
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Long-term digital microfluidic chips for regulating macrophage cellular interactions in inflammation†
Oksana K.
Savchak
ab and
Burcu
Gumuscu
 *abc
*abc
aBiosensors and Devices Lab, Department of Biomedical Engineering, Eindhoven University of Technology, Eindhoven, Netherlands. E-mail: b.gumuscu@tue.nl; burcugumuscu@gmail.com
bInstitute of Complex Molecular Systems, Eindhoven University of Technology, Eindhoven, Netherlands
cEindhoven Artificial Intelligence Systems Institute, Eindhoven University of Technology, Eindhoven, Netherlands
First published on 4th March 2025
Abstract
We introduce a robust multilayer dielectric stack for digital microfluidic chips to withstand the humid conditions of cell culture incubators for at least 60 days. Consisting of a combination of 1 μm polyvinylidene difluoride and 5 μm SU-8 layers, the stack demonstrated high breakdown voltages up to 1600 V and minimal surface currents <30 nA at 100 V. Long-term stability and precision in liquid handling enabled us to study macrophage phenotype modulation, pro-inflammatory response induction in macrophage population with single cell cytokine quantification and testing of a potentially anti-inflammatory drug candidate TCB-2 and its influence on macrophage phenotype, morphology, and cytokine release. The multilayer dielectric stack offers a durable solution for long-term biological assays on digital microfluidic platforms.
Introduction
Inflammation is a fundamental biological response triggered when the body detects foreign materials or damage signals. Regulating inflammation involves a complex interplay of two major contributors: cells and biochemical molecules. Macrophage cells emerge as central to inflammation and tissue repair among the other key players. Upon encountering foreign materials or damage signals, macrophages undergo phenotypic changes that dictate their function. Initially, macrophages secreting biomarkers dominated by pro-inflammatory signals are recruited to clear debris and pathogens, followed by a transition to macrophages secreting biomarkers dominated by anti-inflammatory signals that promote tissue healing and resolution of inflammation.1,2 The inflammatory site undergoes continuous influx of newly recruited macrophages, which are not yet conditioned to a specific phenotype. Understanding the polarization process, as well as the inter-cellular signalling is challenging yet crucial for mitigating immune-related complications.3,4 In addition to cellular responses, biochemical molecules such as immunosuppressants and regulatory drugs play a significant role in managing inflammation. Cost-effective testing of the new drug candidates presents additional challenges due to the sophisticated cell culture models and precise measurement techniques required to study macrophage behaviour. As an interesting example, TCB-2, a selective serotonin 5-HT2A receptor agonist, shows promise as a novel candidate for modulating immune response to reduce inflammation.5,6 While other compounds, such as psilocybin, have been studied as anti-inflammatory agents and are believed to express their activity through the 5-HT2A receptors,7 they also interact with other 5-HT receptors, making it hard to discriminate the effect of 5-HT2A specific role. TCB-2, on the other hand is a highly selective and highly potent agonist of the 5-HT2A receptor. However, research into TCB-2 has been limited so far, being only scarcely studied in anti-inflammatory potential and macrophage polarization modulation for the body immune system.6,8 Exploring the potential of TCB-2 could lead to the development of more targeted and effective therapies, ultimately reducing immune-related complications. Traditional cell culturing well plates often involve laborious, expensive, and complex workflows, when it comes to studying the effects of drug candidates.9 While miniaturized cell cultures can better match biological scale and improve cost-efficiency, they often involve a trade-off between maintaining physiological relevance and ensuring spatial and temporal control in the experiments. This limitation restricts our understanding of macrophage behaviour and the ways to regulate the immune response.10 Digital microfluidic (DMF) chips have emerged as promising tools in biological studies, offering precise manipulation of cells and fluids at the microscale, facilitating the complex, dynamic microenvironments in nanoliter-volume droplets.11,12 Despite these advantages, DMF chips have not been widely used in longer-term cell studies yet. This limited adoption is primarily due to the short lifespan of the chips when exposed to the high humidity and mild temperature conditions necessary for cell culturing.13,14We introduce long-term cell culture-enabling DMF chips, a powerful approach that can bring spatiotemporal control to cell culture experiments for up to 60 days. The electrode design is shown in Fig. 1A, the chip contains 96 working electrodes on which the liquid manipulation and cell culturing are done. This technology leverages dielectric stacks above the electrodes, providing a higher dielectric constant for an increased capacitance of the material that allows to decrease the actuation voltage, as well as functional strength to increase the breakdown voltage (Fig. 1B and C). On DMF chips, we assess the phenotype changes in naïve macrophage cells seeded in a cell culture functionalized area (Fig. 1D). In addition, we examined the macrophage single-cell phenotype response when inducing inflammatory response, as well as treatment with an anti-inflammatory drug candidate TCB-2 of the macrophage cell culture (Fig. 1E). The introduction of long-term cell culture-compatible DMF chips offers new opportunities for spatiotemporally controlled cell culture conditions, providing valuable insights into macrophage behaviour in inflammation and beyond.
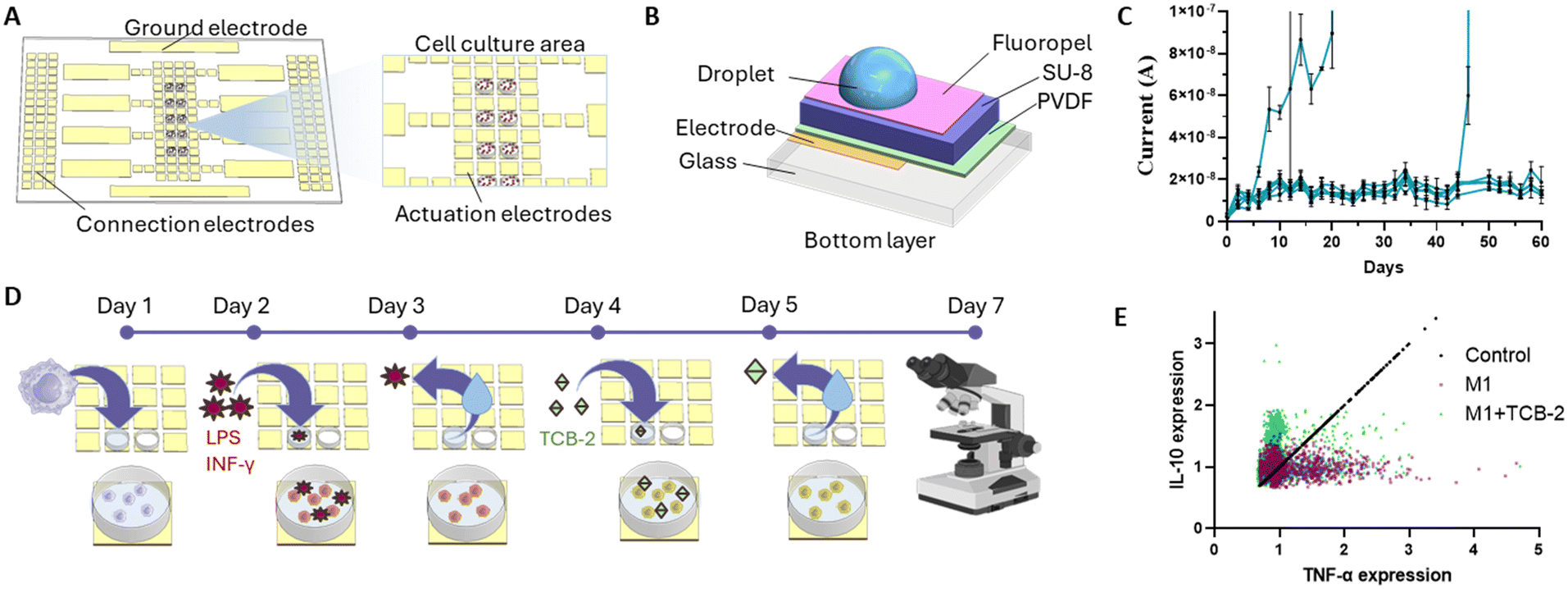 | ||
| Fig. 1 Long-term digital microfluidic platform. (A) Schematic overview of the chip layout and cell culture areas. (B) Cross section of the bottom part of the chip consisting of multiple dielectric layers structured on a glass substrate. (C) Current leakage measurement in PVDF (1 μm) + SU-8 (5 μm) chips over the course of 60 days. The chips were contained in a cell culture incubator during the experiment. Each blue line represents an individual measurement. The spiking current denotes the breakdown of a chip. (D) The workflow of the experiment where naïve macrophage population was treated with pro-inflammatory stimuli in order to undergo inflammatory polarization and later treated with a potentially anti-inflammatory drug TCB-2. (E) Quantitative analysis of macrophage cytokine expression over the course of 7 days based on TNFα and IL-10 expressions (n = 3). | ||
Results and discussion
Fabrication and characterization of cell culture-compatible DMF chips
Conventional DMF chips have a single layer dielectric structure that cannot withstand the warm and humid conditions of cell culture incubators for extended periods.13 Such chips usually consist of an electrode layer, a dielectric layer (Parylene C), and a hydrophobic layer (Teflon) from bottom to top, respectively. Although the dielectric layer prevents voltage dissipation to the top of the multilayer structure, conventional dielectric materials may not always provide adequate protection against humidity, which is crucial for cell culture applications. Polyvinylidene difluoride (PVDF) has been mostly preferred as a dielectric layer due to its high dielectric constant.15 However, the deposition technique and conditions affect the integrity of the PVDF layer, as well as the electrical stability of the material,16 leading to inconsistent performance in cell culturing over extended periods.We fabricated a dielectric stack consisting of PVDF and SU-8 to ensure structurally robust and high dielectric constant layer which can withstand the cell culture environment for extended period of time. To achieve this, we consistently fabricated PVDF and SU-8 with 1 μm and 5 μm thicknesses, respectively (Fig. 2A). Such layer thicknesses were selected based on the dielectric properties and estimated actuation voltages compared to the commercially available chips. These layers were always coated with a 65 nm-thick hydrophobic Fluoropel layer before the measurements (SD = 11 nm, n = 2, Fig. S1†). We quantitatively analysed the performance of each dielectric layer separately and in combination. The surface roughness of the 1 μm-thick PVDF layer was 40 nm (Fig. 2B). We measured no breakdown up until 1500 V (Fig. S2A†) and an actuation voltage of 70 V (Fig. 2C) on this layer. We attribute this to the Young–Lippmann equation,17,18 which indicates that reduced layer thickness results in lower actuation voltages. While the breakdown measurement might not reflect the DMF chip actuation due to the use of copper tape and not direct measurement of current in the solution on the surface of dielectric material, the voltage–current measurement performed in a saline solution droplet also reflected no evident breakdown up to 200 V, accompanied however by a high current output. Thus, applying 70 V actuation voltage resulted in a surface current of 100 μA/4 mm2, showing a moderate conductive behaviour of the material, which could potentially affect cellular response as it exceeds the threshold of 2 μA.19 We also found high variation in the current levels in the surface current, with an average of 21 μA/4 mm2 and 132% standard deviation (Fig. 2D), and decided not to use 1 μm-thick PVDF as sole dielectric. Next, we characterized the SU-8 layer. Once fully polymerized, SU-8 transitions to a glass-like state, providing isolation from humidity.20 The SU-8 layer exhibited a surface roughness of 100 nm without any visible structural defects after fabrication (Fig. 2B). The breakdown voltage and actuation voltage of the SU-8 layer were measured as 800 V (Fig. S2B†) and 90 V (Fig. 2D), respectively. The lower breakdown voltage and higher actuation voltage were expected, because the dielectric strength of SU-8 is 160 V μm−1,21 which is lower than that of PVDF (400–450 V μm−1).22 We also measured the surface current as 2.6 μA/4 mm2 at the actuation voltage, as well as a high increase after 110 V to 8 μA with further dielectric destabilization and high current output between 180 V and 200 V (Fig. 2D). Due to the high current-value with respect to the minimum cell current detection limit, as well as showing steep increase at voltages close to the actuation voltage, we did not use SU-8 as sole dielectric neither.
 | ||
| Fig. 2 Characterization of the operational parameters. (A) Schematic overview of material types and layer thicknesses (not to the scale). (B) Surface profile for each material combination, where σ denotes measured mean roughness of the surface. (C) Droplet actuation voltage determined by water contact angle as a function of voltage (n = 3). (D) The breakdown voltage of the dielectric material characterized by current as a function of voltage (n = 3). All parameters were characterized for SU-8 3005, polyvinylidene fluoride (PVDF), and combination of these materials. The grey shade represents the standard deviation. | ||
We integrated SU-8 and PVDF to leverage on their respective strengths, including PVDF's high dielectric constant and breakdown resistance and SU-8's notable water resistance, while mitigating their limitations—namely, the electrical fragility of SU-8 and the semi-conductive properties of PVDF. This approach enabled us to achieve an optimized dielectric stack with minimal layer thickness. We explored two configurations: PVDF as the base dielectric with SU-8 as the top layer (PVDF + SU-8), and vice versa. Both dielectric stacks showed significant improvements over the individual dielectrics, with no current leakage up to 200 V (Fig. 2D). The surface current output did not surpass 10 nA/4 mm2 for the SU-8 + PVDF dielectric stack and 30 nA/4 mm2 for the PVDF + SU-8 dielectric stack. The breakdown voltage for the SU-8 + PVDF dielectric stack was 1400 V (Fig. S2C†), while that of the PVDF + SU-8 dielectric stack was 1600 V (Fig. S2D†), indicating cumulative dielectric strengths of 233 V μm−1 and 267 V μm−1, respectively. The surface roughness was approximately 100 nm for the SU-8 + PVDF stack and 50 nm for the PVDF + SU-8 stack (Fig. 2B), suggesting that the higher surface roughness of SU-8 can be mitigated by spin-coating it onto the PVDF layer. Both dielectric stacks required approximately 100 V for droplet actuation, with the water contact angle change from the resting state exceeding 30° (Fig. 2C). The higher actuation voltage results from the increased thickness of the dielectric stack compared to the previously characterized single layers. The surface current levels are more than 60 fold lower than a lowest reference value proved to not affect cell viability when cells are stimulated by an electro-conductive surface (2 μA),19 making the stack safe to use with biological samples without influencing the results by the chip actuation. The actuation voltage is also comparable to the commercially available Parylene C, which typically requires 90–100 V for droplet actuation.10 Both samples demonstrated similar performance, showing good electrical stability, no current leakage, and an actuation voltage comparable to commercial DMF chips with Parylene C dielectric layers.
We next validated the humidity resistance of the dielectric material. To achieve this, we placed dielectric stacks in an incubator for 60 days and actuate them for 20 seconds every 24 h with the minimum actuation voltage determined per material earlier in this work. We have chosen 20 seconds in order to have a measurement of the stabilized current output over the course of an average time for which the electrodes could be actuated in a DMF operation. The samples were actuated at three different fixed spots using 0.2 M NaCl and the current in the droplet was monitored using a source measure unit (SMU). Evaporation of the droplets were prevented by placing a wet tissue lining the Petri dish around the DMF chip. Over the entire duration of water resistance assay the chip was kept in a direct contact with the humidified tissue and with three 0.2 M NaCl droplets on the surface. In the first 7 days, 1 μm-thick PVDF samples showed no detectable material breakdown patterns. However, the current output stayed in the range of 100–500 μA/4 mm2 with a standard deviation of 456 μA (Fig. 3A). In alignment with our previous voltage–current measurements, the consistent high current output without irregular spikes (sustained increases in current) indicates the conductive-like nature of PVDF at the studied voltages, attributed to its high dielectric constant. A similar voltage–current trend was observed in the 5 μm-thick SU-8 and SU-8 + PVDF samples as well. Initial current output was recorded as 1.9 μA/4 mm2 for SU-8 and 4.9 nA/4 mm2 for SU-8 + PVDF on day 1, and these values increased gradually over the course of 7 days, reaching at 79 μA/4 mm2 (SD = 100 μA) over 7 days for SU-8 and 40 μA/4 mm2 (SD = 39 μA) over 7 days for SU-8 + PVDF (n = 3, Fig. 3A). For all PVDF, SU-8, and SU-8 + PVDF samples inter-sample deviation was relatively high when compared to PVDF + SU-8 dielectric stack. In a contrast, PVDF + SU-8 dielectric stack maintained the lowest current output of 9 nA/4 mm2 on average and 15 nA/4 mm2 at 7 days, with a low inter-sample deviation (SD = 4.8 nA) over the 7 days. A single layer of 7 μm-thick Parylene C served as a control group as this configuration is commonly used in commercially available DMF chips. Upon fabrication, Parylene C surface was smooth with no observable pinholes or defects and had an average surface roughness of 59 nm (n = 3, STD = 22 nm). After 7 days at incubator, cracks became visible on the material (Fig. 3B), and the surface roughness increased to 2.2 μm when accounting the breakdown cracks (n = 3, STD = 2 μm) (Fig. 3C). Thus, complete breakdown of Parylene C samples occurred within the first 3 days (n = 3). The material appeared observably browned, and the water droplet underwent electrolysis and evaporation while high current output levels were measured in the droplet after the breakdown (Fig. 3A). This observation is consistent with previous findings from Andreas Heid, et al. and Sambit Palit, et al.23,24
 | ||
| Fig. 3 Effect of high humidity and liquid exposure on material electrical and surface properties. (A) Quantitative analysis of current over 7 days for the materials and their combinations. (B) Surface profile of the chips before and after storing chips at 37 °C and 95% humidity and actuating them under these environmental conditions. (C) Quantitative characterization of the surface roughness over time. The chips were stored 37 °C and 95% humidity (n = 3 for each sample) actuated with previously calculated actuation voltage every day for 20 seconds. All parameters were characterized for SU-8 3005, polyvinylidene fluoride (PVDF), and combination of these materials. | ||
Subsequently, we confirmed the current output results with surface profile analysis of the materials (Fig. 3B and C). The surface profilometry allows for a visual comparison of the surface properties of the dielectric before and after exposure to cell culture incubator, where the chips are exposed to liquids and humidity. Upon both fabrication and after 7 days in an incubator, no visible defects were detected on 1 μm-thick PVDF samples, but the average surface roughness increased 5-fold, from an initial value of 52 nm to 273 nm (n = 3, STD = 22 nm at day 0, STD = 85 nm at day 7). SU-8 samples with 5 μm thickness showed fewer changes on the surface after 7 days in the incubator. The initial surface roughness of the SU-8 samples was 93 nm, and it remained the same after 7 days, while the standard deviation increased from 35 nm to 63 nm (n = 3). Therefore, SU-8 facilitated a good resistance to water and humidity-containing environments, yet the current output performance of SU-8 was not consistent due to its dielectric destabilization and high current output near actuation voltages. The surface roughness of SU-8 + PVDF dielectric stack increased 3-fold in the course of 7 days, from an initial value of 42 nm to 145 nm (n = 3, STD = 15 nm at day 0, STD = 69 nm at day 7). We attribute this change to the PVDF top layer, as the PVDF surface characterization in this work also confirm a 5-fold roughness increase after exposure to the incubator environment. As expected, we obtained the most stable surface roughness conditions using PVDF + SU-8 sample. The initial surface roughness increased only 1.6 fold from 40 nm to 64 nm after 7 days in incubator.
Due to promising stability of the PVDF + SU-8 dielectric stack, it was further tested in incubator for 60 days (Fig. 1C). The results show that out of 6 initial samples, 4 remained stable for 60 days, with no dielectric breakdown and maximum current output not surpassing 20 nA. One sample showed early signs of destabilization, inconsistent with the rest of the data, likely due to a fabrication defect or dust contamination at the measured spot. Interestingly, the surface roughness of the PVDF + SU-8 dielectric stack after 60 days exposure reached 112 nm, denoting only a 2.8-fold decrease from the initial value (n = 4, STD = 57 nm) (Fig. 3C). As a result, PVDF + SU-8 dielectric stack showed a significantly improved performance in electrical stability and humidity resistance up to 60 days. DMF chips with the PVDF + SU-8 dielectric stack were finally tested for their performance for controlled droplet manipulation with the DropBot setup. All the basic operations were performed and operated with the fabricated chip, encompassing individual droplet dispensing (Fig. S4A†), droplet fusion (Fig. S4B†), mixing (Fig. S4C and D†) and splitting (Fig. S4E and F†). Consequently, all DMF chips in the remainder of the study, focused on macrophage cell culturing, were fabricated using the PVDF + SU-8 dielectric stack.
Modelling the macrophage inflammatory response in DMF platform
We sought to explore unique performance gains possible in cell culturing using the newly developed DMF chip. We specifically investigated the induction of inflammatory responses and the effects of a potential anti-inflammatory drug in macrophages. This was achieved by culturing non-polarized macrophages on the platform, inducing a pro-inflammatory state, and subsequently treating the inflammation with the candidate drug (Fig. 1D). We started quantifying macrophage cell viability on the DMF chip, where 86%, 92%, and 83% of the cell population was viable across three independent chip experiments, resulting in an average of 87% viable cell culture after 7 days (Fig. 4A and B). The viable cell ratio was higher than in the well-plate control tested under the same conditions, where cell viability was observed to be 72% (Fig. S5A and B†). DMF platform supported a unique cell microenvironment where daily media refreshment through passive dispensing removed dead or damaged cells with compromised attachment, maintaining a cell culture free of cellular debris and associated inflammatory biomarkers.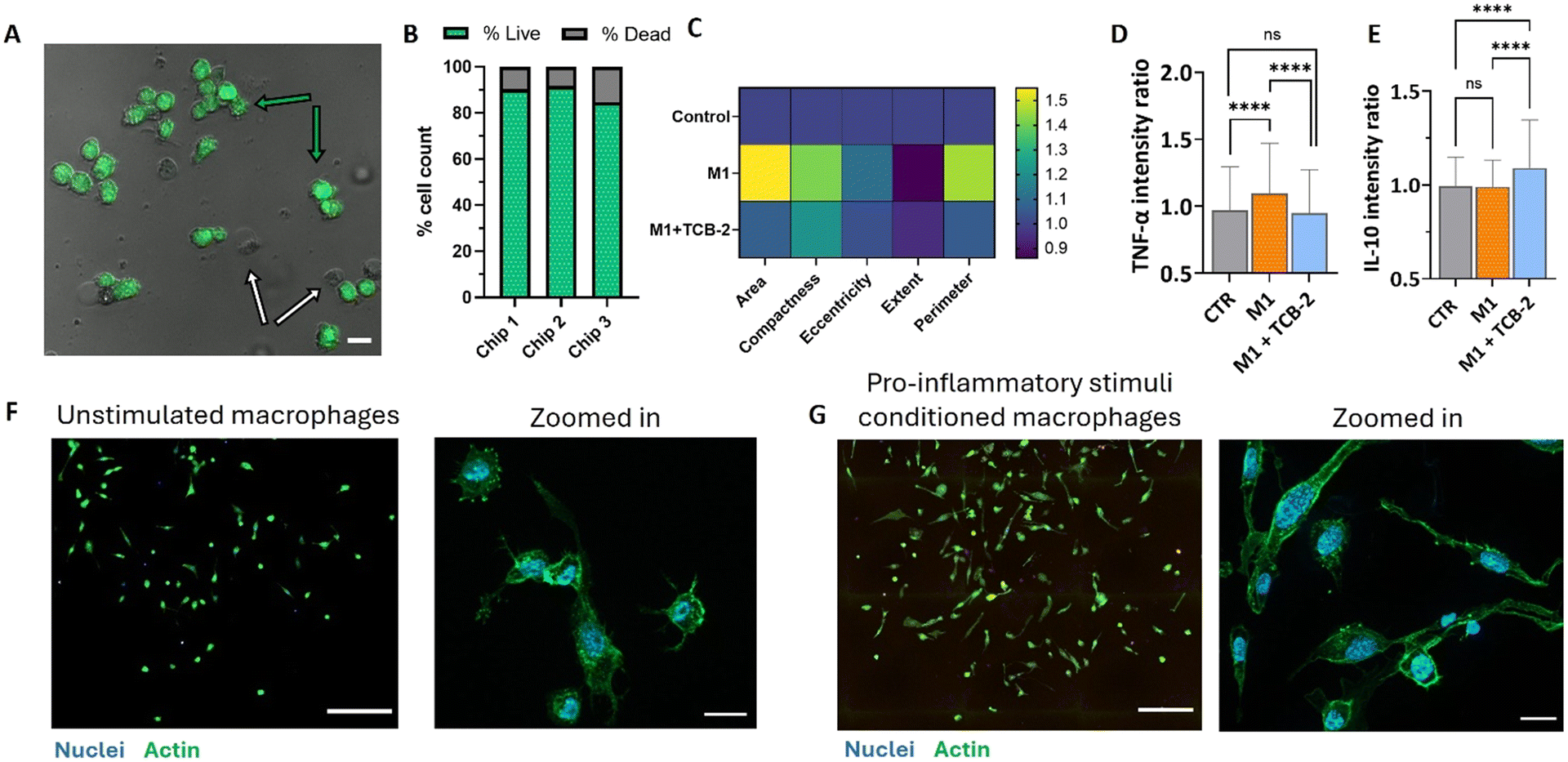 | ||
| Fig. 4 Cell viability on a DMF chip and dose control of potential anti-inflammatory treatment TCB-2. (A) Live-dead imaging of the cells after 7 days of culture on the DMF platform. Green arrows point to examples of live cell and white arrows point to example of dead cells. Scale bar 20 μm. (B) Quantitative analysis of positive and negative controls (n = 3). (C) Cell morphology characterization. All parameters were normalized and represented as fold-change to the control condition (n = 3). (D) TNF-α expression in of TCB-2 stimulated and unstimulated macrophages (n = 3, 100–300 individual cells per condition per n, ns – nonsignificant, **** p < 0.0001). (E) IL-10 expression of TCB-2 stimulated and unstimulated macrophages (n = 3, 100–300 individual cells per condition per n, ns – nonsignificant, **** p < 0.0001). (F) Naïve macrophage population before stimulation and zoomed-in format. Nuclei was stained with DAPI and actin filaments were stained with phalloidin. Scale bar 200 μm (left) and 20 μm (right) (G) pro-inflammatory stimulated macrophages after the stimulation day and the zoomed-in format. Nuclei was stained with DAPI and actin filaments were stained with phalloidin. Scale bar 200 μm (left) and 20 μm (right). | ||
Next, we optimized the cell inflammation protocol using a well plate. Typically, in laboratory conditions inflammatory response in macrophages is induced by the lipopolysaccharide (LPS, endotoxin from Gram-negative bacteria) and interferon-gamma (INF-γ, pro-inflammatory macrophage cytokine).25 A calibration experiment with two distinct inflammatory conditions was tested where we applied 100 ng mL−1 of LPS and 10 ng mL−1 of INF-γ for the first condition and 1 μg mL−1 LPS and 20 ng mL−1 INF-γ for the second condition (Fig. S5C†). Next, we quantified the TNF-α cytokine release. While the lower concentration of LPS and INF-γ did not induce a significant increase in TNF-α release, the higher concentration led to a statistically significant 1.2-fold increase in TNF-α (tumour necrosis factor α, SD = 0.09). Increased production and release of TNF-α by macrophages is generally associated with a pro-inflammatory phenotype. (Fig. S5D†). Therefore, for the following experiments, cells were stimulated with 1 μg mL−1 of LPS and 20 ng mL−1 INF-γ in order to promote cellular polarization into a pro-inflammatory phenotype and increase TNF-α production. The induction of pro-inflammatory phenotype in macrophages was then accessed on a DMF chip cell culture. After 7 days in culture, cells induced into a pro-inflammatory state with 1 μg mL−1 LPS and 20 ng mL−1 INF-γ, without additional treatment (M1 condition – orange bar), were analysed for TNF-α and IL-10 cytokine release. TNF-α quantification showed a 1.1-fold increase (SD = 0.37) (Fig. 4D), consistent with the cytokine release profile characteristic of pro-inflammatory macrophages26 while IL-10 levels showed no significant change (Fig. 4E), aligning with its typical upregulation only in anti-inflammatory macrophages.27 While the standard deviation values vary in between on-chip result and well plate results, it is to note that while DMF platform has a benefit of working with low population of cells that allows for a more individual analysis, it also comes at a cost of in a possibly high average deviation of a heterogeneous population. As the population is positively skewed when the pro-inflammatory phenotype is induced (only small part of the population is seen to have a strong cytokine release response as seen on Fig. 1E), the observed standard deviation is affected by the smaller sample size.
Macrophage phenotype changes were also assessed through morphological analysis. Key factors such as cell area, perimeter (to evaluate membrane rugosity or irregularity), cell body compactness (indicating solidity or spread of the cell body), eccentricity (reflecting roundness or elongation), and extent (a measure of shape regularity) provide insights into cell health and phenotype profile. We confirmed the morphological data through cell profiler analysis (Fig. 4C), which revealed that cells stimulated into a pro-inflammatory phenotype exhibited a 1.6-fold increase in average area and a 1.5-fold increase in perimeter compared to untreated cells. Unstimulated cells displayed a more rounded shape with minor protrusions, which is a characteristic of typical macrophage morphology (Fig. 4F and G). In contrast, the macrophage population stimulated into the pro-inflammatory phenotype showed a more elongated profile, with cells adopting a less circular, spindle-shaped morphology. These cells exhibited higher compactness (1.4-fold increase), increased eccentricity (1.1-fold increase), and reduced extent (0.86-fold decrease), all indicative of a more elongated and less spherical cell morphology consistent with the reported characteristics of pro-inflammatory macrophages. This observation aligns with previous studies by Rostam et al.,28 Vogel et al.,29 and Heinrich et al.,30 which report that unstimulated macrophages generally exhibit a smaller, round morphology, whereas pro-inflammatory-polarized cells display increased cell body elongation and adopt an amoeboid or spindle-shaped morphology. Interestingly, cell area presented a more divided result: some studies (Rostam et al.28) report on pro-inflammatory-stimulated cells are smaller in size with densely packed actin, while others (Lee et al. and Sridharan et al.31,32) observe an increased cell area in pro-inflammatory macrophages compared to the unstimulated phenotype. Altogether, the pronounced morphological alterations of the cell, upregulation of TNF-α cytokine release as well as unaltered IL-10 cytokine release support the conclusion that our model represents a population of pro-inflammatory macrophages.
Evaluating the anti-inflammatory potential of TCB-2 in activated macrophages
The potential anti-inflammatory drug used in this study, TCB-2, is a selective serotonin 5-HT2A receptor agonist which is a receptor consistently expressed in immune cells, including macrophages.33 Traditional activation of the 5-HT2A receptor leads to increased inflammation and has an anxiogenic effect.34 Interestingly, when the same receptor is activated by psychedelic molecules, the effect is reversed, leading to a pronounced anti-inflammatory activity that inhibits the TNF-α pathway.35,36 Although other 5-HT2A agonists have been previously explored, TCB-2, as a potent and selective receptor agonist, remains largely understudied. Its anti-inflammatory potential has primarily been investigated in animal studies, with limited research at the cellular level.To assess the potential anti-inflammatory activity of TCB-2, THP-1 cells were treated with the 5 μM of the drug 24 hours after the pro-inflammatory phenotype induction. The treatment concentration of 5 μM was chosen based on the average dose of psychedelic treatments used in cellular studies.37 The interval was introduced to allow the cells to undergo full polarization, enabling the assessment of TCB-2 activity on an already established inflammatory state rather than its preventive effects. After 24 hours of TCB-2 treatment on the activated macrophages, we replaced the cell media with a stimulus-free medium and allowed the cells to rest and establish a phenotype for an additional 24 hours. At the end of this period, we measured the cytokines expressed by the cell population and found that TCB-2 was able to reduce TNF-α expression to control population levels, with a fold-change of 0.95 in comparison with the control (SD = 0.3, n = 3, Fig. 4D), with a significant decrease of the cytokine expression as compared to the pro-inflammatory state and no significant fold-change from the unstimulated macrophages. Additionally, TCB-2 significantly increased the expression of the anti-inflammatory cytokine IL-10 by 1.1-fold (SD = 0.26, n = 3, Fig. 4E) compared to both the control and pro-inflammatory cell populations. These results indicate that TCB-2 has the potential to treat established inflammation and influence the polarization state of macrophages, possibly driving them toward a more anti-inflammatory phenotype. This result was further validated on a well plate assay, where the inflammatory treatment induced a 1.1 fold increase in TNF-α secretion (SD = 0.09), however, this increase was negated by TCB-2 treatment, with the M1 + TCB-2 condition showing no significant difference (1-fold change) compared to the control condition (SD = 0.06). Additionally, treatment with TCB-2 alone on unstimulated macrophages did not induce any increase in TNF-α release, maintaining a fold-change of 1 compared to the control condition (SD = 0.03) (Fig. S5D†). While the fold-change between conditions may not appear high, it is important to note that the data represent average cytokine release values per cell population. Monocyte and macrophage populations are known to be highly heterogeneous, with only a portion of the population expressing a specific phenotype.38–40 This effect is also seen in the single cell analysis data in Fig. 1E where the bigger part of population does not express any higher amount of either TNF-α, nor IL-10. However, only a subset of the cell population exhibits a pronounced response, showing either an increase in TNF-α release upon inflammatory induction or an anti-inflammatory response following treatment with TCB-2. Additionally, THP-1 cells are known to secrete lower levels of TNF-α when compared to the primary macrophages,41,42 as well as decreasing their capacity of cytokine release rapidly days after the differentiation.43 Since the full assay reported in the study takes 7 days, the cytokine spike is not as pronounced, allowing however to see the differences on a more reactive subset of cell population. It is interesting to note that TCB-2 treatment appears to increase IL-10 cytokine expression not only in inflammation-reactive cells, but also in the cell population that did not show any pronounced pro-inflammatory response by an increase in TNF-α release, possibly showing a potential for the drug use for anti-inflammatory phenotype induction in macrophages instead of only reducing the pro-inflammatory response. Furthermore, on chip treatment with TCB-2 reduces the morphological changes in the cells, decreasing both cell area and perimeter. The cell area and perimeter exhibited a 1.1-fold increase compared to unstimulated cells, with the area and perimeter being reduced by 0.5 and 0.4 units, respectively, relative to pro-inflammatory-polarized macrophages (1.6 times increase in area and 1.5 increase in perimeter from unstimulated cells). The values for compactness and eccentricity also decreased to 1.2-fold and 1-fold compared to the unstimulated cells, indicating differences of 0.2 and 0.1 from the pro-inflammatory-polarized macrophages (1.4 times increase in compactness and 1.1 increase in eccentricity), respectively (Fig. 4C). These findings demonstrate that TCB-2 fundamentally influences the cell response in terms of cytokine expression, while reversing morphological changes to near pre-treatment levels. The TCB-2-treated cells exhibit smaller, more regular, and circular shapes, typical of unstimulated or anti-inflammatory macrophage phenotypes. A similar effect upon macrophage treatment with potentially anti-inflammatory compound was previously reported by Shin, et al.,44 where treatment with cordycepin was able to restore naïve cell shape to the LPS-treated macrophages.
Conclusion
In this study, we introduced long-term cell culture-compatible DMF chips designed to provide spatiotemporal control for macrophage behaviour studies over extended periods. By leveraging dielectric stacks, these chips achieve a more robust DMF system that on the account of used materials and their particular combination allow to enhance the chip stability and durability under cell culture conditions to up to 60 days, opening new perspective for more complex cellular biology studies. This platform enabled us to accurately model and study macrophage inflammatory responses and investigate the immuno-modulating effects of the anti-inflammatory drug candidate TCB-2. Our findings indicate that TCB-2 effectively modulates macrophage polarization through morphological changes and cytokine expression, reducing pro-inflammatory cytokine expression and reversing the morphological changes associated with inflammation after the experimental treatment. These results underscore the potential of TCB-2 as a therapeutic anti-inflammatory agent and highlight the DMF platform's capability to facilitate long-term cell culture studies, potentially offering new avenues for in-depth investigation of immune response.Experimental
Chemicals, reagents, and materials
All photoresists and developers were purchased from MicroChemicals GmbH. Poly(vinylidene fluoride-co-hexafluoropropylene) Mw ∼400![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, Mn ∼130000 in pellets (PVDF-HFP), Pluronic F127 and N,N-dimethylformamide were purchased from Sigma-Aldrich. Fluoropel 1601V was purchased from Cytonix. Glass substrates (75 × 50) were purchased from VWR.
000, Mn ∼130000 in pellets (PVDF-HFP), Pluronic F127 and N,N-dimethylformamide were purchased from Sigma-Aldrich. Fluoropel 1601V was purchased from Cytonix. Glass substrates (75 × 50) were purchased from VWR.
RPMI 1640 media and penicillin–streptomycin (10000 U mL−1) were purchased from Gibco™. Fetal bovine serum was obtained from Serana, origin Brazil. THP-1 cell line was in house and obtained from ATCC. TNF-α secretion assay detection kit and IL-10 secretion assay detection kit were obtained from Miltenyi Biotec. TCB-2 ((4-bromo-3,6-dimethoxybenzocyclobuten-1-yl)methylamine hydrobromide) was purchased from Biotechne Tocris. Fibronectin for surface functionalization, CellTracker dye, propidium iodide and DAPI stains were purchased from Sigma Aldrich.
Fabrication and operation of digital microfluidics chip
The bottom layer of the DMF chip was fabricated by initially patterning electrodes on a glass substrate, followed by the deposition of dielectric layers and a hydrophobic coating. Glass substrates measuring 75 × 50 mm were first cleaned by immersion and sonication in a 2% soap solution in DI water, followed by sequential rinses in acetone and isopropanol for 5 minutes each. Subsequently, the glass substrates were dried at 150 °C for 10 minutes and allowed to cool to room temperature. Next, the substrates underwent treatment with 50 W oxygen plasma for 1 minute using a plasma asher (Emitech K1050X).A 2.5 μm layer of AZ nLOF 2035 photoresist was then spin-coated at 4000 rpm onto the surface and processed according to the manufacturer's instructions to achieve a photoresist pattern with approximately 3 μm height for electrode patterning. The chip was subsequently sputter-coated (Quorum Q300T D) with a 5 nm layer of chromium followed by a 50 nm layer of gold for electrode deposition. The photoresist was then removed using TechniStrip NI555 at 65 °C, followed by acetone to eliminate any residual photoresist. Resulting chips were washed with DI water and dried with a nitrogen gun.
Next, the electrode-patterned chips were coated with dielectric materials. A solution of polyvinylidene fluoride (PVDF) at a concentration of 200 mg mL−1 in N,N-dimethylformamide was spin-coated onto the chip surface at 1000 rpm for 1 minute to form a thin layer. Subsequently, the coating was evaporated for 60 minutes at 60 °C to enhance nucleation and baked for 30 minutes at 110 °C. Following this, the chips were rapidly removed from the hot plate to facilitate rapid cooling. For SU-8 3005 coating, the photoresist was spin-coated onto the chip surface at 4000 rpm to achieve a 5 μm film thickness and processed according to the manufacturer's instructions. Upon completion of the deposition process, SU-8 3005 was hard-baked at 200 °C for 30 minutes, with a gradual heating and cooling rate of approximately 2 °C per minute. For dielectric stacks, each material was fully processed as described above before spin-coating the next layer.
The surface was hydrophobized using Fluoropel PFC1601V. This reagent was spin-coated onto the surface at 1000 rpm with a 500 rpm acceleration for 30 seconds, followed by baking the samples at 180 °C for 10 minutes.
The top plate of the DMF chip was obtained by purchasing commercially available indium tin oxide (ITO)-coated glass slides from Xinyan Technology (China), which were custom-patterned for hydrophilic windows. A chip contained 5 windows with 2 × 4 mm dimensions without ITO coating. These slides were cut to 25 × 50 mm size and coated with Fluoropel using the same procedure as described above. The hydrophilic regions were created by removing Fluoropel using oxygen plasma etching at 100 W for 1 minute to complete the chip fabrication process.
Fabrication of mini samples for characterization of dielectric materials
Mini samples for dielectric characterization were fabricated by depositing a 30 nm layer of chromium onto a 25 × 25 mm glass surface. Subsequently, individual or stacked dielectric materials were deposited on the surface of the electrode, leaving part of the electrode exposed. The dielectric surface was left uncoated for voltage–current characterization and breakdown voltage tests. For actuation voltage and water penetration assays, the surface was coated with Fluoropel as described above.Dielectric material characterization
For breakdown voltage measurements, copper tape with a 0.5 cm2 area was placed onto the dielectric surface, creating a metal-dielectric-metal stack. Iridium probes were used to apply voltage up to 3000 V in 10 V increments. The current output was measured until a current spike of 0.1 mA was detected or 3000 V were reached.
Cell culture and bioassays in DMF platform
THP-1 cells were differentiated into adherent macrophages by transferring 1.5E6 cells to a separate flask and treated with 50 ng mL−1 phorbol 12-myristate 13-acetate (PMA) for 24 hours, followed by a 48 hour resting period. To detach the cells, they were washed once with sterile PBS and incubated with accutase for 10 minutes at room temperature, followed by 20 minutes in an incubator. The dissociation process was halted after detachment by adding an equal volume of supplemented medium and centrifuging the cells at 200g for 5 minutes. The supernatant was discarded, and the cell pellet was resuspended in fresh medium to 5E5 cells per mL, in order to achieve seeding density of 1000 cells per 2 μL. Pro-inflammatory phenotype was induced 24 hours after the seeding on DMF platform (or well plate for well plate controls) by treating the cells with 1 μg mL−1 LPS and 20 ng mL−1 INF-γ for 24 hours, after which the stimuli was removed.
For morphological study, cells were fixated with 3.7% paraformaldehyde solution for 30 minutes at the end of the experiment. Cell nuclei were stained with DAPI, and the cellular cytoskeleton was stained with phalloidin dye.
The chip was transferred from the incubator to the DropBot equipment (SciBots, Canada) every 24 hours. Fluid handling on DMF was facilitated by supplementing the cell media with 0.05% Pluronic F127. Media replenishment involved dispensing a double droplet (∼1.5 μL) from the reservoir onto the hydrophilic spot, followed by removal of excess liquid through a passive dispensing mechanism. This process was repeated three times to ensure a thoroughly rinsed surface for culture sites requiring specific media conditions. Due to the nature of passive dispensing on a DMF chip and multiple wash cycles, the concentrations of LPS, INF-γ and TCB-2 were used as the final reported concentration and introduced to the cells in 3 consecutive passive washes to ensure constant concentration. For the incubation with cell viability dye where no multiple washes protocol was applied, the concentration of the working solution was doubled in order to achieve correct final concentration at the culturing site.
Microscopy and image analysis
For the cytokine production assay, the amount of cytokines produced was accessed by imaging the previously stained cells. Morphological data were obtained by staining fixed cells with phalloidin dye. Imaging was performed using a Nikon microscope (Nikon Eclipse Ti2-D-PD), which scanned the entire cell culture area. High-resolution imaging was performed using a Zeiss Axio Observer Apotome microscope using a 63× objective. Image analysis was performed using Cell Profiler software.Data processing and statistical analysis
Voltage–current measurements were performed by the help of a MATLAB script. Voltage current measurements were performed by applying 2 V increments each 2 seconds and recording the current output.Image analysis was done with ImageJ for manual cell viability counting and Cell Profiler software for cytokine expression (average object intensity measurement) and morphological assessment of cell body through the size and shape module. In size and shape module, parameters for cell area, perimeter, eccentricity, compactness, and extent were analysed as essential for the morphological assessment.
Statistical analysis was performed by GraphPad software. Data distribution was accessed by Shapiro–Wilk normality test to a normal Gaussian distribution with a significance level of 0.05. Statistical analysis in between conditions was performed by non-parametric ANOVA, with Kruskal–Wallis test.
Data availability
The data is currently submitted for an intellectual property application and cannot be shared due to confidentiality requirement. Once the process is finalized, the data will be made available at the 4TU.ResearchData repository.Author contributions
O. K. S. and B. G. conceived the experiments, O. K. S. conducted the experiments, O. K. S. and B. G. analysed the results. All authors reviewed and have given approval to the final version of the manuscript.Conflicts of interest
O. K. S. and B. G. are inventors on Eindhoven University of Technology intellectual property (IP) related to this research and may benefit from royalties' interests if IP is licensed.Acknowledgements
The authors acknowledge the members of the Biosensors and Devices Lab and BioInterface Science group, as well as Jan de Boer and Anthal Smits for fruitful discussions. B. G. and O. K. S. acknowledge the financial support provided by the Irene Curie fellowship funding and the TU/e ICMS Institute Immuno-engineering funding. Microscopy was performed at the LCTE Microscopy Facility, TU/e. The MATLAB script for voltage–current recording was written with help from Dr. Eveline van Doremaele.References
- B. Hoffmann and C. Schäfer, Cell Adhes. Migr., 2010, 4, 190–193 CrossRef PubMed
.
- N. Jain, J. Moeller and V. Vogel, Annu. Rev. Biomed. Eng., 2019, 21, 267–297 CrossRef CAS PubMed
- J. W. Godwin, A. R. Pinto and N. A. Rosenthal, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 9415–9420 CrossRef CAS PubMed
- T. M. Tsarouchas,
et al.
, Nat. Commun., 2018, 9, 4670 CrossRef PubMed
- F. Nau Jr., B. Yu, D. Martin and C. D. Nichols, PLoS One, 2013, 8, e75426 CrossRef PubMed
-
G. Cocco, Effects of TCB-2 on Alzheimer's neuropathology, Undergraduate Honours Thesis, University of Lethbridge, 2022 Search PubMed
- A. Szabo, Front. Immunol., 2015, 6, 358 Search PubMed
- R. Dong, B. Yu, L. Chen and W. Yu, Exp. Ther. Med., 2016, 12, 3583–3588 CrossRef CAS PubMed
- H. Kang, S. H. D. Wong, Q. Pan, G. Li and L. Bian, Nano Lett., 2019, 19, 1963–1975 CrossRef CAS PubMed
- I. Barbulovic-Nad, H. Yang, P. S. Park and A. R. Wheeler, Lab Chip, 2008, 8, 519–526 RSC
- A. H. Ng, B. B. Li, M. D. Chamberlain and A. R. Wheeler, Annu. Rev. Biomed. Eng., 2015, 17, 91–112 CrossRef CAS PubMed
- L. Pang, J. Ding, X. X. Liu and S. K. Fan, TrAC, Trends Anal. Chem., 2019, 117, 291–299 CrossRef CAS
- X. Xu, L. Cai, S. Liang, Q. Zhang, S. Lin and M. Li,
et al.
, Lab Chip, 2023, 23, 1169–1191 RSC
- C. Yang, X. Gan, Y. Zeng, Z. Xu, L. Xu and C. Hu,
et al.
, Biosens. Bioelectron., 2023, 115723 CrossRef CAS PubMed
- X. Min and W. S. Kim, Microfluid. Nanofluid., 2019, 23, 127 CrossRef
- W. Xia and Z. Zhang, IET Nanodielectrics, 2018, 1, 17–31 CrossRef
- H. Liu, S. Dharmatilleke, D. K. Maurya and A. A. O. Tay, Microsyst. Technol., 2010, 6, 449–460 CrossRef
- F. Mugele and J. C. Baret, J. Phys.: Condens. Matter, 2005, 17, R705 CrossRef CAS
- G. Shi, M. Rouabhia, S. Meng and Z. Zhang, J. Biomed. Mater. Res., Part A, 2008, 84, 1026–1037 CrossRef PubMed
- T. Winterstein, M. Staab, C. Nakic, H.-J. Feige, J. Vogel and H. F. Schlaak, Micromachines, 2014, 5, 1310–1322 CrossRef
- MicroCHem (2024, September 3rd), SU-8 Permanent Photoresists, (https://kayakuam.com/wp-content/uploads/2019/09/SU-8-table-of-properties-1.pdf).
- S. Chaipo and C. Putson, J. Phys.: Conf. Ser., 2021, 1719, 012065 CrossRef CAS
- A. Heid, R. von Metzen, A. Stett and V. Bucher, Curr. Dir. Biomed. Eng., 2016, 2, 39–41 CrossRef
- S. Palit, D. Varghese, H. Guo, S. Krishnan and M. A. Alam, IEEE Trans. Device Mater. Reliab., 2015, 15, 308–318 CAS
- M. Orecchioni, Y. Ghosheh, A. B. Pramod and K. Ley, Front. Immunol., 2019, 10, 1084 CrossRef CAS PubMed
- H. L. Lu, X. Y. Huang, Y. F. Luo, W. P. Tan, P. F. Chen and Y. B. Guo, Biosci. Rep., 2018, 38, BSR20171555 CrossRef CAS PubMed
- I. Dhande, W. Ma and T. Hussain, Hypertens. Res., 2015, 38, 21–29 CrossRef CAS PubMed
- H. M. Rostam, P. M. Reynolds, M. R. Alexander, N. Gadegaard and A. M. Ghaemmaghami, Sci. Rep., 2017, 7, 3521 CrossRef PubMed
- D. Y. S. Vogel, J. E. Glim, A. W. D. Stavenuiter, M. Breur, P. Heijnen, S. Amor, C. D. Dijkstra and R. H. J. Beelen, Immunobiology, 2014, 219, 695–703 CrossRef CAS PubMed
- F. Heinrich, A. Lehmbecker, B. B. Raddatz, K. Kegler, A. Tipold, V. M. Stein, A. Kalkuhl, U. Deschl, W. Baumgärtner, R. Ulrich and I. Spitzbarth, PLoS One, 2017, 12, e0183572 CrossRef PubMed
- H.-S. Lee, S. J. Stachelek, N. Tomczyk, M. J. Finley, R. J. Composto and D. M. Eckmann, J. Biomed. Mater. Res., 2013, 101, 203–212 CrossRef PubMed
- R. Sridharan, B. Cavanagh, A. R. Cameron, D. J. Kelly and F. J. O'Brien, Acta Biomater., 2019, 89, 47–59 CrossRef CAS PubMed
- I. Cloëz-Tayarani, A.-F. Petit-Bertron, H. D. Venters and J.-M. Cavaillon, Int. Immunol., 2003, 15, 233–240 CrossRef PubMed
- M. S. Shajib and W. I. Khan, Acta Physiol., 2015, 213, 561–574 CrossRef CAS PubMed
- I. Cloëz-Tayarani, A.-F. Petit-Bertron, H. D. Venters and J.-M. Cavaillon, Int. Immunol., 2003, 15, 233–240 CrossRef PubMed
- F. Nau Jr., B. Yu, D. Martin and C. D. Nichols, PLoS One, 2013, 8, e75426 CrossRef PubMed
- E. Ghasemi Gojani, B. Wang, D. Li, O. Kovalchuk and I. Kovalchuk, Psychoactives, 2024, 3, 48–64 CrossRef
- H. Dueck, J. Eberwine and J. Kim, BioEssays, 2016, 38, 172–180 CrossRef PubMed
- Q. Xue, Y. Lu, M. R. Eisele, E. S. Sulistijo, N. Khan and R. Fan,
et al.
, Sci. Signaling, 2015, 8, ra59 CrossRef PubMed
- L. Li, E. Alsema, N. R. M. Beijer and B. Gumuscu, ACS Biomater. Sci. Eng., 2024, 10(11), 6974–6983 CrossRef CAS PubMed
- A. Schildberger, E. Rossmanith, T. Eichhorn, K. Strassl and V. Weber, Mediators Inflammation, 2013, 2013, 697972 CrossRef PubMed
- T. Hoppenbrouwers, S. Bastiaan-Net, J. Garssen, N. Pellegrini, L. Willemsen and H. Wichers, PharmaNutrition, 2022, 22, 100322 CrossRef CAS
- H. Shiratori, C. Feinweber, S. Luckhardt, B. Linke, E. Resch, G. Geisslinger, A. Weigert and M. J. Parnham, Mol. Immunol., 2017, 88, 58–68 CrossRef CAS PubMed
- S. Shin, S. Moon, Y. Park, J. Kwon, S. Lee, C. K. Lee, K. Cho, N. J. Ha and K. Kim, Immune Netw, 2009, 9, 255–264 CrossRef PubMed
- Q. Lang,
et al.
, ACS Appl. Mater. Interfaces, 2015, 7(48), 26792–26801 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4lc00947a |
| This journal is © The Royal Society of Chemistry 2025 |