
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Modelling of the multicellular tumor microenvironment of pancreatic ductal adenocarcinoma (PDAC) on a fit-for-purpose biochip for preclinical drug discovery†
Alina
Deipenbrock
a,
Ben Eric
Wilmes
a,
Thomas
Sommermann
b,
Nader
Abdo
b,
Kyra
Moustakas
a,
Martin
Raasch
b,
Knut
Rennert
b and
Nicole E.
Teusch
 *a
*a
aInstitute of Pharmaceutical Biology and Biotechnology, Heinrich-Heine-University Düsseldorf, Universitätsstr. 1, 40225 Düsseldorf, Germany. E-mail: Nicole.Teusch@hhu.de
bDynamic42 GmbH, Jena, Germany
First published on 28th February 2025
Abstract
Pancreatic ductal adenocarcinoma (PDAC) is the most common and lethal form of pancreatic cancer. One major cause for a fast disease progression is the presence of a highly fibrotic tumor microenvironment (TME) mainly composed of cancer-associated fibroblasts (CAF), and various immune cells, especially tumor-associated macrophages (TAM). To conclusively evaluate drug efficacy, it is crucial to develop in vitro models that can recapitulate the cross talk between tumor cells and the surrounding stroma. Here, we constructed a fit-for-purpose biochip platform which allows the integration of PDAC spheroids (composed of PANC-1 cells and pancreatic stellate cells (PSC)). Additionally, the chip design enables dynamic administration of drugs or immune cells via a layer of human umbilical vein endothelial cells (HUVEC). As a proof-of-concept for drug administration, vorinostat, an FDA-approved histone deacetylase inhibitor for cutaneous T cell lymphoma (CTCL), subjected via continuous flow for 72 h, resulted in a significantly reduced viability of PDAC spheroids without affecting vascular integrity. Furthermore, dynamic perfusion with peripheral mononuclear blood cells (PBMC)-derived monocytes resulted in an immune cell migration through the endothelium into the spheroids. After 72 h of infiltration, monocytes differentiated into macrophages which polarized into the M2 phenotype. The polarization into M2 macrophages persisted for at least 168 h, verified by expression of the M2 marker CD163 which increased from 72 h to 168 h, while the M1 markers CD86 and HLA-DR were significantly downregulated. Overall, the described spheroid-on-chip model allows the evaluation of novel therapeutic strategies by mimicking and targeting the complex TME of PDAC.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is a highly aggressive cancer type with a five-year survival rate of 13%.1–3 Early metastases, late diagnosis and therapy resistance are known factors of the low survival rates of patients with PDAC. To date, cytotoxic chemotherapy with the combinatory regimen FOLFIRINOX (leucovorin, 5-fluoruracil, irinotecan and oxaliplatin) or, if intolerable, the combination of gemcitabine and capecitabine represents the first line treatment for patients who are not eligible for surgical resection.4In PDAC, the tumor microenvironment (TME) is characterized by a large cellular heterogeneity. The main cell types within the tumor stroma include pancreatic stellate cells (PSC), and a variety of immune cells that form a highly fibrotic and immunosuppressive TME.5,6
During tumorigenesis, PSC convert into cancer-associated fibroblasts (CAF), which exhibit a myofibroblast-like phenotype and constitute approximately 90% of the TME.7,8 CAF secrete large amounts of extracellular matrix (ECM) components such as fibronectins, collagens and hyaluronic acid, responsible for the dense and stiff TME9 forming a mechanical barrier surrounding the tumor lesions.8,10,11 Additionally, activated PSC release factors like interleukin-6 (IL-6), interleukin-10 (IL-10), CXCL12 transforming growth factor-beta (TGF-β), macrophage colony-stimulating factor (M-CSF), and vascular endothelial growth factor (VEGF), which are described for their involvement in immunosuppression, epithelial-mesenchymal transition (EMT), or angiogenesis.12–14 For example, the release of M-CSF promotes the differentiation of monocytes into M2 macrophages.15 The initiation of angiogenesis through the release of proangiogenic factors like VEGF leads to the generation of atypical and multibranched vessels that, however, collapse under the high interstitial pressure within the PDAC TME, resulting in a leaky and dysfunctional vasculature which then ultimately leads to a hypovascular and hypoxic tumour phenotype.16 Importantly, a poor vascular supply also limits the delivery of chemotherapeutic agents into the tumor tissue.17 Overall, the highlighted relevance of CAF in the tumor-stroma crosstalk leading to a highly desmoplastic milieu on one hand, with limited drug delivery on the other hand, underlines the need for novel preclinical 3D TME models.6
In addition to CAF, the PDAC TME encompasses a variety of immune cell types and is abundantly infiltrated by tumor-associated macrophages (TAM). Macrophages are traditionally classified into two phenotypically and functionally distinct subsets: the immunogenic M1 phenotype and the immunosuppressive M2 phenotype. Recent research has highlighted the dynamic and versatile nature of TAM phenotypes18 and in the case of PDAC, TAM are mainly polarized towards a M2 phenotype (i.e. CD163+).19,20 In fact, a high infiltration of CD163+ macrophages is associated with a lower overall survival rate in PDAC patients.21,22 TAM contribute to tumor progression by enhancing both, chemoresistance and immunosuppression, as well as promoting acinar-to-ductal metaplasia through the secretion of tumor necrosis factor α (TNFα) and the activation of matrix metalloproteinases (MMPs).23–26 TAM macrophages release different mediators that lead to the activation of signaling pathways in tumor cells like PI3K/Akt, JAK/STAT or others subsequently contributing to chemoresistance.27–29 Studies in mice showed that depletion of macrophages or blocking macrophage recruitment via the CC-chemokine ligand 2 (CCL2) axis can restore the sensitivity for gemcitabine or FOLFIRINOX, confirming the important role for M2 macrophages with respect to current and future therapeutic strategies.30–32 Characteristic for their immunosuppressive behaviour, M2 macrophages secret anti-inflammatory cytokines and chemokines such as IL-10, IL-6, IL-1-RA, CXCL7 and CCL2 which promote exhaustion of cytotoxic CD8+ T cells on one hand24,33 and drive the differentiation of CD4+ T cells towards Th2, Th17 and regulatory T cells (Treg), widely known for their immunosuppressive and tumour promoting functions.34,35 Furthermore, TAM inhibit the anti-tumour activity of cytotoxic CD8+ T cells by expressing immune checkpoint inhibitor ligands, such as PD-L1 and Dectin-1, on their surface.36,37 Additionally, TAM induce the expression of these checkpoint inhibitors on tumor cells, directly contributing to the immune escape of cancer cells.38
In vitro modelling of the complex cellular interplay within the PDAC TME is technically and scientifically challenging as conventional two-dimensional (2D) tumor cell culture settings are inadequate in mimicking cell–cell and cell–matrix interactions, spatial orientation, physicochemical stiffness as well as nutrient and oxygen gradients.39 This limitation significantly reduces the prediction of therapeutic efficacy in 2D tumor cell monolayers in general and for PDAC in particular. Thus, to achieve more physiologically relevant and conclusive in vitro models, to date, multicellular tumor spheroids (MCTS), patient-derived organoids (PDO), and microfluidic devices represent the cutting edge for preclinical tumor models focussing on TME modulation.40 MCTS were first described by Kunz-Schughart et al. in 2001, who generated spheroids composed of breast cancer cells and fibroblasts.41 Since then, a variety of MCTS, composed of cancer cells combined with one or more stromal cell types, have been established for various malignancies, including pancreatic cancer.42–47 However, these simplistic models were mainly generated with pancreatic cancer cell lines, CAF and one type of ECM components like collagens or Matrigel.42,45,46 A few MCTS also include other important stroma cell types, such as endothelial cells44 or TAM.48 The incorporation of monocytes into PDAC MCTS demonstrates the ability of in vitro spheroids models to differentiate monocytes into macrophages and to polarize them towards an M2 phenotype.48–50 An alternative to MCTS are PDO, which are generated from patient-derived tumor cells offering the opportunity to study molecular/cellular heterogeneity and directly mimic individual patient's biology.51 However, spheroid and organoid models have inherent limitations, including the lack of a functional vasculature prohibiting the investigation of a dynamic drug or immune cells application route.52
Recent advances in microfluidic technologies have facilitated the development of tumor-on-chip systems, which enable the 3D culture of multiple cell types under flow conditions.53 In addition, a microfluidic system enables the dynamic application of drugs. Dynamic drug administration via microfluidic systems surpasses traditional static dosing by enabling controlled release of therapeutics over extended periods or with variable concentrations, thereby more closely mimicking natural pharmacokinetics.54–56
In the context of PDAC, different tumor-on-chip models have been developed over the past decade to investigate various aspects of the TME and its influence on drug response.57–63 Examples for simplified models incorporated PDAC tumor cells and CAF into microfluidic chips to study the crosstalk between stromal and pancreatic cancer cells, but lack the addition of immune and endothelial cells.57,61 Other previously described models allow high throughput drug screening, but still do not include macrophages or a vasculature.55,59 However, these models demonstrate that the dynamic administration of drugs influences their cytotoxic impact on PDAC spheroids.55 This enhances our understanding of drug uptake and distribution in tumor models by providing new insights into how dynamic dosing regimens can improve treatment responses. Other chip models encompass patient-derived pancreatic tumor cells.60,62,63 However, compared to our approach described here, these models also miss either an endothelial layer60 or integrated immune cells.62,63 The most complex PDAC model described so far was set up in a three-compartment chip with patient-derived PDAC organoids at the bottom, PSC in the middle and an endothelial tube at the top, which was perfused with peripheral blood mononuclear cells (PBMC).58 However, this model is missing a continuous flow.
Here, we report a novel PDAC-on-chip model incorporating relevant hallmarks of the desmoplastic and immunosuppressive TME allowing dynamic drug administration as well as immune cell infiltration via a continuous flow. This microfluidic biochip model combines MCTS composed of PANC-1 cells and PSC with a newly designed biochip incorporating an endothelial layer of human umbilical vein endothelial cells (HUVEC). Despite of the existing shear stress, an intact vasculature in the presence of MCTS could be established, allowing drug delivery via the endothelial layer. We previously reported that treatment with vorinostat, an FDA-approved drug for cutaneous T cell lymphoma (CTCL), also known as suberoylanilide hydroxamic acid (SAHA),64 displays a cytotoxic effect on 3D PDAC spheroids generated from PANC-1 cells and PSC.65 Here, we show that treatment with vorinostat delivered via a continuous flow through an endothelial layer composed of HUVEC reduced spheroid viability, while not significantly compromising the integrity of the endothelial layer. Additionally, our system is amenable to study immune cell infiltration as demonstrated by perfused monocytes which successfully infiltrated the spheroids and differentiated into macrophages. The polarization into M2 macrophages started 96 h after the infiltration and lasted for at least 168 h.
Experimental
Biochip design and fabrication
Biochips were manufactured by Dynamic42 GmbH (Jena, Germany) from injection-molded polybutylene terephthalate (PBT) base bodies. The biochip harbors two chambers each consisting of 3 vertically stacked channels (top = TOP, middle = MID, bottom = BOT) equipped with individual inlets and outlets in luer-format. Channels on the top and the bottom side of the biochip are sealed by a poly carbonate (PC) bonding foil and separated on the inside from MID channel by porous membranes which serve as culture surface for cells. The total channel volumes, including inlets and outlets, are 290 μL (TOP), 390 μl (MID) and 270 μL (BOT). The interfacing membrane of top to middle channel is made of 12 μm thick polyethylene terephthalate (PET) with 8 μm pores at a density of 1 × 106 cm−2 (Sabeu, Radeberg, Germany) and has a surface area of 1.32 cm2. The second interfacing membrane from the middle to the bottom channel is made of PC with 2 μm pores at a pore density of 1 × 106 cm−2, and has a surface area of 0.61 cm2 and is imprinted with 25 microcavities with a diameter of 800 μm (300 Microns, Karlsruhe, Germany).Cell culture
The PANC-1 cell line, acquired from the American Type Culture Collection (ATCC), was cultured in Dulbecco's modified Eagle's medium (DMEM) (#41965039, Gibco Grand Island, NY, USA) supplemented with 10% fetal bovine serum (FBS) (#10270-106, Gibco), and 1% penicillin–streptomycin (pen-strep) (#15140122, Gibco). PSC were provided by Dr. Erkan of Koç University Hospital in Turkey. The ethics committee for the biomedical sciences at KOÇ University granted ethical approval, and all patients consented in writing. PSC were obtained from patients diagnosed with ductal adenocarcinoma, and cultured in DMEM/F12 (#11320074, Gibco) with 20% FBS and 1% pen–strep under aseptic conditions. HUVEC (University Hospital, Jena, Germany) were cultured in ECGM (# CC-3156, Lonza, Basel, Switzerland) supplemented with ECGM SingleQuots (#CC-4176) and 1% pen–strep. All cell types were incubated at 37 °C with 5% CO2.Isolation of monocytes
Peripheral blood mononuclear cells (PBMC) were isolated from buffy coats, obtained from anonymized healthy donors (Düsseldorf University Hospital, Germany) approved for in vitro research, using the Ficoll-Paque density gradient method. The buffy coats were mixed with D-PBS and layered on top of Histopaque (#10771, Sigma-Aldrich). After centrifugation at 850 × g for 20 minutes (with 0 acceleration and deceleration) a ring of PBMCs was obtained. PBMCs were collected and incubated for 10 minutes in ammonium chloride solution for red blood cell lysis. After washing with D-PBS the cell number was determined. CD14+ monocytes were isolated from PBMC by magnetic activated cell sorting (MACS) using monocyte-specific anti-CD14 monoclonal antibodies conjugated to paramagnetic microbeads (#130-097-052, Miltenyi Biotec, Bergisch Gladbach, Germany) according to the manufacture's protocol and the MidiMACS separator (#130-042-302, Miltenyi Biotec). Monocytes were cultured in RPMI (#21875-034, Gibco) supplemented with 10% FBS and 1% pen–strep at a concentration of 1 × 106 cells per mL.Spheroid formation
The generation of co-culture spheroids, consisting of PANC-1 and PSC cells, was performed as previously described by Xie et al.45 Briefly, 1 × 104 cells per mL PANC-1 cells and 2 × 104 cells per mL PSC were combined in DMEMF12. Collagen-I (#A1048301, Gibco, Waltham, MA USA) was added to a final concentration of 0.1 mg mL−1. The cell suspension was transferred to an ultra-low attachment coated 96-well plate (#650970, Greiner Bio-One) and cultivated at 37 °C and 5% CO2. For the generation of PANC-1 spheroids 1500 PANC-1 cells in 50 μL DMEM with 2.5% (v/v) Matrigel (#356230, Corning, NY, USA) were added into a well of an ultra-low attachment coated 96-well plate (#650970, Greiner Bio-One) and centrifuged at 1500 rpm for 5 min. Spheroids were transferred into the biochip 4 days after seeding.HUVEC cultivation in and integration of 3D spheroids into the chip
The membrane in the TOP channel was coated with collagen-I (#A1048301, Gibco, Waltham, MA USA). 150![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 HUVEC were seeded on the coated membrane in the TOP channel. Following daily media exchange with 250 μL EGM-2 in the TOP channel, HUVEC were cultivated for 4 days in the chip. 4 days old spheroids were collected in a 1.5 mL tube. A minimum of 20 spheroids were loaded into the microcavities of the MID channel. The spheroids were cultivated on the biochip in 70% EGM-2 mixed with 30% DMEMF12 and 2.5% (v/v) Matrigel.
000 HUVEC were seeded on the coated membrane in the TOP channel. Following daily media exchange with 250 μL EGM-2 in the TOP channel, HUVEC were cultivated for 4 days in the chip. 4 days old spheroids were collected in a 1.5 mL tube. A minimum of 20 spheroids were loaded into the microcavities of the MID channel. The spheroids were cultivated on the biochip in 70% EGM-2 mixed with 30% DMEMF12 and 2.5% (v/v) Matrigel.
Perfusion of the biochip
For perfusion of the biochip the TOP and BOT channel of the biochip were individually connected via tubing to a peristaltic pump (Ismatec™ IPC 4 Peristaltic Pump, # ISM930A, Masterflex, Barrington, IL, USA). Each tubing system includes a PHarMed BPT thermoplastic elastomer 2-stop tube (ISMATEC, Wertheim, Germany), filled with 250 μL medium and a medium reservoir (MoBiTec, Göttingen, Germany), filled with 750 μL of medium. The TOP and the BOT channel were perfused with a flow rate of 25 μL min−1 for 72 h at 37 °C and 5% CO2. The MID channel harbouring tumor spheroids was non-perfused and closed using standard luer-plugs (Mobitec, Göttingen, Germany).Cell viability
The CellTiter-Glo® 3D cell viability assay (#G9681, Promega, Madison, WI, USA) was used to analyze the cell viability of 3D spheroids. Cell viability of HUVEC, PANC-1 and PSC cell, was determined using the PrestoBlue HS Cell Viability Assay (#P50201, Thermo Scientific, Waltham, MA, USA) 10000 HUVEC, 5000 PANC-1 and 2000 PSC were seeded per well in 96-well plates (#655090, Greiner Bio-One). Compound treatment was performed 24 h after seeding, for 72 h. Fluorescence values were recorded using the Tecan SPARK instrument (Tecan Group, Switzerland).
Immunofluorescence (IF) staining of the HUVEC layer
The biochip was cut open on the TOP side and the interfacing membrane between TOP and MID channel was detached from the chip using a standard lab scalpel (#11566, Swann Morton, Sheffiel, United Kingdom) and placed in a well of a 24-well plate prefilled with 500 μL D-PBS. The membrane was fixed for 15 minutes with 4% paraformaldehyde (PFA) (#0964.1, Sigma-Aldrich, Saint Louis, MI, USA) solution (in D-PBS) and then blocked for 30 minutes with 0.1% saponin and 3% normal donkey serum (NDS) in D-PBS at room temperature (RT). The membrane was incubated overnight at 4 °C with 50 μL of primary antibody solution (VE-cadherin (# AF938, R&D Systems), von Willebrand factor (#A0082, Agilent, Santa Clara, CA, USA)). The secondary antibody solution (Alexa Fluor 546 (donkey anti-rabbit antibody, Invitrogen), Alexa Fluor 488 (donkey anti-goat antibody, Invitrogen, Waltham, MA, USA), 2 μg mL−1 DAPI (#D9542, Sigma-Aldrich,)) was prepared in PBS containing 0.1% saponin and 3% NDS, and the membrane was incubated in 50 μL in the dark for 1 hour at RT. The membrane was mounted on a glass slide with fluorescence mounting medium (#GM30411-2, Agilent Technologies, Santa Clara, CA, USA), dried overnight and imaged with the CQ1 High-Content Spinning Disk System (part number 90ZA00673, Yokogawa, Tokyo, Japan). The cell viability staining was performed according to manufacturer instructions using the Viability Kit (#L3224, Invitrogen, Carlsbad, CA, USA). Briefly, HUVEC on the membrane were incubated for 30 min in PBS containing calcein-AM and ethidium homodimer-1. Afterwards, the membrane was washed with PBS and fixed with 4% PFA. The membrane was mounted on a glass slide with fluorescence mounting medium, dried overnight and imaged with the CQ1 High-Content Spinning Disk System (Part Number 90ZA00673, Yokogawa).Immunofluorescence staining of spheroids
Spheroids were collected with a pipette from the biochip after 72 h and stained according to the MACS clearing Kit (#130-126-719, Miltenyi Biotec). Briefly, the spheroids were fixed with 4% PFA for 1 hour at RT. Following 6 h of permeabilization the spheroids were incubated with 100 μL of primary antibody solution (Human Ki67/MKI67 (#MAB7617, R&DSystems, Minneapolis, MN, USA), α-SMA (#19245S, Cell Signal, Danvers, MA, USA), pan-cytokeratin (#130-133-439, Miltenyi Biotec), CD45 (#130-110-638, Miltenyi Biotec), CD163 (#130-112-286, Miltenyi Biotec)) for 24 hours at 4 °C. The secondary antibody solution (Alexa Flour 647 donkey (H + L) anti-mouse (#A-31571, Invitrogen), Alexa Flour 488 donkey (H + L) anti-rabbit (#A-21206, Invitrogen)) including 2 μg mL−1 DAPI (#D9542, Sigma-Aldrich) was incubated at 4 °C for 24 hours. The clarification process was carried out by successive incubation in three ethanol solutions of increasing concentration (50%, 70% and 100%), each containing 2% Tween. The incubation was initially carried out for 2 hours each (for the 50% and 70% ethanol concentrations) and finally in the presence of 100% ethanol overnight. The final step was clearing for 6 h with the clearing solution. Afterwards, the spheroids were transferred to an imaging plate (# 89626, ibidi GmbH) and imaged with the CQ1 High-Content Spinning Disk System (Part Number 90ZA00673, Yokogawa). 3D images were visualized using the 3D viewer plugin66 in in the Fiji Software.67Quantitative real-time PCR (qRT-PCR)
To determine the cytokine expression of PANC-1, PSC, co-culture spheroids and PANC-1 spheroids a qRT-PCR was performed. The following primers were utilized to determine the mRNA levels of TNF-α, IL-8 and IL-6: glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (forward, 5′-TGCACCACCAACTGCTTAGC-3′; reverse, 5′-GGCATGGACTGTGGTCATGAG-3′), TNF- α (forward 5′-CCCTGCTGCACTTTGGAGTG-3′; reverse 5′-CGGGGTTCGAGAAGATGAT-3′), IL-6 (forward 5′-GGCACTGGCAGAAAACAACC-3′; reverse 5′-GCAAGTCTCCTCATTGAATCC-3′), and IL-8 (forward 5′-ACTGAGAGTGATTGAGAGTGGAC-3′; reverse 5′-AACCCTCTGCACCCAGTTTTC-3′). PANC-1, PSC, co-culture spheroids and PANC-1 spheroids were cultivated for 72 h in 3D medium. The qRT-PCR was performed as previously described by us.65 The ΔΔCt method was used to calculate the Ct values. Sample data were normalized to the housekeeping gene GAPDH. Quantitative analysis was conducted using qPCRsoft 4.1 software (Analytik Jena, Jena, Germany).Permeability characterization of the HUVEC layer
Fluorescein isothiocyanate–dextran (FITC–dextran, Merck) with an average molecular weight of 3–5 kDa was used to assess the barrier permeability of the HUVEC layer. Therefore, medium in both channels was replaced by prewarmed PBS. A volume of 250 μL 0.1 mg mL−1 FITC–dextran solution in PBS was added in the TOP channel. The chips were incubated for 1 h at 37 °C and 5% CO2 allowing FITC–dextran to permeate through the cell layer. After the incubation the solutions from the TOP, MID and BOT channel were collected separately and transferred into a 96 well plate. All steps were performed protected from light. The fluorescence values were measured with the Tecan SPARK instrument (Tecan Group, Switzerland). FITC–dextran concentrations were calculated from a standard curve obtained from the 0.1 mg mL−1 stock solution.Drug adsorption
A circular perfusion setup consisting of tubing and a 1 mL reservoir was connected to both the upper and lower channel of an empty, sterilized and coated biochip. All channels, tubing and reservoirs were filled with ECGM containing indicated levels of vorinostat and 1:1000 DMSO. Upper and lower channel were perfused with 25 μl min−1, at 37 °C and 5% CO2. After 24 h media in the perfusion setup was pooled and vorinostat levels were quantified using LCMS. Controls were incubated for 24 h at 37 °C in glass vials.
LCMS analysis
Ultra-high-performance liquid chromatography (LCMS) coupled with high resolution mass spectrometry was carried out using a Thermo (Bremen, Germany) UltiMate HPG-3400 RS binary pump, WPS-3000 auto sampler which was set to 10 °C and which was equipped with a 25 μL injection syringe and a 100 μL sample loop. The column was kept at 25 °C within the column compartment TCC-3200. Chromatography column Thermo Accucore® C-18 RP (100 × 2.1 mm; 2.6 μm) was run at a constant flow rate of 0.4 mL min−1 using a gradient (ESI† Table S1) (time in min/concentration eluent B in %) – 0/0; 0.2/0; 8/100; 11/100; 11.1/0; 12.0/0 (ESI† Table S1). Eluent A was water, with 2% acetonitrile and 0.1% formic acid. Eluent B was pure acetonitrile.Mass spectra were recorded with Thermo QExactive plus orbitrap mass spectrometer coupled to a heated electrospray source (HESI). Column flow was switched at 0.5 min from waste to the MS and at 11.5 min again back to the waste, to prevent source contamination. For monitoring two full scan modes were selected with the following parameters. Polarity: positive; scan range: 100 to 1500 m/z; resolution: 70000; AGC target: 3 × 106; maximum IT: 200 ms. General settings: sheath gas flow rate: 60; auxiliary gas flow rate 20; sweep gas flow rate: 5; spray voltage: 3.0 kV; capillary temperature: 320 °C; S-lens RF level: 90; auxiliary gas heater temperature: 400 °C; acquisition time frame: 0.5–11.5 min. For negative mode, all values were kept instead of the spray voltage which was set to 2.5 kV.
Flow cytometry of infiltrated macrophages
The time course of the polarization of monocytes after infiltration into the co-culture spheroids was analyzed with flow cytometry. For this, co-culture spheroids were collected 24 h, 48 h, 72 h, 96 h, and 168 h after infiltration with PBMC-derived monocytes. After spheroid digestion with Accutase (#A1110501, Gibco), the cells were stained with the following antibodies according to manufactures instructions; CD163-PE (#130-112-286, Miltenyi Biotec), CD11b-VioBlue (#130-110-616, Miltenyi Biotec), CD86-APC (#130-116-161, Miltenyi Biotec), CD45-VioGreen (#130-110-638, Miltenyi Biotec), HLA-DR-PEVio770 (#130-111-944, Miltenyi Biotec). Flow cytometry was performed using the CytoFlex (Beckman-Coulter, Brea, CA, USA) flow cytometer. The data were analysed with the CytExpert software (version 10.10.0, Beckman-Coulter, Brea, CA, USA).Statistical analysis
For statistical and graphical analysis, the GraphPad Prism 8.4.3 software (GraphPad Software, Boston, MA, USA) was used. The IC50 values were determined by non-linear regression to assess cell viability. One-way analysis of variance (ANOVA) and unpaired t-test were used to test the statistical data, with p-values ≤ 0.05 considered statistically significant.Results and discussion
Design of the PDAC on chip model
The Dynamic42 BC003 biochip was developed for organ-on-chip applications integrating spheroids or organoids in a perfused system. Designs were fine-tuned for injection moulding using the CAD software SolidWorks. Each biochip consists of two independent chambers (Fig. 1A). Individual chambers are comprised of three vertically stacked channels which can all be separately loaded and perfused using the respective inlets and outlets (Fig. 1A and B). Channels are separated by two porous membranes with the lower membrane carrying 25 microcavities with a diameter of 800 μm for spheroid insertion. Channel dimensions of MID channel are at least 1 mm throughout the whole biochip to support non-destructive spheroid loading of spheroids up to 800 μm in diameter. In the PDAC setup, a confluent layer of HUVEC is generated on the membrane in the TOP channel and perfused at 25 μL min−1 to mimic the vascular flow (Fig. 1B–D). A minimum of 20 PDAC-spheroids are inserted into the microcavities of the MID channel, which is then sealed with plugs. The microcavities of the MID channel project into the BOT channel. The BOT channel is also perfused at 25 μL min−1 to improve nutrient availability for spheroids. Importantly, spheroids are shielded from flow and shear forces by the porous microcavity membrane. Reservoirs are inserted into both perfused inlets to keep cell culture medium available and to simultaneously serve as bubble traps. | ||
| Fig. 1 Design of the biochip. (A) Exploded view of the chip (B) translucent view of the chip. The upper panel shows the TOP view with underlying channels in dotted lines. Coloured circles indicate the location of ports for TOP (TOP), middle (MID) and bottom (BOT) channels. The lower panel shows a cross section of a chamber at the indicated position. The position of the bonding foil (BF), the membrane pore size of 8 μm (M) and microcavity membrane pore size of 2 μm (MCM) are indicated. Red numbers depict the channel height at the bottlenecks (red arrows) in millimeters. (C) Perfusion setup for the PDAC model. The tubing connects ports of the TOP and the BOT channel to create two independent perfusion circles. The MID channel is sealed. Arrows indicate the media flow direction in the TOP (magenta) and the BOT (blue) channel during perfusion. (D) Schematic illustration of the PDAC model. Arrows depict the flow direction in the TOP (magenta) and the BOT (blue) channel. | ||
The potential of adsorption of small molecules is a challenge in many biochip systems due to their large surface area, additional tubing systems and non-favourable biochip base body material.68 To test drug retention in the biochip made of PBT and described here, media containing 0.5–10 μM of vorinostat was either perfused through the TOP and BOT channel of the biochip or incubated in glass vials. After 24 h, the vorinostat concentration in the perfused chip was determined by LCMS. Notably, irrespective of the starting concentration, perfused and non-perfused controls did not show any significant loss of vorinostat levels over input, confirming that vorinostat is neither degraded in nor adsorbed to the biochip surface (Table 1).
| Starting concentration vorinostat [μM] | Measured vorinostat concentration [μM] after 24 h in the biochip |
|---|---|
| 0.5 | 0.693 ± 0.094 |
| 1 | 1.017 ± 0.191 |
| 5 | 5.131 ± 0.109 |
| 10 | 10.149 ± 0.182 |
Spheroid viability in the biochip
To analyze potential effects on the viability of PDAC spheroids cultivated in the biochip under continuous or controlled perfusion, a total of 25 PDAC mono- or co-culture spheroids were loaded into the respective microcavities within the MID channel (Fig. 2A). Immunofluorescence staining confirmed that PANC-1 mono-spheroids and co-culture spheroids maintain their structure in the biochip, with the latter keeping its subcellular composition of a fibrotic shell of PSC surrounding a core of PANC-1 cells45 (Fig. 2B). Evaluating spheroid viability after 72 h of cultivation without a continuous flow (so-called “static condition”) revealed a decrease in viability by 44% in the co-culture spheroids and by 59% in the PANC-1 spheroids in the biochip compared to a cultivation setting in a 96-well plate (Fig. 2C). Importantly, the viability of the PDAC spheroids significantly increased under flow conditions in the chip from 56% to 78% in co-culture spheroids and from 41% to 91% in PANC-1 spheroids, respectively. Notably, a direct viability comparison of spheroids cultured in the presence of a continuous flow of 25 μL min−1 and spheroids cultured in 96-well plate still revealed a significant decrease in cell viability in co-culture spheroids of 22% but only a slight decrease in PANC-1 spheroids of 9%. The reduced viability observed under static conditions on the chip may be attributed to the spheroid-to-medium volume ratio, the MID channel contains 250 μL of medium for 25 spheroids, equating to 10 μL per spheroid, in contrast to the 100 μL per spheroid provided in the microtiter plate conditions. A total of 1500 μL of medium is supplied through the MID channel and the perfusion system in the BOT channel for 25 spheroids, amounting to 60 μL per spheroid. In conclusion, this indicates that our microfluidic system provides efficient nutrient supply and is suitable for the cultivation of both spheroid types under microfluidic conditions. | ||
| Fig. 2 Cultivation of spheroids on the biochip. (A) Co-culture spheroids and PANC-1 spheroids loaded into the microcavities of the biochip. Images are taken via bright field microscopy after loading of the spheroids. Scale is 1000 μm. (B) If staining of 3D PANC-1 and co-culture spheroids after 72 h cultivation on the biochip shown as maximum intensity projection (MIP) of a Z-stack. DAPI-staining of nuclei (blue); staining of the proliferation marker Ki67 (green); staining of fibroblast marker α-SMA (purple); merging of all three channels. Scale bar is 500 μm. (C) Investigation of cell viability of 3D spheroids after 72 h cultivation on biochip either under flow or static conditions in comparison to the cultivation in a 96-well plate. Error bars indicate the standard errors of the mean of at least three independent experiments, with **** = p ≤ 0.0001. | ||
Cultivation of HUVEC in the biochip
Tumor vascularization plays a pivotal role in facilitating immune cell infiltration, nutrient and oxygen supply and drug delivery.6 Consequently, integrating a perfusable vasculature into in vitro TME models is essential for accurately recapitulating the in vivo vascular transport dynamics of immune cells, and drugs into the tumor.69 As demonstrated in Fig. 3, HUVECs were seeded into the TOP channel in the chip and cultured under static conditions for 4 days until full confluency was achieved. HUVECs were either cultivated for further 72 h in the presence of a continuous flow of 25 μL min−1 or without any flow. After 72 h of cultivation, under static or under flow conditions, a tight endothelial barrier was established, as confirmed by the expression of the endothelial markers Willebrand factor (vWF) and adherence junction marker vascular endothelial (VE-) cadherin (Fig. 3). Furthermore, assessment of the macromolecular permeability of the endothelial layer (ESI† Fig. S1) revealed that the barrier permeability is not influenced by cultivation under flow conditions. In the presence of co-culture spheroids and under flow conditions, the endothelial cells failed to maintain a tight endothelial layer (ESI† Fig. S2) whereas the endothelial layer remained unaffected under static conditions (ESI† Fig. S2) suggesting that the increased viability of the spheroids under flow conditions sensitize endothelial cells to cells stress. Previous studies show that the expression of the proinflammatory cytokines TNF-α, IL-8 and IL-6, which are also secreted by co-culture spheroids (ESI† Fig. S3) might impact endothelial adherence.58 Indeed, the addition of conditioned medium obtained from the supernatant of co-culture spheroids resulted in an increase of loose connections between the HUVEC cells (ESI† Fig. S4), arguing that spheroids secrete soluble factors, like TNF-α, IL-8 and IL-6 (ESI† Fig. S3), which impair the HUVEC layer integrity under flow conditions.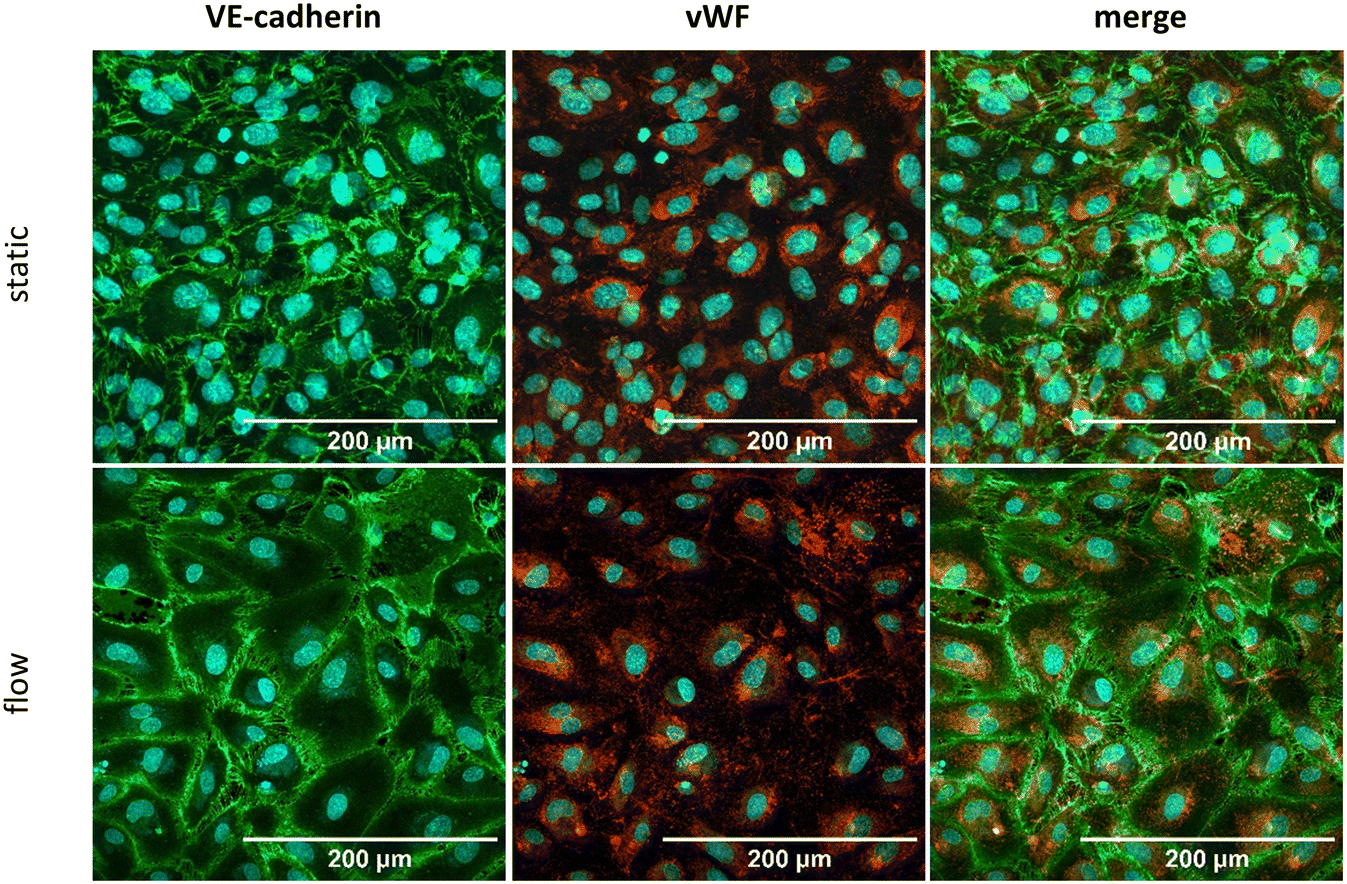 | ||
| Fig. 3 Cultivation of HUVEC on the biochip. HUVEC were cultivated for 72 h under static (upper row) or flow (bottom row) conditions. The endothelial markers VE-cadherin (green), von Willebrand factor (vWF) (red) and the nuclear dye DAPI (blue) were stained. Shown as MIP of a Z-stack. Scale bar is 200 μm. | ||
Massive ECM secretion by CAF within the PDAC TME creates a dense and stiff mechanical barrier around the tumor cells.69 Absence of such barrier might enhance negative effects of the tumor on the endothelium. To recreate this mechanical barrier in our model, 2.5% Matrigel was introduced into the MID channel embedding the spheroids. In addition, as illustrated in Fig. 4, the incorporated Matrigel restored vascular stability under perfusion in the presence of both, co-culture spheroids and PANC-1 spheroids (Fig. 4). Furthermore, a quantitative permeability assessment of the HUVEC layer confirms that the addition of Matrigel significantly restored the barrier permeability under flow conditions (ESI† Fig. S1). These results are in line with other PDAC-on-chip platforms with incorporated HUVEC-based vasculature, which also achieved an intact endothelial layer by embedding the tumor cells or organoids in Matrigel.58,63,70
 | ||
| Fig. 4 Rescue strategy for HUVEC layer in the presence of pdac spheroids. The vasculature remains stable under static (A) and flow (B) with an addition of 2.5% matrigel both in the presence of co-culture spheroids (upper row) as well as in the presence of PANC-1 spheroids (bottom row). Stained the endothelial markers VE-cadherin (green), von Willebrand factor (vWF) (red) and the nuclear dye DAPI (blue) were stained. Shown as MIP of a Z-stack. Scale bar is 200 μm. | ||
Proof-of-concept for a dynamic drug administration
A dynamic application of drugs through a microfluidic system offers an improved simulation of the in vivo drug transport and can significantly influence treatment responses, as initially demonstrated for PDAC by Schuster et al.55 The histone deacetylase inhibitor vorinostat, approved for the treatment of CTCL, exhibits cytotoxic effects on cells that overexpress class I histone deacetylases (HDACs) and HDAC6.64 This effect has been demonstrated across various malignancies, including pancreatic ductal adenocarcinoma (PDAC),71 while sparing healthy cells.64 As demonstrated by our previous work, the histone deacetylase inhibitor vorinostat, also known as suberoylanilide hydroxamic acid (SAHA), exhibits a cytotoxic effect on co-culture spheroids with an IC50 value of 0.85 μM.65 In contrast, an IC50 value of 5.7 μM was determined for HUVECs (ESI† Table S2). This cell type dependent efficacy difference opens a treatment window for targeting 3D PDAC spheroids without affecting the viability of endothelial cells. Thus, concentrations of 1.5, 2.5 or 3.5 μM SAHA were introduced via a flow of 25 μL min−1 in the TOP channel. After a treatment period of 72 h, the vasculature integrity (Fig. 5A) and the co-culture spheroids viability (Fig. 5B) were assessed, respectively. Analysis of the expression of the endothelial markers VE-cadherin and vWF indicated that the integrity of the endothelial layer was unaffected at SAHA concentrations up to 2.5 μM (Fig. 5A). These results were verified by a live dead staining of the HUVEC layer (ESI† Fig. S5) and a barrier integrity test (Fig. 5C), confirming no significant increase in FITC dextran leakage upon treatment with 1.5 or 2.5 μM SAHA, but a slight leakage increase of 11.5% compared to 9.9% for the untreated control after treatment with 3.5 μM SAHA. In contrast to its impact on endothelial cells, SAHA treatment significantly impaired the viability of the co-culture spheroids by 41% or 49% at concentrations of 2.5 μM and 3.5 μM, respectively (Fig. 5B). In conclusion, our model allows administration of drug candidates via an intact vasculature under flow conditions while significantly and selectively effecting spheroid viability. | ||
| Fig. 5 Dynamic administration of the HDAC inhibitor SAHA in the PDAC biochip model. (A) Investigation of the influence of increasing SAHA concentrations on the vasculature integrity. VE-cadherin (green), von Willebrand factor (red) as marker of the endothelial cells, DAPI for staining the cell nuclei (blue). Shown as MIP of a Z-stack. Scale 200 μm. (B) Investigation of the viability of the 3D PDAC spheroids in the biochip in the presence of SAHA after 72 h administration (with **** = p ≤ 0.0001). (C) Permeability assay of HUVEC layer after treatment with SAHA for 72 h. FITC dextran assay was performed to measure the barrier integrity of the HUVEC layer after 72 h treatment with 1.5, 2.5, or 3.5 μm SAHA. FITC dextran solution was added into the top channel. After 1 h the leakage into the mid channel was determined via fluorescence measurement. | ||
Integration and spatial distribution of macrophages
TAM which have been shown to be significantly associated with a poor overall survival rate in PDAC patients72 are described as CD163+ M2-polarized.19 Overall, CD163+ M2 macrophages represent an immunosuppressive and pro-fibrotic cell type19 that is also reported to release mediators which activate different pathways in tumor cells like PI3K/Akt, JAK/STAT or others, subsequently contributing to chemoresistance.27,73,74 Since TAM polarization is the result of the crosstalk between tumor cells and CAF through the secretion of different signalling molecules, like IL-4, TGF-β, or M-CSF,19,25,74 we integrated PBMC-derived monocytes into our PDAC biochip model to investigate the polarization into TAM.To this end, we perfused primary PBMC-derived monocytes for 72 h through the top channel of the biochip. As shown by the positive signal of the leucocyte marker CD45 (Fig. 6A), the monocytes significantly infiltrated the co-culture spheroids (Fig. 6A). Notably, primary monocytes also infiltrate PANC-1 spheroids, where they distribute evenly in the whole spheroid (ESI† Fig. S6). Furthermore, analysis of CD163 expression of integrated monocytes indicate that PDAC spheroids polarize integrated macrophages towards an M2 phenotype. Indeed, when compared to unstimulated monocytes, expression of CD163 increased by 1.9-fold within 72 h of infiltration of co-culture spheroids under flow (Fig. 6B). Notably, polarization persisted in the co-culture spheroids for up to 168 h as indicated by CD163 upregulation (ESI† Fig. S7). Simultaneously, the M1 markers CD86 and HLA-DR are significantly downregulated when compared to monocytes before infiltrating the spheroids (Fig. 6B) indicating the that the polarization is triggered by the integration. These results are in line with the findings from Madsen et al. (2021).49
 | ||
| Fig. 6 Polarization of monocytes after perfusion in the biochip. (A) If staining of co-culture spheroids after perfusion with primary monocytes over the endothelial layer. Co-culture spheroids were seeded into the biochip and primary monocytes were perfused over the endothelial layer in the TOP channel. After 72 h of perfusion the co-culture spheroids were stained for the tumor cell marker pan-cytokeratin (purple), the fibroblast marker α-SMA (blue), the immune cell marker CD45 (green), and the M2 marker CD163 (red); merging of all four channels. Shown as MIP of a Z-stack. Scale bar is 500 μm. (B) Marker characterization of polarized macrophages. Polarization after infiltration into co-culture spheroids was analyzed by flow cytometry 72 h after perfusion with monocytes in the biochip. Macrophages identified as CD11b+ and CD45+ were analyzed for surface expression of CD163, CD206, CD86, and HLA-DR. Infiltrated cells were compared to unstimulated monocytes as controls. Error bars indicate the standard errors of the mean of three independent experiments, with ** = p ≤ 0.01; *** = p ≤ 0.005. | ||
To investigate the spatial distribution of the infiltrated macrophages in the co-culture spheroids, a 3D image of a whole spheroid was generated and digitally divided into two halves (Fig. 7A). As demonstrated in Fig. 7B and in the ESI† Video S1, the infiltrated macrophages preferably localize in the fibrotic shell part of the co-culture spheroids. These findings show, that our in vitro model despite of its simplicity is able to recreate the spatial distribution of macrophages in PDAC patients, where M2 macrophages are significantly enriched in the stromal part of the TME.75 It is currently hypothesized that the secretion of multiple cytokines and chemokines, like IL-10, GM-CSF, IL-4 or IL-33 by pancreatic cancer cells and other cell types of the TME, especially CAF and T helper cells 2 (Th2), can mainly trigger the polarization of TAM towards a M2 phenotype in PDAC.19,25 For example, activation of the epidermal growth factor receptor (EGFR)/AKT/cAMP signalling pathway through cancer cell secreted REG4 enhances the expression of CD163, CD206 and IL-10.76
 | ||
| Fig. 7 Spatial distribution of infiltrated monocytes. (A) 3D-rendered confocal Z-stack image of a monocyte-infiltrated spheroid, consisting of 300 slices each 1 μm per step, visualized using image software Fiji with the 3D viewer plugin. For enhanced clarity, the spheroid was spatially cut in half, allowing better visualization of the fibrotic shell, the core of PANC-1 cells and the spatial distribution of infiltrating macrophages. Created in biorender. (B) Fluorescent 3D image of the halved spheroid. Scale bars in 100 μm steps are provided, and the spatial orientation of the halved section is indicated. If staining for the tumor cell marker pan-cytokeratin (purple), the fibroblast marker α-SMA (blue), the and the M2 marker CD163 (green) merged in one image. White arrows indicate CD163+ signals. | ||
Overall, the reported findings open a path for more in-depth characterization of macrophage polarization in the presence of different drugs screened in the here described PDAC-on-chip model. In conclusion, our microfluidic biochip is applicable to investigate dynamic monocytes infiltration via a vascular layer and the polarization towards M2 in the spheroids. In future studies, the implementation and integration of additional relevant immune cell types present in the TME of PDAC,5 such as Treg, cytotoxic T cells or natural killer cells, appears necessary to investigate the cell-specific influence on drug treatments.
Conclusions
In summary, we developed a novel tumor-on-chip model to mimic the microenvironment of PDAC. Tumor-on-chip models play a crucial role in advancing our understanding of tumor-TME interactions, particularly in evaluating therapeutic efficacy. In this study, we developed an innovative microfluidic PDAC model integrating a perfusable endothelial layer with multicellular tumor spheroids (MCTS) consisting of PANC-1 cancer cells and PSC embedded within an ECM. This model provides an initial proof-of-concept for controlled drug administration and immune cell infiltration.These initial optimizations represent foundational steps in enhancing this approach. By reliably simulating key processes such as vascular transport, immune cell polarization, and drug delivery, this model facilitates a deeper exploration of potential drug synergies, the incorporation of more complex multicellular structures by introducing additional immune and non-immune cell types, and the comprehensive study of the direct and indirect effects on the entire TME. This includes evaluating aspects like spatial immune cell polarization, cytotoxic activity, and drug resistance mechanisms. Ultimately, this advanced PDAC model offers a powerful platform for identifying promising new drug candidates, thereby reducing the reliance on animal models and improving translational potential for clinical applications.
Data availability
Data are available upon request.Conflicts of interest
M. R. and K. R. hold equity in Dynamic42 GmbH. All other authors have no conflict of interest to declare.Acknowledgements
This work was supported by a grant to N. E. T. and M. R. by the Bundesministerium für Bildung und Forschung (grant number 16LW0306K). We express our gratitude to Michael Schell and Fredrik Wieland for their invaluable assistance in devising the CellPathFinder image analysis. Moreover, we would like to thank the working group of Rüdiger Sorg at Heinrich Heine University Düsseldorf and Nico Überschaar at Friedrich Schiller University Jena for providing the Cytoflex flow cytometer and performing the LCMS analysis, respectively.Notes and references
- R. L. Siegel, K. D. Miller, N. S. Wagle and A. Jemal, Ca-Cancer J. Clin., 2023, 73, 17–48 Search PubMed.
- R. L. Siegel, A. N. Giaquinto and A. Jemal, Ca-Cancer J. Clin., 2024, 74, 12–49 CrossRef PubMed.
- F. Bray, M. Colombet, J. F. Aitken, A. Bardot, S. Eser, J. Galceran, M. Hagenimana, T. Matsuda, L. Mery, M. Piñeros, I. Soerjomataram, E. de Vries, C. Wiggins, Y.-J. Won, A. Znaor and J. Ferlay, Cancer Incidence in Five Continents, Vol. XII, https://ci5.iarc.who.int (accessed 01.10.2024, 2024) Search PubMed.
- T. Conroy, P. Pfeiffer, V. Vilgrain, A. Lamarca, T. Seufferlein, E. M. O'Reilly, T. Hackert, T. Golan, G. Prager, K. Haustermans, A. Vogel and M. Ducreux, Ann. Oncol., 2023, 34, 987–1002 CrossRef CAS PubMed.
- M. H. Sherman and G. L. Beatty, Annu. Rev. Pathol., 2023, 18, 123–148 CrossRef CAS PubMed.
- C. Hartupee, B. M. Nagalo, C. Y. Chabu, M. Z. Tesfay, J. Coleman-Barnett, J. T. West and O. Moaven, Front. Immunol., 2024, 15, 1287459 CrossRef CAS PubMed.
- L. N. Barrera, A. Evans, B. Lane, S. Brumskill, F. E. Oldfield, F. Campbell, T. Andrews, Z. Lu, P. A. Perez-Mancera, T. Liloglou, M. Ashworth, M. Jalali, R. Dawson, Q. Nunes, P. A. Phillips, J. F. Timms, C. Halloran, W. Greenhalf, J. P. Neoptolemos and E. Costello, Cancer Res., 2020, 80, 2861–2873 CrossRef CAS PubMed.
- J. Norton, D. Foster, M. Chinta, A. Titan and M. Longaker, Cancers, 2020, 12(5), 1347 CrossRef CAS PubMed.
- Y. Wu, C. Zhang, K. Jiang, J. Werner, A. V. Bazhin and J. G. D'Haese, Front. Oncol., 2020, 10, 621937 CrossRef PubMed.
- A. Morgan, M. Griffin, L. Kameni, D. C. Wan, M. T. Longaker and J. A. Norton, Biology, 2023, 12, 1044 CrossRef CAS PubMed.
- P. P. Provenzano, C. Cuevas, A. E. Chang, V. K. Goel, D. D. Von Hoff and S. R. Hingorani, Cancer Cell, 2012, 21, 418–429 CrossRef CAS PubMed.
- R. Ajina and L. M. Weiner, Pancreas, 2020, 49, 1014–1023 CrossRef CAS PubMed.
- D. Thomas and P. Radhakrishnan, Adv. Exp. Med. Biol., 2020, 1234, 57–70 CrossRef CAS PubMed.
- Y. Ju, D. Xu, M. M. Liao, Y. Sun, W. D. Bao, F. Yao and L. Ma, npj Precis. Oncol., 2024, 8, 199 CrossRef PubMed.
- A. Zhang, Y. Qian, Z. Ye, H. Chen, H. Xie, L. Zhou, Y. Shen and S. Zheng, Cancer Med., 2017, 6, 463–470 CrossRef CAS PubMed.
- M. C. Whittle and S. R. Hingorani, Gastroenterology, 2019, 156(7), 2085–2096 CrossRef CAS PubMed.
- H. Y. Tanaka, T. Nakazawa, A. Enomoto, A. Masamune and M. R. Kano, Cancers, 2023, 15, 724 CrossRef CAS PubMed.
- M. Locati, G. Curtale and A. Mantovani, Annu. Rev. Pathol., 2020, 15, 123–147 CrossRef CAS PubMed.
- S. Yang, Q. Liu and Q. Liao, Front. Cell Dev. Biol., 2020, 8, 607209 Search PubMed.
- H. Hu, J.-J. Hang, T. Han, M. Zhuo, F. Jiao and L.-W. Wang, Tumor Biol., 2016, 37, 8657–8664 CrossRef CAS PubMed.
- G. Atanasov, C. Pötner, G. Aust, K. Schierle, C. Dietel, C. Benzing, F. Krenzien, M. Bartels, U. Eichfeld, M. Schmelzle, M. Bahra, A. Pascher and G. Wiltberger, Onco Targets Ther, 2018, 9, 29715–29726 Search PubMed.
- R. Zhang, Q. Liu, J. Peng, M. Wang, T. Li, J. Liu, M. Cui, X. Zhang, X. Gao, Q. Liao and Y. Zhao, J. Cancer, 2020, 11, 2371–2381 CrossRef CAS PubMed.
- Y. Yang, Z. Guo, W. Chen, X. Wang, M. Cao, X. Han, K. Zhang, B. Teng, J. Cao, W. Wu, P. Cao, C. Huang and Z. Qiu, Mol. Ther., 2021, 29, 1226–1238 CrossRef CAS PubMed.
- A. R. Poh and M. Ernst, Cancers, 2021, 13(12), 2860 CrossRef CAS PubMed.
- H. J. Lin, Y. Liu, K. Caroland and J. Lin, Cancers, 2023, 15(13), 3507 CrossRef CAS PubMed.
- G. Y. Liou, H. Döppler, B. Necela, M. Krishna, H. C. Crawford, M. Raimondo and P. Storz, J. Cell Biol., 2013, 202, 563–577 CrossRef CAS PubMed.
- S. Wang, J. Wang, Z. Chen, J. Luo, W. Guo, L. Sun and L. Lin, Npj Precis. Oncol., 2024, 8, 31 CrossRef PubMed.
- B. Deswal, U. Bagchi and S. Kapoor, Anti-Cancer Agents Med. Chem., 2024, 24, 146–156 CrossRef CAS PubMed.
- J. Zhang, J. Song, S. Tang, Y. Zhao, L. Wang, Y. Luo, J. Tang, Y. Ji, X. Wang, T. Li, H. Zhang, W. Shao, J. Sheng, T. Liang and X. Bai, Cell Rep., 2023, 42, 112620 CrossRef CAS PubMed.
- K. Kuwada, S. Kagawa, R. Yoshida, S. Sakamoto, A. Ito, M. Watanabe, T. Ieda, S. Kuroda, S. Kikuchi, H. Tazawa and T. Fujiwara, J. Exp. Clin. Cancer Res., 2018, 37, 307 CrossRef CAS PubMed.
- T. M. Nywening, B. A. Belt, D. R. Cullinan, R. Z. Panni, B. J. Han, D. E. Sanford, R. C. Jacobs, J. Ye, A. A. Patel, W. E. Gillanders, R. C. Fields, D. G. DeNardo, W. G. Hawkins, P. Goedegebuure and D. C. Linehan, Gut, 2018, 67, 1112–1123 CrossRef CAS PubMed.
- S. M. Buchholz, R. G. Goetze, S. K. Singh, C. Ammer-Herrmenau, F. M. Richards, D. I. Jodrell, M. Buchholz, P. Michl, V. Ellenrieder, E. Hessmann and A. Neesse, Cancers, 2020, 12(7), 1978 CrossRef CAS PubMed.
- C. Zheng, J. Wang, Y. Zhou, Y. Duan, R. Zheng, Y. Xie, X. Wei, J. Wu, H. Shen, M. Ye, B. Kong, Y. Liu, P. Xu, Q. Zhang and T. Liang, Cell Rep., 2024, 43, 114088 CrossRef CAS PubMed.
- D. Daley, V. R. Mani, N. Mohan, N. Akkad, G. Pandian, S. Savadkar, K. B. Lee, A. Torres-Hernandez, B. Aykut, B. Diskin, W. Wang, M. S. Farooq, A. I. Mahmud, G. Werba, E. J. Morales, S. Lall, B. J. Wadowski, A. G. Rubin, M. E. Berman, R. Narayanan, M. Hundeyin and G. Miller, J. Exp. Med., 2017, 214, 1711–1724 CrossRef CAS PubMed.
- F. Pratticò and I. Garajová, Curr. Oncol., 2024, 31, 4241–4260 CrossRef PubMed.
- E. M. Palsson-McDermott, L. Dyck, Z. Zasłona, D. Menon, A. F. McGettrick, K. H. G. Mills and L. A. O'Neill, Front. Immunol., 2017, 8, 1300 CrossRef PubMed.
- D. Daley, V. R. Mani, N. Mohan, N. Akkad, A. Ochi, D. W. Heindel, K. B. Lee, C. P. Zambirinis, G. S. B. Pandian, S. Savadkar, A. Torres-Hernandez, S. Nayak, D. Wang, M. Hundeyin, B. Diskin, B. Aykut, G. Werba, R. M. Barilla, R. Rodriguez, S. Chang, L. Gardner, L. K. Mahal, B. Ueberheide and G. Miller, Nat. Med., 2017, 23, 556–567 CrossRef CAS PubMed.
- M. Tsukamoto, K. Imai, T. Ishimoto, Y. Komohara, Y. I. Yamashita, S. Nakagawa, N. Umezaki, T. Yamao, Y. Kitano, T. Miyata, K. Arima, H. Okabe, Y. Baba, A. Chikamoto, T. Ishiko, M. Hirota and H. Baba, Cancer Sci., 2019, 110, 310–320 CrossRef CAS PubMed.
- B. Gündel, X. Liu, M. Löhr and R. Heuchel, Front. Cell Dev. Biol., 2021, 9, 741162 CrossRef PubMed.
- O. E. Atat, Z. Farzaneh, M. Pourhamzeh, F. Taki, R. Abi-Habib, M. Vosough and M. El-Sibai, Hum. Cell, 2022, 35, 23–36 CrossRef CAS PubMed.
- L. A. Kunz-Schughart, P. Heyder, J. Schroeder and R. Knuechel, Exp. Cell Res., 2001, 266, 74–86 CrossRef CAS PubMed.
- H. Y. Tanaka, T. Kurihara, T. Nakazawa, M. Matsusaki, A. Masamune and M. R. Kano, Biomaterials, 2020, 251, 120077 CrossRef CAS PubMed.
- M. J. Ware, V. Keshishian, J. J. Law, J. C. Ho, C. A. Favela, P. Rees, B. Smith, S. Mohammad, R. F. Hwang, K. Rajapakshe, C. Coarfa, S. Huang, D. P. Edwards, S. J. Corr, B. Godin and S. A. Curley, Biomaterials, 2016, 108, 129–142 CrossRef CAS PubMed.
- G. Lazzari, V. Nicolas, M. Matsusaki, M. Akashi, P. Couvreur and S. Mura, Acta Biomater., 2018, 78, 296–307 CrossRef CAS PubMed.
- B. Xie, J. Hänsel, V. Mundorf, J. Betz, I. Reimche, M. Erkan, I. Büdeyri, A. Gesell, R. G. Kerr and N. P. Ariantari, et al. , Bioengineering, 2020, 7(2), 57 CrossRef CAS PubMed.
- X. Liu, B. Gündel, X. Li, J. Liu, A. Wright, M. Löhr, G. Arvidsson and R. Heuchel, Transl. Oncol., 2021, 14, 101107 CrossRef CAS PubMed.
- G. Giustarini, G. Teng, A. Pavesi and G. Adriani, Front. Oncol., 2023, 13, 1156769 CrossRef CAS PubMed.
- N. Bidan, G. Dunsmore, M. Ugrinic, M. Bied, M. Moreira, C. Deloménie, F. Ginhoux, C. Blériot, M. de la Fuente and S. Mura, Drug Delivery Transl. Res., 2024, 14, 2085–2099 CrossRef CAS PubMed.
- N. H. Madsen, B. S. Nielsen, S. L. Nhat, S. Skov, M. Gad and J. Larsen, Pathogens, 2021, 10, 969 CrossRef CAS PubMed.
- J. Kuen, D. Darowski, T. Kluge and M. Majety, PLoS One, 2017, 12, e0182039 CrossRef PubMed.
- A. Zeöld, G. O. Sándor, A. Kiss, A. Soós, T. Tölgyes, A. Bursics, Á. Szűcs, L. Harsányi, Á. Kittel, A. Gézsi, E. I. Buzás and Z. Wiener, Cell. Mol. Life Sci., 2021, 78, 3005–3020 CrossRef PubMed.
- A. Sontheimer-Phelps, B. A. Hassell and D. E. Ingber, Nat. Rev. Cancer, 2019, 19, 65–81 Search PubMed.
- M. Geyer and K. Queiroz, Front. Cell Dev. Biol., 2021, 9, 761807 CrossRef PubMed.
- H. Xu, J. Wen, J. Yang, S. Zhou, Y. Li, K. Xu, W. Li and S. Li, Cell Commun. Signaling, 2024, 22, 515 CrossRef PubMed.
- B. Schuster, M. Junkin, S. S. Kashaf, I. Romero-Calvo, K. Kirby, J. Matthews, C. R. Weber, A. Rzhetsky, K. P. White and S. Tay, Nat. Commun., 2020, 11, 5271 CrossRef CAS PubMed.
- Y. Zhu, D. Jiang, Y. Qiu, X. Liu, Y. Bian, S. Tian, X. Wang, K. J. Hsia, H. Wan, L. Zhuang and P. Wang, Bioact. Mater., 2024, 39, 59–73 CAS.
- V. Sgarminato, S. L. Marasso, M. Cocuzza, G. Scordo, A. Ballesio, G. Ciardelli and C. Tonda-Turo, Biomater. Sci., 2023, 11, 208–224 RSC.
- M. Geyer, L. M. Gaul, S. L. D Agosto, V. Corbo and K. Queiroz, Front. Immunol., 2023, 14, 1155085 Search PubMed.
- M. Geyer, D. Schreyer, L. M. Gaul, S. Pfeffer, C. Pilarsky and K. Queiroz, Cell Death Discovery, 2023, 9, 20 Search PubMed.
- M. R. Haque, C. R. Wessel, D. D. Leary, C. Wang, A. Bhushan and F. Bishehsari, Microsyst. Nanoeng., 2022, 8, 36 Search PubMed.
- C. R. Drifka, K. W. Eliceiri, S. M. Weber and W. J. Kao, Lab Chip, 2013, 13, 3965–3975 Search PubMed.
- D. Gioeli, C. J. Snow, M. B. Simmers, S. A. Hoang, R. A. Figler, J. A. Allende, D. G. Roller, J. T. Parsons, J. D. Wulfkuhle, E. F. Petricoin, T. W. Bauer and B. R. Wamhoff, Lab Chip, 2019, 19, 1193–1204 RSC.
- F. L. Lai Benjamin, X. Lu Rick, Y. Hu, H. L. Davenport, W. Dou, E. Y. Wang, N. Radulovich, M. S. Tsao, Y. Sun and M. Radisic, Adv. Funct. Mater., 2020, 30(48), 2000545 CrossRef PubMed.
- P. A. Marks and R. Breslow, Nat. Biotechnol., 2007, 25, 84–90 CrossRef CAS PubMed.
- K. Schiedlauske, A. Deipenbrock, M. Pflieger, A. Hamacher, J. Hänsel, M. U. Kassack, T. Kurz and N. E. Teusch, Pharmaceuticals, 2024, 17, 752 CrossRef CAS PubMed.
- B. Schmid, J. Schindelin, A. Cardona, M. Longair and M. Heisenberg, BMC Bioinf., 2010, 11, 274 CrossRef PubMed.
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld, B. Schmid, J. Y. Tinevez, D. J. White, V. Hartenstein, K. Eliceiri, P. Tomancak and A. Cardona, Nat. Methods, 2012, 9, 676–682 CrossRef CAS PubMed.
- S. B. Campbell, Q. Wu, J. Yazbeck, C. Liu, S. Okhovatian and M. Radisic, ACS Biomater. Sci. Eng., 2021, 7, 2880–2899 CrossRef CAS PubMed.
- W. J. Ho, E. M. Jaffee and L. Zheng, Nat. Rev. Clin. Oncol., 2020, 17, 527–540 CrossRef PubMed.
- Y. Bi, V. S. Shirure, R. Liu, C. Cunningham, L. Ding, J. M. Meacham, S. P. Goedegebuure, S. C. George and R. C. Fields, Integr. Biol., 2020, 12, 221–232 CrossRef PubMed.
- X. S. Xiang, P. C. Li, W. Q. Wang and L. Liu, Biochim. Biophys. Acta, Rev. Cancer, 2022, 1877, 188676 CrossRef CAS PubMed.
- M. Yu, R. Guan, W. Hong, Y. Zhou, Y. Lin, H. Jin, B. Hou and Z. Jian, Cancer Manage. Res., 2019, 11, 4041–4058 CrossRef PubMed.
- G. D'Errico, M. Alonso-Nocelo, M. Vallespinos, P. C. Hermann, S. Alcalá, C. P. García, L. Martin-Hijano, S. Valle, J. Earl, C. Cassiano, L. Lombardia, J. Feliu, M. C. Monti, T. Seufferlein, L. García-Bermejo, P. Martinelli, A. Carrato and B. Sainz, Oncogene, 2019, 38, 5469–5485 CrossRef PubMed.
- Z. Hussain, T. Bertran, P. Finetti, E. Lohmann, E. Mamessier, G. Bidaut, F. Bertucci, M. Rego and R. Tomasini, Cell Commun. Signaling, 2024, 22, 1 CrossRef CAS PubMed.
- S. A. Väyrynen, J. Zhang, C. Yuan, J. P. Väyrynen, A. Dias Costa, H. Williams, V. Morales-Oyarvide, M. C. Lau, D. A. Rubinson, R. F. Dunne, M. M. Kozak, W. Wang, D. Agostini-Vulaj, M. G. Drage, L. Brais, E. Reilly, O. Rahma, T. Clancy, J. Wang, D. C. Linehan, A. J. Aguirre, C. S. Fuchs, L. M. Coussens, D. T. Chang, A. C. Koong, A. F. Hezel, S. Ogino, J. A. Nowak and B. M. Wolpin, Clin. Cancer Res., 2021, 27, 1069–1081 CrossRef PubMed.
- X. Ma, D. Wu, S. Zhou, F. Wan, H. Liu, X. Xu, X. Xu, Y. Zhao and M. Tang, Oncol. Rep., 2016, 35, 189–196 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4lc01016g |
| This journal is © The Royal Society of Chemistry 2025 |