
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMicrofluidic loading of verteporfin into extracellular vesicles for neuroblastoma therapy†
Caterina
Piunti
 ab,
Sara
Micheli
ab,
Sara
Giancaterino
c,
Pina
Fusco
b,
Cristiana
Boi
c and
Elisa
Cimetta
*ab
ab,
Sara
Micheli
ab,
Sara
Giancaterino
c,
Pina
Fusco
b,
Cristiana
Boi
c and
Elisa
Cimetta
*ab
aDepartment of Industrial Engineering, University of Padua, Padua, Italy. E-mail: elisa.cimetta@unipd.it
bFondazione Istituto di Ricerca Pediatrica Città della Speranza, Padua, Italy
cDepartment of Civil, Chemical, Environmental, and Materials Engineering, University of Bologna, Bologna, Italy
First published on 26th February 2025
Abstract
Despite contributing to cancer progression, extracellular vesicles (EVs) could serve as potential drug delivery systems in cancer treatment, having the ability to dissolve water-insoluble drugs and facilitate targeted delivery. However, the clinical translation of EVs is still in its infancy. While traditional methods for EV modifications will remain relevant, microfluidic approaches are expected to replace benchtop methods. Taking advantage of lab-on-chip devices, passive cargo loading through microfluidic mixing and incubation may be an important strategy to produce functional engineered EVs. This study focuses on developing a microfluidic device to generate EVs loaded with verteporfin (VP), a hydrophobic porphyrin with potential applications in neuroblastoma (NB) therapy, aiming to enhance its therapeutic effectiveness. The platform ensures perfect mixing and tunable incubation time for mesenchymal stem cell-derived EVs and VP, demonstrating a significantly higher loading efficiency than traditional methods, while operating under gentle conditions that preserve EV integrity and functionality, unlike other microfluidic techniques that involve harsh mechanical or chemical treatments. The VP-loaded EVs (VP-EVs) can then be easily recovered, making them available for subsequent analysis and use. MTT assay confirmed that VP-EVs are more efficient than free VP in reducing the viability of a NB cell line. Finally, immunofluorescence assay and western blot demonstrated a greater reduction in YAP expression after treatment with VP-EVs in an NB cell line when compared to free VP. Being both non-destructive and straightforward, this microfluidic loading technique facilitates its adaptability to a wide spectrum of therapeutic compounds. As a versatile tool, microfluidic technology will help to fully unlock the potential of EVs for speeding up precision medicine and disease treatment.
Introduction
Effective drug delivery systems (DDSs) offer significant potential for advancing cancer treatment by enabling precise delivery of therapeutics to target sites while preserving active compounds. However, challenges such as rapid clearance, poor bioavailability, and unspecific cytotoxicity continue to limit their therapeutic efficacy.1Extracellular vesicles (EVs), nano-sized particles (30–4000 nm, including exosomes, microvesicles, and apoptotic bodies) of endocytic origin, have emerged as promising candidates for drug delivery thanks to their natural role in the intercellular exchange of biomolecules.2 Their therapeutic benefits, particularly in cancer therapy, stem from their ability to cross biological barriers such as the blood–brain barrier, and their intrinsic and target-specific capabilities.3 EVs can naturally reach the tumor microenvironment via enhanced permeability and retention effects, while also protecting biological cargo from degradation in vivo.4 EVs can be purified from various sources, including mammalian and prokaryotic cell cultures, blood plasma, milk, and plants.5 Among these, mesenchymal stem cell-derived EVs (MSC-EVs) are particularly notable for their immunomodulatory properties and regenerative potential, making them highly attractive for cancer therapy.6 Due to their heterogeneity and involvement in various physiological and pathological processes, MSC-EVs have been investigated for applications across a wide range of tissue types and are already under clinical assessment.7
Modern engineering techniques such as exogenous drug loading can further enhance EVs' therapeutic potential.8 The encapsulation of exogenous cargo into EVs is typically achieved through methods such as passive incubation, electroporation, or physical and chemical treatments like freeze–thaw cycles, extrusion, and membrane permeabilizers. These methods, while widely used, often result in low efficiency, structural damage to EVs, and variability across batches.9
Addressing these challenges, microfluidic platforms offer a novel, precise, and reproducible approach for engineering drug-loaded EVs.10 By handling small volumes, microfluidic systems enable continuous and parallel fluid processing, leading to improved automation, steady-state operation, higher throughput, enhanced efficiency, reduced experimental times, and lower overall costs.11 These advancements significantly broadened EVs potential for clinical applications as therapeutic delivery systems.12
In line with these advancements, this study presents the design and development of a microfluidic platform to load MSC-EVs with verteporfin (VP), an FDA-approved drug with anti-cancer properties that acts via inhibition of the YAP/TAZ signaling pathway. While VP shows promise in treating aggressive cancers such as neuroblastoma (NB), its clinical utility has been limited by off-target effects and non-specific toxicity.13
The design of the microfluidic platform for generating drug-loaded EVs was based on the procedure published by Fuhrmann et al., where porphyrins of different hydrophobicities were employed as model drugs and encapsulated into EVs using various passive and active methods (electroporation, saponin, and extrusion).14 Active methods can lead to EV or cargo aggregation, altering the EVs' physicochemical and morphological characteristics. On the other hand, passive methods, e.g. simple incubation of EVs with drugs, result in very low loading efficiency. The microfluidic device was designed to enhance passive loading efficiency by microfluidic mixing and incubation. By integrating AutoCAD®-designed geometries and COMSOL Multiphysics® simulations, our platform achieved perfect mixing and controlled incubation of EVs with VP, resulting in significantly improved loading efficiency compared to traditional methods. Additionally, unlike other published microfluidic approaches that often rely on harsh procedures such as mechanical compression, fluid shear stress, or surfactants, our platform employs passive mixing under gentle conditions, preserving the structural integrity and functionality of EVs.15 The resulting VP-loaded EVs (VP-EVs) demonstrated significantly enhanced therapeutic efficiency compared to free VP, measured by reduced NB cell viability and lower YAP expression levels.
Beyond its immediate application, this versatile microfluidic platform holds great promise for loading diverse therapeutic agents into various EV types, positioning it as a transformative tool for advancing cancer nanomedicine.
Experimental design
Device design and simulation
The microfluidic platform was designed using AutoCAD® in both 2D and 3D environments. The device, characterized by a Y-type inlet channel, consisted of two sections: a mixing unit, where the two fluids entering the platform were homogeneously mixed, and an incubation unit designed to guarantee a residence time of 10 minutes. Once injected through the inlets, the two fluids merged into a 200 μm wide and 1.7 mm long channel connecting to the mixing unit. The micromixer had V-shaped obstacles with a 122° angle and arms 240 μm and 200 μm long. The incubation unit comprised 700 μm wide and 264 mm long delay-lines. The simulation of fluid flow within the device was carried out using COMSOL® Multiphysics and aimed at predicting the mixing between EVs and VP within the mixing unit and ensuring a precise incubation time of 10 minutes.The built-in physics modules were laminar flow to describe fluid motion inside the channels, and the transport of a diluted species to simulate the convective/diffusive behavior.16 The velocity term was calculated by coupling both physics. Key parameters defined included a temperature of 20 °C, the inlet flow rates at 1 μL min−1, outlet pressure at 0 relative (gauge), inflow normalized concentration set to 0 or 1, and diffusion coefficients DEVs = 5.10 10−12 m2 s−1 and DVP = 4.30 10−10 m2 s−1 for the EVs and VP, respectively (refer to S1† for details). A fine mesh with default elements was created (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 002415 elements; average quality: 0.5979) and both steady-state and time-dependent simulations were performed. In the latter case, the platform behavior was investigated between 0 and 30 minutes to verify the correct incubation time. Finally, the mixing index (η) was evaluated through a MATLAB® code (refer to S2† for details).17
002415 elements; average quality: 0.5979) and both steady-state and time-dependent simulations were performed. In the latter case, the platform behavior was investigated between 0 and 30 minutes to verify the correct incubation time. Finally, the mixing index (η) was evaluated through a MATLAB® code (refer to S2† for details).17
Materials and methods
Device fabrication and simulation
The master mold for the microfluidic device was produced through standard photolithography, as described previously.18 Briefly, a silicon wafer was coated with a 100 μm thick layer of SU-82100 (MicroChem) using a spin coater (SPS SPIN150). After baking at 95 °C to remove the solvent and release residual tension in the resin, the wafer was aligned with a photomask and exposed to UV light using a mask aligner. Subsequent baking facilitated the cross-linking of SU-8, and then the wafer was immersed in SU-8 developer. Polydimethylsiloxane (PDMS, Sylgard® 184, Dow Corning) was used for replica molding. In brief, PDMS was cast onto the SU-8 mold and cured at 70 °C for 1 hour. The PDMS replica was cut from the SU-8 mold, and holes for the inlets and the outlet were punched. Finally, plasma treatment (Harrick Plasma) was used to form an irreversible hydraulic seal to a glass slide (Corning™).Experimental fluid dynamic validation was carried out using both colored and fluorescent tracers. Food coloring solutions were used as colored tracers, and the flow was driven by a syringe pump (PHD Ultra, Harvard Apparatus) set to a flow rate of 1 μL min−1. The blue and yellow solutions were connected to separate inlets and injected simultaneously, providing a clear visual of the fluid behavior. Fluorescein isothiocyanate dextrans (Sigma Aldrich, Ex/Em 490/520 nm) were used as fluorescent tracers. A 500 kDa dextran was chosen for its similarity with VP's molecular weight of 718.8 kDa. The solution was prepared by dissolving dextrans in water at a concentration of 10−5 g mL−1. This setup enabled visualization of the platform's geometry under a fluorescent microscope (Invitrogen EVOS FL Cell Imaging System, Thermo Fisher Scientific), using a GFP fluorescence filter at 4× magnification.
Cell culture
Human bone marrow mesenchymal stem cells (MSCs, STEMCELL, #70071) were used as an EV source. MSCs were cultured in MesenCult™ medium (combination of MesenCult™ MSC basal medium and MesenCult™ MSC stimulatory supplement, STEMCELL, #05411) and used up to passage 8.19The SK-N-AS cell line derived from the bone marrow metastasis in a child with poorly differentiated embryonal NB was used as target cells for biological analyses. The SK-N-AS cell line (ATCC, CRL 2137) was grown in DMEM containing 10% FBS (both from ATCC), 2 mM glutamine, 100 U mL−1 penicillin/streptomycin (Life Technologies), and 1 × MEM non-essential amino acids (Biowest). Both cell lines were maintained at 37 °C and 5% CO2.
Isolation and characterization of mesenchymal stem cell-EVs
:100–1:1000) in PBS were analyzed with a Nanosight NS300 (Malvern Panalytical). Each measurement involved the capture of three 60-second videos. Experiments were repeated in triplicate and analyzed with the ZetaView analysis software (ZetaView 8.04.02 SP2). Data were expressed as the mean ± standard error of the mean (SEM).
:1000 in PBS were applied onto a 400-mesh holey film grid. Excess liquid was removed using filter paper (Whatman). Subsequently, the EVs were stained with 1% uranyl acetate for 2 minutes. For immunogold staining, the EVs were incubated with mouse anti-CD81 (1:50; ab59477, Abcam) for 30 minutes, followed by a 30-minute incubation with secondary anti-mouse 10 nm protein A-gold conjugates (Sigma Aldrich). The samples were then observed using a Tecnai G2 (FEI) transmission electron microscope operating at 100 kV, equipped with a Veleta CCD camera (Olympus Soft Imaging System).
The morphology of VP-EVs was analyzed by scanning electron microscopy (SEM), using a field emission gun instrument (Tescan Mira3). VP-EV samples were diluted 1:500 in PBS. A droplet of each EV suspension was deposited onto a 30 kDa PES membrane (5 × 5 mm) and attached with conductive carbon tape to SEM stubs. The samples were then fixed overnight at 4 °C in a solution of 2.5% glutaraldehyde (Sigma Aldrich) and 2% sucrose (Sigma Aldrich) in PBS. Subsequently, samples underwent triple rinsing with ultrapure water, followed by 10 minutes of incubation, and dehydrated using a gradient of ethanol–water solutions (35%, 50%, 70%, 90%, 100% ethanol in water), with each step incubated for 15 minutes. Final dehydration was achieved using pure hexamethyldisilazane (HDMS, Sigma Aldrich) with two sequential 15-minute incubations. Gold coating (thickness <10 nm) was applied using the electrodeposition method in a sputter coater (Quorum Q150R ES) to make the samples conductive. SEM analysis was conducted using a secondary electron (SE) detector with an accelerating voltage of 10 kV, beam intensity set to 3, and working distance of 10 mm.
Loading of verteporfin into EVs
VP (Tocris Biosciences, Mw = 718.79, Fig. S3†) was dissolved in dimethylsulfoxide (DMSO, Sigma Aldrich) at a stock concentration of 10 mM and stored at −20 °C before use. All manipulations and experiments involving VP were carried out protecting it from light. For off-chip loading of VP in EVs a passive incubation method was used. Briefly, VP (100 μM) and EVs (150 μg, corresponding to ≅109 particles) were mixed in a 1.5 mL centrifuge tube and incubated at room temperature for 10 min. For on-chip loading, fluid flow was controlled via a syringe pump (PHD Ultra, Harvard Apparatus). Two 1 mL syringes were used to dispense 0.5 mL of VP (100 μM) and 0.5 mL of EVs (150 μg, corresponding to ≅109 particles) into the inlets at a flow rate of 1 μL min−1 at room temperature. The mixture was collected at the outlet in a 1.5 mL centrifuge tube placed on ice to reduce passive diffusion, ensuring minimal post-loading interaction beyond the intended 10-minute incubation period, and to stabilize EV integrity.After loading, both off-chip and on-chip mixtures were ultrafiltered at 3000g for 10 min using a 100 kDa MWCO Spin-X 500 concentrator (Corning) to separate the VP-EVs from free VP via diafiltration. After ultrafiltration, the final VP-EV solution volume was adjusted back to the starting volume, maintaining the same total volume as the initial condition. The concentration of VP in EVs was indirectly determined using an ultra-violet visible scanning spectrophotometer (Tecan microplate reader Spark) at 690 nm. In brief, standard curves of absorbance vs. concentration were created for the range of 0–70 μM VP (Fig. S4†). Loading efficiency was calculated as the ratio of VP retained in EVs after ultrafiltration to the total VP initially added, expressed as a percentage,
| %Loading efficiency = (Loaded VP)/(Added VP) × 100. |
Cell viability
SK-N-AS cells were seeded in 96-well plates at a density of 1.5 × 104 cells per well, and exposed to increasing concentrations of naïve EVs (1, 100, and 1000 μg mL−1), and both free VP and VP-EVs (0.001–10 μM) for 48 hours. Cell viability was assessed through MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay (Invitrogen). Briefly, 10 μL of 5 mg mL−1 MTT reagent was added to the well and the plate was incubated for 4 h in a humidified 37 °C incubator. Then, 100 μL of DMSO was added to dissolve formazan crystals formed in the cells. The optical density at 540 nm was measured using a spectrophotometer micro-plate reader (Tecan microplate reader Spark). Cell viability was normalized using the following formula:| %Viability = (Sample − Negative control)/(Positive control − Negative control) × 100 |
Immunofluorescence and Immunoblotting
For immunofluorescence analysis, SK-N-AS cells were plated in an 8-well chamber slide at a density of 2.0 × 104 cells per well and treated with either VP or VP-EVs at 1 μM, or an equivalent amount of negative control (i.e., cells treated with either DMSO or EVs alone) for 24 h, fixed in 3.7% formaldehyde and permeabilized with 0.1% Triton-X. After blocking with 5% BSA in PBS, cells were incubated with primary rabbit polyclonal anti-YAP (GTX129151) overnight at 4 °C and secondary antibody Alexa Fluor 488 goat anti-mouse (115-547-185) at room temperature for 1 hour. Nucleus labeling was performed by incubating cells with DAPI (Invitrogen) for 15 min. Imaging was performed on a Zeiss LSM800 Airyscan. Image quantification using ImageJ (National Institutes of Health, Bethesda, MD, USA) measured the green areas of YAP expression. The reported YAP fluorescence intensities corresponded to the fluorescence intensities of 15 cells in at least 3 different images per condition, repeated in triplicate. Data were expressed as mean ± SEM.For immunoblotting analysis of YAP protein levels in SK-N-AS, cells were treated with either VP or VP-EVs at 1 μM at 37 °C for 3 h. Total cellular lysates were prepared by using lysis buffer (FNN0011, Invitrogen) supplemented with phosphatase inhibitor cocktail 2 (P5726, Sigma Aldrich), phosphatase inhibitor cocktail 3 (P044, Sigma Aldrich), protease inhibitor cocktail (Sigma Aldrich), and PMSF serine protease inhibitor (Sigma Aldrich). Based on the BCA estimate of protein concentrations, 10 μg of proteins were loaded on gels. Before loading, the protein samples were diluted in LDS sample buffer (Invitrogen) and heated at 70 °C for 10 min. The denatured samples were loaded alongside 5 μL of protein standard markers (LC5925, Invitrogen) on a Bolt 4–12% Bis-Tris Plus gel (Invitrogen). Then, gels were inserted into an electrophoresis chamber which was filled with SDS running buffer (B0002, Invitrogen). The proteins were fractionated by size at 200 V for approximately 30 min. After separation, proteins were transferred to a PVDF membrane using the same semi-dry blotting technique described for EVs. After the protein transfer step, membranes were blocked with I-Block reagent (Thermo Fisher) and incubated overnight at 4 °C with primary antibodies: rabbit polyclonal anti-YAP1 (GTX129151, Genetex); mouse monoclonal anti-Vinculin (ab130007, Abcam), as a loading control. Finally, the membranes were incubated with the appropriate peroxidase-conjugated secondary antibodies, goat anti-mouse (G-21040, Invitrogen) and goat anti-rabbit (G-21234, Invitrogen) at room temperature for 1 hour. Image acquisition was performed using Westar Hypernova ECL substrate (Cyanagen) and iBright western blot imaging systems (Invitrogen).
Statistical analysis
Graphs and statistical analyses were performed using GraphPad Prism software (GraphPad). Statistical significance was determined using Student's t-test. Asterisks indicate a significant difference between the treated and the control group, unless otherwise specified: **P < 0.01, ****P < 0.0001.Results and discussion
Characterization of mesenchymal stem cell-derived EVs
Once isolated from the conditioned medium (CM), the identity of MSC-EVs (EVs in short) was confirmed using NTA, immunogold TEM, SEM, and immunoblot analysis of EV protein markers according to the minimal criteria defined by the ISEV.21EV samples measured by NTA had a Dmode = 114.3 ± 7.7 nm (Fig. 1A) and mean particle concentration of 2.8 × 1010 ± 3.1 × 109 particles per mL. The mean protein concentration, measured by BCA, was 1.9 ± 0.2 mg mL−1. The ratio of particle number to protein amount was ∼107 particles per μg protein of EVs. The yield, determined calculating the number of particles per unit volume of the processed CM, was 1.3 × 108 particles per mL CM. TEM images of EVs show globular particles with a diameter of ∼100 nm as shown in Fig. 1B. Additionally, the gold nanoparticle, associated with CD81 and shown as a black dot, was associated with the isolated EV, confirming the nature of the vesicles. Immunoblot results also showed the presence of CD81 and CD63, chosen as target protein biomarkers, Alix and GAPDH, chosen as target cytosolic proteins, and the absence of cytochrome C, calnexin, and VEGF, providing additional information on possible intracellular origins of EVs or co-isolates (Fig. 1C). Additionally, EVs showed typical negative charges with a mean zeta potential of −11.4 ± 1.0 mV.
 | ||
| Fig. 1 Characterization of MSC-EVs. (A) Representative size distribution of EVs analyzed by NTA (EVs diluted 1:100). (B) Morphology was assessed by TEM. CD81 positive dots (arrow) of EVs by immunogold-TEM. Scale bar: 100 nm. (C) Representative western blot showing the presence of cell surface antigens CD81 and CD63 and cytosolic proteins Alix and GAPDH and the absence of calnexin, VEGF, and cytochrome C. | ||
Overall, these data indicate that EVs were successfully isolated from MSCs.
A simple microfluidic strategy for efficient verteporfin encapsulation in EVs
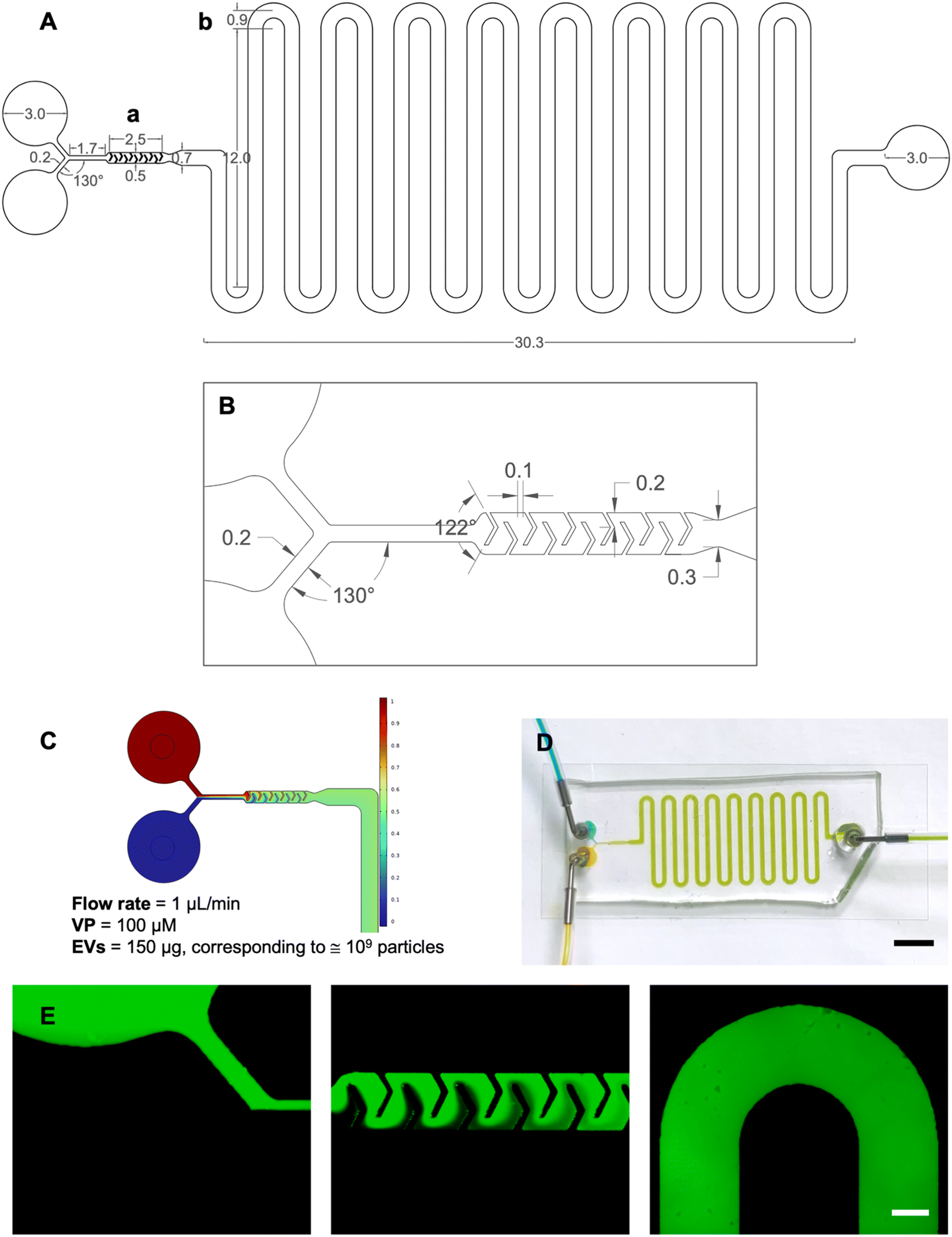 | ||
| Fig. 2 Design, simulation and fluid dynamic validation of the microfluidic device. (A) The platform, designed using AutoCAD®, was formed by two sections: the mixing unit (A-a) and the incubation (A-b) allowing precise residence times as a function of the imposed flow rate. Reported lengths are in mm. (B) Detail of the mixing unit. (C) COMSOL Multiphysics® concentration surface plot. (D) Fluid dynamic validation performed using colored tracers confirms both perfect mixing and incubation times. Scale bar: 1 cm. (E) Fluid dynamic validation performed using fluorescent isothiocyanate-dextrans. Scale bar: 0.4 mm. | ||
COMSOL Multiphysics was used to predict whether EVs and VP could be perfectly mixed (Fig. 2B) and that the correct incubation time required by the protocol was met (Fig. 2C). CFD simulations were conducted to model fluid behavior and mixing efficiency across different flow rates. The optimized flow rate (1 μL min−1) was determined through iterative simulations to achieve consistent and thorough mixing within the confined space. This rate aligned with the incubation period and ensured that the fluid pathways in the delay lines provided the necessary residence time for effective interaction. For the incubation unit, the length of the delay lines was determined based on the required residence time for effective interaction, the dimensions of the structure (height and width), and the flow rate at the micromixer outlet. The flow rate was obtained by using a tool that provides the average velocity across a specified plane.
To assess the effectiveness of mixing, the mixing index was calculated. The computed value was η ≅ 0.99, indicating near-complete mixing. For reference, a mixing index of 0 corresponds to entirely unmixed fluids, while a value of 1 represents fully mixed fluids.
Finally, time-dependent simulations verified the correct incubation times satisfying the protocol requirements.
After a successful modeling validation, the microfluidic device was fabricated via standard photolithography and replica molding in PDMS. The footprint of the device was 48 × 14.4 mm2 and the height was 100 μm.
The fluid dynamic validation was performed using both food coloring and dextrans with the syringe pump set to the desired 1.0 μL min−1 flow rate. After approximately 10 minutes, injecting yellow and blue solutions from the inlets resulted in the obtainment of green fluid at the outlet, demonstrating perfect mixing and incubation times consistent with COMSOL simulations (Fig. 2D). The video in S5† was recorded from time zero corresponding to the initiation of infusion. These observations supported the correct design aimed at microfluidic loading of VP into EVs ensuring perfect mixing and precise incubation times. The use of dextrans allowed for detailed visualization of the device's geometry, including the Y-type inlet channel, the micromixer, and a section of the incubation unit coil (Fig. 2E).
 | ||
| Fig. 3 Characterization of verteporfin-loaded extracellular vesicles (VP-EVs). (A) Representative size distribution of vesicles analyzed by NTA (VP-EVs diluted 1:100). (B) The morphology of VP-EVs was assessed by SEM. Scale bar: 500 nm. (C) Zeta Potential of EVs and VP-EVs. (D) Comparison of VP loading efficiency into EVs using standard off-chip passive loading and our on-chip method (****p < 0.0001). | ||
As mentioned, the loading efficiency of VP into EVs with standard incubation is relatively low.14 The loading efficiency calculated for our on-chip protocol was 37.9 ± 10.4%, significantly higher than the 3.5 ± 2.6% for standard incubation (Fig. 3D, n = 8 and 5, respectively). The platform loading efficiency was comparable to literature data reporting a 31–37% efficiency for doxorubicin (Dox) using mechanical compression and fluid shear stress.22 However, our platform better preserved the quality and integrity of the loaded EVs by combining the advantages of passive mixing without harsh mechanical or chemical treatments. Thakur et al. reported lower efficiencies of 8–32% using saponin for EV loading, further emphasizing the advantages of our method.23
EV-mediated delivery of verteporfin inhibits SK-N-AS cell proliferation
The impact of naïve EVs on the proliferation of NB cells was first assessed. SK-N-AS cells were exposed to varying concentrations of EVs (10, 100, and 1000 μg mL−1 equivalent to 103, 104 and 105 EVs per cell, respectively, where 104–105 is the estimated number of vesicles considered necessary to cover the surface of a cell24) for 48 hours before evaluating cell proliferation via MTT assay. The control groups were cultured in unconditioned medium preprocessed like CM. SK-N-AS cells treated with EVs had a statistically significant decrease in their growth compared to the control group, suggesting an intrinsic inhibitory effect of MSC-EVs on NB cell proliferation (Fig. 4A). | ||
| Fig. 4 VP-EVs reduce cell viability of a neuroblastoma cell line (SK-N-AS). (A) Cell viability was measured by MTT test after 48 h of incubation with different concentrations of mesenchymal stem cell-derived EVs. (B) Cell viability was measured by MTT test after 48 h of incubation with 1 μM VP and 1 μM VP-EVs, normalized on DMSO and EVs (at a concentration approximately corresponding to the amount of EVs present under the 1 μM VP-EV condition), respectively. Data are expressed as mean ± SEM (**p < 0.01; ****p < 0.0001). | ||
The effect of VP-EVs on the viability of NB cells was then examined. SK-N-AS cells were treated with different concentrations of VP and VP-EVs (ranging from 0.001 to 10 μM) and analyzed after 48 hours of incubation (Fig. 4B and S6†).
Viability data for VP-EVs and VP-treated samples were normalized using EVs (at a concentration approximately corresponding to the amount of EVs present under the respective VP-EV condition) and DMSO treatments as controls, respectively.
The findings revealed that the VP-EV treatment was more efficient in inhibiting SK-N-AS proliferation compared to free VP (p < 0.01). This result was also supported by the lower IC50 at 48 hours for VP-EVs (1.33 μM) compared to the IC50 of free VP (3.37 μM). Overall, these results suggest that the concentration of VP required to achieve 50% cell death was higher when free-VP treatment was used, implying a more targeted effect of the VP-loaded EVs. While a full cell viability analysis across the 0.001–10 μM range was not possible due to insufficient VP-EV stock concentrations from off-chip loading, the available data (n = 1 replicate) allowed for a preliminary comparison of relative treatment efficacy up to 1 μM VP. No significant differences were observed between VP-EVs loaded on-chip and off-chip (Fig. S6†), but further validation is needed. Based on these results, 1 μM VP concentration was chosen for further experiments.
EV-mediated delivery of verteporfin inhibits YAP expression in SK-N-AS cells
YAP expression was evaluated via immunofluorescence assay on SK-N-AS cells treated with either 1 μM free-VP or VP-EVs (Fig. 5). First, the correct cellular internalization of VP-EVs was assessed using confocal microscopy demonstrating that VP-EVs were mostly found in the cytoplasm of recipient cells (Fig. 5A). With VP identified as a YAP/TAZ complex inhibitor, the difference in YAP gene expression (green) in treated and untreated SK-N-AS cells was evaluated (Fig. 5B). A semi-quantitative image analysis of YAP signals using ImageJ proved that internalized VP-EVs more significantly reduced YAP expression in recipient cells compared to free VP (Fig. 5C). This result was further confirmed by western blot, with lower YAP expression in samples treated with VP-EVs than in VP alone after just 3 hours of treatment (Fig. 5D). These important findings supported the hypothesis that EVs are highly efficient drug carriers capable of delivering VP into cancer cells to favor the drug's activity more efficiently than the free drug. | ||
| Fig. 5 Extracellular vesicle (EV)-mediated delivery of verteporfin (VP) inhibits YAP expression in SK-N-AS cells. (A-a) Representative fluorescence microscopy images at different magnifications (40 and 100×) of SK-N-AS cell lines (nuclei in blue), after incubation with either VP (left, in red) or VP-EVs (right, in red) at 1 μM at 37 °C for 24 h and (A-b) stained for YAP (green) at 40× magnification. (B) The YAP green fluorescence intensities of SK-N-AS cell lines incubated with either VP or VP-EVs at 1 μM at 37 °C for 24 h are reported. Data are presented as means ± SEM (*** P < 0.001, **** P < 0.0001). (C) Western blot analysis of the protein level of YAP in the SK-N-AS cell line treated with either VP or VP-EVs at 1 μM at 37 °C for 3 h. Vinculin was detected as a loading control. The grouping of blots was cropped from different parts of the same gel. | ||
Conclusions
Microfluidic technologies, highly promising tools for a range of biomedical applications, offer several distinct advantages over traditional large-scale methods, including reduced reagent use, lower costs, high throughput, and precise control over experimental conditions. These attributes make microfluidics particularly suitable for the engineering of EVs, especially in the context of advancing precision medicine.The objective of this study was to design a microfluidic device capable of loading drugs into EVs while preserving their integrity and functionality. The potential of combining MSC-EVs with the hydrophobic porphyrin drug VP for cancer treatment through microfluidic mixing and incubation was explored.
The device ensured perfect mixing between EVs and VP within the mixing unit and the desired incubation times. The results demonstrated a significantly higher loading efficiency of VP in EVs compared to traditional incubation methods, highlighting the device's ability to improve and optimize the loading process. Furthermore, encapsulation of VP in EVs resulted in a significant increase in its therapeutic effect compared to free drugs, as evidenced by the greater inhibition of SK-N-AS cell proliferation and reduced expression of YAP.
Future research will focus on optimizing the device design to accommodate multiple drugs and different EV types, further proving its versatility.
For clinical applications, enhancing device scalability through parallelization strategies should be prioritized, as adapting microfluidic platforms for higher throughput, and conducting pilot-scale studies to ensure that key features such as uniform drug loading and EV integrity are maintained at larger scales. Addressing these areas will determine whether this promising microfluidic approach can transition from laboratory research to practical, scalable solutions for industrial production of therapeutics.
Currently, the described technology may be most applicable for personalized, bedside treatments where small-scale, patient-specific batches can be produced on demand. This approach would enable freshly prepared, tailored EV therapeutics, bypassing the complexities of large-scale manufacturing and storage logistics. Furthermore, incorporating on-chip purification systems would streamline post-loading processes, minimizing the time and effort required to prepare EV-based therapeutics.
The main limiting factor in this research was the low yield of EVs obtained via MSCs. Further studies need to address isolation scalability by using scalable sources (e.g., milk-derived EVs) and scalable isolation methods such as tangential flow filtration (TFF). Additionally, the protocol is optimized for hydrophobic molecules like VP, and it is hypothesized that its applicability may be limited for hydrophilic molecules, which would require more than simple passive loading for effective encapsulation. Finally, the absorption of hydrophobic molecules by PDMS should be investigated, as it restricts the usable concentration of these molecules. This issue may be particularly relevant for drugs other than VP. While this study demonstrates the feasibility of microfluidic-based VP loading into EVs, further investigations are needed to confirm the generalizability of this approach for other drug and EV combinations.
Finally, while the preservation of EV integrity post-loading was assessed, long-term stability studies and in vivo validations are necessary to fully evaluate the therapeutic efficacy of VP-EVs in clinically relevant models. Addressing these limitations in future studies will be essential to fully harness the potential of this microfluidic strategy for EV-based drug delivery.
In conclusion, the method outlined in this study presents a versatile and adaptable strategy for loading a hydrophobic therapeutic molecule into EVs. Its non-destructive nature enables its application with a wide range of EV types, thus enhancing its potential in drug delivery. Moreover, with its solvent-free and straightforward fabrication process, this technology holds promise for the production of drug-loaded EVs for clinical use.
Data availability
The raw data produced and analyzed in this study are available at https://researchdata.cab.unipd.it/1470/.Author contributions
Caterina Piunti: writing – original draft, visualization, methodology, investigation, formal analysis, data curation, conceptualization. Sara Micheli: writing – review & editing, methodology, investigation. Sara Giancaterino: writing – review & editing, investigation. Pina Fusco: formal analysis. Cristiana Boi: writing – review & editing, supervision. Elisa Cimetta: writing – review & editing, supervision, methodology, funding acquisition, conceptualization.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the ERC starting grant (ERC-StG) MICRONEX project (759467, PI E Cimetta). The authors also wish to thank Ing Francesco De Nicolo for his contribution in the preliminary design stages.References
- A. M. Vargason, A. C. Anselmo and S. Mitragotri, Nat. Biomed. Eng., 2021, 5, 951–967 CrossRef PubMed.
- C. Théry, L. Zitvogel and S. Amigorena, Nat. Rev. Immunol., 2002, 2, 569–579 CrossRef PubMed.
- L. Cheng and A. F. Hill, Nat. Rev. Drug Discovery, 2022, 21, 379–399 CrossRef CAS PubMed.
- S. Walker, S. Busatto, A. Pham, M. Tian, A. Suh, K. Carson, A. Quintero, M. Lafrence, H. Malik, M. X. Santana and J. Wolfram, Theranostics, 2019, 9, 8001–8017 CrossRef CAS PubMed.
- S. Giancaterino and C. Boi, Biotechnol. Adv., 2023, 63, 108092 Search PubMed.
- M. Kou, L. Huang, J. Yang, Z. Chiang, S. Chen, J. Liu, L. Guo, X. Zhang, X. Zhou, X. Xu, X. Yan, Y. Wang, J. Zhang, A. Xu, H. f. Tse and Q. Lian, Cell Death Dis., 2022, 13, 580 Search PubMed.
- I. K. Herrmann, M. J. A. Wood and G. Fuhrmann, Nat. Nanotechnol., 2021, 16, 748–759 Search PubMed.
- O. M. Elsharkasy, J. Z. Nordin, D. W. Hagey, O. G. De Jong, R. M. Schiffelers, S. E. Andaloussi and P. Vader, Adv. Drug Delivery Rev., 2020, 159, 332–343 CrossRef CAS PubMed.
- J. Wolfram and M. Ferrari, Nano Today, 2019, 25, 85–98 CrossRef CAS PubMed.
- C. Piunti and E. Cimetta, Biophys. Rev., 2023, 4, 31304 Search PubMed.
- S. Haeberle and R. Zengerle, Lab Chip, 2007, 7, 1094 Search PubMed.
- Q. Zhu, M. Heon, Z. Zhao and M. He, Lab Chip, 2018, 18, 1690–1703 Search PubMed.
- P. Fusco, E. Mattiuzzo, C. Frasson, G. Viola, E. Cimetta, M. R. Esposito and G. P. Tonini, Eur. J. Pharmacol., 2021, 893, 173829 CrossRef CAS PubMed.
- G. Fuhrmann, A. Serio, M. Mazo, R. Nair and M. M. Stevens, J. Controlled Release, 2015, 205, 35–44 CrossRef CAS PubMed.
- C. M. Abreu, B. Costa-Silva, R. L. Reis, S. C. Kundu and D. Caballero, Lab Chip, 2022, 22(6), 1093–1125 Search PubMed.
- S. Micheli, P. Mocellin, M. Sorgato, L. Bova and E. Cimetta, Biochem. Eng. J., 2022, 181, 108415 Search PubMed.
- S. Sarkar, K. K. Singh, V. Shankar and K. T. Shenoy, Chem. Eng. Process.: Process Intesif., 2014, 85, 227–240 CrossRef CAS.
- Y. Xia and G. M. Whitesides, Annu. Rev. Mater. Sci., 1998, 28, 153–184 CrossRef CAS.
- D. B. Patel, K. M. Gray, Y. Santharam, T. N. Lamichhane, K. M. Stroka and S. M. Jay, Bioeng. Transl. Med., 2017, 2, 170–179 CrossRef CAS PubMed.
- R. J. Lobb, M. Becker, S. W. Wen, C. S. F. Wong, A. P. Wiegmans, A. Leimgruber and A. Mö Ller, J. Extracell. Vesicles, 2015, 4, 27031 CrossRef PubMed.
- J. A. Welsh, D. C. I. Goberdhan, L. O'Driscoll, E. I. Buzas, C. Blenkiron, B. Bussolati, H. Cai, D. Di Vizio, T. A. P. Driedonks, U. Erdbrügger, J. M. Falcon-Perez, Q. L. Fu, A. F. Hill, M. Lenassi, S. K. Lim, M. G. Mahoney, S. Mohanty, A. Möller, R. Nieuwland, T. Ochiya and K. W. Witwer, J. Extracell. Vesicles, 2024, 13(2), e12404 Search PubMed.
- R. Hao, Z. Yu, J. Du, S. Hu, C. Yuan, H. Guo, Y. Zhang and H. Yang, Small, 2021, 17, 2102150 Search PubMed.
- A. Thakur, R. K. Sidu, H. Zou, M. K. Alam, M. Yang and Y. Lee, Int. J. Nanomed., 2020, 15, 8331–8343 CrossRef PubMed.
- G. Adamo, D. Fierli, D. P. Romancino, S. Picciotto, M. E. Barone, A. Aranyos, D. Božič, S. Morsbach, S. Raccosta, C. Stanly, C. Paganini, M. Gai, A. Cusimano, V. Martorana, R. Noto, R. Carrotta, F. Librizzi, L. Randazzo, R. Parkes, U. Capasso Palmiero, E. Rao, A. Paterna, P. Santonicola, A. Iglič, L. Corcuera, A. Kisslinger, E. D. Schiavi, G. L. Liguori, K. Landfester, V. Kralj-Iglič, P. Arosio, G. Pocsfalvi, N. Touzet, M. Manno and A. Bongiovanni, J. Extracell. Vesicles, 2021, 10, e12081 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4lc01103a |
| This journal is © The Royal Society of Chemistry 2025 |