
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electronic coupling and photoluminescence anisotropy in van-der-Waals-stacks of tungsten disulphide with molecular single crystals†
Mohammed
Adel Aly
 ab,
Dominik
Muth
c,
Bettina
Wagner
d,
Martin
Koch
*a,
Johanna
Heine
*d and
Marina
Gerhard
*c
ab,
Dominik
Muth
c,
Bettina
Wagner
d,
Martin
Koch
*a,
Johanna
Heine
*d and
Marina
Gerhard
*c
aDepartment of Physics and Marburg Centre for Quantum Materials and Sustainable Technologies, Semiconductor Photonics Group, Philipps-Universität Marburg, Renthof 7a, 35032 Marburg, Germany
bDepartment of Physics, Faculty of Science, Ain Shams University, Cairo 11566, Egypt
cDepartment of Physics and Marburg Centre for Quantum Materials and Sustainable Technologies, Semiconductor Spectroscopy Group, Philipps-Universität Marburg, Renthof 7a, 35032 Marburg, Germany. E-mail: marina.gerhard@physik.uni-marburg.de
dDepartment of Chemistry and Marburg Centre for Quantum Materials and Sustainable Technologies, Philipps-Universität Marburg, Hans-Meerwein-Straße 4, 35032 Marburg, Germany
First published on 15th April 2025
Abstract
Heterostructures comprised of organic semiconductors and transition metal dichalcogenides are promising candidates for device applications. In order to promote efficient charge or exciton transfer, the arrangement of the molecules at the heterointerface is crucial. However, to complement theoretical efforts, there is presently a lack of model systems to comprehensively study electronic coupling phenomena. Here, we explore heterostructures of tungsten disulphide with monocrystalline thin flakes of the organic compound pyrenemethylammonium chloride. The ionic character of the organic compound enables controlled fabrication of heterostructures via exfoliation of single crystals, thereby minimizing structural disorder in the molecular compound. In order to explore the optical properties and the photoexcitation dynamics related to the heterointerface, we employ time and polarization resolved photoluminescence spectroscopy. Evidence for electronic coupling is provided by three experimental findings: first, a faster decay of the luminescence of the organic compound in the region of the heterostructure indicates exciton or charge transfer, which is also surprisingly efficient for the structurally relaxed localized excimer state. Second, we find that the organic compound introduces a polarization anisotropy into the emission of tungsten disulphide, potentially due to the lowering of symmetries in the band structure. Third, we identify a weakly emissive feature at an energy around 1.7 eV, which could be related to an interfacial state. Overall, ionic molecular systems could serve as a versatile platform for combined experimental and theoretical studies of the interactions at the heterointerface.
Introduction
Heterostructures (HS) of organic semiconductors (OSCs) with transition metal dichalcogenides (TMDs) are capable of merging the benefits of two highly complementary material classes:1–3 OSCs offer a broad chemical tunability enabling tailored opto-electronic properties and high absorption coefficients, whereas TMDs demonstrate excellent transport properties due to their higher structural rigidity, as well as large spin–orbit coupling, leading to valley-polarization effects, which could be exploited to transmit binary signals.4–6 Molecules which are either physisorbed or chemisorbed on TMDs could for example be employed as sensitizers, dopants7,8 or defect passivating agents.9 Potential application scenarios for OSC/TMD-based devices include pn-junctions,10 transistors,11–13 chemical sensors14,15 or solar cells.16A particularly high degree of flexibility in tailoring the desired properties is offered by van-der-Waals- (vdW-) bound heterostructures, which can be produced e.g. via molecular beam deposition17–19 or physical stacking20–22 without the boundary conditions imposed by epitaxial growth of covalently bound compounds. Even though the vdW-interactions between the TMD and the physisorbed OSCs are weak compared to covalent bonds, they can strongly influence the excited state dynamics, leading for example to photo-induced exciton or charge transfer from one layer to another.2,23,24 In so-called type-II heterojunctions, the interfacial energetic landscape and the dielectric screening between the photoexcited charges can either promote charge separation or the formation of interlayer excitons with hybrid character.2,11,24–28 Moreover, despite the absence of a ground state charge transfer, the vdW interactions between the compounds can still lead to hybridization effects and modifications of the electronic states at the interface.29
An intriguing challenge for the microscopic understanding of the electronic properties of OSC/TMD-HS is the combination of two contrasting conceptual descriptions, namely the picture of delocalized Wannier-excitons in the covalently bound inorganic compound and the concept of rather localized Frenkel-excitons originating from the comparably weak vdW-interactions within the OSC.30 Merging both materials thus raises questions about the interfacial electronic landscape, the resulting exciton optics and the coupling mechanisms governing the excited state dynamics. While theoretical studies have addressed the coupling between excitonic states in molecular adsorbates and continuum states of inorganic wide-gap semiconductors31,32 and also TMDs,29,33–35 thereby exploring possibilities of exciton and charge transfer as well as hybridization effects, the lack of suitable model systems often impedes a direct comparison between theory and experiment.
A key problem is the availability of molecular compounds which can be deposited on TMDs with defined packing motifs. Controlling the molecular orientation on the substrate would enable a systematic tuning of the interactions between the π-orbitals of the molecular compounds with the TMD and thus provide control over the interfacial electronic structure. Moreover, micrometre-sized oriented domains of molecules would allow for polarization-resolved optical studies, from which the anisotropic properties of the molecular excitons can be thoroughly investigated. Recently, some progress was made regarding the successful fabrication and optical characterization of defined heterostructures comprised of TMDs and pentacene, as well as its perfluorinated derivative,17,36,37 which are particularly challenging to grow in a controlled fashion on TMD monolayers.38 Also for HS of molybdenum disulphide (MoS2) with phthalocyanines, a correlation between the excited state dynamics and the molecular arrangement on the TMD was successfully demonstrated.39 Excitonic coupling was also found for HS of tungsten disulphide (WS2) with single crystals of β-perylene.20
In order to improve the understanding of the structure–property relations of OSC/TMD hybrids, it would be highly desirable to expand the family of molecular compounds for which a controlled orientation on TMDs is possible. In the present study, we therefore explore the potential of ionic molecular crystals for the controlled fabrication of HS. The specific packing of the ionic molecular crystals allows for easy exfoliation and ensures that the HS is created using the (001) crystal facet, a principle that has been previously employed for 2D perovskites.40,41 The resulting thin and smooth layers can be matched with TMDs, either with the organic or the TMD material on top. Here, we investigate heterostructures of mechanically exfoliated pyrenemethylammonium-chloride42 (in the following referred to as PyMACl, structural formula presented in Fig. 1a) with monolayers of the classical TMD material WS2. We focus on polarization-dependent, spatially and time-resolved photoluminescence (TRPL) spectroscopy, which helps us to find evidence for electronic coupling between the OSC and the TMD in terms of charge and/or exciton transfer, as well as induced polarization anisotropy in the TMD and emission signatures, which can be assigned to the heterointerface.
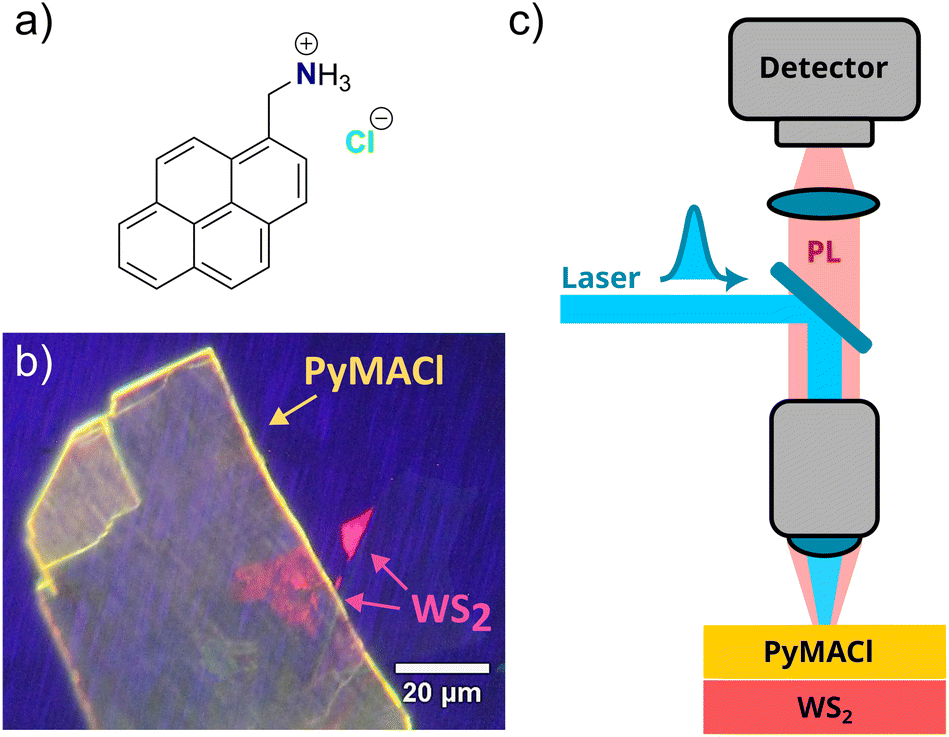 | ||
| Fig. 1 Structural formula of the ionic PyMACl molecular crystals (a). The fabricated HS was inspected with a fluorescence microscope (b) and the detection scheme for steady state and time-resolved spectroscopy is sketched in (c). For the steady state experiments, the PL signal was detected by a CCD camera attached to a spectrometer and for the time-resolved experiments, a streak camera was used as detector. | ||
Methods
Sample fabrication
The studied hybrid organic/inorganic HS is based on a WS2 monolayer and a few layers of PyMACl. The desired layers were fabricated by a micro-mechanical exfoliation process for both materials. The WS2 monolayer was isolated from commercially available bulk crystals obtained from 2D Semiconductors Inc. (USA). The few-layer PyMACl single crystal was isolated from a PyMACl bulk crystal that was grown by recrystallization of the commercially available compound (1-pyrenemethylamine hydrochloride, CAS: 93324-65-3, obtained from Sigma-Aldrich) from hot ethanol solution, as described in a previous literature report.42 The exfoliation of WS2 flakes was performed by breaking down a piece of a WS2 crystal with the assistance of Nitto tape, repeatedly folding and unfolding the tape several times. The exfoliated WS2 flakes were subsequently transferred onto a polydimethylsiloxane (PDMS) gel placed on a glass slide for monolayer identification. Under white light illumination of an optical microscope, the monolayers (MLs) can be distinguished from other flakes of different thicknesses by their optical contrast. Additionally, monolayers were identified through their photoluminescence (PL) in comparison to thicker flakes. As seen in Fig. 1b, the WS2 monolayer exhibits a high-yield red PL, whereas flakes with higher layer numbers show significantly lower emission due to their indirect band gap.The same procedure was followed to obtain the few layers of the PyMACl sample, where exfoliation is possible along the c-axis (see ESI† for details of the crystal structure). Few-layer PyMACl flakes were extracted and their PL was verified, as shown in Fig. 1b. The identified WS2 TMD monolayer was transferred to the target substrate (SiO2/Si) using the viscoelastic stamping technique. Subsequently, the PDMS was slightly lifted, leaving the ML attached to the target substrate. The final HS was produced by stamping a few layers of PyMACl on the top of the WS2 ML. Fig. 1b shows one of the prepared heterostructures under the fluorescence microscope.
Optical characterization
TRPL spectroscopy was conducted using a home-built confocal microscope setup (cf.Fig. 1c) equipped with a Hamamatsu streak camera (C6860) with a time resolution of 5 ps over the employed time window of 460 ps. Spectrally resolved data were obtained with a Bruker imaging monochromator (model 250-30). For excitation, we used a pulsed titanium–sapphire laser with a repetition rate of 80 MHz and 100 fs pulse duration. The emission wavelength was 720 nm, yielding an excitation wavelength of 360 nm (3.44 eV) after frequency doubling. A set of mirrors and a 376 nm long pass dichroic mirror directed the laser beam to a 40x microscope objective (NA = 0.6), which served to focus the beam onto the sample with a spot radius of approximately 2.5 μm. The laser power was set to 50 μW to prevent nonlinear processes such as exciton–exciton annihilation and potential damage to the PyMACl organic layers. The emitted photoluminescence signal was collected by the same microscope objective and directed to the detection path. A long pass 376 nm dichroic mirror filtered out the reflected laser light. Additionally, a 400 nm long pass coloured glass filter was placed in the detection path to block any residuals of laser light from the PL signal. Furthermore, a CMOS camera and a flip mirror were incorporated into the detection path for optical control and imaging of the HS. For temperature-dependent measurements, the samples were mounted in a liquid-helium flow microscopy cryostat (CryoVac) to enable precise control of the temperature within the range from 10 to 300 K. To ensure seamless measurements, the microscope objective correction collar was set to a value of 1.5 mm to minimize aberrations caused by the cryostat's thick glass window (1.5 mm). The recorded TRPL data were corrected for the spatially inhomogeneous sensitivity of the system and for the spectral response using a broadband tungsten light source as a reference. Moreover, a background measurement was subtracted from the data. Displayed PL spectra were converted to the energy scale via a Jacobian transformation.43Polarization-resolved time integrated PL spectra were recorded with a home-built micro-photoluminescence (μ-PL) setup (see ESI†) equipped with a nitrogen-cooled CCD camera attached to an imaging monochromator (Princeton Instruments Acton SP2300). A 532 nm laser (2.33 eV) was used as an excitation source, with the laser power set between 50 and 500 μW. A linear polarizer was added to the excitation path to create a defined polarization of the impinging laser light. The laser beam was guided by a set of optics and focused to a 2.5 μm spot by a 40x microscope objective (NA = 0.6). The HS under investigation was kept at room temperature under vacuum in a microscopy cryostat (CryoVac) to maintain stability and to avoid undesired laser induced thermal degradation. The emitted PL from the sample was collected by the same microscope objective and directed to the detection path. To suppress the laser light from the collected PL data, a 550 nm long-pass filter was placed in the detection path. Additionally, a polarizer was placed in the detection path to analyse the polarization anisotropy of the emitted PL signal from the sample under investigation. The setup was calibrated before performing the measurements using a set of polarizers and a laser source to ensure that the observed phenomena arose from the sample and not from any setup-related effects. This was done by rotating the polarizer in the excitation path and checking the detected intensity on the CCD. Furthermore, the polarizer in the detection path was first fixed to align with the laser polarization, and then it was rotated to check and exclude any contributions from the setup.
Results and discussion
Before providing more detailed insights into the HS, we will first discuss the optical properties of PyMACl in greater detail. Absorbance spectra of the compound dissolved in ethanol and of the crystalline solid are presented in Fig. 2a. Overall, the optical properties of the molecules in solution are very similar to those of dissolved neat pyrene molecules. The oscillator strength of the S0 → S1 transition at 3.31 eV (375 nm) is very weak compared to the higher S0 → Sn transitions, because it is symmetry forbidden.44,45 In the solid, the absorption edge shows a significant red-shift. Assuming a direct band edge, the Tauc method46 yields an absorption onset of the PyMACl crystal of approximately 3.19 eV (389 nm, see ESI†). From the enlarged plot of the spectra close to the absorption edge in Fig. 2b it becomes apparent that the perceived red-shift in the solid mainly originates from a gain in oscillator strength of the transitions close to the absorption edge. The observation is similar to pyrene single crystals or nanoaggregates, where the red-shift was attributed to the admixture of higher electronic states,47,48 which undergo a more pronounced bathochromic shift in pyrene single crystals compared to the energetically lowest optical transition.45 The lowest excitonic resonance in the solid peaks around 3.26 eV (380 nm) and shows a rather moderate red-shift of approximately 50 meV with respect to the molecular S0 → S1 transition. For pyrene single crystals, it was argued that this behaviour is expected due to the more localized character of the lowest electronic transition, whereas intermolecular interactions and a higher degree of delocalization change the energetics of the higher electronic transitions more significantly. Since the first excited state in the solid is rather localized and thus mainly governed by the molecular electronic structure and less by intermolecular interactions, the packing motif should not have a strong influence on the absorption properties close to the optical gap, which could explain the similar absorption properties of crystalline pyrene45,47,48 and PyMACl in this regime. However, due to the presumed admixture of higher electronic transitions we assume that the excitation energy of 3.44 eV does not exclusively address the energetically lowest excitonic transition in our experiments.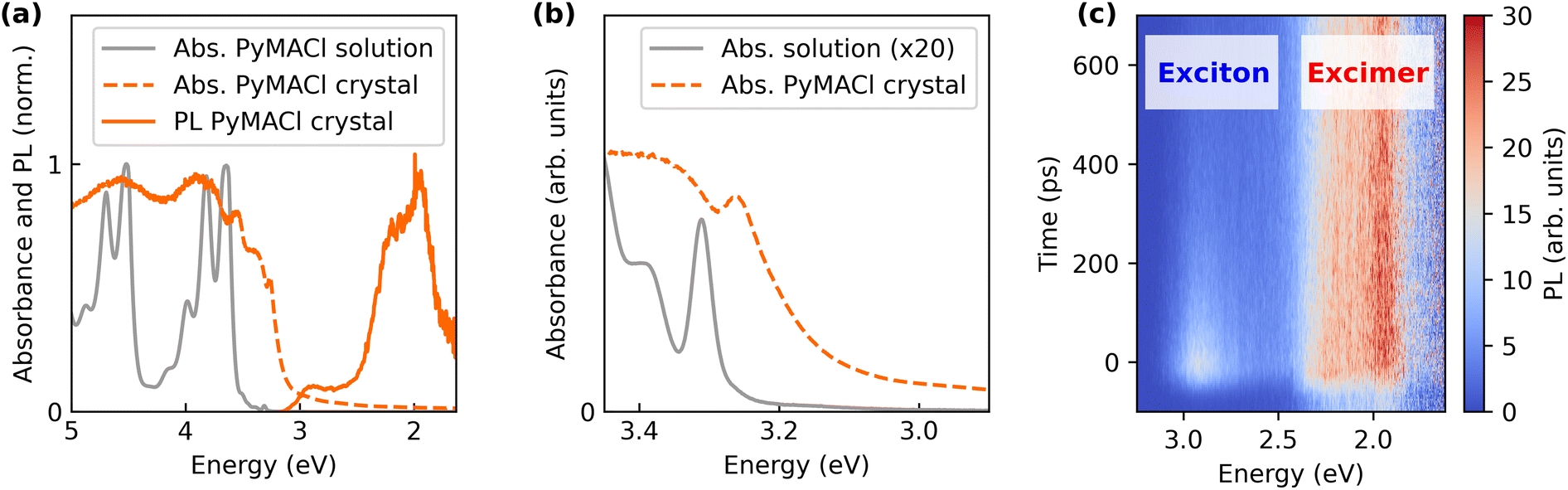 | ||
| Fig. 2 (a) Steady state absorption spectra of PyMACl dissolved in ethanol and in the solid phase. The absorbance spectrum of the solid was obtained from diffuse reflectance data via a Kubelka-Munk-transform.49 The quasi static PL data were obtained by integrating over a 2 ns time window of streak camera data recorded from a PyMACl microcrystal. Panel (b) shows an enlarged plot of the energy range near the absorption edge. For better comparison, the absorbance spectrum of the solution was multiplied by a factor of 20. Spectrally and time-resolved TRPL data highlighting free exciton and excimer emission are presented as false colour plot in (c). The TRPL data was actually stitched from three individual measurements. | ||
The photoluminescence spectrum of PyMACl shows two major features. A comparably faint signature peaks at energies around 2.92 eV (425 nm). The emission energy coincides with the energy range that was reported for monomeric emission in pyrene solution.45,50,51 A weak feature around 3 eV was also reported for pyrene single crystals and nanoaggregates51 and an earlier report on the synthesis of PyMACl mentioned PL in this range.42 Following the arguments of earlier work, we assign the observed PL peak to the emission of the energetically lowest free exciton state which has, as discussed, a strong monomeric character. The pronounced red-shift with respect to the exciton resonance in the absorption spectra could be the result of a filtering effect, leading to re-absorption of the photons emitted at higher energies in the range of the absorption onset.52 On the other hand, the pronounced Stokes shift of the exciton emission could also be caused by a partial structural relaxation or by disorder effects, which cause excitons to migrate into local minima of the density of states.
Besides the presumed free exciton emission, an energetically broader more intense signature is observed in the energy range between 1.9 and 2.4 eV. Neat pyrene crystals are known for their “dimeric” character, which originates from a sandwich-herringbone type packing of the molecules.53 This packing motif intrinsically supports the formation of excimers, because it is comprised of pairs of molecules, which are arranged in a coplanar fashion. When excited, these dimers relax into a configuration with lower inter-molecular spacing, which is associated with the red-shifted emission of excimers.50,54 In neat pyrene, excimer emission was reported at energies around 2.6 eV (480 nm). The long-lived emission observed here for PyMACl is significantly more red-shifted. This discrepancy can be rationalized by the different crystal structure of PyMACl, which possesses a slip-stacked arrangement of the molecules. Such an arrangement enables more extended pi-stacks of larger numbers of molecules compared to the dimer-like arrangement in neat pyrene. This could, in turn, lead to higher dielectric stabilization, similar to one-dimensional aggregates. Structural relaxation could further contribute to this effect. Notably, even though the red-shifted signature is expected from the packing of the molecules, it was not observed in earlier work on PyMACl single crystals.42 Presently, we cannot explain this discrepancy, however, the spatial arrangement of the crystal in optical experiments (detection of edge emission or surface emission) may be critical for the dominant signatures in the optical spectra. Despite this discrepancy, we tentatively assign the observed red-shifted feature to excimer-type emission originating from the pi-stacking and structural relaxation of the slip-stacked molecules.
Our assignments of the PL signatures are further supported by time-resolved experiments. Exemplary TRPL data of the PyMACl emission are presented in Fig. 2c. While the exciton decays within the first 500 ps, the presumable excimer shows long-lived emission, which does not fully decay within the time spacing of 12.5 ns between two laser pulses. This is evidenced by the residual emission before t = 0 in the energy range of excimer emission. The longer-lived decay of the excimer emission likely arises from the comparably weak spatial overlap of the electron and hole wave functions and the localized character of the population. Recombination of free excitons on the other hand exhibits a higher oscillator strength and we expect a population with increased mobility. The latter makes them more likely to reach non-radiative centres, which could explain the overall faster decay of the PL emission.
In order to explore the PL dynamics in greater detail, we have conducted temperature dependent measurements. The temperature of the system has a strong influence on the diffusion of free excitons, the presence of phonons and the activation of certain non-radiative channels. Therefore, varying the temperature provides insight into the kinetic competition of different decay channels of the system, including exciton or charge transfer in the HS with WS2. In the following, we will discuss the energy ranges of the free exciton (2.88–2.95 eV) and the excimer emission (2.29–2.58 eV) separately. When the sample is heated from 20 K to 295 K, the PL dynamics of the free exciton emission become faster (see Fig. 3a and b), which most likely originates from thermal activation of non-radiative processes. Interestingly, the excimer emission (Fig. 3c and d) shows an opposite trend, i.e. the decay becomes slower with increasing temperature. We attribute the observation to thermally activated excimer formation, which is also known from other organic systems, such as perylene or anthradithiophene.55–57 In earlier work, it was suggested that while at low temperatures, thermal energy is not sufficient for excitons to overcome a certain energetic barrier to form the excimer, this process becomes possible at elevated temperatures through vibronic excitations. The observed slower dynamics of the excimer emission with increasing temperature could originate from superposition of the feature with some energetically relaxed excitonic states, which get more and more obscured by the increasing contribution of the longer-lived excimer emission.
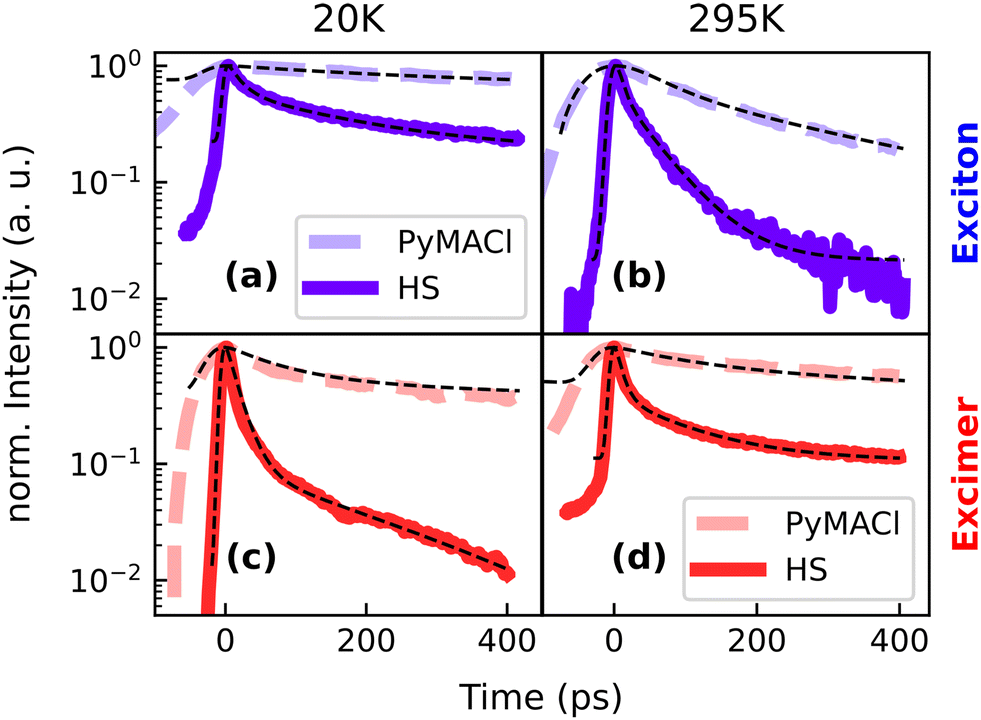 | ||
| Fig. 3 PL dynamics of neat PyMACl and the HS. We used different settings of the streak camera to probe the different time regimes of the PL decay, resulting in a broader instrument response function for the data of neat PyMACl, which was actually much longer-lived than the HS. Panels (a and b) show the decay in the energy range of the free exciton emission (2.88–2.95 eV), whereas (c and d) report the dynamics in the energy range of excimer emission (2.29–2.58 eV). | ||
We find pronounced differences between the PL dynamics of the neat PyMACl compound and the HS. In the latter, the PL originating from PyMACl decays much faster compared to sample regions probed outside the HS. This observation is an indicator for an efficient electronic coupling mechanism between both compounds, which could either lead to exciton or charge transfer from PyMACl into WS2. Experimentally, the pyrene/WS2 interface has achieved little attention so far.58 However, first principles studies based on DFT utilizing band unfolding techniques suggest that the compounds form a type-II heterojunction with hybridization occurring between the molecular orbitals of pyrene and the band structure of WS2.29 As a consequence, electron transfer could emerge from the free excitons which are generated in PyMACl. Alternatively, there is the possibility of exciton transfer from PyMACl to WS2, which could be followed by hole transfer from WS2 to PyMACl. Interestingly, for the excimer state, efficient quenching is observed as well. However, in this case it is challenging to make assumptions about the underlying transfer mechanism, because the energy level arrangement of the involved molecular orbitals with respect to the WS2 band structure is unclear and it has to be considered that the energetics are governed by substantial structural re-arrangement of the organic lattice in the course of excimer formation. It is noteworthy, however, that the range of emission energies of the excimer coincides with the absorption of the excitonic resonance in WS2,59 which is a prerequisite for Förster resonance energy transfer (FRET),54,60 as we will discuss further in connection with the analysis conducted below.
With our experimental approach, we cannot draw definite conclusions about the underlying transfer mechanisms, which could involve both exciton and charge transfer. Notably, it was not possible to obtain TRPL data of sufficient quality for selective excitation of WS2. Therefore, transfer channels for charges and excitons originating from WS2 remain an open topic. However, we point out that in the steady state PL data and the recorded PL images, the emission of WS2 in the region of the HS was weaker compared to the emission outside of this region (see ESI† for details). This is an indication for a hole transfer from WS2 to PyMACl, as expected for the type-II interface. An enhancement of the WS2 emission originating from a potential transfer of excitons from PyMACl on the other hand is not observed, which gives further evidence for the presence of a type-II instead of a type-I heterojunction, similar to other heterostructures of molecular compounds and TMD monolayers.2,24,27,28 Ideally, future work on comparable material systems will also be able to capture the photoexcitation dynamics of WS2. One promising approach is fluorescence lifetime imaging, which is highly sensitive when based on photomultipliers or avalanche photodiodes and which also allows for a more thorough correlation of the PL dynamics with spatial information.61,62 On the other hand, transient absorption or transient reflection spectroscopy opens the possibility to access different photoexcited populations with sub-picosecond time resolution and was already successfully applied to organic/TMD heterostructures.24
In a next step, we further quantify the temperature dependent PL dynamics. Due to the presumed statistic distribution of non-radiative centres into which mobile excitons can diffuse and due to the statistic distribution of transfer times at the heterointerface, the PL is expected to show multiexponential decay kinetics. In the observed time window, however, we find biexponential fits to give a good approximation to the experimental data. As an effective measure for the PL dynamics, we extract the amplitude averaged lifetimes63 from those fits. The resulting decay times extracted from the energy ranges of exciton and excimer emission of neat PyMACl and from the region of the HS are displayed in Fig. 4. The lifetimes of the free exciton emission increase by a factor of about 3, when the material is cooled from room temperature to 20 K. A similar trend is observed for the HS, whereas the quenching of excitons by the WS2 layer accelerates the decay dynamics by almost one order of magnitude. It is likely that the thermally activated faster PL dynamics are caused by thermally activated exciton migration, such that non-radiative centres are reached more quickly. For the presumed excimer emission on the other hand, no thermally activated non-radiative channel is observed, which we attribute to the more localized character of the species. In fact, the decay times of the excimer emission in PyMACl rather indicate the opposite trend, i.e. faster decay at temperatures below 150 K. As discussed, this behaviour could originate from the absence of excimer emission at lower temperatures. An interesting result originating from a comparison of the decay times of neat PyMACl and the HS is, however, that the excimer undergoes efficient quenching in the presence of WS2 for all investigated temperatures, suggesting pronounced electronic coupling between the structurally and energetically relaxed excimer species and the electronic structure of WS2, potentially via FRET, as discussed in the previous section. The trends observed in the lifetimes of the free exciton and excimer emission are also reproduced by the relative intensities of the species (see ESI†), confirming that changes in decay times are indeed caused by non-radiative processes, such as deep trap recombination or transfer processes at the interface of the HS, rather than by an increase of radiative rates.
 | ||
| Fig. 4 Temperature dependent amplitude-averaged lifetimes extracted from the energy ranges of free exciton and excimer emission. | ||
With the acquired exciton dynamics in neat PyMACl and in the HS, it is possible to estimate the time scale of exciton or charge transfer. Following the argumentation above, these transfer times should be rather considered as effective values, originating from a statistical distribution of decay rates and transfer times. Assuming kinetic competition between radiative decay with the time constant τr, non-radiative decay via τnr and a transfer mechanism characterized by τtr, the overall PL dynamics of the excited states in PyMACl stacked with a WS2 monolayer is described by:
| τ−1HS = τ−1r + τ−1nr + τ−1tr | (1) |
| τ−1py = τ−1r + τ−1nr | (2) |
 | (3) |
 | ||
| Fig. 5 Estimated transfer times from PyMACl to WS2 for the energy ranges of free exciton and excimer emission (a), based on eqn (3). Note that the data points at 120 K, which are severe outliers, were omitted from the plot in order not to impair its readability. Panels (b) and (c) show the hypothesized transfer mechanisms after preferential excitation of PyMACl and after excitation of WS2, as discussed in the text. | ||
Type-II heterostructures of organic semiconductors and monolayer TMDs, which promote exciton transfer and/or exciton splitting via interfacial charge transfer, were predicted and experimentally characterized for various material systems.2,24,26,29 Our observation of luminescence quenching, i.e. a faster PL decay in the organic layer in the heterostructure indicates charge or exciton transfer to the TMD, which is consistent with previous findings based on time-resolved photoelectron spectroscopy of heterostructures comprised of zinc-phthalocyanine and molybdenum disulfide.26 Interlayer charge and exciton transfer was also confirmed for heterostructures of tetracene and WS2 by means of transient absorption spectroscopy.24 Thus, type-II energy level arrangement seems to be a property that can be realized for a variety of organic/TMD systems.
Finally, we explore the properties of the system after photoexcitation of WS2. In order to selectively excite WS2, we chose an excitation energy of 2.33 eV (532 nm), which is well below the absorption onset of PyMACl. Surprisingly, even for this low excitation energy, yellow emission of the organic compound is observed, as presented in Fig. 6a. This finding suggests that the organic compound shows weak absorption below the absorption onset, which is in accordance with the weak absorption tail observed in Fig. 2b. This absorption could originate from defect states or from some weak coupling of the excimer state to the ground state. As the signatures of the PyMACl emission coincide with the range of energies that was attributed to excimer emission, it is likely that the excimer state is responsible for the observed emission. We also find a distinct polarization of the yellow emission, which is significantly rotated with respect to the polarization of the free exciton state (see ESI†). Due to the anisotropic nature of the crystal, transition dipole moments with different orientations are in fact expected for the different excitonic species.67,68 On the other hand, the distinct polarization of the yellow emission could also originate from defect states, which are associated with a certain structural configuration in the crystal lattice of the organic compound. At this point, we cannot make a definite statement about the origin of the PL after excitation below the absorption onset of PyMACl. The overlap with the A-exciton emission (XA) of WS2 makes it challenging to extract quantitative information about the dynamics of the comparably weak emission from WS2. This difficulty is potentially exacerbated by hole transfer from WS2 to PyMACl, which could lead to remarkable quenching of the excitonic WS2 emission. However, by using steady state PL spectroscopy, it was possible to obtain some insight into the polarization of the WS2 emission.
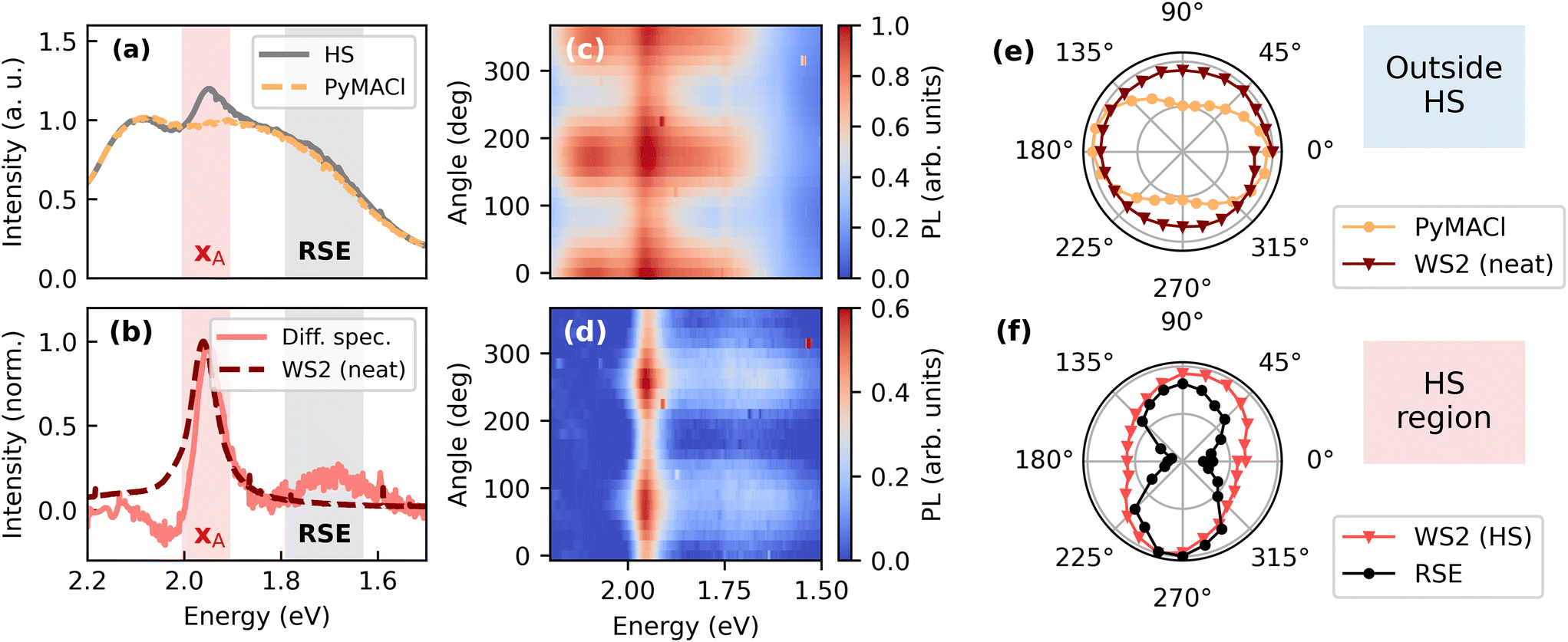 | ||
| Fig. 6 Polarization resolved characterization of the HS. In order to extract the intensity of the WS2 exciton emission (XA), the PL background of PyMACl was recorded outside the HS region, normalized to the PL spectrum of the HS in the energy range between 2.10 and 2.12 eV (a) and subtracted from the HS data, yielding the difference spectrum presented in (b). Beside the XA exciton signature of WS2, the HS shows red-shifted emission (RSE) around 1.7 eV, potentially originating from the interface. Polarization dependent emission spectra and difference spectra are presented as false-colour plots in (c) and (d). Polar plots of the integrated intensities of PyMACl, the XA peak (neat WS2 and HS) and the RSE peak are shown in panels (e) and (f). | ||
In order to disentangle the XA emission of WS2 from the PL underground of PyMACl, we recorded PL spectra in the region of the HS and outside of this region. As a next step, the PyMACl spectrum recorded outside the HS was normalized to the PyMACl emission of the HS and subtracted from the data. A difference spectrum calculated from the exemplary data in Fig. 6a is shown in Fig. 6b. This spectrum gives us an estimate of the intensity of the XA emission. Moreover, we can explore other contributions to the PL spectrum in the region of the HS. Interestingly, the difference spectrum at emission energies slightly above the XA resonance becomes negative, indicating that the relative emission intensity of PyMACl in this regime is reduced compared to the neat PyMACl layer. This finding could originate from exciton transfer from PyMACl to WS2, which is expected to be particularly strong in the spectral region where the PyMACl emission overlaps with the XA absorption.
Another interesting observation is the emergence of a weakly emissive red-shifted emission (RSE) peaking around 1.7 eV, which occurs exclusively in the region of the HS. The origin of this spectrally broad signature could be an emissive defect state that is specific for the PyMACl/WS2 interface or an interlayer excitonic state, which was also reported for other organic/TMD systems with presumed type-II energy level arrangement.11,24,69 In future work, spatially resolved PL microscopy could help to clarify whether the emission is associated with specific structural defects, or whether it occurs homogeneously across the whole HS. Moreover, interfacial excitons should show some mobility, which can be explored by spatiotemporal PL or transient absorption measurements.24 To further distinguish the feature from an underlying defect state, surface sensitive techniques such as time-dependent second harmonic generation70 or time resolved photoelectron spectroscopy can be employed.71 Moreover, time and angular resolved photoelectron spectroscopy is capable of revealing the momentum fingerprint of charge transfer excitons, when dispersive bands are formed.72 Similar to Moiré excitons in TMD heterostructures, systematic modification of the stacking angle between the TMD and the organic layer could lead to variations in the optical properties, if interlayer excitons are responsible for the RSE.73 While the mentioned techniques would be attractive for future studies, interestingly, our PL spectra reveal differences in the RSE, when comparing the investigated HS to an inverted HS discussed in the ESI.† This indicates different electronic couplings between the layers, suggesting that the observed feature is indeed an interface state rather than a defect. However, further investigations with control over the stacking angle are needed and beside different stacking angles, also the different dielectric environments of WS2 in the HS and the inverted HS could play a role.
We find that both the residual PyMACl emission as well as the underlying PL of WS2 reveal distinct polarizations. Emission spectra and difference spectra recorded for different orientations of the analyser are presented as false-colour plots in Fig. 6c and d, respectively. The emission intensity of PyMACl shows a clear polarization dependence when probed inside and outside the region of the HS. The respective integrated intensities are shown in the polar plot in Fig. 6e, together with the integrated intensity of the XA emission of WS2 outside the HS. The emission of neat WS2 is isotropic as expected.74 Interestingly, the WS2 emission in the region of the HS is linearly polarized (cf.Fig. 6f) and approximately perpendicular to the polarization of the yellow emission originating from PyMACl. Also, the RSE signature is linearly polarized in the same direction as the XA emission, suggesting that the RSE is associated with a specific configurational arrangement at the heterointerface, resulting in a specific alignment of the corresponding transition dipole moment. Notably, the polarizations of the XA emission and the RSE emission differ from each other in case of the inverted HS (see ESI†).
For the case of the inverse HS with WS2 on top of PyMACl (see ESI†), we find similar orientations of the polarizations of the yellow excimer- or defect-type PyMACl and the XA emission, which are approximately perpendicular to each other. This finding confirms that the anisotropic emission of WS2 is indeed introduced by the electronic coupling with the organic compound and not just a trivial filtering effect. Since the polarizations of the yellow PyMACl emission and the free exciton emission, on the other hand, form an angle of approx. 45° to each other (see ESI†), this means that the induced polarization anisotropy of the excitons in WS2 is rotated by approximately 45° with respect to the free exciton emission of PyMACl.
We hypothesize that this peculiar rotation of the polarization could be a result of the specific arrangement of the molecules at the interface to WS2 and the strain induced in the TMD monolayer. The transition dipole moment (TDM) of the lowest excitonic state in pyrene is oriented along the long molecular axis.68 In the optical experiments, luminescence is observed from the ab-plane of the molecular crystals. The orientation of the TDM of the pyrene chromophores is tilted with respect to the main axes of the ab-plane, as shown in the ESI.† On the other hand, the molecular crystal lattice potentially acts as a patterned template, on which the thin WS2 layer adapts an energetically favorable commensurate arrangement. This re-arrangement of the WS2 lattice implies anisotropic deformation and the build-up of strain, presumably along the main axes of the supporting PyMACl lattice. Similar effects are known from TMDs deposited on patterned substrates75 or HS of TMDs with anisotropic 2D materials.76 The presence of strain in WS2 leads to a lifting of the threefold rotation symmetry of the lattice, resulting in new excitonic eigenstates, which are linearly polarized along the main axes of the strain tensor.77 For the anisotropy induced by the organic substrate, these axes should coincide with the a and b axis of PyMACl. We would like to stress that without any further theoretical analysis, our considerations remain simplistic and do not take any further effects into account, such as the influence of the methylammonium group on the TDM, coupling effects between different molecules or hybridization of electronic states at the interface.29 Nevertheless, our considerations may provide a qualitative explanation for the observed linear polarization of the WS2 emission and the differing polarization direction of the free exciton in the organic layer.
For the polarization resolved optical studies it is important to note that these can only be carried out for systems, in which monocrystalline and oriented domains of the organic compound are large enough to be resolved by an optical microscope. In this context, an earlier study focused on WS2 stacked with beta-perylene.20 While the authors found polarized emission of the TMD, consistent with our work, their excitation was below the optical gap of the organic substrate, not allowing for a direct comparison of the emission polarizations. The authors attributed the observed polarization response of the TMD to anisotropic coupling with the substrate, caused by the anisotropic refractive index, leading to different reflectivity of both polarization components for a given sample thickness. While such an interference effect could, in principle, explain our experimental observations, we propose that our findings of anisotropic PL emission from WS2 rather originate from the template effect described above, for several reasons: (i) we find evidence that the electronic structure of WS2 is altered, leading to emission from a presumed interfacial state and energetic shifts of the WS2 peak in case of the inverted heterostructure (see ESI†). (ii) Our organic crystals were exfoliated and therefore, their thickness should be on the order of 10 nm. Thus, interference effects should be of minor importance for our samples. (iii) It is well-established that different templates can modify the strain and with this the electronic structure of TMDs and polarization anisotropy.75–77
Conclusions
Our work introduces a promising approach for the controlled fabrication of organic/TMD heterostructures with a well-defined alignment of the molecules in the organic ionic crystal. Such heterostructures are useful to validate theoretical descriptions. Moreover, they can serve as model interfaces to study the impact of the molecular orientation on the interfacial electronic structure and on mechanisms of exciton and charge transfer, which is highly relevant for device applications. Further studies could address different stacking configurations of the same system or they may explore the influence of distinct optical properties, such as chirality, which could be induced into the TMD via specifically tailored organic compounds. We have demonstrated electronic couplings at the PyMACl/WS2 interface by means of time- and polarization resolved optical spectroscopy, presumably leading to exciton and charge transfer at the type-II heterointerface. Interestingly, beside the free excitons formed in PyMACl, also the lower-energetic localized excimer promotes efficient exciton transfer into WS2. This is potentially related to the fact that its emission is isoenergetic with the absorption of the XA exciton in WS2, leading to efficient FRET. From a theoretical point of view, this aspect is challenging to address, because structural distortions of the system after photoexcitation need to be included in the description. At the same time, the observation bears some relevance e.g. for sensing applications, because it demonstrates that the excimer states can serve as efficient sensitizers for exciton transfer to the TMD, which could be a motivation for theoretical efforts in this direction. Beside the observed transfer processes, we also find that the organic compound leads to anisotropic PL emission of WS2 and we identify weakly emissive low energetic PL signatures around 1.7 eV in the heterostructures, potentially emerging from the heterointerface, which can be seen as further indicators for electronic coupling. Overall, our study introduces a new platform for specifically tailored heterostructures comprised of TMDs and ionic molecular single crystals. For a satisfying understanding of these systems and the underlying mechanisms of electronic coupling, we find it particularly important to take the strong electron–phonon-coupling of the organic compounds into account, which should be further addressed in future work.Data availability
Raw data on which our manuscript entitled “Electronic coupling and photoluminescence anisotropy in van-der-Waals-stacks of tungsten disulphide with molecular single crystals” is based are available at figshare via the following link:https://figshare.com/s/adedbbc617991584ec2b
DOI: https://doi.org/10.6084/m9.figshare.27201633
The dataset includes:
• Fluorescence micrographs.
• Static and time-resolved spectroscopic raw data.
• Python-based scripts to analyse the data.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the German Research Foundation DFG via the Collaborative Research Centre No. 223848855-SFB 1083, sub-projects B10, B12 and B15. Moreover, J. H. gratefully acknowledges funding by the DFG through the Heisenberg Programme (505757318). M. A. A. acknowledges support from the Egyptian Ministry of Higher Education and Scientific Research and from the German Academic Exchange Service (DAAD). The authors thank Julia Kessel for her assistance with the polarization-resolved measurements.References
- Y. Li Huang, Y. Jie Zheng, Z. Song, D. Chi, A. T. S. Wee and S. Ying Quek, Chem. Soc. Rev., 2018, 47, 3241–3264 RSC
.
- D. B. Sulas-Kern, E. M. Miller and J. L. Blackburn, Energy Environ. Sci., 2020, 13, 2684–2740 RSC
- J. Ji and J. H. Choi, Nanoscale, 2022, 14, 10648–10689 RSC
- C. Chakraborty, A. Mukherjee, L. Qiu and A. N. Vamivakas, Opt. Mater. Express, 2019, 9, 1479–1487 CrossRef CAS
- P. Rivera, K. L. Seyler, H. Yu, J. R. Schaibley, J. Yan, D. G. Mandrus, W. Yao and X. Xu, Science, 2016, 351, 688–691 CrossRef CAS PubMed
- Y. Li, J. Ludwig, T. Low, A. Chernikov, X. Cui, G. Arefe, Y. D. Kim, A. M. Van Der Zande, A. Rigosi, H. M. Hill, S. H. Kim, J. Hone, Z. Li, D. Smirnov and T. F. Heinz, Phys. Rev. Lett., 2014, 113, 266804 CrossRef PubMed
- M. W. Iqbal, E. Elahi, A. Amin, G. Hussain and S. Aftab, Superlattices Microstruct., 2020, 137, 106350 CrossRef CAS
- B. Han and P. Samorì, Acc. Chem. Res., 2024, 57, 2532–2545 CrossRef CAS PubMed
- Y. Cai, H. Zhou, G. Zhang and Y.-W. Zhang, Chem. Mater., 2016, 28, 8611–8621 CrossRef CAS
- F. Liu, W. L. Chow, X. He, P. Hu, S. Zheng, X. Wang, J. Zhou, Q. Fu, W. Fu, P. Yu, Q. Zeng, H. J. Fan, B. K. Tay, C. Kloc and Z. Liu, Adv. Funct. Mater., 2015, 25, 5865–5871 CrossRef CAS
- S. B. Homan, V. K. Sangwan, I. Balla, H. Bergeron, E. A. Weiss and M. C. Hersam, Nano Lett., 2017, 17, 164–169 CrossRef PubMed
- H. J. Park, C. J. Park, J. Y. Kim, M. S. Kim, J. Kim and J. Joo, ACS Appl. Mater. Interfaces, 2018, 10, 32556–32566 CrossRef CAS PubMed
- D. Jariwala, S. L. Howell, K. S. Chen, J. Kang, V. K. Sangwan, S. A. Filippone, R. Turrisi, T. J. Marks, L. J. Lauhon and M. C. Hersam, Nano Lett., 2016, 16, 497–503 CrossRef CAS PubMed
- F. J. Urbanos, S. Gullace and P. Samorì, ACS Nano, 2022, 16, 11234–11243 CrossRef CAS PubMed
- M. Feierabend, G. Berghäuser, A. Knorr and E. Malic, Nat. Commun., 2017, 8, 14776 CrossRef CAS PubMed
- Y. Zhao, L. Yu and M. Sun, Sol. Energy, 2021, 218, 621–638 CrossRef CAS
- M. Dreher, D. Günder, S. Zörb and G. Witte, Chem. Mater., 2020, 32, 9034–9043 CrossRef CAS
- M. Dreher, P. M. Dombrowski, M. W. Tripp, N. Münster, U. Koert and G. Witte, Nat. Commun., 2023, 14, 1554 CrossRef CAS PubMed
- D. He, Y. Pan, H. Nan, S. Gu, Z. Yang, B. Wu, X. Luo, B. Xu, Y. Zhang, Y. Li, Z. Ni, B. Wang, J. Zhu, Y. Chai, Y. Shi and X. Wang, Appl. Phys. Lett., 2015, 107, 183103 CrossRef
- Z. An, Q. Ai, H. Chen, X. Wang and T. Gao, J. Appl. Phys., 2022, 132, 114302 CrossRef CAS
- A. K. Geim and I. V. Grigorieva, Nature, 2013, 499, 419–425 CrossRef CAS PubMed
- M. A. Aly, E. O. Enakerakpor, M. Koch and H. Masenda, Nanomaterials, 2023, 13, 2769 CrossRef CAS PubMed
- L. Zhang, A. Sharma, Y. Zhu, Y. Zhang, B. Wang, M. Dong, H. T. Nguyen, Z. Wang, B. Wen, Y. Cao, B. Liu, X. Sun, J. Yang, Z. Li, A. Kar, Y. Shi, D. Macdonald, Z. Yu, X. Wang and Y. Lu, Adv. Mater., 2018, 30, 1–8 Search PubMed
- T. Zhu, L. Yuan, Y. Zhao, M. Zhou, Y. Wan, J. Mei and L. Huang, Sci. Adv., 2018, 4, 3104 CrossRef PubMed
- S. H. Amsterdam, T. K. Stanev, Q. Zhou, A. J.-T. Lou, H. Bergeron, P. Darancet, M. C. Hersam, N. P. Stern and T. J. Marks, ACS Nano, 2019, 13, 38 CrossRef PubMed
- T. R. Kafle, B. Kattel, P. Yao, P. Zereshki, H. Zhao and W.-L. Chan, J. Am. Chem. Soc., 2019, 141, 11328–11336 CrossRef CAS PubMed
- P. A. Markeev, E. Najafidehaghani, G. F. Samu, K. Sarosi, S. B. Kalkan, Z. Gan, A. George, V. Reisner, K. Mogyorosi, V. Chikan, B. Nickel, A. Turchanin and M. P. De Jong, ACS Nano, 2022, 16, 16668–16676 CrossRef CAS PubMed
- R. Dziobek-Garrett and T. J. Kempa, J. Chem. Phys., 2024, 160, 200902 CrossRef CAS PubMed
- J. Krumland and C. Cocchi, Electron. Struct., 2021, 3, 044003 CrossRef CAS
- C. Draxl, D. Nabok and K. Hannewald, Acc. Chem. Res., 2014, 47, 3225–3232 CrossRef CAS PubMed
- E. Verdenhalven, A. Knorr, M. Richter, B. Bieniek and P. Rinke, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 235314 CrossRef
- M. P. Ljungberg, O. Vänskä, P. Koval, S. W. Koch, M. Kira and D. Sánchez-Portal, New J. Phys., 2017, 19, 033019 CrossRef
- N. Shen and G. Tao, Adv. Mater. Interfaces, 2017, 4, 1601083 CrossRef
- X. Y. Liu, X. Y. Xie, W. H. Fang and G. Cui, J. Phys. Chem. A, 2018, 122, 9587–9596 CrossRef CAS PubMed
- J. J. P. Thompson, M. Gerhard, G. Witte and E. Malic, npj 2D Mater. Appl., 2023, 7, 69 CrossRef CAS
- S. R. Kachel, P.-M. Dombrowski, T. Breuer, M. Gottfried and G. Witte, Chem. Sci., 2021, 12, 2575–2585 RSC
- D. Günder, K. Watanabe, T. Taniguchi and G. Witte, ACS Appl. Mater. Interfaces, 2020, 12, 38757–38767 CrossRef PubMed
- D. Günder, M. Axt and G. Witte, ACS Appl. Mater. Interfaces, 2024, 16, 1911–1920 CrossRef PubMed
- S. Padgaonkar, S. H. Amsterdam, H. Bergeron, K. Su, T. J. Marks, M. C. Hersam and E. A. Weiss, J. Phys. Chem. C, 2019, 123, 13337–13343 CrossRef CAS
- L. Dou, A. B. Wong, Y. Yu, M. Lai, N. Kornienko, S. W. Eaton, A. Fu, C. G. Bischak, J. Ma, T. Ding, N. S. Ginsberg, L.-W. Wang, A. P. Alivisatos and P. Yang, Science, 2015, 349, 1518–1521 CrossRef CAS PubMed
- A. Yang, J.-C. Blancon, W. Jiang, H. Zhang, J. Wong, E. Yan, Y.-R. Lin, J. Crochet, M. G. Kanatzidis, D. Jariwala, T. Low, A. D. Mohite and H. A. Atwater, Nano Lett., 2019, 19, 4852–4860 CrossRef CAS PubMed
- N. Tokunaga, Y. Fujiki, S. Shinkai and K. Sada, Chem. Commun., 2006, 3617–3618 RSC
- J. Mooney and P. Kambhampati, J. Phys. Chem. Lett., 2013, 4, 3316–3318 CrossRef CAS
-
J. B. Birks, Organic Molecular Photophysics, Wiley, 1973 Search PubMed
- R. D. Pensack, R. J. Ashmore, A. L. Paoletta and G. D. Scholes, J. Phys. Chem. C, 2018, 122, 21004–21017 CrossRef CAS
- P. Makula, M. Pacia and M. Wojciech, J. Phys. Chem. Lett., 2018, 9, 6814–6817 CrossRef CAS PubMed
- J. Ferguson, J. Chem. Phys., 1958, 28, 765–768 CrossRef CAS
- M. K. Chaudhuri and S. C. Ganguly, J. Phys. C: Solid State Phys., 1970, 3, 1791–1796 CrossRef CAS
- P. Kubelka and F. Munk, Z. Tech. Phys., 1931, 12, 593–601 Search PubMed
- X. Feng, X. Wang, C. Redshaw and B. Z. Tang, Chem. Soc. Rev., 2023, 52, 6715–6753 RSC
- B. Manna and D. K. Palit, J. Phys. Chem. C, 2020, 124, 24470–24487 CrossRef CAS
- A. P. Piquette, M. E. Hannah and K. C. Mishra, ECS Trans., 2012, 41, 1–9 CrossRef CAS
- A. Camerman and J. Trotter, Acta Crystallogr., 1965, 18, 636–643 CrossRef CAS
-
A. Köhler and H. Bässler, Electronic Processes in Organic Semiconductors, WILEY-VCH, 2015 Search PubMed
- R. Seyfang, H. Port, P. Fischer and H. C. Wolf, J. Lumin., 1992, 51, 197–208 CrossRef CAS
- M. Gerhard, J. Hausch, N. Hofeditz, J. Bredehöft, K. Broch and F. Schreiber, J. Phys. Chem. C, 2024, 128, 13926–13936 CrossRef CAS
- B. Walker, H. Port and H. C. Wolf, Chem. Phys., 1985, 92, 177–185 CrossRef CAS
- R. Canton-Vitoria, S. Nufer, X. Che, Y. Sayed-Ahmad-Baraza, R. Arenal, C. Bittencourt, A. Brunton, A. B. Dalton, C. P. Ewels and N. Tagmatarchis, Mater. Adv., 2020, 1, 2459–2466 RSC
- M. R. Molas, K. Nogajewski, A. O. Slobodeniuk, J. Binder, M. Bartos and M. Potemski, Nanoscale, 2017, 9, 13128–13141 RSC
- H. Sahoo, J. Photochem. Photobiol., C, 2011, 12, 20–30 CrossRef CAS
- Y. Garcia-Basabe, G. G. Parra, M. B. Barioni, C. D. Mendoza, F. C. Vicentin and D. G. Larrudé, Phys. Chem. Chem. Phys., 2019, 21, 23521–23532 RSC
- Y. Liu, H. Li, C. Qiu, X. Hu and D. Liu, Nano Res., 2020, 13, 661–666 CrossRef CAS
- A. Sillen and Y. Engelborghs, Photochem. Photobiol., 1998, 67, 475–486 CrossRef CAS
- O. V. Mikhnenko, P. W. M. Blom and T. Q. Nguyen, Energy Environ. Sci., 2015, 8, 1867–1888 RSC
- R. S. Swathi and K. L. Sebastian, J. Chem. Sci., 2009, 121, 777–787 CrossRef CAS
- D. Prasai, A. R. Klots, A. Newaz, J. S. Niezgoda, N. J. Orfield, C. A. Escobar, A. Wynn, A. Efimov, G. K. Jennings, S. J. Rosenthal and K. I. Bolotin, Nano Lett., 2015, 15, 4374–4380 CrossRef CAS PubMed
- K. Kolata, T. Breuer, G. Witte and S. Chatterjee, ACS Nano, 2014, 8, 7377–7383 CrossRef CAS PubMed
- H. Stegemeyer, J. Hasse and W. Laarhoven, Chem. Phys. Lett., 1987, 137, 516–520 CrossRef CAS
- J. J. P. Thompson, V. Lumsargis, M. Feierabend, Q. Zhao, K. Wang, L. Dou, L. Huang and E. Malic, Nanoscale, 2023, 15, 1730–1738 RSC
- J. E. Zimmermann, M. Axt, F. Mooshammer, P. Nagler, C. Schüller, T. Korn, U. Höfer and G. Mette, ACS Nano, 2021, 15, 14725–14731 CrossRef CAS PubMed
- X. Zhu, N. R. Monahan, Z. Gong, H. Zhu, K. W. Williams and C. A. Nelson, J. Am. Chem. Soc., 2015, 137, 8313–8320 CrossRef CAS PubMed
- D. Schmitt, J. P. Bange, W. Bennecke, A. AlMutairi, G. Meneghini, K. Watanabe, T. Taniguchi, D. Steil, D. R. Luke, R. T. Weitz, S. Steil, G. S. M. Jansen, S. Brem, E. Malic, S. Hofmann, M. Reutzel and S. Mathias, Nature, 2022, 608, 499–503 CrossRef CAS PubMed
- K. Tran, G. Moody, F. Wu, X. Lu, J. Choi, K. Kim, A. Rai, D. A. Sanchez, J. Quan, A. Singh, J. Embley, A. Zepeda, M. Campbell, T. Autry, T. Taniguchi, K. Watanabe, N. Lu, S. K. Banerjee, K. L. Silverman, S. Kim, E. Tutuc, L. Yang, A. H. Macdonald and X. Li, Nature, 2019, 567, 71–75 CrossRef CAS PubMed
- A. T. Hanbicki, K. M. McCreary, G. Kioseoglou, M. Currie, C. S. Hellberg, A. L. Friedman and B. T. Jonker, AIP Adv., 2016, 6, 055804 CrossRef
- R. Ai, X. Cui, Y. Li and X. Zhuo, Nano-Micro Lett., 2025, 17, 104 CrossRef PubMed
- A. Usman, M. Adel Aly, H. Masenda, J. J. P. Thompson, S. M. Gunasekera, M. Mucha-Kruczyński, S. Brem, E. Malic and M. Koch, Nanoscale, 2022, 14, 10851–10861 RSC
- M. M. Glazov, F. Dirnberger, V. M. Menon, T. Taniguchi, K. Watanabe, D. Bougeard, J. D. Ziegler and A. Chernikov, Phys. Rev. B, 2022, 106, 125303 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4lf00351a |
| This journal is © The Royal Society of Chemistry 2025 |