
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An in vivo mimetic liver-lobule-chip (LLoC) for stem cell maturation, and zonation of hepatocyte-like cells on chip†
Philip
Dalsbecker‡
 a,
Siiri
Suominen‡
b,
Muhammad Asim
Faridi§
a,
Reza
Mahdavi§
ac,
Julia
Johansson
a,
Charlotte Hamngren
Blomqvist
a,
Mattias
Goksör
a,
Katriina
Aalto-Setälä¶
b,
Leena E.
Viiri¶
b and
Caroline B.
Adiels
*a
a,
Siiri
Suominen‡
b,
Muhammad Asim
Faridi§
a,
Reza
Mahdavi§
ac,
Julia
Johansson
a,
Charlotte Hamngren
Blomqvist
a,
Mattias
Goksör
a,
Katriina
Aalto-Setälä¶
b,
Leena E.
Viiri¶
b and
Caroline B.
Adiels
*a
aDepartment of Physics, University of Gothenburg, Origovägen 6B, SE-41258 Gothenburg, Sweden. E-mail: caroline.adiels@physics.gu.se
bHeart Group, Finnish Cardiovascular Research Center and Science mimicking life Research Center, Faculty of Medicine and Health Technology, Tampere University, Tampere, Finland
cBiotechnology Department, Faculty of Chemical Engineering, Tarbiat Modares University, Tehran, Iran
First published on 28th May 2025
Abstract
In vitro cell culture models play a crucial role in preclinical drug discovery. To achieve optimal culturing environments and establish physiologically relevant organ-specific conditions, it is imperative to replicate in vivo scenarios when working with primary or induced pluripotent cell types. However, current approaches to recreating in vivo conditions and generating relevant 3D cell cultures still fall short. In this study, we validate a liver-lobule-chip (LLoC) containing 21 artificial liver lobules, each representing the smallest functional unit of the human liver. The LLoC facilitates diffusion-based perfusion via sinusoid-mimetic structures, providing physiologically relevant shear stress exposure and radial nutrient concentration gradients within each lobule. We demonstrate the feasibility of long term cultures (up to 14 days) of viable and functional HepG2 cells in a 3D discoid tissue structure, serving as initial proof of concept. Thereafter, we successfully differentiate sensitive, human induced pluripotent stem cell (iPSC)-derived cells into hepatocyte-like cells over a period of 20 days on-chip, exhibiting advancements in maturity compared to traditional 2D cultures. Further, hepatocyte-like cells cultured in the LLoC exhibit zonated protein expression profiles, indicating the presence of metabolic gradients characteristic of liver lobules. Our results highlight the suitability of the LLoC for long-term discoid tissue cultures, specifically for iPSCs, and their differentiation in a perfused environment. We envision the LLoC as a starting point for more advanced in vitro models, allowing for the combination of multiple liver cell types to create a comprehensive liver model for disease-onchip studies. Ultimately, when combined with stem cell technology, the LLoC offers a promising and robust on-chip liver model that serves as a viable alternative to primary hepatocyte cultures—ideally suited for preclinical drug screening and personalized medicine applications.
1 Introduction
Within the field of pharmaceutical development, the utilization of in vitro cell cultures is crucial for evaluating drug efficacy and safety, facilitating early-stage screening prior to the initiation of in vivo trials. A key focus in drug development is the scrutiny of hepatic toxicity, necessitating thorough evaluation during the preclinical testing phases. However, the reliability of conventional 2D in vitro hepatic culture systems is inadequate, contributing to late-stage drug failures due to undetected hepatotoxicities.1–4Addressing this gap, there is growing need for more physiologically relevant culture systems. Over the past 15 years, liver-on-a-chip (LoC) and liver microphysiological systems (MPS) have evolved significantly, progressing from micropatterned two-dimensional (2D) coculture systems1,5 to more complex 3D structures.6–9 Such a system could be designed for direct implementation in high-throughput studies or to generate a physiologically relevant environment for in-depth research into cell differentiation, disease development, and pharmacodynamics.10 Additionally, the complexity may vary based on the used cell source, ranging from immortalized cell lines to primary cells and induced pluripotent stem cell (iPSC)-derived cells. Despite being considered the gold standard, primary cells face limitations in availability11,12 and donor variability.13–15 Conversely, iPSC-derived cells provide a substantial advantage, demonstrating adaptability across diverse research objectives and offering unique prospects for disease modeling and personalized medicine. Nevertheless, challenges may emerge, stemming from iPSC-derived cells' sensitivity and specific MPS culture requirements.16
In the pursuit of enhanced hepatic cell culture environments, key physiological traits have been identified to mirror the hepatic niche. These include a controlled microenvironment, e.g., regulated shear stress and nutrient distribution as well as self-organized three-dimensional (3D) culture, all acknowledged for fostering a more physiologically relevant hepatic phenotype in vitro.17–21
Fluid shear stress has consistently shown benefits for hepatocytes in multiple studies, resulting in elevated expression of genes associated with drug metabolism, detoxification,17 cytochrome P450 (CYP) enzyme activity, and serum protein secretion.20,22
Fluid shear stress levels exceeding 10−6 dyn per cm2 have proven advantageous for hepatocytes in a 2D environment, increasing albumin secretion,21 glycogen storage, resistance to alcohol-mediated injury,20 and enhancing the expression of genes related to drug metabolism and detoxification, notably the xenosensor.17,20 However, it's crucial to recognize that an excessive level of fluid shear stress (>5 dyn per cm2) has been observed to have detrimental effects on hepatocyte functionality, as seen in various studies of 2D adherent cultures.20,23–25 In 3D cultures, the situation appears more intricate. Experimental research show significant disparities between 2D and 3D cultures.22 Additionally, theoretical simulations pinpoint that the 3D extracellular matrix contributes to even higher shear stress than suggested by averaged simulations.26 Also matrix stiffness has proven to have an impact on hepatocyte cultures, both by itself and in combination with shear stress modulation.21 Hence, evaluating and adjusting shear stress levels is necessary to provide optimal conditions for cells cultured in LoCs.
Similarly to shear stress, 3D cell–cell interaction have been shown to benefit hepatic polarity, viability, and functionality in vitro.27–30 These benefits have been utilized in LoC cultures through both spheroid-on-a-chip systems,31–33 3D mass cultures,6,7,34,35 and combinations thereof.36 Spheroids in static culture may develop necrotic cores due to the absence of vascularization.37 Such a limitation is partially addressed by spheroid-on-a-chip systems, which facilitate easier access to fresh medium through culture perfusion.31 However, the nutrient transport to the centers of the spheroids is still reliant on diffusion and, therefore, may not always suffice to fully support the cells in the spheroid core, depending on the size and density of the spheroid as well as the metabolic demands of the constituent cells.38,39
In contrast, 3D mass culture systems have the potential to solve or circumvent this issue through structural means. This is exemplified by34 who developed a chip with a 3D cell culture environment fluidically connected to a perfused channel, sustaining 3D high-density cultures of rat primary hepatocytes for 7 days. Similarly,7 created a liver lobule-mimicking chip accommodating either a hepatic cell line or iPSC-derived hepatocytes, both showcasing sustained metabolic activity for over two and three weeks, respectively.
Interestingly, in vivo, hepatocytes exhibit distinct metabolic functions depending on their position within the liver lobule. These functional differences are governed by a continuous gradient of oxygen and nutrient concentrations along the sinusoidal axis, from the periportal to the perivenous region. The periportal region (zone 1) located close to the outer periphery of the liver lobule, receives the most oxygenated and nutrient-rich blood, whereas the perivenous region (zone 3) near the central vein, is exposed to blood with the lowest oxygen and nutrient content.40 The intermediate zone (zone 2) displays metabolic features between those of zones 1 and 3. This physiological gradient not only drives metabolic zonation but also activates Wnt signaling, particularly in the perivenous zone, where Wnt ligands are highly expressed.41 Wnt signaling plays a central role in maintaining zonal gene expression and establishing the functional identity of the three zones within the lobule. Zone 1 hepatocytes are predominantly involved in processes such as gluconeogenesis, β-oxidation, and urea formation, while zone 3 hepatocytes exhibit higher glycolysis, glutamine formation, lipogenesis and xenobiotic metabolism through elevated expression of cytochrome P450 enzymes.42 Recreating liver zonation in 3D in vitro culture systems is increasingly important for modeling organ-specific functions in drug development and disease research. Many liver disorders—including metabolic dysfunction-associated steatotic liver disease (MASLD) and hepatocellular carcinoma—are linked to disruptions in zonation and region-dependent hepatocyte activity, making spatial organization essential for accurate assessment of hepatotoxicity and disease mechanisms.
In this context, we present a microfluidic liver-lobule-chip (LLoC) design, which enables optimal support for non-proliferating iPSC-derived hepatocyte-like cells (HLCs), offering on-chip differentiation and long-term culturing into mature HLCs in 3D. The design echoes those of Lee et al.,34 and Banaeiyan et al.,7 but features larger diffusion channels, individually seeded artificial liver lobules, and a modular top layer for cell culture perfusion flexibility. We provide a comprehensive description of the LLoC design, computational fluid dynamics simulations, and show the successful long-term culturing of functional hepatoma cells. Subsequently, we demonstrate the sustained on-chip differentiation of sensitive iPSC-derived HLCs into mature HLCs on-chip followed by their zonation as indicated by distinct protein expression profiles. Our LLoC, with the potential for co-culturing various liver-specific cell types, is established by these findings as a fundamental and physiologically meaningful in vitro screening tool.
2 Materials and methods
2.1 Liver-lobule-chip characteristics, design and operating principles
Our LLoC is characterized by an array of artificial liver lobules situated within the main flow channel of the device, ensuring homogeneity in nutrient and solute supply, as well as shear stress levels within a physiologically relevant range. The interior of the artificial liver lobules structurally mirrors the original liver lobule-specific sinusoids, partitioned from the main flow channel by boundary walls perforated with diffusion channels. Additionally, the dual-layer design of the LLoC mimics the in vivo microenvironment of liver lobules by guiding the cell culture medium through the 3D cell discoids and away via a central-vein mimetic in each artificial liver lobule.43In the main layer flow channel of the LLoC, 21 perfusable artificial liver lobules are arranged in a hexagonal array mirroring the layout of hepatic lobules in vivo, see Fig. 1. The main flow channel measures 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 μm at its maximum width and gradually narrows towards the inlet and outlet ports. Each artificial liver lobule has a 2500 μm outer diameter, an inner height of 60 μm, a cell culture area of 3.4 mm2, and features a perforated circular structure mimicking the sinusoidal capillaries of hepatic lobules in vivo, connecting the portal triad at the acinus to the central vein. The artificial liver lobules contain 16 such sinusoid-mimetics—eight short (435 μm) and eight long (715 μm)—evenly and intermittently spaced around the artificial liver lobule wall. Both the indentations and the surrounding wall are perforated with 5 μm wide and 34 μm high channels to enable perfusion and diffusion of nutrients. This dual barrier wall serves to shield the hepatocytes from excessive shear stress induced by the medium flow, mimicking the role of the hepatic fenestrated endothelium in vivo. The longer sinusoid-mimetics facilitate more efficient cell culture medium delivery to the 3D cell discoids near the lobule centers, while the shorter ones enhance perfusion in the outer lobule regions.
600 μm at its maximum width and gradually narrows towards the inlet and outlet ports. Each artificial liver lobule has a 2500 μm outer diameter, an inner height of 60 μm, a cell culture area of 3.4 mm2, and features a perforated circular structure mimicking the sinusoidal capillaries of hepatic lobules in vivo, connecting the portal triad at the acinus to the central vein. The artificial liver lobules contain 16 such sinusoid-mimetics—eight short (435 μm) and eight long (715 μm)—evenly and intermittently spaced around the artificial liver lobule wall. Both the indentations and the surrounding wall are perforated with 5 μm wide and 34 μm high channels to enable perfusion and diffusion of nutrients. This dual barrier wall serves to shield the hepatocytes from excessive shear stress induced by the medium flow, mimicking the role of the hepatic fenestrated endothelium in vivo. The longer sinusoid-mimetics facilitate more efficient cell culture medium delivery to the 3D cell discoids near the lobule centers, while the shorter ones enhance perfusion in the outer lobule regions.
 | ||
| Fig. 1 The LLoC design. A) Displayed in a first-angle multi-view (2D), the figure showcases the LLoC and its corresponding flow directions. The LLoC consists of two structured pieces of PDMS, the main layer and the top layer, stacked on top of a glass slide. The large polygonal flow channel, located within the main layer, has an inlet for culture medium (solid red arrow), an outlet for culture medium (dashed red arrow) from the main layer, and an air trap marked as dashed circle. At its widest, the flow channel measures 13600 μm. Within the flow channel are 21 individual artificial liver lobules forming a hexagonal pattern. The incoming culture media flow is coloured red, and the media flow exiting the artificial liver lobules is coloured blue. The top layer collects the medium from all artificial liver lobules via their chimney-like apertures to the common top layer outlet (solid blue arrow). B) The three layers of the LLoC, top layer, main layer and glass slide, are shown in axonometric projection (3D). The air trap is marked as a solid grey cylinder. The scale bar of 1 cm is valid for both A) and B). C) A single artificial liver lobule is shown in semi-transparent axonometric projection (3D). Emerging from the centre of the artificial liver lobule is the chimney-like aperture used for cell seeding and for mimicking the central vein of a liver lobule. Each artificial liver lobule is filled with cells and perfused by cell culture medium. They measure 2500 μm in outer diameter and each one accommodates a total culture area of 3.4 mm2 with a height of 60 μm. D) A close-up view representing 1/16th of an artificial liver lobule reveals more details. Each lobule has 16 sinusoid mimetics, featuring eight short (435 μm) and eight long (715 μm) structures distributed evenly along the circular chamber wall. Both the chamber perimeter and the convoluted interior are intricately perforated with diffusion channels, measuring 5 μm in width and 34 μm in height. | ||
At the core of each artificial liver lobule lies a chimney-like aperture, measuring 1 mm in diameter, which ascends to the top layer of the LLoC (Fig. 1). Serving as both a cell seeding port and a central-vein-mimetic, these apertures guide the cell culture media through and away from the artificial liver lobules and into the top layer channels (34 μm in height). The media then exits the device via a collection outlet port in the top layer. This design closely mirrors the blood flow pattern in hepatic lobules in vivo.
The top layer is reversibly placed on top of the main layer, allowing for adjustment or replacement. When in place, it allows for culture medium efflux through the channels of the top layer as well as through the outlet port on the main layer. Alternatively, during culture, the top layer can be substituted with a flat lid, directing the cell culture medium around the artificial liver lobules and out through the outlet port of the main layer. Conversely, with the top layer in position, the main layer's outlet port can be sealed, channeling all the cell culture medium through the artificial liver lobules and the top layer. During cell seeding, the top layer is absent, and cells are seeded directly into the artificial liver lobules through the aperture. Once seeded, a top layer or plain lid is applied to enable flow of medium through or around the artificial liver lobules and out through the seeding ports and/or main layer outlet.
The wafer mold for the main layer was made using sequential deep reactive ion etching to etch patterns of two different depths into a silicon wafer.43 A SiO2-coated silicon wafer was patterned with AZ1512HS photoresist (MicroChemicals, Ulm, Germany) using direct laser writing, creating a patterned mask protecting the SiO2 layer. The wafer was then wet-etched in buffered hydrofluoric acid to etch away the uncoated areas of SiO2. The photoresist was removed and replaced with a new layer of photoresist, which was patterned with direct laser writing using a different pattern. The wafer was then etched using deep reactive ion etching, first with the new photoresist pattern in place and then again once the photoresist mask had been removed, using the underlying SiO2 layer as a mask. This sequential etching resulted in a mold that is a structural negative of the microfluidic devices. The wafer was post-processed to remove the SiO2 layer and add a silane layer to facilitate PDMS detachment.
The wafer mold for the top layer was produced using patterned photoresist on a silicon wafer. Following the manufacturer's instruction, SU-83035 (Kayaku Advanced Materials, Inc., Westborough, Massachusetts, USA) was spin-coated onto the wafer, baked and exposed to UV light through a patterned photomask (MicroLitho, Chelmsford, UK). Following development, the wafer was post-processed to add a silane layer, facilitating PDMS detachment.
Both layers of the LLoC were cast in PDMS (Sylgard 184, Dow Corning, Midland, Michigan, USA) by mixing polymer base and crosslinking agent in a 5:1 (w/w) ratio, in a fume hood. The mixture was degassed, applied to the wafers, and baked at 60 °C for at least 24 h. Seeding, inlet, and outlet ports on the chamber layer, as well as the outlet port on the top layer, were punched using a 1 mm tissue biopsy puncher (33-31AA-P/25, Miltex, Integra LifeSciences, Princeton, New Jersey, USA). The main layer and the glass microscope slides were cleaned with ethanol and exposed to air plasma (18 W, 30 s; PDC-32G-2, Harrick Plasma, Ithaca, New York, USA) before being bonded to each other by conformal contact and cured at 60 °C for at least 1 h. Neither the top layer nor the plane lid were plasma treated, in order for them to be detachable.
2.2 Simulations of the LLoC configurations
The LLoC was designed using AutoCAD (Autodesk, San Rafael, CA, USA), imported into COMSOL Multiphysics 6.0 (Comsol, Burlington, MA, USA) as a 2D DXF file. It was then refined and converted to 3D using the geometry tools in COMSOL.The simulation model comprises a fluid flow analysis coupled with mass transfer analysis for glucose in parallel. Glucose, being one of the main constituents of cell culture medium, was chosen to model the consumption of nutrients in the medium by the cells. The diffusion coefficient of glucose was chosen to be 6.16 × 10−10 m2 s−1 in culture medium at a concentration of 11 mM, and the glucose consumption rate of HepG2 cells (2.4 × 10−10 mol min per million cells) was used as consumption flux at the culture region inside the artificial liver lobules.44
The computational fluid dynamics (CFD) analysis was performed by coupling Navier–Stokes and continuity equations. The fluid was modelled as an incompressible Newtonian fluid, with a density of 1.009 g cm−3 and a dynamic viscosity of 0.93 mPa s based on the properties of the DMEM culture medium supplemented with 10% FBS and at 37 °C.45 The flow, ranging from 0.25 to 1.0 μL min−1, was modelled using no-slip boundary conditions. The pressure at the outlets was set to zero and the flow rate at the inlet was added accordingly.
To minimize computation time, only one artificial liver lobule was studied to assess flow field and glucose distribution. Given that each artificial liver lobule is radially symmetrical, a slice representing 1/16th of the entire lobule was used as the computation domain. Subsequently, the results were extrapolated to the entire artificial liver lobule. The geometry was meshed using COMSOL's physics-controlled mesh setting, with the mesh element size set to fine.
For the entire LLoC simulation, we used the Brinkman assumption for porous media to reduce the computation demand.46 We considered the diffusion channels around the lobule as a porous structure. Subsequently, we calculated the porosity and permeability of the region containing the diffusion channels. These values were then utilized to model the area surrounding the artificial lobules as a porous medium using the Brinkman equation, enabling us to simulate the entire LLoC.
For the oxygen distribution simulations, we adopted a reduced-scale modeling approach due to the high computational cost of simulating oxygen transport across the entire device—particularly when solving the Navier–Stokes equations under perfused conditions. This approach was consistent with the strategy for glucose and shear stress analyses. Specifically, we modeled a representative 1/16th section of the artificial liver lobule, applying boundary conditions derived from a preliminary full-device simulation conducted under static (no-flow) conditions. This initial simulation provided the steady-state oxygen concentration at the lobule boundaries, which was then used as the input for the smaller-scale model. By avoiding the need to solve fluid dynamics directly in the lobule simulation, we substantially improved computational efficiency. Oxygen distribution within the lobule was then solved using a convection-diffusion equation, incorporating experimentally established oxygen uptake rates for HepG2 cells in 3D culture.
2.3 Cell cultures
In order to validate the LLoC, hepatocytes were cultured on-chip and assayed for key metabolic markers. For initial method development and benchmarking of adherent on-chip culture conditions, immortalized HepG2 cells were chosen for their relative stability and low complexity of handling. To further validate the LLoC for more complex cultures, iPSC-derived definitive endoderm cells were cultured on-chip and differentiated into hepatocyte-like cells (iPSC-HLCs).For maintenance and expansion, the iPSCs were cultured on Geltrex (A1413202, Thermo Fisher Scientific, henceforth Geltrex-A) pre-coated standard six-well plates in mTeSR-1 medium (StemCell Technologies). 10 mM ROCK inhibitor Y-27632 (StemCell Technologies) was added to the medium at the thawing and passaging steps to improve cell survival. Cell lines were maintained at 37 °C in 5% CO2.
Hepatic differentiation was performed based on the differentiation protocol initially published by Kajiwara et al.48 This protocol is an alteration of iPSC-HLC differentiation protocols introduced by Si-Tayeb et al.49 and by Hay et al.50 and consists of three stages (henceforth ST1, ST2 and ST3). Here, ST1 was performed on traditional 2D cultures, while ST2 and ST3 were performed on both 2D and 3D cultures, off-chip and on-chip, respectively.
We substituted the ST1 media described in the protocol of Kajiwara et al.48 with a commercially available STEMdiff Definitive Endoderm (DE) kit (StemCell Technologies, Cat no. 05510). After the iPSC colonies became 80–90% confluent, differentiation to DE cells was initiated by seeding the iPSCs as a single-cell suspension on Geltrex-coated plates on day 0 to reach 90–100% confluency on day 1. The rest of the DE differentiation was performed as per the manufacturer's instructions.
On day 5, hepatic specification (ST2) was initiated by switching to another medium (henceforth ST2 medium) consisting of KnockOut Dulbecco's modified Eagle medium (KO-DMEM) supplemented with 20% KnockOut Serum Replacement (KO-SR, Gibco), 1 mM GlutaMAX (Gibco), 1% nonessential amino acids (NEAA, Lonza), 0,1% 2-mercaptoethanol (2-ME, Lonza), 50 U ml−1 penicillin/streptomycin and 1% dimethyl sulfoxide (DMSO, Sigma) for 7 days until differentiation day 11. From day 11 onward, cells were cultured in ST3 media consisting of Hepatocyte Basal Medium (HBM, Lonza) supplemented with single quotes (CC-4182, Lonza), 25 ng ml−1 hepatocyte growth factor (HGF, Invitrogen) and 20 ng ml−1 oncostatin M (OSM, R&D systems).
2.4 Flow cytometry
To determine the efficiency of the ST1 differentiation of iPSCs, DE cells were harvested for flow cytometry on the fifth day of the iPSC differentiation. Cells were detached by a 5 min incubation with gentle cell dissociation reagent and re-suspended in 5% FBS before being stained with CXCR4-conjugated antibody (R&D systems) in the dark at RT for 15 minutes. The percentage of CXCR4-positive cells was determined using CytoFLEX S (Beckman Coulter) and CytExpert Software (Beckman Coulter).2.5 Microfluidic experimental procedure
Owing to the different requirements of the HepG2 cells and the iPSC-derived cells, certain key differences were found to be necessary in their respective seeding and culture methods.:1 with cold Geltrex-A and 0.5 μl of suspension at approximately 110 to 120 × 106 cells per ml was seeded into each chamber. Each aperture was then topped up with 1 μl of 37 °C Geltrex-B. The apertures were then sealed with a plane lid and ST2 medium was added as previously described. The LLoCs were incubated for 3 hours, during which the media was changed once manually, after which tubes were attached and the flow of medium (1 μl min−1) was initiated. The following day, the plane lids were replaced with the top layers, and a collection tube was attached to its outlet. iPSC-HLC LLoCs were pumped without pump-pause cycles with Ismatec peristaltic pump (IP-N 12, Cole Parmer, Germany). The iPSC-HLC LLoCs were incubated for 20 days, and the output medium was collected every second day.
Two different 2D control cultures were also established for the LLoC iPSC-HLC cultures. Control 1 (Ctrl 1) had iPSC-HLCs (UTA.11304.EURCCs) differentiating from the start of ST1 (day 0) to the end of ST3 (day 26) with no outside interference on a 12-well plate on top of Geltrex-A coating. Control 2 (Ctrl 2) cells underwent a similar (detaching-reseeding) step at the end of ST1 on day 5 of differentiation when compared to the LLoC cultures, where the DE cells were first detached and then seeded on Geltrex-A-coated wells to continue their differentiation on well-plates. We estimated a 20% fraction of the cells to die due to the detaching-reseeding step and plated the cells accordingly.
2.6 Albumin secretion assay
The albumin secretion was measured by using a human albumin ELISA kit (ab108788, Abcam, Cambridge, UK) and analyzed with FLUOstar Omega, BMG LABTECH (Ortenberg, Germany), using the online-tool ELISA INFO from Boster Biological Technology CO LTD, (California, USA) with a 4PL fitting. The mean values and the standard deviations were calculated and visualized using MATLAB R2023a, MathWorks (Massachusetts, USA). The sampled output medium from the LLoCs were diluted 100 and 240 times. The results were normalized according to the seeded cell number (approximated to 0.5 million cells per LLoC), the volume of the collected downstream medium, and the elapsed time. Output media samples from the 3D iPSC-derived HLC LLoCs were not diluted, while 2D samples were diluted twice, and the results were normalized according to the seeding cell number, the volume of the collected medium and the elapsed time.2.7 On-chip immunofluorescence and viability staining
The on-chip staining protocols were carried out with slight variations for the two cell types described below.:2000 in DPBS) three times, each followed by a 15-minute incubation at RT. Lastly, the LLoCs were washed three times with PBS and imaged using Olympus IX51 inverted phase-contrast fluorescence microscope (Olympus Corporation, Hamburg, Germany).
Equivalent immunofluorescence staining was performed for the 2D iPSC-HLC cultures with minor changes. The flushes were omitted and replaced with continuous incubations. As the last step before imaging, the samples were mounted with Vectashield mounting medium (Vector Laboratories) that included DAPI.
To study the zonation of iPSC-HLCs, the above-described protocol for LLoC staining was followed on day 25 of differentiation with two zone-specific primary antibodies arginase 1 (ARG1, Novus Biologicals) and glutamine synthetase (GS, InVitrogen). After completing the staining process, the LLoCs were imaged with Leica DMi 8 inverted wide-field fluorescence microscope.
2.8 Light microscopy
Wide field images of the HepG2 cells were acquired using an LRI Olympus CKX41 in transmission mode with a phase contrast component.Fluorescence z-stack images of fixed HepG2 cells on day 7 (45 sections 0.95 μm apart) were acquired with an LSM 880 Airyscan microscope (Carl Zeiss) equipped with an air immersion Plan-Apochromat 20×/0.8 M27 objective and the Zen Black 2.3 software. Ethidium homodimer-1 and Hoechst were excited by 514 nm and 405 nm lasers, respectively, and imaged sequentially frame-by-frame.
Morphology and viability of the iPSC-HLCs were assessed by imaging using EVOS FL cell imaging system (Thermo Fisher Scientific). After double-fluorescence immunostaining, the cells were imaged with Olympus IX51 inverted phase contrast fluorescence microscope (Olympus Corporation, Hamburg, Germany) using Hamamatsu Orca Flash4.0LT + sCMOS camera and Olympus cellSens Dimension 2.3 software. LLoCs stained with zone-specific primary antibodies were imaged with Leica DMi 8 inverted wide-field fluorescence microscope (Leica Microsystems, Wetzlar, Germany) using LAS X software.
Images were processed by ImageJ/Fiji.
3 Results and discussion
3.1 Computational fluid dynamic analysis of shear stress, glucose consumption and oxygen distribution profiles
Our LLoC aimed to expand upon the principles and concepts of the chip described by Banaeiyan et al.7 (termed the very large-scale liver lobule chip, henceforth VLSLL), utilizing its strengths while improving on its practical handling, utility, and capacity. The LLoC, similar to the VLSLL, consists of a dual-layer polydimethylsiloxane (PDMS) structure wherein cell culture compartments are separated from a central flow path by a perforated barrier. While the VLSLL relied primarily on diffusion for nutrient transport, the LLoC improves upon this by incorporating a sinusoid-mimicking design to enhance perfusion. Within the LLoC, the medium flows through these sinusoid-mimetic channels, passing through a cell tissue compartment before reaching an upper outlet, simulating the central vein in vivo. One additional benefit of these sinusoid-mimetic channels is their ability to protect the artificial liver tissue from shear stress, providing low-resistance pathways for the medium. Consequently, this design ensures consistent delivery of nutrients and solutes to the artificial liver lobules, while also exposing the cells to physiologically relevant levels of shear stress.To evaluate our LLoC design, we conducted CFD simulations to ensure that glucose distribution and shear stress levels align with physiologically relevant ranges. We examined a limited range of flow rates in the simulations: 0.25, 0.5, and 1.0 μL min−1 (Fig. 2). This approach allowed us to determine the influence of flow rate on shear stress and nutrient availability to the initially used HepG2 cells, and to identify the optimal balance between the two. It is preferable to maintain a relatively low total volume output to obtain detectable levels of secreted proteins, while ensuring that hepatocytes are exposed to an adequate level of nutrients and kept within the normal range of shear stress.
 | ||
| Fig. 2 Computational fluid dynamics analysis ensures physiologically relevant and consistent glucose and oxygen distribution as well as shear stress levels. A) Shear stress in the liver lobules was simulated in 1/16th of a lobule for three different flow rates: 0.25, 0.50 and 1.0 μL min−1. For all tested flow rates, the shear stress inside the LLoC is in a range between 10−4 and 10−2 dyn per cm2, and fall well below the detrimental range of shear stress for hepatic cells.17,20,21,23–25 B) Glucose concentration was simulated for the three flow parameters and considered the glucose consumption rate of seeded cells in the culture area of the artificial liver lobule. Starting at 11 mM, glucose concentration decreased along the radius toward the central vein. At all tested flow rates, the glucose concentration decreased radially and also affected the concentration in the diffusion channels. C) Oxygen simulation within a representative 1/16th of a lobule, showing a radial gradient of concentration toward the center of the lobule. Across the range of tested flow rates there was insignificant differences in oxygen distribution, demonstrating diffusion-limited oxygen transport. | ||
Fig. 2A shows the simulated shear stress in 1/16th of a lobule for the three different flow rates. The simulations are conducted using an empty artificial liver lobule, resulting in 3D simulations displaying shear stress at the bottom surface. The shear stress profiles in the artificial liver lobules ranges from a minimum of 10−4 dyn per cm2 (10−5 Pa) for 0.25 μL min−1 flow to a maximum of 10−2 dyn per cm2 (10−3 Pa) for 1.0 μL min−1. The highest shear stress is found in the narrow diffusion channels. These resulting shear stress values are in the range of 10−2 dyn per cm2 to 10−1 dyn per cm2 at the chosen flow rates.
The three explored flow rates all yield shear stress levels well within the range known to be acceptable to hepatocyte-like cells in 2D studies, i.e., 10−6 dyn per cm2 to 5 dyn per cm2.17,20,21,23–25 However, since the LLoC regards 3D cell cultures, it is difficult to foresee the actual forces generated due to the cell bodies themselves and the presence of extracellular matrix components.26 The selected flow rates were intentionally set at the lower end of the acceptable range, with the expectation that the actual shear stress experienced by the cells would likely be higher than the simulated values. In our results it is anticipated that the majority of each artificial liver lobule will encounter a shear stress level closer to 10−3 dyn per cm2. The higher end of the simulated results (approximately 10−2 dyn per cm2) is mainly concentrated at the edges of the artificial lobules, where the diffusion channels meet the culture chambers. Ultimately, the shear stress values must be considered in relation to the required flow rate for nutrient distribution within the artificial liver lobule, as well as downstream assessments of cell viability and functionality.
Glucose, serving as the primary nutritional component in the culture media, was employed in the simulations as a representative for overall nutrient distribution. The glucose distribution was simulated in the artificial liver lobules, considering the number of seeded HepG2 cells (24000 cells per artificial liver lobule) and their known glucose consumption rate when cultivated in a 3D setting (see section 2.2). The result is a radial gradient of glucose concentration, exemplified for 1/16th of a lobule in Fig. 2B. This gradient mirrors the nutrient concentration pattern observed in in vivo liver lobules, depicting decreasing nutrient access for cells in the mid-lobule (zonation).51,52 The LLoC received cell culture media with an initial glucose concentration of 11 mM. At a flow rate of 0.25 μL min−1, there was a 50% reduction in glucose concentration across the radius of the artificial liver lobule. This decline raises concerns, as proliferating cells can face nutrient deficiency at this rate, given their increasing consumption of glucose and other nutrients. Conversely, a higher flow rate of 1.0 μL min−1 showed no significant change in glucose concentration across the radius of the artificial liver lobule compared to the 0.5 μL min−1 flow rate.
We reasoned that, for a physiologically relevant mimicry of nutrient gradients resembling liver zonation, a flow rate of 0.5 μL min−1 to 1.0 μL min−1 should be chosen. This decision aims to balance the nutrient demands of proliferating cells while preserving a resemblance to the natural nutrient distribution found in the liver and minimized total output volume. It is important to note that this scenario is specific to HepG2 cells and their known metabolic glucose consumption rate in 3D, considering the initial seeded cell number. Consequently, different cell types may likely prefer slightly different flow rates, reflecting their nutrient demands over time.
To further characterize the microenvironment within the artificial liver lobules, we simulated oxygen distribution by modeling a representative 1/16th section of the artificial liver lobule (see section 2.2). As illustrated in Fig. 2C, the simulated oxygen concentration forms a physiologically relevant radial gradient across the lobule section, decreasing toward the central zone where cell density and oxygen consumption are highest. To validate the assumption that oxygen distribution is largely governed by cellular uptake rather than perfusion at the examined flow rates, we repeated the simulation using the same boundary data at flow rates of 0.25, 0.5, and 1.0 μL min−1. Across all scenarios, the resulting oxygen profiles showed no significant differences, indicating that perfusion rate had minimal impact on oxygen availability within the artificial liver lobule (data not shown). These results validate our initial modeling assumption and align with prior observations that oxygen transport in dense 3D hepatocyte cultures is largely diffusion-limited rather than flow-dependent.
3.2 Functional HepG2 3D cultures within the LLoC
The HepG2 cells were cultured in the LLoC with continuous perfusion (0.5 μL min−1) for a duration of up to 14 days, resulting in the formation of 3D discoid tissue structures that filled the artificial liver lobules, as illustrated in Fig. 3A. An optimized number of HepG2 cells (24000 cells per artificial liver lobule) was evenly distributed inside each artificial liver lobule allowing for cell proliferation throughout the experiment.
 | ||
| Fig. 3 HepG2 viability and functionality in the liver-lobule-on-chip (LLoC). A) The cell proliferation in the LLoC illustrated by a time series of one single artificial liver lobule over the course of 14 days, imaged using conventional bright field microscopy. During this time the cells gradually filled the space and formed a dense tissue-like structure. The scale bar is 700 μm. B) The LLoC connected to inlet and outlet tubes. The cell medium is pumped in through the inlet (1), and the supernatant is initially exiting the device through the main layer outlet (2). After 24 hours post cell attachment, the plane lid is exchanged for the patterned top layer and the media is redirected via the chimney-like apertures and exiting through the top layer outlet (3). Supernatant from either outlets is collected into separate tubes within the stand (4). C) The albumin secretion in HepG2-filled LLoCs was determined using human albumin ELISA assays. The plot shows the mean value of the albumin collected from twelve separate LLoCs (n = 12) in three different experiments of 14 days, with the standard deviation presented as error bars. The data points are two days apart. After the first media collection on day two, the lower outlet of the LLoC was clamped. D) Z-stack using confocal imaging of an example subsection of an artificial liver lobule, displayed as maximum intensity projections. The cells were imaged seven days after seeding and nuclei of living and dead cells stained with Hoechst and ethidium homodimer-1 (EthD-1), respectively. At the day of investigation, the majority of cells are healthy, but a fraction of them are disrupted and, hence, are stained by the EthD-1. The scale bar is 100 μm. | ||
Around one week into the experiment, the discoid liver tissues started to completely fill the artificial liver lobules. As the cell cultivation continued throughout the entire experimental period, the tissue further compacted, as indicated by the darker area observed on days 8 and 14 in Fig. 3A. This occurrence is ascribed to the limited space available for cell occupancy. Any cells that perish remain confined within the artificial liver lobule and the developed discoid structure. This discovery strongly indicates the necessity of utilizing non-proliferating or limited-proliferating cell types in combination with the LLoC.
The dynamic functionality of discoid tissues was assessed by measuring the concentration of secreted albumin in the collected media, as depicted in Fig. 3B and C. The albumin secretion, normalized to the initially seeded HepG2 cell number, showed an noticeable twofold increase from day 2 to day 4 resulting in an albumin production reaching ∼400 ng h per million seeded cells. Following this, the albumin production seemed to stabilize, settling at around 500 ng h per million seeded cells by day 14 (Fig. 3C). This is likely attributed to a near equilibrium reached between the cell proliferation and cell mortality rates. This assumption is reinforced by the immunofluorescent cell staining using EthD-1 and Hoechst, revealing that while the majority of cells are not adversely affected, some cells exhibit membrane rupture at day 7 (Fig. 3D).†† The evaluation was also performed at the final time point, but the dense structure of the discoids hindered complete penetration of the dye, resulting in inconclusive results (data not shown). The compact nature of the discoids was already apparent by day 7 (ESI† Movie S1) and has previously been commonly observed in spheroid cultures. On a positive note, this characteristic provides evidence of close cell contacts and 3D tissue formation. Furthermore, thanks to the LLoC design for enhanced perfusion and smaller tissue dimensions, the risk of the liver discoid tissue developing necrotic cores as may occur in larger spheroids37,38 is eliminated. Albumin data were collected from a total of twelve individual LLoCs, in which HepG2 cells were cultured under flow conditions for 14 days. The experiments were conducted across three independent runs. Fig. 3C demonstrates the variations within individual experiments and across different experiments. The results suggest that while HepG2 cells, serving as a foundational model for liver tissue, can form functional 3D structures inside the LLoC, the device holds potential for more advanced cell models, such as human-derived iPSCs. This particular cell model is anticipated to overcome limitations associated with the finite volume, given that iPSCs generally do not proliferate. Furthermore, given the theoretical nutritional and oxygen gradients present in the LLoC, we hypothesize that exposing iPSCs to this environment during their differentiation process will mimic the in vivo conditions that induce zonation.
3.3 On-chip maturation of iPSC-derived hepatocyte-like cells (HLCs)
Culturing human iPSCs in 3D is seen as the development of an in vitro tissue model with substantial potential. In order to assess our ability to replicate in vivo-like microenvironmental conditions within our LLoC, iPSC-derived HLCs were chosen for on-chip differentiation in extended 3D cell cultures. Two iPSC lines were used for the LLoC experiments: UTA.10211.EURCAs and UTA.11304.EURCCs. On day 5 of their differentiation, iPSC-derived cells exhibited a cobblestone-like morphology typical for definitive endoderm stage.53,54 The DE differentiation efficiency was further studied by flow cytometry. The number of CXCR4-positive cells was between 80–95% of all cells on day 5 of differentiation, indicating successful DE differentiation.53,54To generate the 3D LLoC cultures, the cells were detached on day 5 of differentiation, combined with Geltrex-B and seeded into the artificial liver lobules of LLoCs. A bright field image captured 24 hours after the cell seeding reveals uniformly filled lobules (Fig. 4A). The viability of the differentiating HLCs was assessed 5 days after cell seeding, and the fluorescent image display predominately live cells, affirming the success of cell seeding and culture (Fig. 4B).
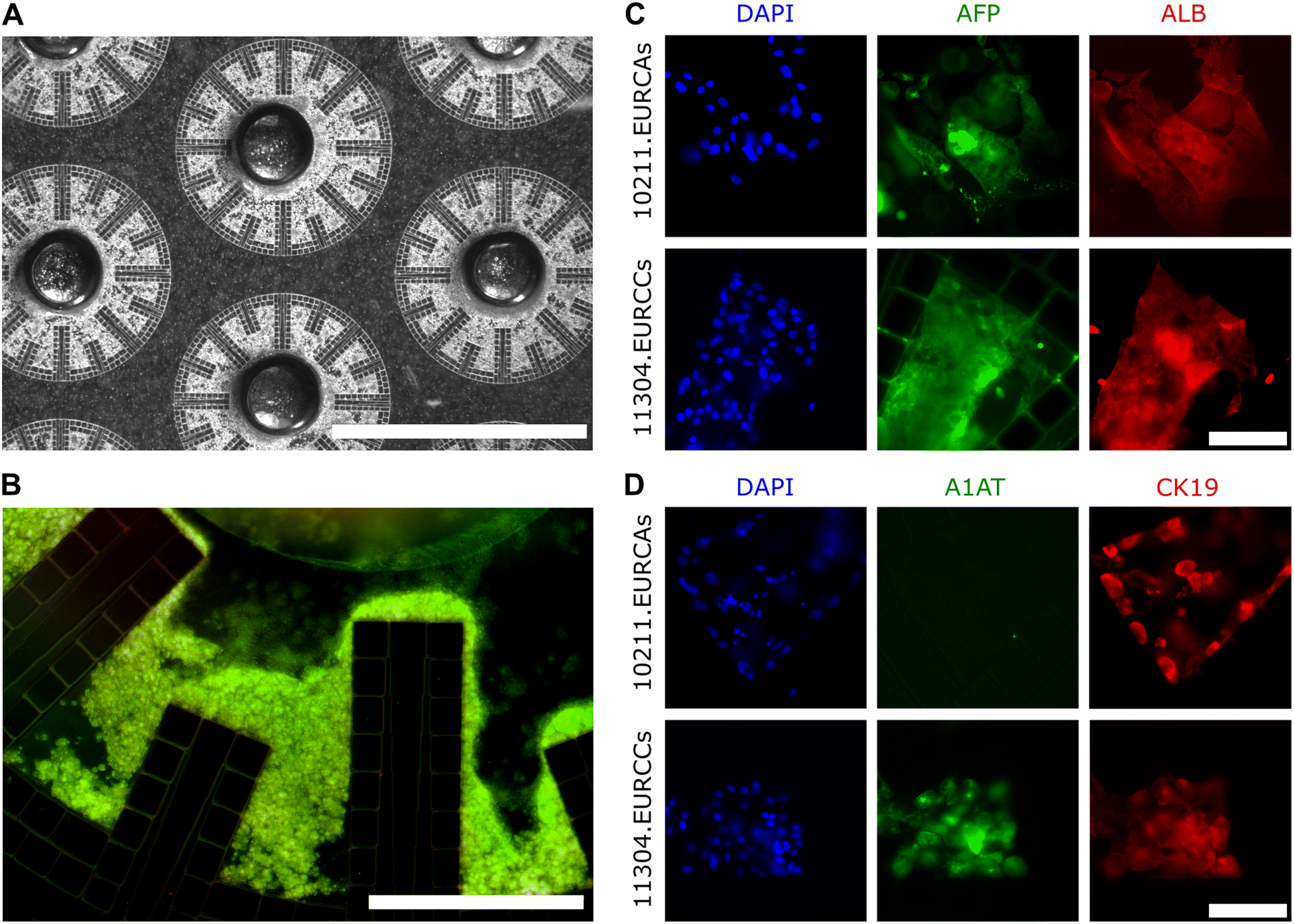 | ||
| Fig. 4 On-chip differentiated iPSC-derived HLC viability and cell line variability. A) A representative transmission microscopy image depicting differentiating iPSC-derived HLCs, specifically definitive endoderm cells, 24 hours after cell seeding into the LLoC. The scale bar is 2000 μm. B) A fluorescence image displaying differentiating iPSC-derived HLCs within a subsection of a LLoC lobule stained with calcein-AM (green) and ethidium-1 (red) for live and dead cells, respectively, on day 5 of differentiation. The scale bar is 400 μm. C and D) Two iPSC lines, UTA.10211.EURCAs (top row) and UTA.11304.EURCCs (bottom row) were differentiated towards HLCs for 25 days (of which 20 days on-chip), after which they were fixed and stained for hepatic markers and imaged with Olympus 1X51 fluorescence microscope. Nuclei were stained with DAPI and are shown in blue. C) Both cell lines express alpha-fetoprotein (AFP, green), and albumin (ALB, red). The scale bar is 100 μm D) while 10211.EURCAs cell line does not express alpha-1 antitrypsin (A1AT, green), its expression in 11304.EURCCs cell line is prominent. Both cell lines express liver progenitor marker cytokeratin 19 (CK19, red). The scale bar is 100 μm. | ||
After 20 days of LLoC culture, the devices were stained with immunofluorescence targeting liver-specific markers. We detected strong expression of AFP and albumin in both cell lines on day 20 on-chip (Fig. 4C) which indicates that the cells had successfully differentiated towards mature hepatocytes when the differentiation procedure is carried out on-chip. The expression of liver specific A1AT was observed predominantly in the UTA.11304.EURCCs cell line, while the expression of liver progenitor marker CK19 was similar between the two cell lines (Fig. 4C and D).
The differences in A1AT expression could be due to donor-dependent variation in the hepatic differentiation, a common attribute of iPSC-derived cells.48 Based on these results, UTA.11304.EURCCs was chosen to be used in the subsequent experiments.
To compare 3D iPSC-HLC cultures differentiated in LLoCs with 2D control cultures, cells underwent one of three conditions after reaching the DE stage on day 5 of differentiation (Fig. 5A): 1) Ctrl 1: uninterrupted growth and differentiation on the same well-plate, 2) Ctrl 2: detachment and re-seeding on similar well-plates to continue differentiation in 2D, or 3) LLoC: detachment and combination with Geltrex-B, followed by seeding into the liver lobules of LLoCs for a 3D culture.
 | ||
| Fig. 5 Comparison between differentiated 2D and 3D iPSC-derived HLC cultures. A) The experimental outline of comparing LLoC and 2D control cultures using iPSC-derived HLCs. The differentiation protocol for iPSC-derived HLCs comprises three stages. At the end of the first stage, when the cells have reached the definitive endoderm (DE) stage, they undergo one of three conditions: 1) Ctrl 1: allowed to grow without disruption, 2) Ctrl 2: detached and re-seeded on well plates, or 3) LLoC: detached, combined with hydrogel (HG), and seeded into the LLoCs. The first stage is executed on well plates for all conditions. The circles on the timeline indicate the days for the albumin secretion assays depicted in B). B) Albumin secretion was evaluated using human albumin ELISA assays. The iPSC-derived HLCs in the LLoCs exhibited a progressive increase in albumin secretion. Although the secretion peaked higher in the Ctrl 1 cultures compared to the LLoC cultures, production started to decrease from day 19 onwards and reached a lower level compared to the LLoC cultures on day 25. Ctrl 2 cultures consistently maintained a lower level throughout the experimental time frame. C) LLoC, Ctrl 1, and Ctrl 2 cultures were stained for the mature hepatic marker alpha-1 antitrypsin (A1AT, green) and the liver progenitor marker cytokeratin 19 (CK19, red) on day 15 and day 20 of LLoC culture, corresponding to day 20 and day 25 of differentiation, respectively. The LLoC culture displayed the most prominent stain of A1AT at the later time point. Nuclei are shown in blue. The scale bar is 100 μm. D) LLoC, Ctrl 1, and Ctrl 2 cultures were stained for the hepatic marker alpha-fetoprotein (AFP, green) and mature hepatic marker albumin (ALB, red) on day 15 and day 20 of LLoC culture, corresponding to day 20 and day 25 of differentiation, respectively. The LLoC culture displayed the most prominent stain of ALB at the later time point. Nuclei are shown in blue. The scale bar is 100 μm. | ||
Cell functionality was evaluated by measuring albumin production into the conditioned media. iPSC-derived HLCs in the LLoCs showed a continuous rise in albumin secretion throughout the entire experimental period (Fig. 5B). While albumin production peaked higher in Ctrl 1 cultures compared to LLoC cultures, it started declining from day 19 onwards and reached a lower level compared to LLoC cultures by the end of the experiment. Additionally, Ctrl 2 cultures consistently maintained a lower level throughout the experimental time frame.
Both LLoC and control iPSC-HLC cultures underwent immunofluorescence staining on days 20 and 25 to identify changes in hepatic markers over time and between culture conditions. On day 25, around 60% of the cells in 2D cultures, both Ctrl 1 and Ctrl 2, had died and detached from the well bottoms. Consequently, the immunofluorescence images represent the remaining cultures.
When comparing the expression of liver-specific markers on days 20 and 25, LLoC cultures exhibited higher expression of the mature hepatocyte marker albumin (Fig. 5C), aligning with the albumin ELISA results. Additionally, the expression of another mature hepatic marker, A1AT, was more pronounced at the later time point, indicating ongoing maturation in the LLoC cultures at least until day 25 (Fig. 5D). Concurrently, the expression of CK19, a marker of liver progenitor cells, decreased over time in the LLoC cultures (Fig. 5D), probably due to a more progressed state of maturation. In contrast, in both iPSC-HLC control cultures, the expression of both A1AT and albumin decreased, and the expression of CK19 remained constant over the same time frame, suggesting that the best maturation was reached around day 20 in the 2D cultures. (Fig. 5C and D).
Based on these results, which are in line with the hypothesis, the LLoC support the iPSC-HLCs cultures better than traditional 2D platforms. The impact of detachment on cells is apparent as indicated by the lower expression of albumin and CK19 in Ctrl 2 cells on day 20 compared to Ctrl 1 cultures. Additionally, the albumin production levels remain low for the duration of the experiment in the Ctrl 2 cultures. Notably, the LLoC environment provides sufficient support for the cells to overcome these challenges, enabling them to surpass the levels observed in Ctrl 1 cultures.
When compared to the HepG2 cultures, the iPSC-derived HLC cultures exhibit an approximately 50-fold lower production of albumin. This outcome was anticipated, as it has been previously observed,55 owing to the presence of clonal heterogeneity and inherent developmental stage of the cells, where iPSC-HLCs may not have fully matured to produce albumin. Moreover, unlike HepG2 cells, iPSC-HLCs do not proliferate, which means that the initial cell count remains static or may even decrease due to hydrogel disintegration.
To further validate the in vivo-like environment within the LLoC, we assessed protein expression profiles characteristic of hepatic zonation. Arginase I, the final enzyme in the urea cycle, is primarily expressed in the periportal region (zone 1) of the liver lobule.56 Immunofluorescence staining (Fig. 6) revealed increased arginase I expression near the outer regions of the lobule mimetics, suggesting zone 1-like functionality. Conversely, glutamine synthetase–responsible for converting ammonia and glutamic acid into glutamine—is typically expressed in the perivenous (zone 3) region.57 In our cultures, this marker was predominately found in cells near the LLoC central aperture, mimicking the central vein, thereby reflecting the in vivo spatial distribution.
 | ||
| Fig. 6 On-chip differentiated iPSC-derived HLC zonation in artificial liver lobules. Representative fluorescence microscope images depicting the expression of the periportal marker arginase 1 (ARG1, red) and the pericentral marker glutamine synthetase (GS, green) in iPSC-derived HLCs on day 20 of LLoC culture. Nuclei are stained with DAPI and shown in blue. A) A subsection of a lobule showing a typical zonation pattern. B) A subsection of a lobule depicting zonation with areas of high GS expression on cells not directly in contact with the perfusion channels. The scale bars are 200 μm. | ||
This study presents two key outcomes: (1) enhanced hepatic maturation and long-term viability of iPSC-derived HLCs in 3D cultures within our LLoC, and (2) successful induction of physiologically relevant zonation in these cells. First, the LLoC supports extended culture and functional maturation of iPSC-derived HLCs beyond what is typically observed in standard 2D systems. While most 2D HLC cultures deteriorate after approximately 25 days, cells in the LLoC remain viable and continue to mature over the same period. This is particularly important given that iPSC-HLCs commonly retain fetal-like characteristics in conventional formats.58,59 In our system, prolonged culture led to increased albumin production and sustained hepatic marker expression (Fig. 5C and D), indicating improved functional maturation. These findings highlight the LLoC's potential for overcoming a long-standing limitation in iPSC-derived liver models.
Second, the LLoC facilitates the emergence of hepatic zonation, a key feature of liver physiology. The gradient-like and opposing distribution of arginase I and glutamine synthetase suggests that the LLoC successfully recapitulates native zonation patterns. Interestingly, glutamine synthetase was also detected in deeper regions of high cell density extending toward the periportal area, further supporting a model in which metabolic consumption, in concert with the chip geometry, drives zonation. Hence, the formation of these gradients can be explained by the LLoC's architecture and material properties. Fabricated from PDMS, the chip allows oxygen diffusion through both the culture medium and device surfaces. However, outer cell layers intercept much of the diffused oxygen, limiting availability to inner, deeper-located cells. Cells at the bottom of the lobule chamber, primarily in contact with the glass substrate and surrounded by neighboring cells, rely more heavily on oxygen delivered via medium perfusion (Fig. 2C). Since perfusion channels reach only halfway up the lobule mimetics, lower outer cells are the first to receive oxygen and nutrients, while deeper regions are perfused last. These regions, therefore, experience nutrient and oxygen depletion but accumulate signaling molecules secreted by upstream cells. This is further supported by two observations: (1) cells cultured under static (no-flow) conditions do not remain viable,‡‡ and (2) immunostaining confirms spatially resolved expression of zonation markers.
Many earlier hepatic in vitro models rely on gradual conditioning of medium by upstream cells, enabling polarization to be enforced not only by the oxygenation but by signaling factors secreted by the upstream cells into the medium as well. One example is the device used by Prodanov et al., a PDMS-based chip consisting of two superimposed channels separated by a porous membrane.60,61 Hepatocytes are cultured in a monolayer in the lower channel below a collagen gel containing stellate cells, and the medium only flows in the upper channel, and thus medium must pass through the layer of endothelial cells and the porous membrane before reaching the hepatocytes. The endothelial cells thus impact the oxygenation and composition of the medium further below. Another chip by Xie et al.,61 made from folded polyimide tape, contains a static-medium hepatocyte 2D culture in the middle, bounded on either side by a porous membrane separating the middle channel from a flow channel on either side. The hepatocytes are grown on the static side of the two membranes, with endothelial cells grown on the flow-channel side of the membranes. Thus, it creates a similar situation as in the chip used by Prodanov et al., albeit surrounded by a different material than PDMS, which affects the oxygenation of the medium. Both models rely on the medium being primarily conditioned by sinusoidal cells on the flow side. The hepatocytes may secrete signaling substances into the medium in the static channel, but this can diffuse upstream as well as downstream, potentially weakening the zonation impact.
Alternative designs rely on flow systems exposing the hepatocytes to shear and placed adjacent to gas channels. For example, Tonon et al.62 designed a PDMS-based device with parallel gas and flow channels to create a lateral oxygen gradient. Their system successfully modulated the expression of liver-specific genes in 2D cultures of differentiated human embryonic stem cells, including albumin, GLUL, and CPS1. Mahdavi et al.63 advanced this concept by embedding interwoven gas channels beneath the culture chamber to produce a tri-zonal pattern of albumin expression biased toward the periportal region of 2D HepG2 cell cultures.
In the context of these prior liver-on-a-chip designs, the LLoC offers a distinct advantage. Unlike these externally controlled approaches, it generates physiologically relevant gradients internally, through 3D tissue architecture and intrinsic cellular metabolism. The inclusion of sinusoid-mimetic channels breaks radial symmetry, aligning nutrient and oxygen consumption with the actual spatial distribution of cells. Although the observed zonation does not perfectly mirror in vivo patterns, it emerges from biologically plausible mechanisms that reflect the natural behavior of hepatocytes in hepatic tissue. Yet another promising application of the LLoC is its potential to support and perfuse 3D spheroids or hepatic organoids with minor design modifications. A recent study by Wesseler et al.36 demonstrated organoid zonation by engineering structural features directly into the chip. Their device consists of 3D-printed hydrogel fibers arranged in three adjacent chambers, with perfusion sequentially applied through fibers in one chamber at a time. This configuration establishes serial gradients of oxygen, nutrients, and signaling molecules via diffusion between the fibers and the surrounding organoid culture. As a result, each chamber experiences distinct micro environmental conditions, effectively mimicking zones 1–3 of the hepatic sinusoid. Although our approach differs—relying on intrinsic gradients shaped by chip geometry and cellular metabolism rather than forced structural patterning—similar zonation patterns may emerge in spheroids cultured within the LLoC. The current 3D structures formed in our device span approximately 5–6 cell layers,§§ which appears to be a sufficient diffusion distance to generate physiologically relevant metabolic gradients. This suggests that the LLoC could be adapted to promote zonation in spheroid-based liver models, expanding its applicability to a broader range of tissue-engineered systems.
4 Conclusions
The advancement of sophisticated in vitro culture systems plays a pivotal role in expanding the horizons of research in cell differentiation, disease development, and pharmacodynamics. This progression signifies a paradigm shift from conventional micropatterned two-dimensional (2D) coculture systems to more intricate liver-on-a-chip and liver microphysiological systems, as observed in the evolution over the past 15 years. Such trajectory offers a nuanced selection of culture complexity tailored to specific applications, accommodating both high-throughput studies and the creation of physiologically relevant environments for detailed investigations.Our LLoC emulates the architecture of the in vivo liver lobule, providing a 3D environment for cell culture along with a physiologically relevant perfusion pattern that supports hepatocyte function. The successful on-chip differentiation and zonation of definite endoderm cells, alongside cultures of HepG2 cells, suggest that the perfusion and shear stress levels within the device are favorable for hepatic maturation–laying the foundation for more advanced functional studies.
The LLoC's seeding strategy allows for precise control over the number of cells introduced into each lobule chamber, avoiding the use of branched seeding channels that can result in uneven cell distribution and unwanted deposition in inlet regions. The design minimizes variability between replicates and ensures consistency across experiments. When identical seeding conditions are applied, the device promotes uniform flow rates across all artificial lobules, and flow parameters can be readily optimized for specific applications. Additionally, the platform supports both compartmentalized seeding of different cell types into separate lobule chambers and sequential seeding within the same chamber, enabling flexible co-culture configurations.
Future work will focus on incorporating co-cultures of parenchymal and non-parenchymal cells—such as stellate cells and liver sinusoidal endothelial cells—to better replicate the in vivo liver microenvironment. This will also support the development of disease models that, e.g., reflect various stages of metabolic dysfunction-associated steatotic liver disease (MASLD), while preserving key intercellular signaling interactions. Exploring different combinations of cells types may further enhance tissue functionality; for example, a recent study has shown that iPSC-HLCs co-cultured with HUVECs and human adipose stem cells in microfluidic systems exhibit improved albumin secretion and enhanced maturity compared to monocultures,64 indicating a promising direction for further investigation.
In our investigations, the LLoC demonstrates a distinct gradient of glucose and medium-borne solutes throughout the artificial liver lobule. This feature aims to mimic the in vivo phenomenon of metabolic zonation,51,52 resulting in spatial heterogeneity of cellular phenotypes forming a gradient along the sinusoids. Our LLoC design replicates this gradient within the artificial lobules, where cells closer to the perforated walls are exposed to the medium and its nutrients earlier than deeper-lying cells. Consequently, deeper-lying cells receive medium with reduced concentrations of oxygen and nutrients but enriched with secreted metabolites and signaling substances from the outer cells, influencing their phenotype.
In summary, these findings demonstrate that the LLoC enables both advanced hepatic maturation of iPSC-derived cells and the emergence of spatially organized, zonated function within a 3D culture environment. By supporting on-chip differentiation and self-organized zonation in a controlled setting, the LLoC offers a reproducible and scalable alternative to primary hepatocyte cultures, which are often limited by donor variability, availability, and cost. This makes it particularly well-suited for applications requiring low inter-experimental variability, reinforcing its value as a physiologically relevant platform for disease modeling, drug screening, and personalized medicine applications.
Data availability
Data supporting this article, including the CAD files and corresponding documentation for usage are available either at DORIS – SND's data organisation and information system at [URL – https://doi.org/10.5878/9x7f-j603] or have been included as part of the ESI.†Author contributions
Philip Dalsbecker: conceptualization, methodology, software, validation, investigation, writing – original draft. Siiri Suominen: conceptualization, methodology, validation, investigation, writing – original draft and visualization. Muhammad Asim Faridi: methodology, writing – original draft. Reza Mahdavi: methodology, software, validation, formal analysis, investigation, writing – original draft, and visualization. Julia Johansson: validation, formal analysis, investigation, writing – original draft, data curation. Charlotte Hamngren Blomqvist: methodology, validation, investigation, writing – original draft, and visualization. Mattias Goksör: conceptualization, methodology, resources, writing – original draft, funding acquisition, and supervision. Katriina Aalto-Setälä: writing – review & editing, funding acquisition, project administration and supervision. Leena E. Viiri: conceptualization, writing – original draft, project administration and supervision. Caroline Adiels: conceptualization, methodology, resources, writing – original draft, visualization, funding acquisition, project administration and supervision.Conflicts of interest
The authors have no conflicts of interests to declare.Acknowledgements
We acknowledge the Centre for Cellular Imaging at the University of Gothenburg and the National Microscopy Infrastructure, NMI (VR-RFI 2019-00217) for providing assistance in microscopy (Fig. 3). We acknowledge Tampere facility of iPSC core, Biocenter Finland (BF), CellTech Laboratory and Tampere Imaging Facility (TIF) (Fig. 4 and 5) for their service. We also acknowledge Kaisa Tornberg and Lassi Sukki for providing assistance with pump set-up and device holders. The project received funding from Swedish Foundation for Strategic Research (ITM17-0384) and the European Union's Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie grant agreement No. 766181. Additionally funding for this project was obtained from the Research Council of Finland's Center of Excellence Body-on-Chip project, the Finnish Cardiovascular Research Foundation, the Päivikki and Sakari Sohlberg Foundation, Sigrid Juselius Foundation and the Ella and Georg Ehrnrooth Foundation.We acknowledge Stefano Romeo and coworkers for kindly providing the HepG2 cell line. Further, we also thank Leo van Grunsven and colleagues, especially Nuša Krivec, for kindly providing the mNeonGreen-expressing HepG2 cell line, and Jesús Antunez Dominguez for assisting with the two-photon movie compilation.
Notes and references
- S. R. Khetani, D. R. Berger, K. R. Ballinger, M. D. Davidson, C. Lin and B. R. Ware, J. Lab. Autom., 2015, 20, 216–250 CrossRef PubMed.
- E. W. Esch, A. Bahinski and D. Huh, Nat. Rev. Drug Discovery, 2015, 14, 248–260 CrossRef PubMed.
- H. Dirven, G. E. Vist, S. Bandhakavi, J. Mehta, S. E. Fitch, P. Pound, R. Ram, B. Kincaid, C. H. C. Leenaars, M. Chen, R. A. Wright and K. Tsaioun, Sci. Rep., 2021, 11(1), 6403 CrossRef PubMed.
- L. Ewart, A. Apostolou, S. A. Briggs, C. V. Carman, J. T. Chaff, A. R. Heng, S. Jadalannagari, J. Janardhanan, K.-J. Jang, S. R. Joshipura, M. M. Kadam, M. Kanellias, V. J. Kujala, G. Kulkarni, C. Y. Le, C. Lucchesi, D. V. Manatakis, K. K. Maniar, M. E. Quinn, J. S. Ravan, A. C. Rizos, J. F. K. Sauld, J. D. Sliz, W. Tien-Street, D. R. Trinidad, J. Velez, M. Wendell, O. Irrechukwu, P. K. Mahalingaiah, D. E. Ingber, J. W. Scannell and D. Levner, Commun. Med., 2022, 2(1), 154 CrossRef PubMed.
- S. N. Bhatia and D. E. Ingber, Nat. Biotechnol., 2014, 32, 760–772 CrossRef PubMed.
- S. Ya, W. Ding, S. Li, K. Du, Y. Zhang, C. Li, J. Liu, F. Li, P. Li, T. Luo, L. He, A. Xu, D. Gao and B. Qiu, ACS Appl. Mater. Interfaces, 2021, 13, 32640–32652 CrossRef PubMed.
- A. A. Banaeiyan, J. Theobald, J. Paukstyte, S. Woelfl, C. B. Adiels and M. Goksor, Biofabrication, 2017, 9, 015014 CrossRef PubMed.
- J. Liu, C. Feng, M. Zhang, F. Song and H. Liu, Front. Oncol., 2022, 5(12), 959299 CrossRef PubMed.
- L. Docci, N. Milani, T. Ramp, A. A. Romeo, P. Godoy, D. O. Franyuti, S. Kraehenbuehl, M. Gertz, A. Galetin, N. Parrott and S. Fowler, Lab Chip, 2022, 22, 1187–1205 Search PubMed.
- P. Dalsbecker, C. Beck Adiels and M. Goksör, Am. J. Physiol., 2022, 323, G188–G204 Search PubMed.
- J. Nulty, H. Anand and A. Dhawan, Stem Cells Transl. Med., 2024, 13, 204–218 CrossRef.
- K. Zhang, L. Zhang, W. Liu, X. Ma, J. Cen, Z. Sun, C. Wang, S. Feng, Z. Zhang, L. Yue, L. Sun, Z. Zhu, X. Chen, A. Feng, J. Wu, Z. Jiang, P. Li, X. Cheng, D. Gao, L. Peng and L. Hui, Cell Stem Cell, 2018, 23, 806+ Search PubMed.
- P. Godoy, N. J. Hewitt, U. Albrecht, M. E. Andersen, N. Ansari, S. Bhattacharya, J. G. Bode, J. Bolleyn, C. Borner, J. Boettger, A. Braeuning, R. A. Budinsky, B. Burkhardt, N. R. Cameron, G. Camussi, C.-S. Cho, Y.-J. Choi, J. C. Rowlands, U. Dahmen, G. Damm, O. Dirsch, M. Teresa Donato, J. Dong, S. Dooley, D. Drasdo, R. Eakins, K. S. Ferreira, V. Fonsato, J. Fraczek, R. Gebhardt, A. Gibson, M. Glanemann, C. E. P. Goldring, M. Jose Gomez-Lechon, G. M. M. Groothuis, L. Gustavsson, C. Guyot, D. Hallifax, S. Hammad, A. Hayward, D. Haeussinger, C. Hellerbrand, P. Hewitt, S. Hoehme, H.-G. Holzhuetter, J. B. Houston, J. Hrach, K. Ito, H. Jaeschke, V. Keitel, J. M. Kelm, B. K. Park, C. Kordes, G. A. Kullak-Ublick, E. L. LeCluyse, P. Lu, J. Luebke-Wheeler, A. Lutz, D. J. Maltman, M. Matz-Soja, P. McMullen, I. Merfort, S. Messner, C. Meyer, J. Mwinyi, D. J. Naisbitt, A. K. Nussler, P. Olinga, F. Pampaloni, J. Pi, L. Pluta, S. A. Przyborski, A. Ramachandran, V. Rogiers, C. Rowe, C. Schelcher, K. Schmich, M. Schwarz, B. Singh, E. H. K. Stelzer, B. Stieger, R. Stoeber, Y. Sugiyama, C. Tetta, W. E. Thasler, T. Vanhaecke, M. Vinken, T. S. Weiss, A. Widera, C. G. Woods, J. J. Xu, K. M. Yarborough and J. G. Hengstler, Arch. Toxicol., 2013, 87, 1315–1530 CrossRef PubMed.
- S. P. den Braver-Sewradj, M. W. den Braver, N. P. E. Vermeulen, J. N. M. Commandeur, L. Richert and J. C. Vos, Toxicol. In Vitro, 2016, 33, 71–79 Search PubMed.
- M. Niemeijer, W. Wiecek, S. Fu, S. Huppelschoten, P. Bouwman, A. Baze, C. Parmentier, L. Richert, R. S. Paules, F. Y. Bois and B. van de Water, Environ. Health Perspect., 2024, 132(3), 37005 CrossRef PubMed.
- X. Chen, Y. S. Zhang, X. Zhang and C. Liu, Bioact. Mater., 2021, 6, 1012–1027 Search PubMed.
- B. Vinci, C. Duret, S. Klieber, S. Gerbal-Chaloin, A. Sa-Cunha, S. Laporte, B. Suc, P. Maurel, A. Ahluwalia and M. Daujat-Chavanieu, Biotechnol. J., 2011, 6, 554–564 CrossRef PubMed.
- C. C. Bell, A. C. A. Dankers, V. M. Lauschke, R. Sison-Young, R. Jenkins, C. Rowe, C. E. Goldring, K. Park, S. L. Regan, T. Walker, C. Schofield, A. Baze, A. J. Foster, D. P. Williams, A. W. M. van de Ven, F. Jacobs, J. van Houdt, T. Lahteenmaki, J. Snoeys, S. Juhila, L. Richert and M. Ingelman-Sundberg, Toxicol. Sci., 2018, 162, 655–666 CrossRef PubMed.
- M. Vinken, Curr. Opin. Toxicol., 2021, 25, 1–5 CrossRef.
- W. Li, Y. Wu, W. Hu, J. Zhou, X. Shu, X. Zhang, Z. Zhang, H. Wu, Y. Du, D. Lu, S. Lu, N. Li and M. Long, JHEP Rep., 2023, 5(12), 100905 CrossRef PubMed.
- W. Li, P. Li, N. Li, Y. Du, S. Lu, D. Elad and M. Long, Am. J. Physiol., 2020, 320, G272–G282 Search PubMed.
- H. Rashidi, S. Alhaque, D. Szkolnicka, O. Flint and D. C. Hay, Arch. Toxicol., 2016, 90, 1757–1761 CrossRef PubMed.
- A. Tilles, H. Baskaran, P. Roy, M. Yarmush and M. Toner, Biotechnol. Bioeng., 2001, 73, 379–389 Search PubMed.
- Y. Tanaka, M. Yamato, T. Okano, T. Kitamori and K. Sato, Meas. Sci. Technol., 2006, 17, 3167–3170 Search PubMed.
- J. Park, Y. Li, F. Berthiaume, M. Toner, M. L. Yarmush and A. W. Tilles, Biotechnol. Bioeng., 2008, 99, 455–467 Search PubMed.
- J. A. Pedersen, F. Boschetti and M. A. Swartz, J. Biomech., 2007, 40, 1484–1492 CrossRef PubMed.
- F. Pampaloni, E. G. Reynaud and E. H. K. Stelzer, Nat. Rev. Mol. Cell Biol., 2007, 8, 839–845 Search PubMed.
- C. C. Bell, D. F. G. Hendriks, S. M. L. Moro, E. Ellis, J. Walsh, A. Renblom, L. F. Puigvert, A. C. A. Dankers, F. Jacobs, J. Snoeys, R. L. Sison-Young, R. E. Jenkins, A. Nordling, S. Mkrtchian, B. K. Park, N. R. Kitteringham, C. E. P. Goldring, V. M. Lauschke and M. Ingelman-Sundberg, Sci. Rep., 2016, 6, 25187 CrossRef.
- R. M. Tostoes, S. B. Leite, M. Serra, J. Jensen, P. Bjorquist, M. J. T. Carrondo, C. Brito and P. M. Alves, Hepatology, 2012, 55, 1227–1236 CrossRef PubMed.
- V. M. Lauschke, R. Z. Shafagh, D. F. G. Hendriks and M. Ingelman-Sundberg, Biotechnol. J., 2019, 14(7), e1800347 CrossRef PubMed.
- L.-D. Ma, Y.-T. Wang, J.-R. Wang, J.-L. Wu, X.-S. Meng, P. Hu, X. Mu, Q.-L. Liang and G.-A. Luo, Lab Chip, 2018, 18, 2547–2562 RSC.
- J.-Y. Kim, D. A. Fluri, J. M. Kelm, A. Hierlemann and O. Frey, J. Lab. Autom., 2015, 20, 274–282 CrossRef PubMed.
- D. Jiao, L. Xie and W. Xing, Analyst, 2024, 149, 4675–4686 RSC.
- S. Y. Lee, D. Kim, S. H. Lee and J. H. Sung, APL Bioeng., 2021, 5, 041505 CrossRef.
- L. J. Y. Ong, L. H. Chong, L. Jin, P. K. Singh, P. S. Lee, H. Yu, A. Ananthanarayanan, H. L. Leo and Y.-C. Toh, Biotechnol. Bioeng., 2017, 114, 2360–2370 CrossRef.
- M. F. Wesseler, N. Taebnia, S. Harrison, S. Youhanna, L. C. Preiss, A. M. Kemas, A. Vegvari, J. Mokry, G. J. Sullivan, V. M. Lauschke and N. B. Larsen, Acta Biomater., 2023, 171, 336–349 CrossRef PubMed.
- K. Funatsu, H. Ijima, K. Nakazawa, Y. Yamashita, M. Shimada and K. Sugimachi, Artif. Organs, 2001, 25, 194–200 CrossRef.
- R.-Z. Lin and H.-Y. Chang, Biotechnol. J., 2008, 3, 1172–1184 CrossRef.
- G. H. Underhill and S. R. Khetani, Cell. Mol. Gastroenterol. Hepatol., 2018, 5, 426+ CrossRef PubMed.
- K. Jungermann and T. Kietzmann, Hepatology, 2000, 31, 255–260 CrossRef.
- L. McEnerney, K. Duncan, B.-R. Bang, S. Elmasry, M. Li, T. Miki, S. K. Ramakrishnan, Y. M. Shah and T. Saito, Exp. Mol. Med., 2017, 49(12), e413 CrossRef.
- K. Jungermann, Semin. Liver Dis., 1988, 8, 329–341 CrossRef.
- P. Dalsbecker, PhD thesis, University of Gothenburg, Gothenburg, Sweden, 2021 Search PubMed.
- D. Bavli, S. Prill, E. Ezra, G. Levy, M. Cohen, M. Vinken, J. Vanfleteren, M. Jaeger and Y. Nahmias, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E2231–E2240 CrossRef.
- C. Poon, J. Mech. Behav. Biomed. Mater., 2022, 126, 105024 CrossRef.
- H. C. Brinkman, Appl. Sci. Res., 1949, 1, 27–34 Search PubMed.
- M. Kiamehr, L. Viiri, T. Vihervaara, K. Koistinen, M. Hilvo, K. Ekroos, R. Käkelä and K. Aalto-Setälä, Dis. Models Mech., 2017, 10, 1141–1153 Search PubMed.
- M. Kajiwara, T. Aoi, K. Okita, R. Takahashi, H. Inoue, N. Takayama, H. Endo, K. Eto, J. Toguchida and S. Uemoto, et al. , Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 12538–12543 CrossRef.
- K. Si-Tayeb, F. K. Noto, M. Nagaoka, J. Li, M. A. Battle, C. Duris, P. E. North, S. Dalton and S. A. Duncan, Hepatology, 2010, 51, 297–305 CrossRef.
- D. C. Hay, D. Zhao, J. Fletcher, Z. A. Hewitt, D. McLean, A. Urruticoechea-Uriguen, J. R. Black, C. Elcombe, J. A. Ross and R. Wolf, et al. , Stem Cells, 2008, 26, 894–902 CrossRef.
- S. Ben-Moshe and S. Itzkovitz, Nat. Rev. Gastroenterol. Hepatol., 2019, 16, 395–410 CrossRef PubMed.
- A. Soto-Gutierrez, A. Gough, L. A. Vernetti, D. L. Taylor and S. P. Monga, Exp. Biol. Med., 2017, 242, 1605–1616 CrossRef PubMed.
- K. D'Amour, A. Agulnick, S. Eliazer, O. Kelly, E. Kroon and E. Baetge, Nat. Biotechnol., 2005, 23, 1534–1541 Search PubMed.
- R. Siller, E. Naumovska, S. Mathapati, M. Lycke, S. Greenhough and G. J. Sullivan, Sci. Rep., 2016, 6, 37178 CrossRef PubMed.
- M. Kiamehr, A. Alexanova, L. E. Viiri, L. Heiskanen, T. Vihervaara, D. Kauhanen, K. Ekroos, R. Laaksonen, R. Kakela and K. Aalto-Setala, J. Cell. Physiol., 2019, 234, 3744–3761 CrossRef PubMed.
- A. Moorman, J. Vermeulen, R. Charles and W. Lamers, Hepatology, 1989, 9, 367–372 CrossRef PubMed.
- A. Castegna and A. Menga, Genes, 2018, 99(2), 108 CrossRef PubMed.
- R. Gupta, Y. Schrooders, D. Hauser, M. van Herwijnen, W. Albrecht, B. ter Braak, T. Brecklinghaus, J. V. Castell, L. Elenschneider, S. Escher, P. Guye, J. G. Hengstler, A. Ghallab, T. Hansen, M. Leist, R. Maclennan, W. Moritz, L. Tolosa, T. Tricot, C. Verfaillie, P. Walker, B. van de Water, J. Kleinjans and F. Caiment, Arch. Toxicol., 2021, 95, 573–589 CrossRef PubMed.
- M. Baxter, S. Withey, S. Harrison, C.-P. Segeritz, F. Zhang, R. Atkinson-Dell, C. Rowe, D. T. Gerrard, R. Sison-Young, R. Jenkins, J. Henry, A. A. Berry, L. Mohamet, M. Best, S. W. Fenwick, H. Malik, N. R. Kitteringham, C. E. Goldring, K. P. Hanley, L. Vallier and N. A. Hanley, J. Hepatol., 2015, 62, 581–589 CrossRef.
- L. Prodanov, R. Jindal, S. S. Bale, M. Hegde, W. J. McCarty, I. Golberg, A. Bhushan, M. L. Yarmush and O. B. Usta, Biotechnol. Bioeng., 2016, 113, 241–246 CrossRef PubMed.
- X. Xie, S. Maharjan, C. Kelly, T. Liu, R. J. Lang, R. Alperin, S. Sebastian, D. Bonilla, S. Gandolfo, Y. Boukataya, S. M. Siadat, Y. S. Zhang and C. Livermore, Adv. Mater. Technol., 2022, 7(5), 2100677 CrossRef PubMed.
- F. Tonon, G. G. Giobbe, A. Zambon, C. Luni, O. Gagliano, A. Floreani, G. Grassi and N. Elvassore, Sci. Rep., 2019, 9, 13557 CrossRef PubMed.
- R. Mahdavi, S. Hashemi-Najafabadi, M. A. Ghiass, S. Valaskivi, H. Valimaki, J. Kreutzer, C. H. Blomqvist, S. Romeo, P. Kallio and C. B. Adiels, Biomed. Microdevices, 2025, 27, 8 Search PubMed.
- S. Suominen, T. Hyypijev, M. Venaelaeinen, A. Yrjaenaeinen, H. Vuorenpaeae, M. Lehti-Polojaervi, M. Raesaenen, A. Seppaenen, J. Hyttinen, S. Miettinen, K. Aalto-Setaelae and L. E. Viiri, Cell, 2023, 12, 2368 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4lc00509k |
| ‡ Co-first authors. |
| § Co-second authors. |
| ¶ These authors contributed equally. |
| || See ESI† for further information about cell culture media equilibration. |
| ** Details about the utilized antibodies are available in ESI.† |
| †† A corresponding confocal Z-stack video is available via the ESI† S4. |
| ‡‡ A mortality staining under static conditions is available in ESI.† |
| §§ A discoid Z-stack movie of fluorescent HepG2 cells is available via the ESI† S6. |
| This journal is © The Royal Society of Chemistry 2025 |