
O) bond stretch at 1654 cm−1.39
Of note, the above studies were conducted in buffer with a single triggering agent (H2O2), but in a complex biological environment there may be other triggers that can lead to H2S generation, such as cysteine, which has been shown to diffuse into micelles and release H2S.20 However, the location of the arylboronate/thiocarbamate group in the hydrophobic portion of the vesicle is expected to provide some protection, especially to more polar activators. The observed slower rate of H2S-release for the mPEG49-BBOT24 (10) vesicles (Fig. 4E) compared to the monomer methacrylate BBOT (1) (Fig. 3) could also indicate that diffusion20 of H2O2 is rate-determining in H2S release from the vesicles.
Encapsulation of drug cargo in H2S-producing particles
Finally, the COS/H2S-producing nanoparticles were examined for their potential as a drug delivery system. Such a system could be used to deliver a dual payload (whereby a therapeutic effect is exerted by H2S and cargo in a synergistic fashion23,50) or to limit off target toxicities.28 To increase loading potential self-assembly was performed with 2 mg mL−1 polymer concentration, resulting in larger particles of 164 ± 8 nm when unloaded or 217 ± 4 nm when loaded (Tables S2 and S3†).Self-assembled mPEG49-BBOT24 (10) particles were passively loaded with doxorubicin·HCl (Table S3†) and achieved a drug loading content of 2.3%.30 Higher loading was achieved for the mPEG49-BBOT24 (10) > mPEG49-BBOC23 (12) > mPEG49-FBOT38 (11), which is speculated to be related to the relative hydrophilicities of the monomeric components (ClogP 4.9, 4.2 and 3.5, respectively). Triggering with H2O2 resulted in release of the model drug (Fig. 5). As particles are not lysed (Fig. 4) this suggests bilayers become permeable to small molecules and provides evidence these systems could have a dual function.
 | ||
| Fig. 5 Release of doxorubicin·HCl from mPEG49-BBOT24 (10) particles in response to PBS control conditions (blue), 0.1 mM (orange) or 1 mM (red) H2O2 at 37 °C. Mean ± SD, n = 3. | ||
Conclusions
Polymeric nanoparticles containing caged carbonyl sulfide (COS) in the thiocarbamate bond of the polymer backbone were formulated from mPEGylated polymers. The thiocarbamate polymer backbone was functionalised with repeating arylboronate monomers that were synthesized via ARGET-ATRP. The polymer BBOT (1) and the polymeric nanoparticles mPEG49-BBOT24 (10) were oxidised by H2O2, activating the release of COS and generation of H2S via carbonic anhydrase hydrolysis. The released H2S caused a reduction of the azide group in pro-fluorophore AzMC and a measurable fluorescent output. The control, non-COS/H2S generating monomers FBOT (2) and BBOC (3), and the mPEG24-FBOT38 (11)/mPEG49-BBOC23 (12) nanoparticles did not reduce/activate the pro-fluorophore AzMC, providing a strong case for future in vivo work using the mPEG49-BBOT24 (10) particles for azido-prodrug activation strategies.Overall, these larger donors complement the current arsenal of small molecule H2S donors and could have therapeutic potential in diseases with low levels of H2S or provide a targeted method for aryl azide reduction and activation of prodrugs and drug release.
Author contributions
D. A. P. designed and conducted most of the experiments, supervised J. D., and contributed to project ideas. A. L. contributed to the design and synthesis and provided expertise in synthetic chemistry. J. D. conducted some of the synthesis and formulation experiments with D. A. P. S. H. provided supervision and guidance on the formulation of nanoparticles and contributed to project ideas. A. B. G. conceived the project idea and provided overall supervision of the project and experiments. D. A. P. and A. B. G. wrote the manuscript with contributions to the final version from all the authors.Data availability
The data, including complete experimental methods, supporting this article have been included as part of the ESI.† The following additional references can be found in the ESI.†51,52
Conflicts of interest
There are no conflicts to declare.Acknowledgements
D. P. would like to thank the University of Otago for Doctoral Scholarship and Otago Micro and Nanoscale Imaging (OMNI) for Studentship grant. We also thank the Department of Chemistry, University of Otago, for use of NMR and high-resolution mass spectrometry instruments. This work was funded in part by a University of Otago Grant (UORG) and a Lottery Health Research Grant from the NZ Lottery Grants Board, New Zealand (2017). Graphical abstract created in BioRender. Gamble, A. (2025) https://BioRender.com/u21a178. Fig. 1 Created in BioRender. Gamble, A. (2025) https://BioRender.com/a62d458.References
- R. Wang, FASEB J., 2002, 16, 1792–1798 CrossRef CAS PubMed.
- J. L. Wallace and R. Wang, Nat. Rev. Drug Discovery, 2015, 14, 329–345 CrossRef CAS PubMed.
- M. D. Hartle and M. D. Pluth, Chem. Soc. Rev., 2016, 45, 6108–6117 RSC.
- J. Bełtowski, Pharmacol. Rep., 2015, 67, 647–658 CrossRef PubMed.
- H. Zhu, V. Dronamraju, W. Xie and S. S. More, Med. Chem. Res., 2021, 30, 305–352 Search PubMed.
- Y. Chen, F. Zhang, J. Yin, S. Wu and X. Zhou, J. Cell Physiol., 2020, 235, 9059–9070 CrossRef CAS PubMed.
- Y.-Z. Wang, E. E. Ngowi, D. Wang, H.-W. Qi, M.-R. Jing, Y.-X. Zhang, C.-B. Cai, Q.-L. He, S. Khattak, N. H. Khan, Q.-Y. Jiang, X.-Y. Ji and D.-D. Wu, Int. J. Mol. Sci., 2021, 22, 2194 CrossRef CAS PubMed.
- B. Liu, S. Wang, M. Xu, Y. Ma, R. Sun, H. Ding and L. Li, Front. Pharmacol., 2022, 13, 899859 CrossRef CAS PubMed.
- Z.-L. Song, L. Zhao, T. Ma, A. Osama, T. Shen, Y. He and J. Fang, Med. Res. Rev., 2022, 42, 1930–1977 CrossRef CAS PubMed.
- C. Szabo, C. Coletta, C. Chao, K. Módis, B. Szczesny, A. Papapetropoulos and M. R. Hellmich, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 12474–12479 Search PubMed.
- C. Munteanu, M. A. Turnea and M. Rotariu, Antioxidants, 2023, 12, 1737 CrossRef CAS PubMed.
- H. Lu, Y. Chen and P. Hu, Adv. Ther., 2023, 6, 2200349 Search PubMed.
- Y. Zheng, B. Yu, L. K. De La Cruz, M. R. Choudhury, A. Anifowose and B. Wang, Med. Res. Rev., 2018, 38, 57–100 CrossRef CAS PubMed.
- B. Szczesny, K. Módis, K. Yanagi, C. Coletta, S. Le Trionnaire, A. Perry, M. E. Wood, M. Whiteman and C. Szabo, Nitric Oxide, 2014, 41, 120–130 CrossRef CAS PubMed.
- E. E. Ngowi, A. Afzal, M. Sarfraz, S. Khattak, S. U. Zaman, N. H. Khan, T. Li, Q.-Y. Jiang, X. Zhang, S.-F. Duan, X.-Y. Ji and D.-D. Wu, Int. J. Biol. Sci., 2021, 17, 73–88 CrossRef CAS PubMed.
- E. R. DeLeon, G. F. Stoy and K. R. Olson, Anal. Biochem., 2012, 421, 203–207 CrossRef CAS PubMed.
- M. C. Urquhart, N. V. Dao, F. Ercole, B. J. Boyd, T. P. Davis, M. R. Whittaker and J. F. Quinn, ACS Macro Lett., 2020, 9, 553–557 CrossRef CAS PubMed.
- C. R. Powell, J. C. Foster, S. N. Swilley, K. Kaur, S. J. Scannelli, D. Troya and J. B. Matson, Polym. Chem., 2019, 10, 2991–2995 RSC.
- K. Kaur, R. J. Carrazzone and J. B. Matson, Antioxid. Redox Signal., 2019, 32, 79–95 CrossRef PubMed.
- J. C. Foster, R. J. Carrazzone, N. J. Spear, S. C. Radzinski, K. J. Arrington and J. B. Matson, Macromolecules, 2019, 52, 1104–1111 CrossRef CAS PubMed.
- S.-S. Qi, J.-H. Sun, H.-H. Yu and S.-Q. Yu, Drug Delivery, 2017, 24, 1909–1926 CrossRef CAS PubMed.
- Y. S. Loo, N. I. Zahid, T. Madheswaran and I. D. M. Azmi, J. Drug Delivery Sci. Technol., 2022, 71, 103300 CrossRef CAS.
- J. C. Foster, S. C. Radzinski, X. Zou, C. V. Finkielstein and J. B. Matson, Mol. Pharm., 2017, 14, 1300–1306 CrossRef CAS PubMed.
- N. V. Dao, F. Ercole, L. M. Kaminskas, T. P. Davis, E. K. Sloan, M. R. Whittaker and J. F. Quinn, Biomacromolecules, 2020, 21, 5292–5305 CrossRef CAS PubMed.
- S. H. Yu, L. Esser, S. Y. Khor, D. Senyschyn, N. A. Veldhuis, M. R. Whittaker, F. Ercole, T. P. Davis and J. F. Quinn, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 1982–1993 CrossRef CAS.
- X. Dong, R. K. Brahma, C. Fang and S. Q. Yao, Chem. Sci., 2022, 13, 4239–4269 RSC.
- A. Xie, S. Hanif, J. Ouyang, Z. Tang, N. Kong, N. Y. Kim, B. Qi, D. Patel, B. Shi and W. Tao, EBioMedicine, 2020, 56, 102821 Search PubMed.
- K. M. Dillon, R. J. Carrazzone, Y. Wang, C. R. Powell and J. B. Matson, ACS Macro Lett., 2020, 9, 606–612 CrossRef CAS PubMed.
- N. Ma, Y. Li, H. Ren, H. Xu, Z. Li and X. Zhang, Polym. Chem., 2010, 1, 1609–1614 RSC.
- E. Jäger, O. Ilina, Y. Dölen, M. Valente, E. A. W. van Dinther, A. Jäger, C. G. Figdor and M. Verdoes, Biomacromolecules, 2024, 25, 1749–1758 CrossRef PubMed.
- K. E. Broaders, S. Grandhe and J. M. J. Fréchet, J. Am. Chem. Soc., 2011, 133, 756–758 Search PubMed.
- C. R. Powell, K. M. Dillon and J. B. Matson, Biochem. Pharmacol., 2018, 149, 110–123 CrossRef CAS PubMed.
- C. M. Levinn, M. M. Cerda and M. D. Pluth, Acc. Chem. Res., 2019, 52, 2723–2731 CrossRef CAS PubMed.
- C. M. Levinn, A. K. Steiger and M. D. Pluth, ACS Chem. Biol., 2019, 14, 170–175 CrossRef CAS PubMed.
- A. K. Steiger, S. Pardue, C. G. Kevil and M. D. Pluth, J. Am. Chem. Soc., 2016, 138, 7256–7259 CrossRef CAS PubMed.
- C. M. Levinn, M. M. Cerda and M. D. Pluth, Antioxid. Redox Signal., 2019, 32, 96–109 CrossRef PubMed.
- Y. Zhao and M. D. Pluth, Angew. Chem., Int. Ed., 2016, 55, 14638–14642 CrossRef CAS PubMed.
- C. Li, J. Hu, T. Liu and S. Liu, Macromolecules, 2011, 44, 429–431 CrossRef CAS.
- D. A. Paterson, W.-K. Fong, S. Hook and A. B. Gamble, Biomacromolecules, 2021, 22, 4770–4782 CrossRef CAS PubMed.
- A. K. Steiger, M. Marcatti, C. Szabo, B. Szczesny and M. D. Pluth, ACS Chem. Biol., 2017, 12, 2117–2123 CrossRef CAS PubMed.
- J. S. Robbins, K. M. Schmid and S. T. Phillips, J. Org. Chem., 2013, 78, 3159–3169 CrossRef CAS PubMed.
- E. A. Garcia, D. Pessoa and M. Herrera-Alonso, Soft Matter, 2020, 16, 2473–2479 RSC.
- M. K. Thorson, T. Majtan, J. P. Kraus and A. M. Barrios, Angew. Chem., Int. Ed., 2013, 52, 4641–4644 CrossRef CAS PubMed.
- K. Min, H. Gao and K. Matyjaszewski, Macromolecules, 2007, 40, 1789–1791 CrossRef CAS.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- J. Y. T. Chong, X. Mulet, L. J. Waddington, B. J. Boyd and C. J. Drummond, Soft Matter, 2011, 7, 4768–4777 RSC.
- J. Zhai, R. B. Luwor, N. Ahmed, R. Escalona, F. H. Tan, C. Fong, J. Ratcliffe, J. A. Scoble, C. J. Drummond and N. Tran, ACS Appl. Mater. Interfaces, 2018, 10, 25174–25185 Search PubMed.
- Z. Deng, Y. Qian, Y. Yu, G. Liu, J. Hu, G. Zhang and S. Liu, J. Am. Chem. Soc., 2016, 138, 10452–10466 CrossRef CAS PubMed.
- X. Wang, G. Liu, J. Hu, G. Zhang and S. Liu, Angew. Chem., Int. Ed., 2014, 53, 3138–3142 CrossRef CAS PubMed.
- K. Chegaev, B. Rolando, D. Cortese, E. Gazzano, I. Buondonno, L. Lazzarato, M. Fanelli, C. M. Hattinger, M. Serra, C. Riganti, R. Fruttero, D. Ghigo and A. Gasco, J. Med. Chem., 2016, 59, 4881–4889 Search PubMed.
- R. Glaser, R. Hillebrand, W. Wycoff, C. Camasta and K. S. Gates, J. Org. Chem., 2015, 80, 4360–4369 CrossRef CAS PubMed.
- S. Modi and B. D. Anderson, Mol. Pharm., 2013, 10, 3076–3089 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5lp00040h |
| This journal is © The Royal Society of Chemistry 2025 |