
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRapid screening of CO2 capture fluids†
Yaohao
Guo
ab,
Feng
Li
b,
Sepehr
Saber
b,
Mohammad
Zargartalebi
b,
Siyu Sonia
Sun
b,
Yurou Celine
Xiao
 b,
Bo
Bao
*a,
Zhi
Xu
*a and
David
Sinton
*b
b,
Bo
Bao
*a,
Zhi
Xu
*a and
David
Sinton
*b
aSchool of Chemical Engineering, East China University of Science and Technology, Shanghai, 200237, China
bDepartment of Mechanical & Industrial Engineering, University of Toronto, Toronto, M5S 3G8, Canada. E-mail: sinton@mie.utoronto.ca
First published on 19th May 2025
Abstract
The evaluation of CO2 capture fluids is crucial for the advancement of carbon capture technologies. Recent advancements in amine-based carbon capture fluids motivate a broad search for high-performance fluids and the development of methods capable of exploring a large chemical space. Here, we present a microfluidic approach paired with automated image processing and density functional theory simulations that enables comprehensive rapid screening of capture fluids. The principle of measurement leverages the ability to monitor phase expansion and contraction in fixed-volume dead-end channels. This approach enables fast comparative assessments of reaction kinetics and thermodynamic parameters, including CO2 absorption rate (∼30 s), desorption rate (∼30 s), absorption capacity (∼20 min), and vapor pressure (∼5 min), exceeding the speed of conventional methods by two orders of magnitude. The method is broadly applicable, effective for primary, secondary, and tertiary amine types. Rapid screening of capture fluids holds promise for the accelerated discovery of improved CO2 capture processes and an opportunity for the microfluidics community to contribute to decarbonization efforts.
1. Introduction
Increasing emissions of CO2 underscore the imperative need for CO2 capture technologies as a strategy towards achieving carbon neutrality.1,2 Among the major types of CO2 capture solutions developed to date – amine solutions, alkaline solutions, ionic liquids, and ammonia – amines are the most developed.3–5 With decades of research and development, aqueous amine solvents are now employed to capture CO2 from a wide variety of post-combustion flue gases.6,7 The typical requirements for a CO2 capture fluid are high absorption rate and capacity, selectivity, and energy efficiency in desorption, as well as exhibiting low vapor pressure, corrosiveness, and toxicity.8,9 However, current capture solutions do not meet these requirements in full. For instance, the widely used 30 wt% MEA aqueous solution exhibits rapid reaction kinetics but exhibits low absorption capacity and suffers high desorption energy demand.10,11 Recent advancements in material science, combined with in-depth investigations of CO2 capture systems, have yielded a diverse array of novel capture fluids and hybrid systems.12,13 These efforts broaden the spectrum of promising chemistries and present a testing bottleneck.14 Blends of capture solutions are the norm, and with over 150 common capture solvents in use and more under development,15 the chemical space is vast, with over 107 potential combinations to explore.Current approaches to evaluate capture fluid performance involve, in effect, smaller versions of the capture system.16–18 To determine absorption and desorption rates, for instance, bubble column reactors, wetted wall columns, and pack towers are used. These approaches are batch and low-throughput, with test times on the order of hours.19–21 Furthermore, the substantial sample volumes needed, ranging from tens of milliliters to a few liters, contribute to inefficiency and cost.22 These coupled time- and volume-requirement limitations are analogous to the biomedical laboratory processes that have been disrupted by lab-on-chip technologies over the last few decades.23,24 Microfluidic technology has demonstrated exceptional versatility and broad applicability in CO2-related studies, spanning capture, conversion, and monitoring applications.25–27 Adapting the lab-on-chip approach to advance decarbonization technology is needed for the decades ahead.28,29
Herein, we introduce a microfluidic approach for the comprehensive and high-throughput screening of CO2 capture fluids, integrating automated image analysis and density functional theory (DFT) simulations for enhanced efficiency and accuracy. The microfluidic approach leverages dead-end channels as gas–liquid reactors to assess CO2 absorption/desorption rates, absorption capacity, and vapor pressure simultaneously. This approach circumvents the complexities associated with dynamic gas–liquid segmented flows and provides a stable and controllable gas–liquid interface. Notably, variations in fluid properties may introduce deviations in capture performance evaluation. The DFT simulations are used to calculate the Gibbs free energy profiles, offering theoretical insights into the intrinsic reaction kinetics between CO2 and capture fluid. Additionally, comparative analysis with microfluidic data helps distinguish genuine performance from experimental artifacts. This combined approach enables an assessment of both the potential of a given capture fluid and how it would perform in practice, and provides an understanding of the underlying mechanisms that can inform the next test or formulation.
2. Methodology and experimental design
The microfluidic assay was based on the 1-D fixed-volume dead-end channels that have been proven robust in many lab-on-chip applications. Examples included assessing CO2-oil interactions,30,31 thousands of such channels applied to assess phase properties of mixtures,32 and colloidal particle sorting.33 The channels here were 300 μm wide and 250 μm deep, and distributed in a parallel array. These channels functioned as miniaturized gas–liquid reactors, as illustrated in Fig. 1a. Initially, CO2 gas was introduced into these channels under a predefined pressure, followed by the infusion of the capture fluid, thus triggering the reaction under test. Additionally, we monitored the pressure during the introduction of the capture fluid to gain insights into the thermodynamic conditions within the channel. During the subsequent absorption process, the pressure was assumed to remain constant due to the small volume of trapped CO2. The structure ensured the constancy of the gas and liquid segments' diameter, with volume variations restricted to the channels' longitudinal axis. Taking amine solutions as an example, at room temperature, the gas slug in the dead-end shrank gradually as the solvent efficiently absorbs CO2. Conversely, elevated temperatures facilitated the reverse reaction, promoting amine regeneration and liberation of CO2, ultimately resulting in an enlargement of the gas slug. These temperature-dependent reactions and interface dynamics served as indicators for assessing the absorption and desorption rates. The absorption capacity measurement shared similarities with that for determining CO2 absorption rates. However, to assess capacity a finite-length absorbent slug was introduced into the test channel and then segmented by a gas phase, leading to an excess of CO2 and a deficiency of absorbent within the microchannels (see Fig. 1a). As the absorbent within the slug becomes depleted, the final position of the slug reflected its absorption capacity. To assess vapor pressure measurements, we invoked the proportional relationship between vapor pressure and evaporation rate.34 Each experiment was conducted three times, and for each run, the reported result represents the average across all channels imaged simultaneously. | ||
| Fig. 1 Microfluidic-driven comprehensive and fast screening of CO2 capture fluids. (a) Operational principle and schematic representation of microfluidic measurement for CO2 absorption rate, desorption rate, absorption capacity and vapor pressure. Different colors on the chip denote distinct temperature regions, with blue indicating the low-temperature region at 35 °C and red signifying the high-temperature region at 80 °C. The cartoon diagram illustrates the movement of the interface and the reaction process during different parameter measurements, where the intensity of absorbent color reflects the concentration distribution of reaction products. (b) Microfluidic measurements are integrated with automated image analysis and DFT simulations to establish an accelerated platform for CO2 capture fluids screening. (c) Schematic diagram of experimental setup. The microfluidic device consists of a stainless-steel manifold that secures the silicon-glass chip, providing enclosed fluid pathways. Temperature control is achieved through a copper block and an electric heater. The chip features multiple dead-end channels arranged for the comparison of amine solutions' performance by monitoring the movement of gas–liquid interface. (d) Automated image analysis and performance parameters calculation. The CO2-amine interface is detected via intensity changes using the Sobel edge detection method. Interface position and velocity are calculated along a predefined coordinate axis. The absorption rate, desorption rate, and absorption capacity are then determined through a gas equation of states. | ||
Microfluidic measurements were integrated with automated image analysis and DFT simulations to expedite and enrich the evaluation of CO2 capture fluids (Fig. 1b). Specifically, DFT simulations were applied to ascertain the Gibbs free energy changes along the reaction pathways, which — together with adsorption and desorption measurements — indicated the potential of the capture fluid and its applicability. These experimental measurements were implemented using a microfluidic device (Fig. 1c), where real-time interfacial dynamics during CO2 absorption/desorption cycles were tracked via automated imaging (Fig. 1d, ESI† Appendix section S1) and analyzed in conjunction with the gas state equation.
Fig. 2 shows the schematic diagram of the experimental setup for high-throughput screening of CO2 capture fluids. The central component of our microfluidic screening platform is a customized microfluidic chip (75.2 mm × 35 mm × 1.5 mm). Its fabrication starts with creating patterns on a chromium mask, which are transferred onto a silicon substrate via UV lithography, followed by deep reactive ion etching (RIE) to achieve 250 μm channel depths. The silicon substrate is then anodically bonded to a borosilicate glass cover, forming tightly sealed microchannels that allow direct optical observation of fluidic behaviors. This silicon-glass construction provides excellent chemical resistance and can withstand high temperatures (∼200 °C) and pressures (∼200 bar). A high-pressure syringe pump (Teledyne ISCO 200DX) delivers CO2 or N2 gas to maintain experimental pressure, while a high-precision syringe pump (Harvard Apparatus, Pump 11 Elite) continuously injects the capture solution at a constant rate. Before experiments, a vacuum pump evacuates the system to ensure gas purity. Pressure sensors (Omega, PX409) monitor gas and liquid pressures at the inlet and outlet channels.
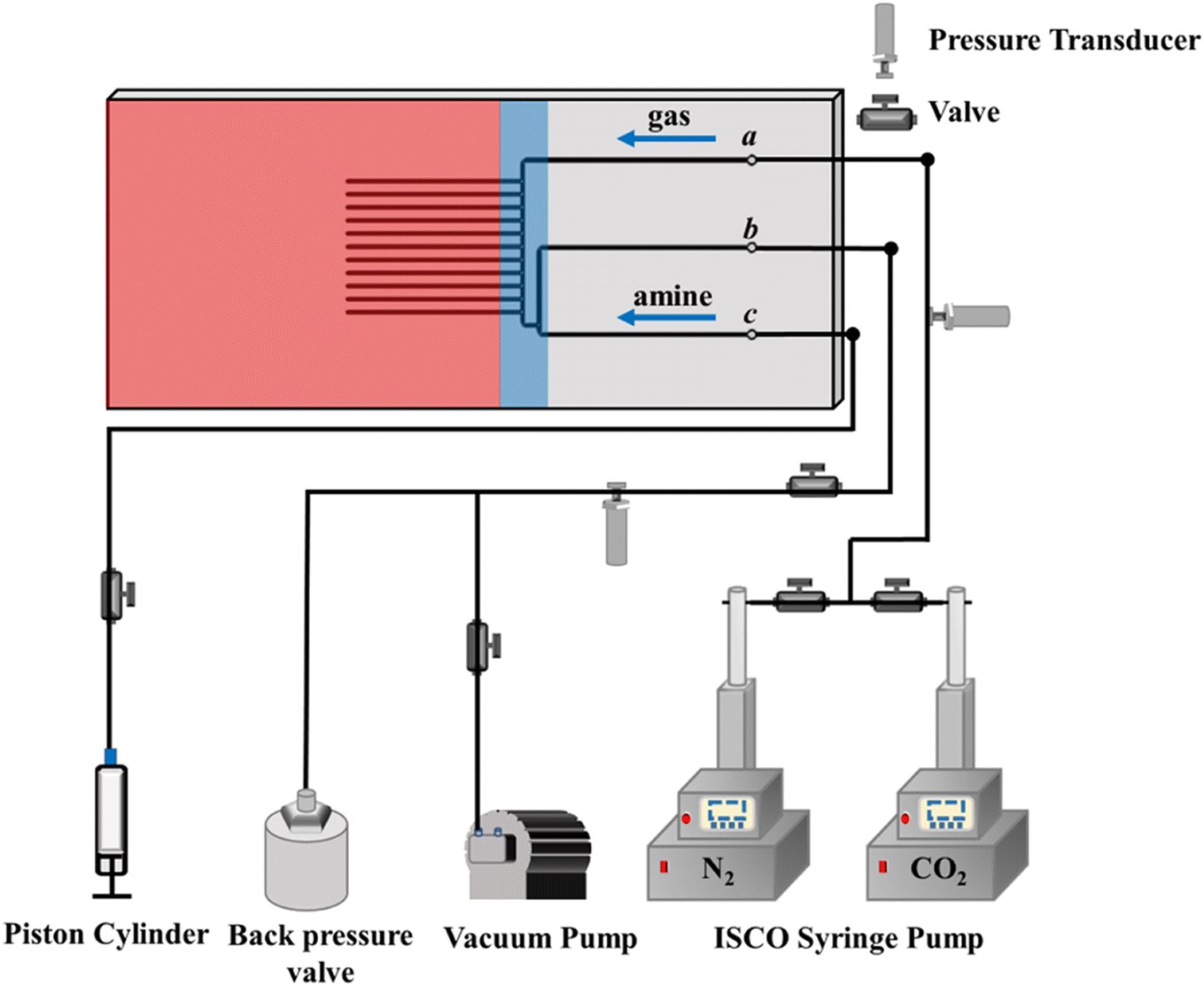 | ||
| Fig. 2 Schematic diagram of experimental setup for CO2 capture fluids screening. | ||
The experimental procedures for CO2 capture performance evaluation comprise four key tests: absorption rate, desorption rate, absorption capacity, and vapor pressure comparison.
2.1. Absorption rate test
CO2 is injected into the chip through port a and maintained at a target pressure (∼2.15 bar), while a 3 mol L−1 aqueous amine solution (35 °C) is introduced through port c. A T-shaped junction upstream of the testing channels diverts pre-reacted fluid to port b, ensuring only fresh solution enters the dead-end channels. Once unreacted fluid is confirmed at the T-junction, port b is closed to direct the solution into the testing channels. To ensure consistency and robustness of the reported results, we present the averaged response across multiple channels.2.2. Desorption rate test
Following the absorption rate protocol, capture solution injection ceases when the gas–liquid interface reaches the appropriate position. N2 is then injected through port a to purge residual solution from the main channel. Ports a and c are vented to atmospheric pressure, and the chip is heated to 70–80 °C to initiate CO2 release from the amine solution.2.3. Absorption capacity test
After replicating the absorption rate procedures, N2/CO2 is injected through port a to flush residual solution from the main channel, forming a finite liquid slug in the testing channel. Subsequently, port c is closed, and N2/CO2 is maintained at target pressure. CO2 occupies the slug's left side, while N2/CO2 fills the right side. Tests use 1 mol L−1 amine solution at 25 °C.2.4. Vapor pressure comparison
The microfluidic system is evacuated, followed by the injection of capture fluid to saturate the dead-end testing channels. N2 purges residual solution from the main channel via port a. The chip is heated to 35 °C or 80 °C to initiate evaporation, with N2 continuously supplied at ∼1.1 bar to accelerate evaporation and minimize atmospheric CO2 interference.3. Results and discussion
3.1. Microfluidic results
We selected six different types of amines (monoethanolamine, pentylamine, diethylamine, diethanolamine, N-methyldiethanolamine, triethanolamine) as model CO2 capture solutions. This set includes the most commonly used amines and covers primary, secondary, and tertiary amine types (details in Table S1 of the ESI† Appendix). | (1) |
 | ||
| Fig. 3 CO2 absorption and desorption tests at 35 °C and 80 °C, respectively. (a) Dynamic interface movement during CO2 absorption: the red dashed line outlines the interface, with amine solution on the right and CO2 on the left; (b) temporal evolution of interface displacement in absorption tests, with gray shading indicating standard deviation from at least 3 experimental sets; (c) calculation of average CO2 absorption rate based on the real gas state equation (Van der Waals equation, a = 3.66 L2 mol−2, b = 0.0429 L mol−1) and interface displacement within 10s (T = 35 °C, P = 2.15 bar); (d) dynamic interface movement during CO2 desorption: the red dashed line outlines the interface, with amine solution on the right and CO2 on the left; (e) temporal evolution of interface displacement in desorption tests; (f) calculation of average CO2 desorption rate within 10s (T = 80 °C, P = 1 bar). | ||
The CO2 desorption test follows the CO2 absorption test. Here, rapid heating on the microchip is employed to contain the diffusion of reaction products within the test channels. The system can elevate the chip's temperature to ∼70 °C within ∼6 s and further to 80 °C after ∼16 s (refer to section S3 of ESI† Appendix). Within a time frame of less than 100 seconds and the absence of convection, the diffusion distance remains under 1 mm (section S4 of ESI† Appendix). This limited diffusion ensures that the absorption and desorption reactions occur in close proximity to the gas–liquid interface (see Fig. 1a), approximating a closed system within the test channel. The heating of the chip instigates the release of absorbed CO2, leading to an increase in pressure within the residual gas slug and a gradual shift of the gas–liquid interface towards the right (Fig. 3d).
Fig. 3e shows the interface movement, indicating the thermal response associated with CO2 desorption in various amine solutions. The expansion of the gas segment is not solely influenced by CO2 release, but also by the combined effects of thermal expansion and vapor pressure changes. With regard to vapor pressure, the capture fluids (3 mol L−1 aqueous solutions) possess a solute molar fraction below 0.1. According to Raoult's law, the vapor pressure of the solution is primarily determined by water, provided that the vapor pressure of the solute is lower than that of water (See section S5 of ESI† Appendix). Consequently, the contribution of vapor pressure to the expansion of the gas slug is largely consistent across different test fluids, rendering it negligible in the comparative analysis of desorption rates. The effect of thermal expansion can be compensated for using the gas state equation. Desorption rate is defined as the average rate over 10 seconds, and detailed outcomes are presented in Fig. 3f. Notably, the desorption rate trend generally inverses that of absorption, with tertiary amines exhibiting more rapid CO2 release than primary and secondary amines. This difference is primarily attributed to the simpler decomposition of bicarbonate (main reaction product between tertiary amines and CO2) compared to that of carbamate (main reaction product between primary/secondary amines and CO2).19,38,39 An anomalous case is observed with diethylamine (DA), which exhibits the highest desorption rate among all examined amine solutions, deviating from expected behaviour.40 This anomaly is due to its relatively high vapor pressure at 80 °C, DA contributes a substantial partial pressure (∼16 kPa) despite its low mole fraction, making its effect on gas slug expansion during desorption non-negligible compared to other amines (see ESI† section S5).
 | ||
| Fig. 4 CO2 absorption capacity and vapor pressure comparison tests. (a) Dynamic movement process of absorbent slug, with the final position reflecting the absorption capacity for CO2; (b) absorption capacities of DEA, MDEA and MEA at a CO2 partial pressure of 2.65 bar, along with the variation in MEA absorption capacity under different CO2 partial pressures; (c) dynamic interface movement in vapor pressure tests. The evaporation process of the amine occurs within an N2 environment; (d) average evaporation rate of different amines. Experiments for amines with higher vapor pressures (i.e., DA, MEA, PA) are conducted at a lower temperature (35 °C), while experiments for amines with lower vapor pressures (i.e., DEA, MDEA, TEA) are conducted at a higher temperature (80 °C) to shorten the testing duration. | ||
3.2. DFT simulation
The microfluidic measurements leverage the dynamics of the gas–liquid interface to evaluate the reaction kinetics and thermodynamic parameters involved in CO2 capture. Within the quasi-closed dead-end channels, the movement of the interface is primarily governed by the absorption or desorption of CO2. However, the interface movement it is also influenced by fluid properties such as interfacial tension and viscosity. To further corroborate that the experimental data accurately reflects the reaction rates, DFT simulations were employed to assess CO2 and absorbent molecule interactions, with further details available in section S8 of ESI† Appendix. The changes in Gibbs free energy and transition states during CO2 adsorption on six amine variants are illustrated in Fig. 5. | ||
| Fig. 5 DFT calculated energy barrier diagram of the CO2 and amine reaction in aqueous solution. H, white; C, gray; N, blue; O, red. | ||
The energy barriers for CO2 absorption and desorption are represented by ΔGTs-IS and ΔGTs-FS, respectively. DFT calculations reveal a CO2 absorption kinetic sequence of DA ≈ PA ≈ MEA > DEA ≈ MDEA > TEA, while exhibiting an inverse trend in desorption. This ordering matches that of the experimental data. However, limitations in each methodology lead to some discrepancies. The theoretical prediction for desorption kinetics for DA is slow, in stark contrast to the experimental result showing the fastest CO2 desorption rate. This discrepancy stems from the significant influence of DA's vapor pressure on experimental outcomes (see section S5 of ESI† Appendix). Similarly, DFT predicts comparable absorption or desorption kinetics for some capture fluids, in contrast to the differences observed in the experimental results. This may be attributed to the influence of fluid properties and mass transfer processes in the liquid phase – aspects not captured by DFT. Thus, the microfluidic experiments assess apparent reaction rates while DFT calculations reveal intrinsic kinetics. The combination enables an assessment of both the potential of a given capture fluid and how it would perform in practice.
4. Conclusion
We introduce a microfluidic-driven framework for comprehensive and rapid screening of CO2 capture fluids, integrated with automated image processing and DFT simulations for accelerated analysis. The DFT simulations elucidate intrinsic reaction kinetics between capture fluids and CO2, offering theoretical insights that augment the experimental findings. The reliable, and high-throughput experimental data enriches theoretical prediction models with empirical evidence.The microfluidic system employs dead-end channels that circumvent challenges of two-phase dynamic flow reactors. The quasi-static nature of measurement facilitates a stable and controllable interface and improves the versatility of the platform. This design enables a rapid comparative assessment of capture fluid performance in terms of CO2 absorption rate (∼30 s), desorption rate (∼30 s), absorption capacity (∼20 min), and vapor pressure (∼5 min), achieving a speed that surpasses traditional methodologies by two orders of magnitude. The integration of microfluidic measurements with automated image processing and DFT simulation provides a means to screen, develop, and ultimately discover CO2 capture fluids. When extending this approach to novel fluids, practical implementation may benefit from parameter optimization (e.g., flow rates, pressure) under extreme conditions (such as exceptionally high viscosity or low surface tension) and supplementary characterization (e.g., viscosity, vapor pressure) to contextualize performance deviations, while the demonstrated success with diverse amine solutions confirms its foundational reliability for scalable CO2 capture fluids discovery.
Data availability
The data and code supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to express their sincere gratitude for the financial backing provided by the Natural Sciences and Engineering Research Council of Canada (NSERC), and the Canada Research Chairs Program (CRC-2021-00316). D. Sinton holds a Canada Research Chair in Fluids and Energy.References
- N. MacDowell, N. Florin, A. Buchard, J. Hallett, A. Galindo, G. Jackson, C. S. Adjiman, C. K. Williams, N. Shah and P. Fennell, Energy Environ. Sci., 2010, 3, 1645–1669 RSC.
- D. Y. Shu, S. Deutz, B. A. Winter, N. Baumgärtner, L. Leenders and A. Bardow, Renewable Sustainable Energy Rev., 2023, 178, 113246 CrossRef CAS.
- F. Shakerian, K.-H. Kim, J. E. Szulejko and J.-W. Park, Appl. Energy, 2015, 148, 10–22 CrossRef CAS.
- T. N. Borhani and M. Wang, Renewable Sustainable Energy Rev., 2019, 114, 109299 CrossRef CAS.
- F. O. Ochedi, J. Yu, H. Yu, Y. Liu and A. Hussain, Environ. Chem. Lett., 2021, 19, 77–109 CrossRef CAS.
- F. Meng, Y. Meng, T. Ju, S. Han, L. Lin and J. Jiang, Renewable Sustainable Energy Rev., 2022, 168, 112902 CrossRef CAS.
- S. Laribi, L. Dubois, G. De Weireld and D. Thomas, Int. J. Greenhouse Gas Control, 2019, 90, 102799 CrossRef CAS.
- O. Aschenbrenner and P. Styring, Energy Environ. Sci., 2010, 3, 1106–1113 RSC.
- A. Gautam and M. K. Mondal, Fuel, 2023, 334, 126616 CrossRef CAS.
- L.-C. Lin, A. H. Berger, R. L. Martin, J. Kim, J. A. Swisher, K. Jariwala, C. H. Rycroft, A. S. Bhown, M. W. Deem and M. Haranczyk, Nat. Mater., 2012, 11, 633–641 CrossRef CAS PubMed.
- B. Aghel, S. Sahraie and E. Heidaryan, Energy, 2020, 201, 117618 CrossRef CAS.
- W. Yu, T. Wang, A.-H. A. Park and M. Fang, Nanoscale, 2019, 11, 17137–17156 RSC.
- P. K. Chhotaray, S. K. Biswal and S. Pandey, J. Mol. Liq., 2020, 312, 113477 CrossRef CAS.
- X. Zhang, X. Zhang, H. Dong, Z. Zhao, S. Zhang and Y. Huang, Energy Environ. Sci., 2012, 5, 6668–6681 RSC.
- J. L. McDonagh, S. Zavitsanou, A. Harrison, D. Zubarev, T. van Kessel, B. H. Wunsch and F. Cipcigan, Digital Discovery, 2024, 3, 528–543 RSC.
- J.-G. Lu, A.-C. Hua, L.-L. Bao, S.-X. Liu, H. Zhang and Z.-W. Xu, Sep. Purif. Technol., 2011, 82, 87–92 CrossRef CAS.
- H. Karlsson and H. Svensson, Energy Procedia, 2017, 114, 2009–2023 CrossRef CAS.
- C. Nwaoha, P. Tontiwachwuthikul and A. Benamor, Int. J. Greenhouse Gas Control, 2019, 82, 218–228 CrossRef CAS.
- B. Lv, B. Guo, Z. Zhou and G. Jing, Environ. Sci. Technol., 2015, 49, 10728–10735 CrossRef CAS PubMed.
- I. M. Bernhardsen and H. K. Knuutila, Int. J. Greenhouse Gas Control, 2017, 61, 27–48 CrossRef CAS.
- R. Rivera-Tinoco and C. Bouallou, J. Cleaner Prod., 2010, 18, 875–880 CrossRef CAS.
- G. Puxty, R. Rowland and M. Attalla, Chem. Eng. Sci., 2010, 65, 915–922 CrossRef CAS.
- J. Ma, S. M.-Y. Lee, C. Yi and C.-W. Li, Lab Chip, 2017, 17, 209–226 RSC.
- H. Shi, K. Nie, B. Dong, M. Long, H. Xu and Z. Liu, Chem. Eng. J., 2019, 361, 635–650 CrossRef CAS.
- T. Cubaud, M. Sauzade and R. Sun, Biomicrofluidics, 2012, 6(2), 022002 CrossRef PubMed.
- D. Voicu, M. Abolhasani, R. Choueiri, G. Lestari, C. Seiler, G. Menard, J. Greener, A. Guenther, D. W. Stephan and E. Kumacheva, J. Am. Chem. Soc., 2014, 136, 3875–3880 CrossRef CAS PubMed.
- M. Abolhasani, A. Günther and E. Kumacheva, Angew. Chem., Int. Ed., 2014, 53, 7992–8002 CrossRef CAS PubMed.
- S. S. Datta, I. Battiato, M. A. Fernø, R. Juanes, S. Parsa, V. Prigiobbe, E. Santanach-Carreras, W. Song, S. L. Biswal and D. Sinton, Lab Chip, 2023, 23, 1358–1375 RSC.
- T.-H. M. Ho, J. Yang and P. A. Tsai, Lab Chip, 2021, 21, 3942–3951 RSC.
- A. Sharbatian, A. Abedini, Z. Qi and D. Sinton, Anal. Chem., 2018, 90, 2461–2467 CrossRef CAS PubMed.
- Y. Guo, F. Liu, J. Qiu, Z. Xu and B. Bao, Energy, 2022, 256, 124524 CrossRef CAS.
- Y. Xu, J. Riordon, X. Cheng, B. Bao and D. Sinton, Am. Ethnol., 2017, 129, 14150–14155 Search PubMed.
- M. Nooryani, A. M. Benneker and G. Natale, Lab Chip, 2023, 23, 2122–2130 RSC.
- D. Mackay and I. van Wesenbeeck, Environ. Sci. Technol., 2014, 48, 10259–10263 CrossRef CAS PubMed.
- S. P. De Visser, J. Phys. Chem. B, 2011, 115, 4709–4717 CrossRef CAS PubMed.
- G. Sartori and D. W. Savage, Ind. Eng. Chem. Fundam., 1983, 22, 239–249 CrossRef CAS.
- J. Gomes, S. Santos and J. Bordado, Environ. Technol., 2015, 36, 19–25 CrossRef CAS PubMed.
- M. Kumari, F. Vega, L. M. G. Fernández, K. P. Shadangi and N. Kumar, J. Mol. Liq., 2023, 122288 CrossRef CAS.
- Z. Wang, M. Fang, Y. Pan, S. Yan and Z. Luo, Chem. Eng. Sci., 2013, 93, 238–249 CrossRef CAS.
- F. A. Chowdhury, H. Yamada, T. Higashii, K. Goto and M. Onoda, Ind. Eng. Chem. Res., 2013, 52, 8323–8331 CrossRef CAS.
- T. W. de Haas, B. Bao, H. Acosta Ramirez, A. Abedini and D. Sinton, Energy Fuels, 2021, 35, 7866–7873 CrossRef CAS.
- J. I. Lee, F. D. Otto and A. E. Mather, J. Chem. Eng. Data, 1972, 17, 465–468 CrossRef CAS.
- A. M. Bhairi, Experimental equilibrium between acid gases and ethanolamine solutions, PhD, Oklahoma State University, 1984 Search PubMed.
- J. I. Lee, F. D. Otto and A. E. Mather, J. Appl. Chem. Biotechnol., 1976, 26, 541–549 CrossRef CAS.
- W. Chen, A. J. Haslam, A. Macey, U. V. Shah and C. Brechtelsbauer, J. Chem. Educ., 2016, 93, 920–926 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4lc00772g |
| This journal is © The Royal Society of Chemistry 2025 |