
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRheokinetics of ε-caprolactam anionic-ring polymerization applied to the rapid production of thermoplastic composites†
Karima
Ben Hamou
a,
Ralf
Brüning
b,
Gabriel
La Plante
 c,
Marie-Hélène
Thibault
a,
Jacques
Robichaud
a and
Yahia
Djaoued
*a
c,
Marie-Hélène
Thibault
a,
Jacques
Robichaud
a and
Yahia
Djaoued
*a
aLaboratoire de Recherche en Matériaux et Micro-spectroscopies Raman et FTIR, Université de Moncton – Campus de Shippagan, Shippagan, NB, Canada E8S 1P6. E-mail: yahia.djaoued@umoncton.ca
bPhysics Department, Mount Allison University, Sackville, NB, Canada E4L1E6
cFaculté d'ingénierie, Université de Moncton – Campus de Moncton, Moncton, NB, Canada E1A 3E9
First published on 7th April 2025
Abstract
The present study focuses on the synthesis and structural analysis of poly-ε-caproamide (PA6), produced through anionic polymerization of ε-caprolactam in bulk, utilizing mono and bifunctional activators. The research investigates the physical properties of PA6 synthesized under various polymerization conditions, aiming to understand how these conditions influence the polymer's behavior. The polymerization kinetics were monitored via dynamic rheology, offering insights into the progression of ε-caprolactam's conversion into PA6. Microstructural changes in the PA6 samples, including variations in the degree of crystallinity and the formation of α and γ crystalline structures, were systematically studied. These transformations were dependent on both the type and concentration of the activator used, as well as the specific polymerization parameters applied. The interplay between these factors significantly impacted the resulting chemical and physical structure of the PA6 samples. In the latter part of the study, hybrid composites were fabricated by reinforcing poly-ε-caproamide with two distinct types of fiber fabrics by reactive processing, achieving a 25% weight fraction of reinforcement. Scanning electron microscopy (SEM) revealed excellent interfacial adhesion between the fibers and the polymer matrix, confirming the effectiveness of the fabrication process and the potential of these composites for advanced material applications.
Introduction
Key sustainable green chemical process parameters include solvent-free reactions, quantitative conversion without by-products, and minimal energy consumption. The industrial method of bulk anionic ring-opening polymerization (AROP) of lactams aligns well with these principles, offering an environmentally friendly and cost-effective approach for polyamide synthesis. This method generates no by-products and achieves near-quantitative yields.1–9 While anionic polymerization of lactams is not a new research area, it has gathered renewed interest in recent years due to its increasing relevance in advanced technological applications. Examples of these applications include reactive vacuum infusion,10–13 reactive pultrusion,14–16 reactive resin transfer moulding,17–19 and reactive rotational moulding.20–22 Recent reviews highlight advancements in lactam ring-opening polymerization, particularly in catalysis and kinetics, which open new pathways for innovation in this field.2,23 This research aims to continue the development of poly-ε-caproamide (PA6) by the anionic polymerization of ε-caprolactam. The purpose is to provide an efficient and economical technology that facilitates reactive processing, particularly for thermoplastic composites. In this study, the homopolymerization of ε-caprolactam was initiated using N,N-dicyclohexylcarbodiimide (DCCI) or N-acetylcaprolactam (ACL) as activators, with dicaprolactamato-bis-(2-methoxyethoxo)-aluminate (DL) serving as the catalyst. Despite numerous studies on anionic lactam polymerization since Joyce and Ritter's seminal work in 1941,24 this specific combination of activators and catalyst had not been explored previously. The present study investigates the impact of various processing parameters on the polymerization kinetics of caprolactam. Key factors include polymerization temperature, catalyst/activator combinations, and concentrations. The primary focus is understanding the rheological behaviour of the anionically initiated ring-opening polymerization of caprolactam, where PA6 is synthesized. These rheological characteristics are crucial as they define the polymers’ end properties. This research represents a concerted effort to optimize bulk polymerization conditions and establish a simple yet robust polymerization technique suitable for reactive moulding operations. By refining this process, the work aims to provide a more practical and scalable solution for industrial applications of PA6, with potential implications for developing advanced thermoplastic composites.Experimental
Materials
The monomer used in this work was ε-caprolactam (ε-CL) (99%) purchased from TCI AMERICA. The material was dried at 60 °C for 24 h. The catalyst sodium dicaprolactamato-bis-(2-methoxyethoxo)-aluminate (DL) (82 wt% in toluene) was acquired from Katchem, Czech Republic, and used without further treatment. As polymerization activators, N-acetylcaprolactam (ACL) (99%) as monofunctional activator have been bought from Alfa Aesar and N,N-dicyclohexylcarbodiimide (DCCI) (95%) as bifunctional activator was purchased from Thermo Fisher Scientific Chemicals, Inc. Methanol, and sulfuric acid (98%), were purchased from Millipore Sigma. All products were used without further purification.Two types of fibers were used to produce the hybrid composite samples: Hexcel HexForce 7781 E-glass 8-Harness Satin fabric (299 g m−2), manufactured by Hexcel Corporation (Stamford, CT) and generously supplied by Innov (Shippagan, New Brunswick, Canada), and hemp fibers (Cannabis sativa L.) provided by GEMS Consultants Inc. (New Brunswick, Canada). The hemp fibers had an average length of 7 ± 0.5 cm and a diameter of 25 ± 5 μm. To enhance their properties, the hemp fibers underwent a chemical treatment using a 2 wt% aqueous NaOH solution at room temperature.25 They were immersed in the NaOH solution for 24 hours and then thoroughly washed with distilled water to remove any excess alkali. Acetic acid was applied to neutralize the residual sodium hydroxide.26 Both sodium hydroxide and acetic acid, used during the treatment, were procured from Millipore Sigma with a purity of 99%.
Polymerization runs
At room temperature, an amount of ε-CL in white granules was melted in a double-necked flask at 100–110 °C. The required amount of catalyst (DL) was added under a nitrogen atmosphere to keep moisture and oxygen out. Because the catalyst concentration significantly impacts the polymer's solidification time, the weight of the compound was carefully controlled. When the temperature reached 140 °C ≤ Tpoly ≤ 170 °C, the activator (ACL or DCCI) was added to the above mixture to form the polymer. The polymerization reaction began as soon as the activator was injected into the mixture, and after a period, the viscosity rose rapidly, and the solid PA6 was formed. Various formulations were explored, labeled as TX-Catalyst/Activator-Y, where X represents the polymerization temperature, and Y indicates the activator concentration. The catalyst is DL, while the activator is ACL or DCCI. A summary of the formulations examined is provided in Table 1.| DL/ACL | DL/DCCI | ||||||
|---|---|---|---|---|---|---|---|
| Sample designation | Catalysta (%) | Activatora (%) | Temperature (°C) | Sample designation | Catalysta (%) | Activatora (%) | Temperature (°C) |
| a (mol% per 1 mol caprolactam). | |||||||
| T140-DL/ACL-1.5 | 1.5 | 1.5 | 140 | T140-DL/DCCI-0.75 | 1.5 | 0.75 | 140 |
| T160-DL/ACL-1.5 | 1.5 | 1.5 | 160 | T160-DL/DCCI-0.75 | 1.5 | 0.75 | 160 |
| T170-DL/ACL-1.5 | 1.5 | 1.5 | 170 | T170-DL/DCCI-0.75 | 1.5 | 0.75 | 170 |
| T140-DL/ACL-3 | 3 | 3 | 140 | T140-DL/DCCI-1.5 | 3 | 1.5 | 140 |
| T160-DL/ACL-3 | 3 | 3 | 160 | T160-DL/DCCI-1.5 | 3 | 1.5 | 160 |
| T170-DL/ACL-3 | 3 | 3 | 170 | T170-DL/DCCI-1.5 | 3 | 1.5 | 170 |
Manufacturing of the hybrid composites
A hybrid composite was prepared using 1.5 mol% of a monofunctional activator (ACL) and 1.5 mol% of a DL catalyst at 170 °C. The premix was made at 100–110 °C following the detailed protocol described in the Polymerization Runs section. The experimental design incorporated two types of fibers: glass fibers (GF) and alkali-treated hemp fibers (tHF). The filler content was set at 25 wt% for all composite laminate samples. The hybrid glass/treated hemp fiber composites were produced via compression molding in a rectangular steel mold using a hydraulic hot-plate press (Wabash Genesis Model G100-18, Wabash, IN). After homogenization, the premix was quickly transferred into a preheated rectangular mold (190 × 190 × 5 mm), which was then positioned horizontally between the plates of the hot press heated to 170 °C. The composite was cured for 60 minutes under a pressure of 10 tons. Once the polymerization was complete, the mold was cooled, and the composite was removed. Fig. S1† illustrates the flowchart of the fabrication process of the hybrid glass/treated hemp fiber composite samples. Besides the matrix, each composite sample contained a total of five layers with varying glass-to-treated hemp fiber weight fraction ratios of 0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :25, 10:15, 15:10, 20:5, and 25:0.
:25, 10:15, 15:10, 20:5, and 25:0.
Analysis methods
FTIR Spectra analyses were performed by Agilent Cary 630 FTIR operated in Attenuated Total Reflection mode. A total of 32 scans were performed to obtain the spectrum within the wavelength range of 4000 to 400 cm−1, with a resolution of 4 cm−1.The degree of conversion (DC) of the synthesized samples was determined for various resin formulations at several temperatures. The polymerized samples were ground, weighed to determine the initial mass mi, and refluxed overnight in methanol. After drying, the final mass mf was obtained. While the caprolactam monomer dissolves easily in methanol, its polyamide does not, and the degree of conversion was determined according to eqn (1):27
 | (1) |
Differential Scanning Calorimeter (DSC) measurements were conducted using a TA Q200 V24.11 Build 124 thermal analyzer (New Castle, DE, USA) to determine the melting, glass transition, and crystallization temperatures Tm, Tg, and Tc, respectively. The temperature was scanned from −20 to 300 °C at a heating rate of 10 °C min−1 with the sample in a nitrogen atmosphere. The degree of crystallinity (χDSCc) was calculated according to eqn (2):
 | (2) |
 is the melting enthalpy of fully crystalline poly-ε-caproamide (PA6) (190 J g−1),28 and ΔHm is it's heat of fusion.
is the melting enthalpy of fully crystalline poly-ε-caproamide (PA6) (190 J g−1),28 and ΔHm is it's heat of fusion.
Thermogravimetric analysis (TGA) was performed on a TA Q500 V20.13 Build 39 thermal analysis instrument (New Castle, DE, USA) under a nitrogen atmosphere at a 10 °C min−1 heating rate from room temperature up to 700 °C.
Following the procedure outlined in reference,29 an approximation of the viscosity average molar mass (![[M with combining overline]](https://www.rsc.org/images/entities/i_char_004d_0305.gif) v) for each sample was obtained by employing diluted solution viscometry with a suspended-level Ubbelohde viscometer maintained at 25 °C in aqueous H2SO4 (98%) at a concentration of 0.2 g dl−1. The molar mass of PA6 was determined using the Mark–Houwink equation.10,30,31K = 4 × 10−3 dl g−1 and α = 0.7 are the relevant parameters in this context.32 The average flow times were calculated from five separate runs.
v) for each sample was obtained by employing diluted solution viscometry with a suspended-level Ubbelohde viscometer maintained at 25 °C in aqueous H2SO4 (98%) at a concentration of 0.2 g dl−1. The molar mass of PA6 was determined using the Mark–Houwink equation.10,30,31K = 4 × 10−3 dl g−1 and α = 0.7 are the relevant parameters in this context.32 The average flow times were calculated from five separate runs.
To know the evolution of the viscosity of the reactive systems according to the reaction time in isothermal mode. Viscosities were measured dynamically at various temperatures on a stress-controlled rheometer DHR (Discovery Hybrid Rheometer, TA Instruments, USA). The premix was quickly introduced into the preheated parallel plate geometry (θ = 25 mm) at the polymerization temperature. The variations in complex viscosity during the polymerization step were then monitored by time sweep oscillatory experiments under a strain amplitude of 10% and an angular frequency of 100 rad s−1. The rheometer chamber was purged with nitrogen for one hour, preventing thermal oxidation and polymerization inhibition and ensuring that the moisture level in the chamber was kept to a minimum. Before introducing a sample, the instrument was first equilibrated at the desired temperature, and the gap was set.
X-ray diffraction (XRD) was carried out with a custom-built θ–θ diffractometer equipped with a pyrolytic graphite monochromator and analyzer crystals. Cu-Kα radiation with wavelength λ = 0.154 nm was used for the measurements. The crystallinity (χXRDc) was calculated according to:
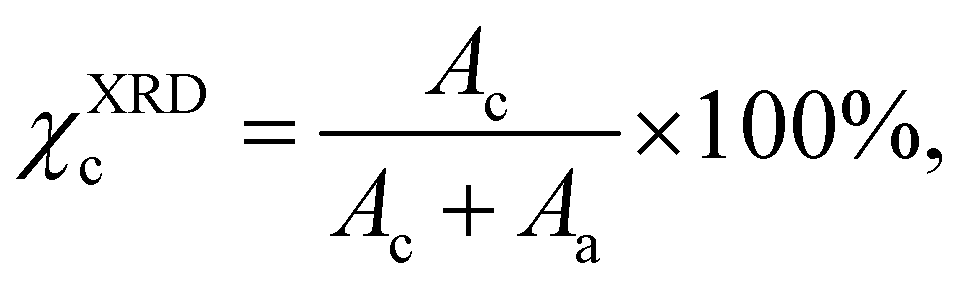 | (3) |
Results and discussion
Chemical features of anionic ring-opening ε-caprolactam polymerization
The anionic ring-opening polymerization process has a complicated chemical mechanism.33–35 Reversible and irreversible processes consume and regenerate the active species during the anionic ring-opening polymerization of ε-caprolactam.36 Several side reactions have been reported during polymerization, especially at elevated temperatures.22,36 The reaction strategy for the anionic ring-opening polymerization of caprolactam is presented in Fig. 1. Two transacylation processes, initiation and propagation, are pivotal in the reaction mechanism of ring-opening polymerization of ε-caprolactam. Initiation occurs when the activator and catalyst engage in an addition, a crucial step that sets the reaction in motion. Repetition of addition and hydrogen abstraction reactions leads to propagation, a process that builds the polymer chain. These processes, along with deactivation, branching, and reversible transacylation, contribute to the formation of yield-side reaction products, heterogeneities in the final polymer structure, and a broad molecular weight distribution.36 The intricate interplay of the activator and catalyst in these processes underscores the complexity of polymerization. | ||
| Fig. 1 Illustration of the initiation and propagation steps in the anionic ring-opening polymerization (AROP) of lactams using N-acyllactam as an activator. | ||
Impact of activator type and concentration on polymerization rate, monomer conversion, and molecular weight
Anionic polymerization of ε-caprolactam involves a series of exothermic reactions (polymerization) and processes (crystallization). The exothermic nature of polymerization can be used to determine the polymerization rate. Polymerization data are listed in Table 2, where the polymerization time, degree of conversion (DC), and the viscosity average molar mass (v) are given as functions of both the chosen activator and the initial reaction temperature ranged between 140 °C and 170 °C. In this study, both mono and bifunctional activators, N-acetylcaprolactam and N,N′-dicyclohexylcarbodiimide ([A]), were analyzed and the amount of the catalyst [C] ranged from 1.5 to 3 mol%. The amount of activator [A] was chosen to satisfy the following relation, [C] = [A] × N, where N is the functionality of the activator.
| Sample designation | Polymerization time (min) | DC (%) |
v (g mol−1) |
|---|---|---|---|
| T140-DL/ACL-1.5 | 10 | 97 | 24287 |
| T140-DL/ACL-3 | 6 | 96 | 27171 |
| T160-DL/ACL-1.5 | 7 | 99 | 44509 |
| T160-DL/ACL-3 | 1.5 | 95 | 31344 |
| T170-DL/ACL-1.5 | 3 | 98 | 45223 |
| T170-DL/ACL-3 | 0.75 | 95 | 31647 |
| T140-DL/DCCI-0.75 | 40 | 99 | 41036 |
| T140-DL/DCCI-1.5 | 16 | 97 | 27171 |
| T160-DL/DCCI-0.75 | 13 | 99 | 70381 |
| T160-DL/DCCI-1.5 | 4 | 98 | 35394 |
| T170-DL/DCCI-0.75 | 7 | 99 | 76892 |
| T170-DL/DCCI-1.5 | 3 | 98 | 37926 |
As mentioned above, the reaction consists of three basic steps: (1) dissociation of the catalyst or anion formation, (2) complex formation between the catalyst and the activator, and (3) polymerization through the anions, during which an anion is regenerated after every monomer addition.37 The combination of activator and catalyst primarily determines the reaction rate but is also controlled by the initial polymerization temperature, which will be highlighted in detail in the following section. Fig. S2† shows the change in temperature for two catalyst-activator combinations polymerized at initial temperatures ranging from 140–170 °C. Compared to the bifunctional activator, the polymerization time for complete conversion with the monofunctional activator was very impressive. This is because the DL/DCCI combination does not immediately form a complex, which explains the slow linear conversion increase at the start of the reaction. The carbamoylcaprolactam group is replaced with an acetylcaprolactam group following a single monomer addition. As a result of this group's ability to form a complex with the catalyst, the reaction rate increases over time (after 40 min for T140-DL/DCCI-0.75 and 16 min for T140-DL/DCCI-1.5). The monomer is consumed faster as the catalyst concentration increases because more anions are released, and complexes can form. More activators mean more chain growth in start locations. Thus, Fig. S2† shows that the time needed to consume all monomers up to the equilibrium conversion level decreases, whereas Table 2 shows that the final degree of conversion decreases. Because each catalyst molecule contributes a cation (see Fig. 1), a caprolactam anion must neutralize its positive charge. As the concentration of the catalyst increases, more anions are liberated, and more complexes can form; as a result, the monomer is consumed more quickly. Increasing the amount of activator increases the number of chain growth initiation points. Therefore, the time necessary to consume all monomers up to the equilibrium conversion level lowers, as seen in Fig. S2,† while the final degree of conversion drops, as indicated in Table 2. Each catalyst molecule introduces a cation (see Fig. 1), whose positive charge must be neutralized by a caprolactam anion throughout the reaction. Not all caprolactam polymerizes to compensate for these cations, limiting the highest degree of conversion possible. Increasing the activator concentration increases the proportion of oligomers with low molecular weight in the final polymer.38 Being soluble in methanol, these oligomers decrease the conversion rate as assessed by the previously stated approach. Table 2 demonstrates that the measured conversion decreases as the activator and catalyst concentrations increase, and the activator's functionality notably affects the final conversion of the reacted polymers. For the best results, the bifunctional activator (DCCI), should be used to maintain a high and nearly constant degree of conversion. In contrast, the monofunctional activator exhibits a noticeable drop in conversion. But in absolute terms, these conditions result in higher monomer conversion of polycaprolactam compared to conventional methods of ε-caprolactam polymerization.39
According to Table 2, the viscosity average molar mass (v) decreases with increasing activator (i.e., catalyst) concentration in the presence of both monofunctional and bifunctional activator in the polymerization mixture. For a given catalyst concentration, the v of the sample obtained with a monofunctional activator is significantly less than in the case of a bifunctional activator. This occurs because branching reactions can produce species with extremely high molecular weights.37 Although these types of reactions occur for monofunctional and bifunctional activators, branching is enhanced by bifunctional activators.40 Van Rijswijk et al.10 suggested that using a slower catalyst-activator combination (i.e., a longer time during which branching can take place), can produce a significant degree of branching, even at lower temperatures. The dependence of the degree of branching on the activator concentration and the reaction rate has also been reported by Mateva et al.40
The impact of initial polymerization temperature on polymerization rate, monomer conversion, molecular weight, and complex viscosity
There are two ways in which the initial polymerization temperature can affect the reaction rate.30 On the one hand, at high temperatures, it increases the polymerization rate, whereas on the other hand, it decreases the crystallization rate. As seen in Table 2, the polymerization rate reduced as the initial polymerization temperature increased. This occurred because, according to the Arrhenius equation,37 the reaction rate constant, k, increased as the polymerization temperature increased. This indicates that, as previously observed,30,37,41 the polymerization rate increased with increasing polymerization temperature, resulting in a shorter polymerization time. As a result of increasing the initial polymerization temperature, the reaction rate and the degree of monomer conversion increased. According to the literature, the amount of residual CL in PA6 depends on the ring-chain equilibrium characteristic of CL at various temperatures.36Table 2 shows the effect of the initial polymerization temperature on thev of the synthesized polymer. The v has been enhanced by increasing the initial polymerization temperature, as is evident. This trend is consistent with what has been reported in the literature.10 One possible explanation is the high energy available during polymerization at higher temperatures, which leads to condensation reactions (branching) between polymer chains.42
Rheokinetic investigation of the poly-ε-caproamide reactions
The anionic polymerization of lactams is a typical example of the ionic polymerization path in polymer synthesis. Because the reaction path and kinetics of this reaction vary significantly depending on the initial monomers and activators used, it presents fruitful opportunities for analyses of major rheokinetic patterns in these processes. To understand better the time-dependent growth in complex viscosity during the anionic polymerization of ε-caprolactam using DL/DCCI and DL/ACL combinations, here, we present the evolution of the complex viscosity as a function of time for various isothermal conditions over the range of 140–170 °C while rheological measurements are made. Fig. 2 depicts the evolution of the complex viscosity as a function of time under different isothermal conditions for the DL/ACL system. According to the results of the viscosity profiles, the molecular change caused by reaction effects led to notable variations in rheological behaviour. As shown in Fig. 2, under isothermal conditions, the complex viscosity increased dramatically as the reaction progressed. Indeed, the polymerization-induced increase in molecular weight resulted in a higher complex viscosity.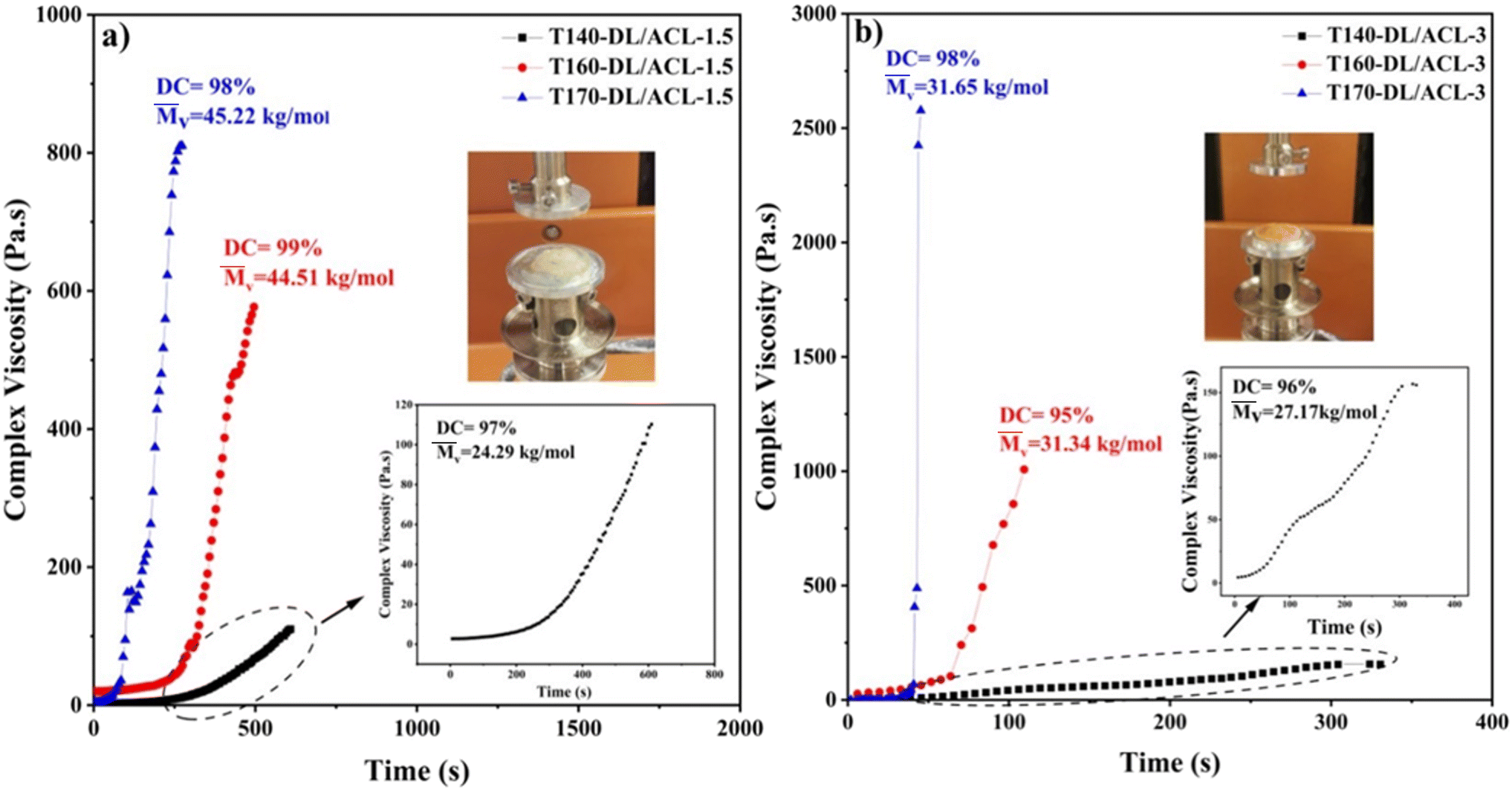 | ||
| Fig. 2 Isothermal complex viscosity evolution versus time during anionic polymerization of ε-caprolactam using DL/ACL as the catalyst/activator system at two concentrations: (a) 1.5/1.5 and (b) 3/3 at 140 °C, 160 °C and 170 °C. The degree of conversion and the molar mass are also shown. | ||
We examined the temperature's impact on the induction time and viscosity behaviour of the DL/ACL system at two concentrations. The data shows that increasing the temperature significantly shortened the induction time. In contrast, when looking at the effect of catalyst/activator concentration across all three temperatures, it was found that the DL/ACL-1.5 composition required about 10 minutes at 140 °C and 3 minutes at 170 °C to reach the maximum viscosity value, while the DL/ACL-3 composition required about 3 minutes at 140 °C and 0.75 minutes at 170 °C. As mentioned above, a higher quantity of activators resulted in a more significant number of chain development initiation locations. Moreover, more anions were liberated with a higher catalyst concentration, and more complexes could form. The acquired results matched those reported in the literature. Fig. 3 illustrates the complex viscosity-time curve for the DL/DCCI system. The time required to achieve the maximum permissible torque on the instrument was varied from about 40 minutes at 140 °C to 7 minutes at 170 °C for the composition DL/DCCI-0.75 and from 20 minutes at 140 °C to 3 minutes at 170 °C for the composition DL/DCCI-1.5. This time is very close to the time required to achieve near-infinite complex viscosity, also known as total solidification.
 | ||
| Fig. 3 Isothermal complex viscosity evolution versus time during anionic polymerization of ε-caprolactam using DL/DCCI as the catalyst/activator system at two concentrations: (a) 1.5/0.75 and (b) 3/1.5 at 140 °C, 160 °C and 170 °C. The degree of conversion and the molar mass are also shown. | ||
When the DCCI activator was used, the polymerization seemed to begin slowly. This is evidenced by the viscosity remaining stable for short durations, despite the absence of an initial complex formation.10 However, after a single monomer addition, the carbamoylcaprolactam group was replaced by an acetylcaprolactam group. Since the latter was able to form a complex with the catalyst, the reaction rate increased after a while. In this regard, it is worth noting that the induction time decreased with increasing polymerization temperature and DL/DCCI concentration. The reaction was also affected by the amount of the reactive species utilized and the temperature,10,22 in addition to the catalyst and activator types. Increasing the polymerization temperature and concentration were demonstrated to boost the polymerization rate.
The results reported so far lead to the conclusion that the combination of DL/ACL led to complex formation, resulting in rapid polymerization and an exponentially increasing viscosity with time. A reduced rate of viscosity increase was seen while using a DL catalyst in combination with DCCI since no complex was formed initially, explaining the slower increase in viscosity. The critical time increase was about 10 minutes for the DL/ACL system at 140 °C, whereas it was about 40 minutes for the DL/DCCI system at the same temperature. These critical times are calculated using the tangents method when the viscosity grows drastically. The present results also highlight that a higher degree of conversion and molecular weight were obtained with DCCI as the activator. Lastly, this work demonstrates that the reaction time was faster when DL was associated with ACL as the activator.
Thermal properties
| Sample designation | DSC data | TGA data | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Melting process | Crystallization process | ||||||||
| T m (°C) | ΔHma (J g−1) | χ cDSC (%) | T c (°C) | ΔHca (J g−1) | T donset (°C) | T dmax (°C) | T d10% (°C) | T d50% (°C) | |
| a The values in the top and bottom rows correspond to the first and second scans, respectively. | |||||||||
| T140-DL/ACL-1.5 | 221 | 107 | 56 | 185 | 56 | 351 | 422 | 362 | 409 |
| 210 | 69 | 36 | 183 | 65 | |||||
| T140-DL/ACL-3 | 217 | 98 | 52 | 176 | 69 | 344 | 417 | 357 | 407 |
| 213 | 70 | 37 | 176 | 68 | |||||
| T160-DL/ACL-1.5 | 218 | 98 | 52 | 175 | 72 | 326 | 411 | 336 | 397 |
| 215 | 60 | 32 | 175 | 70 | |||||
| T160-DL/ACL-3 | 214 | 88 | 46 | 172 | 72 | 320 | 407 | 333 | 396 |
| 211 | 62 | 33 | 172 | 73 | |||||
| T170-DL/ACL-1.5 | 216 | 100 | 53 | 173 | 70 | 353 | 410 | 334 | 400 |
| 214 | 59 | 31 | 172 | 67 | |||||
| T170-DL/ACL-3 | 212 | 85 | 45 | 170 | 71 | 331 | 405 | 334 | 394 |
| 210 | 59 | 31 | 169 | 67 | |||||
| T140-DL/DCCI-0.75 | 221 | 111 | 58 | 170 | 69 | 309 | 408 | 324 | 383 |
| 212 | 59 | 31 | 168 | 70 | |||||
| T140-DL/DCCI-1.5 | 217 | 99 | 52 | 173 | 70 | 269 | 325 | 289 | 322 |
| 211 | 60 | 31 | 168 | 68 | |||||
| T160-DL/DCCI-0.75 | 217 | 93 | 50 | 176 | 67 | 328 | 432 | 359 | 420 |
| 215 | 56 | 29 | 176 | 67 | |||||
| T160-DL/DCCI-1.5 | 216 | 113 | 59 | 168 | 71 | 304 | 426 | 321 | 379 |
| 210 | 64 | 34 | 166 | 70 | |||||
| T170-DL/DCCI-0.75 | 213 | 95 | 50 | 173 | 64 | 341 | 457 | 385 | 443 |
| 214 | 55 | 28 | 171 | 63 | |||||
| T170-DL/DCCI-1.5 | 216 | 103 | 54 | 173 | 68 | 332 | 437 | 348 | 423 |
| 211 | 61 | 32 | 170 | 69 | |||||
It is also important to note that the second heat traces of all produced samples feature two peaks (as shown in Fig. S3 and S4†). After a second heating, a lower and new peak is seen at the typical melting point. The polymerization of caprolactam at initial temperatures between 140 and 170 °C (i.e., below the melting point of poly-ε-caproamide) may result in a material with a combination of α and g crystalline structures. Puffr et al.36 stated that as many as five melting endotherms may occur in samples of PA6 with the mixed α and g structures: the peaks of original and recrystallized γ structures; the peaks of original and recrystallized α structures (higher by several degrees); and also a small peak approximately 10 °C above crystallization temperature (Tc), assigned to the most defective crystals formed by secondary crystallization. In Fig. S3 and S4,† the two peaks seen on the DSC thermograms, possibly arising from the original α and γ structures formed during polymerization because the sample was not exposed to thermal history for recrystallization.
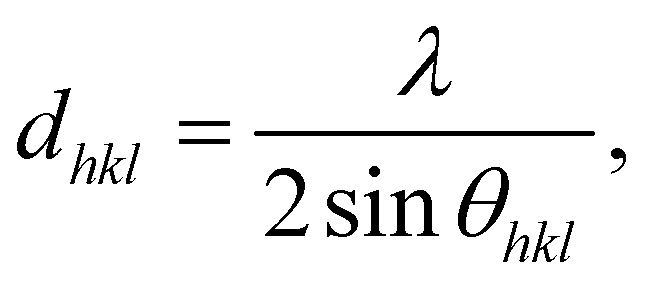 | (4) |
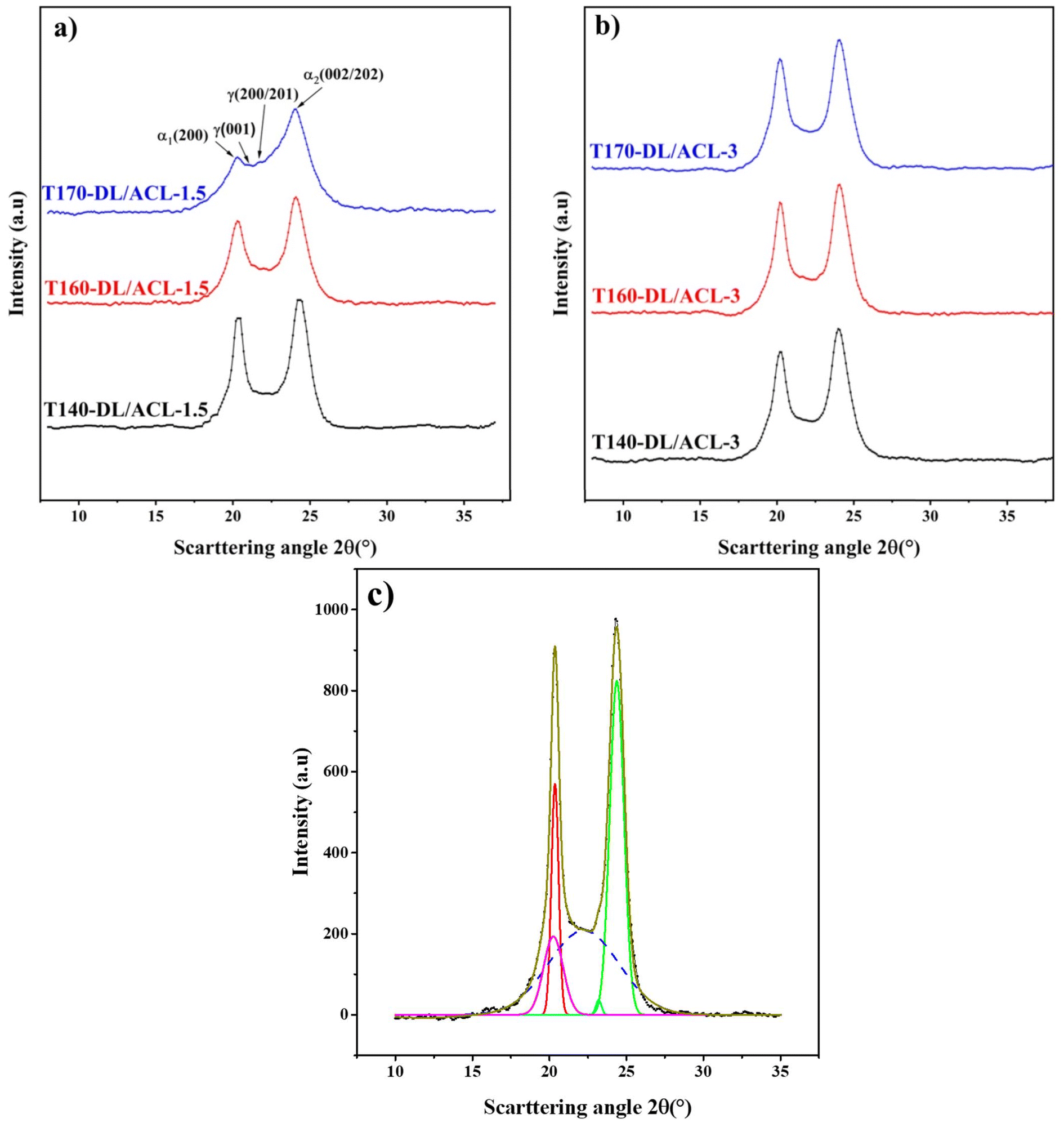 | ||
| Fig. 4 1D WAXD intensity profiles of the poly-ε-caproamide (PA6) using DL/ACL as the catalyst/activator system at two concentrations, (a) DL/ACL-1.5, (b) DL/ACL-3 at different polymerization temperatures and (c) Selected WAXS pattern of T140-DL/ACL 1.5 sample and its fitting with Gaussian peaks: crystalline peaks are represented by solid lines, while dashed lines represent the amorphous halo. | ||
| Sample designation | Crystallinity Index (%) | α/γ | d-Spacing (Å) | ICP (Å) |
|
|||
|---|---|---|---|---|---|---|---|---|
| α-Phasea | γ-Phasea |
χ
cXRDb |
α 1(200) | α 2(002/202) | ||||
a
 ; ;  .
b
χ
cXRD is determined using the deconvolution method (Gaussian fitting with fitting coefficients r2 ≈ 0.99). .
b
χ
cXRD is determined using the deconvolution method (Gaussian fitting with fitting coefficients r2 ≈ 0.99).
|
||||||||
| T140-DL/ACL-1.5 | 46.00 | 11.5 | 57.5 | 4.0 | 4.477 | 3.748 | 0.728 | 0.837 |
| T140-DL/ACL-3 | 39.4 | 12.6 | 52.0 | 3.1 | 4.505 | 3.799 | 0.706 | 0.843 |
| T160-DL/ACL-1.5 | 40.2 | 8.9 | 49.1 | 4.5 | 4.492 | 3.790 | 0.702 | 0.844 |
| T160-DL/ACL-3 | 35.2 | 9.3 | 44.5 | 3.8 | 4.507 | 3.800 | 0.707 | 0.843 |
| T170-DL/ACL-1.5 | 29.4 | 17.8 | 47.2 | 1.6 | 4.496 | 3.794 | 0.702 | 0.844 |
| T170-DL/ACL-3 | 29.6 | 11.9 | 41.5 | 2.5 | 4.507 | 3.802 | 0.705 | 0.843 |
| T140-DL/DCCI-0.75 | 41.5 | 16.7 | 58.2 | 2.4 | 4.464 | 3.748 | 0.715 | 0.840 |
| T140-DL/DCCI-1.5 | 25.0 | 11.0 | 36.0 | 2.3 | 4.453 | 3.765 | 0.688 | 0.845 |
| T160-DL/DCCI-0.75 | 32.8 | 16.3 | 49.1 | 2.0 | 4.429 | 3.756 | 0.673 | 0.848 |
| T160-DL/DCCI-1.5 | 24.7 | 6.3 | 31.0 | 3.9 | 4.446 | 3.794 | 0.652 | 0.853 |
| T170-DL/DCCI-0.75 | 34.4 | 8.6 | 43.0 | 4.0 | 4.453 | 3.800 | 0.652 | 0.853 |
| T170-DL/DCCI-1.5 | 17.7 | 13.3 | 31.0 | 1.3 | 4.477 | 3.748 | 0.728 | 0.837 |

According to previous research,56,58 the two peaks with 2θ at about 20° and 24° were assigned to the (200) and (002/202) crystallographic planes of the monoclinic unit cell of the α-PA6 polymorph. Additionally, two Gaussian peaks corresponding to (001) and (200) crystalline planes with 2θ between 22 and 23° were discovered, indicative of the PA6 γ-crystalline form with a pseudo-hexagonal unit cell.
It is well-known that FTIR spectroscopy is one of the most well-established methods for the characterization of nylons and that it provides information about the crystalline phase of polyamide samples.59,60Fig. 5 illustrates the FTIR spectra of poly-ε-caproamide (PA6) samples at several polymerization conditions, along with characteristic infrared bands and their roles. The bands at 3196 cm−1 and 1654 cm−1 in the spectra of the monomer ε-caprolactam correspond to the stretching vibrations of the N–H and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds, respectively,61 and are coupled with the self-associated hydrogen bonds produced between CO and N–H that define the amide II mode. The bands in the 1465–1150 cm−1 range correspond to the methyl alkyl group (CH2) from cyclic lactam related to the bands in the 3500 and 2750 cm−1 region. The bands at 1486 and 1438 cm−1 correspond to the scissor vibration in the (CH2) group, whereas the band at 1417 cm−1 refers to bending vibrations in the same group. Moreover, the bands at 1365, 1312, 1290, 1258, and 1198 cm−1 correspond to the amide group covalently bound to the alkyl group (NH–CH2). The lactam CO bond was detected at 1654 cm−1 and confirmed by the 822 and 693 cm−1 bands. However, the spectra of poly-ε-caproamides (PA6) display two bands at 3290 and 1535 cm−1, ascribed to hydrogen bond stretching and bending vibration in amide II, respectively. The 1635 cm−1 band is associated with mode I amide bond bending vibrations. Torsion in amide mode I and mode II hydrogen bonds are attributed to the 683 cm−1 and 574 cm−1 bands, respectively. Also, the presence of two bands at 1199 and 1169 cm−1 is according to amide mode III. The carbonyl (CO) band is located at 1635 cm−1 and overlaps with the mode I band of the amide. As previously stated, the (OCN) bond related to the caprolactam ring was broken (by ring opening) due to the catalyst, and the sodium–caprolactam was formed; this is confirmed since a band is located at 3074 cm−1. In addition, the ring opening is confirmed by the band's presence at 1263 cm−1, which corresponds to the C–N aliphatic bond. Finally, in the poly-ε-caproamides (PA6) CH2 rocking band area, there are three bands. Sandeman and Keller assigned the 928 cm−1 band of the α-form of PA6 to the rocking mode CH2.62 Asai et al.63 and Miyake64 ascribe the remaining two bands at 730 and 683 cm−1 to the NH out-of-plane deformation mode (the amide V band). All phases are produced during polymerization, regardless of the raw materials, monomers, and synthesis conditions. In the process of re-crystallization, the γ-phase is eliminated for material exposure. The primary peak in the DSC analysis at around 221–212 °C and the main peaks in the diffraction patterns at 2 = 20° and 24°, as stated previously, indicate that the phase (monoclinic crystal) is the predominant and stable crystalline phase in the synthesized PA6.
O bonds, respectively,61 and are coupled with the self-associated hydrogen bonds produced between CO and N–H that define the amide II mode. The bands in the 1465–1150 cm−1 range correspond to the methyl alkyl group (CH2) from cyclic lactam related to the bands in the 3500 and 2750 cm−1 region. The bands at 1486 and 1438 cm−1 correspond to the scissor vibration in the (CH2) group, whereas the band at 1417 cm−1 refers to bending vibrations in the same group. Moreover, the bands at 1365, 1312, 1290, 1258, and 1198 cm−1 correspond to the amide group covalently bound to the alkyl group (NH–CH2). The lactam CO bond was detected at 1654 cm−1 and confirmed by the 822 and 693 cm−1 bands. However, the spectra of poly-ε-caproamides (PA6) display two bands at 3290 and 1535 cm−1, ascribed to hydrogen bond stretching and bending vibration in amide II, respectively. The 1635 cm−1 band is associated with mode I amide bond bending vibrations. Torsion in amide mode I and mode II hydrogen bonds are attributed to the 683 cm−1 and 574 cm−1 bands, respectively. Also, the presence of two bands at 1199 and 1169 cm−1 is according to amide mode III. The carbonyl (CO) band is located at 1635 cm−1 and overlaps with the mode I band of the amide. As previously stated, the (OCN) bond related to the caprolactam ring was broken (by ring opening) due to the catalyst, and the sodium–caprolactam was formed; this is confirmed since a band is located at 3074 cm−1. In addition, the ring opening is confirmed by the band's presence at 1263 cm−1, which corresponds to the C–N aliphatic bond. Finally, in the poly-ε-caproamides (PA6) CH2 rocking band area, there are three bands. Sandeman and Keller assigned the 928 cm−1 band of the α-form of PA6 to the rocking mode CH2.62 Asai et al.63 and Miyake64 ascribe the remaining two bands at 730 and 683 cm−1 to the NH out-of-plane deformation mode (the amide V band). All phases are produced during polymerization, regardless of the raw materials, monomers, and synthesis conditions. In the process of re-crystallization, the γ-phase is eliminated for material exposure. The primary peak in the DSC analysis at around 221–212 °C and the main peaks in the diffraction patterns at 2 = 20° and 24°, as stated previously, indicate that the phase (monoclinic crystal) is the predominant and stable crystalline phase in the synthesized PA6.
 | ||
| Fig. 5 FTIR spectra of the studied poly-ε-caproamide (PA6) synthesized utilizing a DL/ACL system at varying polymerization temperatures. (a) 4000–400 cm−1, (b) 1500–500 cm−1. | ||
The findings of XRD measurements, presented in Table 4, show that the degree of crystallinity of poly-ε-caproamide (PA6) samples decreased as the amount of catalyst/activator increased, in agreement with the degree of crystallinity determined by DSC analysis listed in Table 3. Increasing the catalyst/activator concentration results in an elevation in polymerization temperature because of a rise in polymerization rate (i.e., exothermic polymerization). The temperature at which polymerization occurs affects the process in two different ways. When heated to high temperatures, it has the opposite effect of slowing the crystallization rate and speeding up the polymerization rate.65 However, the crystallization rate is very high at low temperatures, and reactive groups can become caught inside developing crystals before they can polymerize. This can only happen when the temperature is very low. When polymerization occurs at temperatures higher than 170 °C, the crystallization rate is reduced, although the polymerization rate remains high.30
Table 4 illustrates the effect of initial polymerization temperature on the degree of crystallinity of the synthesized polymer. As the initial polymerization temperature rose from 140 °C to 170 °C, the degree of crystallinity diminished. Because of the competitive nature of polymerization and crystallization in this process, the higher thermal motion of the polymer chains, and also the enhanced rate of branching at higher temperatures,37,46 the reduction in crystallinity of synthesized poly-ε-caproamide (PA6) that can be achieved by increasing the initial temperature of polymerization is caused by a combination of these factors. The fraction of α-PA6 predominates in all poly-ε-caproamide (PA6) samples, with α/γ > 1 for both DL/ACL-1.5 and DL/ACL-3 formulations. The d-spacings of α(200), α(002)/(202) of α-PA6 using DL/ACL as the catalyst/activator system at two concentrations and varying polymerization temperatures are shown in Table 4. A crystalline perfection index (ICP) is derived from the relationship between crystalline perfection and crystallinity to quantify variations in crystalline perfection. The index of chain packing (ICP) is defined as the difference of the d-spacings for both the (200), (002)/(202) peaks,66 ICP = Δd = d200 − d002/202.
Fig. 6 demonstrates that the crystal lattice parameters of poly-ε-caproamide (PA6) are impacted by both the catalyst/activator concentration and the polymerization temperature, as indicated by the rise in d-spacings with increasing polymerization temperature and catalyst/activator concentration. With increasing catalyst/activator incorporation into the PA6 chain, the resulting changes in crystal lattice properties become statistically significant. This leads to significant morphological alterations and significant disruption of the crystal structure. The Δd of the synthesized poly-ε-caproamide (PA6) is around 0.702–0.728 Å for DL/ACL: 1.5/1.5, and around 0.705–0.707 Å for DL/ACL: 3/3 depending on the polymerization temperature.
 | ||
| Fig. 6 Change in d-spacing of some (hkl) diffraction of the studied poly-ε-caproamide (PA6) using DL/ACL as the catalyst/activator system at two concentrations at different polymerization temperatures. | ||
With the DL/ACL-1.5 formulation, T140-DL/ACL-1.5 exhibited ICP = 0.728 Å, which was greater than poly-ε-caproamides synthesized at 160 °C (T160-DL/ACL-1.5) and 170 °C (T170-DL/ACL-1.5). Murthy et al.66 reported that a structure with a higher ICP corresponds to an energetically lower and more stable PA6 lattice. However, when comparing samples of the DL/ACL-3 formulation, we found that the ICP was roughly constant from one to the next, with no clear correlation to the synthesis conditions. Poly-ε-caproamides (PA6) chain packing index does not appear to be affected by polymerization temperature. For the DL/ACL-1.5 formulation, both T160-DL/ACL-1,5 and T170-DL/ACL-1.5 have da200/da002/202 of about 0.844, which represents the ratio c/a of unit cell.67 This is a little bit greater than T140-DL/ACL-1.5 (0.837), and it suggests that the distance between hydrogen-bonded sheets (i.e. dα002/202) in the unit cell is slightly larger than that of T140-DL/ACL-1.5. A comparison of samples of the DL/AC-3 formulation reveals that the distance between hydrogen-bonded sheets is constant over all three polymerization temperatures, with a dα200/dα002/202 ratio of around 0.843.
 | ||
| Fig. 7 1D WAXD intensity profiles of the poly-ε-caproamide (PA6) using DL/DCCI as the catalyst/activator system at two concentrations, (a) DL/DCCI-0.75, (b) DL/DCCI-1.5 at different polymerization temperatures and (c) selected WAXS pattern of T160-DL/DCCI-0.75 sample and its fitting with Gaussian peaks: crystalline peaks are represented by solid lines and dashed lines represent the amorphous halo. | ||
This is consistent with the findings of the IR investigation presented below. As seen in Fig. 8. The IR spectrum of the obtained homopolymer presents strong absorption bands at 3290 cm−1 (N–H stretch vibration), 1635 cm−1 (amide I, CO), and 1536 cm−1 (amide II, N–H deformation) which are characteristic of the amide groups existing in the trans planar conformation. As indicated earlier, the amide II band is extremely sensitive to the crystalline structure corresponding to the α-phase.59,60 Furthermore, the out-of-plane bends of the NH (amide V) and CO (amide VI) groups, which appear at 684 and 575 cm−1, respectively, are polymorph sensitive and indicate that the form of the crystalline phase of homopolymer is α-type.59,60,68 As stated before, the results show that monoclinic crystal is the most common and stable crystalline phase in the synthesized PA6.
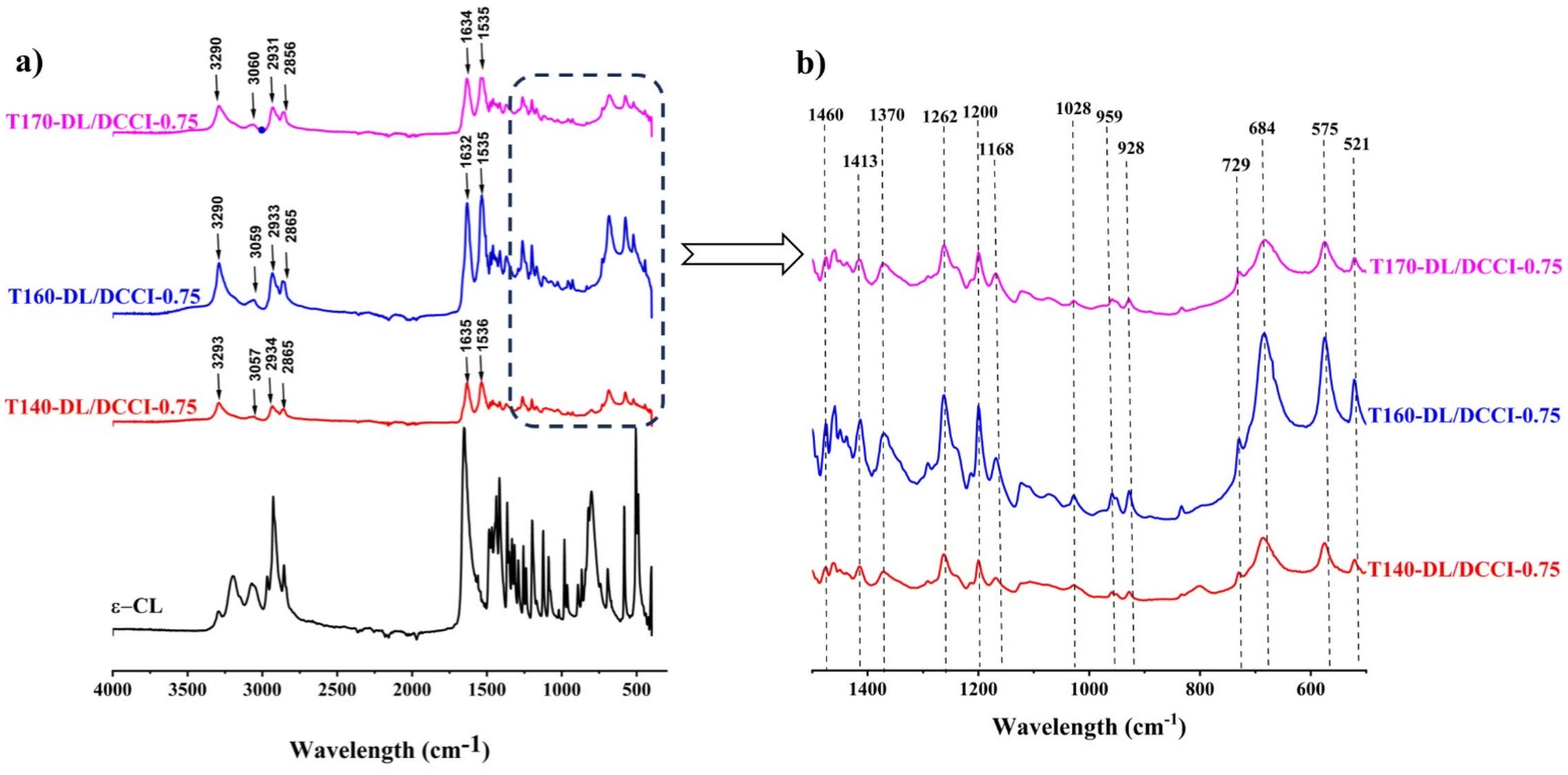 | ||
| Fig. 8 FTIR spectra of poly-ε-caproamide (PA6) synthesized utilizing a DL-DCCI system at varying polymerization temperatures. (a) 4000–400 cm−1, (b) 1500–500 cm−1. | ||
Based on the XRD data reported in Table 4, it is clear that the degree of crystallinity of poly-ε-caproamide (PA6) samples is dependent on both the catalyst/activator concentration and polymerization temperature. Increasing the amounts of bifunctional activator and catalyst reduced the crystallinity. DSC analysis shows that the degree of crystallinity tends to go the same way. Since the activator N,N-dicyclohexylcarbodiimide (DCCI) has two functions, two or less acyllactam functions can start the polymerization process. The macromolecular chains cannot reorganize into a perfect crystalline form due to changes in chain length and, most likely, increased chain mobility due to diminished protonic contact between NH and CO groups.
The effect of initial polymerization temperature on the degree of crystallinity of the produced polymer is depicted in Table 4. As shown before for the system DL/ACL, the degree of crystallinity decreased when the initial polymerization temperature increased from 140 °C to 170 °C. Due to the competitive nature of polymerization and crystallization in this process, the higher thermal motion of the polymer chains, and the enhanced rate of branching at higher temperatures,37,38 increasing the initial temperature of polymerization can reduce the crystallinity of synthesized poly-ε-caproamide (PA6).
The d-spacings of α(200), α(002)/(202) and the index of chain packing (ICP) of α-PA6 using DL/DCCI as the catalyst/activator system at two concentrations at varying polymerization temperatures are shown in Table 4.
Fig. 9 demonstrates that the crystal lattice parameters of poly-ε-caproamide (PA6) is impacted by both the catalyst/activator concentration and the polymerization temperature, as indicated by the rise in d-spacings with increasing polymerization temperature and catalyst/activator concentration. With increasing catalyst/activator incorporation into the PA6 chain, the resulting changes in crystal lattice properties become statistically significant. This leads to significant morphological alterations and significant disruption of the crystal structure.
 | ||
| Fig. 9 Change in d-spacing of some (hkl) diffraction of the studied formulations of poly-ε-caproamide (PA6) using DL/DCCI as the catalyst/activator system at two concentrations at different polymerization temperatures. | ||
With the DL/DCCI-0.75 formulation, T140-DL/DCCI-0.75 exhibited ICP = 0.715 Å, which was greater than poly-ε-caproamides synthesized at 160 °C (T160-DL/DCCI-0.75) and 170 °C (T170-DL/DCCI-0.75). Comparing samples of the DL/DCCI-1.5 formulation, we noticed that T170-DL/DCCI-1.5 had a higher ICP than T140-DL/DCCI-1.5 and T170-DL/DCCI-0.75. Murthy et al.66 reported that a structure with a higher ICP corresponds to an energetically lower and more stable PA6 lattice.
For the DL/DCCI-0.75 formulation, the dα200/dα002/202 ratio, which represents the ratio c/a of unit cell,67 of T170-DL/DCCI-0.75, it has a ratio of about 0.853. This is a little bit greater than that of T140-DL/DCCI-0.75 and T160-DL/DCCI-0.75 (0.840), and it suggests that the distance between hydrogen-bonded sheets (i.e. dα002/202) in the unit cell is slightly greater than that of T170-DL/DCCI-0.75. Comparison of samples of the DL/DCCI-1.5 formulation, shows that T160-DL/DCCI-1.5 has a larger distance between hydrogen-bonded sheets with a dα200/dα002/202 ratio around 0.853 compared to T140-DL/DCCI-1.5 and T170-DL/DCCI-1.5.
In brief, the effect of the activator on the structure of poly-ε-caproamides (PA6) is governed by two factors: (a) changes in the rate of the polymerization and crystallization process and, (b) changes in the chemical structure due to the incorporation of the activator into the polymer chain.69 In fact, the reported data suggest that the type of catalyst-activator combination exerts a significant influence on the polymerization and subsequent crystallization of PA6. By varying the amount of appropriately chosen activators, it is possible to control the crystallinity level throughout a wide range. According to the findings of the present experiments, both the type and quantity of activating additives have a substantial impact on the bulk structural development of poly-ε-caproamides (PA6).
 | ||
| Fig. 10 Cross-sectional SEM images illustrating (A) the edge and (B) the middle sections of a hybrid composite composed of E-glass (20 wt%) and hemp fibre (5 wt%). | ||
Conclusions
The bulk anionic polymerization of ε-caprolactam was investigated with focus on the influence of two catalyst-activator systems: sodium dicaprolactamato-bis-(2-methoxyethoxo)-aluminate (DL) with N-acetylcaprolactam (ACL), and DL with N,N′-dicyclohexylcarbodiimide (DCCI). The study identifies the polymerization temperature, as well as the type and concentration of the activators, as critical parameters influencing the polymerization dynamics and polymer properties. Increasing the initial polymerization temperature reduces polymerization time and concomitantly decreases crystallinity. However, as the polymerization temperature rises, the average molar mass of the polymer increases. Furthermore, a combination of higher polymerization temperature and lower catalyst-activator concentration yields a higher degree of monomer conversion. On the other hand, higher concentrations of catalyst and activator accelerate polymerization but reduce the average molar mass. A comparative analysis of the catalyst-activator combinations reveals that the DL/ACL system is the most effective for achieving rapid polymerization. Material characterization was performed using DSC, WAXD, and FTIR. These techniques confirm that the α-phase, a monoclinic crystalline structure, is the predominant and thermodynamically stable phase in the synthesized polyamide 6 (PA6). The results underscore the critical role of processing conditions in tailoring polymer properties and the significance of the DL/ACL system for efficient and controlled polymerization. E-glass/treated hemp fibers hybrid composites using polyamide 6 through anionic polymerization have been fabricated successfully with no observable inhibition of the polymerization process by the presence of these fibers and strong adhesion between the fibers and matrix. Future research will examine the influence of fiber content, processing parameters, and polymerization conditions on these composites’ physical, thermomechanical, and microstructural properties.Author contributions
Karima Ben Hamou: conceptualization, data curation, investigation, methodology, writing – original draft. Ralf Brüning: data curation, investigation, calculation, writing – reviews & editing. Jacques Robichaud: data curation, formal analysis, writing – reviews & editing. Marie-Hélène Thibault: data curation, formal analysis, funding acquisition, writing – reviews & editing. Gabriel LaPlante: data curation, funding acquisition, writing – reviews & editing. Yahia Djaoued: conceptualization, data curation, supervision, project administration, funding acquisition, writing – review & editing.Data availability
The data supporting this article have been included as part of the ESI.†The FTIR, DSC, TGA, DTG, XRD, and rheological data supporting this study's findings are provided within the manuscript and ESI.†
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The financial support from the Atlantic Canada Opportunities Agency (grant #212225), the New Brunswick Innovation Fund (grant # RIF2019-025), the National Sciences and Engineering Research Council (NSERC) of Canada (grant #2023-05942), the Regional Development Corporation of New Brunswick (grant #11266), the John R. Evans Leaders Fund (Canada Foundation for Innovation) (grant #27741), the Mitacs Accelerate (grant #IT16139/FR48699/NB-ISDE).References
- S. Russo, S. Maniscalco and L. Ricco, Polym. Adv. Technol., 2015, 26, 851–854 CrossRef CAS.
- M. Wilhelm, R. Wendel, M. Aust, P. Rosenberg and F. Henning, J. Compos. Sci., 2020, 4, 1–19 Search PubMed.
- T. Ageyeva, I. Sibikin and J. Karger-Kocsis, Polymers, 2018, 10, 357 CrossRef PubMed.
- C. W. Choi, J. W. Jin, H. Lee, M. Huh and K. W. Kang, Fibers Polym., 2019, 20, 1021–1028 CrossRef CAS.
- J. un Jang, H. S. Lee, J. W. Kim, S. Y. Kim, S. H. Kim, I. Hwang, B. J. Kang and M. K. Kang, Chem. Eng. J., 2019, 373, 251–258 CrossRef.
- E. Rusu, J. Compos. Mater., 2020, 54, 345–362 CrossRef CAS.
- M. Jiang, H. Tan, L. Jiang, J. Duan, X. Gu, L. Feng and C. Zhang, Ind. Eng. Chem. Res., 2024, 63, 19476–19485 CrossRef CAS.
- K. Ben Hamou, R. Bruning, G. LaPlante, M. Thibault, J. Robichaud and Y. Djaoued, J. Appl. Polym. Sci., 2024, 141, 1–15 Search PubMed.
- Z. Gao, R. Zhao and X. Ning, Chem. Eng. J., 2025, 504, 158758 CrossRef CAS.
- K. van Rijswijk, H. E. N. Bersee, A. Beukers, S. J. Picken and A. A. van Geenen, Polym. Test., 2006, 25, 392–404 CrossRef CAS.
- K. van Rijswijk, A. A. van Geenen and H. E. N. Bersee, Composites, Part A, 2009, 40, 1033–1043 CrossRef.
- J. J. E. Teuwen, A. A. Van Geenen and H. E. N. Bersee, J. Thermoplast. Compos. Mater., 2012, 25, 965–986 CrossRef.
- S. Kashani Rahimi and J. U. Otaigbe, Polym. Compos., 2019, 40, 1104–1116 Search PubMed.
- K. van Rijswijk and H. E. N. Bersee, Composites, Part A, 2007, 38, 666–681 CrossRef.
- S. Epple and C. Bonten, AIP Conf. Proc., 2014, 1593, 454–457 CrossRef CAS.
- K. Chen, M. Jia, H. Sun and P. Xue, Materials, 2019, 12, 463 CrossRef CAS PubMed.
- M. Dkier, M. Yousfi, K. Lamnawar and A. Maazouz, Eur. Polym. J., 2019, 120, 109227 Search PubMed.
- A. Maazouz, K. Lamnawar and M. Dkier, Composites, Part A, 2018, 107, 235–247 CrossRef CAS.
- Z. Kovács, Á. Pomázi and A. Toldy, Polym. Degrad. Stab., 2022, 195, 109797 CrossRef.
- E. Harkin-Jones and R. J. Crawford, Polym. Eng. Sci., 1996, 36, 615–625 CrossRef CAS.
- G. H. Rusu, M. Rusu, E. Rusu, A. Stoleriu and C. A. Teaca, Polym.-Plast. Technol. Eng., 2000, 39, 233–247 CrossRef CAS.
- N. Barhoumi, A. Maazouz, M. Jaziri and R. Abdelhedi, eXPRESS Polym. Lett., 2013, 7, 76–87 CrossRef CAS.
- S. Russo and E. Casazza, Ring-Opening Polymerization of Cyclic Amides (Lactams), Elsevier B.V., 2012, vol. 4 Search PubMed.
- R. M. Joyce and D. M. RitterU. S. Pat.2251519, 1941 Search PubMed.
- A. Bledzki and J. Gassan, Prog. Polym. Sci., 1999, 24, 221–274 CrossRef CAS.
- M. Le Troedec, D. Sedan, C. Peyratout, J. P. Bonnet, A. Smith, R. Guinebretiere, V. Gloaguen and P. Krausz, Composites, Part A, 2008, 39, 514–522 CrossRef.
- K. van Rijswijk, S. Lindstedt, D. P. N. Vlasveld, H. E. N. Bersee and A. Beukers, Polym. Test., 2006, 25, 873–887 CrossRef CAS.
- N. V. Dencheva, J. F. B. Braz and Z. Z. Denchev, J. Appl. Polym. Sci., 2022, 139, 1–17 Search PubMed.
- W.-M. Kulicke, Viscosimetry of Polymers and Polyelectrolytes, Springer Science & Business Media, 2004 Search PubMed.
- K. Van Rijswijk, H. E. N. Bersee, W. F. Jager and S. J. Picken, in Composites Part A: Applied Science and Manufacturing, Elsevier Ltd, 2006, vol. 37, pp. 949–956 Search PubMed.
- B. Lu, K. Lamnawar, A. Maazouz and H. Zhang, Soft Matter, 2016, 12, 3252–3264 RSC.
- R. Mateva and O. Delev, Polym. J., 1995, 27, 449–460 CrossRef CAS.
- R. Puffr and J. Šebenda, Eur. Polym. J., 1972, 8, 1037–1044 CrossRef.
- R. S. Davé, R. L. Kruse, L. R. Stebbins and K. Udipi, Polymer, 1997, 38, 939–947 CrossRef.
- R. Mateva and N. Dencheva, J. Polym. Sci., Part A: Polym. Chem., 1992, 30, 1449–1462 CrossRef CAS.
- R. Puffr and V. Kubanek, Lactam-Based Polyamides, CRC Press, 1991, vol. I Search PubMed.
- K. Udipi, R. S. Davé, R. L. Kruse and L. R. Stebbins, Polymer, 1997, 38, 927–938 CrossRef CAS.
- K. Ueda, M. Hosoda, T. Matsuda and K. Tai, Polym. J., 1997, 30, 186–191 CrossRef.
- S. Russo, A. Imperato, A. Mariani and F. Parodi, Macromol. Chem. Phys., 1995, 196, 3297–3303 CrossRef CAS.
- R. Mateva, P. Petrov, S. Rousseva, R. Dimitrov and G. Zolova, Eur. Polym. J., 2000, 36, 813–821 CrossRef CAS.
- S. Davtyan, H. Zakaryan, A. Tonoyan and G. Varderesyan, e-Polym., 2007, 7, 1–8 Search PubMed.
- K. J. Kim, D. S. Hong and A. R. Tripathy, J. Appl. Polym. Sci., 1997, 66, 1195–1207 CrossRef CAS.
- R. Mateva, O. Delev and E. Kaschcieva, J. Appl. Polym. Sci., 1995, 58, 2333–2343 CrossRef CAS.
- B. G. Risch, G. L. Wilkes and J. M. Warakomski, Polymer, 1993, 34, 2330–2343 CrossRef CAS.
- J. R. Ebdon, Polym. Int., 1992, 27, 207–208 CrossRef.
- L. Ricco, S. Russo, G. Orefice and F. Riva, Macromolecules, 1999, 32, 7726–7731 CrossRef CAS.
- S.-P. Rwei, P. Ranganathan and Y.-H. Lee, Polymers, 2019, 11, 472 CrossRef PubMed.
- C. L. Zhang, L. F. Feng and G. H. Hu, J. Appl. Polym. Sci., 2006, 101, 1972–1981 CrossRef CAS.
- D. R. Holmes, C. W. Bunn and D. J. Smith, J. Polym. Sci., 1955, 17, 159–177 CrossRef CAS.
- H. Arimoto, M. Ishibashi, M. Hirai and Y. Chatani, J. Polym. Sci., Part A: Gen. Pap., 1965, 3, 317–326 CrossRef CAS.
- N. S. Murthy, S. M. Aharoni and A. B. Szollosi, J. Polym. Sci., Polym. Phys. Ed., 1985, 23, 2549–2565 CrossRef CAS.
- N. S. Murthy, R. G. Bray, S. T. Correale and R. A. F. Moore, Polymer, 1995, 36, 3863–3873 CrossRef CAS.
- Y. Liu, L. Cui, F. Guan, Y. Gao, N. E. Hedin, L. Zhu and H. Fong, Macromolecules, 2007, 40, 6283–6290 CrossRef CAS PubMed.
- X. Liu and Q. Wu, Polymer, 2002, 43, 1933–1936 CrossRef CAS.
- C. A. Taylor, Acta Crystallogr., Sect. A, 1970, 26, 700–701 CrossRef.
- J. M. Samon, J. M. Schultz and B. S. Hsiao, Polymer, 2000, 41, 2169–2182 CrossRef CAS.
- N. Dencheva, T. G. Nunes, M. J. Oliveira and Z. Denchev, J. Polym. Sci., Part B: Polym. Phys., 2005, 43, 3720–3733 CrossRef CAS.
- N. Dencheva, T. Nunes, M. J. Oliveira and Z. Denchev, Polymer, 2005, 46, 887–901 CrossRef CAS.
- M. I. Kohan, Nylon Plastics Handbook, Hanser, Munich, Germany, 1995 Search PubMed.
- S. M. Aharoni, n-Nylons: their synthesis, structure, and properties, 1997 Search PubMed.
- J. C. Farias-Aguilar, M. J. Ramírez-Moreno, L. Téllez-Jurado and H. Balmori-Ramírez, Mater. Lett., 2014, 136, 388–392 CrossRef CAS.
- I. Sandeman and A. Keller, J. Polym. Sci., 1956, 19, 401–435 CrossRef CAS.
- M. Asai, M. Tsuboi, T. Shimanouchi and S. I. Mizushima, J. Phys. Chem., 1955, 59, 322–325 CrossRef CAS.
- A. Miyake, J. Polym. Sci., 1960, 44, 223–232 CrossRef CAS.
- R. M. Hedrick, J. D. Gabbert and M. H. Wohl, in ACS Symposium Series, 1985, pp. 135–162.
- N. S. Murthy, H. Minor and R. A. Latif, J. Macromol. Sci., Part B: Phys., 1987, 26, 427–446 CrossRef.
- N. S. Murthy, Polym. Commun., 1990, 32, 301–305 Search PubMed.
- J. L. Yeh, J. F. Kuo and C. Y. Chen, Mater. Chem. Phys., 1994, 37, 161–169 CrossRef CAS.
- R. Mateva, O. Delev and S. Rousseva, Eur. Polym. J., 1997, 33, 1377–1382 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5lp00015g |
| This journal is © The Royal Society of Chemistry 2025 |