
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Simultaneous determination of Cl, Br and I by aerosol-assisted PVG-ICP-MS†
Gustavo R.
Bitencourt
 ab,
Paola A.
Mello
a,
Patricia
Grinberg
*b and
Ralph E.
Sturgeon
b
ab,
Paola A.
Mello
a,
Patricia
Grinberg
*b and
Ralph E.
Sturgeon
b
aDepartamento de Química, Universidade Federal de Santa Maria, Santa Maria, RS 97105-900, Brazil
bMetrology Research Centre, National Research Council Canada, Ottawa, ON K1A 0R6, Canada. E-mail: Patricia.Grinberg@nrc-cnrc.gc.ca
First published on 16th May 2025
Abstract
Simultaneous determination of Cl, Br and I by aerosol-assisted photochemical vapor generation (PVG) with detection by inductively coupled plasma mass spectrometry (ICP-MS) was investigated. The photoreactor comprised a modified cyclonic spray chamber fitted with a central UV source for irradiation of pneumatically generated sample aerosol. A systematic evaluation of variables was conducted, focusing on achieving compromise optimal PVG conditions suitable for simultaneous Cl, Br and I generation. By using a sample medium comprising 1% v/v acetic acid containing 20 mg L−1 Cu2+ as mediator, the signal intensities for Cl, Br and I were enhanced by 3-, 40- and 30-fold, respectively, compared to those obtained by conventional pneumatic nebulization (PN). LODs of 4.2 ng mL−1, 6.3 pg mL−1 and 1.9 pg mL−1 were achieved for Cl, Br and I, respectively, with corresponding estimated overall PVG efficiencies of 10, 99 and 90%. In addition to the halides, halate species, i.e., ClO3−, BrO3− and IO3−, were also examined, but poor PVG efficiencies (lower than 5%) were encountered. However, addition of 20 mg L−1 SO32− to the generation medium enhanced response for BrO3− and IO3−, achieving similar values to those obtained from Br− and I−. The impact of NO3− (as both HNO3 and KNO3) and NH4OH on generation efficiencies was also investigated. The method was tested by the successful simultaneous determination of total Cl, Br and I in a variety of certified reference materials (CRMs) digested by microwave-induced combustion (MIC), including NRCC DORM-5 – Fish Protein, NIST SRM 1632c – Coal, NIST SRM 1515 – Apple Leaves and NIST SRM 1549 – Non-fat Milk Powder.
1 Introduction
Chlorine, bromine and iodine are present in a wide variety of food, biological, clinical, industrial and environmental samples. Their concentration range and chemical species define the benefits or toxicity for humans, their impact on the environment, as well as use in industry.1 Chlorine is considered an essential element for humans (as Cl−) and has an important role in living cells and is often present at high concentrations in most samples. High concentrations of Cl− in water can cause corrosion or clogging of metallic pipes and even at trace levels may damage device fabrication in the electronics and semiconductor industries.2,3 Bromine is present at trace levels in the human body, but its role remains uncertain. While some benefits have been demonstrated, such as in the development of tissue and collagen structure,4 or as an antiepileptic agent,5 bromine can competitively reduce iodine accumulation in the thyroid and skin when present at high levels, in addition to increasing iodine excretion by the kidneys.6 Iodine is a micronutrient essential for human metabolism, crucial to synthesis of thyroid hormones. Its deficiency or excess exposure can induce a range of disorders, including hypo- or hyperthyroidism, as well as other clinical abnormalities.7It is clear that routine determination of Cl, Br and I in a variety of matrices is essential, requiring analytical techniques capable of addressing trace level concentrations. Several techniques have been used, including ion chromatography (IC) with conductivity detection,8–10 gas chromatography mass spectrometry (GC-MS),11–13 X-ray fluorescence spectrometry (XRF),14,15 high-resolution continuum source atomic absorption spectrometry (HR-CS-AAS),16–18 inductively coupled plasma optical emission spectrometry (ICP-OES)19,20 and inductively coupled plasma mass spectrometry (ICP-MS),21,22 among others.23 Of these techniques, ICP-MS is particularly noteworthy due to its superior limits of detection (LODs).1 However, several shortcomings impair halogen detection by ICP-MS, including their high ionization potentials (12.9, 11.8 and 10.5 eV for Cl, Br and I, respectively) with consequent low degree of ionization, severe memory effects, and potential spectral interferences from both sample matrix concomitants and typical plasma ions (e.g., 18O16O1H+, 38Ar40Ar1H+ and 40Ar40Ar1H+ generating isobaric interferences on 35Cl, 79Br and 81Br, respectively). Furthermore, the poor sample nebulization efficiency (typically 2–3%) provided by conventional pneumatic nebulization (PN) further limits potential LODs.1,24
To overcome some such shortcomings, use of photochemical vapor generation (PVG) for analyte introduction has been the focus of significant research over the past two decades. Production of volatile analyte species induced by UV photolysis of aqueous solutions typically containing added low-molecular-weight organic acids (e.g., acetic acid), has been successfully utilized for the determination of numerous metals and non-metals, including the halogens.25–27
Iodine is recognized as the halogen most readily amenable to PVG.28,29 In the presence of 5% v/v acetic acid and under UV-C irradiation, an estimated generation efficiency of 94% is achieved,29 yielding a 40-fold enhancement of the LOD compared to that obtained by PN. PVG methodology was validated through analysis of several different sample matrices.28 In another study, the ethanol content in alcoholic beverages was used to produce radicals through UV irradiation, thereby enabling direct photochemical generation of iodine, enhancing sensitivity 64% over that arising from PN.30 Photochemical generation of Br was successfully achieved from a 2% v/v acetic acid solution containing 3 mg L−1 NH4Cl. The NH4+ ions were hypothesized to serve as electron scavengers, enhancing PVG efficiency to yield a 17-fold improvement in the LOD compared to that with PN, and a generation efficiency of 95%. Determination of Br in IRMM BCR-611 (Low Level Bromide in Groundwater), NIST SRM 1568b (Rice Flour), and SRM 1632 (Bituminous Coal) reference materials was successfully demonstrated.31
PVG of Cl− is not possible based on the conditions utilized for I− and Br−. Under UV-C irradiation, photochemical transformation of X− (X = Br or I) occurs via a charge transfer-to-solvent process (CTTS), resulting in the formation of a cage complex comprising X˙ and solvated electrons (e−(aq)). Subsequently, X˙ interacts with H3C˙ arising from the photolysis of acetic acid present in solution, generating volatile CH3X.26 Emission from the UV source, limited to 185 nm, cannot excite a CTTS process for chloride due to its short UV absorption line (177.6 nm). However, a metal-ion assisted/mediated PVG reaction can be successfully conducted.26 Prior studies have demonstrated their impact in enhancing PVG efficiencies of the halogens, including the use of added Cu2+ for F, Cl and Br,2,32,33 Co2+ for Br32 and Fe3+ for Cl and Br.34,35 The added metal ions alter the mechanism of the reaction by formation of a Cu–X complex (X = Cl or Br), which can be excited by UV-B (longer wavelength) to undergo an efficient ligand-to-metal charge transfer process, yielding X˙ to permit subsequent formation of CH3X.32,36 Using 1% v/v acetic acid and 7.5 μg g−1 Cu2+, a 74-fold enhancement in sensitivity for Cl− was obtained compared to PN.2 For Br−, generation efficiencies of 92% and 94% were achieved using a flow-through lamp (emitting at 185 and 254 nm) and a simple germicidal lamp (emitting at 254 nm), respectively, from a 2% v/v acetic acid medium in the presence of 10 mg L−1 Cu2+.32 Generation of volatile Cl− and Br− (in addition to F−) species was also reported from a simple copper acetate solution, resulting in their effective PVG from a so-called “organic acid-free” medium.37
Typically, PVG hardware comprises a photoreactor and a tandem gas–liquid phase separator (GLS) to ensure efficient release of generated volatile species from the irradiated liquid medium, which are then directed to the detection system by a carrier gas.26 This setup has been used in most of the aforementioned studies of halogen generation, wherein the photoreactor is a high efficiency flow-through low pressure mercury discharge lamp emitting both 254 and 185 nm Hg lines. Nevertheless, other photoreactors have been shown to be well-suited for PVG systems, including simpler germicidal lamps. Of particular interest for this study is a relatively unexplored combined UV-assisted spray chamber38 introduced nearly two decades ago, consisting of a standard cyclonic spray chamber modified to accommodate a central UV-C pen lamp. In addition to the absence of any perturbation to operation of the spray chamber using conventional PN sample introduction, this photoreactor offers the advantage of a direct ICP-MS compatible assembly that simplifies PVG operation.
The sparse number of studies using the UV-assisted spray chamber28,29,38,39 is likely linked to a number of significant shortcomings inherent to the system, such as the elimination of the gas–liquid phase separation feature which serves to minimize introduction of sample matrix (a notable advantage of vapor generation techniques), the relatively brief analyte residence time within the spray chamber, yielding UV irradiation times on the order of only 3 s, and the need to generate a fine aerosol, which limits solution uptake rates and eliminates the advantages of high vapor flux processing (i.e., solution flow rates limited to <1 mL min−1). While potential detrimental matrix effects impacting the photochemical reactions and analyte detection by ICP-MS can be mitigated by various means, such as simple sample dilution, use of reaction/collision cells, reliance on internal standards and application of isotope dilution, or the employment of an appropriate sample preparation method,1 the limited irradiation time represents an inherent constraint that must be overcome through reliance on optimized PVG conditions. Noteworthy in this context is that the interaction between analytes, reducing species and UV radiation within aerosolized microdroplets generated by PN could potentially accelerate the kinetics of photochemical (and other) reactions,40–42 thereby offsetting the detrimental impact of short irradiation times.
The purpose of the present study was to develop a suitable method for the simultaneous determination of chlorine, bromine and iodine by aerosol-assisted PVG-ICP-MS using the convenience of a UV-assisted spray chamber. A systematic evaluation of PVG conditions was undertaken, focused on identifying suitable compromise conditions. The resultant fit-for-purpose methodology was demonstrated through the analyses of a variety of certified matrix reference materials (CRMs) digested by microwave-induced combustion (MIC).
2 Experimental
2.1 Instrumentation
A schematic diagram of the PVG system used in this work is illustrated in Fig. 1. The photoreactor consisted of a 50 mL internal volume water-jacketed cyclonic spray chamber (Glass Expansion, Australia) in which the standard waste removal line was adapted to accommodate a 6 W Analamp mercury UV-C source emitting at 254 and 185 nm (model 81-1057-51, BHK Inc., Canada).38 Sample solution was delivered at 1 mL min−1 through a glass concentric nebulizer (Meinhard, USA) via a peristaltic pump. As the vertical dimension of the photoreactor was larger than the stand-alone spray chamber, its direct coupling to the base of the injector torch necessitated use of a short ball-and-socket glass transfer line (length of 20 cm and i.d. of 5 mm). This line was maintained at 70 °C through the use of a heating tape in order to prevent moisture condensation. All measurements were undertaken using an Elan® DRC II inductively coupled plasma mass spectrometer (PerkinElmer-Sciex, Canada) fitted with conventional Ni sampler/skimmer cones. The ICP-MS operating conditions are summarized in Table 1. | ||
| Fig. 1 Schematic of the PVG-ICP-MS system. | ||
| Parameter | ICP-MS |
|---|---|
| RF power, W | 1300 |
| Nebulizer gas flow rate, L min−1 | 1.15 |
| Auxiliary gas flow rate, L min−1 | 1.1 |
| Plasma gas flow rate, L min−1 | 17.5 |
| Sample flow rate, mL min−1 | 1.0 |
| Measurement mode | Peak hopping |
| Dwell time, ms | 50 |
| Replicates | 5 |
| Isotopes monitored, m/z | 35Cl, 79Br, 81Br, 127I |
A Multiwave 5000 microwave sample preparation system (Anton Paar, Austria) was used for sample digestion by MIC. The microwave system was operated at its maximum power, temperature and pressure of 1500 W, 280 °C and 80 bar, respectively. The system is equipped with eight high-pressure quartz vessels each of 80 mL internal volume. Commercial quartz sample holders (Anton Paar) were used to accommodate sample pellets for combustion.
All statistical evaluations and data manipulation were performed using GraphPad InStat software (GraphPad InStat Inc, Version 3.06, 2007); confidence levels of 95% were accepted.
2.2 Reagents, standards and certified reference materials
Ultrapure water obtained from a Milli-Q Advantage system (18.2 MΩ cm, Millipore Sigma, USA) was used for preparation of all working solutions. Both ACS-grade (≥99.7%, Fisher Scientific, Canada) and TAMA Pure AA-100 (TAMA Chemical Inc., Japan) acetic acid were used. Ammonium hydroxide, ACS grade (28 to 30%), was obtained from Fisher Scientific (Canada). For MIC procedures, a 3.0 mol L−1 solution of NH4NO3 was prepared from its respective salt (Merck, Germany) and used as the combustion igniter. Oxygen (99.5%, White Martins, Brazil) was used to pressurize the digestion vessels to 20 bar. Small discs of filter paper (15 mm diameter, about 12 mg) with low ash content (Black Ribbon Ashless, Schleicher and Schuell, Germany) were used to aid the combustion process. The filter paper was previously cleaned with ethanol (Merck, Brazil) for 20 min in an ultrasonic bath, rinsed with water and dried in a class 100 laminar bench (CSLH-12, Veco, Brazil) before use. Nitric acid (Fisher Scientific, Canada), in-house double-distilled by a sub-boiling system, and potassium nitrate (Anachemia Chemicals, Canada) were used to evaluate potential interferences from NO3−. Sodium sulfite (Caledon Laboratories Ltd., Canada) was evaluated for reduction of ClO3−, BrO3− and IO3− species.Stock solutions of 1000 mg L−1 of Cl−, Br− and I− were prepared by dissolving their sodium salts (Anachemia Chemicals, all analytical-grade) in ultrapure water. Additionally, stock solutions (1000 mg L−1) of ClO3−, BrO3− and IO3− were prepared by dissolving potassium chlorate, sodium bromate and sodium iodate (Anachemia Chemicals, all analytical-grade), respectively, in ultrapure water. These reagents were dried at 105 °C in an air convection oven for 2 h and cooled in a desiccator prior to gravimetric preparation of individual nominal 1000 mg L−1 stock solutions. Working solutions were prepared daily by diluting the stock solutions with appropriate concentrations of acetic acid.
A stock solution of nominal 5000 mg L−1 of Cu2+ was prepared by dissolving a high-purity copper wire certified reference material (NRC CRM HICU-1, 99.999%, National Research Council, Canada) in 30% v/v double-distilled nitric acid. In order to minimize the final NO3− concentration, the copper solution was carefully evaporated in a class 100 clean hood and the salt reconstituted with 1% v/v acetic acid.
Four certified reference materials (CRMs) were selected to evaluate the performance of the proposed procedure: NRCC CRM DORM-5 Fish Protein (National Research Council Canada, Canada), NIST SRM 1632c Coal, NIST SRM 1515 Apple leaves and NIST SRM 1549 Non-fat milk powder (National Institute of Standards and Technology, USA). In accordance with their certificates, dry weight corrections for moisture content were determined by drying separate subsamples of each material at 60 °C in a conventional oven (Nova Ética, Brazil) until constant weight was achieved. For MIC digestion, samples were pressed for 3 min into pellets (diameter of 13 mm) using a hydraulic press (Specac, UK) set at 5 ton.
2.3 Analytical procedures
Unless otherwise specified, optimization of the PVG parameters was conducted using multielement test solutions containing 5 mg L−1 Cl−, 10 μg L−1 Br− and 2 μg L−1 I− to ensure adequate precision of measurement. These same concentrations were used when experiments with ClO3−, BrO3− and IO3− species were undertaken. As the purpose of this work was to develop a method for simultaneous determination of Cl, Br and I using PVG sample introduction, all parameters were evaluated using both individual and mixed solutions of the analytes to ascertain whether cross-element interactions influenced their generation. For the PVG parameters, the effect of the acetic acid concentration in the range 0.25 to 10% v/v and of the amount of added Cu2+ mediator (in range from 5 to 100 mg L−1) were evaluated. Potential interferences induced by NO3− present in the range 1 to 100 mmol L−1 (as HNO3 or KNO3) and by NH4OH (0.1 to 200 mmol L−1) on PVG efficiencies were also investigated. Use of SO32− for the reduction of ClO3−, BrO3− and IO3− was examined in the range 5 to 100 mg L−1 in 1% v/v acetic acid containing 20 mg L−1 Cu2+.
2.4 Safety considerations
The full range and identity of volatile compounds produced during PVG is unknown. Standard safety precautions should be taken during all experiments and an adequate ventilation/exhaust system should be used.3 Results and discussion
3.1 Optimization of PVG conditions using acetic acid and Cu2+ as ion mediator
Preliminary investigations were devoted to evaluation of the signals arising from PN of an aqueous multi-standard solution generated in the physical presence and absence of the UV pen lamp. No statistical differences between signal intensities were detected (t test, 95% confidence level). Furthermore, all experiments performed during method optimization were also undertaken using both single and mixed solutions of the halogens with any differences being less than 10% (data not shown). Thus, results obtained based on a multi-standard solution were used for all subsequent studies.An acetic acid medium is commonly used for PVG of the halogens as the production of water-soluble acids (HX) and poorly volatile species (C2H5X) impairs the use of formic and propionic acids, respectively.2,26,31 Acetic acid was thus solely evaluated as the generation medium in this study.
Fig. 2 illustrates the time evolution of signal profiles obtained from solutions containing 5 mg L−1 Cl−, 10 μg L−1 Br− and 2 μg L−1 I− in 1% v/v acetic acid (dotted lines without Cu2+ and solid lines with 20 mg L−1 Cu2+). Conventional PN intensities are recorded for 50–60 s before the UV lamp is powered on and serve to illustrate the reference signals arising in the absence of photochemical processes. As evident in Fig. 2A, there is no difference in the steady-state Cl− signal intensity obtained in only acetic acid whether the aerosol is subjected to UV irradiation or not (compare dotted line intensities before and after the lamp is powered on). As noted earlier, the short UV absorption line of Cl− (177.6 nm band maximum) precludes its overlap with any emission lines from the UV source and hence no photochemical reaction develops. For Br− and I−, enhancement factors of 5- and 30-fold are evident in Fig. 2B and C, respectively, when the UV lamp is powered on. Although the UV pen lamp emits at 185 nm, the poor overlap with the absorption band maximum for Br− (i.e., 197.8 nm), and the relatively short irradiation time (approximately 3 s) in the spray chamber, limits the extent of photochemical reactions leading to CH3Br generation. Iodine is the most readily responsive halogen for PVG as its absorption band maximum is at 226 nm. Its 30-fold signal enhancement factor is in agreement with that reported in previous studies.28,29
 | ||
| Fig. 2 PVG-ICP-MS response from a multi-element solution of (A) 5 mg L−1 Cl−, (B) 10 μg L−1 Br− and (C) 2 μg L−1 I− in 1% v/v acetic acid (dotted line) and 1% acetic acid containing 20 mg L−1 Cu2+ (solid line). A 5% v/v NH4OH solution was used to rinse the system to minimize carryover. | ||
The presence of Cu2+ has earlier been shown to enhance the PVG efficiency for Cl− and Br−.2,32 It is clear from Fig. 2A and B that when the UV lamp is powered on, PVG responses for Cl− and Br− are enhanced 3- and 40-fold, respectively. No additional PVG enhancement is evident for I− in the presence of Cu2+ (Fig. 2C) because CH3I is already efficiently generated from the 1% v/v acetic acid medium. The impact of added Cu2+ on PVG response from Cl− is significantly lower than that earlier reported by Hu et al.2 (74-fold; 43-fold normalized to 1.0 mL min−1), highlighting the consequence of the brief aerosol UV irradiation time within the spray chamber (≈3 s) compared to that in a flow-through lamp (45 seconds at a sample flow rate of 1.7 mL min−1).
Noteworthy is that in all cases, PVG signal intensities increase the moment the solution is irradiated, but it is evident that this occurs at different rates for each halogen. A pronounced spike occurs for I− and was attributed to the intense irradiation of species adsorbed on the cool UV pen lamp surface prior to powering it.29 This spiking was not evident if the UV lamp was permitted to stabilize for 20 min before analytical measurements were recorded, supporting this supposition. For reasons unknown, the spike was only observed when Cu2+ was present in the solution. Signals for Br− and Cl− increased more slowly, possibly a result of the warming of the UV lamp leading to temporally increasing intensities of the shorter 185 nm UV radiation required to effect PVG of these elements.45
Optimization studies were conducted to evaluate the effect of varying concentrations of acetic acid and Cu2+ on PVG response; results are shown in Fig. 3 and 4 for processing multielement solutions. Optimal response for Br− was obtained using 0.25% v/v acetic acid, whereas 1% v/v was evident for Cl− and I−. Higher concentrations led to a notable decrease in all signals, possibly due to spectral shadowing in the presence of this absorber, leading to a decreasing depth of penetration of short UV photons into the liquid medium at higher concentrations of acetic acid.25 A concentration of 1% v/v acetic acid was selected as a compromise for further evaluation.
 | ||
Fig. 3 Effect of acetic acid concentration (in the presence of 20 mg L−1 Cu2+) on the normalized response from a multi-element solution containing Cl− (5 mg L−1,  ), Br− (10 μg L−1, ), Br− (10 μg L−1,  ) and I− (2 μg L−1, ) and I− (2 μg L−1,  ). Error bars presenting the standard deviation (n = 3) are typically buried within the signal marker. ). Error bars presenting the standard deviation (n = 3) are typically buried within the signal marker. | ||
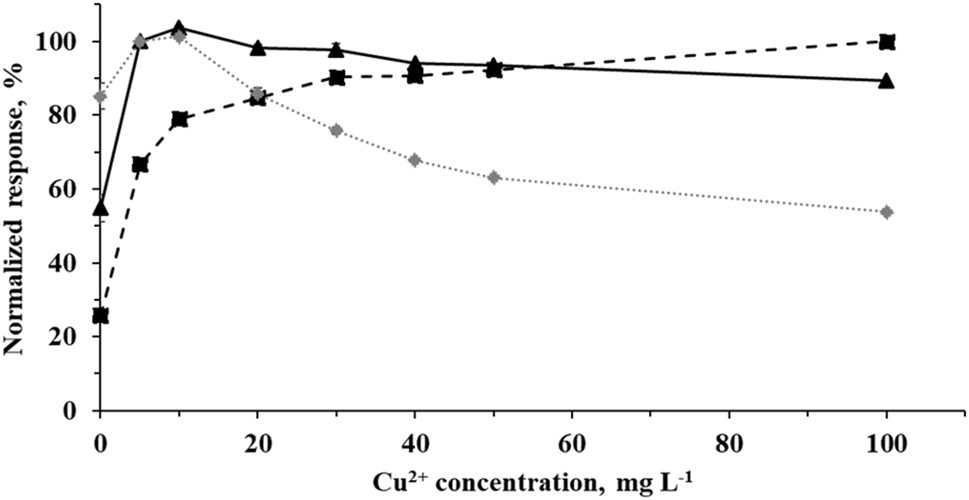 | ||
Fig. 4 Effect of Cu2+ concentration (in the presence of 1% v/v acetic acid) on the normalized response from a multi-element solution containing Cl− (5 mg L−1,  ), Br− (10 μg L−1, ), Br− (10 μg L−1,  ) and I− (2 μg L−1, ) and I− (2 μg L−1,  ). Error bars presenting the standard deviation (n = 3) are typically buried within the signal marker. ). Error bars presenting the standard deviation (n = 3) are typically buried within the signal marker. | ||
Fig. 4 shows that addition of increasing concentrations of Cu2+ enhances the response for Cl−, demonstrating its crucial role as a mediator for CH3Cl generation.2 Highest signal intensity for Br− was obtained in the presence of 10 mg L−1 Cu2+, and no significant decline in response occurred even at concentrations exceeding 20 mg L−1 (maximum signal suppression at 100 mg L−1 Cu2+ was about 10%). The intensity of the signal for I− obtained with 5 and 10 mg L−1 Cu2+ was only 15% higher than that generated in its absence, indicating that Cu2+ may also serve to slightly enhance PVG efficiency of I− when using an aerosol-assisted process, kinetically compensating for the reduced UV exposure time. At 20 mg L−1 Cu2+, response from I− was identical to that observed using only 1% v/v acetic acid. On the other hand, a remarkable decrease was noted with higher Cu2+ concentrations, suggesting that it may interfere with the generation of CH3I due to a possible further spectral shadowing effect (i.e., decrease in photon penetration depth). As such, 20 mg L−1 Cu2+ was selected as the optimal compromise concentration for further evaluations, based on a sacrifice in response from I− due to its intrinsically higher generation efficiency.
No memory effects were evident following rinsing of the system with 5% v/v NH4OH for 2 min between samples. However, it is noteworthy that use of the rinse solution resulted in a slower recovery of the Cl signal intensity compared to its more rapid stabilization achieved when the system had not been previously exposed to NaOH, as shown in Fig. 5. This may be attributed to the time required for the interior surface of the spray chamber to become equilibrated with the sample (cf. Section 3.3, consistent with interferences from NH4OH on PVG of Cl−). Data acquisition was thus started following a minimum 2 min of sample introduction or until a signal precision better than 2% for all analytes was attained.
 | ||
| Fig. 5 Effect of the 5% v/v NH4OH rinse solution on the signals from 2 μg L−1 I− (solid black line), 5 mg L−1 Cl− (dashed black line) and 10 μg L−1 Br− (dotted gray line) in 1% v/v acetic acid + 20 mg L−1 Cu2+. Note that 5% v/v NH4OH was introduced as a rinse between the successive replicate sample introductions. | ||
3.2 Interferences during simultaneous PVG of halogens
Development of a method for simultaneous PVG of multiple elements is a challenge, especially considering halogens and their species specific generation efficiencies.2 Although HNO3 is less commonly used for solubilization and stabilization of halogens, its effects can be problematic and must be investigated. The photolysis of NO3− generates multiple oxidizing species that consume reductive radicals (e(aq)−, H˙ and R˙) that are often required for the PVG process.46 In this context, the influence of both NO3− (a possible concomitant present in sample digests) and NH4OH (a common solution selected for halogen extraction in several matrices) were evaluated.Firstly, the influence of NO3− on the photochemical generation of Cl−, Br− and I− was undertaken using HNO3 present at concentrations ranging from 1 to 500 mmol L−1 in a medium of 1% acetic acid and 20 mg L−1 Cu2+. As illustrated in Fig. 6A, Cl− demonstrated good tolerance up to 500 mmol L−1 HNO3, with only 7% signal suppression. PVG of Br− was affected at HNO3 concentrations exceeding 100 mmol L−1, with a notable 53% suppression at 500 mmol L−1. On the other hand, the addition of only 5 mmol L−1 HNO3 led to a 14% reduction of the I− signal. These findings differ from those of previous studies2,32 in which Cl− and Br− generation efficiencies were reduced even in the presence of 15 mmol L−1 HNO3 (Cu2+ was used as mediator in both cases) using a flow-through lamp as the photochemical reactor. The brief residence time within the UV-assisted spray chamber must be considered a possible factor in mitigating the effects of HNO3 on their generation efficiencies. When selecting the sources of NO3− it is important to consider that the preparation of the Cu2+ solution from high purity copper involved its dissolution in 5% v/v HNO3, yielding a 3 mmol L−1 HNO3 concentration in the working solutions (20 mg L−1 Cu2+). At this concentration, a negligible effect of HNO3 on the generation of Cl− and Br− is expected. However, the higher susceptibility of I− to the presence of HNO3 required evaporation of the Cu2+ solution to near dryness and its subsequent reconstitution in 1% v/v acetic acid to readily avoid this source of interference.
 | ||
Fig. 6 Effects of NO3− added as HNO3 (A) or KNO3 (B), and NH4OH (C) on PVG of 5 mg L−1 Cl− ( ), 10 μg L−1 Br− ( ), 10 μg L−1 Br− ( ) and 2 μg L−1 I− ( ) and 2 μg L−1 I− ( ). Secondary ordinate represents solution pH ( ). Secondary ordinate represents solution pH ( ). Error bars presenting the standard deviation (n = 3) are typically buried within the signal marker. ). Error bars presenting the standard deviation (n = 3) are typically buried within the signal marker. | ||
The presence of higher concentrations of HNO3 also decreases the pH of the test solutions ( in Fig. 6A). To assess the isolated impact of NO3− ions, further information was obtained by examining interference from NO3− derived from KNO3 (Fig. 6B). The effect of NO3− was significantly more pronounced in the form of added KNO3 compared to that for HNO3. Even at a concentration of 1 mmol L−1 KNO3, severe interference was evident (i.e., 11% suppression for I−). Comparing the effects of the two sources of NO3− (HNO3 and KNO3) suggests that the pH of the solution plays a significant role in PVG of the halides. As expected, addition of KNO3 to the reaction media had no impact on solution pH. Minimizing excess H+ through use of KNO3 instead of HNO3 may reduce formation of H˙ which may lead to “unproductive” generation of XH species, reducing analyte response.
in Fig. 6A). To assess the isolated impact of NO3− ions, further information was obtained by examining interference from NO3− derived from KNO3 (Fig. 6B). The effect of NO3− was significantly more pronounced in the form of added KNO3 compared to that for HNO3. Even at a concentration of 1 mmol L−1 KNO3, severe interference was evident (i.e., 11% suppression for I−). Comparing the effects of the two sources of NO3− (HNO3 and KNO3) suggests that the pH of the solution plays a significant role in PVG of the halides. As expected, addition of KNO3 to the reaction media had no impact on solution pH. Minimizing excess H+ through use of KNO3 instead of HNO3 may reduce formation of H˙ which may lead to “unproductive” generation of XH species, reducing analyte response.
The performance of the developed PVG methodology was examined by the analysis of various matrix CRMs subjected to prior MIC digestion (Section 3.6), utilized in an effort to minimize residual nitrate. As NH4OH is used as the absorbing solution, its impact on subsequent halide generation was investigated (Fig. 6C). Solutions of 1% v/v acetic acid and 20 mg L−1 Cu2+ containing NH4OH ranging from 0.1 to 200 mmol L−1 were examined. As with HNO3, the pH of the solutions is strongly influenced by the NH4OH concentration. At 200 mmol L−1, the pH was higher than 8, and the solution exhibited a light blue color. At alkaline pH, Cu2+ forms a Cu(OH)2 precipitate, even at low concentrations of NH4OH. Excess NH4OH forms a dark blue [Cu(NH3)4]2+ complex.47 A substantial decrease in Cl− PVG signal (the halide most dependent on the added Cu2+) occurs in the presence of 5 mmol L−1 NH4OH (pH 3). Also dependent on Cu2+, PVG response for Br− is suppressed in solutions containing more than 25 mmol L−1 NH4OH. In the case of PVG of I−, which efficiently occurs even in the absence of Cu2+, suppression was evident only in the range of 200 mmol L−1 NH4OH and likely due to uncharacterized effects on the photochemical reactions that occur under alkaline conditions.
3.3 PVG of ClO3−, BrO3− and IO3− species
Photochemical reactions involving chlorate (ClO3−), bromate (BrO3−) and iodate (IO3−) anions present some kinetic limitations, resulting in different generation rates when compared with their corresponding halides.36 The impact of these species on their PVG using the UV-assisted spray chamber was investigated using earlier optimized conditions (i.e., 1% v/v acetic acid + 20 mg L−1 Cu2+). Poor generation efficiencies for all the investigated halate species arise, with only 3- and 2-fold enhancements obtained for BrO3− and IO3− compared to their conventional solution PN, whereas chlorate provides no PVG response. Although prior studies have reported higher generation efficiencies for ClO3−,2 BrO3− (ref. 31 and 32) and IO3−,28,29 the majority have employed a flow-through lamp providing a significantly longer irradiation time than that possible in the UV-assisted spray chamber, highlighting the slow kinetics of their stepwise photochemical reactions. Additionally, the previous studies comprising PVG of I from IO3− did not employ any metal mediator, which functions as an interference in this case. This is evidenced in Fig. S3 (ESI†), wherein no difference was observed for the signals obtained for I− and IO3− in the 1% v/v acetic acid medium whereas a significant decrease occurs for IO3− in the presence of 20 mg L−1 Cu2+.The potential use of UV/SO32−-based advanced reduction processes (ARPs) has been widely explored for the degradation of a range of environmental contaminants, including BrO3−.48,49 Under UV irradiation, SO32− and HSO3− give rise to the formation of e(aq)−, H˙ and SO3−˙ radicals. In addition to the generation of successively reduced Br species from BrO3− (bromite, hypobromite and bromide) mediated by e(aq)− and H˙, H2SO3 and HSO3− may also be involved in BrO3− reduction, as illustrated by eqn (1)–(5).48
| BrO3− + H2SO3 → SO3−˙ + BrO2 + H2O | (1) |
| 2BrO2 + HSO3− + H2O → 2HBrO2 + SO42− + H+ | (2) |
| 2BrO2 + H2O → HBrO2 + BrO3− + H+ | (3) |
| HBrO2 + HSO3− → HOBr + SO42− + H+ | (4) |
| HOBr + HSO3− → Br− + SO42− + 2H+ | (5) |
Noteworthy is that Br− and SO42− are the final products that remain following the treatment of BrO3− by UV/SO32−. Although not yet studied, it is reasonable to hypothesize that such reactions may also apply to ClO3− and IO3− species. Thus, the potential of SO32− as an adjuvant for the generation of methyl halides from their halate forms (either with the addition of Cu2+ or not) was investigated. The impact of UV/SO32− treatment on PVG of Cl−, Br− and I− species was concomitantly evaluated to ensure that no new interferences were encountered.
As can be seen in Fig. S1–S3 (ESI†), the presence of 100 mg L−1 SO32− in a 1% v/v acetic acid medium did not affect signals from Cl− and Br−, but a detrimental effect on PVG of I− and IO3− was clear. Significant suppression of PVG response for all the halides was encountered when SO32− was present in solutions also containing 20 mg L−1 Cu2+. On the other hand, an enhancement in generation efficiency from BrO3− and IO3− was achieved in the presence of added SO32−, resulting in signal intensities comparable to those observed for Br− and I−. Unfortunately, response from ClO3− remains essentially unaltered despite the presence of added SO32−.
The influence of the concentration of SO32− added to the acetic acid/Cu2+ generation medium was evaluated over a range from 1 to 100 mg L−1 with the objective of seeking an optimal condition exhibiting minimal suppression effects on the halides in conjunction with the greatest enhancement in PVG response for the halates. Results are presented in Fig. 7.
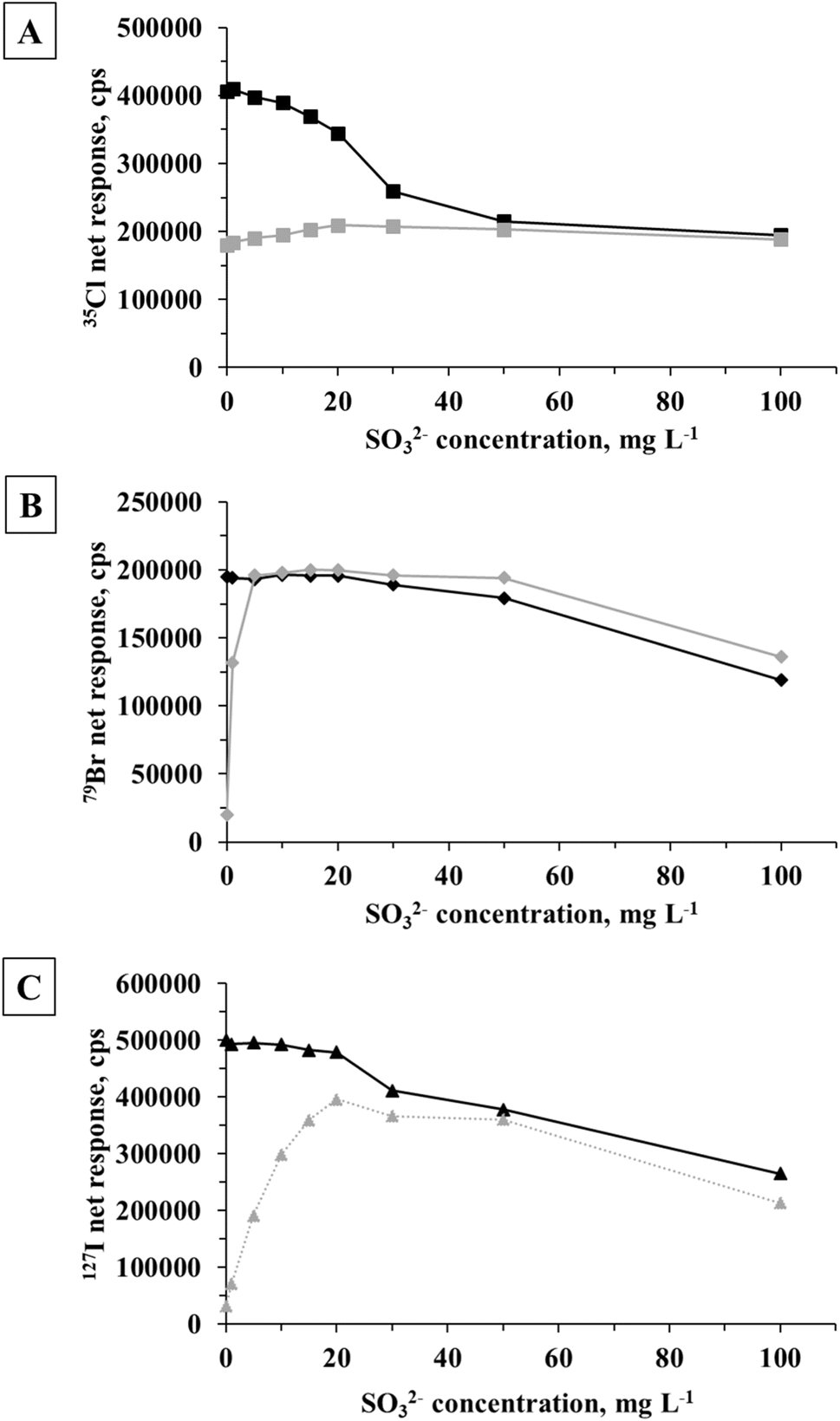 | ||
Fig. 7 Influence of the SO32− concentration on PVG response from: (A) 5 mg L−1 Cl− ( ) or ClO3− ( ) or ClO3− ( ); (B) 10 μg L−1 Br− ( ); (B) 10 μg L−1 Br− ( ) or BrO3− ( ) or BrO3− ( ); and (C) 2 μg L−1 I− ( ); and (C) 2 μg L−1 I− ( ) or IO3− ( ) or IO3− ( ) in a medium of 1% v/v acetic acid/20 mg L−1 Cu2+. Error bars presenting the standard deviation (n = 3) are typically buried within the signal marker. ) in a medium of 1% v/v acetic acid/20 mg L−1 Cu2+. Error bars presenting the standard deviation (n = 3) are typically buried within the signal marker. | ||
In the case of ClO3−, a maximum 16% signal increase was achieved in the presence of 20 mg L−1 SO32− (Fig. 7A). Although degradation of ClO4−via a ClO3− intermediate to ultimately yield Cl− is feasible with bulk radiolysis,48 use of SO32− is apparently not efficient for the rapid reduction of ClO3− when using the UV-assisted spray chamber. It was also evident that the baseline for 35Cl in the presence of 100 mg L−1 SO32− increased about 3-fold, likely due to the generation of a 34S1H+ polyatomic ion.
Generation efficiency is significantly enhanced for BrO3− in the range 5 to 50 mg L−1 of added SO32−, providing a response similar to that obtained with Br− (Fig. 7B). Furthermore, the addition of SO32− did not interfere with generation of Br− up to a concentration of 50 mg L−1. In the presence of 100 mg L−1 SO32−, 40% and 30% signal suppressions occurred for Br− and BrO3−, respectively.
Maximum response from IO3− was obtained using 20 mg L−1 of SO32− (differing by 20% compared to the signal obtained from I−, Fig. 7C). At concentrations exceeding 40 mg L−1 SO32−, a decline in signal intensity was observed for both I species.
Although enhanced PVG generation efficiencies achieved for BrO3− and IO3− in an acetic acid/Cu2+/SO32− medium render the PVG-ICP-MS methodology a feasible approach for the determination of total Br and I in samples wherein both their halide and halate species may be present, this approach remains entirely unsatisfactory for total Cl. Noteworthy from Fig. 7 is that at a SO32− concentration of 50 mg L−1 it would appear that each of the tested halates is converted to the corresponding halide to provide the same response for both species, albeit at an overall suppressed response in each case.
3.4 Figures of merit
Figures of merit for the halides, summarized in Table 2, were evaluated under optimized compromise conditions employing a solution of 1% v/v acetic acid containing 20 mg L−1 Cu2+ as the PVG medium. Comparative performance metrics with conventional sample PN are also presented. Calibration curves for both PVG and PN sample introduction were constructed in the ranges 0.1 to 7.5 mg L−1 for Cl−, 0.1 to 20 μg L−1 for Br− and 0.01 to 5 μg L−1 for I−, resulting in coefficients of determination (R2) higher than 0.999 for all elements using either sample introduction system. The PVG method demonstrated an approximate 3-, 40- and 30-fold increase in sensitivity for Cl−, Br− and I−, respectively, compared to PN. This resulted in similar enhancements in their limits of detection (LODs). Although the net sensitivity for Br− at m/z 81 was indistinguishable from that observed at m/z 79, the elevated background from the rising edge of 81Ar2H+ and from the tail of 80Ar2+ resulted in 4-fold degradation in the LOD for 81Br and elevated signal intensities evident for both PN and PVG blanks.| Parameter | 35Cl | 79Br | 81Br | 127I | ||||
|---|---|---|---|---|---|---|---|---|
| PVG | PN | PVG | PN | PVG | PN | PVG | PN | |
| a Slope of the calibration function, expressed as cps ppm−1 for Cl and cps ppb−1 for Br and I. b LOD = 3s/m, where m is the slope of the calibration function and s is the standard deviation of ten replicate measurements of the 1% v/v ACS-grade acetic acid + 20 mg L−1 Cu2+ blank. c Relative standard deviation of measurement of 10 consecutive samples containing 5 mg L−1 for Cl−, 10 μg L−1 for Br− and 1 μg L−1 for I− in 1% v/v ACS-grade acetic acid + 20 mg L−1 Cu2+. d PVG efficiency, estimated from an analysis of the residual Cl, Br and I in the waste solution carefully collected from the UV-assisted spray chamber following sample irradiation. | ||||||||
| Sensitivitya (×10−5) | 1.2 | 0.37 | 0.25 | 0.007 | 0.25 | 0.007 | 3.5 | 0.11 |
| Blank, cps | 21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 500 |
16500 |
6000 | 4000 | 31500 |
32400 |
8800 | 4900 |
| LODb, pg mL−1 | 4200 | 9500 | 6.3 | 270 | 27 | 1300 | 1.9 | 30 |
| Precisionc, % | 5 | 4 | 2 | 3 | 3 | 2 | 3 | 3 |
| PVG efficiencyd, % | 10 ± 3 | — | 99 ± 2 | — | 99 ± 2 | — | 90 ± 3 | — |
The relatively high blank levels for the halides present in the acetic acid used in this study compromises the achievable LODs when employing PVG methodology. Method blanks (arising from PVG matrix constituents) corresponded to concentrations of 0.18 mg L−1 for 35Cl, 0.24 μg L−1 for 79Br and 0.03 μg L−1 for 127I. In an effort to reduce their magnitude, TAMA AA-100 ultrapure acetic acid was tested. Although a reduction of approximately 40% in the blank value for I− was achieved, a significant contamination for Br−, estimated at 30 μg L−1, persisted. Furthermore, no difference in blanks were evident for Cl− compared to the ACS-grade acetic acid. Thus, given the demands for achieving simultaneous determination of the halides using this method, the ACS-grade acetic acid was retained for application to subsequent analyses.
Overall efficiencies (generation and transport) were estimated following an analysis of the residual Cl, Br and I content in the waste solution carefully collected from the UV-assisted spray chamber. Once a steady-state signal for a solution containing 5 mg L−1 Cl−, 10 μg L−1 Br−, and 2 μg L−1 I− in 1% v/v acetic acid and 20 mg L−1 Cu2+ had been achieved, the generated waste was collected and re-analyzed by PVG-ICP-MS.
Sample introduction efficiency achieved with PN was estimated to be between 2–3% using a mass balance difference between that of solution uptake and waste recovery. For PVG, the 10 ± 3% efficiency estimated for Cl− is improved relative to PN but notably lower than the earlier reported value of 80% obtained using a flow-through lamp as the photoreactor,2 highlighting the importance of the sample irradiation time on PVG synthesis of CH3Cl (approximately 3 s within the UV-assisted spray chamber vs. 45 s within the flow-through lamp). Despite this, the LOD achieved for Cl− can be considered fit-for-purpose (4.2 μg L−1), providing a wide range of applicability due to the often-elevated chlorine concentrations typically found in many different types of real samples.
Efficiencies of 99 ± 2 and 90 ± 3% estimated for Br− and I−, respectively, represent a significant improvement over standard PN introduction and are consistent with those reported in previous studies, in which both the flow-through lamp and the UV-assisted spray chamber were employed.29,32 These results indicate that the short sample irradiation time with the UV-assisted spray chamber does not impair the generation efficiency of these two elements.
PVG efficiencies for the generation of Cl, Br and I from their respective halates were no better than 5% in the absence of added SO3−. As discussed earlier, addition of 20 mg L−1 of SO3− to the generation medium enhanced efficiency for BrO3− and IO3− without affecting response from Br− and I−. As a result, 40- and 26-fold increases in sensitivity compared to conventional PN sample introduction were obtained for Br and I detection following their generation from BrO3− and IO3− using 1% v/v acetic acid containing 20 mg L−1 Cu2+ and 20 mg L−1 SO32−. This resulted in an overall PVG efficiency of 99 ± 1% for BrO3− and 86 ± 2% for IO3−. As noted earlier, use of SO32− did not prove efficacious for ClO3−.
Precision of measurement was reflected in the relative standard deviation of the mean signals derived from 10 consecutive samples containing 5 mg L−1 Cl−, 10 μg L−1 Br− and 1 μg L−1 I−. Results are in the range of 2 to 5% for both the PVG and PN analyte introduction approaches.
A short justification of the relatively high flow rate for PVG sample introduction used in this study is warranted at this point. Although plasma-based analyses are often developed with low flow applications in mind, which are beneficial to sample consumption and alleviation of issues relating to matrix deposition on interface components, the highest sample flow rate consistent with generation of a stable aerosol is desirable for PVG work. Aerosols permitting efficient photolysis reactions on a short time scale and rapid separation of synthesized analyte from the liquid phase to enhance product transport to the plasma are desirable since signal response is proportional to flow rate (all other conditions being satisfied), yielding optimal analytical metrics.
3.5 Simultaneous determination of total Cl, Br and I in real samples by aerosol-assisted PVG-ICP-MS
Application of PVG-ICP-MS for the analysis of real samples often remains a significant challenge due to severe interferences induced by common reagents used for their preparation, particularly HNO3. For the determination of the halogens, the use of MIC has been widely explored due to a number of attractive factors, including the elimination of the risk of their loss as volatile HCl, HBr and HI, rapid sample throughput, capacity to digest relatively high sample masses, enhanced digestion efficiencies (yielding digests with low residual carbon content), and the low blank levels achieved through the use of dilute reagents or even water as the absorbing medium.1,24 These characteristics render MIC particularly appealing as a sample preparation method for subsequent halogen determinations by PVG-ICP-MS. The methodology developed herein was thus evaluated by analysis of four different CRM matrices digested using MIC: NIST SRM 1515 (Apple Leaves), NIST SRM 1549 (Non-fat Milk Powder), NIST SRM 1632c (Coal), and NRCC CRM DORM-5 (Fish Protein).The use of alkaline reagents as absorber solutions is recommended for the quantitative recovery and stabilization of halogens following MIC, and dilute solutions of NH4OH are typically used for this purpose.24 However, as discussed in Section 3.3, NH4OH induces signal suppressions for PVG of Cl−, Br− and I− when present at concentrations above 5, 25, and 100 mmol L−1, respectively. In this work, a 50 mmol L−1 NH4OH solution was used to absorb the analytes, giving rise to a final concentration of 12 mmol L−1 NH4OH in the prepared digests. As a consequence of poor PVG of the analytes at such a NH4OH concentration, a minimum final dilution factor of at least 4-fold is required to ensure an interference-free response. Considering the impact of nitrate on PVG, and the typical use of 6 mol L−1 NH4NO3 as the combustion aid for MIC, a final digest containing 12 mmol L−1 NO3− would be expected. This contributes to significant interferences (Section 3.3). In an effort to minimize the final NO3− concentration, the NH4NO3 igniter was reduced to 3 mol L−1 (providing a concentration of 6 mmol L−1 in the final sample digests). Despite the expected longer sample ignition time required using 3 mol L−1 NH4NO3 (vs. 6 mol L−1),50 effective combustion appeared evident for all samples without significant undigested material or sample residues. In this way, a maximum 4-fold dilution factor is sufficient to mitigate interferences induced by both NH4OH and NO3−. Furthermore, dilution should not compromise the quantification of the elements in the tested samples, given their expected analyte concentrations and method LODs.
Analytical results are summarized in Table 3. Calibration was accomplished against external matrix free multielement halide standards prepared in the same PVG medium. With the notable exception of iodine in NIST SRM 1549 (Milk powder), there is generally good agreement between determined and certified values (t test, confidence level of 95%) or information/reference values for the halogens in these materials, supporting the accuracy of the proposed method. Results for I in NIST SRM 1549 are biased significantly low compared to the certified value. Subsequent studies were performed by adding 20 mg L−1 SO3− as a second mediator (data not presented in Table 3), but similar results were obtained, indicating that the lower concentration is not related to the presence of IO3− endogenous to the sample or arising from the MIC process. This finding was consistent with the results of a previous study44 reporting that halate species are not formed during the MIC process. This conclusion was further corroborated by subsequent analysis of NRCC GSEA-1 (Ground Seaweed) CRM in the laboratory of the authors at UFSM. Statistical agreement of results generated by ion chromatography coupled to PN ICP-MS indicated the presence of only halide species, permitting its accurate quantitation against external halide-based standards.
| Samplea, μg g−1 | Cl | Br | I | |||
|---|---|---|---|---|---|---|
| Determined | Certified value | Determined | Certified value | Determined | Certified value | |
| a Values expressed for dry weight basis as mean ± standard deviation, n = 3. b Information value. c Values expressed in % w/w. d Reference value. | ||||||
| NIST SRM 1515 | 564 ± 12 | 582 ± 15 | 1.77 ± 0.12 | 1.8b | 0.318 ± 0.010 | 0.3b |
| NIST SRM 1549 | 1.02 ± 0.03c | 1.09 ± 0.02c | 12.1 ± 0.9 | 12b | 2.84 ± 0.08 | 3.38 ± 0.02 |
| NIST SRM 1632c | 0.117 ± 0.002c | 0.1139 ± 0.0041c | 16.2 ± 0.3 | 18.7 ± 0.4d | — | — |
| NRCC DORM-5 | 14100 ± 700 |
12220b |
52.6 ± 4.4 | 50.7b | 7.13 ± 1.63 | 7.5 ± 1.4d |
Attention then focused on the earlier decision to minimize the final concentration of NO3− in the sample digests by utilizing 3.0 mol L−1 NH4NO3 as the ignitor for MIC instead of the recommended 6.0 mol L−1. For NIST SRM 1549, values of 2.73 ± 0.14 and 3.21 ± 0.17 μg g−1 were generated for I by calibration with PN ICP-MS on samples prepared in 3 and 6 mol L−1 NH4NO3, respectively, completely accounting for the discrepancy with the certified value of 3.38 ± 0.02 μg g−1.
4 Conclusion
A rapid, accurate, precise and simple method for the simultaneous determination of Cl, Br and I in real samples was developed based on use of a UV-assisted spray chamber as photoreactor for PVG sample introduction and ICP-MS detection. Using Cu2+ as mediator (in 1% v/v acetic acid medium), LODs were enhanced 3-, 40- and 30-fold compared to conventional PN for Cl−, Br− and I−, respectively. Most noteworthy is that PVG efficiency for only Cl− is severely limited by the short sample irradiation time within the photoreactor as compared to that with use of a flow through lamp. The generally high efficiency may be supportive of recent findings regarding the unique environments produced within microdroplets by sample aerosolization, in which reaction rates may be accelerated due to altered physical properties relative to the bulk medium.40–42,51 A full understanding of the impact of such an environment on PVG reactions is currently unknown. In this context, it may be instructive to undertake further studies to elucidate the influence of the microdroplet environment on these photochemical reactions by, for example, using means to alter the droplet diameter distributions.Although interferences from NO3− are inherent to many PVG reactions and have impact even at low concentrations, they could be minimized using the UV-assisted spray chamber. A low solution pH may be beneficial in reducing the impact of NO3− with this system. Also noteworthy was that the alkaline pH obtained at concentrations of NH4OH higher than 5 mmol L−1 significantly influenced PVG from halides, with a pronounced decrease in response from Cl−, possibly attributed to availability of free Cu2+ (due to formation of Cu(OH)2 or [Cu(NH3)4]2+). This affected PVG of Br− to a lesser extent whereas PVG of I− was suppressed only at higher concentrations of NH4OH (>100 mmol L−1).
Photochemical generation of ClO3−, BrO3− and IO3− introduced unresolved issues with the use of the UV-assisted spray chamber that could only be partly improved by addition of SO32− as a secondary modifier. Response from BrO3− and IO3− ultimately achieved 40- and 26-fold enhancements in sensitivity, respectively, compared to PN. This approach should prove useful when both Br− and BrO3− or I− and IO3− are present in the sample, enabling the use of the aerosol-assisted PVG-ICP-MS as a detection technique when coupled with ion chromatographic separation for rigorous speciation analysis. However, it is important to consider potential interferences caused by SO32− on their PVG generation. Further investigations are required to improve the photochemical reduction of ClO3− in the presence of SO32−, such as by use of a flow-through photoreactor.
Despite the above identified difficulties, the determination of total halogen content in samples subjected to sample preparation by MIC yields results with fit-for-purpose accuracy for a wide variety of sample matrices based on use of a simple PVG medium. Fig. 7 suggests that in samples where halite species are suspected, this methodology should still be capable of accurate analyses of total halogen content in the presence of SO32− at an added concentration of 50 mg L−1 if a loss of 50% in generation efficiency can be tolerated.
Data availability
Data supporting the experimental findings of this study are presented within the article and ESI;† those mentioned by way of a simple summary statement are available from the corresponding author upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES, Finance Code 001) for the sandwich PhD fellowship process number 8887.898440/2023-00 (CAPES PrInt) awarded to Gustavo Rossato Bitencourt. This study was financed in part by the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, grant 314254/2023-4, by P. A. Mello).References
- E. M. M. Flores, P. A. Mello, S. R. Krzyzaniak, V. H. Cauduro and R. S. Picoloto, Challenges and trends for halogen determination by inductively coupled plasma mass spectrometry: A review, Rapid Commun. Mass Spectrom., 2020, 34, e8727 CrossRef CAS PubMed.
- J. Hu, R. E. Sturgeon, K. Nadeau, X. Hou, C. Zheng and L. Yang, Copper Ion Assisted Photochemical Vapor Generation of Chlorine for Its Sensitive Determination by Sector Field Inductively Coupled Plasma Mass Spectrometry, Anal. Chem., 2018, 90, 4112–4118 CrossRef CAS PubMed.
- ASTM D5127-13, Standard Guide for Ultra-Pure Water Used in the Electronics and Semiconductor Industries, ASTM International, West Conshohocken, PA, 2013 Search PubMed.
- A. S. McCall, C. F. Cummings, G. Bhave, R. Vanacore, A. Page-McCaw and B. G. Hudson, Bromine Is an Essential Trace Element for Assembly of Collagen IV Scaffolds in Tissue Development and Architecture, Cell, 2014, 157, 1380–1392 CrossRef CAS PubMed.
- R. C. Woody, Bromide Therapy for Pediatric Seizure Disorder Intractable to Other Antiepileptic Drugs, J. Child Neurol., 1990, 5, 65–67 CrossRef CAS PubMed.
- S. Pavelka, Metabolism of Bromide and Its Interference with the Metabolism of Iodine, Physiol. Res., 2004, 53, 81–90 Search PubMed.
- L. Patrick, Iodine: Deficiency and therapeutic considerations, Alternative Med. Rev., 2008, 13, 116–127 Search PubMed.
- L. S. F. Pereira, M. F. Pedrotti, P. D. Vecchia, J. S. F. Pereira and E. M. M. Flores, A simple and automated sample preparation system for subsequent halogens determination: Combustion followed by pyrohydrolysis, Anal. Chim. Acta, 2018, 1010, 29–36 CrossRef CAS PubMed.
- S. R. Krzyzaniak, R. F. Santos, F. M. Dalla Nora, S. M. Cruz, E. M. M. Flores and P. A. Mello, Determination of halogens and sulfur in high-purity polyimide by IC after digestion by MIC, Talanta, 2016, 158, 193–197 CrossRef CAS PubMed.
- A. L. H. Müller, C. C. Müller, F. G. Antes, J. S. Barin, V. L. Dressler, E. M. M. Flores and E. I. Müller, Determination of Bromide, Chloride, and Fluoride in Cigarette Tobacco by Ion Chromatography after Microwave-Induced Combustion, Anal. Lett., 2012, 45, 1004–1015 CrossRef.
- A. D'Ulivo, E. Pagliano, M. Onor, E. Pitzalis and R. Zamboni, Vapor Generation of Inorganic Anionic Species After Aqueous phase Alkylation with Trialkyloxonium Tetrafluoroborates, Anal. Chem., 2009, 81, 6399–6406 CrossRef.
- E. Pagliano, J. Meija, J. Ding, R. E. Sturgeon, A. D'Ulivo and Z. Mester, Novel Ethyl-Derivatization Approach for the Determination of Fluoride by Headspace Gas Chromatography/Mass Spectrometry, Anal. Chem., 2013, 85, 877–881 CrossRef CAS PubMed.
- Z. Gajdosechova, Z. Mester and E. Pagliano, A rapid and sensitive method for the determination of inorganic chloride in oil samples, Anal. Chim. Acta, 2019, 1064, 40–46 CrossRef CAS PubMed.
- D. Desideri, M. A. Meli and C. Roselli, Determination of essential and non-essential elements in some medicinal plants by polarised X ray fluorescence spectrometer (EDPXRF), Microchem. J., 2010, 95, 174–180 CrossRef CAS.
- S. Yamasaki, A. Takeda, T. Watanabe, K. Tagami, S. Uchida, H. Takata, Y. Maejima, N. Kihou and N. Tsuchiya, Bromine and iodine in Japanese soils determined with polarizing energy dispersive X-ray fluorescence spectrometry, Soil Sci. Plant Nutr., 2015, 61, 751–760 CrossRef CAS.
- U. Heitmann, H. Becker-Ross, S. Florek, M. D. Huang and M. Okruss, Determination of non-metals via molecular absorption using high-resolution continuum source absorption spectrometry and graphite furnace atomization, J. Anal. At. Spectrom., 2006, 21, 1314–1320 RSC.
- F. Cacho, L. Machynak, M. Nemecek and E. Beinrohr, Determination of bromide in aqueous solutions via the TlBr molecule using high-resolution continuum source graphite furnace molecular absorption spectrometry, Spectrochim. Acta, Part B, 2018, 144, 63–67 CrossRef CAS.
- M. Schneider, B. Welz, M.-D. Huang, H. Becker-Ross, M. Okruss and E. Carasek, Iodine determination by high-resolution continuum source molecular absorption spectrometry – A comparison between potential molecules, Spectrochim. Acta, Part B, 2019, 153, 42–49 CrossRef CAS.
- T. Taflik, F. A. Duarte, É. L. M. Flores, F. G. Antes, J. N. G. Paniz, É. M. M. Flores and V. L. Dressler, Determination of bromine, fluorine and iodine in mineral supplements using pyrohydrolysis for sample preparation, J. Braz. Chem. Soc., 2012, 23, 488–495 CrossRef CAS.
- T. S. Nunes, C. C. Muller, P. Balestrin, A. L. H. Muller, M. F. Mesko, P. de A. Mello and E. I. Muller, Determination of chlorine and sulfur in high purity flexible graphite using ion chromatography (IC) and inductively coupled plasma optical emission spectrometry (ICP OES) after pyrohydrolysis sample preparation, Anal. Methods, 2015, 7, 2129–2134 RSC.
- F. G. Antes, J. S. F. Pereira, M. S. P. Enders, C. M. M. Moreira, E. I. Müller, E. M. M. Flores and V. L. Dressler, Pyrohydrolysis of carbon nanotubes for Br and I determination by ICP-MS, Microchem. J., 2012, 101, 54–58 CrossRef CAS.
- J. T. P. Barbosa, C. M. M. Santos, L. dos Santos Bispo, F. H. Lyra, J. M. David, M. das G. A. Korn and E. M. M. Flores, Bromine, Chlorine, and Iodine Determination in Soybean and its Products by ICP-MS After Digestion Using Microwave-Induced Combustion, Food Anal. Methods, 2013, 6, 1065–1070 CrossRef.
- E. Tjabadi and N. Mketo, Recent developments for spectrometric, chromatographic and electroanalytical determination of the total sulphur and halogens in various matrices, TrAC, Trends Anal. Chem., 2019, 118, 207–222 CrossRef CAS.
- M. F. Mesko, V. C. Costa, R. S. Picoloto, C. A. Bizzi and P. A. Mello, Halogen determination in food and biological materials using plasma-based techniques: challenges and trends of sample preparation, J. Anal. At. Spectrom., 2016, 31, 1243–1261 RSC.
- R. E. Sturgeon, Photochemical vapor generation: A radical approach to analyte introduction for atomic spectrometry, J. Anal. At. Spectrom., 2017, 32, 2319–2340 RSC.
- R. E. Sturgeon, in Vapor Generation Techniques for Trace Elements Analysis, ed. A. D'Ulivo and R. E. Sturgeon, Elsevier, Amsterdam, 2022, pp. 213–263 Search PubMed.
- R. E. Sturgeon and P. Grinberg, Some speculations on the mechanisms of photochemical vapor generation, J. Anal. At. Spectrom., 2012, 27, 222–231 RSC.
- P. Grinberg and R. E. Sturgeon, Ultra-trace determination of iodine in sediments and biological material using UV photochemical generation-inductively coupled plasma mass spectrometry, Spectrochim. Acta, Part B, 2009, 64, 235–241 CrossRef.
- P. Grinberg and R. E. Sturgeon, Photochemical vapor generation of iodine for detection by ICP-MS, J. Anal. At. Spectrom., 2009, 24, 508–514 RSC.
- R. M. Oliveira, B. S. Soares and D. L. G. Borges, A simple dilute-and-shoot approach using UV photochemical vapor generation for the determination of iodine in alcoholic beverages by ICP-MS, J. Food Compos. Anal., 2021, 95, 103655 CrossRef CAS.
- R. E. Sturgeon, Detection of bromine by ICP- oa -ToF-MS following photochemical vapor generation, Anal. Chem., 2015, 87, 3072–3079 CrossRef CAS PubMed.
- R. M. De Oliveira, D. L. G. Borges, P. Grinberg, Z. Mester and R. E. Sturgeon, Copper-ion assisted photochemical vapor generation of bromide and bromate, J. Anal. At. Spectrom., 2021, 36, 1235–1243 RSC.
- R. E. Sturgeon and E. Pagliano, Evidence for photochemical synthesis of fluoromethane, J. Anal. At. Spectrom., 2020, 35, 1720–1726 RSC.
- A. Bakač, Effects of halide ions and carbon monoxide on the reactions of iron(III) with alkyl radicals, Croat. Chem. Acta, 2001, 74, 633–640 Search PubMed.
- J. M. Carraher, O. Pestovsky and A. Bakac, Transition metal ion-assisted photochemical generation of alkyl halides and hydrocarbons from carboxylic acids, Dalton Trans., 2012, 41, 5974–5980 RSC.
- D. Leonori and R. E. Sturgeon, A unified approach to mechanistic aspects of photochemical vapor generation, J. Anal. At. Spectrom., 2019, 34, 636–654 RSC.
- J. Hu, H. Chen, X. Jiang and X. Hou, Photochemical Vapor Generation of Halides in Organic-Acid-Free Media: Mechanism Study and Analysis of Water Samples, Anal. Chem., 2021, 93, 11151–11158 CrossRef CAS PubMed.
- R. E. Sturgeon, S. N. Willie and Z. Mester, UV/spray chamber for generation of volatile photo-induced products having enhanced sample introduction efficiency, J. Anal. At. Spectrom., 2006, 21, 263–265 RSC.
- H. Matusiewicz, M. Ślachciński, P. Pawłowski and M. Portalski, Evaluation of Five Phase Digitally Controlled Rotating Field Plasma Source for Photochemical Mercury Vapor Generation Optical Emission Spectrometry, Anal. Sci., 2015, 31, 987–995 CrossRef CAS PubMed.
- J. K. Lee, D. Samanta, H. G. Nam and R. N. Zare, Micrometer-Sized Water Droplets Induce Spontaneous Reduction, J. Am. Chem. Soc., 2019, 141, 10585–10589 CrossRef CAS PubMed.
- H. Xiong, J. K. Lee, R. N. Zare and W. Min, Strong Electric Field Observed at the Interface of Aqueous Microdroplets, J. Phys. Chem. Lett., 2020, 11, 7423–7428 CrossRef CAS PubMed.
- M. Girod, E. Moyano, D. I. Campbell and R. G. Cooks, Accelerated bimolecular reactions in microdroplets studied by desorption electrospray ionization mass spectrometry, Chem. Sci., 2011, 2, 501–510 RSC.
- R. S. Picoloto, M. Doneda, E. L. M. Flores, M. F. Mesko, E. M. M. Flores and P. A. Mello, Simultaneous determination of bromine and iodine in milk powder for adult and infant nutrition by plasma based techniques after digestion using microwave-induced combustion, Spectrochim. Acta, Part B, 2015, 107, 86–92 CrossRef CAS.
- É. M. M. Flores, M. F. Mesko, D. P. Moraes, J. S. F. Pereira, P. A. Mello, J. S. Barin and G. Knapp, Determination of Halogens in Coal after Digestion Using the Microwave-Induced Combustion Technique, Anal. Chem., 2008, 80, 1865–1870 CrossRef PubMed.
- H. Coutinho de Jesus, P. Grinberg and R. E. Sturgeon, System optimization for determination of cobalt in biological samples by ICP-OES using photochemical vapor generation, J. Anal. At. Spectrom., 2016, 31, 1590–1604 RSC.
- G. S. Lopes, R. E. Sturgeon, P. Grinberg and E. Pagliano, Evaluation of approaches to the abatement of nitrate interference with photochemical vapor generation, J. Anal. At. Spectrom., 2017, 32, 2378–2390 RSC.
- L. Velásquez-Yévenes and R. Ram, The aqueous chemistry of the copper-ammonia system and its implications for the sustainable recovery of copper, Clean. Eng. Technol., 2022, 9, 100515 CrossRef.
- L. Yang, L. He, J. Xue, Y. Ma, Y. Shi, L. Wu and Z. Zhang, UV/SO32− based advanced reduction processes of aqueous contaminants: Current status and prospects, Chem. Eng. J., 2020, 397, 125412 CrossRef CAS.
- B. D. Fennell, S. P. Mezyk and G. McKay, Critical Review of UV-Advanced Reduction Processes for the Treatment of Chemical Contaminants in Water, ACS Environ. Au, 2022, 2, 178–205 CrossRef CAS PubMed.
- M. F. Pedrotti, L. S. F. Pereira, C. A. Bizzi, J. N. G. Paniz, J. S. Barin and E. M. M. Flores, Microwave-induced combustion: Thermal and morphological aspects for understanding the mechanism of ignition process for analytical applications, Talanta, 2017, 174, 64–71 CrossRef CAS PubMed.
- Q. He, N. Zhang, Y. Qiao, C. Li and J. Zhang, Vapor generation of mercury and methylmercury in aqueous microdroplets produced by pneumatic nebulization, J. Anal. At. Spectrom., 2022, 37, 1894–1901 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ja00079c |
| This journal is © The Royal Society of Chemistry 2025 |