
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAutomated electrochemical oxygen sensing using a 3D-printed microfluidic lab-on-a-chip system†
Daniel
Kaufman
 a,
Steffen
Winkler
bc,
Christopher
Heuer
bc,
Ahed
Shibli
a,
Alexander
Snezhko
a,
Gideon I.
Livshits
a,
Janina
Bahnemann
*bc and
Hadar
Ben-Yoav
*a
a,
Steffen
Winkler
bc,
Christopher
Heuer
bc,
Ahed
Shibli
a,
Alexander
Snezhko
a,
Gideon I.
Livshits
a,
Janina
Bahnemann
*bc and
Hadar
Ben-Yoav
*a
aNanobioelectronics Laboratory (NBEL), Department of Biomedical Engineering, Ilse Katz Institute for Nanoscale Science and Technology, Ben-Gurion University of the Negev, 8410501 Beer Sheva, Israel. E-mail: benyoav@bgu.ac.il
bInstitute of Physics, University of Augsburg, Universitätsstraße 1, 86159 Augsburg, Germany. E-mail: janina.bahnemann@uni-a.de
cCentre for Advanced Analytics and Predictive Sciences (CAAPS), University of Augsburg, 8615, Augsburg, Germany
First published on 28th December 2024
Abstract
Dissolved oxygen is crucial for metabolism, growth, and other complex physiological and pathological processes; however, standard physiological models (such as organ-on-chip systems) often use ambient oxygen levels, which do not reflect the lower levels that are typically found in vivo. Additionally, the local generation of reactive oxygen species (ROS; a key factor in physiological systems) is often overlooked in biology-mimicking models. Here, we present a microfluidic system that integrates electrochemical dissolved oxygen sensors with lab-on-a-chip technology to monitor the physiological oxygen concentrations and generate hydrogen peroxide (H2O2; a specific ROS). This microfluidic lab-on-a-chip system was fabricated using high-resolution 3D printing technology in a one-step process. It incorporates a micromixer, an on-chip bubble-trap, an electrochemical cell with fabricated gold or platinum black-coated working electrodes as well as an Ag/AgCl reference electrode, and a commercial optical oxygen sensor for validation. This device enables an automated variation of the oxygen levels as well as sensitive electrochemical oxygen monitoring (limit of detection = 11.9 ± 0.3 μM), with a statistically significant correlation with the optical sensor. The proposed system can serve as a tool to characterize and evaluate custom-made electrodes. Indeed, we envision that in the future it will be used to regulate dissolved oxygen levels and oxygen species in real time in organ-on-chip systems.
Introduction
The dissolved oxygen (dO2) concentration is one of the most critical parameters in biological systems, in part because it greatly influences metabolism and growth, but also because it impacts a host of other even more complex processes, for example, blood vessel formation (angiogenesis).1 However, in standard cell culture systems the oxygen level is often set at ambient levels of 21% (215 μM dO2), which does not accurately reflect physiological conditions in the human body.2 In fact, dO2 levels in vivo are typically much lower, ranging from 2 to 9% dO2 in most tissues and microbiomes. This discrepancy highlights that cell culture environments frequently use dO2 concentrations that are much higher than those found in natural physiological conditions, which may detrimentally impact both the relevance and the accuracy of in vitro experiments.1,3,4 As a result, the precise dO2 concentration should be assessed and monitored locally in the area of interest, and then adjusted accordingly – a requirement that we addressed here by developing a microfluidic lab-on-a-chip (LOC) system suitable for automated dO2 sensing.Analytical biomaterial-integrated LOC devices are an emerging technology that is increasingly being used to study and monitor the conditions in natural microbial communities or cell culture environments.5,6 Importantly, they offer researchers a host of versatile applications ranging from biomedical diagnostics6–9 to bioprocess development10–12 and organ-on-chip micro-systems.13–16 These microfluidic systems offer significant advantages including precise control17,18 and manipulation of small fluid volumes16–19 that enhance accuracy and reduce the degree of reagent and sample consumption in experimental settings. These systems can integrate multiple laboratory functions into a miniaturized chip, facilitating more automation,20–22 with improved reproducibility23 and greater portability for in-field applications.24
Oxygen sensing in microenvironments is typically achieved using optical and electrochemical sensors integrated into LOC technology. Optical sensors (which typically rely on fluorescent dyes) are more precise in sensing low dO2 levels25,26 and are generally less degradable than electrochemical sensors.26–28 Moreover, integrating optical sensors into microfluidic systems is often quite straightforward (for example, using sensor spots29,30) and many studies have reported the use of these sensors to measure the dO2 concentrations in microfluidic channels.29,31–35 In contrast, electrochemical sensors are based on the oxygen reduction reaction (ORR) and thus have a higher sensitivity to fluctuations in dO2 levels,27 a much faster response time,36–38 and they also tend to be more cost-effective in practice.36,39,40 These properties make electrochemical sensors more suitable for LOC and organ-on-chip platforms, where local and fast-response monitoring is important for tracking heterogeneous and dynamic conditions.41,42 Moreover, under specific oxygen concentration conditions, electrodes can generate reactive oxygen species (ROS) such as superoxides (O2−) and hydrogen peroxides (H2O2) via the oxygen reduction pathway,43–46 which is a useful capability that facilitates more accurate mimicking of organ or tissue physiological conditions (for example, in microfluidic systems). Yet despite these intriguing and useful characteristics, although several studies have implemented electrochemical sensors in bioreactors to sense dO2 levels,47–49 their integration into microfluidics remains challenging due to difficulties in miniaturization, integration, and stability.50,51 Hence, examples of label-free electrochemical dO2 sensors in such devices are scarce52 and, to the best of our knowledge, there is no report of physiologically relevant dO2 levels in situ in a microfluidic device using label-free electrodes.
Improvements in resolution of 3D printing technology offers new opportunities to fabricate microfluidic systems for sensor integration with considerably reduced development times. These improvements enable fabrication of most complex 3D structures at the micrometer scale in a single-step process. However, challenges can be faced regarding the high surface roughness, which compromises leak-tight sealing and leads to an increased adherence of bubbles. Furthermore, for cell culture applications (e.g. organ-on-a-chip) the biocompatibility must be considered, which are not only dependent on the material itself but also on the post-processing, sterilization method and cell type.53
Here, we present a micro-system that combines electrochemical dO2 sensors with microfluidic LOC technology to monitor the physiological concentrations of dO2. This device combines different functional units (e.g., a micromixer, bubble trap, working (WE), and reference electrodes (RE), as well as a commercial dO2 sensor used as a reference). Moreover, it strategically leverages the advantages offered by microfluidics (e.g., enhanced control overflow dynamics and reduced reagent and sample volumes) as well as those advantages of electrochemical sensors (e.g., response time, sensitivity, and cost-effectiveness). By using two different types of electrodes, namely, gold (Au) as a control electrode for testing and platinum black (PtB) as the main sensing material, we demonstrated that this system can enable researchers to automatically adjust and accurately measure physiologically relevant dO2 concentrations (R2 > 0.997) with limits of detection as low as 11.9 ± 0.3 μM dO2 (equivalent to 1.16 ± 0.02% dO2, lower concentration than in most physiological tissues).1,3
Experimental
Chemicals and materials
Methanol (001368052100, Bio-Lab, Ltd.), acetone (376, Bio-Lab, Ltd.), potassium chloride (11595, Alfa Aesar), and 2-propanol (1301221, Bio-Lab, Ltd.), sulfuric acid (258105, Bio-Lab, Ltd.), potassium hydroxide (KOH, 1307471, BioLab, Ltd.), hydrogen peroxide (H2O2, 7722841, ThermoFisher), potassium hexacyanoferrate(II) trihydrate (‘Ferrocyanide’, 1.04984.0100, Merck), potassium hexacyanoferrate(III) (‘Ferricyanide’, 1.04973.0100, Merck), dihydrogen hexachloroplatinate(IV) hexahydrate (chloroplatinic acid; 011051, Alfa aesar), 99% lead(II) acetate trihydrate (lead acetate; A11746, Alfa Aesar), and sodium chloride (1.06404.1000, Merck) were used without further purification. Phosphate buffered saline (PBS) tablets (BP2944100, ThermoFisher) were used to prepare a 0.01 M PBS solution. All aqueous solutions were prepared with ultra-pure water (resistivity 18.2 MΩ cm at 25 °C) obtained from an ELGA PureLab water purification system (Veolia Water Technologies, United Kingdom).Lithography and fabrication of microelectrodes
Circular single-disk electrodes (1 mm radius) were patterned onto a 4-inch diameter glass wafer using Clewin4 (Clewin 6.1, Clewin, Inc.) layout software. The fabrication process involved photolithography and thin-film deposition to create an Au electrode on a prime-grade glass substrate (100 mm diameter, 500 μm thickness, double-side polished, University Wafer, Ltd.) (see Fig. S1A†). The glass substrate was first cleaned with acetone, isopropanol, and deionized (DI) water, followed by 2 minutes of oxygen plasma (Plasma Asher Diener, 192 W) treatment to remove organic residues. The substrate was then dehydrated on a contact hot plate at 110 °C for 10 minutes, and allowed to cool down to room temperature for 10 minutes. Next, an AZ-nLOF (product no. 97, Micro Chemicals) photoresist was spin-coated onto the substrate using a Universal Spin-Coating system (80RCDelta, SUSS MicroTec) ((1) 500 RPM with an acceleration of 100 RPM s−1 for 20 seconds, (2) 3000 RPM with an acceleration of 300 RPM s−1 for 40 seconds, and (3) 0 RPM with an acceleration of 100 RPM s−1 for 1 second). The coated substrate was left to settle at room temperature for 5 minutes, then pre-baked on a contact hot plate at 110 °C for 1 minute. After having been cooled to room temperature for 10 minutes, the substrate was exposed through a transparency mask (light flux of 8.7 mW cm−2 for 18 seconds using a Karl Suss Mask Aligner MA6 system, SUSS MicroTec). Following another 5-minute cooling period, a post-exposure bake was performed on the contact hot plate at 110 °C for 1 minute, with a subsequent 10-minutes cooling period. The exposed photoresist was then developed in AZ-726 MIF developer (Micro Chemicals) for 73 seconds, rinsed with distilled water for 1 minute, and dried with nitrogen gas before undergoing a final 2-minute oxygen plasma cleaning.Subsequently, 25 nm of chrome and 250 nm of Au were deposited onto the developed substrate using an E-gun deposition system (VST Service, Ltd.). The substrate was then immersed in a 70 °C NMP solution for 30 minutes, rinsed with distilled water, and dried with nitrogen gas. To define the microelectrode chamber, SU8-3005 (Gas Technologies & Chemicals) was used, enabling the chamber to be cleaned with an AMI (acetone, methanol, and isopropanol sequentially) solution without damage. First, the wafer was cleaned with acetone, isopropanol, and DI water, and then dehydrated on a contact hot plate at 110 °C for 5 minutes, and finally spin-coated with SU8-3005 ((1) 500 RPM with an acceleration of 100 RPM s−1 for 10 seconds, (2) 3000 RPM with an acceleration of 300 RPM s−1 for 30 seconds, and (3) 0 RPM with an acceleration of 100 RPM s−1 for 1 second). After the spin-coating, the substrate was allowed to settle at room temperature for 5 minutes, followed by a soft bake on a hot plate at 95 °C for 8 minutes, and then cooled to room temperature for 10 minutes. Next, the photoresist was exposed to light using a hard contact exposure of 8.7 mW cm−2 for 45.1 seconds with a Mask Aligner (MA6, SUSS MicroTec). Following a 5-minute cooling period, a post-exposure bake was performed at 95 °C for 4 minutes, with a subsequent 10-minute cooling period. The wafer was developed in PGMA/EBR developer (MetalChem Ltd.) solution for 6 minutes, washed in isopropanol for 10 seconds, and then hard-baked on a contact hot plate at 150 °C for 5 minutes to remove any hydration. After having been cooled to room temperature for 10 minutes, oxygen plasma cleaning (Plasma Asher Diener, 192 W, 2 minutes) was performed to eliminate residues or impurities. Finally, the processed substrate was diced into individual microchamber chips using a Dicer ADT-7100 (ADT) (see Fig. S1B†).54,55
Electrochemical measurements
All electrochemical measurements were performed using a PalmSens4 and MUX16 multiplexer plug-in (Palmsens, Ltd.) and a three-electrode cell configuration consisting of a microfabricated Au single electrode (working electrode; ‘WE’), a microfabricated Au single electrode (counter electrode; ‘CE’), and an externally introduced commercial Ag/AgCl electrode (CHI111P, CH Instruments, reference electrode; ‘RE’). All electrochemical potential values were measured versus the Ag/AgCl half-cell potential.Prior to the electrochemical testing, the microfabricated electrodes were cleaned via rinsing with AMI, and then cleaned again by dipping them in a solution containing 50 mM KOH and 25% v/v H2O2 for 10 min. Finally, the electrodes were rinsed with distilled water to remove the organic residues and dried using nitrogen gas.55,56
For systems validation and effective surface area measurements, cyclic voltammetry in the presence of 5 mM ferrocyanide (Fcn(R)) and 5 mM ferricyanide (Fcn(O)) redox couple solution was performed with a potential sweep between −0.2 V and 0.65 V vs. Ag/AgCl, at a scan rate of 0.1 V s−1, 3 voltammetric cycles for each electrode and flow state (1 mL min−1 in total). For dO2 sensing measurements, chronoamperometry (−0.5 V vs. Ag/AgCl, 1 minute) was performed for each electrode and similar flow rate.
Platinum black electrode modification
A platinum black (PtB) electrodeposition solution was prepared by mixing 1% dihydrogen hexachloroplatinate(IV) hexahydrate and 0.05% lead(II) acetate trihydrate in DI water, followed by stirring and adding 0.0025% of 32% hydrochloric acid. The solution was then covered with aluminum foil and stored at room temperature. Prior to modification, the microfabricated au electrode was cleaned with AMI solution, rinsed with DI water, and dried with nitrogen gas. PtB was electrodeposited onto the Au electrode by dipping it into the electrodeposition solution and applying chronopotentiometry (a current density of +230 A m−2 for 450 seconds) with a 30-second cooling period in the solution.57Atomic force microscopy imaging
Atomic force microscopy [AFM, NanoWizard 4, JPK Bruker, tip: HQ:NSC15/Cr-Au BS 325 kHz 40 N m−1], scanning electron microscopy [SEM, Thermo Fisher Verios 460 L field-emission scanning electron microscope, HV – 500 kV, current – 25 pA, det – TLD, mode - SE], and a stylus profilometer (Veeco Dektak 150) were used to study the PtB/Au morphology and surface coverage (Fig. S2 and S3†).Fabrication of the 3D-printed microfluidic lab-on-a-chip system
3D models of the 3D-printed parts were created using SOLIDWORKS CAD software (2022 edition). All parts (except for the gasket) were fabricated with the high-precision MultiJet 3D printer MJP 2500 Plus (3D Systems). This printer offers a resolution of 800 × 900 × 790 DPI in the xyz axes, with a layer thickness of 32 μm. For the model material, polyacrylate VisiJet M2S-HT90 Plus (3D Systems) was used, with VisiJet M2-SUP Plus wax (3D Systems) serving as the support material. After printing, the parts were post-processed to remove the support material as follows: first, the printed components were detached from the build plate by incubating them at −18 °C for 10 minutes. Then, the parts were placed in a preheated steam bath using the EasyClean system (3D Systems) for 45 minutes to melt the wax support material. Next, they were further treated using another EasyClean system by immersing them in paraffin oil at 65 °C for 30 minutes. To ensure that any remaining support material was fully removed from the microfluidic channels, the parts were processed in an ultrasonic paraffin oil bath (Bandelin electronic) at 65 °C for another 30 minutes. To remove any traces of the oil, the parts were washed in an ultrasonic bath (Bandelin electronic) with DI and 1% (v/v) detergent (Fairy Ultra Plus, Procter and Gamble). The process was repeated three times for 30 minutes each at 65 °C. Finally, the parts were rinsed in clean DI water to remove any detergent residues and then air-dried. During each cleaning step, the microfluidic channels were flushed with 5 mL of liquid using a standard syringe to refresh the fluid inside.The gasket was fabricated using a different high-resolution MultiJet printer, AGILISTA-3200 W (Keyence Deutschland GmbH), which has a resolution of 635 × 400 dots per inch and a layer thickness of 15 μm. For printing, the soft silicone material AR-G1L (Keyence Deutschland GmbH) was used as model material and AR-S1 (Keyence Deutschland GmbH) was used as support material. After printing, objects were incubated twice in an ultrasonic water bath (Bandelin electronic) for 30 minutes at 60 °C with detergent (Fairy Ultra Plus, Procter and Gamble), and then once more with DI water (Arium® Sartorius Stedim Biotech GmbH).
Lab-on-a-chip assembly and experimental setup
Prior to each experiment, the 3D-printed microfluidic lab-on-a-chip was assembled and connected to its peripheral equipment. Technical drawings of all printed parts are summarized in the ESI† in Fig. S4–S7. To create the on-chip bubble trap, a 10 × 10 mm2 sized sheet of an 80 μm thin standard polytetrafluoroethylene (PTFE; Avantor, Inc) membrane was attached to the chip by using a piece of double-sided medical grade adhesive tape (3 M 9877, 3 M Medical Solutions Division, Healthcare Business Group). Au/PtB electrodes were integrated using a 3D-printed lid, four manually tightened M3 screws, and a 3D-printed gasket. To integrate the Ag/AgCl RE (RE-1B, ALS Electrochemistry & Spectroelectrochemistry), a 3.5 × 1.5 mm o-ring (Fluorine Kautschuk Material (FKM), Shore A 75) was placed in its designated position above the RE chamber. Then, the RE was rinsed with DI water and plugged into the chip. Next, the electrochemical cell (WEs, CE, and RE) was connected to a potentiostat to record the electrochemical signal. The commercial oxygen SensorPlug (PreSens Precision Sensing GmbH) was also plugged into its port and tightened using a 3D-printed screw cap. The inlets and outlet of the assembled chip were then connected via PEEK 1/4 28′′ threaded fittings (IDEX Health & Science LLC) to standard PTFE tubing (ID 0.5 mm, OD 1.59 mm), and finally, the inlet PTFE tubing was connected via PEEK peristaltic tubing adapters for 1/16′′ OD PTFE tubing (IDEX Health & Science LLC) to 3-stop Tygon® Masterflex® Ismatec® LMT-55 pump tubing (ID: 0.51 mm; Masterflex SE). The PTFE and Tygon® tubing at the inlets had a length of 30 cm and 40 cm, respectively.After the lab-on-a-chip and the fluidic connections were assembled, the tubing, electrodes, and SensorPlug were all connected to their respective electronic devices. The pump tubing was installed on a Masterflex® Ismatec® Reglo ICC digital peristaltic pump (Masterflex SE) that features four independent pump channels, each equipped with twelve rollers. Prior to an experiment, the appropriate inner diameter of the tubing, the correct flow direction, and the flow rate unit in μL min−1 were all adjusted in the peristaltic pump settings. The electrodes were connected via crocodile clips to the potentiostat and controlled using PStrace version 5.9 software (Palmsens B.V.). To enable the automated measurement of three distinct WEs, an EmStatMUX8-R2 (Palmsens B.V.) multiplexer was used in between the electrodes and the potentiostat. The commercial dO2 sensor was connected to an OXY-1 ST oxygen transmitter (PreSens Precision Sensing GmbH) via an optical fiber 1 mm in diameter (PreSens Precision Sensing GmbH) and controlled via PreSens Measurement Studio 2 version 4.0.0.2353 software (PreSens Precision Sensing GmbH). The dO2 sensor was calibrated by using the intrinsic calibration function of the software; a deoxygenated PBS solution was set at 0%, and a PBS solution in equilibrium with the environment at 21%. All electronic devices were connected to a standard desktop computer via a universal serial bus (USB). The complete setup was controlled via custom Python scripts (see data availability statement – link for code).
Experimental procedure and data analysis for lab-on-a-chip validation and electrode surface determination
The experimental aim was to successfully detect changes in the redox-active analyte concentration using the developed LOC platform. To that end, we employed two reservoirs: (1) a 0.01 mM PBS solution (PBS, control), and (2) 5 mM Fcn(R) with 5 mM Fcn(O) in a 0.01 mM PBS solution (‘Fcn(O/R)’, which is a stable and electrochemically reversible redox couple). In the LOC, four single Au disk electrodes (r = 1 mm) were arranged in a row; the first electrode was designated as the CE and electrodes #2–4 were designed as WEs. For measurements using PtB-coated electrodes, WEs #2–4 were replaced by the PtB-coated electrodes. The concentration of Fcn(O/R) was varied by adjusting the flow rate ratios of the reservoirs (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0, 8:2, 6:4, 4:6, 2:8, and 0:10 sequentially), with a combined flow rate of 1 mL min−1 for both pumps. In each pumping step, a volume of 0.5 mL (pumping for 1 minute) was pumped into the chip, thereby ensuring a complete exchange of the present liquid inside the sensor chambers. The electrode's effective surface area was calculated using cyclic voltammetry and the Randles–Sevcik equation (eqn (1)):
:0, 8:2, 6:4, 4:6, 2:8, and 0:10 sequentially), with a combined flow rate of 1 mL min−1 for both pumps. In each pumping step, a volume of 0.5 mL (pumping for 1 minute) was pumped into the chip, thereby ensuring a complete exchange of the present liquid inside the sensor chambers. The electrode's effective surface area was calculated using cyclic voltammetry and the Randles–Sevcik equation (eqn (1)): | (1) |
Experimental procedure for automated dissolved oxygen sensing and the electrochemical oxygen reduction reaction
The experimental procedure for determining electrochemical ORR at different dO2 levels and continuous dO2 monitoring included two reservoirs: (1) an oxygen-rich 0.01 mM PBS solution (O2, reservoir open to air), and (2) an oxygen-poor 0.01 mM PBS solution (N2, nitrogen gas bubbled into the reservoir for 1 hour). Three WEs, one CE, and one RE were integrated as described in “Experimental: Lab-on-a-chip assembly and experimental setup”. In this setup, dO2 was continuously measured during an experiment using an oxygen SensorPlug. The concentration of dO2 was varied by adjusting the flow rate ratios of the reservoirs (10:0, 8:2, 6:4, 4:6, 2:8, and 0:10 sequentially) with a combined flow rate of 1 mL min−1 for both pumps. Following the chronoamperometry technique, we calculated the total charge sensed by the electrode by summing the cathodic current from 10 to 60 seconds (excluding the first 10 seconds during system stabilization) and normalized this charge by determining the electrode's effective surface area. The charge density was subsequently plotted as a function of the dO2 concentration detected by the optical sensor. The limit of detection (LOD) was calculated using the following equation (eqn (2)):58 | (2) |
Results & discussion
Design and manufacturing of the 3D-printed electrochemical lab-on-a-chip platform
The 3D-printed LOC for automating dO2 level control and sensing was fabricated from a biocompatible 3D printing material;53 the LOC includes several custom-made design features (as illustrated in Fig. 1). Starting from the left to right (Fig. 1A), the chip incorporates two inlets suitable for feeding different ratios of oxygen-rich and oxygen-poor PBS solutions and for generating different predefined dO2 levels. Since laminar flow is present at the scale of the microchannels, which prevents complete axial mixing, a 3D-printed micromixer59 is integrated. A bubble-trap is embedded prior to the sensor chambers using a PTFE membrane and a thin adhesive tape. This membrane allows air bubbles to escape from the channel system, thus preventing them from entering the sensor chambers and potentially disturbing the electrochemical and optical measurements. The LOC platform itself includes four Au electrodes: one that serves as a CE and three as WEs. These electrodes are sealed to the chip using a 3D-printed lid and gasket; thus, a 2.7 μL large sensor chamber is formed. The RE is plugged into the chip and sealed by an o-ring (as during the experiments, it must be possible to plug the electrode in and out while avoiding degradation of the electrode and maintaining a stable reference potential call). The RE port includes a cylindrical attachment that ensures the exact alignment of this electrode with the top of its sensor chamber. Finally, the use of the commercially available oxygen SensorPlug enables online monitoring of dO2 concentrations and validates the performance of the electrochemical sensor. The tip of the SensorPlug includes a sensor foil with a fluorescent dye. By exciting the dye with light via an optical fiber, the oxygen-dependent phase shift can be measured. A screw cap is used to press the plug into the chip; the geometry of the sensor port helps to ensure that the sensor foil is properly aligned with the top of the chamber. Fig. 1B presents the assembled LOC design (with the placed CE and WEs), and Fig. 1C shows the LOC as it is attached to the microfluidic, electrochemical, and optical apparatus during an experiment.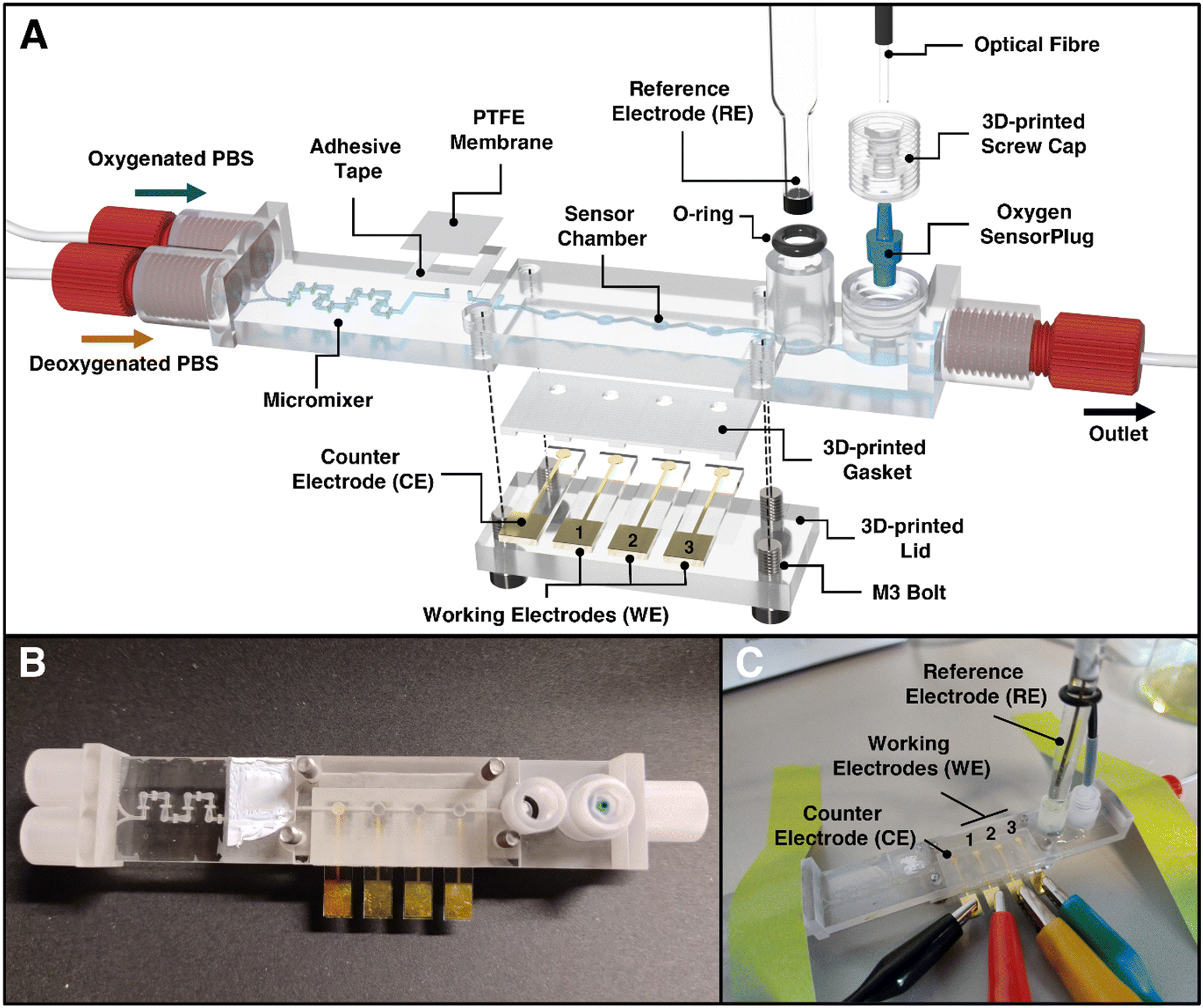 | ||
| Fig. 1 Illustration of the chip design of the 3D-printed microfluidic LOC for control and quantification of dissolved oxygen. (A) Rendered image of the LOC in exploded view illustrating its features and assembly. The LOC includes key components such as (1) HC-shaped micromixer from Enders et al.59 for rapid mixing of oxygenated and deoxygenated PBS, (2) a bubble-trap including a gas-permeable PTFE-membrane for the removal of bubbles, (3) the sensor chambers for gold and platinum black coated electrodes, (5) a glass reference electrode and (6) an opto-chemical oxygen sensor as reference. (B) Photograph of the assembled LOC in top view (RE not included). (C) Photograph of the assembled device during experiments. The device is assembled by simply (1) connecting the gold/platinum black coated electrodes to crocodile clamps, (2) plugging of the reference electrode into its o-ring sealed port and (3) inserting an oxygen SensorPlug, that is fixed using a screw cap and connected to an optical fiber for readout. For the dimensions of the LOC, see the technical drawings depicted in Fig. S4–S7.† | ||
Characterization of the lab-on-a-chip performance and the electrode's effective surface area
To ensure accurate reagent mixing and stable electrochemical signals using the LOC, we validated the system using the commonly used electro-active Fcn(O/R). A modified setup, with PBS and Fcn(O/R) reservoirs, without dO2 detection, was used (Fig. 2A). The setup illustrates the observed Fcn(O/R) levels of six different Fcn(O/R) ratios (10:0, 8:2, 6:4, 4:6, 2:8, 0:10). Each individual ratio was introduced into the system according to a predefined sequence: (1) feeding a desired mixture into the channel for 2 minutes, (2) no flow for 10 seconds to release residual pressure, (3) a cyclic voltammetry measurement of a single WE, and (4) 1 minute of flow (the same ratio). Steps 2–4 were repeated for each WE, and all steps were repeated once for every Fcn(O/R) ratio. For Au WEs, we obtained stable voltammograms with E1/2 = 0.21 V vs. Ag/AgCl – results that agree with the electrochemical behavior of Fcn(O/R) (Fig. 2B). We observed an increase in the cyclic voltammograms and anodic/cathodic current peaks as a function of the Fcn(O/R) solution flow rate (Fig. 2B1). When we plotted the anodic peak current as a function of the expected Fcn(O/R) concentration, we obtained a clear positive linear relationship (linear fitting; R2 = 0.996) (Fig. 2B2) that is in agreement with the Randles–Sevcik equation60 (eqn (1)). Using this equation, we could calculate the effective surface area of the WE and we arrived at values similar to the theoretical calculation (3.62 ± 0.12 mm2 and 3.14 mm2, respectively).
 | ||
| Fig. 2 LOC platform validation experiment. (A) Experimental setup where (Fcn(O/R); ‘FF’) and PBS (‘P’) are introduced by a peristaltic pump using different ratios and the electrodes synchronously sense the electro-active analyte, evaluating the Fcn(O/R) concentration and the electrode's effective surface area. The blue arrows indicate the fluid path, whereas the black lines indicate the electrical connections. (CE: Counter electrode; WE: working electrode; RE: reference electrode). (B) Cyclic voltammograms using Au electrodes as WEs. (1) Cyclic voltammograms using different flow ratios (P:FF) and (2) the anodic peak current as a function of the expected Fcn(O/R) concentration showing a positive linear relationship (R2 = 0.996), in agreement with the Rendles–Sevcik equation. (C) Cyclic voltammograms using coated platinum black (PtB) electrodes as WEs. (1) Cyclic voltammograms using different flow ratios (P:FF) and (2) the anodic peak current as a function of the expected Fcn(O/R) concentration showing a linear relationship (R2 = 0.997), in agreement with the Rendles–Sevcik equation. The error bars represent the mean (marker) and standard deviation (caps) of three WEs in the LOC platform. | ||
Then, we performed validation testing using the PtB-coated electrodes (Fig. 2C). We conducted cyclic voltammetry with similar parameters and obtained stable voltammograms with E1/2 = 0.2 V vs. Ag/AgCl, reflecting the electrochemical behavior of Fcn(O/R) (Fig. 2C1). As with the Au electrodes, we observed an increase in the cyclic voltammogram and the anodic/cathodic current peaks as a function of the Fcn(O/R) solution flow rate. A positive linear relationship (linear fitting; R2 = 0.997), consistent with the Randles–Sevcik equation (Fig. 2C2), was also obtained for the PtB-coated electrodes. The effective surface area of the PtB-coated electrodes was calculated as 7.58 ± 0.19 mm2 (2.1 times larger than the bare Au electrode surface). To validate the PtB effective surface area, we performed surface analysis including AFM, profilometer testing, and SEM imaging. A 1.47 ± 0.02 PtB/Au linear surface area ratio was detected using AFM on a 40 μm × 40 μm area (Experimental and Fig. S2 and S3†).
Calculating the surface area based on this ratio yielded 7.51 ± 0.01 mm2, a result similar to the electrochemically calculated value, and one that underscores the sensitivity of the given LOC design.
The obtained results demonstrate the capability of this system to both effectively measure and alter the concentration of electro-active analytes in an LOC microchannel using automated microfluidic pumping and electrochemical sensors.
Dissolved oxygen electrochemical detection in a concentration-monitored microfluidic lab-on-a-chip
We aimed to detect changes in the dO2 concentration in our LOC platform using the ORR mechanism.43–45 Here, the setup included an oxygen-rich (O2; open to air) and an oxygen-poor (N2; nitrogen gas bubbled into the reservoir for 1 hour) PBS solution, both of which were pumped into the system in varied ratios (Fig. 3A). The dO2 levels were monitored inside the microchannel using the oxygen SensorPlug and a corresponding oxygen transmitter. The signal of the dO2 sensor during one complete experiment is illustrated in Fig. 3B. | ||
| Fig. 3 Dissolved oxygen electrochemical detection in the microfluidic LOC. (A) Experimental setup where oxygen-rich (O2) and oxygen-poor (N2) are fed by a peristaltic pump at different ratios and the oxygen concentrations are measured by chronoamperometric quantification of the ORR reaction and the commercial oxygen SensorPlug. The blue arrows indicate the fluid path, whereas the black lines indicate electrical connections. (CE: Counter electrode; WE: working electrode; RE: reference electrode). (B) Oxygen signal of SensorPlug during a complete experiment illustrating decreased oxygen levels at higher fractions of the oxygen-poor solution. The purple circles emphasize the sequences of the electrochemical measurements. The zoom-in graph to a 20-minute time frame illustrates a measurement sequence performed for every ratio with (1) 10 minutes of flushing followed by sequence, (2) no flow for 10 seconds to release any residual pressure, (3) 1-minute measurement of a single WE, and (4) another 1 minute of flushing. Steps 2–4 were repeated for every WE. The black arrow indicates the stoppage of the pump and the blue arrow indicates the start of pumping. | ||
It illustrates the observed dO2 levels of six different dO2 ratios (10:0, 8:2, 6:4, 4:6, 2:8, 0:10). Each individual ratio is fed into the system according to a predefined sequence: (1) feeding a desired mixture into the channel for 10 minutes, (2) no flow for 10 seconds to release residual pressure, (3) a chronoamperometry measurement of a single WE, and (4) 1 minute of flow (the same ratio). Steps 2–4 were repeated for each WE, and all steps were repeated once for every oxygen ratio. As expected, the dO2 levels decreased when there was a higher proportion of the N2-gasified solution; however, a relatively long flush time of 10 minutes was required for each curve to saturate.
Furthermore, stopping the pump for electrochemical measurements was found to lead to a relatively fast increase in the dO2 levels in the absence of flow, especially with low dO2 concentrations – as shown in the magnified graph in Fig. 3B. Both of these observations can be attributed to the presence of residual oxygen (e.g., air micro-bubbles trapped in the relatively rough 3D-printing material or inside the material itself) diffusing into the system and moving towards the dO2 sensor.61,62 Consequently, the pumping sequence includes an additional minute of pumping (step 4) to reduce the increased dO2 levels observed from the measurement with the first WE to the last. The primary reason for the prolonged flushing, extended saturation phase and increasing dO2 signals after stopping the flow is likely the presence of residual oxygen within the material. This effect might be investigated and mitigated by flushing the system with deoxygenated solutions for several hours. Particularly, a 1% sulfite solution could be used to chemically remove oxygen from the solution and the whole system.63,64 Diffusion of oxygen through the tubing as another explanation for the prolonged saturation time is unlikely, as the oxygen level rises sharply immediately after the pump is stopped, and the diffusion rate of oxygen in water over distances of several centimeters is too low for this to occur. Additionally, axial dispersion has been identified as another reason for extended flushing times, consistent with observations in experiments with Fcn(O/R), where 500 μL were needed to flush the system prior to a measurement point and to reach a desired concentration. Axial dispersion arises due to the spreading of the solute band along the direction of flow due to velocity gradients of the laminar flow. This effect is amplified by longer channel networks, increased volumes in front of the sensor, and the presence of chambers with higher volumes or dead zones, such as bubble traps. Mitigation of axial dispersion could be achieved by reducing the system's length and volume or by introducing gases via a membrane positioned in close proximity to the sensor chambers, as demonstrated for chromatography65 or in alveoli-inspired lung-on-a-chip.66
The results of chronoamperometry measurements (step 3) are exemplarily shown for a single Au electrode in Fig. 4A1. As expected, a reduction in the cathodic current, along with decreased dO2 levels, was observed. To determine the amount of dO2via the ORR, we calculated the total charge sensed by the electrode and normalized it against the electrode's effective surface area, minimizing differences and accounting for electrode geometry. The resulting charge density as a function of the dO2 concentration measured by the optical sensor displayed a clear linear relationship (linear fitting; R2 = 0.997) and an LOD of 15.4 ± 0.4 μM of dO2 (Fig. 4A2). To test the stability of the LOC system, we repeated the experiment after 24 hours. For that matter, we kept PBS solution in the channel for 24 hours, followed by repetition of the same experimental steps. The system was open to air and the electrochemical and optical sensors were not disassembled from the LOC. We replicated the data analysis on the new results and received similar fitted equations of −14.3–0.1 × dO2 (day 1) and −14.48–0.12 × dO2 (day 2), respectively, as shown in Fig. 4A3 and S8B.†
 | ||
| Fig. 4 Results of ORR-based oxygen sensing using (A) Au WEs and (B) PtB-coated WEs. (1) ORR chronoamperograms using different flow rate ratios of oxygen-poor and oxygen-rich solutions, (2) charge density as a function of the optical oxygen sensor readings, showing a clear linear relationship (Au: −14.3–0.11 × dO2, R2 = 0.997; LOD = 15.4 ± 0.4 μM; PtB: −13.7–0.05 × dO2, R2 = 0.998; LOD = 11.2 ± 0.3 μM), and (3) a stability test of the electrochemical LOC showing the change in the charge density as a function of the oxygen concentration on day 1 (orange) and time day 2 (blue) showing no significant difference between the calls. All error bars represent the mean (marker) and standard deviation (caps) of 3 WEs using the LOC platform. LOD is calculated by eqn (2) and the error represents the standard deviation. | ||
Then, electrochemical measurements of oxygen-rich/oxygen-poor solutions using PtB-coated WEs were performed. The generated current decreased with lower dO2 levels (Fig. 4B1). Normalization and plotting of the charge density revealed a clear linear relationship to the optically measured dO2 levels of up to 225 μM dO2 (linear fitting; R2 = 0.997), along with a low LOD of 11.9 ± 0.3 μM dO2 (Fig. 4B2). At higher dO2 concentrations (>230 μM), we observed a significantly higher cathodic charge density likely due to the high sensitivity of platinum to elevated dO2 levels (Fig. S8A†). To test the stability of the LOC system, we repeated the experiment after 24 h. For that matter, we repeated the conditions executed for the experiment with Au electrodes.
The data analysis once again produced similar fitted equations of −13.7–0.05 × dO2 (day 1) and −12.1–0.04 × dO2 (day 2), respectively, as shown in Fig. 4B3 and S8C.†
Conclusions
This work demonstrates, for the first time, an automated LOC platform suitable for electrochemical monitoring of dO2 levels in a microfluidic environment. This microfluidic device consists of an innovative mixer unit, a simplistic and effective bubble trap, three WEs, CE and RE, as well as an optical oxygen SensorPlug that serves as a reference to the electrochemical dO2 sensing. This assembled device was observed to synchronously control the flow of two fluid inlets, mixing the solution using different flow rates and accurately measuring the redox active analyte concentration at a specific location in the channel. In addition, we developed a unified computer code to automatically control both the flow rate ratios introduced by a peristaltic pump and the electrochemical and optical sensors, making this system fully operational without any human intervention.This device was first evaluated using a common electrochemically reversible analyte Fcn(O/R) and was found to demonstrate precise concentration detection. Subsequently, we demonstrated the ability of this device to introduce various dO2 concentration solutions by mixing oxygenated and non-oxygenated buffer solution at different flow rate ratios, whereas the electrochemical and optical sensors in the LOC locally monitored the dO2 concentration along various points within the platform.
The results of our experiments demonstrate the system ability to automatically control the dO2 levels while also synchronizing optical sensing and electrochemical measurements. Both the Au and PtB WEs are reusable, and provided a clear linear calibration curve with low limits of detection, showing higher sensitivity than other electrochemical in situ oxygen sensing platforms.47 The approach of coating biocompatible materials to increase the selectivity of the electrochemical sensor to the target analyte can be expended to other biochemical molecules with physiological relevance, such as antioxidants and reactive oxygen species. We also believe that simultaneous use of optical and electrochemical sensors in our device may enhance data collection, improve information accuracy, and allow for error correction, resulting in more precise sensing of biochemical analytes.
Using 3D printing, the fabrication of the LOC, including post-processing, was completed within a few hours, significantly reducing development time compared to traditional fabrication methods (e.g., lithography, molding, milling). However, challenges were encountered due to high surface roughness, which compromised leak-tight sealing and increased bubble adhesion. Additionally, for future cell culture applications (e.g., organ-on-a-chip), biocompatibility must be carefully considered, as it depends not only on the material but also on post-processing, sterilization methods, and the specific cell type used.53
The ability of the system to present 0% dO2 can enable the full range of hypoxia and normoxia conditions that are present in the human body. The low LOD (equivalent to 1.16 ± 0.02% dO2) enables tracking and control of physiological and pathological environment in most of the tissues/microbiome studies. However, the implementation of cells/microbiota in our device may alter the sensing capabilities of electrochemical and optical sensors. Another challenge may be presented as biologically induced fast changes in oxygen concentrations67,68 and creation of radial oxygen gradient in the microchannel.68,69
Owing to the nature of 3D printing, enabling rapid prototyping, it should be noted that this LOC system can potentially be customized to incorporate various sensor types (e.g., custom-made electrodes) and their performance at varying conditions can be tested. As such, the LOC holds significant promise for use within biosensor development research (in terms of oxygen control and monitoring, ROS generation and tracking as well as other oxygen-dependent physiological processes, generally or locally evoked) and aid precise mimicking of oxygen dependent human physiology and pathologies. In terms of oxygen monitoring, we envision that this system can be applied to regulate the dO2 levels in microphysiological systems; however, it is essential that the dO2 levels and the dO2 local gradients match the relevant physiological conditions.
Data availability
The data supporting this article have been included as part of the (ESI†). The python code and instructions for automated flow and electrochemistry experiments can be found at: https://github.com/NBELbenyoav/Automated-Electrochemical-Oxygen-Sensing.Author contributions
Daniel Kaufman: conceptualization, methodology, data analysis, validation, and writing the original draft. Steffen Winkler: methodology, validation, 3D design, and printing. Christopher Heuer: methodology, discussion, and revision. Ahed Shibli: coding and designing the LOC. Alexander Snezhko: designing and fabricating the electrodes. Gideon I. Livshits: atomic force microscopy imaging and image processing. Janina Bahnemann & Hadar Ben-Yoav: conceptualization, project administration, funding acquisition, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Ilse Katz Institute for Nanoscale Science & Technology and the Nano-Fabrication Center for the fabrication equipment and help with the fabrication process. We also wish to thank the Israel Innovation Authority (grant #75700) and the Kreitman School for Advanced Studies Doctoral Scholarship for funding this work. We also want to thank the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation, SFB/TRR-298-SIIRI – Project-ID: 426335750) for funding this work. Several panels for Fig. 2 and 3 were created in https://Biorender.com.Notes and references
- A. Mohyeldin, T. Garzón-Muvdi and A. Quiñones-Hinojosa, Cell Stem Cell, 2010, 7, 150–161 CrossRef PubMed.
- K. R. Rivera, M. A. Yokus, P. D. Erb, V. A. Pozdin and M. Daniele, Analyst, 2019, 144, 3190–3215 RSC.
- S.-P. Hung, J. H. Ho, Y.-R. V. Shih, T. Lo and O. K. Lee, J. Orthop. Res., 2012, 30, 260–266 CrossRef.
- M. D. Brennan, M. L. Rexius-Hall, L. J. Elgass and D. T. Eddington, Lab Chip, 2014, 14, 4305–4318 RSC.
- B. K. Nahak, A. Mishra, S. Preetam and A. Tiwari, ACS Appl. Bio Mater., 2022, 5, 3576–3607 CrossRef PubMed.
- S. Preetam, B. K. Nahak, S. Patra, D. C. Toncu, S. Park, M. Syväjärvi, G. Orive and A. Tiwari, Biosens. Bioelectron.:X, 2022, 10, 100106 Search PubMed.
- Z. Razavi, M. Soltani, H. Pazoki-Toroudi and P. Chen, Sens. Actuators, A, 2024, 376, 115625 CrossRef.
- C. Heuer, J.-A. Preuss, M. Buttkewitz, T. Scheper, E. Segal and J. Bahnemann, Lab Chip, 2022, 22, 4950–4961 RSC.
- K. Awawdeh, M. A. Buttkewitz, J. Bahnemann and E. Segal, Microsyst. Nanoeng., 2024, 10, 100 CrossRef PubMed.
- J. Schellenberg, M. Dehne, F. Lange, T. Scheper, D. Solle and J. Bahnemann, Bioengineering, 2023, 10, 656 CrossRef PubMed.
- M. P. Marques and N. Szita, Curr. Opin. Chem. Eng., 2017, 18, 61–68 CrossRef.
- A. Enders, A. Grünberger and J. Bahnemann, Mol. Biotechnol., 2024, 66, 365–377 CrossRef.
- D. Kaufman and H. Ben-Yoav, Curr. Opin. Electrochem., 2024, 44, 101442 CrossRef.
- S. Jalili-Firoozinezhad, F. S. Gazzaniga, E. L. Calamari, D. M. Camacho, C. W. Fadel, A. Bein, B. Swenor, B. Nestor, M. J. Cronce, A. Tovaglieri, O. Levy, K. E. Gregory, D. T. Breault, J. M. S. Cabral, D. L. Kasper, R. Novak and D. E. Ingber, Nat. Biomed. Eng., 2019, 3, 520–531 Search PubMed.
- P. Shah, J. V. Fritz, E. Glaab, M. S. Desai, K. Greenhalgh, A. Frachet, M. Niegowska, M. Estes, C. Jäger, C. Seguin-Devaux, F. Zenhausern and P. Wilmes, Nat. Commun., 2016, 7, 11535 CrossRef.
- H. J. Kim, H. Li, J. J. Collins and D. E. Ingber, Proc. Natl. Acad. Sci. U. S. A., 2015, 113(1), E7–E15 Search PubMed.
- G. M. Whitesides, Nature, 2006, 442, 368–373 CrossRef PubMed.
- S. Xu, Z. Nie, M. Seo, P. Lewis, E. Kumacheva, H. A. Stone, P. Garstecki, D. B. Weibel, I. Gitlin and G. M. Whitesides, Angew. Chem., Int. Ed., 2005, 44, 724–728 CrossRef PubMed.
- D. B. Weibel and G. M. Whitesides, Curr. Opin. Chem. Biol., 2006, 10, 584–591 CrossRef.
- J. Kang, D. Lee, Y. J. Heo and W. K. Chung, Lab Chip, 2017, 17, 3891–3897 RSC.
- J. R. Goodell, J. P. McMullen, N. Zaborenko, J. R. Maloney, C.-X. Ho, K. F. Jensen, J. A. Porco and A. B. Beeler, J. Org. Chem., 2009, 74, 6169–6180 CrossRef.
- N. Zaborenko, M. W. Bedore, T. F. Jamison and K. F. Jensen, Org. Process Res. Dev., 2011, 15, 131–139 CrossRef.
- S. K. Sia and G. M. Whitesides, Electrophoresis, 2003, 24, 3563–3576 CrossRef PubMed.
- E. Samiei, M. Tabrizian and M. Hoorfar, Lab Chip, 2016, 16, 2376–2396 RSC.
- X. Wang and O. S. Wolfbeis, Chem. Soc. Rev., 2014, 43, 3666–3761 RSC.
- X. Wu, N. Zhou, Y. Chen, J. Sun, L. Lu, Q. Chen and C. Zuo, Light:Sci. Appl., 2024, 13, 237 CrossRef PubMed.
- Y. Wei, Y. Jiao, D. An, D. Li, W. Li and Q. Wei, Sensors, 2019, 19, 3995 CrossRef PubMed.
- X. Qi, D. Shu, F. Qiu and J. Chen, IEEE Sens. J., 2013, 13, 4279–4286 Search PubMed.
- B. Müller, P. Sulzer, M. Walch, H. Zirath, T. Buryška, M. Rothbauer, P. Ertl and T. Mayr, Sens. Actuators, B, 2021, 334, 129664 CrossRef.
- H. Zirath, M. Rothbauer, S. Spitz, B. Bachmann, C. Jordan, B. Müller, J. Ehgartner, E. Priglinger, S. Mühleder, H. Redl, W. Holnthoner, M. Harasek, T. Mayr and P. Ertl, Front. Physiol., 2018, 9, 815 CrossRef.
- J. W. Grate, B. Liu, R. T. Kelly, N. C. Anheier and T. M. Schmidt, ACS Sens., 2019, 4, 317–325 CrossRef PubMed.
- S. Matsumoto, A. R. Safitri, M. Danoy, T. Maekawa, H. Kinoshita, M. Shinohara, Y. Sakai, T. Fujii and E. Leclerc, Biotechnol. Prog., 2019, 35, e2854 CrossRef.
- L. Orcheston-Findlay, A. Hashemi, A. Garrill and V. Nock, Microelectron. Eng., 2018, 195, 107–113 CrossRef.
- Y. Ando, H. P. Ta, D. P. Yen, S.-S. Lee, S. Raola and K. Shen, Sci. Rep., 2017, 7, 15233 CrossRef.
- S.-H. Huang, K.-S. Huang and Y.-M. Liou, Microfluid. Nanofluid., 2016, 21, 3 CrossRef.
- E. Bakker and M. Telting-Diaz, Anal. Chem., 2002, 74, 2781–2800 CrossRef.
- O. Simoska and K. J. Stevenson, Analyst, 2019, 144, 6461–6478 RSC.
- J. Baranwal, B. Barse, G. Gatto, G. Broncova and A. Kumar, Chemosensors, 2022, 10, 363 CrossRef.
- U. Guth, W. Vonau and J. Zosel, Meas. Sci. Technol., 2009, 20, 042002 CrossRef.
- P. Bergveld, Sens. Actuators, B, 2003, 88, 1–20 CrossRef.
- S. K. Srivastava, G. W. Foo, N. Aggarwal and M. W. Chang, Biotechnol. Notes, 2024, 5, 8–12 CrossRef.
- Z. Li, J. Hui, P. Yang and H. Mao, Biosensors, 2022, 12, 370 CrossRef PubMed.
- R. Ma, G. Lin, Y. Zhou, Q. Liu, T. Zhang, G. Shan, M. Yang and J. Wang, npj Comput. Mater., 2019, 5, 1–15 CrossRef.
- S. Li, L. Shi, Y. Guo, J. Wang, D. Liu and S. Zhao, Chem. Sci., 2024, 15, 11188–11228 Search PubMed.
- J. Zhao, C. Fu, K. Ye, Z. Liang, F. Jiang, S. Shen, X. Zhao, L. Ma, Z. Shadike, X. Wang, J. Zhang and K. Jiang, Nat. Commun., 2022, 13, 685 CrossRef PubMed.
- Y. Guo, M. Yang, R.-C. Xie and R. G. Compton, Chem. Sci., 2021, 12, 397–406 RSC.
- J. M. Stine, L. A. Beardslee, R. M. Sathyam, W. E. Bentley and R. Ghodssi, Sens. Actuators, B, 2020, 320, 128381 CrossRef.
- M. Doering, L. L. Trinkies, J. Kieninger, M. Kraut, S. J. Rupitsch, R. Dittmeyer, G. A. Urban and A. Weltin, ACS Omega, 2024, 9, 19700–19711 CrossRef PubMed.
- F. Liebisch, A. Weltin, J. Marzioch, G. A. Urban and J. Kieninger, Sens. Actuators, B, 2020, 322, 128652 CrossRef.
- M. A. Buttkewitz, C. Heuer and J. Bahnemann, Curr. Opin. Biotechnol., 2023, 83, 102978 CrossRef PubMed.
- V. F. Annese and C. Hu, Micromachines, 2022, 13, 1923 CrossRef PubMed.
- O. Estrada-Leypon, A. Moya, A. Guimera, G. Gabriel, M. Agut, B. Sanchez and S. Borros, Bioelectrochemistry, 2015, 105, 56–64 CrossRef CAS PubMed.
- S. Winkler, K. V. Meyer, C. Heuer, C. Kortmann, M. Dehne and J. Bahnemann, Eng. Life Sci., 2022, 22, 699–708 CrossRef CAS PubMed.
- R. P. Shukla, R. Cazelles, D. L. Kelly and H. Ben-Yoav, Talanta, 2020, 209, 120560 CrossRef CAS PubMed.
- D. Kaufman, C.-Y. Chen, C.-Y. Tsao, Z. Zhao, A. Lavon, G. F. Payne, W. E. Bentley and H. Ben-Yoav, Biosens. Bioelectron., 2024, 262, 116546 CrossRef CAS PubMed.
- D. Kaufman and H. Ben-Yoav, Chem. Eng. J., 2023, 469, 143901 CrossRef CAS.
- R. P. Shukla, R. H. Belmaker, Y. Bersudsky and H. Ben-Yoav, J. Neural Transm., 2020, 127, 291–299 CrossRef CAS.
- G. L. Long and J. D. Winefordner, Anal. Chem., 1983, 55, 712A–724A CrossRef CAS.
- A. Enders, I. G. Siller, K. Urmann, M. R. Hoffmann and J. Bahnemann, Small, 2019, 15, 1804326 CrossRef.
- P. Zanello, Inorganic electrochemistry: theory, practice and application, Royal Society of Chemistry, Cambridge, 2003 Search PubMed.
- Y. Ying, M. P. Browne and M. Pumera, Sustainable Energy Fuels, 2020, 4, 3732–3738 RSC.
- S. Sarvari, D. McGee, R. O'Connell, O. Tseytlin, A. A. Bobko and M. Tseytlin, Mol. Imaging Biol., 2024, 26, 511–524 Search PubMed.
- G. T. F. Wong and L.-S. Zhang, Mar. Chem., 1992, 38, 109–116 CrossRef.
- B. Jiang, C. Ren, Y. Li, Y. Lu, W. Li, Y. Wu, Y. Gao, P. J. Ratcliffe, H. Liu and C. Zhang, JBIC, J. Biol. Inorg. Chem., 2011, 16, 267–274 CrossRef.
- W. Frenzel and I. Markeviciute, J. Chromatogr. A, 2017, 1479, 1–19 CrossRef PubMed.
- A. De, S. Paul, Y. N. Reddy, V. Sharma, J. Bhaumik and V. K. Tippavajhala, J. Drug Delivery Sci. Technol., 2022, 78, 103930 CrossRef.
- I. Vacca, Nat. Rev. Microbiol., 2017, 15, 574–575 CrossRef PubMed.
- L. Albenberg, T. V. Esipova, C. P. Judge, K. Bittinger, J. Chen, A. Laughlin, S. Grunberg, R. N. Baldassano, J. D. Lewis, H. Li, S. R. Thom, F. D. Bushman, S. A. Vinogradov and G. D. Wu, Gastroenterology, 2014, 147, 1055–1063.e8 CrossRef PubMed.
- R. Singhal and Y. M. Shah, J. Biol. Chem., 2020, 295, 10493–10505 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4lc00962b |
| This journal is © The Royal Society of Chemistry 2025 |