
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An efficient and stable sample preparation and calibration strategy for nanoparticle analysis using laser ablation single particle-ICP-MS
Laura
Kronlachner
 *a,
Zuzana
Gajarska
a,
Pascal
Becker
b,
Detlef
Günther
b and
Andreas
Limbeck
*a
*a,
Zuzana
Gajarska
a,
Pascal
Becker
b,
Detlef
Günther
b and
Andreas
Limbeck
*a
aTU Wien, Institute of Chemical Technologies and Analytics, Getreidemarkt 9/164, 1060 Vienna, Austria. E-mail: laura.kronlachner@tuwien.ac.at; andreas.limbeck@tuwien.ac.at
bETH Zurich, Laboratory of Inorganic Chemistry, Department of Chemistry and Applied Biosciences, ETH Zurich, Vladimir-Prelog-Weg 1, 8093 Zürich, Switzerland
First published on 3rd January 2025
Abstract
Nanoparticles are used in various fields, such as material manufacturing, catalysis and medicine, due to their unique physical and chemical properties. Accurate characterization of nanoparticles is essential for manufacturing purposes as well as for assessing their impact on the environment and human health. To achieve this, single particle inductively coupled plasma mass spectrometry (sp-ICP-MS) has become an essential analytical technique for nanoparticle analysis. It can also be used with laser ablation as a sampling method to overcome challenges related to introducing nanoparticles in liquid suspension. Similar to conventional sp-ICP-MS, laser ablation sp-ICP-MS requires standards for signal calibration, which is challenging as the availability of standard reference materials is limited for all different kinds of nanoparticles. In this work, nanoparticles embedded in polymer thin films are analyzed using laser ablation sp-ICP-MS, whereby the laser is used to sample and transport the intact particles to the plasma. For creating a calibration for mass and size investigations, defined amounts of the element of interest were introduced into the ICP-MS by quantitatively ablating polymer thin film spiked with a defined amount of liquid element standard with different laser spot sizes. The method was developed and optimized using gold nanoparticles with certified sizes that were analyzed using a quadrupole-ICP-MS in single-element mode. The nanoparticles were sized using the proposed calibration approach with a deviation of ≤2.5% from the certified diameter value. Using the calibration approach, a limit of detection for gold of 3 × 10−7 ng was calculated, which translates to a particle size of approximately 15.5 nm, comparable to values in the literature for liquid-suspension-based approaches. Multi-element nanoparticles in the form of gadolinium-doped cerium oxide (GDC) nanoparticles with two elements of interest were analyzed using an ICP-TOFMS utilizing the thin-film-based calibration approach. Comparative measurements of the material confirmed the investigated sizes and composition of the particles. This developed alternative approach circumvents the need for certified particulate standard materials by using in-house-produced spiked polymer thin films as storage-stable calibration standards. Moreover, changing the laser spot size makes it straightforward to alter the number of particles introduced into the ICP-MS.
1 Introduction
Nanotechnology is a rapidly emerging field of science due to the unique chemical and physical properties of materials in the nanometer size range that differ significantly from those of bulk materials. The novel properties demonstrated by nanoparticles are put to use in a wide range of including industrial applications such as material manufacturing,1 catalysis,2 energy harvesting and electronics,3 as well as in life sciences like bioanalytical and biomedical applications,4,5 medicine, including drug delivery, and biology.6,7 Furthermore, nanoparticles have an increasingly significant role in consumer products and household items and can be found in cosmetics such as sunscreens, textiles, sports items, paints, food and food packaging.1,8,9 This widespread use of nanoparticles requires reliable and detailed analysis methods for comprehensive characterization of the materials.For the engineering, development and quality control of nanomaterials and the implementation of new applications, knowledge of the characteristics of the nanoparticles is necessary. This includes obtaining information about the elemental compositions of the particles as well as the average size, size distribution and particle number concentration,10 which is not only of interest in the manufacturing and engineering of nanoparticles but also in studies of their impact on the environment, including human health.3,11 The widespread use in industry applications and consumer goods results in the increasing occurrence of nanoparticles in the environment, making a risk assessment of engineered nanoparticles vital.12
Several analysis techniques have reportedly been used for investigating nanoparticles, which have their individual strengths and weaknesses regarding the capabilities of characterizing the nanoparticle samples. These techniques include light scattering methods,12 fractionation methods with chromatography and MS,12 as well as various microscopy techniques such as atomic force microscopy (AFM) as well as types of electron microscopy like transmission electron microscopy (TEM), scanning electron microscopy (SEM) and energy dispersive X-ray spectroscopy (EDX).12–14 Atomic force microscopy is an ideal tool to gain information about the morphology and topography of a sample in sub-nanometer resolution by scanning the surface of a sample with an oscillating probe.13 The resulting images are created by the topography and can thus give information about the nanoparticles on the sample's surface. However, no information about the elemental constituents of the nanoparticles can be gathered.12 With scanning electron microscopy and transmission electron microscopy, the interaction of the sample with a focused electron beam is utilized. Depending on the method and measurement mode, the secondary electrons, backscattered electrons or transmitted electrons are monitored, which results in an image of the sample.13 The electron microscopy techniques have the vital benefit of providing access to information about the elemental composition of the sample nanoparticles using energy dispersive X-ray spectroscopy but also require a more tedious sample preparation such as conductive coatings on the sample or the requirement for conductive grids to allow transmission.12,13
When analyzing nanoparticle samples with microscopic methods, data about individual particles can be gained. However, for a meaningful characterization of a nanoparticle sample, a high number of particles has to be sized and counted to obtain reliable counting statistics and thus create representative results for the entirety of the sample. This makes the use of microscopy techniques laborious when measuring particle populations.12,14
Recently, single-particle inductively coupled plasma mass spectrometry (sp-ICP-MS) has gained more and more importance as an analytical tool for the characterization of nanoparticles.10 This is due to the combination of two essential advantages: the analysis is performed nanoparticle after nanoparticle, enabling particle counting, while the ICP-MS gives information about the intensity detected for the monitored elements. By using a calibration, this leads to the mass of the individual particles.15 A quadrupole-based ICP-MS allows only single-element-analysis whereas, in contrast to an ICP-TOFMS, all isotopes can be measured quasi-simultaneously, which provides the opportunity to measure transient events of each element and thus analyze the composition of multicomponent nanoparticles.
Even though conventional single-particle ICP-MS using liquid suspension as samples provides valuable analytical information for both natural and engineered nanoparticles,10,15 the measurement in suspension comes with challenges. Problems include the limited stability of nanoparticles in suspension over time, the low transport efficiency of standard pneumatic nebulizers as well as spectral interferences stemming from the suspension medium.16,17
Hence, laser ablation as the sample introduction system for nanoparticle analysis of solid samples has been proposed as a method to overcome the disadvantages mentioned above. The benefits of using laser ablation include the direct analysis of nanoparticles in the solid sample and the intact particle transport (with high transport efficiency as shown by Garcia et al.18), which also makes spatial information available.
However, one drawback of laser ablation in bulk and imaging applications is the need for matrix-matched calibration standards19 that can be solid certified reference materials or in-house prepared standards using various techniques,20 which tends to be tedious and complex. Nevertheless, a large number of trace elements in minerals have been determined accurately using non-matrix matched calibration and internal standardization.21
In the field of LA-sp-ICP-MS, the use of nanoparticles in suspension and liquid introduction thereof22 and nanoparticles in solidified gelatine17 have been reported for assessment of particle size and number concentrations.
Both approaches require nanoparticles with a defined size to create the calibration, which might not be commercially available for many types of nanoparticles.
In this study, nanoparticles are embedded in polymer thin films, creating storage-stable calibration material and samples. The created samples can also be analyzed using other methods, such as electron microscopy or secondary ion mass spectrometry. By ablation of known amounts of material with defined concentrations of the element of interest using different spot sizes and subsequent determination of the laser crater dimensions, exact masses of analyte can be introduced into the ICP-MS. This enables calibration without the need for certified nanoparticles and produces storage-stable calibration materials. Laser ablation is then used for sample introduction of the prepared nanoparticle sample via soft ablation, where nanoparticles are transported without being destroyed. This methodology was successfully applied for the characterization of Au nanoparticles, and further applicability was demonstrated by the analysis of gadolinium-doped cerium oxide (GDC) nanoparticles.
2 Experimental
2.1 Instrumentation
The method development was carried out using single-element Au nanoparticles, thus, a quadrupole ICP-MS could be used to monitor Au. The ablation of the sample material was carried out using an imageGEO193 ArF excimer laser ablation system operating at a wavelength of 193 nm equipped with a TwoVol3 sample chamber and an analytical cup (Elemental Scientific Lasers, USA). An iCAP Qc ICP-MS system (ThermoFisher Scientific, Germany) was coupled to the laser ablation system. The introduction into the ICP-MS was done via a dual concentric injector (DCI, Elemental Scientific, United States). The carrier gas used to constantly flush the laser chamber and transport the ablated material to the ICP-MS was helium (purity 99.999%). Argon (purity 99.999%) was used for the ICP-MS and as a make-up gas. Both gases were obtained from Messer Austria (Austria).Capabilities for multi-element analysis were demonstrated using GDC nanoparticles. For this application, an ArF excimer laser (193 nm, GeoLas C, Lambda Physik, Göttingen) equipped with a modified parallel flow ablation cell (PFAC, opening widened from 1.5 to 3.5 mm)23,24 was used. The ablation cell was operated using helium and argon (both 99.999%, PanGas AG, Dagmersellen, Switzerland) and coupled to an ICP-TOFMS (icpTOF2R, TOFWERK AG, Thun, Switzerland) as the simultaneous monitoring of two elements of interest was necessary.
A KLM Spin-Coater SCV-10 (Schaefer-Tec, Germany) was employed for spin-coating the polymer solutions. To determine the thickness of the polymer thin films, a Dektak XT stylus profilometer (Bruker Corporation, MA, USA) was used to measure the film thickness and the ablation crater geometries, providing crucial information for the quantification. Comparative size distribution measurements were carried out using a ZetaView PMX 120 (ParticleMetrix, Meerbusch, Germany), and bulk analysis of the GDC nanoparticles was carried out using a radial view Agilent ICP-OES 5110 (Agilent Technologies, USA).
2.2 Chemicals and sample materials
Polymers used for creating thin films were polyvinylpyrrolidone (PVP) (molecular weight 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300000 K85–95) and poly(methyl methacrylate) (PMMA) (molecular weight 35000). Both polymers were obtained from Acros Organics (Belgium). For the preparation of polymer solutions, acetonitrile (purity 99.9%) (Honeywell, NC, USA), acetone (purity 99.9%) (Honeywell, NC, USA) and high-purity water (resistivity 18.2 MΩ cm) dispensed from a Barnstead EASYPURE II water system (ThermoFisher Scientific, OH, USA) were used as solvents.
300000 K85–95) and poly(methyl methacrylate) (PMMA) (molecular weight 35000). Both polymers were obtained from Acros Organics (Belgium). For the preparation of polymer solutions, acetonitrile (purity 99.9%) (Honeywell, NC, USA), acetone (purity 99.9%) (Honeywell, NC, USA) and high-purity water (resistivity 18.2 MΩ cm) dispensed from a Barnstead EASYPURE II water system (ThermoFisher Scientific, OH, USA) were used as solvents.
The dissolved Au, Gd and Ce standards were ICP-MS standard stock solutions with a concentration of 1000 mg L−1 purchased from VWR Chemicals (Belgium). The Au nanoparticles used for our investigation (80 nm diameter, stabilized in citrate buffer, optical density of 1) were purchased from Sigma-Aldrich (MO, USA). For multi-element nanoparticles, GDC (GDC20, nominal stoichiometry and composition: Ce0.8Gd0.2O1.95, 80 mol% Ce and 20 mol% Gd) was used. This sample was a technical material obtained from Treibacher (Althofen, Austria).
2.3 Preparation of standards and samples
For preparing the polymer thin films, solutions of 2.5% poly(methyl methacrylate) in acetonitrile/acetone and 3% polyvinylpyrrolidone in high-purity water were prepared by combining and shaking until complete dissolution was reached.To create the element standards, first, a 10 μg g−1 solution was prepared by diluting the 1000 μg g−1 Au/Gd/Ce standard with high-purity water. A mass of 0.05 g of the diluted liquid standards was added to 1.00 g of polymer solution, shaken vigorously and put in an ultrasonic bath for 30 minutes to ensure homogeneity. The derived contents in the prepared standard polymer films were between 8.3 and 8.6 μg g−1 each for Au, Ce and Gd.
For the Au nanoparticle samples, the nanoparticle suspension was diluted by a factor of 10, pipetted into the polymer solution and homogenized, creating an original concentration in the polymer suspension of approximately 106 particles per mL.
The sample of GDC nanoparticles was created by combining the dry nanoparticles with the PVP polymer solution and subsequent dilution until again a concentration of approximately 106 particles per mL was reached.
The spin-coating substrate was a silicon wafer with a dimension of 10 mm × 10 mm. 50 μL of the polymer solutions containing liquid element standard or nanoparticles were dropped onto the spinning wafer and spun for 90 seconds at room temperature. The optimized rotation speed for creating even thin and uniform layers was determined to be 65 rounds per second.
For the standards and samples prepared in PMMA in acetonitrile, the solvent vaporized completely during the spin coating process, creating 74 nm ± 1 nm (n = 50) thin films. The derived PVP films had to be cured after the spin coating process, which was achieved by placing them onto a 60 °C hot plate, resulting in quasi-instantly evaporation of the water and curing. The samples were then kept on the heating plate for 5 minutes to ensure complete evaporation of remaining water. The thin films created out of PVP resulted in a thickness of 253 ± 2 nm (n = 50).
2.4 Analysis setup and method parameters
The Au nanoparticle ablation experiments were conducted with a laser energy of 0.3 J cm−2, a square spot with a side length of 20 μm and a laser frequency of 5 Hz. The low laser energy was chosen to ensure non-destructive ablation of the particles, as suggested by Metarapi et al.,25 to avoid misreporting the nanoparticle size due to degradation during the laser ablation process. The ablation was carried out at a new sample position for each spot, and lines with a length of 5 mm were ablated.
A calibration standard containing a defined amount of Au was ablated using single laser shots to calibrate the system. The same laser energy as for the particle ablation of 0.3 J cm−2 was employed which still enabled the complete ablation of the entire polymer layer. For calibration of the Au measurement, square laser spots with side lengths ranging from nominally 20 μm to 55 μm were used for ablation. For each laser spot size, the single spots were repeated ten times, and a delay of 5 seconds was maintained between each spot to ensure a complete washout of the previous ablation. Subsequently, different, precisely defined amounts of Au were detected. The amounts were calculated using information about the crater areas, the thickness of the thin films and their concentration of the investigated element. The profilometer measurement showed uncertainties of <± 0.3 μm for the ten repeats for the sides of each of the sizes. The geometrical deviations result in an uncertainty for the crater volume uncertainty of <± 3.7%.
The ablation of the GDC nanoparticles was carried out with the same optimized laser energy of 0.3 J cm−2, a round spot with a 24 μm diameter and a laser frequency of 5 Hz. An overlap of 25% of the round spots was used for the ablation, and lines of 5 mm were ablated.
Two calibration standards containing defined amounts of Gd and Ce, respectively, were ablated using single laser shots for calibration. Again, a laser energy of 0.3 J cm−2 was employed for complete ablation of the polymer layer. Round laser spots with diameters from nominally 24 μm to 60 μm were used, introducing a defined amount of Gd and Ce. Ablation with each laser spot size was repeated ten times with a delay of 5 seconds between each spot. The uncertainty of the diameters of the spots was <± 0.4 μm, resulting in a crater volume uncertainty of <± 4.2%.
2.5 Signal evaluation
The signal obtained from the ablation of individual laser spots on the calibration standards resulted in a peak of approximately 60 ms for each shot. Every peak was integrated and then averaged over the ten replicates. By considering the element concentration in the polymer, the thickness of the thin film, and the size of the laser spot, it was possible to calculate the mass of the element introduced into the ICP-MS per individual laser pulse. The mean peak area was then correlated with the mass of the element in the ablated volume, resulting in a linear calibration curve.The obtained signal structure of the measured polymer samples consisted of a background and randomly distributed spikes representing the NP embedded in the polymer matrix. The baseline signal originated from a dissolved content present in the nanoparticle suspension, while the spikes were the result of the nanoparticles themselves, which formed an ion packet within the plasma. The width of this spike peak is shorter than the time resolution of 0.5 ms for the ICP-Q-MS and 3.04 ms for the ICP-TOFMS and can thus only be estimated. To integrate the spikes in the signal, the baseline was first fitted using a median filter applied to 10 neighboring points. The threshold for peak detection was then determined by multiplying the median-fitted baseline by a factor of 5. Peaks exceeding this threshold were identified and integrated to obtain the peak areas. The diameter of the analyzed nanoparticles could be calculated by using the determined mass obtained by the previously established linear calibration curve and considering the density, stoichiometry and spherical shape of the nanoparticle material. This allowed for the determination of the nanoparticle size distribution, resulting in a range of diameters centered around the most abundant particle size.
3 Results and discussion
3.1 Optimization of the sample preparation
The method was developed by means of the analysis of 80 nm Au nanoparticles.When using a film created from PMMA solved in acetonitrile, there was a high abundance of smaller peak areas, which do not stem from the spikes in the nanoparticles in the signal but rather from the dissolved background. After applying background correction measures with 20 times the median fitted background for the threshold, the histogram shown in Fig. 1a was obtained. However, despite employing various thresholds, the histogram failed to exhibit a differentiation between background intensities and intensities stemming from the nanoparticles. Although initially expected to yield a single maximum, the applied Kernel smoothing revealed two local maxima due to the overlapping nature of the distributions. Additional measurements using acetone as an alternative solvent resulted in identical results to those using acetonitrile.
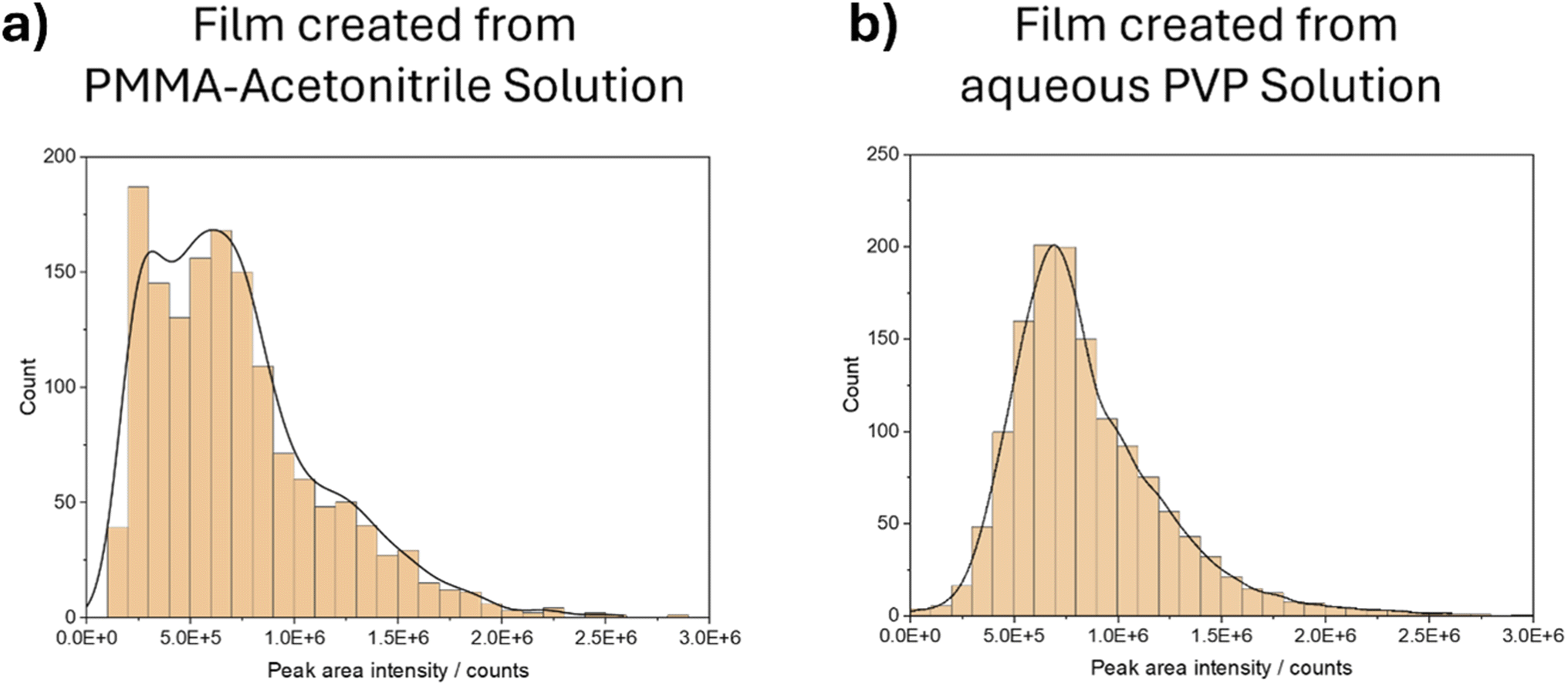 | ||
| Fig. 1 Histogram of the intensities of integrated spikes after background correction in the signal using film created from PMMA-acetonitrile-solution (a) and using film created from aqueous PVP solution (b); Kernel smoothing shown in black solid line. | ||
Using an aqueous solution of polyvinylpyrrolidone (PVP) to create the samples and standards enabled a separation between the peak areas originating from the background and those stemming from the nanoparticle spikes, resulting in the corrected intensity histogram in Fig. 1b. A complete distinction from the background peaks was possible by applying a threshold of 5 times the median fit. Moreover, this differentiation is also evident in the Kernel fitting analysis, which exhibits a single, distinct maximum.
The challenging distinguishability when using the organic solvents required for the preparation of PMMA thin films is likely attributed to the high dissolved Au content in the sample, resulting in a high background and, thus, a low signal-to-noise ratio. This phenomenon may be attributed to certain chemical changes and interactions between the Au nanoparticles and the acetonitrile and acetone, respectively.
To overcome this challenge, it is possible to reduce the crater size and, thus, the mass of the polymer matrix introduced into the ICP. This would lead to a reduced background originating from the dissolved Au species while the signals stemming from the NPs would remain constant. Reducing the side lengths of the square laser spot by half would reduce the ablated area and, thus, the signal by a factor of 4. However, when choosing an optimum for the crater size, more parameters have to be considered, as a smaller spot size also leads to a prolonged analysis time for the same sample area.
The NPs are sold in an aqueous suspension (with citrate buffer for stabilization), which suggests limited chemical interaction between the Au NPs and the water used as the suspension medium. Fabrication of PVP thin films is possible from this aqueous solution, thus, no additional solvent is required. This is confirmed by the outcomes when using an aqueous polymer solution to create the samples and standards, which leads to a reduced background, meaning a lower dissolved Au content is present. Consequently, the water-based suspension medium can be used to overcome the challenges associated with background peak areas.
3.2 Method development for Au nanoparticles
The standard and sample approach using an aqueous PVP solution was used to investigate the influence of parameters concerning the sample preparation and laser ablation analysis with the experimental conditions described in 2.4. The calibration was set in a range of 2 × 10−6 ng to 6 × 10−6 ng mass of Au with an R2 of 0.998 and a Limit of Detection (LOD) of 3 × 10−7 ng calculated according to the calibration approach using 3.3 times the standard error of the regression divided by the slope. This translates to a particle size of approximately 15.5 nm, which is in accordance with the literature values reporting size LODs for Au nanoparticles of 11 to 20 nm using liquid-suspension-based approaches on comparable hardware.26,27Different dilutions of nanoparticles in aqueous PVP solution were created by diluting the originally created PVP suspension 1:10, 1:20 and 1:50, resulting in four different concentrations. The signals for the analysis of the originally created PVP suspension and the 1:10 dilution are shown in Fig. 2.
 | ||
| Fig. 2 Raw signals of an Au nanoparticle measurement showing the transient signal detected on m/z = 197 containing the spikes stemming from the individual nanoparticles for the originally created PVP suspension (a) and the 1:10 dilution of it (b). | ||
The signal for the different dilutions showed isolated spikes with similar intensities corresponding to the individual nanoparticles being introduced into the ICP-MS, suggesting that the particles on the substrate were non-agglomerated. As expected, the frequency of the spikes correlated with the dilution and, thus, the number of nanoparticles in the investigated area.
In identical timespans, the number of detected spikes of the 1:10 dilution with n = 19 is approximately one-tenth of the number of spikes of the original PVP suspension with n = 168.
By applying the previously determined appropriate calibration function, it was possible to determine the mass distribution of the nanoparticles based on the signal obtained from the ICP-MS analysis. By assuming a spherical shape, it could subsequently be transformed into the diameter distribution (Fig. 3).
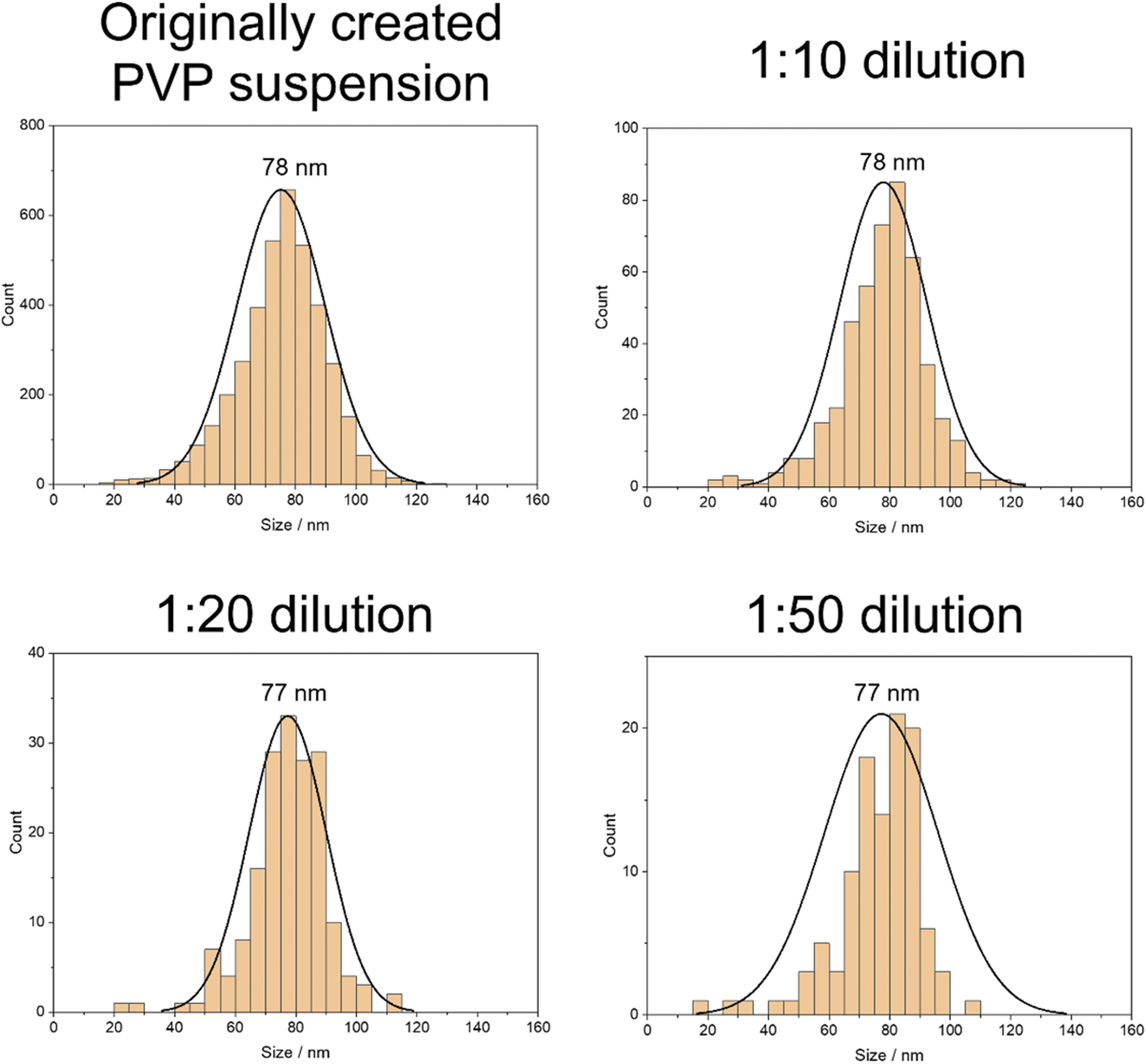 | ||
| Fig. 3 Histograms of the calculated average diameters of the nanoparticles in aqueous PVP solution in different concentrations, including their standard distribution curves. | ||
The investigated size of the nanoparticles did not show any significant variation with the dilution factor. This outcome aligns with expectations and confirms that non-agglomerated nanoparticles were successfully analyzed in all concentrations.
Moreover, no significant differences in the determined size of the nanoparticles in regard to the position on the sample were observed.
Since the same area on the sample was ablated for all the concentrations, the count of the particles decreased linearly with the increased dilution and, thus, lower nanoparticle concentration. Consequently, an increased sample area would need to be analyzed for diluted samples to achieve robust counting statistics.
However, this outcome underscores the importance of finding a compromise between obtaining a sufficient number of particles for statistical significance and avoiding agglomeration or other issues, such as the detection of double events that may arise with higher concentrations. Moreover, a higher concentration and, thus, a higher number of particles per area allows short acquisition times, as more particles are ablated in the same time span.
For a sample with a given NP concentration, this could be obtained by adjusting the sample area investigated per laser shot. Increasing or decreasing the employed laser spot size results in an enhanced or reduced number of nanoparticles introduced into the ICP-MS without altering the size determination. It can thus be considered as an alternative to conventional “diluting” or “concentrating” of the nanoparticle content of the sample and can be accomplished without further sample treatment. In contrast, when using the conventional liquid suspension approach, the sample has to be diluted again if it is too concentrated. However, problems related to the agglomeration of NP could not be solved with this approach.
The investigation was carried out for spot sizes between 10 μm × 10 μm and 60 μm × 60 μm for a sample with identical nanoparticle concentration. Since the number of laser shots was kept constant for all experiments, a smaller spot size corresponds to a smaller ablated area and, consequently, a reduced number of detected nanoparticles. Fig. 4 illustrates the size histograms corrected for double and multi-events in orange overlayed on the non-corrected histograms in blue. The double events were detected based on the integrated peaks in the transient signal, removing peaks with areas that are double or higher than the maximum of the peak area distribution.
 | ||
| Fig. 4 Overlayed histograms (orange: corrected for double and multi-events; blue: non-corrected) including normal distribution curves of the calculated average diameters of the nanoparticles in aqueous PVP solution ablated with different laser spot sizes. | ||
For spot sizes ranging from 10 μm × 10 μm to 30 μm × 30 μm, no observable changes in the form and maximum of the size distributions appear, suggesting that the distribution of the particles is sufficiently sparse for measurements using 30 μm × 30 μm without considerable occurrences of multiple particle events. This is an advantage as it allows for faster scanning of larger areas by using larger spot sizes.
However, with a spot size of 60 μm × 60 μm, the uncorrected size distribution shows a considerable amount of apparently larger particles, which results in a shift towards a higher average size of the blue histogram. This shift is caused by the increased abundance of coinciding nanoparticles when two or more particles are introduced into the ICP-MS simultaneously and thus appear as one larger particle. The inference of two coinciding 80 nm Au nanoparticles results in the detection of one particle with an estimated size of 101 nm, as the Au mass is identical for both cases. Consequently, a higher abundance of particles with an apparent size of 100 to 105 is detected, as indicated by the blue size histogram, whereas the orange one shows the size distribution after correction for double and multi-events.
This finding indicates that the likelihood of detecting two particles as a single larger particle increases when a certain number of nanoparticles are ablated simultaneously. In such cases, it becomes necessary to either dilute the sample or decrease the spot size to mitigate this interference and ensure accurate detection and analysis. The results highlight the importance of carefully selecting the laser spot size and considering the potential for interference when interpreting the size distributions of nanoparticles.
3.3 Application for the evaluation of GDC nanoparticles
Likewise, to the determination of the Au nanoparticles, the signal for the Ce and Gd isotopes monitored in the experiments performed with GDC showed spikes in the signal corresponding to the particles being ablated and introduced sequentially.Additionally, the use of ICP-TOFMS allowed for the identification of potential contaminations in the substrate and polymer. Analysis of intensity across the entire mass range revealed that only sodium was present as a contaminant. However, the background on the elements of interest in the substrate and polymer was under the limit of detection. This indicates a high purity of the silicon wafer substrate, PVP, and solvent. The detected contamination with sodium could either be inherently present in one of the materials or introduced during handling. Furthermore, besides sodium, the sample preparation, including spin coating, did not introduce any additional contamination.
The calibration was set in a range of 2 × 10−6 ng to 8 × 10−6 ng mass of introduced Ce and a mass of 2 × 10−6 ng to 6 × 10−6 ng for Gd. The Ce calibration had an R2 of 0.999, while the calibration for Gd had an R2 of 0.993. The LODs calculated using the calibration approach as described in 3.2 were at 2 × 10−7 ng for Ce and 5 × 10−7 ng for Gd.
The simultaneous monitoring of the two elements in GDC allowed the concurrent determination of the size and composition of the individual nanoparticles. The results indicated a close similarity between both isotopes of the two respective elements that were evaluated. The more abundant isotope was chosen for the determination because of the higher sensitivity. Fig. 5 shows the results for the size distribution. The histogram in Fig. 5a displays the distribution when the particle diameter is evaluated using 158Gd, while the histogram in Fig. 5b shows the size distribution when evaluated with 140Ce. In both cases, a stoichiometry of Ce0.8Gd0.2O1.95, meaning 80 mol% Ce and 20 mol%, was assumed. This makes it a de facto single-element analysis where the size of the multi-element nanoparticle is determined via extrapolation. The blue histograms show the results without correction for double and multi-events, while the orange histograms show the corrected results.
 | ||
| Fig. 5 Histograms of the calculated average diameters of the GDC nanoparticles using 158Gd (a) and 140Ce (b) for the size evaluation, with blue being the uncorrected results and orange the corrected results. | ||
The histograms for nanoparticle diameter in Fig. 5 exhibit a wide distribution with a broad peak ranging from 80 to 140 nm. A slight difference in mean particle size between the two elements used for the evaluation occurs, with Gd (Fig. 5a) at 90 to 140 nm and Ce (Fig. 5b) showing the maximum at 80 to 130 nm. The comparison between corrected and uncorrected results indicates that even though double and multi-events do appear, the position of the histogram maximum remains unchanged. A triple event would show as a particle with a diameter of approx. 180 nm, which is within the uncorrected histogram, making a definitive distinction between the size histogram and the multi-events not feasible. The progression and form of the distribution, however, contradicts triple-events.
As the vital advantage of the ICP-TOF-MS is the quasi-simultaneous analysis of more than one element, the molar portions of both of the elements of interest in the individual nanoparticles could be determined and are given in histograms in Fig. 6.
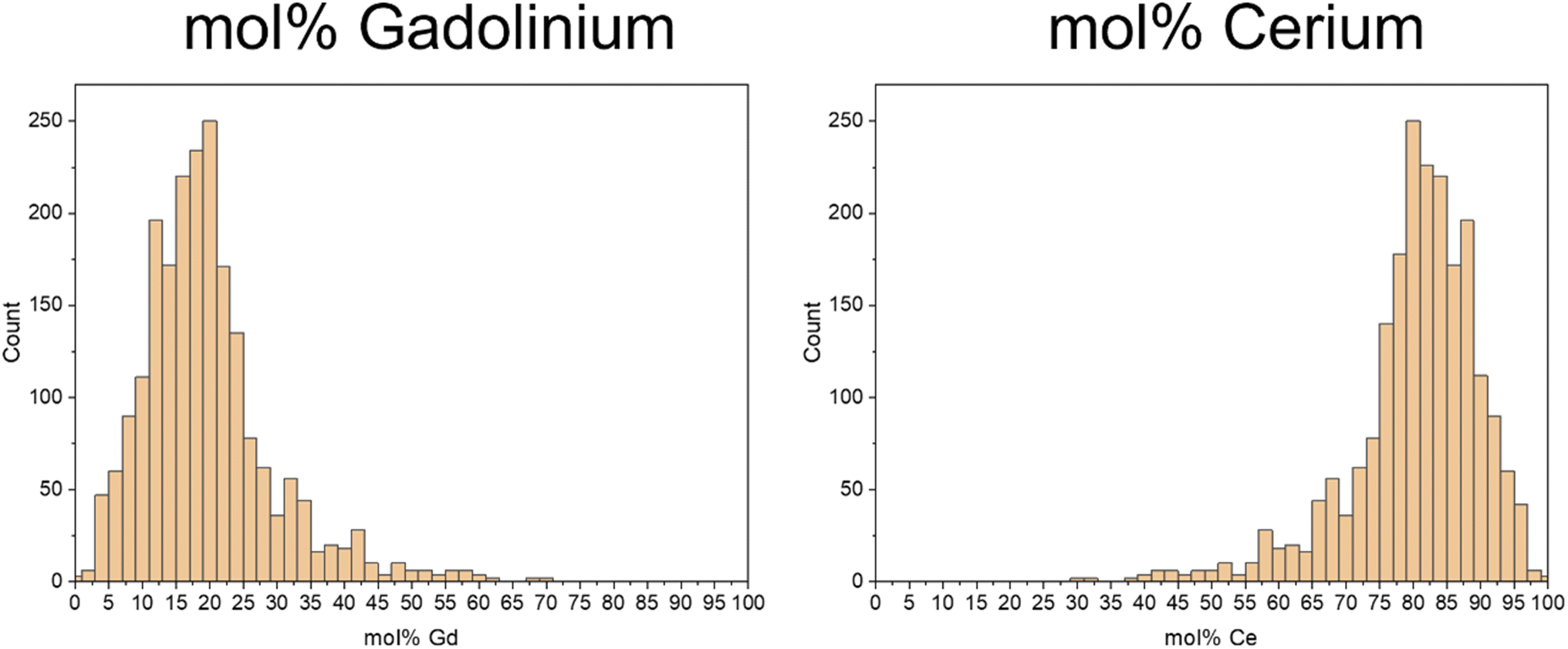 | ||
| Fig. 6 Histograms of mol% of Gd (a) and Ce (b) in the individual analyzed nanoparticles. | ||
In the composition of the individual nanoparticles, the Gd results (Fig. 6a) exhibit a distribution with a broad maximum of 80 to 85 mol%, while for Ce (Fig. 6b), the distribution shows a wide maximum of 15 to 20 mol%.
The operating principle of the TOF mass analyzer also allows the calculation of the masses of the two elements and combining them for the calculation of each individual particle. This results in the histogram given in Fig. 7, which shows the broad distribution between 80 and 140 nm again and demonstrates that with an ICP-TOF-MS, the size determination is possible even without the known molar concentration, however, it is necessary to know the density of the “mixture material”.
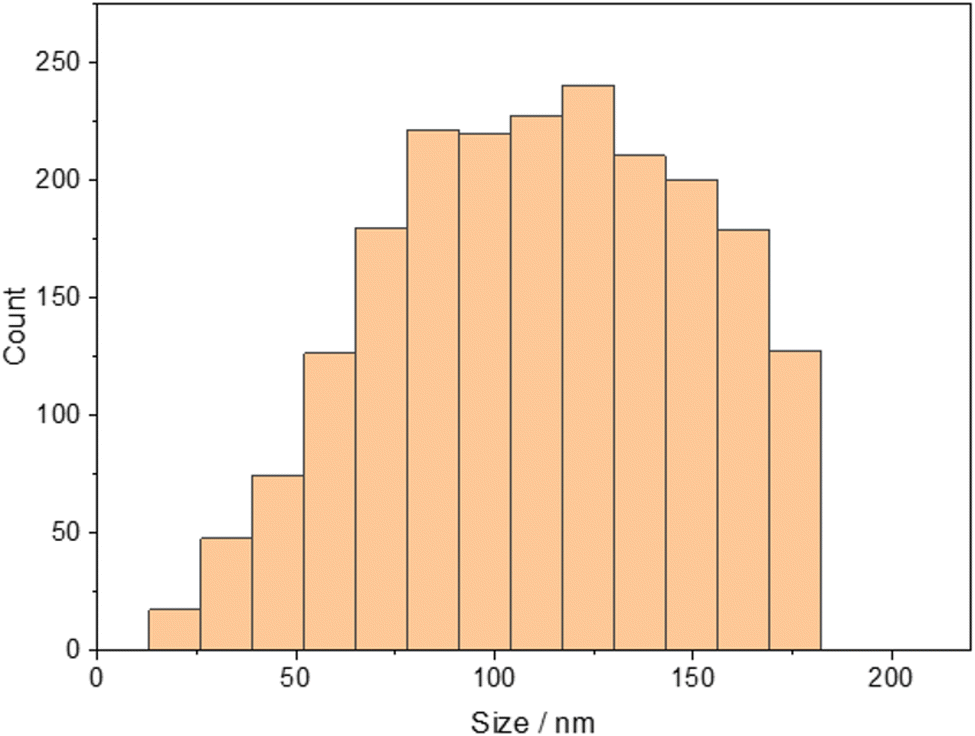 | ||
| Fig. 7 Histogram of the calculated average diameters of the GDC nanoparticles using the combined mass of 158Gd and 140Ce for the size evaluation. | ||
As no reference data regarding the size distribution and molar composition is available, comparative measurements with other methods were conducted. To validate and compare the results, the nanoparticles' size distribution was analyzed in an aqueous suspension with a concentration of 1.5 × 107 particles per mL using a nanoparticle tracking analyzer, revealing a maximum of 121 nm and a standard deviation of ±52 nm. These findings align with the LA-ICP-MS analysis, depicted in Fig. 5.
The composition of the individual nanoparticles was validated by analyzing a liquid suspension of the GDC nanoparticles using ICP-OES. The concentration of the suspension (∼1010 particles per mL) was chosen in a way that a continuous signal could be registered during analysis, which was then calibrated using liquid standards of Gd and Ce. This revealed proportions of 22 ± 3.3 mol% Gd and 78 ± 2.0 mol% Ce in the nanoparticle sample, closely matching the nominal concentrations of 20 mol% Gd and 80 mol% Ce.
This result can be regarded as the verification that the distribution of the molar percentages of the individual nanoparticles can indeed be determined using the proposed LA-sp-ICP-MS procedure.
4 Conclusion
This study presents an efficient and stable calibration and measurement approach developed for the analysis of nanoparticles with sp-ICP-MS using laser ablation for sampling that allows the determination of particle sizes and size distributions.Our approach involved the preparation of polymer thin films with a defined added concentration of the target element using spin coating, allowing for the introduction of precise amounts of the elements into the plasma through laser ablation. By correlating the known amount of the introduced element with the corresponding signal, a calibration could be established. The nanoparticle samples were also prepared in polymer thin films by adding nanoparticles to the polymer solution and subsequent spin coating. The signals stemming from the nanoparticles can be used to determine the corresponding mass of the element using the calibration function based on solution-doped polymer samples with the same matrix. Subsequently, the diameter can be calculated using information about the density, stoichiometry and spherical geometry of the particles.
The procedure was developed and verified using Au nanoparticles with well-defined sizes and allowed accurate determination of the sizes without the need for certified particle reference materials using a quadrupole-ICP-MS. For single-element NPs, Q-ICP-MS delivers accurate results as no problems with the sequential nature of the mass analyzer appear when monitoring only one m/z ratio. We also demonstrated the impact of dilution on the sampled number of particles for a measurement, as well as the influence of the laser spot size and the potential for double events. The approach was successfully transferred to ICP-TOFMS and applied to the technical material GDC in the form of nanoparticles ranging from 100 to 150 nm. Apart from nanoparticle sizing, the methodology allowed the determination of the elemental composition of the individual particles as the ICP-TOFMS enabled simultaneous analysis of a multitude of isotopes.
The achieved size LOD value for Au nanoparticles is in accordance with conventional approaches discussed in the literature, which indicates that the laser ablation approach transports the NP intact into the plasma and, therefore, processes between solution nebulization and LA transport are identical. The developed approach employing laser ablation sampling has the advantage of straightforwardly adjusting laser spot sizes to increase or decrease the number of sampled nanoparticles and to adjust the NP signal to the matrix background. Furthermore, the created nanoparticle samples can be stored over a long period of time, are reusable and would allow the use of any additional analysis technique to be applied together with LA-ICP-MS measurements.
Data availability
The data supporting the findings of this study are available within the article. Other relevant data can be shared upon reasonable request.Author contributions
Laura Kronlachner: conceptualization, data curation, investigation, methodology, writing – original draft, visualization, validation; Zuzana Gajarska: software, visualization, data curation, investigation; Pascal Becker: conceptualization, investigation, writing – review & editing; Detlef Günther: conceptualization, methodology, resources, supervision, writing – review & editing; Andreas Limbeck: conceptualization, funding acquisition, methodology, project administration, resources, supervision, writing – review & editing.Conflicts of interest
The authors declare that they have no conflicts of interest.Acknowledgements
The authors acknowledge TU Wien Bibliothek for financial support through its Open Access Funding Program and the “KUWI Stipendium” of the TU Wien for the financial support for academic research abroad.References
- W. J. Stark, P. R. Stoessel, W. Wohlleben and A. Hafner, Chem. Soc. Rev., 2015, 44, 5793–5805 RSC.
- D. Astruc, Nanoparticles and Catalysis, John Wiley & Sons, 2008 Search PubMed.
- J. Lead and E. Smith, Environmental and Human Health Impacts of Nanotechnology, 2009 Search PubMed.
- A. Scheffer, C. Engelhard, M. Sperling and W. Buscher, Anal. Bioanal. Chem., 2008, 390, 249–252 CrossRef CAS PubMed.
- L. Dykman and N. Khlebtsov, Chem. Soc. Rev., 2012, 41, 2256–2282 RSC.
- O. V Salata, J. Nanobiotechnol., 2004, 6, 1–6 Search PubMed.
- M. De, P. S. Ghosh and V. M. Rotello, Adv. Mater., 2008, 20, 4225–4241 CrossRef CAS.
- M. Das, N. Saxena and P. D. Dwivedi, Nanotoxicology, 2009, 3, 10–18 CrossRef CAS.
- I. Ijaz, E. Gilani, A. Nazir and A. Bukhari, Green Chem. Lett. Rev., 2020, 13, 59–81 Search PubMed.
- D. Mozhayeva and C. Engelhard, J. Anal. At. Spectrom., 2020, 35, 1740–1783 RSC.
- V. Srivastava, D. Gusain and Y. C. Sharma, Ind. Eng. Chem. Res., 2015, 54, 6209–6233 CrossRef CAS.
- M. Hassellöv, J. W. Readman, J. F. Ranville and K. Tiede, Ecotoxicology, 2008, 17, 344–361 CrossRef PubMed.
- D. Mavrocordatos, W. Pronk and M. Boller, Water Sci. Technol., 2004, 50, 9–18 CrossRef CAS PubMed.
- D. Mavrocordatos, D. Perret and G. G. Leppard, IUPAC Ser. Anal. Phys. Chem. Environ. Syst., 2007, 10, 345 CAS.
- F. Laborda, E. Bolea and J. Jiménez-Lamana, Trends Environ. Anal. Chem., 2016, 9, 15–23 CrossRef CAS.
- I. Benešová, K. Dlabková, F. Zelenák, T. Vaculovič, V. Kanický and J. Preisler, Anal. Chem., 2016, 88, 2576–2582 CrossRef PubMed.
- D. Metarapi, M. Šala, K. Vogel-Mikuš, V. S. Šelih and J. T. Van Elteren, Anal. Chem., 2019, 91, 6200–6205 CrossRef CAS PubMed.
- C. C. Garcia, H. Lindner and K. Niemax, Spectrochim. Acta, Part B, 2007, 62, 13–19 CrossRef.
- D. Guenther and B. Hattendorf, TrAC, Trends Anal. Chem., 2005, 24, 255–265 CrossRef CAS.
- R. E. Russo, X. Mao, J. J. Gonzalez, V. Zorba and J. Yoo, Anal. Chem., 2013, 85, 6162–6177 CrossRef CAS PubMed.
- D. Günther, A. V. Quadt, R. Wirz, H. Cousin and V. J. Dietrich, Mikrochim. Acta, 2001, 136, 101–107 CrossRef.
- S. Yamashita, Y. Yoshikuni, H. Obayashi, T. Suzuki, D. Green and T. Hirata, Anal. Chem., 2019, 91, 4544–4551 CrossRef CAS PubMed.
- C. Neff, P. Becker and D. Günther, J. Anal. At. Spectrom., 2022, 37, 677–683 RSC.
- P. Becker, J. Koch and D. Günther, J. Anal. At. Spectrom., 2022, 37, 1846–1854 RSC.
- D. Metarapi, J. T. Van Elteren and M. Šala, J. Anal. At. Spectrom., 2021, 36, 1879–1883 RSC.
- S. Lee, X. Bi, R. B. Reed, J. F. Ranville, P. Herckes and P. Westerhoff, Environ. Sci. Technol., 2014, 48, 10291–10300 CrossRef CAS PubMed.
- F. Laborda, E. Bolea and J. Jimenez-Lamana, Anal. Chem., 2014, 86, 2270–2278 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |