
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Uranium enrichment measurements using tunable laser spectroscopy coupled with fiber-optic laser-induced breakdown spectroscopy
Yichen
Zhao
 *a,
Haori
Yang
a and
Sivanandan S.
Harilal
*bc
*a,
Haori
Yang
a and
Sivanandan S.
Harilal
*bc
aSchool of Nuclear Science & Engineering, Oregon State University, 3451 SW Jefferson Way, Corvallis, Oregon 97330, USA. E-mail: ych.zhao@hotmail.com
bPacific Northwest National Laboratory, 902 Battelle Boulevard, Richland, Washington 99352, USA. E-mail: sivanandan.harilal@pnnl.gov; hari@pnnl.gov
cDepartment of Chemistry, Washington State University, Pullman, Washington 99164, USA
First published on 8th April 2025
Abstract
Tunable laser absorption spectroscopy (LAS) and laser induced fluorescence (LIF) were combined with fiber-optic laser-induced breakdown spectroscopy (FOLIBS) to characterize 238U and 235U isotopes from a natural uranium (NU) sample. Time-resolved absorption spectroscopy (TRAS) and two-dimensional fluorescence spectroscopy (2D-FS), derived from LAS and LIF respectively, were utilized to visualize uranium isotope shifts. These techniques clearly differentiated the isotopic shift of the U I 394.382 nm transition (≈8.7 GHz) and identified partially resolved hyperfine structures of 235U. Isotope ratio analysis for NU was conducted under 10 torr and 30 torr N2 background gas, and TRAS measurement showed good agreement. However, strong reabsorption in 2D-FS hindered accurate isotope ratio measurements. These rapid, accurate, non-contact laser spectroscopic techniques are promising for in situ measurement of uranium isotopes in hazardous environments, applicable to nuclear energy, safeguards, and forensics.
1 Introduction
Uranium detection and characterization are of great interest in many areas, such as nuclear energy, nuclear security, nuclear safeguards, and nuclear forensics.1 One vital feature is the isotopic enrichment analysis of 235U, which provides useful information for tracing the sample, verifying the declared enrichment level, and detecting undeclared proliferation activities at nuclear facilities. Therefore, uranium isotope characterization techniques play a significant role in such applications. Traditional mass spectrometry-based techniques (e.g., inductively coupled plasma – mass spectrometry, thermal ionization – mass spectrometry, secondary ionization – mass spectrometry, resonance ionization – mass spectrometry and atom probe tomography) involve extensive sample preparation, which is usually time-consuming and requires specialized laboratories.2,3 Such measurements generally cannot be carried out onsite; samples must be shipped to a spectrometry lab for analysis, causing significant delays. Methods based on gamma spectroscopy can also measure the enrichment level of 235U, but they require substantial sample preparation time, suffer from self-shielding in bulk materials, and experience severe spectral interferences in highly radioactive samples.4 In addition, these methods have limitations when applied to nuclear fuel reprocessing facilities due to the extremely high temperature and high radiation levels of the measurement environment. For example, pyro-processing typically maintains an operational temperature higher than 800 °C inside an electro-refiner.5 Therefore, there is a demand for a rapid uranium isotopic enrichment characterization technique that can be implemented in extreme environments and in the fields.Optical-spectroscopic tools such as emission, absorption, and fluorescence, combined with laser-produced plasma (LPP) as an atomic reservoir, have been studied for stand-off measurements of actinides, explosives, and chemical and biological threats.6–8 One of the widely used LPP techniques is laser-induced breakdown spectroscopy (LIBS), which combines LPP and optical emission spectroscopy (OES).7,9,10 LIBS utilizes spontaneous emission from the LPP that can be used for obtaining elemental/isotopic information about the sample from a standoff distance.7,11 Numerous studies have been reported using LIBS to measure uranium isotopes under ambient air or a controlled gas environment.7,12–17 However, significant challenges still exist for techniques that rely on OES to resolve small isotope shifts in optical transitions, which usually require a spectroscopic system with ultra-high resolution to prevent the overlap of adjacent isotope peaks.18 Note that the average isotopic shift of U I and U II transitions in the visible spectral range is approximately 7.5 pm.7,19–21
Compared to LIBS, absorption-based techniques, such as Laser Absorption Spectroscopy (LAS), Laser-Induced Fluorescence (LIF), Dual Comb Spectroscopy (DCS) could achieve extremely high resolving power needed to separate closely spaced isotope peaks of U and other materials.22–40 Absorption-based methods use populations of lower-energy levels, including ground level, and hence provide narrower spectral profiles with high sensitivity.41,42 Two-dimensional fluorescence spectroscopy (2D-FS), a relatively new method, that gives a two-dimensional contour plot of measured emission intensity over a wide range of excitation and emission wavelengths, has been demonstrated with LPP.25,43,44 2D-FS is also capable for characterizing the plasma from a standoff distance.45 Absorption spectroscopy employing tunable lasers and broadband sources are also useful for late-time characterization of LPP and quantification of various species in the plume.31,34,36,41,46–49 Uranium isotope ratio measurements via absorption spectroscopy with different types of uranium-containing solids were previously performed by researchers.23,24 Liu et al.50 used two diode lasers to measure the absorbance of plasma produced by an Nd:YAG laser and determine the uranium isotope ratios in solid samples. Uranium isotope ratios in uranium oxide samples are measured by Quentmeier et al.51 employing diode laser absorption spectroscopy.
Typically, an open-path scheme is commonly used for the laser propagation of both pump and probe beams and/or spontaneous emission data collection. Fiber optics can be utilized for ablation laser pulse and emission signal transport in LIBS, a method known as fiber-optic laser-induced breakdown spectroscopy (FOLIBS).52–54 FOLIBS offers significant advantages due to its potential for remote measurements in extreme environments.11 In this work, we report the LIBS, LAS, and LIF measurements of U spectral and isotopic analysis on plasmas produced with laser pulses transferred through fiber optics. The laser beam profile is smoothed during fiber transport, leading to a flat-top temporal profile. Therefore, the plasma plumes in FOLIBS, compared to conventional LIBS, usually have lower temperatures and lower electron density. Furthermore, the ablation in FOLIBS is more uniform and reproducible with lower variability, making FOLIBS more favorable for quantitative analysis.53,54
The major aim of the present work is to demonstrate uranium isotope measurement via 2D-FS and TRAS combined with FOLIBS. A natural uranium (NU) sample was used for U enrichment analysis. These techniques clearly differentiated the isotopic shift of the U I 394.382 nm transition (≈8.7 GHz) and partially resolved hyperfine structures of 235U. Spectral fitting was applied to both 2D-FS and TRAS results to obtain the isotope ratio of the NU sample, with an accurate isotope ratio estimation of 235U and 238U achieved via TRAS. However, the reabsorption in LIF caused errors in isotopic ratio measurements with 2D-FS. Overall, the results demonstrated that 2D-FS and TRAS combined with FOLIBS have the potential for uranium isotope characterization.
2 Experimental setup
The schematic of the experimental system used in this work is shown in Fig. 1a. Pulses with ≈6 ns duration (FWHM) from a Q-switched Nd:YAG laser (Quantel Q-Smart 450) were utilized to generate plasma on the surface of NU sample. The NU sample (Goodfellow, Inc.) was a pure uranium metal that contains ≈0.71% 235U and ≈99.28% 238U. The laser provided a maximum energy of ≈450 mJ at the fundamental wavelength (1064 nm). The laser energy was controlled by a customized beam attenuation module, which consists of a polarized beam splitter, a half-wave plate, and a thin film polarizer. The attenuated laser pulses were focused and injected into a 1000 μm multimode optical fiber (FT1000EMT, Thorlabs) using a 150 mm focal length antireflection (AR)-coated plano-convex lens. The fiber entrance was placed slightly behind the focal point of the lens to prevent optical breakdown in the fiber.54 The Nd:YAG laser energy was moderated to a relatively safe level to prevent optical fiber damage and reduce the probability of air breakdown before coupling.54 Although rare, air breakdown still happens with relatively low frequency (approximately 11 out of 1000 shots) and it significantly reduces the coupling efficiency, leading to a less dense and weak ablation plasma, which results in data of unsatisfactory quality. Thus, a photodiode (Thorlabs PDA36A2) with a filter mounted is placed near the focal point of the laser-fiber coupling lens to monitor the generation of air breakdown plasma. The data from the monitoring photodiode is utilized to filter out air breakdown events and clean up LIBS, LAS, and LIF data.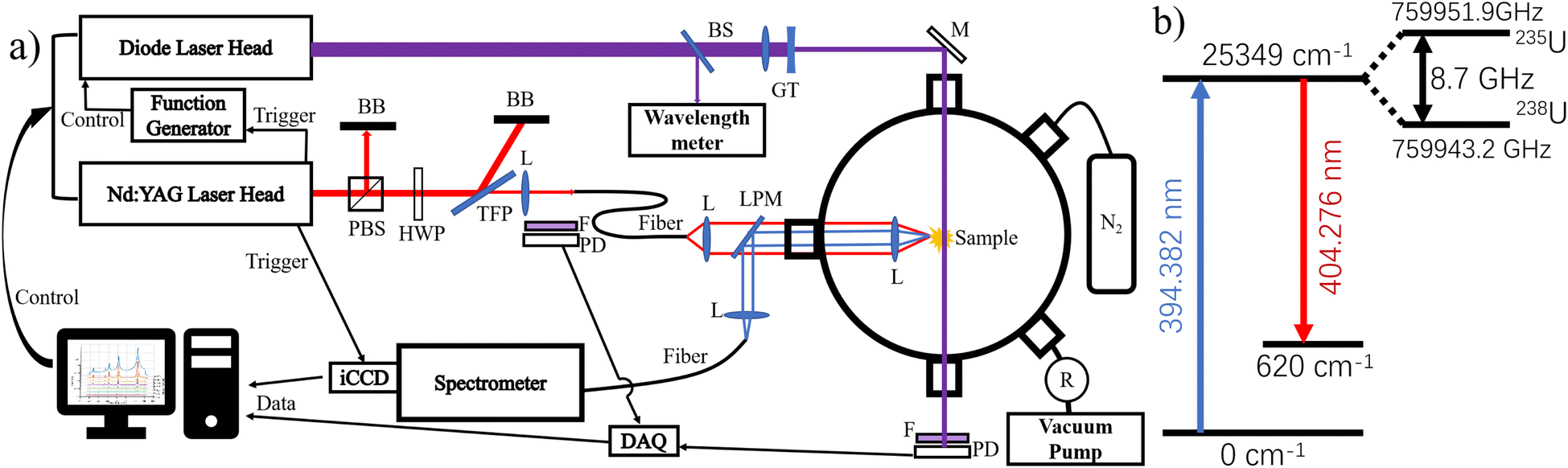 | ||
| Fig. 1 (a) Schematic diagram of the experimental setup. PBS, polarizing beam splitter; BB, beam blocks; HWP, half-wave plate; TFP, thin film polarizer; L, lens; F, filter; PD, photodiode; BS, beam splitter; GT, Galilean Telescope; LPM, long-pass mirror; M, mirror; R, regulator; ICCD, intensified CCD; BB, beam block; R, vacuum or gas regulator; and DAQ, data digitizer. (b) Partial energy diagram of uranium transitions used for LAS and LIF is given. | ||
Laser pulses transferred through the optical fiber are first collimated by a f = 75 mm achromatic lens, then passed through a long-pass mirror, and focused by a f = 30 mm achromatic lens onto the surface of the sample to generate plasma plumes inside a vacuum chamber. The NU sample was positioned in the vacuum chamber to restrain uranium particles generated in the ablation process, preventing environmental contamination. The vacuum chamber was pumped using a roughing vacuum pump (Welch 8912A) with a filter in the gas line between the chamber and the pump. N2 gas was flowed through the chamber to avoid gas-phase oxidation. A vacuum regulator (SMC IRV20) was used in the gas line behind the filter to maintain a specified pressure inside the chamber. The sample, attached to a holder, was mounted on a two-axis translation stage (Zaber X-LSM050A) to provide a surface scanning feature. Several cleaning laser shots were used to remove any oxide layer on the sample surface before collecting any spectral measurements.
A continuous wave (CW) diode laser (DL) (Toptica DL PRO), with a tunable wavelength range of 393–395.5 nm and a 20 GHz mode-hop free tuning range (MHFTR), is used to excite 235U and 238U in the plasma with the selected transition as shown in Fig. 1b. The DL is tuned across the whole 20 GHz (≈10 pm) MHFTR while centered at 394.38 nm to cover both 235U and 238U resonance frequencies with an ≈8.7 GHz (≈4.5 pm) isotopic shift. The change of diode current and voltage are used in combination to provide stable tuning over 20 GHz. A function generator (Tektronix AFG3021C) is used to control the output wavelength during the DL scan. The DL beam is split by a beam sampler and sent into a wavelength meter (HighFinesse WS-7) to ensure the beam is free of mode-hop. The DL beam is reduced to ≈1 mm diameter using a Galilean Telescope and guided through the plasma with a path parallel to the sample surface. The distance between the DL beam and the sample surface is ≈1 mm. The power of the DL beam at the plasma is ≈9 mW and varies slightly during wavelength tuning. An amplified Si photodiode with a 12 MHz bandwidth (Thorlabs PDA36A2) is used to monitor the power of the transmitted DL laser beam after a bandpass filter with a 390 nm center wavelength and 20 nm bandwidth. The signal from the photodiode is digitized by a digitizer (NI PXIe-5122) with a sampling rate of 100 M s−1 and sent to a computer by a remote control module (NI PXIe-8370). Both the digitizer and remote control modules are housed in an NI PXIe-1071 chassis for experiment use.
Emission light from both FOLIBS and LIF is collected by a f = 30 mm achromatic focus lens, reflected by a long-pass mirror, coupled by a f = 25 mm plano-convex lens into a 1000 μm optical fiber, and guided into a 0.75 m Czerny–Turner spectrograph (Andor Shamrock 750A). The collected light is dispersed with a 2400 grooves per mm grating and recorded by an intensified charge-coupled device (ICCD) camera (Andor iStar 340T). The resolution of the spectrograph used is ≈0.03 nm. The Q-switch signal from the Nd:YAG laser is used to trigger the ICCD camera to capture the signal at a selected delay time during a specified gate period. The thermal excitation emissions, which contain the properties of the LPP, provide a practical method to normalize the fluorescence signal.25
DL transmission data and fluorescence data are collected while the tunable laser is scanned across the selected transition with over 161 frequency steps and a 0.12 GHz (0.06 pm) step size. For each frequency step, the time-resolved absorption signal from the photodiode and plasma fluorescence signal from ICCD are recorded, and the average is taken over 8 ablation pulses. The linear stage that holding the sample is translated to the next measurement position after 8 shots at each frequency step to minimize cratering on the sample surface and provide a fresh surface for the following ablation laser pulses.
The absorbance for TRAS is calculated using
| A(f, t) = −ln[I(f, t)/I0(f)] | (1) |
3 Results
3.1 FOLIBS
The NU sample used for this study was positioned in the vacuum chamber with 10 torr flowing N2. The laser energy used was ≈13 mJ, resulting in a power density of ≈1.64 GW cm−2 with a spot size of ≈400 μm. An example of thermally excited emission from the NU sample using the FOLIBS system is shown in Fig. 2 in the spectral range 421–428 nm. The gate delay and width used for the spectral measurement are 2 μs and 50 μs, respectively. According to the Palmer Atlas,55 there are numerous U I and U II transitions within this spectral range. Considering the various broadening mechanisms present in LPP and the moderate resolution spectrograph used in the current study, the overlapping of the U spectral lines is inevitable. | ||
| Fig. 2 Uranium spectrum in the wavelength range 421–428 nm is given (black). The emission measurements are carried out with a gate delay and width of 2 μs and 50 μs respectively. The simulated spectral features with a temperature of 4500 K for U I (red) and U II (blue) are also given. | ||
Spectral modeling was employed to infer the excitation temperature of the U plasma.25,56 For the modeling, a U number density of 1 × 1015 cm−3 and a temperature of 4500 K were used. The results are also depicted in Fig. 2. A comparison between the experimental and modeled spectra reveals that, while there is overall agreement in the spectral features, significant differences in emission intensity are observed. These discrepancies could be attributed to self-absorption, time and space-averaged measurements, and uncertainties in the spectroscopic constants. It is also important to note that the spectral modeling did not account for radiative transport effects in the plasma.
The spectral resolution of the spectroscopic system used in the present study is ≈30 pm, which is significantly larger than the average 7.5 pm isotope shift of 235U and 238U.7 Most researchers take advantage of the relatively large isotope shift of the U II 424.437 nm transition (≈25 pm) for isotopic analysis,13,15,16 which is still unresolvable with our current FOLIBS system due to the limitations imposed by the instrumental resolution and various line broadening mechanisms.7
3.2 2-Dimensional fluorescence spectroscopy
2D-FS measurements were carried out by stepping the probe laser wavelength across the resonance U I transition at 394.382 nm and collecting the emission from the directly coupled U I at 404.276 nm. Such a LIF light collection scheme is beneficial for avoiding light scattering, which may become an issue if the LIF emission at the probe wavelength is monitored. 2D-FS results for the NU sample are shown in Fig. 3a and b. The measurements were taken with a delay of 500 ns and a gate width of 1 ms. The results provide an overall (time-integrated) emission intensity variation for changes in excitation frequency in Fig. 3a and showcase the emission signal of 235U on a zoomed-in scale in Fig. 3b. A strong dependence on excitation frequency can be observed from the results, with 235U and 238U appearing at the expected excitation frequency for NU sample. Overall, 238U showcased a much stronger emission intensity than 235U.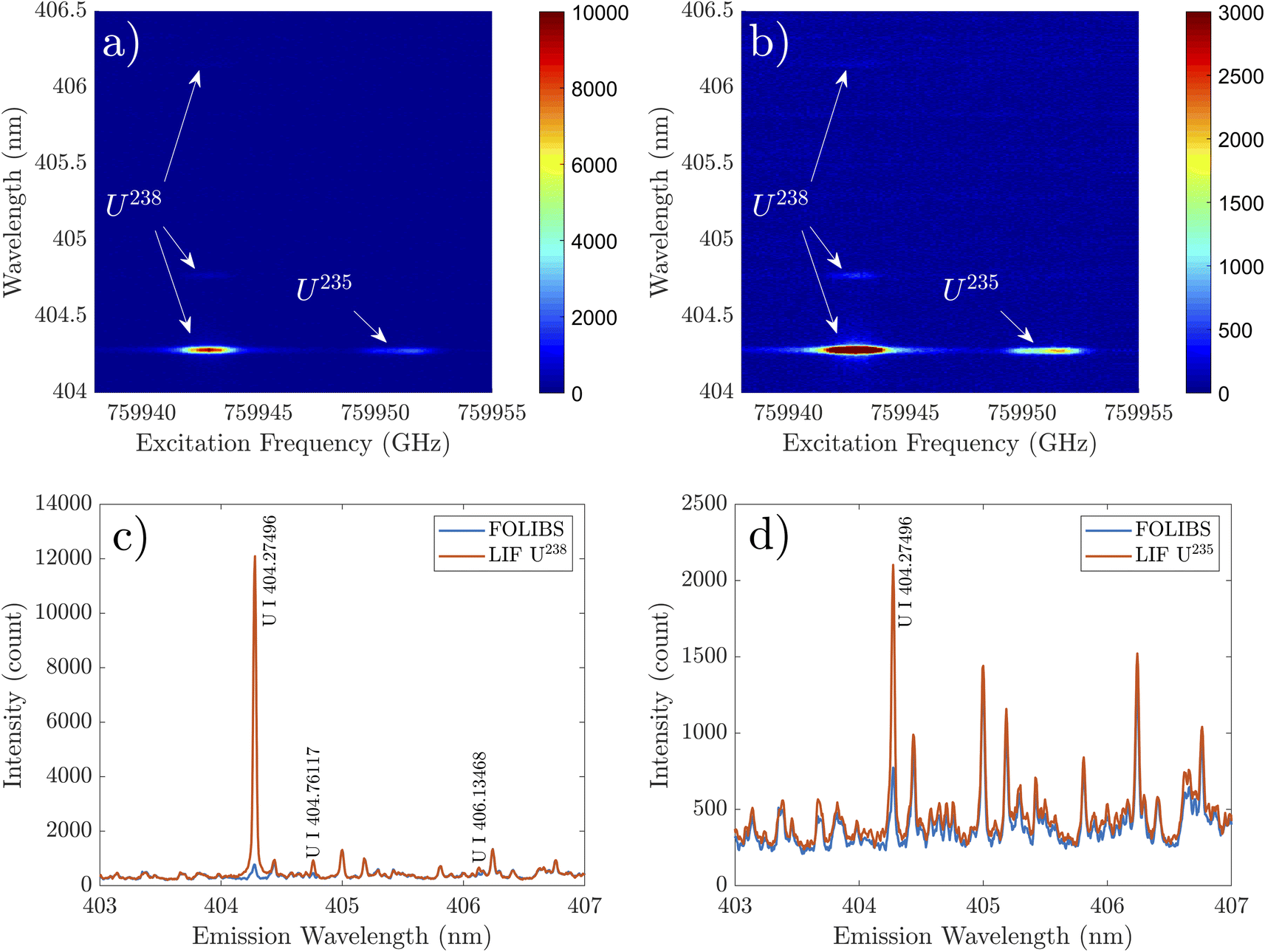 | ||
| Fig. 3 (a) 2D-FS of selected U transition in 10 torr N2 showing LIF and LIBS emission intensity. (b) Shows 2D-FS image with zoomed in scale for emphasizing emission from 235U. Extracted emission spectra at on-resonance (LIF) and off-resonance for (c) 238U I and (d) 235U I. | ||
The 2D-FS map provided in Fig. 3a and b highlights the overlap between absorption and emission signatures. To emphasize the enhancement of the LIF signal at resonance frequencies, emission features are extracted at both resonance and non-resonance frequencies. Unlike 238U, 235U has an asymmetric shape due to the partially resolved hyper-fine structures (HFS).26Fig. 3c and d show the emission spectra recorded with excitation frequency on-resonance and off-resonance with 238U and 235U transitions, respectively. Significant emission intensity enhancement at 404.28 nm can be observed while on-resonance for both 235U and 238U. The enhancement of emissions due to 238U is more significant than 235U as expected. Besides the resonance emission at 404.28 nm in 2D-FS, small increases in emission intensities for U I at 404.76 nm (620–25319 cm−1) and U I at 406.13 nm (620–25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 235 cm−1) are also observed at the 238U resonance frequency. Both these transitions' upper energy levels are very close to the upper energy level of the resonantly pumped U I at 394.382 nm (25349 cm−1). This suggests that collisional transfer may induce fluorescence due to the closely spaced energy levels of the transition of interest.25,57
235 cm−1) are also observed at the 238U resonance frequency. Both these transitions' upper energy levels are very close to the upper energy level of the resonantly pumped U I at 394.382 nm (25349 cm−1). This suggests that collisional transfer may induce fluorescence due to the closely spaced energy levels of the transition of interest.25,57
The nature and pressure of the ambient medium affect the collisional excitation and hence may influence the spectral line broadening. Therefore, additional 2D-FS measurements are taken under 30 torr flowing N2 to investigate the ambient pressure effects on fluorescence signatures. The excitation spectra obtained from the 2D-FS maps are given in Fig. 4 for (a) 10 torr and (b) 30 torr N2 pressure levels, respectively. Overall, strong absorption/emission can be observed when the probe laser is tuned to the resonance frequency corresponding to the major isotope 238U, while the 235U region shows absorbance/emission with less intensity. The partially resolved HFS of the 235U at 394.38 nm, which leads to the asymmetrically broadened peak shape, is also observed in the excitation spectra.
 | ||
| Fig. 4 Excitation spectra of selected U transition at (a) 10 torr and (b) 30 torr obtained from the 2D-FS are given. The circular points correspond to experimental data and the orange smooth curves correspond spectral fits. The R2 values of spectral fits are also given. | ||
Due to the existence of the HFS of 235U, a specified Voigt function with the 235U HFS taken into consideration is built for fitting the excitation spectra. One Voigt peak is used to fit the 238U peak, while 22 Voigt peaks describing the HFS of 235U are used to build the fitting function for 235U spectra. All 22 peaks of the 235U HFS are set to have equal Gaussian and Lorentzian width, while the frequency and relative intensity of each peak are fixed according to calculated values provided by previous reports.25,58–61 The resonance frequency of 238U is used as the reference to calculate the isotope shift data. The Voigt function is used to fit the fluorescence data, and the results are shown in Fig. 4a and b for 10 torr and 30 torr N2 pressure levels. The Voigt function shows a good fit to the experimental data for both the 238U peak and the broadened 235U peak due to partially resolved HFS.
The LIF emission from both isotopes is slightly stronger at 10 torr than at 30 torr. The measured FWHMs from the fitting parameters are 1.55 ± 0.2 GHz (0.8 ± 0.1 pm) for 10 torr and 1.3 ± 0.13 GHz (0.67 ± 0.07 pm) for 30 torr, which are both narrow enough to resolve the isotope shift (≈8.7 GHz) for the U I transition studied here. However, isotope fractions calculated from the areas of the two peaks obtained by the fitting parameters are 82.8 ± 4.1% 238U, 17.2 ± 3.4% 235U for 10 torr, and 94.6 ± 2.7% 238U, 3.7 ± 3.9% 235U for 30 torr, which is inconsistent with the expected isotope ratio for the NU sample. This skewed isotopic ratio provided by the LIF signal is due to the nonlinearity of the LIF signal with atomic number density which results at high values of absorbance.26 The net effect of the nonlinearity leads to an apparent increase in LIF intensity of the 235U peak relative to the 238U peaks in the LIF excitation spectrum. In addition, the bulk isotope in the NU sample, 238U, may also suffer from self-absorption.
3.3 Time-resolved absorption spectroscopy
U isotopic analysis employing 2D-FS showed skewed results due to the nonlinearity of the LIF signal with atomic number density and self-absorption. LAS is immune to self-absorption, and spectral measurements were carried out under similar experimental conditions to those used in the 2D-FS with time steps of 500 ns after laser plasma onset. Fig. 5a and b show the TRAS measurements of the NU sample recorded at 10 torr N2, with Fig. 5a showing the overall absorbance variation and Fig. 5b showing the absorbance signal of 235U on a zoomed-in scale. Strong absorbance can be observed near the resonance frequency of 238U. The peak absorbance for 238U reached very high values (≥5), which indicates more than 99.4% of light being absorbed. A weak absorbance signal due to 235U can also be observed near 759951 GHz.
 | ||
| Fig. 5 (a) TRAS map of U I absorbance for NU in 10 torr N2. (b) Shows TRAS image with absorbance zoomed in for showing 235U hyperfine structures. Transmitted signal intensity over time using NU with diode laser off-resonance and on-resonance are given for (c) 238U and (d) 235U. | ||
Fig. 5c and d show the changes in probe laser transmission intensity over time after the onset of LPP, with the excitation frequency on-resonance and off-resonance for 238U and 235U transitions, respectively. The transmission intensity dropped significantly and saturated when tuned to the 238U resonance frequency, while a moderate but obvious intensity drop could be observed when tuned to the 235U resonance frequency. The intensity eventually recovered to the original level as expected, indicating the decay of the LPP. A slight absorption dip was also observed at early times when wavelength tuned off-resonance, which could be due to the presence of shock waves propagating away from the sample.62
Absorbance spectra at different delay times for both pressures are fitted with the modeled Voigt function as described in the previous subsection. Absorbance data ≥3 are excluded from fitting by setting the weight to zero for those data points. Examples of the Voigt fits for various times and for 10 torr and 30 torr pressures are shown in Fig. 6a and b. These results show a good fit, including the 238U peak and the HFS of 235U. The measured FWHMs of 238U spectral profiles using LAS for 10 torr at 30 μs and 60 μs are 1.02 ± 0.01 GHz and 0.99 ± 0.02 GHz. For 30 torr, the measured values are 0.86 ± 0.01 GHz and 0.93 ± 0.03 GHz at 20 μs and 70 μs respectively.
 | ||
| Fig. 6 Representative spectral fits at indicated times for selected U transition in (a) 10 torr and (b) 30 torr. The R2 values of spectral fits are also given in the bracket. | ||
The measured enrichment levels of 235U from the spectral fits are given in Fig. 7a and b, where the orange dashed line represents the expected value for 235U (0.71%). 235U percentages are found to be higher than the expected value at early delay times and reach the expected value at later times after the plasma onset. The isotope percentage estimation becomes unreliable at very late delay times due to a deteriorated signal-to-noise ratio (SNR). For 10 torr, the enrichment measurement showed the expected value (0.71%) at times approximately 30–60 μs. For 30 torr, the estimated isotope ratio drops more quickly than at 10 torr and reaches the expected value at 20 μs, staying at this level until 70 μs before noise becomes non-negligible.
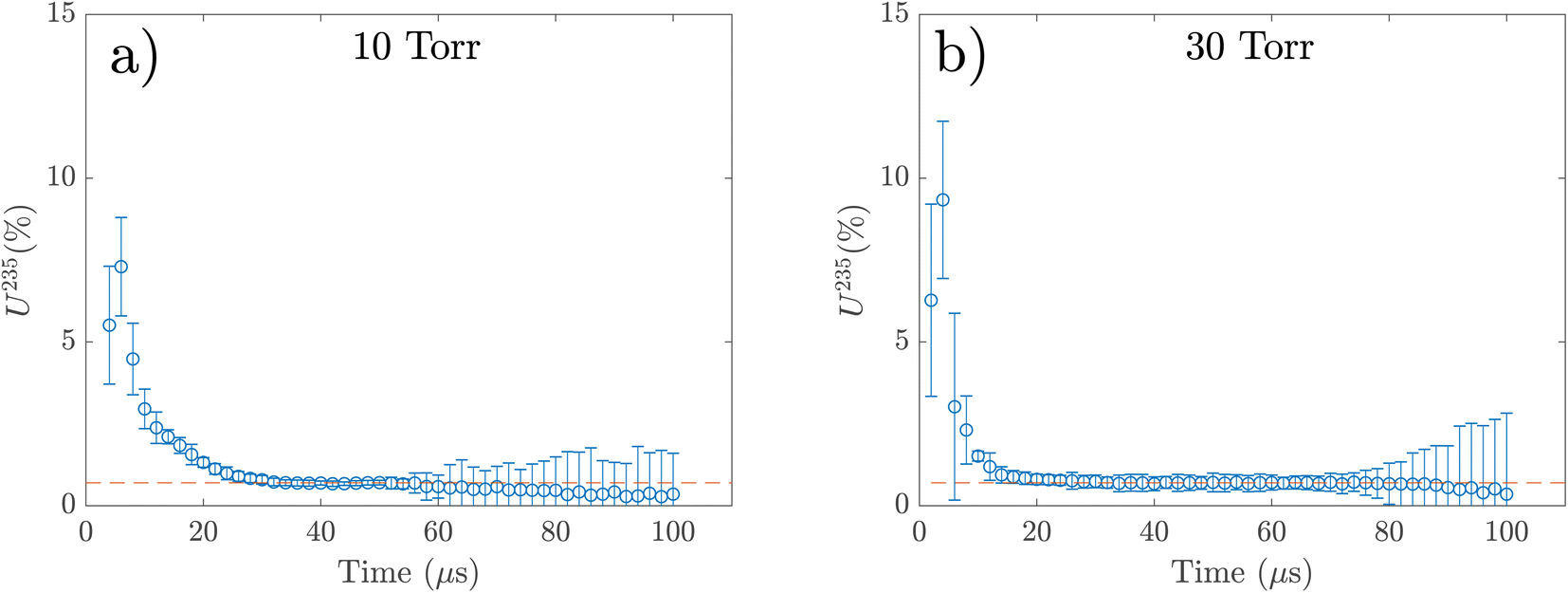 | ||
| Fig. 7 Estimated enrichment level of 235U for NU at various delay times in (a) 10 torr and (b) 30 torr N2 background pressure. The orange dash line represents the expected value for 235U. | ||
4 Discussion
The present study compared the optical spectroscopic signatures (emission, absorption, and fluorescence) from a U LPP for enrichment analysis. A natural U sample was used for the case study. LAS and LIF were combined with FOLIBS to characterize uranium isotopes. For generating plasmas, ns laser pulses were transported through a fiber cable, which is useful for remote measurements in hazardous environments.The thermal emission from the U plasma showed very congested spectral features. Although extensive atomic databases exist for U I and U II transitions,55,63,64 the U database is still incomplete. According to Palmer Atlas,55 there are approximately 125 U I and U II transitions within the spectral range given in Fig. 2. The average isotopic shift for U I and U II transitions is around 7 pm.7 The LIBS spectrum given in Fig. 2 also contains the U II 424.437 nm transition, which possesses a relatively large isotopic shift (≈25 pm) and has been used by several researchers for isotopic analysis employing emission spectroscopy.13,15,16 However, the resolution of the spectroscopic system used in the present study is limited (≈30 pm), making it difficult to observe isotopic splitting using LIBS. Furthermore, the U transitions in the plasma are broadened by various line broadening mechanisms (Stark, Doppler, van der Waals, etc.).7 Since U bulk target is used for the present study, self-absorption in the spectral lines may also be present, contributing additional spectral broadening.
The comparison between the experimental and modeled spectra showed significant differences in spectral intensities. The spectral fits are generated for a temperature of 4500 K. However, the spectral measurements are carried out in a time- and space-integrated manner, representing the weighted average of spectral features. Given the large gradients in temperature and density with space and time for LPP,42 the disagreement in spectral intensities is not unexpected. Thus, the discrepancies in spectral intensities could be attributed to self-absorption, time- and space-averaged measurements, and uncertainties in the spectroscopic constants. Additionally, the spectral modeling did not account for the radiative transport effects in the plasma.
The LIBS spectral features given in Fig. 2 cannot resolve the isotopic shifts because of instrumental and other spectral broadening constraints. LAS and LIF utilize the ground/lower state population, in contrast to LIBS, which monitors the excited level population. As a result, LAS and LIF signals persist longer than the thermally excited population, allowing signals to be collected when the plasma is cooler, thus minimizing line broadening associated with LPP. Furthermore, the instrumental broadening in LAS and LIF is dictated by the linewidth of the probe laser, which is insignificant compared to that in LIBS measurements. Consequently, LAS and LIF inherently provide extremely high spectral resolution.
The 2D-FS, which combines both absorption and emission, is a useful technique for reducing shot-to-shot measurement variability and improving the SNR in the excitation spectrum.43,65 Unlike LIBS measurements, 2D-FS is capable of measuring the isotopic shifts of selected U I transitions (e.g., 394.382 nm) that exhibit an isotopic splitting of approximately 8.7 GHz. Previous results have also highlighted that LIF provides improved detection limits.66 LIF is utilized by several groups to boost LIBS signal levels.67,68 2D-FS maps also showed small increases in the emission intensities of other lines whose upper energy levels are close to resonance pumping, indicating collisional energy transfer. However, the enrichment analysis using LIF showed significant deviations from the expected values and varied with background pressure. For example, the isotope ratios measured at 10 torr and 30 torr demonstrated values of 82.8 ± 4.1% 238U and 17.2 ± 3.4% 235U for 10 torr, and 94.6 ± 2.7% 238U and 3.7 ± 3.9% 235U for 30 torr. These results are inconsistent with the expected isotope ratio for the NU sample. This discrepancy can be explained by the nonlinearity of the LIF signal with atomic number density at high values of absorbance and the presence of self-absorption.26
The LIF emission signal is generated by the excitation laser. Typically, the LIF signal varies linearly with the excitation laser intensity, assuming there is no optical saturation or optical pumping effects. Under these ideal conditions, the LIF signal is proportional to the atomic number density. However, in cases of high absorbance, the relationship between LIF intensity and atomic number density becomes nonlinear and tends toward a maximum limit.26 As depicted in Fig. 6, the absorbance of 235U is low (≤0.2) throughout the plasma evolution, suggesting that the LIF excitation spectra remain nearly linear in relation to the 235U number density and are proportional to the absorbance spectra. Conversely, the 238U exhibits very high absorbance near the line center, resulting in the distorted LIF excitation spectra visible in Fig. 6. To optimize the LIF signal for 235U, a probing laser power of ≈9 mW was utilized; however, absorption saturation cannot be ruled out during the 2D-FS measurements for 238U. Recent studies have demonstrated that absorption saturation can happen at very low power levels for strong transitions.69
Compared to 2D-FS, the isotope ratio measurement from TRAS measurements showed good agreement. However, the isotope ratio estimation varies with delay time and pressure. The enrichment measurement showed the expected values at specific times after the laser plasma onset, and this timing window varied with background pressure. For example, the enrichment analysis showed excellent agreement for times approximately 30–60 μs at 10 torr pressure and approximately 20–70 μs at 30 torr. At early times, due to the high absorbance signals (≥3), the fitting accuracy is poor because of anomalous spectral features. Additionally, the temporal variations in isotopic ratios at early stages could also be contributed by plume transient effects.70 At late delay times, the spectral fits become unreliable due to poor SNR. These results show that time-resolved measurement is also required for isotope ratio analysis due to the temporal variation in plasma properties. Additionally, the line-of-sight averaging, which is inherent to any TRAS measurement, affects the accuracy due to spatial variations in plasma properties. Hence, improved accuracy may be obtained by using spectral models that include spatial variations in plasma properties. It should be noted that the 2D-FS measurements are conducted in a time-integrated manner. The isotopic accuracy in the 2D-FS measurements could be improved by optimizing the detection delay and gate parameters. Additionally, using samples with lower uranium concentrations could help avoid saturation and enhance accuracy.
The change in ambient pressure leads to differences in LIF and TRAS signal levels and persistence. It is well known that the nature and pressure of the buffer gas present during laser-plasma generation can significantly affect the plume dynamics and physical conditions of the LPP.32,39,42 Previous results highlighted that U undergoes gas-phase oxidation in the presence of an oxygen-containing ambient medium, which greatly affects the analytical signal.71 To avoid gas-phase oxidation, the present experiment used a flowing N2 gas. Typically, LAS and LIF of LPP are carried out at moderate pressure levels. At very low pressures (≤100 mTorr), due to reduced density caused by the rapid expansion of the plasma, lower signal levels and reduced persistence can be expected. Doppler splitting has also been reported at lower pressure levels.33,72 At moderate pressures, the plasma becomes collisional due to the confinement caused by the ambient medium, leading to signal stability and enhanced persistence. At high background gas pressures (e.g., 1 atm), flicker noise becomes significant due to large variations in ablated atomic number densities and/or excitation temperatures.25
The measured linewidths of 238U using 2D-FS are higher than those measured with LAS at both 10 torr and 30 torr background pressures. Note that 2D-FS measurements were carried out in a time-integrated manner. The increase in excitation spectral linewidths can be attributed to nonlinearity in the LIF signal with atomic number density at high absorbance levels and self-absorption. The linewidths measured with LAS reach ≤1 GHz (≤0.55 pm), and it provides the inherent linewidth of the selected U transition in the LPP. The linewidth of a transition in an LPP is primarily governed by Stark, Doppler, and van der Waals broadening. Typically, the electron density of ns LPPs is significantly reduced at times ≥5 μs,73,74 making the contribution from the Stark effect negligible at later times. At later times during plasma evolution, and with the background pressure used in the present study (10–30 torr), the Doppler effect is the main contributor to the linewidth.
Miyabe et al.70 investigated the use of LA-LAS for isotopic analysis of Pu employing ns lasers, focusing on its spectroscopic properties and analytical performance, particularly in terms of sensitivity, precision, accuracy, and detection limits. Their results indicated that the linewidths of various Pu I transitions ranged between approximately 350–800 MHz. However, the measured linewidths of U I in the present work were slightly broader, ranging from 0.8 to 1 GHz. This discrepancy is attributed to differences in experimental conditions, such as the use of higher laser energy for ablation and higher background pressures, which may lead to increased Doppler broadening.
Phillips et al.25 applied 2D-FS and TRAS to measure the enrichment level of a uranium sample using fs laser ablation plumes under varying Ar pressures (1–760 torr) and noticed that better isotopic precision was obtained at 10 torr Ar pressure compared to lower and higher pressures. Also, the measurement time was limited to ≤10 μs due to the reduced persistence of fs LPP compared to ns LPP. The measured linewidths are also found to be narrower in the present experiments conducted in N2 ambient compared to Phillips et al.25 in Ar ambient. This is consistent with prior works which reported that molecular ambient gas (e.g., air, N2) more efficiently cools the LPP in comparison to atomic gas (e.g., Ar).75,76 The present results also highlight that better isotopic ratio agreement can be seen for a longer duration of plasma decay due to the increased persistence of ns LPPs compared to fs LPPs.25 This enhanced persistence allows for more extended temporal measurement windows and potentially more stable and reliable isotope ratio determinations.
Comparing the various optical spectroscopic tools used for U isotopic analysis, LIBS offers broadband analysis capability. However, isotopic analysis using LIBS requires high-resolution spectroscopic equipment. Additionally, the close proximity of atomic peaks in the U LIBS spectra can lead to overlaps due to resolution constraints. The LIBS spectral features given in Fig. 2 contain U II 424.4 nm which exhibits a high isotopic shift (≈25 pm). However, this shift could not be resolved due to the inadequate spectral resolution of the instrument (≈30 pm). In contrast, 2DFS and TRAS offer higher spectral resolution owing to their narrowband nature. This attribute allows for greater selectivity of atomic transitions. Nevertheless, LA-LIF faces limitations for high analytical concentrations due to the nonlinear relationship between signal intensity and analyte number density, in addition to self-absorption issues. Despite these challenges, LA-LIF can effectively enhance the signal from LIBS. LA-LAS can provide quantitative information but is not suitable for standoff measurements. Therefore, while each technique has its strengths and limitations, the choice of method depends on the specific requirements of the isotopic analysis and measurement conditions.
The transition selected for LIF and LAS was based on the tunability of the ECDL, which is 394.25 nm ± 1.25 nm, as well as the availability of directly coupled transitions for LIF analysis to prevent any scattering events. With better line selection, appropriate resonance wavelength selection, and the use of instruments with higher spectral resolution (LIBS), some of these technique limitations could be mitigated. Additionally, utilizing a sample with lower uranium concentration, instead of pure uranium, could help reduce absorption saturation and self-absorption effects. By integrating three optical spectroscopic techniques and leveraging the unique advantages of each method, we can assess the strengths and limitations of each and develop a more comprehensive analytical approach.
5 Conclusions
2D-FS and TRAS measurements were carried out on FOLIBS ablation plasma to measure the isotope ratios of 235U and 238U in a natural uranium sample. Isotopic shifts were clearly distinguished, demonstrating sufficient resolving power to separate the uranium isotopic peaks with a relatively small isotopic shift of ≈4.5 pm. Such a small isotopic splitting is difficult to resolve using LIBS due to the presence of instrumental broadening and other broadening mechanisms (Doppler and Stark) that contribute significantly at early times of plasma evolution when thermal excitation dominates.2D-FS and TRAS measurements with the NU sample were conducted under 10 torr and 30 torr of flowing nitrogen, and uranium isotopes were clearly resolved under both pressures. Voigt fitting was applied to LIF emission data for enrichment measurements, but the estimated isotope ratios for both pressures were not at the expected levels due to the nonlinearity of the LIF signal with atomic number density and reabsorption from 238U. TRAS measurements were also analyzed to provide additional information. Absorbance analysis based on TRAS explained the incorrect enrichment level estimation from 2D-FS measurements due to the absorbance saturation effect of the major isotope. Isotope ratio analysis from TRAS measurements shows that the estimated values vary with delay time and operating pressure, but a relatively stable region was discovered that provided a good agreement with the expected enrichment value for both pressures.
This work demonstrates the ability of 2D-FS and TRAS to measure uranium isotope ratios with FOLIBS ablation plasmas. The accuracy of enrichment estimation depends on the conditions of the plasma, delay time, and the modeled fitting function. These techniques provide a feasible method to resolve small isotope shifts using a spectroscopic system. By incorporating the high resolving power of atomic absorption and emission techniques with the flexibility of FOLIBS, the detection system can be implemented for in situ isotope analysis.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Dr Mark Phillips, University of Arizona, for useful discussions. This research is being performed using funding received from the DOE Office of Nuclear Energy's Nuclear Energy University Program No. DE-NE0009166 and No. DE-NE0008756. and U.S. Nuclear Regulatory Commission Grant No. NRC-HQ-84-15-G-0014. SSH acknowledges funding support from the Defense Threat Reduction Agency (HDTRA1-20-2-0001). The content of the information does not necessarily reflect the position or the policy of the federal government, and no official endorsement should be inferred. Pacific Northwest National Laboratory is a multi-program national laboratory operated by Battelle for the U.S. DOE under Contract DE-AC05-76RL01830.Notes and references
- D. LaGraffe, in Nuclear Security Science, Springer International Publishing, 2022, ch. 15, pp. 795–827 Search PubMed.
- J. S. Becker, Spectrochim. Acta, Part B, 2003, 58, 1757–1784 CrossRef.
- M. R. Savina, D. Z. Shulaker, B. H. Isselhardt and G. A. Brennecka, J. Anal. At. Spectrom., 2023, 38, 1205–1212 CAS.
- A. Berlizov, R. Gunnink, J. Zsigrai, C. Nguyen and V. Tryshyn, Nucl. Instrum. Methods Phys. Res., 2007, 575, 498–506 CrossRef CAS.
- M. F. Simpson, Developments of Spent Nuclear Fuel Pyroprocessing Technology at Idaho National Laboratory, Idaho National Laboratory Technical Report INL/EXT-12-25124, 1044209, 2012 Search PubMed.
- S. A. Kalam, E. N. Rao and S. V. Rao, Laser Focus World, 2017, 53, 24–28 Search PubMed.
- S. S. Harilal, B. E. Brumfield, N. L. LaHaye, K. C. Hartig and M. C. Phillips, Appl. Phys. Rev., 2018, 5, 021301 Search PubMed.
- J. L. Gottfried, F. C. De Lucia, C. A. Munson and A. W. Miziolek, Appl. Spectrosc., 2008, 62, 353–363 CAS.
- D. A. Cremers and L. J. Radziemski, Handbook of Laser-Induced Breakdown Spectroscopy, John Wiley and Sons, Chichester, West Sussex, England, Hoboken, NJ, 2006 Search PubMed.
- J. P. Singh and S. N. Thakur, Laser-induced Breakdown Spectroscopy, Elsevier, Amsterdam Kidlington, Oxford Cambridge, MA, 2nd edn, 2020 Search PubMed.
- C. Davies, H. Telle, D. Montgomery and R. Corbett, Spectrochim. Acta, Part B, 1995, 50, 1059–1075 Search PubMed.
- G. C.-Y. Chan, X. Mao, I. Choi, A. Sarkar, O. P. Lam, D. K. Shuh and R. E. Russo, Spectrochim. Acta, Part B, 2013, 89, 40–49 CAS.
- D. A. Cremers, A. Beddingfield, R. Smithwick, R. C. Chinni, C. R. Jones, B. Beardsley and L. Karch, Appl. Spectrosc., 2012, 66, 250–261 CAS.
- F. R. Doucet, G. Lithgow, R. Kosierb, P. Bouchard and M. Sabsabi, J. Anal. At. Spectrom., 2011, 26, 536 CAS.
- W. Pietsch, A. Petit and A. Briand, Spectrochim. Acta, Part B, 1998, 53, 751–761 Search PubMed.
- G. C.-Y. Chan, I. Choi, X. Mao, V. Zorba, O. P. Lam, D. K. Shuh and R. E. Russo, Spectrochim. Acta, Part B, 2016, 122, 31–39 CAS.
- R. E. Russo, J. J. González, D. Oropeza, C. Liu, J. Chirinos and G. C.-Y. Chan, Spectrochim. Acta, Part B, 2024, 216, 106928 CAS.
- P. K. Morgan, J. R. Scott and I. Jovanovic, Spectrochim. Acta, Part B, 2016, 116, 58–62 CrossRef CAS.
- J. Blaise and L. Radziemski, J. Opt. Soc. Am., 1976, 66, 644–659 Search PubMed.
- C. H. Corliss, J. Res. Natl. Bur. Stand., Sect. A, 1975, 80, 1 Search PubMed.
- C. H. Corliss, J. Res. Natl. Bur. Stand., Sect. A, 1976, 80, 429 CrossRef PubMed.
- G. Hull, E. D. McNaghten, C. A. Sharrad and P. A. Martin, Spectrochim. Acta, Part B, 2022, 190, 106378 CrossRef CAS.
- B. W. Smith, A. Quentmeier, M. Bolshov and K. Niemax, Spectrochim. Acta, Part B, 1999, 54, 943 CrossRef.
- M. Miyabe, M. Oba, H. Iimura, K. Akaoka, Y. Maruyama, H. Ohba, M. Tampo and I. Wakaida, Appl. Phys. A, 2013, 112, 87–92 CrossRef CAS.
- M. C. Phillips, B. E. Brumfield, N. LaHaye, S. S. Harilal, K. C. Hartig and I. Jovanovic, Sci. Rep., 2017, 7, 3784 CrossRef PubMed.
- S. Harilal, C. Murzyn, M. Phillips and J. Martin, Spectrochim. Acta, Part B, 2020, 169, 105828 CrossRef CAS.
- J. Merten, E. Nicholas, S. Ethridge, H. Bariola, S. Chestnut, A. Anders, J. Brees and M. Foster, Spectrochim. Acta, Part B, 2023, 200, 106600 CrossRef CAS.
- J. Merten and B. Johnson, Spectrochim. Acta, Part B, 2018, 139, 38 CrossRef CAS.
- N. LaHaye, S. S. Harilal and M. Phillips, Spectrochim. Acta, Part B, 2021, 179, 106096 CrossRef CAS.
- N. Taylor and M. Phillips, Opt. Lett., 2014, 39, 594–597 CrossRef CAS PubMed.
- R. R. D. Weeks, M. C. Phillips, Y. Zhang, S. S. Harilal and R. J. Jones, Spectrochim. Acta, Part B, 2021, 181, 106199 CAS.
- E. J. Kautz, E. N. Weerakkody, M. S. Finko, D. Curreli, B. Koroglu, T. P. Rose, D. G. Weisz, J. C. Crowhurst, H. B. Radousky, M. DeMagistris, N. Sinha, D. A. Levin, E. L. Dreizin, M. C. Phillips, N. G. Glumac and S. S. Harilal, Spectrochim. Acta, Part B, 2021, 185, 106283 CAS.
- M. Miyabe, M. Oba, H. Iimura, K. Akaoka, Y. Maruyama, H. Ohba, M. Tampo and I. Wakaida, J. Appl. Phys., 2012, 112, 123303 CrossRef.
- E. N. Weerakkody and N. G. Glumac, J. Phys. D Appl. Phys., 2021, 54, 125201 CrossRef CAS.
- E. N. Weerakkody, D. G. Weisz, J. Crowhurst, B. Koroglu, T. Rose, H. Radousky, R. L. Stillwell, J. R. Jeffries and N. G. Glumac, Spectrochim. Acta, Part B, 2020, 170, 105925 CrossRef CAS.
- C. Ursu, P. Nica, G. B. Rusu, C. Vitelaru, G. Popa and C. Focsa, Spectrochim. Acta, Part B, 2022, 196, 106510 CrossRef CAS.
- S.-U. Choi, Y. Jo and J.-I. Yun, Anal. Chem., 2024, 96, 10134–10139 CrossRef CAS PubMed.
- J. J. McCauley, M. C. Phillips, R. R. Weeks, Y. Zhang, S. S. Harilal and R. J. Jones, Optica, 2024, 11, 460–463 CrossRef CAS.
- M. Miyabe, M. Oba, H. Iimura, K. Akaoka, Y. Maruyama, H. Ohba, M. Tampo and I. Wakaida, Hyperfine Interact., 2013, 216, 71–77 CrossRef CAS.
- R. G. Wala, M. P. Polek, S. S. Harilal, R. J. Jones and M. C. Phillips, Spectrochim. Acta, Part B, 2025, 107142 CrossRef CAS.
- J. Merten, Spectrochim. Acta, Part B, 2022, 189, 106358 CrossRef CAS.
- S. S. Harilal, M. C. Phillips, D. H. Froula, K. K. Anoop, R. C. Issac and F. N. Beg, Rev. Mod. Phys., 2022, 94, 035002 CrossRef CAS.
- S. S. Harilal, N. L. LaHaye and M. C. Phillips, Opt. Express, 2017, 25, 2312 CrossRef CAS PubMed.
- S. S. Harilal, E. J. Kautz, R. J. Jones and M. C. Phillips, Plasma Sources Sci. Technol., 2021, 30, 045007 CrossRef CAS.
- E. J. Kautz, M. C. Phillips and S. S. Harilal, J. Appl. Phys., 2021, 130, 203302 CrossRef CAS.
- M. P. Polek, M. C. Phillips, F. N. Beg and S. S. Harilal, AIP Adv., 2024, 14, 025043 CrossRef CAS.
- M. C. Phillips, E. J. Kautz and S. S. Harilal, Opt. Lett., 2023, 48, 1942–1945 Search PubMed.
- A. Kuwahara, K. Murakami, H. Tomita, K. Sawada and Y. Enokida, J. Anal. At. Spectrom., 2022, 37, 2033–2041 CAS.
- S. S. Harilal, E. J. Kautz and M. C. Phillips, Phys. Rev. E, 2021, 103, 013213 CrossRef CAS PubMed.
- H. Liu, A. Quentmeier and K. Niemax, Spectrochim. Acta, Part B, 2002, 57, 1611–1623 CrossRef.
- A. Quentmeier, M. Bolshov and K. Niemax, Spectrochim. Acta, Part B, 2001, 56, 45–55 CrossRef.
- A. Matsumoto, H. Ohba, M. Toshimitsu, K. Akaoka, A. Ruas, T. Sakka and I. Wakaida, Spectrochim. Acta, Part B, 2018, 142, 37–49 CrossRef CAS.
- X. Xiao, S. Le Berre, D. Fobar, M. Burger, P. Skrodzki, K. Hartig, A. Motta and I. Jovanovic, Spectrochim. Acta, Part B, 2018, 141, 44–52 CrossRef CAS.
- Y. Zhao, H. Yang and S. S. Harilal, Nucl. Technol., 2024, 1–12 CrossRef.
- B. A. Palmer, R. A. Keller and R. Engleman Jr, Atlas of Uranium Emission Intensities in a Hollow Cathode Discharge, LA-8251-MS Report, Los Alamos Scientific laboratory, 1980 Search PubMed.
- S. S. Harilal, E. J. Kautz, B. E. Bernacki, M. C. Phillips, P. J. Skrodzki, M. Burger and I. Jovanovic, Phys. Chem. Chem. Phys., 2019, 21, 16161–16169 RSC.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, New York, 3rd edn, 2006 Search PubMed.
- O. Axner, J. Gustafsson, N. Omenetto and J. D. Winefordner, Spectrochim. Acta, Part B, 2004, 59, 1–39 CrossRef.
- W. J. Childs, O. Poulsen and L. S. Goodman, Opt. Lett., 1979, 4, 35 CrossRef CAS PubMed.
- W. J. Childs, O. Poulsen and L. S. Goodman, Opt. Lett., 1979, 4, 63–65 CAS.
- P. Schumann, K. Wendt and B. Bushaw, Spectrochim. Acta, Part B, 2005, 60, 1402–1411 Search PubMed.
- S. S. Harilal, E. J. Kautz and M. C. Phillips, J. Appl. Phys., 2022, 131, 063101 CrossRef CAS.
- R. L. Kurucz, Astrophysical Applications of Powerful New Databases, 1995, p. 205 Search PubMed.
- Y. Ralchenko, A. E. Kramida, J. Reader, et al., NIST atomic spectra database, National Institute of Standards and Technology, Gaithersburg, MD, 2008 Search PubMed.
- S. S. Harilal, N. L. LaHaye and M. C. Phillips, Opt. Lett., 2016, 41, 3547–3550 CAS.
- X. K. Shen and Y. F. Lu, Appl. Opt., 2008, 47, 1810–1815 CrossRef CAS PubMed.
- J. M. Li, M. L. Xu, Q. X. Ma, N. Zhao, X. Y. Li, Q. M. Zhang, L. Guo and Y. F. Lu, Talanta, 2019, 194, 697–702 CrossRef CAS PubMed.
- G. Nicolodelli, P. R. Villas-Boas, C. R. Menegatti, G. S. Senesi, D. V. Magalhaes, D. de Souza, D. Milori and B. S. Marangoni, Appl. Opt., 2018, 57, 8366–8372 CAS.
- M. P. Polek, M. C. Phillips, F. Beg and S. S. Harilal, Spectrochim. Acta, Part B, 2025 DOI:10.2139/ssrn.5107113.
- M. Miyabe, M. Oba, K. Jung, H. Iimura, K. Akaoka, M. Kato, H. Otobe, A. Khumaeni and I. Wakaida, Spectrochim. Acta, Part B, 2017, 134, 42–51 CrossRef CAS.
- P. Skrodzki, N. Shah, N. Taylor, K. Hartig, N. LaHaye, B. Brumfield, I. Jovanovic, M. Phillips and S. Harilal, Spectrochim. Acta, Part B, 2016, 125, 112–119 CrossRef CAS.
- B. A. Bushaw and M. L. Alexander, Appl. Surf. Sci., 1998, 127, 935–940 CrossRef.
- J. Freeman, S. Harilal, P. Diwakar, B. Verhoff and A. Hassanein, Spectrochim. Acta, Part B, 2013, 87, 43–50 CrossRef CAS.
- M. Singh and A. Sarkar, Plasma Sci. Technol., 2018, 20, 125501 CrossRef CAS.
- J. Hermann, C. Gerhard, E. Axente and C. Dutouquet, Spectrochim. Acta, Part B, 2014, 100, 189–196 CrossRef CAS.
- I. Urbina, F. Bredice, C. Sanchez-Aké, M. Villagrán-Muniz and V. Palleschi, Spectrochim. Acta, Part B, 2022, 195, 106489 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |