
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA biosensor-integrated filtration device for nanoparticle isolation and label-free imaging†
Leyang
Liu
 ab,
Takhmina
Ayupova
bc,
Saurabh
Umrao
bde,
Lucas D.
Akin
bd,
Han-Keun
Lee
ab,
Joseph
Tibbs
bc,
Xing
Wang
bcde,
Utkan
Demirci
*f and
Brian T.
Cunningham
*abcdeg
ab,
Takhmina
Ayupova
bc,
Saurabh
Umrao
bde,
Lucas D.
Akin
bd,
Han-Keun
Lee
ab,
Joseph
Tibbs
bc,
Xing
Wang
bcde,
Utkan
Demirci
*f and
Brian T.
Cunningham
*abcdeg
aDepartment of Electrical and Computer Engineering, University of Illinois at Urbana-Champaign, Urbana, IL 61801, USA. E-mail: bcunning@illinois.edu
bNick Holonyak Jr. Micro and Nanotechnology Laboratory, University of Illinois at Urbana-Champaign, Urbana, IL 61801, USA
cDepartment of Bioengineering, University of Illinois at Urbana-Champaign, Urbana, IL 61801, USA
dDepartment of Chemistry, University of Illinois at Urbana-Champaign, Urbana, IL 61801, USA
eCarl R. Woese Institute for Genomic Biology, University of Illinois at Urbana-Champaign, Urbana, IL 61801, USA
fBioAcoustic MEMS in Medicine BAMM Laboratory, Canary Center at Stanford for Cancer Early Detection, Stanford School of Medicine, Department of Radiology, Stanford University, Palo Alto, CA 94304, USA. E-mail: utkan@stanford.edu
gCancer Center at Illinois, Urbana, IL 61801, USA
First published on 14th March 2025
Abstract
Rapid, efficient, simple approaches for biological nanoparticle recovery from bodily fluids are required for translating detection strategies from lab diagnostics to low-resource settings, where expensive sample processing instruments such as an ultracentrifuge are not accessible. In this work, we characterize an alternative approach in which intact nanoparticles are filtered from plasma with a nanoporous filtration device that separates particulates within a 100–200 nm diameter range followed by detection on a photonic crystal (PC) biosensor with a portable photonic resonator interferometric scattering microscopy (PRISM) instrument. The biosensor-integrated recovery device's (BIRD) collection efficiency is initially characterized using gold nanoparticles and fluorescent nanobeads suspended in buffer solution and plasma, followed by spiking intact HIV pseudovirus into the same media. We demonstrate a recovery rate of 55.0% for 100 nm diameter AuNP and HIV spiked into the buffer and 11.9% for 100 nm diameter FluoSpheres spiked in human plasma. Using PRISM, we observed the Brownian motion of filtered nanoparticles and virions eluted into the detection compartment, with concentration-dependent counting of transient contact events between the nanoparticles and the PC surface.
Introduction
Sample purification is the first of many steps required for effective biosensing applications, where the initial sample obtained from a patient undergoes multiple stages of interfering substance removal and analyte recovery.1 Common analyte recovery and enrichment procedures involve using expensive instruments and require trained professionals, which is unsuitable for low-resource settings. For example, the isolation of exosomes, which are nanometer-sized (30–180 nm) extracellular vesicles (EVs),2–4 are generally conducted using ultracentrifugation (UC) in the research community,5,6 despite being costly, labor-intensive, time-consuming. As they contain important biomarkers for tumor-specific proteins7 and cancer progression,8,9 exosomes have received great research interest in their purification, recovery, and enrichment from various bodily fluids. Such methods, including multistep filtration,10 immuno-magnetic bead capture,5 acoustic separation,11 and lateral displacement,12 provide alternatives to UC, but at the cost of low throughput and undesired recovery rate. Another example of sample purification is HIV (∼110 nm)13 recovery from human blood, where various interfering biological substances will inevitably reduce the specificity and sensitivity of the downstream detection and analysis.14,15 Low detection limits and high sensitivity are vital to early HIV diagnosis and are traditionally accomplished by nucleic acid-based viral load (VL) testing16 in combination with centrifugation and viral RNA extraction. These VL testing strategies are limited to laboratory use due to complex sample preparation, precise temperature control, and the need for highly trained personnel.17–19 Novel HIV detection strategies, for example, piezoelectric20 and electrochemical-based21 biosensing, also require sophisticated preliminary sample treatment steps such as filtration and precipitation. Therefore, there is a need for a rapid, high-recovery, and simple approach for nanoparticle (NP) isolation from the complex sample matrix, providing an alternative route toward effective biosensing applications in low-resource settings.We report a filtration device that employs a sieving filtration approach, in which nanoporous membrane filters capture target particles based on their size. This technique has been previously applied to isolate extracellular vehicles from plasma,22 urine,23 and virions from whole blood.24 For instance, the exosome total isolation chip technology isolates EVs from plasma using a multi-layer poly(methyl methacrylate) (PMMA) main body, combined with a nanoporous polycarbonate membrane filter (30 nm pore size).22 This device achieves >90% recovery of EVs from culture media and outperforms traditional isolation methods, such as ultracentrifugation. Similarly, the Exodisc technology is a lab-on-a-disc platform that uses a polycarbonate body, one polycarbonate nanoporous membrane filter (600 nm pore size), and one anodized aluminum oxide (AAO) nanoporous membrane filter (20 nm pore size) to recover EVs from urine.23 Exodisc can achieve >95% recovery of EVs from cell culture supernatant through a spinning motion. Additionally, sieving filtration has been used to isolate virions from blood samples with a multi-layer PMMA device and a 2 μm pore-size polycarbonate membrane filter, achieving >75% recovery efficiency from HIV-spiked whole blood.24 Despite these successes, most of these techniques are hindered by long processing times or the need for specialized equipment, limiting their suitability for point-of-care (POC) applications. In this work, we design and characterize the performance of a multi-filter microfluidic cartridge that separates NPs from buffer solution or plasma in the 100–200 nm diameter range, with a rapid 15-minute processing time and operation requiring only a syringe pump. The cartridge is designed to provide optical access to the PC biosensor through transparent windows to facilitate the detection and counting of NPs by a portable photonic resonator interferometric scattering (PRISM) instrument that was reported earlier.25–28 We term the cartridge the biosensor integrated nanoparticle recovery device (BIRD), and the filter pore size can be selected to facilitate pre-concentration of NPs of many materials and size ranges for downstream detection/characterization by a wide variety of sensing transducers or analysis techniques. Here, we demonstrate the effectiveness of the BIRD in the context of gold nanoparticles, polymer fluorescent nanobeads, inactivated HIV, and HIV pseudovirus.
The design and working principle of the BIRD are shown in Fig. 1. The starting sample (for example, plasma) is introduced to a collection cup and is drawn through the filters with pneumatic pressure applied through the pulling of a manual syringe. The device incorporates three membrane filters with decreasing pore sizes of 450 nm, 200 nm, and 100 nm. In the case of HIV isolation, a typical virion diameter is approximately 110 nm,29 resulting in capture upon the surface of the 100 nm pore-size membrane filter. Larger plasma components will be retained at the entrance side surface of the 450 nm or 200 nm pore size filters, while smaller molecules, such as free nucleic acid and protein, will pass through the 100 nm pore filter and flow to the waste reservoir. While this filtration scheme is widely used in exosome isolation,22,23 it has not been previously demonstrated for virus isolation from plasma. Moreover, unlike other filtering schemes that require a separate pre-filtration step,22 the 450 nm pore size prefilter is integrated inside BIRD to simplify the workflow. After the sample is drawn through the filters, the feed and filtrate ports are blocked with plugs to prevent sample interaction with the external environment. The elution port is then connected to a syringe filled with a wash buffer, and a syringe pump is used to push the material that resides in the gap between the 100 nm and 200 nm filters forward, and into the sensing compartments. Once the detection is finished, the stream carries the particles towards the waste port, where the biohazard-containing liquid is contained inside a waste reservoir. The BIRD is comprised of 3D-printed components for its body, commercially available nanoporous filter materials, acrylic double-sided adhesives, PC biosensors, and glass windows. The cost of a BIRD is $32.5 (Table. S1†) without the PC biosensor and can be further reduced with bulk manufacturing. The device utilizes leak-free Luer Lock fittings for the syringe, sample reservoir, and plug, and withstands internal pressures up to approximately −68.9 kPa. We ensured the sterility of the device by choosing sterile membrane filters and syringes, and the 3D printing post-processing steps eliminated potential contamination.
 | ||
| Fig. 1 Schematic diagram and the working principle of the BIRD. (a) Exploded diagram of the BIRD. Intact HIVs are isolated on filter 3, while free proteins, nucleic acid, and debris are filtered or washed through. (b) A photo of the BIRD. Four sensing compartments are prepared for the filtered samples (P1 & P2, filled with red dye) and negative controls (N1 & N2, filled with green dye). (c) A cross-sectional view of BIRD through the central line, showing how the O-rings are used to seal the inner chamber. | ||
We experimentally validated the performance of the BIRD with 100 nm AuNP, 100 nm fluorescent polystyrene beads, inactivated HIV, and HIV pseudovirus. We obtained a recovery rate of nearly 55.0% for AuNP and HIV spiked in PBS buffer, 11.9% for fluorescent beads in 0.03× diluted human plasma, 7.87% for HIV pseudovirus in 0.03× diluted human plasma, with a flow rate of 0.5 mL min−1 and a total sample volume of 10 mL across all experiments. The recovery result for HIV pseudovirus in plasma was verified by detection of HIV-specific nucleic acid sequence in the filtrate by RT-qPCR, and validation of AuNP and FluoroSphere nanoparticle recovery rates were measured by nanoparticle tracking analysis (NTA). In addition, filtered particles that are washed into the PC biosensor detection compartment could be visualized and quantified by PRISM. In this work, the eluted nanoparticles are not specifically captured upon the biosensor. Rather, we use PRISM to record the number of transient nanoparticle-PC contact events within a ten-second time window to simply show the relationship between initial nanoparticle (or virus) concentration in the starting test sample and the number of transient contact events. Using this simple nonspecific label-free detection approach for HIV virions, a detection limit of 1.01 × 107 virus per mL was observable.
Overall, we demonstrate that the BIRD provides a high recovery rate that is comparable to commercially available virus isolation kits, while the entire virus isolation and detection process is held within an enclosed system to minimize the potential for disease transmission from infected samples. When combined with label-free, single-step, rapid, room temperature direct counting of viruses by a portable PRISM instrument, the approach outlined in this report provides an effective potential alternative to conventional pathogen assays that require viral extraction, viral lysis, enzymatic amplification of nucleic acids, thermal cycles, and fluorescence-based detection.
Results and discussion
Working principle of BIRD
The BIRD is designed to simplify the isolation and detection of HIV from human plasma in research and clinical point-of-care settings in this context, but the device is adaptable to other applications such as exosome extraction. It employs sieving filtration, where a series of three membrane filters with decreasing pore sizes are used to remove interferents and isolate nanoparticles within a specific diameter range. The BIRD consists of multiple 3D-printed parts that are hand-assembled into one leak-proof cartridge (Fig. 1a). It features four ports that allow the user to manipulate the direction of liquid flow by blocking specific ports with a plug. Three filters are used in this study with the objective of virus purification from human plasma, namely a 450 nm-pore-size cellulose acetate (CA) membrane filter (filter 1 in Fig. 1a), a 200 nm-pore-size AAO membrane filter (filter 2 in Fig. 1a), and a 100 nm-pore-size AAO membrane filter (filter 3 in Fig. 1a). However, the BIRD is fully modular, and the user can install custom membrane filters to meet their isolation requirements. A PC biosensor and glass observing window are fixed to the main body of BIRD by laser-cut acrylic double-sided adhesive rectangular frames for sample liquid confinement. The filtered sample is transferred downstream into two sensing compartments that lie between the glass window and the PC biosensor. Two extra negative control compartments are used to ensure the specificity of the experiment. The fully assembled BIRD has a dimension of 40 × 90 × 50 mm, (Photo in Fig. 1b). We indicate the two sets of sensing compartments by filling the sample compartment with red dye (labeled N1 & N2) and the control compartment with green dye (P1 & P2) in Fig. 1b, demonstrating that there is no cross flow between the sample compartments and the control compartments.The virus isolation and detection protocol consist of the following three steps:
Step 1 filtration: unfiltered plasma samples enter the feed port and are processed by filter 1–3 (Fig. 2a). The liquid movement is driven by negative pressure created by pulling a syringe that is connected to the filtrate port at a constant flow rate. Filter 1 acts as a prefilter to eliminate larger particles and debris to prevent clogging of filter 2 by substantial volumes of plasma. Filters 2 and 3 act as the limits for the size exclusion filtration process. Since HIV has a mean diameter of approximately 110 nm,29 it will be retained on the top surface of filter 3. Smaller substances with a diameter of less than 100 nm will be washed through filter 3 into the filtrate port.
 | ||
| Fig. 2 Workflow of the BIRD filtration process. (a) Step 1 filtration: the sample runs through all three filters from top to bottom. (b) Step 2 elution: a stream of elution buffer carries the purified HIV virions into the sensing compartments. (c) Step 3 detection: HIV virion is imaged and counted by PRISM. | ||
Step 2 elution: once the viruses are isolated on filter 3, a stream of elution buffer is injected into the elution port to carry the viruses into the sensing compartment (Fig. 2b). During this process, both the feed port and the filtrate port are blocked, and the waste port is opened.
Step 3 detection: the BIRD is transferred to the PRISM instrument for imaging and digital counting (Fig. 2c). The PRISM imaging observes one single field of view (FOV) at a frame rate of approximately 190 frame per second for a ten-second measurement duration once the free HIVs are carried into the compartments. The image processing script will generate a count per second rate of transient virus-to-PC surface contact events to represent the comparative concentration of the stock solution. A video demonstrating the isolation of 100 nm AuNP from the buffer can be found in Video S1.†
A key aspect of the BIRD design is ensuring a leak-proof inner chamber while preventing the sample liquid from bypassing any membrane filter layers. Fig. 1c provides a cross-sectional view through the center of the BIRD along its long edge, illustrating how each component integrates with the main body. Additional engineering details, including inner channels and dimensions, are shown in Fig. S1.† To ensure unidirectional liquid flow and a secure air seal, we used 1.5 mm thick O-rings with various inner diameters to seal the contact surfaces. For instance, an O-ring secures filter 3 to the main body, aided by a retainer that supports the brittle AAO filter and a tightener that applies pressure, as depicted in Fig. 1c. The retainer's radial pattern supports the membrane while allowing liquid to flow through its openings. The tightener, operated with a custom wrench, functions similarly to locking a portafilter on an espresso machine. As the liquid moves downward toward the filtrate port, it must pass through filter 3, enabling effective interaction between the nanoporous filter and the target nanoparticles.
Working principle of PRISM
Previous publications describe the working principle and detection performance of the PRISM instrument,26,27 and a portable version of the instrument.30 The portable PRISM is implemented as a desktop-based system that does not require stringent vibration and stray light control, compared to other precision microscopies. The total cost of the system is slightly under $20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 as reported earlier and could serve as a detection instrument in a clinic or a diagnostic facility. As this work focuses on developing biosensing methods for detecting HIV in low-resource settings, all biosensing experiments were conducted using the portable PRISM. Since our published reports,28,30 we further improved the portable PRISM to increase imaging quality and signal stability. The optical setup used in this work is shown in Fig. 3a. Specifically, a fiber-coupled laser diode replaces the uncoupled laser diode as the illumination source, a high-precision axial motorized stage is added to the setup, and a smaller enclosure (22 × 22 × 16 in3) is designed for housing the instrument. These upgrades reduce illumination speckles, increase focal accuracy, and decrease the system's overall footprint.
000 as reported earlier and could serve as a detection instrument in a clinic or a diagnostic facility. As this work focuses on developing biosensing methods for detecting HIV in low-resource settings, all biosensing experiments were conducted using the portable PRISM. Since our published reports,28,30 we further improved the portable PRISM to increase imaging quality and signal stability. The optical setup used in this work is shown in Fig. 3a. Specifically, a fiber-coupled laser diode replaces the uncoupled laser diode as the illumination source, a high-precision axial motorized stage is added to the setup, and a smaller enclosure (22 × 22 × 16 in3) is designed for housing the instrument. These upgrades reduce illumination speckles, increase focal accuracy, and decrease the system's overall footprint.
 | ||
| Fig. 3 Schematic diagrams of PRISM and SEM images of the nanostructures. (a) Schematic diagrams of the optical components and light paths of PRISM. The red and blue beams represent illumination and scattering light paths, respectively. (b) Magnified view near the focal plane of PRISM. Due to the resonance of the PC, most of the illumination light is reflected, leaving only 0.5% transmittance. The weakly transmitted light interferes with the scattered light, forming an interferometric image at the sensor. (c–f) SEM images of the PC nanostructure, the 450 nm-pore-size CA membrane filter, the 200 nm-pore-size AAO membrane filter, and the 100 nm-pore-size AAO membrane filter. | ||
The working principle of PRISM can be briefly summarized as follows: the illumination light (633 nm) reaches the PC biosensor after polarization tuning and is significantly (>99.5%) reflected due to the PC's guided mode resonance.31 The nanoparticles on the PC's top surface scatter a portion of the incoming photons. The forward scattered photons interfere with the weakly (<0.5%) transmitted light from the laser to form an interferometric image at the camera (Fig. 3b). The calculated magnification is 55.6× and the physical size of the field of view (FOV) is 57.9 × 57.9 μm2 measured with a USAF resolution target under a partial pixel utilization (1024 × 1024 pixels). To image nanoparticles inside BIRD, the device is placed on an aluminum custom-machined holding fixture that sits between two objective lenses. The PC's dielectric grating nanostructure (Fig. 3c) is critical to the performance of PRISM and enhancement of the interferometric signal, and the working mechanisms are described in our previous reports.27,30
Membrane filter characterization
For our application of isolating HIVs from human plasma samples, we identified three suitable filters, namely 450 nm-pore-size CA filter (Fig. 3d), 200 nm-pore-size AAO filter (Fig. 3e), and 100 nm-pore-size AAO filter (Fig. 3f). The large pore-size CA filter acts as a prefiltration medium to remove larger substances, such as protein aggregates and cell debris. Its performance was characterized by feeding BIRD with human plasma and taking scanning electron microscope (SEM) images of the CA filter before and after the filtration. As shown in Fig. 4a, the CA filter becomes clogged after processing approximately 0.3 mL of human plasma compared to the clean CA filter. Nanoparticle-tracking analysis also indicates that the prefiltration process removes particles that are larger than 450 nm and some particles in the size range of 150 to 450 nm (Fig. 4b). The multiple peaks presented in the NTA data are partially caused by analysis defects for polydisperse samples that consist of NPs with a size distribution spanning over a few hundreds of nanometers. To test the performance of the two AAO filters, which are used for size exclusion due to their well-defined pore sizes, we feed a mixture of 100 nm and 400 nm AuNPs with equal concentration (∼108 particles per mL) into BIRD (Fig. 4c) and took SEM images of the filters. Gold nanoparticles are suitable for filter performance evaluation because of their consistent dimensions, chemical inactivity, and lack of pathogenicity compared to HIV. Moreover, the 100 nm AuNPs have a size that resembles that of HIVs and can be used to substitute the viruses for preliminary testing without considerations for biosafety restrictions. The SEM image (Fig. 4c) suggests that most of the 400 nm AuNPs are intercepted by the 200 nm AAO filter, as indicated by red arrows. However, we note that some 100 nm AuNPs are also found inside the pores of the 200 nm AAO filter, representing a loss mechanism responsible for the non-unity recovery rate. The blue arrows in the SEM image of the 100 nm AAO filter indicate 100 nm AuNPs retained on the surface of the filter. We also performed NTA on the two AAO filters individually, and this was accomplished by feeding a mixture of different sizes of AuNPs into a BIRD with only the respective AAO filter installed. We evaluate the size exclusion performance of the 200 nm AAO filter by comparing the filtrate of the mixture of 100 nm and 400 nm AuNPs with the unfiltered mixture (Fig. 4d). For polydisperse samples with large size differences, the NTA results are slightly shifted, with the peak centered around 350 nm indicates the 400 nm AuNP population. The 400 nm AuNP population is greatly reduced after the filtration (Fig. 4d), suggesting the effective removal of particles sized greater than the AAO filter pore size, 200 nm. A mixture of 40 nm and 100 nm AuNPs is used for the NTA of the 100 nm AAO filter, and the eluted retentate is compared with the reference sample. The results in Fig. 4e indicate that only the 100 nm AuNPs are eluted, while the 40 nm AuNPs pass the filter and enter the filtrate port. Another loss mechanism is that a portion of the 100 nm AuNPs can be retained inside the pores (Fig. S2†) or pass through the filter completely. Ideally, the pore size of the filtering material should be at least two times smaller than the size of the particle of interest for effective filtration and recovery of targeted particles. We finalized our membrane filter selection based on the limited choices of commercially available high-performance filters and the complexity of the human plasma environment. A more detailed discussion of the filter material selection can be found in the ESI.†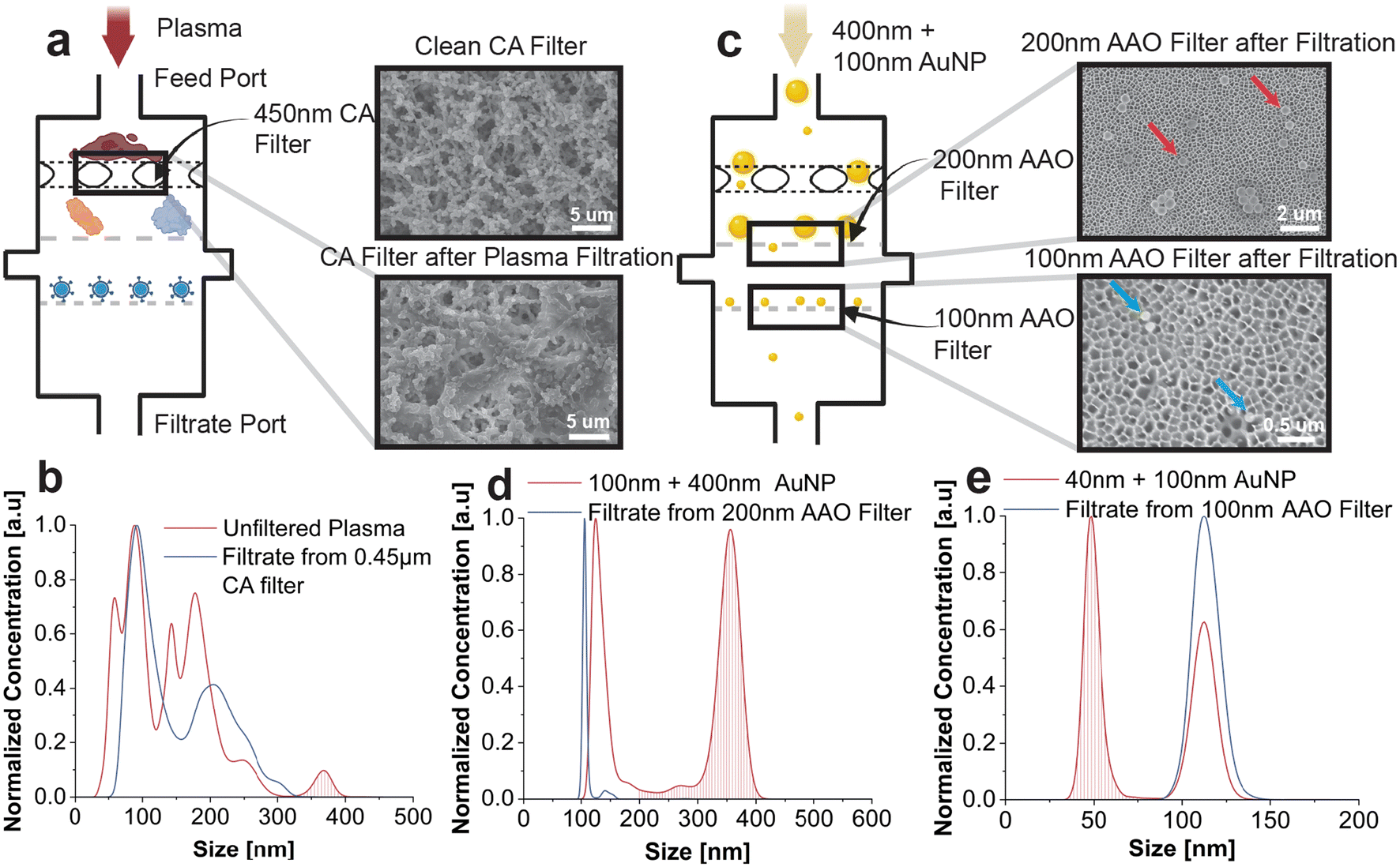 | ||
| Fig. 4 Membrane filter characterization. (a) Performance tests of the 450 nm-pore-size CA membrane filter with human plasma. Two SEM images before and after filtering the plasma sample indicate that the CA membrane removes large interferents. (b) Performance tests of the 200 nm-pore-size and 100 nm-pore-size AAO membrane filters with a mixture of 400 nm and 100 nm-diameter AuNPs. Two SEM images of the two AAO membrane filters with red arrows indicating 400 nm AuNPs and blue arrows indicating 100 nm AuNPs. (c–e) NTA data that evaluates the size exclusion performance of the three filters. The filtered portion of the size distribution is highlighted with drop lines. | ||
BIRD characterization
Once the design and the filter selection were finalized, we initialized the device characterization to evaluate its performance at different operation settings. One of the most critical parameters for a point-of-care filtration device is its flow rate, which directly relates to how rapidly the sample is processed. However, an excessively high flow rate may lead to increased pressure inside the device because forced convection created by the syringe dominates the movement of the nanoparticles and further causes particle caking on the membrane filter surface. To determine the optimal operating flow rate, we measured the negative pressure (vacuum) inside BIRD with a digital differential manometer, using a syringe pump to control the pull rate. The vacuum is measured at the filtrate port and is compared to atmospheric pressure to generate a relative readout (measurement setup shown in Fig. S5†), and the maximum vacuum that BIRD can withstand is approximately −68.9 kPa. A control sample of DI water was used as a reference and the resulting vacuum readout converged to only −1.25 kPa due to the lack of particulates in the medium. The goal of measuring the vacuum over different syringe pull rates is to investigate the relationship between pore availability and flow rate, as a faster flow rate tends to cause NP clogging issues. We started with a sample volume of 5 mL of 0.1× diluted human plasma and operated BIRD at different pull rates while recording the vacuum readings. Considering the ∼3 mL dead volume inside BIRD, the total pull volume is 8 mL for the syringe pump. As the curves in Fig. 5a suggest, the vacuum reaches a maximum of −24.4 kPa for the pull rates of 1, 0.5, and 0.25 mL min−1 after 8 mL of pull volume. This negative pressure is small compared to the busting pressure of typical AAO filters;32,33 however, we would still like to minimize the pressure to reduce the likelihood of damaging intact virions. The envelope of HIVs is comprised of lipid bilayers and is known to deform under force,34 causing virions to either rupture or squeeze through the pores of the membrane filter. In either case, we would expect fewer HIVs to recover, reducing the performance of the subsequent detection step. We then tried a pull rate of 0.1 mL min−1 with the same amount of diluted human plasma sample, and the measured maximum vacuum reduced significantly to −9.73 kPa. However, a slow pull rate will inevitably extend the filtration period; in this case, 80 minutes is needed to process 5 mL of sample. There is, therefore, a tradeoff between the syringe pump pull rate, recovery rate, and the total filtered plasma volume.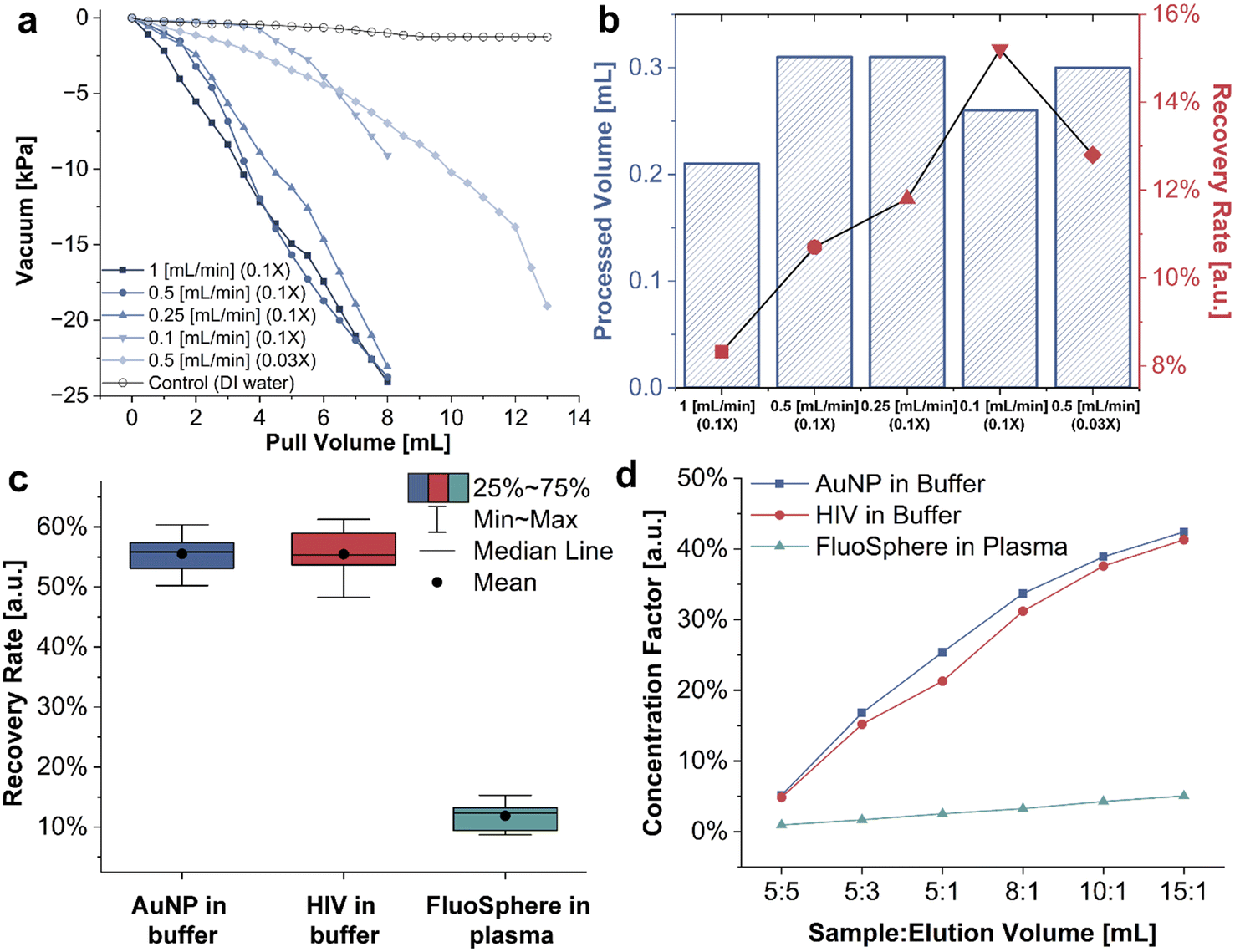 | ||
| Fig. 5 BIRD characterization. (a) Vacuum tests at different operating flow rates and dilution factors. The blue curves indicate 0.1× diluted plasma and the red curve indicates 0.03× diluted plasma. (b) The pre-diluted plasma volume that BIRD can process (blue bars) and the corresponding recovery rate (red points) of different operating flow rates and dilution factors. The recovery rate is calculated with 100 nm FluoSpheres in diluted human plasma, and it is comparable to the green box in Fig. 5c. (c) The recovery rates of 100 nm AuNP in buffer, intact HIV in buffer, and 100 nm FluoSpheres in diluted human plasma. (d) The concentration factors (compared to undiluted samples) of 100 nm AuNP in buffer, intact HIV in buffer, and 100 nm FluoSpheres in diluted human plasma at different sample-to-elution volume ratios. | ||
We determine the concentration of the filtered nanoparticles using NTA, which is a dark-field microscope with particle tracking algorithms and pre-calibrated concentration calculations. Because NTA cannot distinguish a virus from similarly sized bio-substances in plasma (for example, EVs), we use fluorescence-tagged polystyrene beads (FluoSpheres) of 100 nm diameter to simulate HIV in human plasma. It is important to note, however, that the size uniformity, deformability, and zeta potential of FluoSpheres are not identical to HIV. We measured the filtered plasma volume and calculated the recovery rate of FluoSpheres from plasma under the different settings. FluoSpheres were selected as representative nanoparticles for characterizing the BIRD due to their size similarity to HIV, combined with their high signal-to-noise detection in the NTA instrument. The total filtered plasma volume before dilution ranged from 0.2 mL to 0.3 mL after the entire pull volume is processed by the syringe pump, with 1 mL min−1 pull rate yielding the least amount due to shortened interaction time between the filter and the sample, as indicated by the blue bars in Fig. 5b. Human plasma is rich in nanometer-scale biocomponents, such as plasma protein (<20 nm),35 lipoprotein (30–1000 nm),36 small EVs (20–200 nm),8 and large EVs (200–500 nm).6,8 Combined, they create obstacles to the isolation of HIVs by fouling the membrane filters prematurely. We observed a significant increase in negative pressure once approximately 0.35 mL of undiluted plasma was processed (Fig. S6†) and, therefore, we chose our final plasma sample volume to be 0.3 mL given the filter material diameter of 25 mm.
The recovery rates of FluoSpheres from diluted plasma samples are calculated as a ratio of the number of recovered particles to the number of total particles in the original sample. The recovery rate for different pull rates and dilution factors are shown in Fig. 5b as red points, where the highest observed recovery rate was 15.3%. Considering all the factors, we chose a middle point of 0.5 mL min−1 pull rate with 10 mL of 0.03× diluted human plasma sample. The larger sample volume and slower flow rate allow sufficient wetting to the membrane filters and increase the filtration quality.37
Once our sample volume and syringe pump pull rate were selected, we challenged BIRD with three different nanoparticles in various media, namely 100 nm AuNP in 0.01× PBS buffer, inactivated HIV in 1× PBS buffer, and 100 nm FluoSpheres in 0.03× human plasma diluted with 1× PBS buffer. Each combination was repeated ten times, and the recovery rates are plotted in Fig. 5c. The average recovery rate for the AuNP in buffer and HIV in buffer is approximately 55%. However, the distribution of HIV in the buffer is slightly wider due to the viruses' lack of uniformity in size. The recovery rate of FluoSpheres in diluted plasma is significantly lower than the other two, likely because the biological components of plasma compete with the FluoSpheres by fouling the membrane filter pores. To verify the results obtained with the NTA method, we also conducted RT-qPCR tests on HIV pseudovirus spiked in human plasma and obtained an average recovery rate of 7.87% over ten samples (Materials and methods). This number is slightly lower than, but still comparable to, the results for the FluoSpheres spiked in plasma. Uncertainties, such as pipette error, virus degradation due to freeze–thaw cycles, and size nonuniformity, could explain the mismatch between PCR and NTA results.
We also investigated the concentration factor of the three types of samples by altering the sample-to-elution volume ratio. The concentration factor is calculated as a ratio of the eluted concentration to the undiluted sample concentration. It is a good indicator of the concentration change after elution and helpful for determining the elution volume since the PC biosensor is sensitive to the concentration of the analyte, not the absolute number. For all three combinations, the sample is diluted 10-fold before feeding into BIRD, and the isolated particles are diluted into different volumes of elution buffer. From Fig. 5d, we can observe that the concentration of the particle of interest increases as the sample-to-elution ratio increases; however, the trend is not linear and tends to saturate for high sample-to-elution ratios. The diminishing marginal benefit suggests that the elution buffer volume should also increase for large sample volumes that introduce large quantities of isolated nanoparticles to support sufficient recovery.
The AuNP (citrate-capped) and FluoSpheres (carboxylate-modified) are selected to exhibit negative zeta potential under a pH value of 7.4. The particles are surrounded by negative charges, emulating the behavior of HIV and EVs in biological environments.38,39 The AAO membrane filter also demonstrates a negative zeta potential near neutral pH.40 Therefore, the adsorption between our testing NPs and the filter surface is not significant compared to other mechanisms that reduce the recovery rate. Although there is a disparity in mechanical rigidity between our selected NPs and that of biological NPs, the inner vacuum level was carefully selected during characterization to prevent the rupture of virions.
Comparison with other methods
The operating conditions and HIV recovery results of BIRD were compared with two other commercially available virus isolation kits, namely DynaBeads Intact Virus Enrichment and Intact Virus Precipitation Reagent. These kits are not designed for virus isolation from plasma; therefore, the comparison is performed with inactivated HIV spiked in 1× PBS buffer. The comparison detail is shown in Table 1, where BIRD demonstrated the highest recovery rate among the three with a minimum total time of 20 minutes. NTA suggests that the size distribution of the isolated viruses is similar between the three methods, with the main lobe centered around 100 nm (Fig. S7†). The BIRD requires the least amount of equipment, and the viruses are contained within the device for the entire isolation and detection steps, making it ideal for point-of-care diagnostic applications.| BIRD | DynaBeads Intact Virus Enrichment | Intact Virus Precipitation Reagent | |
|---|---|---|---|
| Recovery rate | 55.5% | 44.8% | 23.8% |
| Sample volume | 10 mL | 1 mL | 10 mL |
| Elution volume | 1 mL | 1 mL | 1 mL |
| Total time | 20 minutes | 4 hours | 2.5 hours (incubated overnight) |
| Equipment required | Syringe pump | Magnets, vortex mixer, pipettes | Refrigerator, vortex mixer, pipettes, and centrifuge |
| Are viruses contained during the process? | Yes | No | No |
POC PRISM detection of AuNP and HIV
To demonstrate the idea of detecting BIRD-isolated particles by PRISM, we experimented with 100 nm AuNPs and inactivated HIVs at various concentrations to obtain the dose–response curves. Ten milliliters of each sample were processed by BIRD, and the isolated particles were eluted with one milliliter of buffer. The buffer carries the particles into the sensing compartment, and the particles are free to move during imaging. We selected ten arbitrary locations for each concentration to record a ten-second PRISM video. The raw video was treated with a series of video processing algorithms, such as temporal moving window filtering, Gaussian smoothing, and Laplacian of Gaussian particle localization, to obtain an averaged particle count per FOV. A detailed description of our image processing algorithms is included in the Materials and methods section. Fig. 6a shows the calibration curves for AuNP and HIV detection events (in units of particles per FOV during the ten-second time window). As the sample concentration decreases, we observe a decreased particle count per FOV. The standard deviation for HIV is larger than that of AuNP due to its weaker PRISM contrast signal for HIV (Fig. 6b). Since the particles are free in solution, they encounter the PC surface randomly by Brownian motion, and there is no mechanism for selective recognition or capture. Particle counting events were observable for filtered HIVs from the buffer when the measured NTA concentration in the initial sample was 1 × 106 particles per mL, where we observed 1.27 ± 0.51 particles per FOV HIV-to-PC contact events in 10 seconds. We estimate the number of free particles per FOV to be approximately 5.53 for a sample concentration of 1 × 108 particles per mL through calculations (see ESI†). This number is within the same order of magnitude as the experimental data (7.52 ± 3.91 for 1 × 108 particles per mL). Discrepancies come from the recovery rate, estimated imaging volume, and nonlinear response of the detection algorithm. | ||
| Fig. 6 PRISM detection of 100 nm AuNP and HIV isolated by BIRD. (a) The calibration curve of the digitally detected AuNP (black) and inactivated HIV (red) at various pre-filtration concentrations. The FOV size is 57.9 × 57.9 μm2. (b) Exemplary processed images of isolated AuNP and HIV viruses at various concentrations obtained from PRISM. | ||
The limit of detection (LOD) is calculated using the following formula:
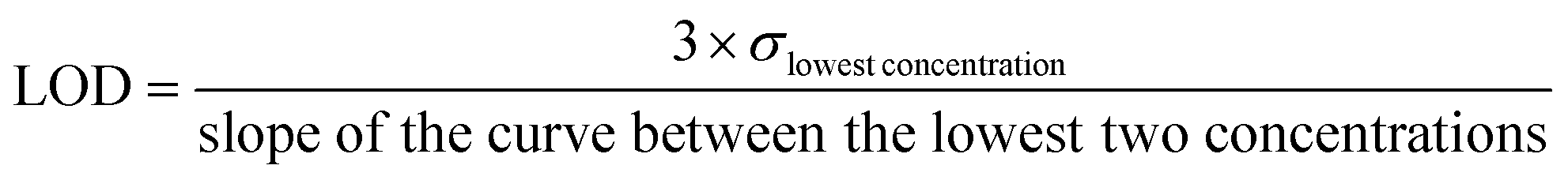 | (1) |
Conclusions
This work presents a nanoparticle isolation device based on sieving filtration for point-of-care diagnosis with simple but robust operation procedures. The BIRD can achieve a recovery rate of 55.0% for AuNP and HIV spiked in buffer, and an estimated recovery rate of 10% for HIV spiked in human plasma within 20 minutes without the need for specialized equipment. BIRD is superior to commercially available virus extraction kits as a consequence of its higher recovery rate, rapid protocol, low equipment requirement, and virus containment during the entire process. The device can be combined with multiple detection methodologies, and in this work, a combination of BIRD and PRISM is demonstrated to observe individual HIV with a LOD of 1.01 × 107 particles per mL. In this work, label-free detection of filtered HIV spiked into buffer was nonspecific, although the PRISM detection technology enabled us to observe the rate of HIV-to-PC contact events within a small ∼60 × 60 μm2 FOV. In future work, we plan to immobilize selective capture molecules upon the PC biosensor surface, such as aptamers and DNA “Nets”41 to convert virus-to-PC contact events into virus-to-PC capture events. Such a selective virus-capturing approach is extremely important, as plasma samples are known to hold a high density (∼1010 particle per mL (ref. 42)) of EVs that are similar in size to HIV and will be present in numbers much greater than HIV. Based upon the observed HIV-to-PC contact rate at an HIV concentration of 1 × 106 virus per mL, we can estimate a 16.76 virus per minute rate of contact events within the FOV when the virus concentration is reduced to 1 × 103 virus per mL. Our intended future strategy is to extend the incubation time to ∼30 minutes to provide greater opportunity for virus capture during Brownian motion in the detection compartment, while also tiling multiple (∼10–20) PRISM FOVs together for endpoint enumeration of virus, rather than the real time, single FOV approach used in this work. The successful particle isolation of BIRD paves the way toward achieving the goal of a simple, rapid, quantitative, and sensitive VL assay, unlocking numerous possibilities for point-of-care processing of human materials for early diagnosis and ART effectiveness monitoring.Materials and methods
BIRD fabrication
All parts of BIRD were fabricated with resin 3D printing (Form 3+; Formlabs, Somerville, MA, USA) using Clear Resin (Formlabs). The geometries of the entities were designed with computer-aided design software (SolidWorks, Waltham, MA, USA) to our specifications. Once the parts were printed, they were cleaned with isopropyl alcohol for 15 minutes to remove the excess resin on the outer surface (Form Wash; Formlabs). All parts were fully cured under ultraviolet light in Form Cure (Formlabs) at 60 °C for 15 minutes for better mechanical properties. O-rings (The O-Ring Store, Clarkston, WA, USA) of various dimensions were used to ensure the contact surfaces between each part were airtight and prevent liquid leakage. Similar to fixing a portafilter to an espresso machine, we employed a turn-to-tight fixing scheme to secure the small parts to the main body. The membrane filters were also secured inside BIRD in similar fashion, where the 450 nm-pore-size CA filter (25 mm diameter; Sterlitech, Auburn, WA, USA) and the 200 nm-pore-size AAO filter (Anodisc, 25 mm diameter with support ring; Whatman, Maidstone, UK) were installed from the top opening and the 100 nm-pore-size AAO filter (Anodisc, 25 mm diameter with support ring; Whatman) was installed from the bottom opening. The PC biosensor was fabricated by Moxtek (Orem, UT, USA) to our design specifications in rectangular pieces (10.2 × 12.7 × 0.7 mm3). The glass cover glass (18 × 18 mm2) (Fisherbrand Square Cover Glasses; Thermo Fisher Scientific, Waltham, MA, USA) and the PC biosensor were attached to the BIRD's main body with acrylic double-sided adhesives (VHB; 3M, Saint Paul, MN, USA). The syringes (10 mL Fisherbrand Sterile Syringes for Single Use; Thermo Fisher Scientific) used in this experiment have female Luer Lock connections and can be easily connected to BIRD's male Luer Lock ports.Device operation
The assembled BIRD was held upright by fixing the syringe to a syringe pump (NE-1600; New Era Pump Systems, Farmingdale, NY, USA). Ten milliliters of sample liquid were transferred to the sample cup connected to the feed port. We adopted a flow rate of 0.5 mL min−1 for our applications, considering the recovery rate and operation time. During the vacuum tests, a monometer (Fisherbrand Traceable Manometer; Thermo Fisher Scientific) was connected to the elution port via tubing during step 1 filtration, giving us a readout of the vacuum inside BIRD. Step 2 elution requires switching ports; the feed port and the filtrate port were blocked, and the elution port and waste port were opened. One milliliter of elution buffer is injected into the elution port, carrying the isolated particles toward the sensing compartment. These liquid manipulations were performed outside the PRISM instrument for the first two steps. Once BIRD is ready for biosensing, it is transferred inside PRISM for imaging. Ten FOVs were saved for the sample, and the data was saved as individual frames for downstream analysis.NTA
The concentration of AuNPs, intact HIVs, and fluorospheres was determined with an NTA system (Nanosight NS300; Malvern Instruments, Malvern, UK). The standard continuous flow protocol was adopted, and five repetitions were performed for each sample to reduce error.Conventional methods for HIV isolation
The HIV isolation performance of BIRD was compared with two conventional methods available on the market. The first method was to precipitate suspended intact HIV from the sample using the Intact Virus Precipitation Reagent (Invitrogen, Thermo Fisher Scientific). Inactivated HIVs were suspended in 10 mL 1× phosphate-buffered saline (PBS) and mixed with 5 mL of Intact Virus Precipitation Reagent. The mixed solution was vortexed and then incubated at 5 °C overnight. The sample was centrifuged at 4000 × g at 5 °C for 30 minutes, and the supernatant was aspirated and discarded, leaving only the precipitated intact virus. The viruses were then resuspended with 1× PBS to a volume of 1 mL.The second method was intact HIV isolation via affinity-based Dynabeads (Invitrogen, Thermo Fisher Scientific). A hundred microliters of Dynabeads were resuspended in 2 mL of wash buffer (10 mM NaCl + 20 mM triethanolamine, pH 6) and were mixed thoroughly by vortexing. The tube containing the Dynabeads was transferred to a magnetic stand, and the beads were attracted to the bottom of the tube. After 1 minute, the supernatant was removed. This washing step was repeated once again. Inactivated intact HIVs were transferred into the tube containing washed beads. The tube was then placed on a rotating shaker that rotates at 15 RPM for 10 minutes at room temperature. Once the viruses bonded to the Dynabeads, the tube was transferred to a magnetic stand for 1 minute, and the supernatant was removed. One milliliter of washing buffer was used to resuspend the beads, and the washing step was repeated once more. Two hundred microliters of release buffer (250 mM KI + 20 mM triethanolamine·HCl, pH 6) were injected into the tube containing the Dynabeads with viruses. The tube was then placed on a rotating shaker that rotates at 20 RPM for 10 minutes at room temperature. After mixing, the tube was transferred to a magnetic stand and allowed to rest for 1 minute. The supernatant containing isolated virus particles was aspirated into a new tube and diluted with 1× PBS to a total volume of 1 mL.
Sample preparation
Inactivated HIVs (NATrol intact HIV-1; Zeptometrix, Buffalo, NY, USA) were spiked into diluted human plasma (with 1× PBS) (Pooled Human Plasma; Innovative Research, Novi, MI, USA) or 1× PBS for different trials. Fluorospheres (F8803; Invitrogen, Thermo Fisher Scientific) were used to obtain recovery results in plasma samples and were mixed with diluted human plasma (with 1× PBS) to reach an initial concentration of ∼108 particles per mL. Gold nanoparticles (100 nm Stabilized Gold Nanoparticles, 1OD; Cyto Diagnostics, Burlington, ON, Canada) were diluted with 0.05× PBS to reach an initial concentration of ∼108 particles per mL. HIV-1 pseudovirus (PV) was used for RT-qPCR results for their genome integrity compared to inactivated HIV. PVs are artificial viruses built upon lentivirus cores with the same surface protein expression as the actual viruses.RT-qPCR
To quantify PV, we first thawed the HIV-1 PV samples with the appropriate standard (VR-3245SD, ATCC) at 4 °C. We sterilized all working surfaces and pre-chilled the centrifuge to 4 °C. Next, we transferred 100 μL of each PV sample into RNase-free microfuge tubes and added 750 μL of cold Trizol-LS solution (Cat No. 10296010, Thermo Fisher) and 150 μL of ultrapure water. We briefly vortexed each sample to mix, then incubated it at room temperature for 5 minutes. Following this, we added 200 μL of chloroform, vortex to mix, incubated for 3 minutes, and centrifuged at 12000g for 15 minutes at 4 °C. We carefully transferred 350 μL of the aqueous phase to a chilled tube, added 600 μL of isopropyl alcohol, and incubated for 10 minutes before centrifuging again at 12000g. After removing the supernatant, we added 750 μL of 75% ethanol and repeated centrifugation. Following the removal of the ethanol supernatant, we air-dried the samples, then added 50 μL of ultrapure water and dissolved the pellet by vortexing, followed by incubation at 55 °C. For reverse transcription, we prepared a master mix containing ultrapure water, RT buffer, random primers, RNase inhibitor, dNTP Mix, and Multiscribe Reverse Transcriptase (Cat No. K1622, Thermo Fisher). We added the master mix to each sample in a PCR plate and ran the thermocycler with a protocol of 25 °C, 37 °C, 85 °C, and 4 °C. Next, we prepared the TaqMan probe master mix (Cat No. 4304437, Thermo Fisher), set up the qPCR thermocycler (Quantstudio 3, Applied Biosystems), and added sample replicates to a 96-well plate. Finally, we analyzed the data against the standard using the delta–delta CT method, ensuring consistency in freeze–thaw cycles and RNA integrity throughout the protocol. The CT value-derived concentration is used to calculate the recovery rate of BIRD.
Image processing and particle localization
In summary, a rolling-window averaging technique was applied to each frame temporally to eliminate the constant background while accounting for dynamic measurements. Each video consists of approximately 2500 consecutive frame images. The resulting video highlights the shot-noise-limited signals from the 3D motion of nanoparticles, making it suitable for single-particle localization. A Laplacian-of-Gaussian filter was subsequently employed to identify local minima, followed by a 2D Gaussian function fit to model the point spread function. The algorithm records the number of detected particles and divides the number by the number of frames in the image series to generate an averaged particle per FOV.Data availability
The data supporting this article has been included as part of the ESI.†Author contributions
Leyang Liu: conceptualization, data curation, formal analysis, investigation, methodology, visualization, and writing – original draft. Takhmina Ayupova: conceptualization, data curation, investigation, and writing – original draft. Saurabh Umrao: data curation, formal analysis, investigation, and writing – original draft. Lucas D. Akin: conceptualization, investigation, and writing – original draft. Han-Keun Lee: conceptualization, and methodology. Joseph Tibbs: conceptualization. Xing Wang: conceptualization. Utkan Demirci: conceptualization. Brian T. Cunningham: conceptualization, funding acquisition, supervision, and writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for financial support from NIH (R01AI159454). J. T. acknowledges support from the NSF Graduate Research Fellowship. H.-K. L. acknowledges support from the Army GI Bill. The SEM and NTA results were partly carried out in the Material Research Laboratory Central Research Facilities, University of Illinois.References
- Y. Dai and C. C. Liu, Angew. Chem., Int. Ed., 2019, 58, 12355 CrossRef CAS PubMed.
- E. G. Trams, C. J. Lauter, N. Salem Jr. and U. Heine, Biochim. Biophys. Acta - Biomembr., 1981, 645, 63–70 CrossRef CAS PubMed.
- B. T. Pan, K. Teng, C. Wu, M. Adam and R. M. Johnstone, J. Cell Biol., 1985, 101, 942–948 CrossRef CAS PubMed.
- R. A. Dragovic, C. Gardiner, A. S. Brooks, D. S. Tannetta, D. J. P. Ferguson, P. Hole, B. Carr, C. W. G. Redman, A. L. Harris, P. J. Dobson, P. Harrison and I. L. Sargent, Nanomed.: Nanotechnol., Biol. Med., 2011, 7, 780–788 CrossRef CAS PubMed.
- C. Théry, S. Amigorena, G. Raposo and A. Clayton, Curr. Protoc. Cell Biol., 2006, 30, 3.22.1–3.22.29, DOI:10.1002/0471143030.cb0322s30.
- L. Muller, C.-S. Hong, D. B. Stolz, S. C. Watkins and T. L. Whiteside, J. Immunol. Methods, 2014, 411, 55–65 CrossRef CAS PubMed.
- H. Shao, J. Chung, L. Balaj, A. Charest, D. D. Bigner, B. S. Carter, F. H. Hochberg, X. O. Breakefield, R. Weissleder and H. Lee, Nat. Med., 2012, 18, 1835–1840 CrossRef CAS PubMed.
- G. Raposo and W. Stoorvogel, J. Cell Biol., 2013, 200, 373–383 CrossRef CAS PubMed.
- K. C. Vickers, B. T. Palmisano, B. M. Shoucri, R. D. Shamburek and A. T. Remaley, Nat. Cell Biol., 2011, 13, 423–433 CrossRef CAS PubMed.
- M. L. Heinemann, M. Ilmer, L. P. Silva, D. H. Hawke, A. Recio, M. A. Vorontsova, E. Alt and J. Vykoukal, J. Chromatogr. A, 2014, 1371, 125–135 CrossRef CAS PubMed.
- K. Lee, H. Shao, R. Weissleder and H. Lee, ACS Nano, 2015, 9, 2321–2327 CrossRef CAS PubMed.
- B. H. Wunsch, J. T. Smith, S. M. Gifford, C. Wang, M. Brink, R. L. Bruce, R. H. Austin, G. Stolovitzky and Y. Astier, Nat. Nanotechnol., 2016, 11, 936–940 CrossRef CAS PubMed.
- Y. Zhuo, H. Hu, W. Chen, M. Lu, L. Tian, H. Yu, K. D. Long, E. Chow, W. P. King, S. Singamaneni and B. T. Cunningham, Analyst, 2014, 139, 1007–1015 RSC.
- J. K. Cornett and T. J. Kirn, Clin. Infect. Dis., 2013, 57, 712–718 CrossRef PubMed.
- M. S. Cohen, C. L. Gay, M. P. Busch and F. M. Hecht, Braz. J. Infect. Dis., 2010, 202, S270–S277 CrossRef PubMed.
- E. D. Weld, J. Appl. Lab. Med., 2020, 6, 324–326 CrossRef PubMed.
- G. L. Damhorst, C. Duarte-Guevara, W. Chen, T. Ghonge, B. T. Cunningham and R. Bashir, Engineering, 2015, 1, 324–335 CrossRef PubMed.
- E. Valera, A. Jankelow, J. Lim, V. Kindratenko, A. Ganguli, K. White, J. Kumar and R. Bashir, ACS Nano, 2021, 15, 7899–7906 CrossRef CAS PubMed.
- A. M. Jankelow, H. Lee, W. Wang, T.-H. Hoang, A. Bacon, F. Sun, S. Chae, V. Kindratenko, K. Koprowski, R. A. Stavins, D. D. Ceriani, Z. W. Engelder, W. P. King, M. N. Do, R. Bashir, E. Valera and B. T. Cunningham, Analyst, 2022, 147, 3838–3853 RSC.
- F. Narita, Z. Wang, H. Kurita, Z. Li, Y. Shi, Y. Jia and C. Soutis, Adv. Mater., 2021, 33, 2005448 CrossRef CAS PubMed.
- M. A. M. Rodrigo, Z. Heger, N. Cernei, A. M. J. Jinemez, O. Zitka, V. Adam and R. Kizek, Int. J. Electrochem. Sci., 2014, 9, 3449–3457 CrossRef.
- F. Liu, O. Vermesh, V. Mani, T. J. Ge, S. J. Madsen, A. Sabour, E.-C. Hsu, G. Gowrishankar, M. Kanada, J. V. Jokerst, R. G. Sierra, E. Chang, K. Lau, K. Sridhar, A. Bermudez, S. J. Pitteri, T. Stoyanova, R. Sinclair, V. S. Nair, S. S. Gambhir and U. Demirci, ACS Nano, 2017, 11, 10712–10723 CrossRef CAS PubMed.
- H.-K. Woo, V. Sunkara, J. Park, T.-H. Kim, J.-R. Han, C.-J. Kim, H.-I. Choi, Y.-K. Kim and Y.-K. Cho, ACS Nano, 2017, 11, 1360–1370 CrossRef CAS PubMed.
- S. Wang, D. Sarenac, M. H. Chen, S. H. Huang, F. F. Giguel, D. R. Kuritzkes and U. Demirci, Int. J. Nanomed., 2012, 7, 5019–5028 Search PubMed.
- S. Ghosh, N. Li, Y. Xiong, Y.-G. Ju, M. P. Rathslag, E. G. Onal, E. Falkiewicz, M. Kohli and B. T. Cunningham, Biomed. Opt. Express, 2021, 12, 4637–4650 CrossRef PubMed.
- N. Li, X. Wang, J. Tibbs, C. Che, A. S. Peinetti, B. Zhao, L. Liu, P. Barya, L. Cooper, L. Rong, X. Wang, Y. Lu and B. T. Cunningham, J. Am. Chem. Soc., 2022, 144, 1498–1502 CrossRef CAS PubMed.
- N. Li, T. D. Canady, Q. Huang, X. Wang, G. A. Fried and B. T. Cunningham, Nat. Commun., 2021, 12, 1744 CrossRef CAS PubMed.
- L. Liu, S. Bhaskar and B. T. Cunningham, Appl. Phys. Lett., 2024, 124(23), 234101 CrossRef CAS.
- P. Zhu, J. Liu, J. Bess, E. Chertova, J. D. Lifson, H. Grisé, G. A. Ofek, K. A. Taylor and K. H. Roux, Nature, 2006, 441, 847–852 CrossRef CAS PubMed.
- L. Liu, J. Tibbs, N. Li, A. Bacon, S. Shepherd, H. Lee, N. Chauhan, U. Demirci, X. Wang and B. T. Cunningham, Biosens. Bioelectron., 2023, 228, 115197 CrossRef CAS PubMed.
- V. Karagodsky, F. G. Sedgwick and C. J. Chang-Hasnain, Opt. Express, 2010, 18, 16973–16988 CrossRef PubMed.
- S. Manzoor, F. Qasim, M. W. Ashraf, S. Tayyaba, N. Tariq, A. L. Herrera-May and E. Delgado-Alvarado, Sensors, 2023, 23(24), 9792 CrossRef CAS PubMed.
- Aminullah, A. K. Kasi, J. K. Kasi and M. Bokhari, Microelectron. Eng., 2018, 187–188, 95–100 CrossRef CAS.
- W. A. Haseltine, FASEB J., 1991, 5, 2349–2360 CrossRef CAS PubMed.
- R. J. Benjamin and L. S. McLaughlin, Transfusion, 2012, 52, 9S–19S CAS.
- K. Bai, G. V. Barnett, S. R. Kar and T. K. Das, Pharm. Res., 2017, 34, 800–808 CrossRef CAS PubMed.
- T. Sparks and G. Chase, in Filters and Filtration Handbook, ed. T. Sparks and G. Chase, Butterworth-Heinemann, Oxford, 6th edn, 2016, pp. 383–413, DOI:10.1016/B978-0-08-099396-6.00012-5.
- N. Arjmandi, W. Van Roy, L. Lagae and G. Borghs, Anal. Chem., 2012, 84, 8490–8496 CrossRef CAS PubMed.
- G. Midekessa, K. Godakumara, J. Ord, J. Viil, F. Lättekivi, K. Dissanayake, S. Kopanchuk, A. Rinken, A. Andronowska, S. Bhattacharjee, T. Rinken and A. Fazeli, ACS Omega, 2020, 5, 16701–16710 CrossRef CAS PubMed.
- B. J. Pedimonte, T. Moest, T. Luxbacher, C. von Wilmowsky, T. Fey, K. A. Schlegel and P. Greil, Acta Biomater., 2014, 10, 968–974 CrossRef CAS PubMed.
- H. Lee, W. Wang, N. Chauhan, Y. Xiong, N. Magazine, O. Valdescruz, D. Y. Kim, T. Qiu, W. Huang, X. Wang and B. Cunningham, Rapid, sensitive detection of intact SARS-CoV-2 using DNA nets and a smartphone-linked fluorimeter, SPIE, 2023 Search PubMed.
- K. B. Johnsen, J. M. Gudbergsson, T. L. Andresen and J. B. Simonsen, Biochim. Biophys. Acta - Rev. Cancer, 2019, 1871, 109–116 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5lc00089k |
| This journal is © The Royal Society of Chemistry 2025 |