
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Functionalization of siliceous materials, part 4: immobilization of fluorinated dyes for optical chemical sensor applications
Saskia
Czihal
ad,
Frank
Bauer
ad,
Marko
Bertmer
 b,
Axel
Kahnt
c,
Sergej
Naumov
c,
Matthias
Lau
*d and
Dirk
Enke
*a
b,
Axel
Kahnt
c,
Sergej
Naumov
c,
Matthias
Lau
*d and
Dirk
Enke
*a
aInstitute of Chemical Technology, Universität Leipzig, D-04103 Leipzig, Germany. E-mail: dirk.enke@uni-leipzig.de
bFelix Bloch Institute for Solid State Physics, Universität Leipzig, D-04103 Leipzig, Germany
cLeibniz Institute of Surface Engineering (IOM), D-04318 Leipzig, Germany
dSentronic GmbH, 01217 Dresden, Germany. E-mail: m.lau@sentronic.de
First published on 9th May 2025
Abstract
Porous glass (PG) particles are ideal supports in developing optical gas sensors as they combine fast mass transfer in macropores with large specific surface areas which are predestined for the deposition of indicator molecules. The well-established PG material is chemically stable and can be post-synthetically functionalized for introducing specific surface properties. Thus, surface functionalization with 3-mercaptopropyltrimethoxysilane (MPTMS) or 3-aminopropyl-triethoxysilane (APTES) provides the basis for the covalent coupling of the indicator molecule platinum(II)-5,10,15,20-meso-tetrakis-(2,3,4,5,6-pentafluorophenyl)-porphyrin (PtTFPP) to the sensor matrix, reducing the risk of migration, aggregation, and leaching of the fluorescent dye. Nucleophilic substitution of a fluorine atom of the pentafluorophenyl PtTFPP groups by amino (APTES) and thiol (MPTMS) groups enables a covalent linkage of the oxygen indicator PtTFPP to the PG surface. The spectroscopic detection of the PtTFPP-silica bonding by solid-state 13C CP MAS NMR spectroscopy has turned out to be rather difficult due to very low indicator amounts. As an inexpensive alternative for PtTFPP, hexafluorobenzene C6F6 was used successfully for the spectroscopic proof of covalent bonds between fluorophenyl groups of an indicator dye and amino- or mercapto-silane cross-linker molecules. The PtTFPP/MPTMS- and the PtTFPP/APTES-modified PG sensor particles showed no leaching in organic solvents and have been applied for oxygen sensing up to oxygen pressures of 400 mbar pO2. The PG sensor particles show non-linear Stern–Volmer calibration plots and it has been found that the covalent bonding of PtTFPP via APTES linkage onto silica surfaces is better suited for various sensor applications.
Introduction
Compared to the most common Clark electrode (based on amperometry), optical oxygen sensors using the principle of fluorescence quenching offer numerous advantages such as fast response, accurate detection, simple operation, and high sensitivity.1 Modern materials for trace oxygen sensing rely on highly photostable platinum(II) and palladium(II) porphyrin complexes, e.g. the commercially available platinum(II)-5,10,15,20-meso-tetrakis-(2,3,4,5,6-pentafluorophenyl)-porphyrin (PtTFPP), embedded into suitable polymeric or organically modified silica materials with high oxygen permeability.2–4 But, a simple physical entrapment of fluorescent dyes into polymeric or silica-gel materials does not completely prevent the indicator molecules from migration, aggregation, and/or leaching. Thus, these dye displacement processes reduce the sensor's accuracy and long-term stability and complicate calibration transfer. In order to prevent such undesired effects, covalent immobilization of the indicator molecules within the support matrix is strongly preferred.2,5 Hence, dye molecules have been covalently bonded via vinyl-6 and styryl-7 functionalization, and propanedithiol-derivatization8 within the polymer host or covalently embedded within a silane-modified sol–gel matrix.2,9–11 In the case of the porphyrin complex PtTFPP, the covalent bonding to a fluoropolymer or silica matrix can be performed via the nucleophilic substitution of the fluorine atom located in the para-position of the pentafluorophenyl part of PtTFPP by alkyl-amines and alkyl-thiols used as cross-linkers.2,5 Irrespective of the specific application of the oxygen sensor, the polymer or silica support impacts the sensing ability by its oxygen permeability and chemical stability.It is evident that indicator dyes embedded in diverse polymers can be a good choice for reliable measurement of oxygen concentrations dissolved in liquids, even under harsh conditions.12 But, the limited diffusion rate of gases through the polymeric support may be unfavorable with respect to a fast response of such polymeric sensors, especially in cases where the oxygen concentrations in the gas phase can rapidly change.13
In comparison, supports made from silica-gel and related porous silica materials come with the benefits of high accessibility to gas molecules from the medium to be analyzed, fast diffusion of the gas molecules within the pore system for practical instantaneous detection by the indicator molecules and a high chemical stability. In the case of silica-gel as a porous matrix, however, the reproducible fabrication of identical sensor materials is not as simple as often assumed due to shrinking processes and crack formations.14,15
In the present study, optical oxygen sensors based on porous glass (PG) are presented as interesting and reproducible silica supports and as an alternative to the well-known silica-gel matrices. Controlled pore glasses are versatile matrix materials with high mechanical, chemical and thermal stability as well as tunable surface area and pore volume as well as adjustable meso/macro pore size distribution.16,17 They are available as monoliths with different shapes including spheres and were already successfully used as supports for applications in catalysis, adsorption, separation, and optical chemosensing.18–20 Our contribution focuses primarily on the spectroscopic verification of the covalent immobilization of the PtTFPP indicator on the silica surface via an amino- or mercapto-silane linkage. In addition, possible effects of the preceding surface silanization by 3-mercaptopropyltrimethoxysilane (MPTMS) and 3-aminopropyltriethoxysilane (APTES) will be discussed.
Experimental section
Materials and methods
Glass beads with a diameter of 50–60 μm and a composition of 70 wt% SiO2, 23 wt% B2O3 and 7 wt% Na2O were first annealed at 650 °C for 24 hours to achieve phase separation, treated at 80 °C with hydrochloric acid to extract the sodium-rich borate phase, washed with deionized water, and finally dried at 120 °C.16 To remove highly dispersed, colloidal silica deposits remaining in the pore system after acidic leaching, the glass beads were stirred in 0.5 N NaOH at 30 °C for 3 hours, washed with 0.1 N HCl and deionized water and dried at 120 °C for 24 h. Furthermore, the pore diameter and the pore volume of the glass beads were adjusted by stirring in 0.2 N NaOH at 30 °C for a total of 10 hours, washing with 0.1 N HCl and deionized water and finally drying at 120 °C for 24 hours. On the basis of the weight loss up to 1000 °C and the BET surface of 24 m2 g−1, a surface density between 4.6 and 6.0 OH groups per nm2 has been obtained.For amino-silane surface silanization, 1 g of PG beads were dispersed in an ethanol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :water mixture (2.05:1 v/v), 0.229 ml 3-aminopropyltriethoxysilane (APTES, Merck Schuchardt OHG, Germany) was added and the suspension was stirred for 1 h at room temperature. After solvent evaporation and drying overnight at 100 °C, the surface modified glass beads were washed three times with an ethanol:water mixture, filtered and dried at 100 °C overnight. The silanization of the silica wafer used as a carrier in the AFM study was done in the same way.
:water mixture (2.05:1 v/v), 0.229 ml 3-aminopropyltriethoxysilane (APTES, Merck Schuchardt OHG, Germany) was added and the suspension was stirred for 1 h at room temperature. After solvent evaporation and drying overnight at 100 °C, the surface modified glass beads were washed three times with an ethanol:water mixture, filtered and dried at 100 °C overnight. The silanization of the silica wafer used as a carrier in the AFM study was done in the same way.
In the case of mercapto-silane surface functionalization, 1 g of PG beads were dispersed in an ethanol:water mixture (2.05:1 v/v), 0.229 ml 3-mercaptopropyltrimethoxysilane (MPTMS, Merck KGaA, Germany) was added, acidified by 0.05 ml 0.1 M HCl and the suspension was stirred for 1 h at room temperature. After drying overnight at 100 °C, the surface modified glass beads were washed three times with an ethanol:water mixture, filtered off and finally dried at 100 °C overnight.
The immobilization of the PtTFPP indicator on both the APTES- and MPTMS-modified PG beads (dispersed in dimethylformamide) has been carried out at 75 °C and a reaction time of 1 h as well as with the addition of triethylamine in accordance with the literature.2,5 The dye-doped beads were dried overnight at 120 °C and subsequently washed with toluene and dichloromethane. The first washing solution showed a small leaching of PtTFPP, whereas further washing steps revealed no leaching of the reddish PtTFPP indicator molecules from the dye-loaded, colored PG. Determined by ICP-OES, the platinum content of the sensor particles has been found to be between 0.093 wt% and 0.086 wt%.
Characterization techniques
For scanning electron microscopy a LEO GEMINI 1530 from Zeiss with an Everhart–Thornley detector (ETD) was used. The samples were attached to the sample carrier by means of an adhesive carbon foil and then vapor-deposited with gold. Measurements were performed at an accelerating voltage of 10 kV and a working distance of 5 mm.Nitrogen sorption and mercury intrusion measurements were performed on Autosorb iQ apparatus (Quantachrome) and on a Quantachrome PoreMaster porosimeter, respectively. Elemental analyses were accomplished with a vario Max CHN (Elementar Analysensysteme GmbH) instrument. The amount of organic species grafted on the porous glass was measured by thermogravimetric analysis (Netzsch STA 409) in air with a heating rate of 10 K min−1.
AFM measurements were done with a Dimension 3000 AFM stage (Bruker, former Digital Instruments) in Tapping Mode™. Standard silicon cantilevers were used with cantilever frequencies around 300 kHz.
13C and 29Si CP MAS solid state NMR experiments were recorded on a Bruker Avance 750 spectrometer. A more detailed experimental description can be found elsewhere.21 For the estimation of the platinum content of PG samples after PtTFPP anchoring, ICP-OES analysis was performed using an OPTIMA 8000 (Perkin Elmer).
For photophysical studies in solution, the absorption spectra of PtTFPP were obtained on a UV-2101PC (Shimadzu) double beam UV-vis spectrometer. Fully corrected fluorescence spectra were measured using a Fluoromax 4 (Horiba). Nanosecond transient absorption measurements and phosphorescence lifetime determinations were performed using a Quanta-Ray GCR-11 Nd:YAG laser (Spectra Physics). Pulse widths of 5 ns with an energy of 5 mJ per pulse at 355 nm were selected. The optical detection was based on a pulsed (pulser MSP 05 Müller Elektronik Optik) xenon lamp (XBO 150, Osram), a monochromator (Spectra Pro 275, Acton Research), an R955 photomultiplier tube (Hamamatsu Photonics) and a 1 GHz digital oscilloscope (TDS 684 A, Tektronix).
Finally, the oxygen-sensing PG particles have been dispersed in a thin layer of silicone rubber (known for its very high oxygen diffusion coefficients) or sprinkled onto the tacky surface of UV polymerizable acrylate coatings (in order to prevent any barriers for oxygen diffusion by an additional polymeric layer, e.g. silicone rubber, for embedding the sensor particles) and then firmly sticked by UV curing under nitrogen flushing.22 The sensory features of the PtTFPP loaded porous glass beads were obtained by means of the sensor getOtwo LAB L3207 WA (GetAMO Ltd., Germany) under various oxygen pressures (pO2 = 0–400 mbar). Unlike the often used intensity measurement techniques with disadvantages in the case of signal fluctuations or photobleaching of the dye, the samples were excited with sinusoidally modulated light and the phase shift between the excitation and luminescence emission has been measured.23
Quantum chemical modeling
Density functional theory (DFT) calculations were carried out using the M06-2X-D3 functional.24 It includes physically and chemically important London dispersion interactions,25 as implemented in Jaguar 9.6 (ref. 26) program packages. The molecular geometries and energies were calculated at the M06-2X-D3/LACVP** level of theory. The LACVP** basis set uses the standard 6-31G(d,p) basis set for light elements and the LAC pseudopotential27 for heavier elements, such as Pt in this case. The electronic transition spectra were calculated with the time-dependent (TD)28 TD M06-2X-D3/LACVP** method. The Maestro 9.7 program29 was used for the visualization of the calculated UV-vis spectra.Results and discussion
Silanization of siliceous materials
For porous silica particles, surface functionalization with silane coupling agents is a critical process in developing highly sensitive and selective biosensors,30 sustained drug delivery materials,31 and solid-base catalysts or adsorbents.32 Despite the multitude of relevant publications on the silanization process of hydroxyl groups (–OH) possessing surfaces, especially with 3-aminopropyltriethoxysilane (APTES), it is uncertain if a universally applicable surface functionalization protocol for silanization can be achieved.33Often, the formation of a stable silane monolayer34 is expected to be more desirable than the formation of a multilayer because thick silane layers could have a very fragile structure yielding a reduced hydrolytical stability. On the other hand, a higher density of silane grafting provides more functionalization sites for the immobilization of dyes and/or biomolecules as well as more crosslinking sites to polymeric host materials interacting with the silanized surface of silica nanoparticles.35
Therefore, it will be essential to understand the silanization processes for our specific application to immobilize the luminescent dye PtTFPP via chemical bonding to APTES or MPTMS grafted on silica and to determine the best silanization protocol for a durable oxygen sensor based on oxygen fluorescence quenching (Fig. 1). Preliminary silanization experiments of silica materials21 indicated that the formation of silane oligomers grafted on the surface of porous glass is to be expected rather than the attachment of monomeric silane species.30
 | ||
| Fig. 1 Schematic preparation of oxygen-sensitive porous glass beads via a two-stage covalent grafting process with amino- or mercaptosilanes and the luminescent dye PtTFPP using ethanol (EtOH), dimethylformamide (DMF), and triethylamine (TEA) as solvents. | ||
Sorption measurements and thermogravimetry
The porous structure formed within PG beads (formed via thermally induced SiO2/B2O3 phase separation and acid extraction of the borate phase) can be made visible by taking SEM pictures (Fig. 2). According to mercury porosimetry, the pore size distribution curves for pristine and APTES-modified PG are shown in Fig. 3. It is evident that both samples exhibited a narrow monomodal pore distribution showing predominantly macropores in the range of 90–150 nm. The average pore diameter of pristine PG is about 130 nm and no significant changes in the pore structure were observed after silanization by APTES (loading of 21 wt%) and by MPTMS (loading of 24 wt%). | ||
| Fig. 2 Scanning electron-microscopic (SEM) image of PG spheres after leaching procedures to create porosity. Inset: Light microscope image of PG beads sticked on the surface of an acrylate coating. | ||
 | ||
| Fig. 3 Pore width distribution function for pristine (black line) and APTES-modified (red line) porous glass beads by mercury porosimetry measurements showing relative pore volumes. Inset: Nitrogen sorption isotherm of the pristine PG sample. | ||
The specific surface areas of PG materials were determined via nitrogen sorption isotherms (see the Fig. 3 inset). All samples exhibit the common characteristics of a type II isotherm for nonporous or macroporous materials. For the pristine PG sample, the specific BET surface area is 24 m2 g−1 and the pore volume is 0.75 cm3 g−1. Modification with APTES and MPTMS reduces the surface area to about 17 m2 g−1 (71%) and 19 m2 g−1 (79%), respectively. Meyer et al.20 and Wenzel et al.36 reported similar reductions of textural parameters after surface modification for comparable porous micro glass beads.
Fig. 4 shows the TGA profiles of pristine PG, after modification with APTES, and finally the silylated PG sample after PtTFPP grafting. The initial weight loss (T < 150 °C) is associated with the evaporation of physically adsorbed water molecules. After functionalization with APTES, the initial weight loss has notably increased pointing to a higher water uptake from the ambient atmosphere due to an enhanced hydrophilicity of the aminosilane-functionalized sample. Conversely, the final PtTFPP anchoring yields a hydrophobic surface with a reduced water uptake. At higher temperatures, the APTES-modified sample exhibits a substantial weight loss between 250 and 600 °C of approximately 3.7 wt% due to the combustion/decomposition of the grafted organosilane. A similar thermogravimetric pattern with a weight loss of 6.5 wt% has been obtained for the MPTMS-modified PG (not shown). Analogous TGA curves after functionalization with APTES have been reported for SBA-type silica nanoparticles37,38 as well as for amorphous precipitated silica after functionalization with MPTMS39 and mesoporous zeolite.40 Based on the BET surface area of 24 m2 g−1, a very high surface coverage of ≈10 APTES species per nm2 and ≈15 MPTMS species per nm2 has been calculated and should be compared with the assumption of about five hydroxyl groups per nm2. Obviously, the often assumed tri-dental grafting mode of trialkoxysilanes within a silane monolayer on silica (very frequently related to the T3 signals in 29Si NMR spectra41) cannot explain these findings. Typical for surface modification using trialkoxysilanes in organic or aqueous media, however, the solution-phase silanization technique usually involves simultaneous condensation of alkoxysilanes with themselves and can lead to the formation of a grafted organosilane network or multilayer at the substrate's surface.42,43
 | ||
| Fig. 4 TGA profiles of pristine PG (gray line), after grafting of APTES (black line), and after PtTFPP anchoring (red line). | ||
Finally, thermal studies performed on PtTFPP/APTES-modified PG show an additional weight loss at temperatures above 300 °C (Fig. 4). Please note, to avoid incorrect TGA result due to any PtTFPP molecules physisorbed on the PG surface, the PtTFPP/APTES-modified sample has been thoroughly washed with toluene and dichloromethane. For comparison, thermal studies performed on platinum phthalocyanine (structurally similar to PtTFPP) by Lokesh et al.44 showed an exceptional thermal stability of such platinum complexes up to 350 °C and a one step degradation in the temperature range 420–480 °C. The final residual weights in the TGA curves (T < 600 °C) corresponded to platinum oxide. Based on the additional weight loss after PtTFPP anchoring of 2.5 wt% for APTES-modified PG (Fig. 4) and 3.0 wt% for MPTMS-modified PG (not shown) a surface coverage of 0.65 PtTFPP species per nm2 and 0.75 PtTFPP species per nm2 has been calculated. Determined by ICP-OES, the platinum content of the samples has been found to be between 0.093 wt% and 0.086 wt%.
FTIR measurements, 29Si CP MAS NMR spectroscopy, and surface topology
The successful modification of PG with APTES and MPTMS was confirmed by FTIR spectroscopy. As shown in Fig. 5, the spectrum of APTES-functionalized PG shows characteristic bands at 2930 cm−1 and 2863 cm−1 corresponding to the C–H stretching vibration of the CH2 groups of APTES. The less pronounced bands at 3370 cm−1 and 3310 cm−1 indicate the symmetric and asymmetric stretching vibrations of the free amine –NH2.45,46 The evidence of both C–H and N–H bands on APTES-modified PG is a strong indication of stable covalent bonds between APTES and silica silanol groups as previously reported for various siliceous materials.47–50 | ||
| Fig. 5 Comparison of DRIFT spectra of pristine PG (black line), after APTES grafting (red line), and after PtTFPP anchoring (blue line) to the ATR-IR spectrum of liquid APTES (gray line) in the wavenumber range between 3900 cm−1 and 2500 cm−1. | ||
However, a reliable proof of condensation reactions between silica silanol and alkoxy groups of trialkoxysilanes can only exist in the disappearance of those bands assigned to the two functional groups involved in the grafting reaction.51,52 Please note, the APTES-functionalized PG material does not exhibit bands at 2972 cm−1 (related to CH3 vibrations of the hydrolysable CH3CH2O– groups of neat APTES) and at 3740 cm−1 (isolated terminal Si–OH silanol groups on silica). These findings indicate both: i) APTES species grafted on porous glass are completely hydrolyzed and ii) chemical bonding of APTES moieties occurs on silanol groups of PG. Please note, the broad band around 3600 cm−1 relates to water adsorbed on the surface of porous glass.
For the final PtTFPP/APTES grafting complex, N–H stretching vibrations in the region 3400–3250 cm−1 (see Fig. 5) can be attributed partially to a primary amine surface species (RNH2), i.e. chemisorbed APTES with two bands in this region at 3370 cm−1 and 3310 cm−1, and a secondary amine surface species R2NH (formed by chemical bonds between APTES and PtTFPP with only one band in the region 3350–3300 cm−1).
But, the comparison between the IR spectra of APTES-functionalized PG and PtTFPP/APTES-PG reveals a clear shift of the peak maximum towards lower wavenumbers after PtTFPP anchoring. We consider these findings as a substantial indication that a covalent bonding between APTES groups and the PtTFPP dye has been carried out by our modification process.
In addition to the FTIR measurements, the covalent grafting of both trialkoxysilanes MPTMS and APTES onto the surface of porous glass has been studied by 29Si CP MAS NMR spectroscopy. The spectrum of the untreated PG (Fig. 6) shows two significant signals at −102.8 (Q3) and −110.7 (Q4) ppm which can be assigned to isolated silanols and siloxane groups,41 respectively. Signals of geminal silanols (Q2) are indicated by the small peak shoulder at −91.8 ppm. As to be expected, the 29Si NMR signal intensity of isolated silanol groups at −102.8 ppm is significantly reduced after MPTMS grafting as well as after the subsequent bonding of PtTFPP dye molecules.
 | ||
| Fig. 6 29Si CP MAS NMR spectra of pristine PG (black line), after MPTMS grafting (gray line), and after PtTFPP anchoring (red line). | ||
For MPTMS modified PG surfaces (see Fig. 6), NMR signals between −45 ppm and −75 ppm indicate the presence of condensed trialkoxysilanes and correspond to different Ti structures either bound to the surface or being connected to each other.41 As expected, the Ti species remain present after PtTFPP anchoring with a dominating T3 content at about −68.1 ppm (Fig. 6). But, the ratio between T2:T3 species is shifted towards a higher degree of condensation. These findings may be caused by the higher drying temperature of 120 °C for the PtTFPP/MPTMS reaction process resulting in additional condensation reactions within the grafted trialkoxysilanes oligomers.
To visualize the changes in the surface topography due to the silylation process with trifunctional silanes, AFM investigations were carried out on silica wafers as previously described.42 Whereas the root-mean-square (Rrms) surface roughness of the native wafer (Rrms of 0.5 nm) indicates a rather smooth surface, the APTES-modified silica wafer (Rrms of 5.3 nm) contains noticeable silane clusters (Fig. 7). These clusters are randomly distributed and separated with a minimum distance of about 50 nm. The AFM images show structures, visually similar to hillocks, with 3–15 nm in height and up to 50 nm in diameter. For comparison, Yadav et al.53 (who studied the solution-phase and vapor-phase deposition of aminosilanes on silica) observed large silane agglomerates of a height larger than ∼300 nm for APTES deposited from toluene solution.
 | ||
| Fig. 7 AFM picture of silica wafer after surface modification by APTES (revealing randomly distributed aminosilane agglomerates with a height up to 15 nm). | ||
Similar nanosized clusters due to surface silylation using trifunctional silanes have also been observed in the case of vinyltrimethoxysilane54 and methacryloxypropyltrimethoxysilane,42 as well as octadecyltrichlorosilane and APTES.53,55 These clusters are believed to be local polysilane networks grown on (or near at) the surface, resulting from the intermolecular condensation of neighboring trifunctional silane molecules. Because all of the cluster sizes are typically larger than the chain length of the silane molecule used as grafting agents, trifunctional chloro- and alkoxy-silanes do not form a uniform monolayer on solid surfaces in solution-phase deposition methods as it is often assumed, but rather a vertical or 3D polymerization of trifunctional silanes has to be expected under typical silanization conditions.56 Thus, it must therefore be assumed that a thin adsorbate layer of water (normally covering silica surfaces under ambient conditions) hydrolyzes the silanes directly at the silica surfaces,57,58 particularly when the grafting is done in an anhydrous solution. Obviously, without any special preparation conditions, the tendency towards a 3D polymerization of trialkoxysilanes is much stronger than that for the formation of smooth uniform layers.53,55,59
Regarding the composition of the polysilane clusters, MALDI-TOF MS studies on methacryloxypropyltrimethoxysilane-modified silica nanoparticles43,60 and ESI-MS measurements on APTES silanization solutions21 suggest the formation of well-ordered ladder-like polysiloxane chains attached onto the solid surface rather than the existence of randomly 3D-polymerised trialkoxysilanes originated via a grafting-from polymerization process. Nevertheless, whatever genuine structure the grafted polysilane has its functional head-group (e.g., vinyl, amino or thiol group) makes the whole polysilane cluster a kind of a multiple linker for the immobilization of a desired sensor molecule onto a solid surface controlling the spatial and electronic isolation of the grafted PtTFPP molecules.
To visualize the final structure of the PtTFPP/APTES grafting complex, DFT studies have been performed. Fig. 8 shows the covalently anchored platinum porphyrin dye in its optimized molecular structure at a distance of about 2.5 nm to the surface of anchored silane oligomers. It is obvious that the four fluorophenyl substituents in meso positions of PtTFPP can more or less freely rotate in relation to the rigid porphyrin plane. It has to be pointed out that metallopophyrins with aromatic substituents in the β-pyrrole positions yield a clear out-of-plane distorted, i.e. nonplanar, porphyrin framework exhibiting red-shifted and broadened optical spectra.61 Nevertheless, the size of all the porphyrin derivatives is usually negligible compared to the macropore diameter ranging between 90 and 150 nm in the PG beads applied, i.e. no spatial restrictions regarding the optimal arrangement of PtTFPP molecules on the pore surface are to be imposed and excellent gas permeability is likewise given.
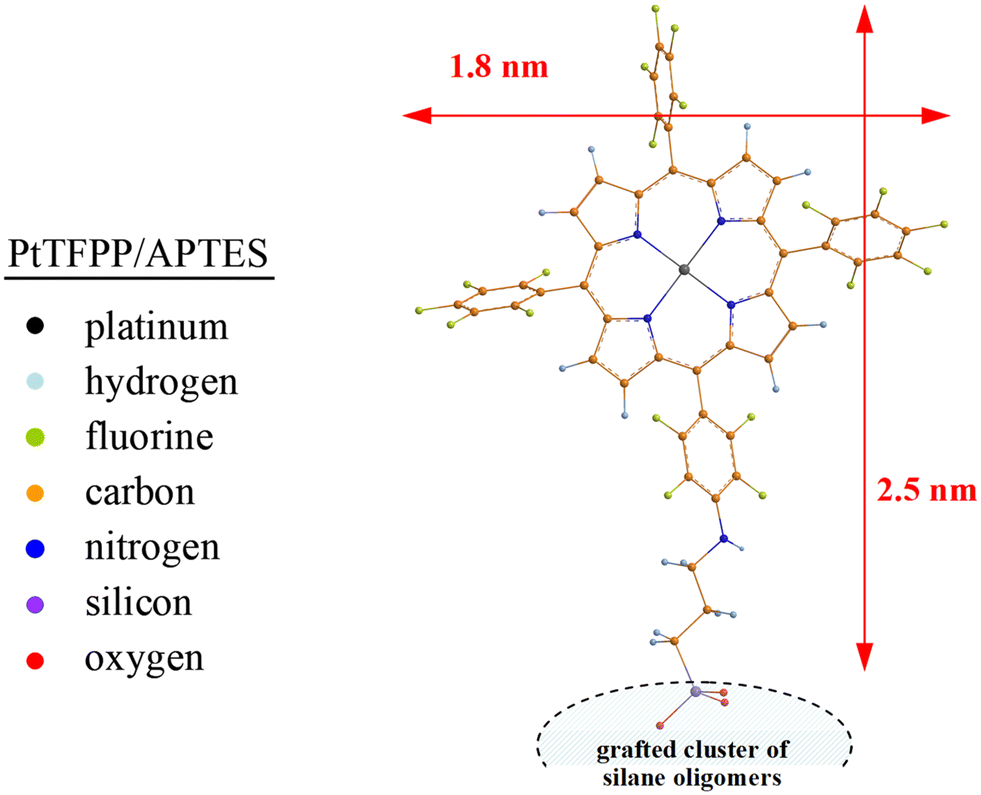 | ||
| Fig. 8 Optimized molecular structure for the PtTFPP/APTES grafting complex at the B3LYP/6-31(d,p) level of theory (revealing the free-rotating fluorophenyl groups of the covalently bound PtTFPP). | ||
Anchoring of fluorophenyl functionalized dyes on silanized silica surfaces
Generally, luminescent dyes are incorporated in polymeric materials or immobilized onto the surface of solid supports. In any case, it has to prevent the leaching, the migration, and the aggregation of sensor molecules.62 These issues can be solved by a covalent attachment of the sensor molecule to its support; exclusive physical entrapment of sensitive dyes into a polymeric or sol–gel matrix9,11,63 has to be characterized in fact as being insufficient. Particularly for the oxygen-sensitive PtTFPP, nucleophilic substitution especially of the labile fluorine atom in the para position of the pentafluorophenyl groups allows covalent coupling via a thiol or amino group linkage to both polymeric materials5,7,64 and silica supports.2,5 As expected, sensing materials based on covalently coupled indicators did not show any migration or leaching of the dye even in organic solvents and at high temperature: a simple and clear evidence of a stable attachment of the sensor molecule to its support. Nevertheless, the successful spectroscopic detection of a dye-support linkage is a desirable objective.Thus, Koren et al.5 studied the individual steps of the PtTFPP-fluoropolymer coupling process by liquid-state 19F NMR spectroscopy using soluble copolymers of pentafluorostyrene/styrene and propanedithiol (as a coupling and crosslinking agent). In the final polymeric sensor material, six different 19F NMR peaks were observed. Because the para-fluorine peak of the introduced PtTFPP molecules decreased by roughly 25%, it has been concluded that the indicator dye is coupled to the fluoropolymer via the dithiol linkage. Unfortunately, the authors do not provide any NMR information on the also accomplished covalent coupling of PtTFPP onto an organically modified, sol–gel matrix (Ormosil) using MPTMS to modify the dye molecule.
A similar, still pending spectroscopic proof concerns the covalent bonding of PtTFPP molecules onto the surface of aminosilane-modified silica-gel particles. Borisov et al.2 compared the quenching behaviour of those indicator/silica-gel particles (having a nanoporous substructure5 which ensures fast diffusion of gases) with that of the conventional oxygen sensors based on polystyrene materials (exhibiting substantially lower oxygen diffusion). The authors observed linear Stern–Volmer calibration plots for the sensor material based on silica-gel (pointing to no influence of the carrier material on the sensor behaviour), but significant deviations from linearity in the case of polystyrene (a possible indication of PtTFPP localization in different polymeric environments).
Following the approach to PtTFPP immobilization on silica-gel via an amino- or mercaptosilane linkage,2 we obtained oxygen-sensitive PG materials which showed no leaching of the luminescent indicator into organic solvents (toluene, dichloromethane). But, the small amount of PtTFPP loaded on the PG sensors revealed solid-state 13C CP MAS NMR spectra with unfavourable signal-to-noise ratios which have been unfortunately unsuitable for the desired proof of covalent PtTFPP bonding. Therefore, larger quantities of hexafluorobenzene C6F6 (chemically similar to the pentafluorophenyl groups of PtTFPP) have been linked to MPTMS- and APTES-modified PG samples for solid-state NMR measurements instead of the more expensive PtTFPP molecules.
Evidence for the existence of a covalent bond between PtTFPP and the MPTMS-modified surface comes from different chemical shifts of the carbon atoms of MPTMS before and after the C6F6 grafting reaction (see Fig. 9). In the case of mercaptosilane-modified PG, the 13C CP MAS NMR signal at 12 ppm and the more intense signal at 27.5 ppm can be assigned to the carbon atom (b), and a combination of the two carbon atoms (c) and (d) of the propyl group of MPTMS, respectively. These solid-state NMR shifts have to be compared with the liquid-state NMR spectrum of MPTMS which indicates both the absence of the hydrolysable methoxy group (a) after silanization and the difficult separation of the signal (c) and (d) after silanization due to line broadening. After C6F6 immobilization onto the MPTMS-modified surface, the signal of the carbon atom (d), which is nearest to the mercapto (SH) group, has been shifted from 27.5 ppm to 37 ppm. These findings indicate that the shielding effect of the original –SH group has changed into –SC6F5 groups due to covalent bonding of hexafluorobenzene which yields the observed downfield shift. Please note, the signal at 27.5 ppm (indirectly characterizing the original mercapto (–SH) group) is completely missing in the NMR spectrum of the C6F6/MPTMS-modified sample, i.e. the nucleophilic substitution of an aromatic fluorine atom by MPTMS (–SH ⇔ C6F6) has proceeded to completion.
 | ||
| Fig. 9 Comparison of solid-state 13C CP MAS NMR spectra of MPTMS-modified (black line) porous glass beads and after C6F6 anchoring (red line) to the liquid-state NMR spectrum of MPTMS (gray line). | ||
Comparative spectroscopic investigations of covalent PtTFPP bonding with an aminosilane (APTES) linkage to the silica surface are shown in Fig. 10. Here again, larger quantities of hexafluorobenzene have been applied to obtain meaningful 13C CP MAS NMR results. After silanization with aminosilane, three different peaks were observed and clearly assigned to the carbon atoms (c), (d), and (e) of the propyl group of APTES; apparently, the hydrolysable ethoxy groups (carbon atoms (a) and (b) in the liquid-state NMR spectrum of APTES) were not observed in the solid-state NMR spectrum of APTES-modified PG. After C6F6 bonding onto the APTES-modified surface, the signal at about 44 ppm assigned to the carbon atom (e), which is nearest to the amino (NH2) group, is split up in two parts (almost equal in size) at 43 ppm and 48 ppm. It may be assumed that the original shielding effect of the –NH2 group has been partially changed by conversion to –NHC6F5 groups yielding the observed downfield shift to 48 ppm. Thus, it is almost certain that only about half of the grafted aminosilane (NH2) groups are involved in the intended nucleophilic substitution of aromatic fluorine atoms by APTES (–NH2 ⇔ C6F6).
 | ||
| Fig. 10 Comparison of solid-state 13C CP MAS NMR spectra of APTES-modified (black line) porous glass beads and after C6F6 anchoring (red line) to the liquid-state NMR spectrum of APTES (gray line). | ||
In addition to changes in the chemical shift of the aliphatic carbon atoms for the coupling agents MPTMS and APTES, the spectroscopic evidence for covalent bonds between fluorophenyl functionalized dyes and amino- or mercaptosilane linker molecules can also be given by appropriate changes in the chemical shift of the aromatic carbon atoms (see Fig. 11).
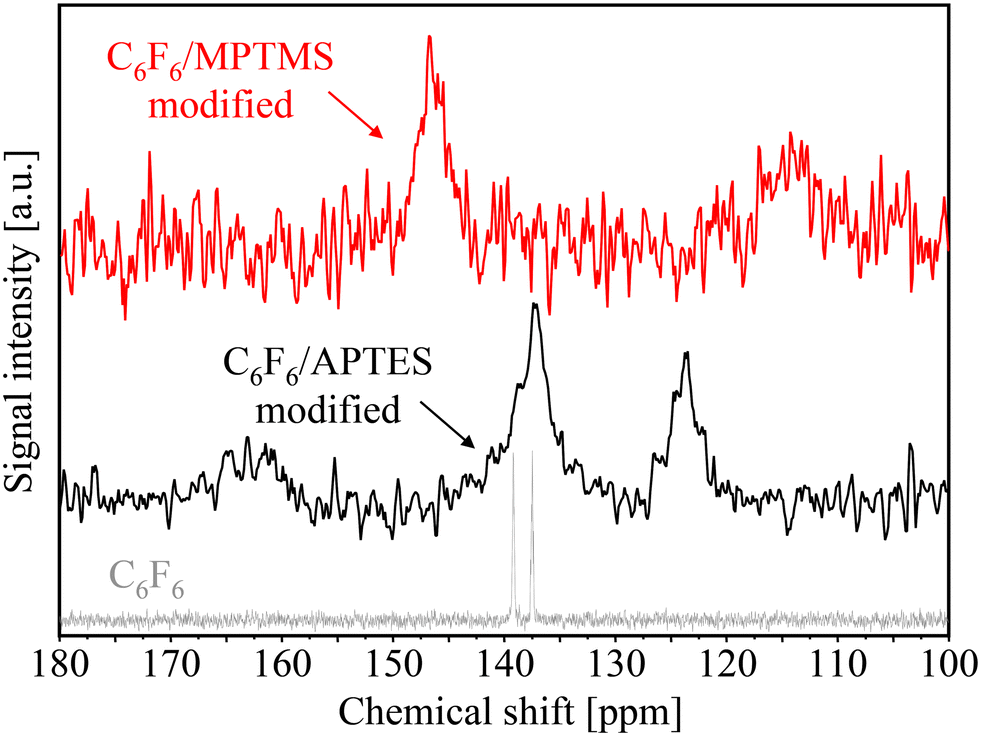 | ||
| Fig. 11 Comparison of solid-state 13C CP MAS NMR spectra of C6F6/APTES-modified (black line) and C6F6/MPTMS-modified (red line) porous glass beads to the liquid-state NMR spectrum of C6F6 (gray line). | ||
Thus, the liquid-state NMR spectrum of the highly symmetrical C6F6 molecules reveals two signals (located very close together) at 137 ppm and 139 ppm while a substantial peak splitting can be observed after nucleophilic substitution of fluorine atoms by MPTMS and APTES linker molecules.
For the C6F6/MPTMS-modified sample, the broad signal at 146 ppm can be assigned to the pentafluorophenyl carbon ring atoms and the weak signal at 114 ppm to the aromatic carbon atom in the neighborhood of the sulfur binding site, respectively. The observed downfield shift of the peak at 146 ppm may be associated with the interaction of sulfur d-orbitals and the aromatic pi-electron system. In the case of C6F6/APTES-modified surfaces, the relevant 13C CP MAS NMR signals are located at 137 ppm (fluorine-containing aromatic carbon atoms) and 124 ppm (aromatic carbon atom near to the NH– binding site). In summary, the loss of the original C6F6 symmetry (indicated by its liquid-state NMR spectrum) after the reaction with MPTMS (upfield shift to 114 ppm) and APTES (upfield shift to 124 ppm) is an indication towards the formation of covalent bonds between fluorophenyl functionalized luminescent dyes and amino- or mercaptosilane linker molecules. Of course, the immobilization of various fluorophenyl-derivatized, π-extended porphyrins65–68 onto silica surfaces can be demonstrated in the same way.
Application of immobilized dyes for optical chemical sensors
Like all porphyrins, the absorption spectrum of PtTFPP (in nitrogen saturated toluene solutions) exhibits Soret- and Q-band absorptions.69 In detail, PtTFPP reveals a strong absorption in the violet/blue region of the optical spectrum with a maximum at 395 nm, which is dedicated to the Soret band, and is flanked by a set of (compared to the Soret band) moderately intense Q-bands absorbing in the green/yellow region with maxima at 509 and 541 nm (see Fig. 12). These findings correspond to optical spectra that have been documented in the literature.70,71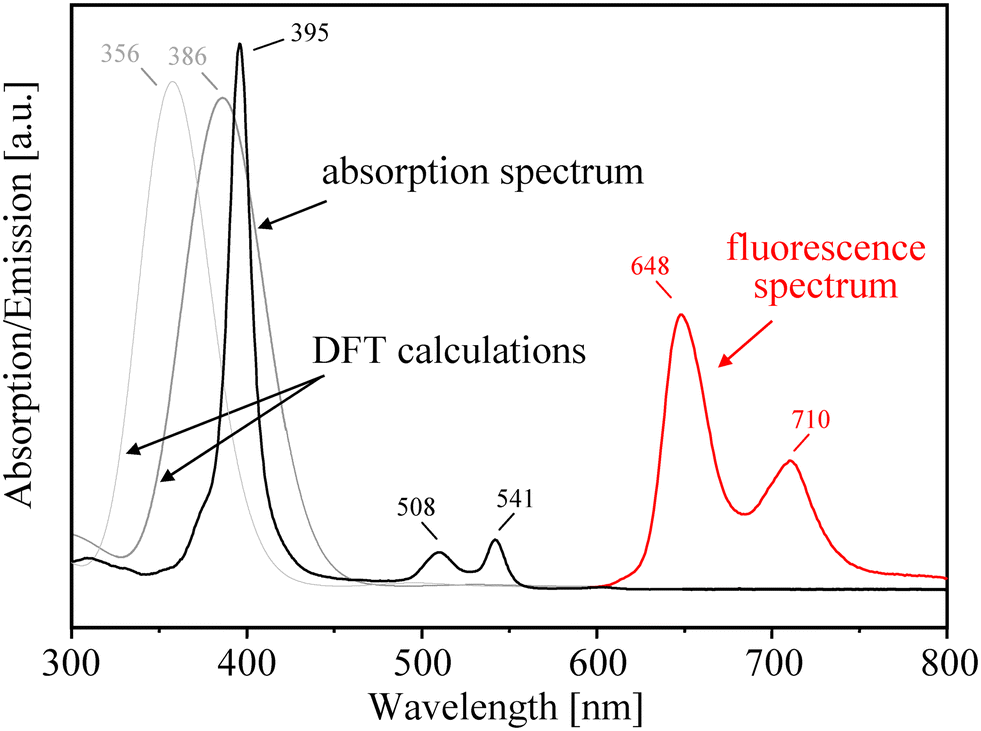 | ||
| Fig. 12 Absorption and emission spectra of PtTFPP (in deoxygenated toluene). The fluorescence was obtained by photoexcitation at 510 nm. In addition, DFT results on the position of the Soret band (typically centered at 395 nm) for various (F1 and F5)-fluorophenyl groups substituted at the meso-position. | ||
Upon photoexcitation of PtTFPP (in deoxygenated toluene solutions) at 510 nm, it exhibits emission bands in the range between 600 and 800 nm with two maxima at 649 nm and 710 nm. The positions of the emission bands are quite similar to values previously reported in the literature.72–75 The phosphorescence lifetime was determined by photoexciting deoxygenized toluene solutions of PtTFPP with 355 nm nanosecond laser pulses, monitoring the phosphorescence at 780 nm and fitting the obtained phosphorescence time profile to a mono-exponential function obtaining a phosphorescence lifetime of 9.6 μs. With the increase of oxygen concentration, the emission at 649 nm severely decreased, indicating the enhanced oxygen sensitivity of PtTFPP molecules and the phosphorescence/triplet character of this emission.
In addition to optical measurements, the absorption spectrum of PtTFPP has been simulated by DFT calculations to study the effect of various substituents on the photophysical properties of porphyrin platinum(II) complexes. It has early become apparent that the introduction of electron withdrawing fluorinated substituents in the porphyrin complex yields a good photostability of PtTFPP which has been explained by reducing the electron density at the metal center.76,77 But, the effect of appropriate electron donating or withdrawing substituents depends significantly on the meso- and/or β-pyrrolic positions of the functionalization.78,79 On the basis of our DFT results, the introduction of fluorinated aromatic groups at the meso-position of the porphyrin periphery seems to have only a minimal impact on the electronic structure of the Pt-porphyrin complex because the calculated positions of Soret bands are lying close together. This statement is supported by optical measurements of Arunkumar et al.,74 who showed that the substitution of various fluorophenyl groups at the meso-position had really a low influence on the positions of the Soret bands which were centered on 395–402 nm. Likewise, the effect of the amino- or mercaptosilane linker groups (attached at the para position of the pentafluorophenyl groups) on the spectrum of covalently grafted PtTFPP is very small.
For the functionalization at the β-positions of the porphyrin periphery, however, our DFT calculations indicate that there should be a notable red-shift of Soret bands, especially, in the case of olefinic or aromatic substituents as already reported by Borisov et al.80 and Ishida et al.81 Obviously, such β-pyrrole functionalized olefinic/aromatic substituents effectively perturb the electronics of the porphyrin core and result in an extension of the π-conjugation porphyrin framework, thereby shifting the absorption bands into lower energy regions.66,82
The quenching behavior of PtTFPP molecules by molecular oxygen can be quantitatively described by the Stern–Volmer equation:
| I0/I = τ0/τ = 1 + KSV [O2] | (1) |
| τ = tanθ/(2πf) | (2) |
For PtTFPP enclosed in polymeric materials, often linear Stern–Volmer curves were observed,77,86–88 suggesting the dominance of a single quenching process. Deviations from linearity are reported, for example, in the case of oxygen sensor molecules embedded in silica gel71,89,90 and are explained by the combination of different quenching mechanisms.91 This two-site model92 is based on the assumption that the indicator molecules are distributed in the host matrix at least in two distinct microenvironments within the matrix and each of the indicator fraction (f1, f2) shows a different accessibility to the quencher and a different quenching constant (KSV1, KSV2).
 | (3) |
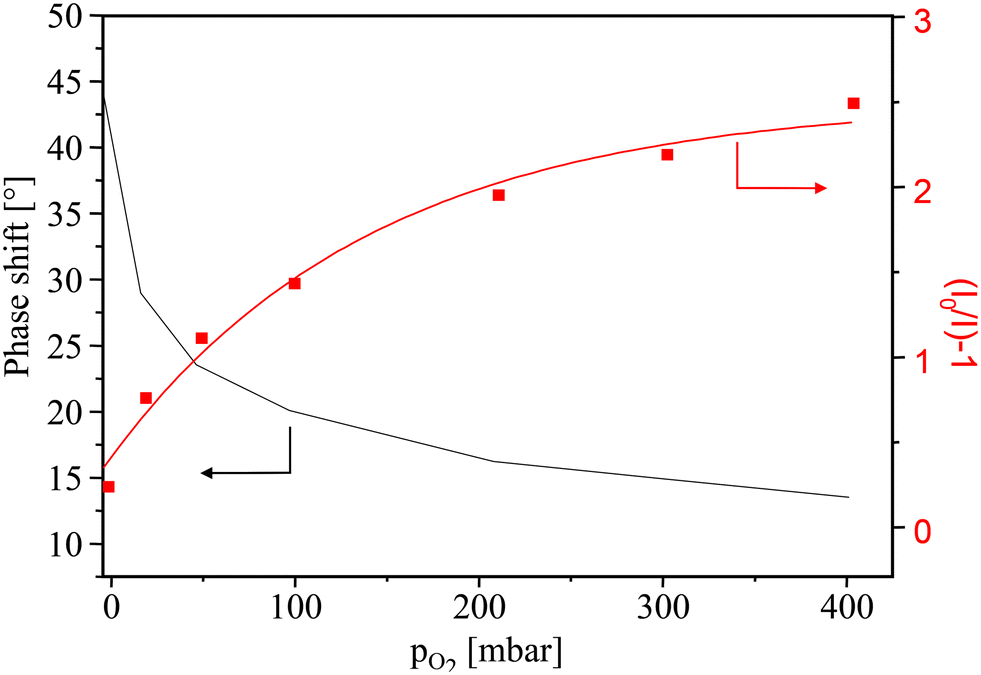 | ||
| Fig. 13 Calibration curves for PtTFPP/APTES-based oxygen-sensing PG materials (left, phase shift plot; right, Stern–Volmer plot). | ||
 | ||
| Fig. 14 Calibration curves for PtTFPP/MPTMS-based oxygen-sensing PG materials (left, phase shift plot; right, Stern–Volmer plot). | ||
For PtTFPP/APTES-modified PG sensor particles (Fig. 13), a phase shift θ of 44° in the absence of oxygen has been observed which is exponentially decreased to 14° at an oxygen pressure of 400 mbar. With a sensor dynamic of 30° and the sharp decrease in the range up to an oxygen pressure of 50 mbar, the PtTFPP/APTES-modified PG materials were found to be highly sensitive to oxygen which makes them promising especially for trace oxygen sensing. The two Stern–Volmer constants KSV1 and KSV2 have been determined to be 1.06 × 10−3 mbar−1 and 6.02 × 10−2 mbar−1, respectively. The KSV1 is a very low value and may only make a very small contribution compared with KSV2.
As shown in Fig. 14, the PtTFPP/MPTMS-modified PG sensor particles exhibit a phase shift θ of 17° in the absence of oxygen which is reduced to 4° at an oxygen pressure of 400 mbar. Such a low sensor dynamic of 13° points to a very low concentration of quenchable indicator molecules interacting with oxygen, although oxygen should really diffuse without restriction throughout the modified porous glass matrix. However, an uneven silane distribution of the first anchored MPTMS linker can lead to an unequal surface distribution of the finally bound PtTFPP indicator species within the PG matrix yielding under certain circumstances to different interactions of oxygen with the indicator sites. Furthermore, the low sensor dynamic of PtTFPP/MPTMS-modified PG may be due to unfavourable consequences of the mercapto (SH) linker group on PtTFPP phosphorescence as supposed by Borisov et al.2 Because the Stern–Volmer calibration plot is again not linear and, as already written above, the two-site model has been applied for an adequate description of the PtTFPP/MPTMS/PG calibration plot where KSV1 with 1.3 × 10−3 mbar−1 and KSV2 with 7.5 × 10−2 mbar−1 are the two different Stern–Volmer constants. These values of the quenching constants (KSV2 is more than 50 times greater) point to quite different quenchable indicator fractions and are an indicator of different micro-heterogeneous environments of the indicator molecules within the porous glass matrix.
In summary, the indicator dye PtTFPP covalently bound to the surface of porous glass has been found to be a more optical trace sensor. To eliminate dye migration and leaching from the silica matrix, appropriated trialkoxysilanes, such as APTES and MPTMS, have been proven to yield a durable chemical bond between PtTFPP and the silica support without a negative impact on the PtTFPP phosphorescence. Furthermore, it has been found that the covalent bonding of PtTFPP via APTES linkage is better suited for oxygen sensor applications. The chosen sensor examples show that reproducible porous glasses with tunable specific surface area, pore volume and adjustable meso/macropore size distribution are versatile matrix materials for optical sensors.
Conclusions
For long-term application of optical oxygen sensors, the spectral properties of luminescent dyes, e.g. the platinum(II) complex of tetrakis(pentafluorophenyl)-porphyrin (PtTFPP), should not be substantially affected by processes such as dye leaching and migration, aggregation and photostability. To avoid some shortcomings of sensors where the indicator is only physically trapped in polymeric or silica-gel matrices, covalent binding of the indicator dye to the matrix material is imperative. Fluorinated porphyrins applied as luminescent dyes come with the advantage that they can easily form covalent bonds with thiol or amino linker groups attached on the surface of matrix materials and retain their photophysical properties essential for the application.Porous glass (PG), having an average pore diameter of about 130 nm and therefore possessing high oxygen permeability, was first modified by MPTMS or APTES, and then the indicator PtTFPP has been firmly grafted on the modified surface of silica. The oxygen-sensitive PG materials showed no leaching of the luminescent indicator into organic solvents (toluene, dichloromethane) and have been characterized by sorption measurements, thermogravimetry, FTIR and 29Si CP MAS NMR spectroscopy, and determination of the surface topology by atomic/scanning force microscopy. However, due to the very small concentration of PtTFPP anchored on PG, larger quantities of hexafluorobenzene C6F6 (chemically similar to the pentafluorophenyl groups of PtTFPP) have been linked to MPTMS- and APTES-modified PG samples for 13C CP MAS NMR measurements. Both the NMR signals of the aliphatic carbon atoms of the MPTMS- and APTES-linking groups and the aromatic carbon atoms of C6F6 have been clearly assigned and prove significantly the covalent coupling of fluorphenyl porphyrin groups to the mercapto/aminosilane-modified PG surface.
A non-linear relationship between oxygen concentration and phase shift, i.e. a non-linear Stern–Volmer calibration plot, was observed for both PtTFPP/APTES- and PtTFPP/MPTMS-modified PG. This nonlinearity of the sensor materials can serve as evidence that the grafted PtTFPP indicator molecules are located in non-uniform microenvironments inside the silica pore system. The observed heterogeneity is probably related with the surface functionalization with trialkoxysilanes rather than the PG formation process. Although both amino- and mercaptosilane linker molecules yield permanent anchoring of PtTFPP indicator molecules on silica surfaces, it has been found that the covalent bonding of PtTFPP via APTES linkage is better suited for oxygen sensor applications, especially for measurements in the trace range.
Data availability
The authors confirm that the data supporting the findings of this study are only available within the article.Author contributions
Conceptualization, investigation, writing – original draft: SC. Conceptualization, methodology, writing – review & editing: FB. Investigation: MB. Investigation, editing: AK. Investigation, visualization: SN. Conceptualization, methodology, editing: ML. Supervision, writing – review & editing: DE. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
The authors declare no conflict of interest. SC, FB and ML are employees in the Company Sentronic GmbH (Dresden) and declare this had no role in the design of the study; in the collection, analysis, or interpretation of data; in the writing of the manuscript, and in the decision to publish the results.Acknowledgements
This research received no external funding. FB wishes to thank Mr. Dietmar Hirsch (Leibniz Institute of Surface Engineering, Leipzig) for recording and processing of the AFM data.References
- O. S. Wolfbeis, BioEssays, 2015, 37, 921–928 CrossRef CAS PubMed.
- S. M. Borisov, P. Lehner and I. Klimant, Anal. Chim. Acta, 2011, 690, 108–115 CrossRef CAS PubMed.
- C. S. Chu and Y. L. Lo, Sens. Actuators, B, 2010, 151, 83–89 Search PubMed.
- S. Medina-Rodriguez, M. Marin-Suarez, J. F. Fernandez-Sanchez, A. de la Torre-Vega, E. Baranoff and A. Fernandez-Gutierrez, Analyst, 2013, 138, 4607–4617 RSC.
- K. Koren, S. M. Borisov and I. Klimant, Sens. Actuators, B, 2012, 169, 173–181 CrossRef CAS PubMed.
- J. Ma, L. Lv, G. Zou and Q. Zhang, ACS Appl. Mater. Interfaces, 2015, 7, 241–249 CrossRef CAS PubMed.
- L. H. Hutter, B. J. Muller, K. Koren, S. M. Borisov and I. Klimant, J. Mater. Chem., 2014, 2, 7589–7598 Search PubMed.
- P. Lehner, C. Staudinger, S. M. Borisov and I. Klimant, Nat. Commun., 2014, 5, 4460 CrossRef CAS PubMed.
- B. J. Basu, Sens. Actuators, B, 2007, 123, 568–577 CrossRef CAS.
- T. W. Sung and Y. L. Lo, Sens. Actuators, B, 2012, 173, 406–413 CrossRef CAS.
- C. S. Chu, T. W. Sung and Y. L. Lo, Sens. Actuators, B, 2013, 185, 287–292 CrossRef CAS.
- C. Staudinger, M. Strobl, J. P. Fischer, R. Thar, T. Mayr, D. Aigner, B. J. Müller, B. Müller, P. Lehner, G. Mistlberger, E. Fritzsche, J. Ehgartner, P. W. Zach, J. S. Clarke, F. Geissler, A. Mutzberg, J. D. Müller, E. P. Achterberg, S. M. Borisov and I. Klimant, Limnol. Oceanogr.: Methods, 2018, 16, 459–473 CrossRef CAS.
- F. Seichter, E. Tütüncü, L. T. Hagemann, J. Vogt, U. Wachter, M. Gröger, S. Kress, P. Radermacher and B. Mizaikoff, J. Breath Res., 2018, 12, 036018 CrossRef CAS PubMed.
- L. L. Hench and J. K. West, Chem. Rev., 1990, 90, 33–72 CrossRef CAS.
- I. Klimant, F. Ruckruh, G. Liebsch, C. Stangelmayer and O. S. Wolfbeis, Microchim. Acta, 1999, 131, 35–46 CrossRef CAS.
- D. Enke, F. Janowski and W. Schwieger, Microporous Mesoporous Mater., 2003, 60, 19–30 CrossRef CAS.
- A. Inayat, B. Reinhardt, J. Herwig, C. Küster, H. Uhlig, S. Krenkel, E. Raedlein and D. Enke, New J. Chem., 2016, 40, 4095–4114 RSC.
- R. Müller, N. Anders, J. Titus and D. Enke, Talanta, 2013, 107, 255–262 CrossRef PubMed.
- M. Goepel, H. Kabir, C. Küster, E. Saraçi, P. Zeigermann, R. Valiullin, C. Chmelik, D. Enke, J. Kärger and R. Gläser, Catal. Sci. Technol., 2015, 5, 3137–3146 RSC.
- R. Meyer, K. Mueller, S. Naumov, F. Bauer and D. Enke, Front. Chem., 2023, 11, 1084046 CrossRef CAS PubMed.
- F. Bauer, S. Czihal, M. Bertmer, U. Decker, S. Naumov, S. Wassersleben and D. Enke, Microporous Mesoporous Mater., 2017, 250, 221–231 CrossRef CAS.
- F. Bauer, R. Flyunt, K. Czihal, H. Ernst, S. Naumov and M. R. Buchmeiser, Nucl. Instrum. Methods Phys. Res., Sect. B, 2007, 265, 87–91 CrossRef CAS.
- C. S. Chu, K. Z. Lin and Y. H. Tang, Sens. Actuators, B, 2016, 223, 606–612 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 1–20 CrossRef PubMed.
- Jaguar, version 9.6, Schrodinger, Inc, New York, NY, 2017 Search PubMed.
- W. R. Wadt and P. J. Hay, J. Chem. Phys., 1985, 82, 284–298 CrossRef CAS.
- R. Bauernschmitt and R. Ahlrichs, Chem. Phys. Lett., 1996, 256, 454–464 CrossRef CAS.
- Schrödinger, Release 2017-2: Maestro, Schrödinger, LLC, New York, NY, 2017 Search PubMed.
- M. Sypabekova, A. Hagemann, D. Rho and S. Kim, Biosensors, 2023, 13, 36 CrossRef CAS PubMed.
- E. Ahmadi, N. Dehghannejad, S. Hashemikia, M. Ghasemnejad and H. Tabebordbar, Drug Delivery, 2014, 21, 164–172 CrossRef CAS PubMed.
- T. Yokoi, Y. Kubota and T. Tatsumi, Appl. Catal., A, 2012, 421, 14–37 CrossRef.
- N. B. Arnfinnsdottir, C. A. Chapman, R. C. Bailey, A. Aksnes and B. T. Stokke, Sensors, 2020, 20, 3163 CrossRef CAS PubMed.
- M. J. Zhu, M. Z. Lerum and W. Chen, Langmuir, 2012, 28, 416–423 CrossRef CAS PubMed.
- F. Bauer, H. J. Gläsel, U. Decker, H. Ernst, A. Freyer, E. Hartmann, V. Sauerland and R. Mehnert, Prog. Org. Coat., 2003, 47, 147–153 CrossRef CAS.
- M. Wenzel, L. Eckert, K. Muller, D. Solonenko, C. Wiebeler, D. R. T. Zahn, D. Enke and J. Matysik, Phys. Chem. Chem. Phys., 2022, 24, 14488–14497 Search PubMed.
- A. L. B. de Barros, K. S. de Oliveira Ferraz, T. C. S. Dantas, G. F. Andrade, V. N. Cardoso and E. M. B. d. Sousa, Mater. Sci. Eng., C, 2015, 56, 181–188 Search PubMed.
- M. Kamble, H. Salvi and G. D. Yadav, J. Porous Mater., 2020, 27, 1559–1567 Search PubMed.
- M. Castellano, E. Marsano, A. Turturro, L. Conzatti and G. Busca, Adsorption, 2012, 18, 307–320 CrossRef CAS.
- S. Mitchell, A. Bonilla and J. Perez-Ramirez, Mater. Chem. Phys., 2011, 127, 278–284 CrossRef CAS.
- K. Albert, R. Brindle, J. Schmid, B. Buszewski and E. Bayer, Chromatographia, 1994, 38, 283–290 Search PubMed.
- F. Bauer, H. Ernst, D. Hirsch, S. Naumov, M. Pelzing, V. Sauerland and R. Mehnert, Macromol. Chem. Phys., 2004, 205, 1587–1593 Search PubMed.
- L. Nothdurft, T. Glueck, W. Dempwolf and G. Schmidt-Naake, Macromol. Mater. Eng., 2008, 293, 132–139 CrossRef CAS.
- K. S. Lokesh, N. Uma and B. N. Achar, Polyhedron, 2009, 28, 1022–1028 CrossRef CAS.
- O. G. Nik, B. Nohair and S. Kaliaguine, Microporous Mesoporous Mater., 2011, 143, 221–229 CrossRef CAS.
- A. L. Xue, S. Y. Zhou, Y. J. Zhao, X. P. Lu and P. F. Han, J. Hazard. Mater., 2011, 194, 7–14 CrossRef CAS PubMed.
- E. Vansant, P. Van Der Voort and K. Vrancken, Characterization and chemical modification of the silica surface, Elsevier Science, Amsterdam, 1995 Search PubMed.
- T. Borrego, M. Andrade, M. L. Pinto, A. R. Silva, A. P. Carvalho, J. Rocha, C. Freire and J. Pires, J. Colloid Interface Sci., 2010, 344, 603–610 CrossRef CAS PubMed.
- N. Majoul, S. Aouida and B. Bessais, Appl. Surf. Sci., 2015, 331, 388–391 CrossRef CAS.
- D. J. Sullivan, T. F. O'Mahony, M. C. Cruz-Romero, E. Cummins, J. P. Kerry and M. A. Morris, ACS Omega, 2021, 6, 30376–30385 CrossRef CAS PubMed.
- F. Bauer, H. Ernst, U. Decker, M. Findeisen, H. J. Gläsel, H. Langguth, E. Hartmann, R. Mehnert and C. Peuker, Macromol. Chem. Phys., 2000, 201, 2654–2659 CrossRef CAS.
- Z. H. Luan, J. A. Fournier, J. B. Wooten and D. E. Miser, Microporous Mesoporous Mater., 2005, 83, 150–158 CrossRef CAS.
- A. R. Yadav, R. Sriram, J. A. Carter and B. L. Miller, Mater. Sci. Eng., C, 2014, 35, 283–290 CrossRef CAS PubMed.
- V. Nguyen, W. Yoshida and Y. Cohen, J. Appl. Polym. Sci., 2003, 87, 300–310 CrossRef CAS.
- T. Baumgartel, C. von Borczyskowski and H. Graaf, Beilstein J. Nanotechnol., 2013, 4, 218–226 CrossRef PubMed.
- V. Palanivel, D. Q. Zhu and W. J. van Ooji, Prog. Org. Coat., 2003, 47, 384–392 CrossRef CAS.
- C. P. Tripp and M. L. Hair, Langmuir, 1992, 8, 1120–1126 CrossRef CAS.
- A. Simon, T. Cohen-Bouhacina, M. C. Porte, J. P. Aime and C. Baquey, J. Colloid Interface Sci., 2002, 251, 278–283 CrossRef CAS PubMed.
- F. Bauer, V. Sauerland, H. Ernst, H. A. Gläsel, S. Naumov and R. Mehnert, Macromol. Chem. Phys., 2003, 204, 375–383 CrossRef CAS.
- F. Bauer, H. J. Glasel, U. Decker, H. Ernst, A. Freyer, E. Hartmann, V. Sauerland and R. Mehnert, Prog. Org. Coat., 2003, 47, 147–153 CrossRef CAS.
- A. Y. Lebedev, M. A. Filatov, A. V. Cheprakov and S. A. Vinogradov, J. Phys. Chem. A, 2008, 112, 7723–7733 Search PubMed.
- O. S. Wolfbeis, Anal. Chem., 2008, 80, 4269–4283 Search PubMed.
- T. S. Yeh, C. S. Chu and Y. L. Lo, Sens. Actuators, B, 2006, 119, 701–707 CrossRef CAS.
- K. Koren, L. Hutter, B. Enko, A. Pein, S. M. Borisov and I. Klimant, Sens. Actuators, B, 2013, 176, 344–350 Search PubMed.
- S. M. Borisov, G. Nuss, W. Haas, R. Saf, M. Schmuck and I. Klimant, J. Photochem. Photobiol., A, 2009, 201, 128–135 CrossRef CAS.
- F. Niedermair, S. M. Borisov, G. Zenkl, O. T. Hofmann, H. Weber, R. Saf and I. Klimant, Inorg. Chem., 2010, 49, 9333–9342 CrossRef CAS PubMed.
- P. W. Zach, M. Maierhofer, S. D. Pilschmann, I. Klimant and S. M. Borisov, Dyes Pigm., 2018, 159, 610–618 Search PubMed.
- G. Schwendt and S. M. Borisov, Sens. Actuators, B, 2023, 393, 134236 CrossRef CAS.
- A. Kahnt, in Applications of Porphyrinoids as Functional Materials, ed. H. Lang and T. Rüffer, The Royal Society of Chemistry, 2021, pp. 220–251 Search PubMed.
- C. Arunkumar, F. R. Kooriyaden and S. Sujatha, J. Porphyrins Phthalocyanines, 2017, 21, 622–631 CrossRef CAS.
- C. Y. Liu, A. S. Sadhu, R. Karmakar, C. S. Chu, Y. N. Lin, S. H. Chang, G. K. Dalapati and S. Biring, Biosensors, 2022, 12, 774 CrossRef CAS PubMed.
- S. W. Lai, Y. J. Hou, C. M. Che, H. L. Pang, K. Y. Wong, C. K. Chang and N. Y. Zhu, Inorg. Chem., 2004, 43, 3724–3732 CrossRef CAS PubMed.
- D. Peng, L. Jiao and Y. Liu, presented in part at the 32nd AIAA Aerodynamic Measurement Technology and Ground Testing Conference, Washington, D.C., 2016, AIAA 2016-3405 Search PubMed.
- C. Arunkumar, F. R. Kooriyaden, X. C. Zhang, S. Sujatha and J. Z. Zhao, New J. Chem., 2017, 41, 4908–4917 RSC.
- S. Biring, A. S. Sadhu and M. Deb, Sensors, 2019, 19, 5124 CrossRef CAS PubMed.
- S. K. Lee and I. Okura, Anal. Commun., 1997, 34, 185–188 RSC.
- Y. Amao, T. Miyashita and I. Okura, J. Fluorine Chem., 2001, 107, 101–106 CrossRef CAS.
- Y. Q. Zhu and R. B. Silverman, J. Org. Chem., 2007, 72, 233–239 CrossRef CAS PubMed.
- B. Sekaran and R. Misra, Coord. Chem. Rev., 2022, 453, 214312 CrossRef CAS.
- S. M. Borisov, G. Nuss and I. Klimant, Anal. Chem., 2008, 80, 9435–9442 CrossRef CAS PubMed.
- M. Ishida, S. W. Park, D. Hwang, Y. B. Koo, J. L. Sessler, D. Y. Kim and D. Kim, J. Phys. Chem. C, 2011, 115, 19343–19354 CrossRef CAS.
- C. Borek, K. Hanson, P. I. Djurovich, M. E. Thompson, K. Aznavour, R. Bau, Y. R. Sun, S. R. Forrest, J. Brooks, L. Michalski and J. Brown, Angew. Chem., Int. Ed., 2007, 46, 1109–1112 CrossRef CAS PubMed.
- M. E. Lippitsch and S. Draxler, Sens. Actuators, B, 1993, 11, 97–101 CrossRef CAS.
- C. McDonagh, C. Kolle, A. K. McEvoy, D. L. Dowling, A. A. Cafolla, S. J. Cullen and B. D. MacCraith, Sens. Actuators, B, 2001, 74, 124–130 CrossRef CAS.
- M. Valledor, J. Carlos Campo, I. Sanchez-Barragan, J. Carlos Viera, J. M. Costa-Fernandez and A. Sanz-Medel, Sens. Actuators, B, 2006, 117, 266–273 CrossRef CAS.
- J. Z. Li, Y. Qiao, T. T. Pan, K. Zhong, J. X. Wen, S. S. Wu, F. Y. Su and Y. Q. Tian, Sensors, 2018, 18, 3752 CrossRef PubMed.
- Y. Qiao, T. T. Pan, J. Z. Li, C. Yang, J. X. Wen, K. Zhong, S. S. Wu, F. Y. Su and Y. Q. Tian, Appl. Sci., 2019, 9, 4404 CrossRef CAS.
- H. Valimaki, T. Hyvarinen, J. Leivo, H. Iftikhar, M. Pekkanen-Mattila, D. K. Rajan, J. Verho, J. Kreutzer, T. Ryynanen, J. Pirhonen, K. Aalto-Setala, P. Kallio, S. Narkilahti and J. Lekkala, Biomed. Microdevices, 2020, 22, 41 CrossRef CAS PubMed.
- J. Lin, J. A. Siddiqui and R. M. Ottenbrite, Polym. Adv. Technol., 2001, 12, 285–292 CrossRef CAS.
- K. Koren, S. M. Borisov, R. Saf and I. Klimant, Eur. J. Inorg. Chem., 2011, 1531–1534 CrossRef CAS PubMed.
- E. R. Carraway, J. N. Demas and B. A. Degraff, Langmuir, 1991, 7, 2991–2998 CrossRef CAS.
- E. R. Carraway, J. N. Demas and B. A. Degraff, Anal. Chem., 1991, 63, 332–336 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |