
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSidechain engineering in poly(2,3-alkylthieno[3,4-b]pyrazine)s via GRIM polymerization: solubility, film formation, and device performance†
Spencer J.
Gilman
a,
Nicolas C.
Nicolaidis
 b,
Tomas J.
Marsh
b,
Paul C.
Dastoor
b and
Seth C.
Rasmussen
*a
b,
Tomas J.
Marsh
b,
Paul C.
Dastoor
b and
Seth C.
Rasmussen
*a
aDepartment of Chemistry and Biochemistry, North Dakota State University, NDSU Dept. 2508, P.O. Box 6050, Fargo, North Dakota 58108, USA. E-mail: seth.rasmussen@ndsu.edu
bCentre for Organic Electronics, University of Newcastle, Callaghan, NSW 2308, Australia
First published on 4th March 2025
Abstract
The effects of branched vs. linear alkyl sidechains were investigated in poly(2,3-dialkylthieno[3,4-b]pyrazine)s via GRIM polymerization, focusing on molecular weight, processibility, and device performance. New 2-ethylhexyl derivatives exhibited enhancement in nearly all aspects, with photovoltaic devices exhibiting photoresponse out to 1300 nm and competitive specific detectivity values for NIR photodetectors.
Conjugated polymers have grown to receive considerable interest as they combine the electronic and optical properties of inorganic semiconductors with the physical properties of organic plastics, including flexibility and their lightweight nature.1 Although commonly believed to be advanced materials of the modern era, the synthesis and study of these materials dates back to the 1830s.2 Still, it was the first reports of their conductive nature in the early 1960s that stimulated a focused interest in conjugated polymers, particularly after the first report of metallic conductivities in the 1970s. Since that time, the study of conjugated polymers has continued to grow, ultimately leading to the current field of organic electronics, with various demonstrated applications including electrochromics, organic field effect transistors (OFETs), organic light-emitting diodes (OLEDs), and organic photovoltaics (OPVs).3–9
An important advantage of conjugated polymers is the ability to tune their optical, electronic, and mechanical properties on a molecular level via molecular design.1,10–14 In particular, considerable focus has been placed on tuning the polymer bandgap (Eg), defined as the energetic separation between the filled valence and empty conduction bands in the bulk solid state material.1,11,14,15 This is particularly critical as the bandgap determines properties like the absorption onset, the energy of any potential emission, and conductivity.
An important landmark in the controlled synthesis of conjugated polymers was the ability to synthesize regioregular poly(3-alkylthiophene)s (rr-PATs) via catalytic cross-coupling.1,10 This began with the 1992 report of rr-PATs via Kumada coupling from McCullough,16 followed by a related study via Negishi coupling from Rieke later that same year.17 McCullough then reported a simplified method in 1999 that involved the room temperature reaction of a dibromothiophene with an alkyl Grignard reagent, followed by Kumada coupling.18 Based on the critical reaction with the Grignard reagent, this method was named Grignard metathesis (GRIM) polymerization.18–20 This then became the basis for the development of conditions allowing chain-growth polycondensations, now known by the general description catalyst-transfer polymerization.1
Although the initial focus of GRIM polymerization was the production of rr-PATs, McCullough expanded its application to the preparation of regioregular poly(3-alkoxythiophene)s in 2005, resulting in low bandgap materials (Eg < 1.5 eV) with Eg values of ∼1.4 eV.21 This was then followed with a 2008 report of GRIM-polymerized poly(2,3-dihexylthieno[3,4-b]pyrazine) (PC6TP) with an Eg of ∼0.93 eV (Scheme 1).22 Although polythieno[3,4-b]pyrazines had been previously produced via both chemical oxidative polymerization with FeCl3 and electropolymerization,23,24 the GRIM-polymerized material exhibited a lower Eg and enhanced solution stability in comparison to the FeCl3 polymerized analogue, as well as enhanced processability over all previous materials, allowing the first application of these materials to photovoltaic devices.22
 | ||
| Scheme 1 Poly(2,3-dialkylthieno[3,4-b]pyrazine)s via GRIM polymerization. | ||
Although bulk heterojunction (BHJ) OPV devices of PC6TP exhibited low external quantum efficiency (EQE) values, the device photoresponse extended out to 1250–1300 nm, one of only two reported materials capable of this at the time.22 As such, these held promise for applications in NIR photodetectors,25–30 with further NIR device characterization reported in 2013.31 Still, it was felt that the GRIM-polymerized PC6TP was limited by its relatively low molecular weights (Mn = 4800–4900), which accounted for the higher Eg in comparison to the electropolymerized material (0.93 vs. 0.7 eV).22
As the molecular weight was essentially independent of the various conditions applied to the GRIM polymerizations, while also only slightly higher than that produced via FeCl3-based oxidative polymerization, it was concluded that this probably indicated the solubility limit of these materials.22 In an attempt to increase solubility, the GRIM polymerization of thieno[3,4-b]pyrazines (TPs) with longer alkyl chain lengths (R = decyl, dodecyl) were then investigated. Unfortunately, it was found that the Mn decreased with the length of the sidechain, which was concluded to be due to reduced solubility resulting from side chain crystallization.32
An obvious next step would be to investigate the application of branched alkyl sidechains, as such side chains generally result in enhanced solubility due to steric and thermodynamic effects.33,34 Unfortunately, although the 2-ethylhexyl function-alized TP (EHTP) and its dibromo derivative were first reported in 2010,35 the products of these reported methods were not of suitable purity for a meaningful polymerization study. However, after optimized methods were developed in 2018 for the production of analytically pure EHTP,36 it was felt time to revisit these GRIM-polymerized materials once again.
As outlined in Scheme 1, treatment of EHTP with NBS at low temperatures in DMF gave the brominated derivative Br2EHTP in 67% yield. A challenge in purifying Br2EHTP is that the conformational disorder of the sidechains gives the product as an oil, rather than the solid products of the linear alkyl analogues. As such, the precipitation methods used for initial purification were not possible.22,32 As purification was thus fully dependent on chromatography, some product was lost in order to obtain suitable purity, resulting in lower yields compared to Br2C6TP.
GRIM polymerization was then carried out via the reflux methods found optimal for the previous linear alkyl TPs.22,32 As shown in Scheme 1, Br2EHTP and methylmagnesium bromide (1.05 equiv.) were heated at reflux for 1 h to promote Grignard metathesis. The nickel pre-catalyst was then added, followed by heating at reflux for another hour. The resulting PEHTP was added to MeOH, isolated, and purified via Soxhlet extraction with MeOH for 24 h. Extraction with CHCl3 then isolated the dark-purple polymer in 68% yield, identical to that obtained for PC6TP. While PC6TP was only partially soluble in CHCl3,22 PEHTP was found to be completely CHCl3 soluble. Furthermore, while saturated CHCl3 solutions of PC6TP occurred at ca. 5–10 mg mL−1, PEHTP solutions were still not saturated at 10 mg mL−1.
Polymer samples were then analyzed by high temperature gel permeation chromatography (GPC) at 110 °C. Under these conditions, PC6TP was determined to have a Mn of 6700, with a PDI of 1.56. While this Mn is a noticeable increase in comparison to the previous low temperature GPC data (4900 vs. 6700),22 the PDI also exhibited an increase from 1.48 to 1.56. This new data would correspond to n = 22. In comparison, PEHTP was found to have a Mn of 7400 and a PDI of 1.39. While this would appear to indicate a longer chain length, the EHTP repeat is also higher molecular weight than C6TP. As such, the PEHTP data actually corresponds to a lower value of n = 21, with the branched side chains not providing longer chain lengths as hoped.
In terms of the TP homopolymers via GRIM polymerization, the chain length of PEHTP was found to be essentially the same as that produced for PC6TP. At the same time, while solubility may have been a limitation for PC6TP, this was no longer the case for PEHTP. As such, one must conclude that solubility is not the limiting factor in terms of molecular weights obtained via this method as previously thought.22,32
In a 2014 study focused on catalyst-transfer polymerization of TP, Koeckelberghs and coworkers concluded that the low Mn was due to a termination reaction where the catalyst dissociated from the polymer, consistent with the weaker binding of the Ni catalyst by electron-deficient polymers.37 While such dissociation would stop the chain-growth process, this would not necessarily stop polymerization as the polymer would still have an active aryl-bromide terminus. Oxidative addition of the dissociated catalyst into this C–Br bond could thus still further chain growth.1 Furthermore, to assume dissociation is due to the electron-deficient nature of the TP polymer reveals a lack of understanding of these materials. While the pyrazine ring of TP is electron-deficient, the TP thiophene is quite electron rich and has been shown to be similar to 3,4-ethylenedioxythiophene (EDOT) in that regard.38,39 It is for this reason that TPs are classified as ambipolar units, as they simultaneously act as both donors and acceptors.38,39 As the catalyst association with the polymer occurs along the polymer backbone,1 this would correspond with the electron-rich nature of the polymer, not the electron-poor.
As the polymerization of TPs seems to completely stop at a particular point, this appears to be more consistent with a case of catalyst poisoning, which typically occurs when a substance binds to the active sites of the catalyst resulting in deactivation. As the TP nitrogens are known to be good Lewis bases, with pKa values nearly identical to both pyrazine and quinoxaline,40 the TP itself could bind the catalyst via nitrogen coordination. However, as the polymerization initiates with a large excess of TP, this does not account for the observed reactivity. Conversely, as the polymer chain grows, trimeric units provide a cleft of suitable size and orientation to bind transition metals via a tridentate chelation (Scheme 2). Such tridentate clefts have been previously proposed to account for the iron binding of TP homopolymers found during FeCl3 polymerization. Furthermore, computational modelling of this Fe-bound cleft revealed comparable strength and structure to binding by the tridentate ligand 2,2′:6′,2′′-terpyridine.23 The strong metal binding of TP units is further supported by the recent report of a TP capable of bidentate binding, along with X-ray structures of its Ru complexes.41 As ligand binding increases significantly with increased denticity, the tridentate clefts produced by the polymer backbone would provide much stronger binding that could realistically poison the catalyst. Thus, if the catalyst binding by the polymer is viewed as an equilibrium process (Scheme 2), a greater number of binding clefts would be produced with polymer growth, thus shifting the equilibrium to the bound form via Le Chatelier's principle. Once shifted completely to the bond form, the catalyst would be inactive.
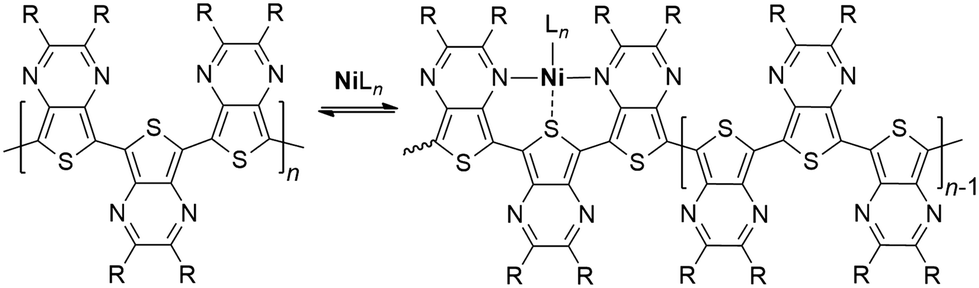 | ||
| Scheme 2 Catalyst binding by poly(2,3-dialkylthieno[3,4-b]pyrazine)s. | ||
Solution and thin-film UV-vis-NIR absorption spectra are given in Fig. 1 for both PC6TP and PEHTP. Both materials exhibit a lower energy transition in the NIR which is formally assigned as an intramolecular charge transfer (ICT) from a HOMO localized on the thiophene-based polymer backbone to a LUMO localized along the pyrazine rings.31 Higher energy transitions at wavelengths below 600 nm are assigned as simple π–π* excitations.
 | ||
Fig. 1 Visible-NIR spectra of poly(2,3-dialkylthieno[3,4-b]pyrazine)s (solution and film spectra are normalized to each other within each polymer pair, with the relative absorption intensities of the two polymers determined from the analysis of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 blends of the two polymers). :1 blends of the two polymers). | ||
As can be seen in Fig. 1, compared to PC6TP, PEHTP exhibits a narrower spectral profile and a higher energy absorption onset, corresponding to an Eg of ca. 1.06 eV. The difference in spectral profile can be at least partially attributed to the lower PDI of PEHTP, resulting in less contribution to absorption from species providing the highest and lowest conjugation lengths. This is further supported by the study of blend films of 1:1 PC6TP:PEHTP that confirm that while PEHTP exhibits less absorption on both the high and low energy sides of the ICT transition, the absorption around 1100 nm is stronger in PEHTP.
The difference in Eg can be attributed to a number of factors, beginning with the fact that the low Eg of 0.93–0.95 eV for PC6TP are only obtained after annealing at 100 °C. Unannealed films of PC6TP exhibit an Eg = ca. 1.0 eV. In comparison, DSC analysis of PEHTP failed to reveal any clear Tg and all attempts to thermally anneal films resulted in no changes in the thin-film spectra. The lack of increased order with annealing is believed to be due to the greater disorder of the branched sidechains. Other possible factors could include the slightly shorter chain lengths of PEHTP as determined by GPC, as well as the fact that the bulkier 2-ethylhexyl sidechains likely contribute to additional steric interactions that limit the extent of backbone planarity.
Using cyclic voltammetry, the frontier molecular orbital energy levels of PEHTP were estimated to give HOMO and LUMO energy levels of ca. −5.1 and −3.8 eV, respectively. This corresponds to an electrochemical Eg of 1.3 eV, with the difference between the electrochemical and optical Eg considered to be the exciton binding energy.15 The frontier orbital levels of PEHTP are in relatively good agreement with those previously determined for PC6TP,22 and the somewhat destabilized HOMO levels are consistent with the electron-rich nature of the TP thiophene.
BHJ OPV devices were fabricated from blends of PEHTP and phenyl-C61-butyric acid methyl ester (PCBM) in order to compare to the previous BHJ devices of PC6TP. Devices were optimized by varying the PEHTP:PCBM ratio, processing solvent, and annealing time, with the best properties obtained from unannealed 1:1 PEHTP:PCBM devices spun from chlorobenzene. The resulting J–V plot and external quantum efficiency (EQE) for a 1:1 PEHTP:PCBM device is shown in Fig. 2.
 | ||
| Fig. 2 External quantum efficiency (EQE) and J–V plot of a 1:1 PEHTP:PCBM device. | ||
Such 1:1 PEHTP:PCBM devices provide a photoconversion efficiency (PCE) of 0.229%. While still very low, this is nearly double that of optimized PC6TP:PCBM devices, which exhibited a maximum PCE of 0.13%.31 This increased PCE is due to higher JSC and fill factor values compared to PC6TP-based devices, which is currently believed to be the result of the enhanced processability of PEHTP, thus resulting in improved interfaces in both the active layer and between the active layer and transport layers. Such interfaces are known to play a significant role on device performance, particularly on the resulting fill factor.42,43 The EQE response of the device exhibits a high-energy maximum at ca. 330 nm, with a low-energy maximum at 1100 nm. This low energy maximum has both a higher EQE and occurs at a longer wavelength than that found for PC6TP devices (1110 vs. 925 nm).22 PEHTP also contributes to the device response down to ca. 1300 nm, similar to the previous response of PC6TP:PCBM devices. This is noteworthy because of the various low Eg polymers applied to photonic devices, the number of such materials capable of NIR response below 1000 nm still encompasses only about 20 polymers.26
To evaluate the ability of these devices to act as NIR photo-detectors, the noise current of the BHJ device was determined so that the specific detectivity (D*(λ)) could be calculated. Such D*(λ) values reflect the ability to detect signals of weak irradiation intensity and are considered the most critical parameter for NIR photodetectors,26–29 with values >1011 Jones (cm Hz½ W−1) considered competitive with inorganic photodetectors. The respective responsivity (R(λ)) and D*(λ) values for the PEHTP device are shown in Fig. 3. Within the NIR region, maximum values of 0.033 A W−1 and 7.7 × 109 Jones are found at 1110 nm, with D*(λ) remaining above 109 Jones out to 1210 nm. Furthermore, much like with the photoresponse shown in Fig. 2, the detectivity of these NIR photodetectors extend beyond 1000 nm, with measurable values beyond 1300 nm.
 | ||
| Fig. 3 Specific detectivity (D*(λ)) and responsivity (R(λ)) for a 1:1 PEHTP:PCBM device. | ||
As NIR photodetectors based on low Eg polymers capable of response below 1000 nm have now achieved D*(λ) values >1013 Jones, the values here can seem modest. Still, it must be noted that most D*(λ) values are determined solely from the dark current,27–30 rather than relative to the total noise current as done here for the PEHTP device. As such, D*(λ) is generally overestimated, as the measured noise current is usually much larger than the theoretical shot noise limit.27,29 Thermal noise contributions to the total noise current are thought to be significant in organic NIR photodetectors, particularly those based on low Eg materials,28,30 with the overestimation of D*(λ) predicted to be greater than an order of magnitude when based only on dark current.30 To illustrate this effect on D*(λ), the value of D*(λ) for the PEHTP device at 1110 nm was also determined using the dark current only, resulting in the considerably higher value of 3.0 × 1012 Jones. This value compares very well with the best reported organic NIR photodetectors, placing it among the top six reported devices capable of detection beyond 1000 nm. At the same time, this also highlights the very real problem with determining these values via dark current alone, as this significantly overestimates the actual detectivity of these devices. Nevertheless, the results here exhibit significant promise and reflect the potential of TP homopolymers for the future development of NIR photodetectors, particularly as these materials have much lower synthetic complexity than other materials targeted for these applications.
Conclusions
Poly(2,3-bis(2-ethylhexyl)thieno[3,4-b]pyrazine) was prepared by GRIM polymerization and compared to its analogue utilizing linear hexyl sidechains. The shift from linear to branched sidechains resulted in the expected enhancement of solubility and processibility, although it did not provide higher molecular weight material as hoped. As such, it is now clear that the molecular weights of these materials are not solubility limited as previously believed. To explain the limited molecular weights of these polymers, a new catalyst poisoning mechanism is proposed, based on the ability of TP homopolymers to bind transition metals via chelation. Finally, the application of the new polymer to BHJ OPV devices revealed a significant improvement in device performance compared to the previous hexyl analogue. Most importantly, these devices are capable of photoresponse out to 1300 nm and NIR photodetectors based on this material exhibit specific detectivities that place them among the best reported devices capable of detection beyond 1000 nm.Author contributions
Conceptualization, S.C.R.; methodology, S.C.R., N.N., and P.C.D.; formal analysis, S.C.R., N.N., and S.J.G.; investigation, S.J.G and T.M.; data curation, S.J.G, N.N., and T.M.; writing – original draft preparation, S.J.G and S.C.R.; writing – review and editing, S.C.R. and S.J.G; visualization, S.C.R.; supervision, S.C.R. and P.C.D.; funding acquisition, S.C.R. and P.C.D.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors wish to thank the National Science Foundation (CHE-2002877) and North Dakota State University for support of this research and Daniel Elkington for discussions around the experimental determination of the noise floor of the devices. Portions of this work was performed in part of the materials node of the Australian National Fabrication Facility, a company established under the National Collaborative Research Infrastructure Strategy to provide nano- and micro-fabrication facilities to Australia's researchers.References
- S. C. Rasmussen, S. J. Gilman and W. D. Wilcox, Conjugated Polymers: Synthesis & Design, ACS in Focus, American Chemical Society, Washington, D.C., 2023 Search PubMed.
- S. C. Rasmussen, ChemPlusChem, 2020, 85, 1412–1429 CrossRef CAS PubMed.
- Handbook of Conducting Polymers, ed. J. R. Reynolds, T. A. Skotheim and B. Thompson, CRC Press, Boca Raton, FL, 4th edn, 2019 Search PubMed.
- Handbook of Thiophene-based Materials, ed. I. F. Perepichka and D. F. Perepichka, John Wiley & Sons, Hoboken, 2009 Search PubMed.
- C. B. Nielsen and I. McCulloch, Prog. Polym. Sci., 2013, 38, 2053–2069 CrossRef CAS.
- A. C. Grimsdale, K. L. Chan, R. E. Martin, P. G. Jokisz and A. B. Holmes, Chem. Rev., 2009, 109, 897–1091 Search PubMed.
- S. C. Rasmussen, S. J. Evenson and C. B. McCausland, Chem. Commun., 2015, 51, 4528–4543 Search PubMed.
- S. Günes, H. Neugebauer and N. S. Sariciftci, Chem. Rev., 2007, 107, 1324–1338 Search PubMed.
- M. C. Scharber and N. S. Sariciftci, Prog. Polym. Sci., 2013, 38, 1929–1940 CrossRef CAS PubMed.
- R. M. Pankow and B. C. Thompson, Polymer, 2020, 207, 122874 CrossRef CAS.
- S. C. Rasmussen, in Encyclopedia of Polymeric Nanomaterials, ed. K. Muellen and S. Kobayashi, Springer, Heidelberg, 2015, pp. 1155–1166 Search PubMed.
- M. C. Scharber and N. S. Sariciftci, Adv. Mater. Technol., 2021, 6, 2000857 CrossRef CAS.
- C. Liu, K. Wang, X. Gong and A. Heeger, J. Chem. Soc. Rev., 2016, 45, 4825–4846 RSC.
- J. Roncali, Rapid Commun., 2007, 28, 1761–1775 CrossRef CAS.
- J.-L. Bredas, Mater. Horiz., 2014, 1, 17–19 RSC.
- R. D. McCullough and R. D. Lowe, J. Chem. Soc., Chem. Commun., 1992, 70–72 RSC.
- T.-A. Chen and R. D. Rieke, J. Am. Chem. Soc., 1992, 114, 10087–10088 CrossRef CAS.
- R. S. Loewe, S. K. Khersonsky and R. D. McCullough, Adv. Mater., 1999, 11, 250–253 CrossRef CAS.
- R. S. Loewe, P. C. Ewbank, J. Liu, L. Zhai and R. D. McCullough, Macromolecules, 2001, 34, 4324–4333 CrossRef CAS.
- M. C. Iovu, E. E. Sheina, R. R. Gil and R. D. McCullough, Macromolecules, 2005, 38, 8649–8656 CrossRef CAS.
- E. E. Sheina, S. M. Khersonsky, E. G. Jones and R. D. McCullough, Chem. Mater., 2005, 17, 3317 CrossRef CAS.
- L. Wen, B. C. Duck, P. C. Dastoor and S. C. Rasmussen, Macromolecules, 2008, 41, 4576–4578 CrossRef CAS.
- D. D. Kenning and S. C. Rasmussen, Macromolecules, 2003, 36, 6298–6299 CrossRef CAS.
- S. C. Rasmussen, R. L. Schwiderski and M. E. Mulholland, Chem. Commun., 2011, 47, 11394–11410 RSC.
- L. Dou, Y. Liu, Z. Hong, G. Li and Y. Yang, Chem. Rev., 2015, 115, 12633–12665 CrossRef CAS PubMed.
- S. C. Rasmussen, S. J. Gilman, E. W. Culver and W. D. Wilcox, Gen. Chem., 2021, 7, 200019 Search PubMed.
- X. Liu, Y. Lin, Y. Liao, J. Wu and Y. Zheng, J. Mater. Chem. C, 2018, 6, 3499–3513 RSC.
- Q. Li, Y. Guo and Y. Liu, Chem. Mater., 2019, 31, 6359–6379 CrossRef CAS.
- P. C. Y. Chow and T. Someya, Adv. Mater., 2020, 32, 1902045 CrossRef CAS PubMed.
- Z. Wu, Y. Zhai, H. Kim, J. D. Azoulay and T. N. Ng, Acc. Chem. Res., 2018, 51, 3144–3153 CrossRef CAS PubMed.
- B. C. Duck, B. Vaughan, L. Wen, C. L. Heth, S. C. Rasmussen, X. Zhou, W. J. Belcher and P. C. Dastoor, Sol. Energy Mater. Sol. Cells, 2013, 110, 8–14 CrossRef CAS.
- M. E. Mulholland, L. Wen and S. C. Rasmussen, Topol. Supramol. Polym. Sci., 2015, 2, 18–29 Search PubMed.
- T. Lei, J.-Y. Wang and J. Pei, Chem. Mater., 2014, 26, 594–603 CrossRef CAS.
- J. Mei and Z. Bao, Chem. Mater., 2014, 26, 604–615 CrossRef CAS.
- C.-H. Chen, C.-H. Hsieh, M. Dubosc, Y.-J. Cheng and C.-S. Hsu, Macromolecules, 2010, 43, 697–708 CrossRef CAS.
- E. W. Culver, T. E. Anderson, J. T. L. Navarrete, M. C. R. Delgado and S. C. Rasmussen, ACS Macro Lett., 2018, 7, 1215–1219 CrossRef CAS PubMed.
- P. Willot, D. Moerman, P. Leclère, R. Lazzaroni, Y. Baeten, M. Van der Auweraer and G. Koeckelberghs, Macromolecules, 2014, 47, 6671–6678 CrossRef CAS.
- L. Wen, C. L. Heth and S. C. Rasmussen, Phys. Chem. Chem. Phys., 2014, 16, 7231–7240 RSC.
- T. E. Anderson, E. W. Culver, I. Badía-Domínguez, W. D. Wilcox, C. E. Buysse, M. C. R. Delgado and S. C. Rasmussen, Phys. Chem. Chem. Phys., 2021, 23, 26534–26546 RSC.
- D. D. Kenning, K. A. Mitchell, T. R. Calhoun, M. R. Funfar, D. J. Sattler and S. C. Rasmussen, J. Org. Chem., 2002, 67, 9073–9076 CrossRef CAS PubMed.
- K. L. Konkol, W. D. Wilcox and S. C. Rasmussen, Dalton Trans., 2024, 53, 16685–16692 RSC.
- D. Gupta, S. Mukhopadhyay and K. S. Narayan, Sol. Energy Mater. Sol. Cells, 2010, 94, 1309–1313 CrossRef CAS.
- B. Qiab and J. Wang, Phys. Chem. Chem. Phys., 2013, 15, 8972–8982 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, synthetic methods, device fabrication and characterization. See DOI: https://doi.org/10.1039/d5lp00023h |
| This journal is © The Royal Society of Chemistry 2025 |