
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Deactivation mechanisms of Cu–Zn–Al2O3 in CO2 hydrogenation induced by SO2 exposure†
Xuan
Bie
a,
Ruoyu
Wu
a,
Bocheng
Yu
a,
Xuelong
Quan
a,
Shiyu
Zhang
a,
Qinghai
Li
ab,
Yanguo
Zhang
ab and
Hui
Zhou
 *ab
*ab
aKey Laboratory for Thermal Science and Power Engineering of Ministry of Education, Beijing Key Laboratory of CO2 Utilization and Reduction Technology, Department of Energy and Power Engineering, Tsinghua University, Beijing 100084, P.R. China. E-mail: huizhou@tsinghua.edu.cn
bShanxi Research Institute for Clean Energy, Tsinghua University, Shanxi, Taiyuan 030000, P.R. China
First published on 27th May 2025
Abstract
The presence of sulfur compounds, particularly SO2, is known to significantly degrade the performance of metal-based catalysts, posing a significant challenge in CO2 hydrogenation reactions. In this study, we systematically investigate the impact of SO2 on Cu–ZnO–Al2O3 catalysts for CO2 hydrogenation to elucidate the deactivation mechanisms. Our findings reveal that SO2 adsorption leads to the formation of surface sulfate and sulfite species, which effectively block active sites, impeding the adsorption and activation of reactants. Moreover, SO2 exposure inhibits CO desorption, further compromising catalytic efficiency. In parallel, progressive sulfidation of Cu and ZnO results in the formation of catalytically inactive CuS, Cu2S, and ZnS phases, ultimately leading to complete catalyst deactivation. These results highlight the dual role of sulfur species in both surface passivation via sulfates/sulfites deposition and irreversible structural transformation via sulfidation. Our study provides new insights into the SO2-induced catalyst deactivation in CO2 hydrogenation and offers a theoretical foundation for enhancing CO2 hydrogenation reactions, with implications for optimizing environmentally sustainable catalytic systems in industrial applications.
Keywords: CO2 hydrogenation; The role of SO2; Deactivation; Phase transition; RWGS.
1 Introduction
The increasing global concern over greenhouse gas emissions has led to significant interest in technologies aimed at reducing CO2 concentrations in the atmosphere.1–3 CO2 hydrogenation has emerged as a promising pathway for the utilization of CO2, enabling its conversion into valuable chemicals such as CO, methanol, and other hydrocarbons.4–8 These processes not only offer a sustainable route to produce fuels and chemicals but also contribute to reducing the overall CO2 footprint.9,10 However, the practical implementation of CO2 hydrogenation, particularly in industrial settings, faces several challenges due to the presence of impurities in feed gases, including sulfur-containing compounds such as sulfur dioxide (SO2).11–13SO2 is a major contaminant commonly found in flue gases from coal-fired power plants, especially those utilizing pulverized coal combustion.14,15 The flue gas typically contains a mixture of gases such as CO2, nitrogen oxides (NOx), and SO2, which can have detrimental effects on catalytic processes.14,16,17 In CO2 hydrogenation, the presence of SO2 is particularly concerning, as it is known to poison metal catalysts by binding to active sites and forming inactive sulfur compounds, leading to catalyst deactivation.18–20 Understanding the impact of SO2 on catalyst performance is critical for optimizing CO2 hydrogenation processes, especially when considering the integration of these processes with industrial CO2 capture systems, where SO2 contamination is inevitable.
Previous studies have highlighted the negative impact of sulfur compounds on various catalytic systems, including those based on transition metals such as Cu, Ni, and Fe.18,21–24 For the Cu-SSZ-13 catalyst, SO2 poisoning under standard NH3-SCR conditions resulted in mostly ammonium sulfate formation at 200 °C, whereas copper sulfates were predominant after poisoning at 400 °C under the conditions of 30 ppm SO2, 8% O2, 5% H2O, 400 ppm NH3 and 400 ppm NO.25 In the integrated CO2 capture and utilization (ICCU) process, exposure to SO2 leads to the formation of thermally stable CaSO4 product which, in turn, drastically reduces CO2 capture capacity due to increased mass transfer resistance.26 Despite these findings, while the negative impact of SO2 on catalytic performance is well-established, comprehensive studies that investigate the specific mechanisms by which SO2 affects catalytic activity in CO2 hydrogenation remain limited.
This study aims to systematically investigate the influence of SO2, derived from pulverized coal boiler flue gas, on the performance of industrial catalyst Cu–ZnO–Al2O3 catalysts during CO2 hydrogenation. By combining advanced characterization techniques such as X-ray photoelectron spectroscopy (XPS), scanning transmission electron microscopy (STEM), operando diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS), and operando X-ray diffraction (XRD), we aim to elucidate the poisoning and deactivation mechanisms of SO2 on the catalyst. The results reveal that exposure to SO2 induces irreversible catalyst deactivation, with both Cu and Zn species fully converted into metal sulfides (Cu2S, CuS, and ZnS). Additionally, sulfide and sulfate species accumulate on the catalyst surface. These findings offer valuable insights into the mechanisms driving catalyst deactivation, paving the way for strategies to enhance the long-term stability and performance of CO2 hydrogenation processes under industrial conditions.
2 Results and discussion
2.1 Catalytic performance
The CO2 hydrogenation was conducted in a quartz tube reactor. The Cu–ZnO–Al2O3 catalyst demonstrated high catalytic performance over a temperature range of 300–700 °C with a H2/CO2 ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (Fig. 1a). However, the introduction of SO2 into the reaction stream induced a significant decrease in catalytic activity. At 300 °C, CO2 conversion decreased from 4.1% to 0.16%, while CO selectivity slightly increased from 91.1% to 94.0% upon addition of 0.1% SO2 (Fig. 1a and b). As the SO2 concentration increased to 0.5% and 2.0%, the catalyst underwent complete deactivation (Fig. 1c), suggesting that higher SO2 concentrations accelerate catalyst deactivation.
:1 (Fig. 1a). However, the introduction of SO2 into the reaction stream induced a significant decrease in catalytic activity. At 300 °C, CO2 conversion decreased from 4.1% to 0.16%, while CO selectivity slightly increased from 91.1% to 94.0% upon addition of 0.1% SO2 (Fig. 1a and b). As the SO2 concentration increased to 0.5% and 2.0%, the catalyst underwent complete deactivation (Fig. 1c), suggesting that higher SO2 concentrations accelerate catalyst deactivation.
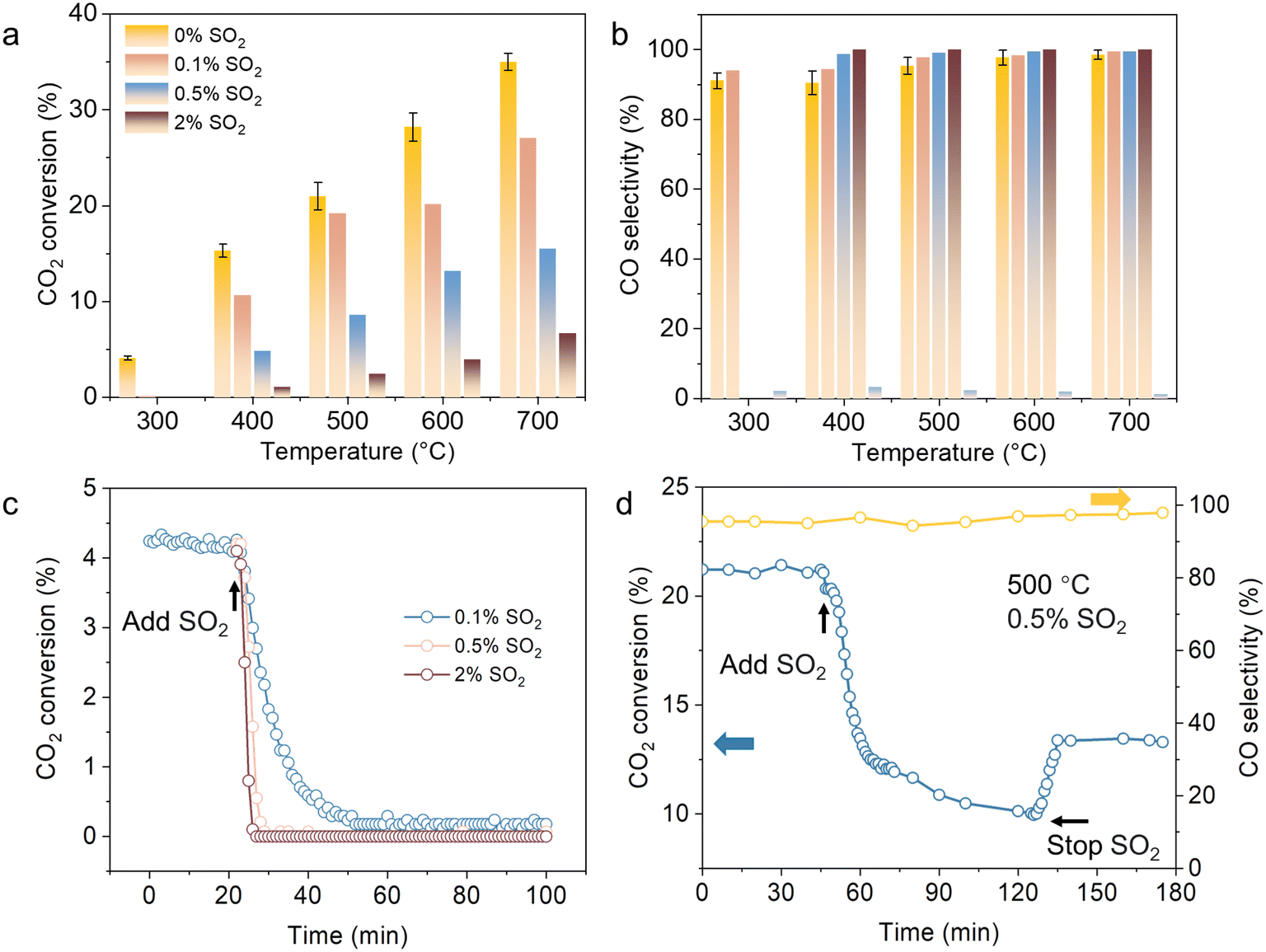 | ||
| Fig. 1 Role of SO2 on the catalyst performance. (a) CO2 conversion and (b) CO selectivity over Cu–ZnO–Al2O3; (c) CO2 conversion under different SO2 concentrations over Cu–ZnO–Al2O3 at 300 °C; (d) changes in CO2 conversion and CO selectivity when switching the gas composition (reaction conditions: 1 bar, H2:CO2:N2 = 1:1:1, contact time: 0.05 s g mL−1, WHSV: 72000 cm3 g−1 h−1). Error bars were obtained from three repeated measurements. | ||
Higher temperatures exacerbated the deactivation of the catalyst. At 400 °C, CO2 conversion decreased from 15.3% to 10.7% with the introduction of 0.1% SO2, while CO selectivity increased from 91.5% to 94.3%. Increasing the SO2 concentration to 0.5% further decreased CO2 conversion to 4.9%, whereas CO selectivity was enhanced to 98.7%. These results suggest that SO2 exposure primarily affects the formation of CH4, with a more pronounced impact on CO2 conversion and a shift in product distribution towards higher CO selectivity.
The CO2 hydrogenation catalyst underwent significant deactivation upon SO2 exposure, indicating partial irreversibility (Fig. 1d). At 500 °C and 0.5% SO2, the CO2 conversion initially decreased from 21.2% to 12.6%, while CO selectivity increased slightly from 95.3% to 96.7%. Furthermore, CO2 conversion continued to decrease rather than stabilize at 12.6%. After 90 min of testing, the CO2 conversion remained at 9.95%, while CO selectivity was 97.1%. Upon removal of SO2 from the reaction stream, CO2 conversion partially recovered to 13.4%, while CO selectivity remained around 97.3%. These results suggest that the deactivation is not fully reversible, implying partial permanent deterioration of the catalyst.
The experiment was extended until complete catalyst deactivation occurred (Fig. 2). Under SO2-free conditions, CO2 conversion remained stable at 21.3% throughout the 400 min. In contrast, under 0.5% SO2 conditions, CO2 conversion steadily declined until full deactivation was observed (Fig. 2a). After the cessation of SO2 flow, no recovery in CO2 conversion was observed, indicating that deactivation was caused by irreversible poisoning. Regarding CO selectivity, it remained constant at 95.8% throughout the reaction under SO2-free conditions (Fig. 2b). However, under 0.5% SO2, CO selectivity initially increased slightly for the first 150 min, reaching a maximum of 99.5%. This increase was attributed to the rapid deactivation of CH4 formation. Once SO2 was introduced, the CH4 formation rate decreased to 1.7 mmol h−1 gcat−1 and dropped below 1 mmol h−1 gcat−1 after 150 min (Fig. 2c). Subsequently, CO selectivity sharply declined to 65.3% due to the significant reduction in CO formation rate. The CO formation rate decreased from 657.8 to 212.8 mmol h−1 gcat−1 after 150 min, and the continuous decline in CO formation contributed to the decline of the CO selectivity (Fig. 2d).
 | ||
| Fig. 2 SO2-induced deactivation of the catalyst during CO2 hydrogenation. Changes in (a) CO2 conversion, (b) CO selectivity, (c) CH4 formation rate, and (d) CO formation rate when switching the gas composition from H2/CO2 to H2/CO2 with 0.5% SO2 (reaction conditions: 500 °C, 1 bar, H2:CO2:N2 = 1:1:1, contact time: 0.05 s g mL−1, WHSV: 72000 cm3 g−1 h−1). | ||
2.2 Deactivation mechanisms caused by SO2
 | ||
| Fig. 3 Characterization of fresh and used Cu–ZnO–Al2O3 catalysts. (a) Cu LMM Auger spectra; (b) Zn LMM Auger spectra; (c) XPS spectra of S 2p. CuZnAlSO2-free: Cu–ZnO–Al2O3 exposed to H2/CO2/N2 (1:1:1) for 400 min at 500 °C. CuZnAl0.5%SO2: Cu–ZnO–Al2O3 exposed to H2/CO2/N2 (1:1:1) with 0.5% SO2 for 400 min at 500 °C. | ||
Cu LMM Auger spectra were further analyzed to distinguish between Cu0 and Cu+ species (Fig. 3a). For the fresh catalyst, the Cu LMM spectra exhibited only Cu0 (568.3 eV) species, with no evidence of Cu+ (570.3 eV).28 For the used catalysts under SO2-free conditions and under 0.5% SO2 conditions, both Cu0 (568.3 eV) and Cu+ (570.3 eV) species were present. We calculated the Cu+/(Cu0 + Cu+) ratio and found that for the used catalyst under SO2-free conditions, Cu+ constituted 18% of the surface copper (Table S1†). In contrast, for the used catalyst under 0.5% SO2 conditions, Cu+ constituted 58% of the surface copper. This indicates that SO2 promoted the oxidation of metallic copper to Cu+ state.
The Zn 2p spectra exhibited two peaks at binding energies of 1045.3 eV and 1022.2 eV, indicating that zinc predominantly exists in the Zn2+ state (Fig. S2†).29 Further analysis of the Zn LMM spectra also revealed the presence of both Zn2+ and Znδ+ species (Fig. 3b). All catalysts showed two peaks at 499.5 eV and 496.0 eV, indicating the presence of Zn2+ and Znδ+ species on the catalysts, respectively.30 The ratios of Znδ+/(Znδ+ + Zn2+) were calculated to be 37%, 37%, and 33% for the fresh catalyst, and for the used catalysts under SO2-free and 0.5% SO2 conditions, respectively (Table S2†). These results suggest that both fresh and used catalysts maintain similar amount of Znδ+, indicating that zinc remains predominantly in its oxidized state.
The S 2p spectra revealed the presence of sulfide (S 2p1/2 at 163.0 eV and S 2p3/2 at 162.1 eV), sulfates (at 169.2 eV) and sulfites (at 168.0 eV) species (Fig. 3c).31,32 Notably, the content of sulfides was 67%, suggesting the majority of sulfur species on the surface of the used catalyst under 0.5% SO2 condition were in the form of metal sulfides. Additionally, the Al 2p spectra displayed consistent signals across all three catalysts, indicating the stability of aluminum within the catalyst (Fig. S3†). The peak at 75.4 eV corresponded to Al 2p, while the shoulder peak at 78.1 eV was attributed to Cu 3p.33–35
Transmission electron microscopy (TEM) was used to investigate the changes in catalyst morphology (Fig. 4). The fresh catalyst exhibited a uniform distribution of Cu and ZnO nanoparticles, suggesting good dispersion and potential for high catalytic activity (Fig. 4a and S4†). For the catalyst used under SO2-free conditions, no significant changes in particle size or morphology were observed, indicating that the catalyst remained stable under these conditions (Fig. 4b and S4†). However, upon exposure to 0.5% SO2, sulfur was detected on the catalyst surface, indicating sulfur-induced sintering or poisoning effects (Fig. 4c and S4†). The observed changes in sulfur deposition could be linked to the decrease in catalytic activity under SO2 exposure, as the sulfur species may interact with active sites and hinder CO2 hydrogenation.
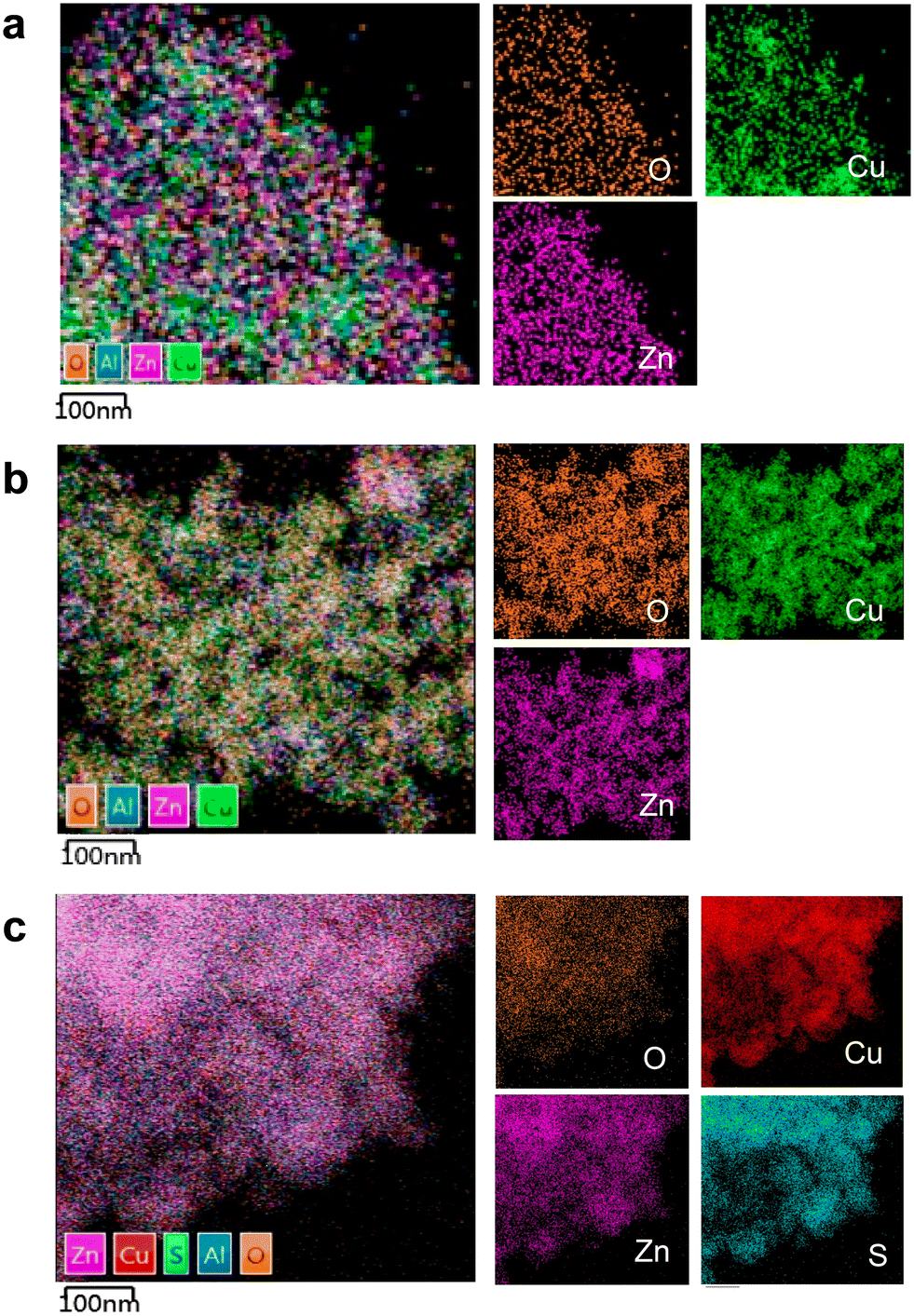 | ||
| Fig. 4 STEM analysis and EDX mapping of Cu–ZnO–Al2O3. (a) Fresh catalyst; (b) the used catalyst exposed to H2/CO2/N2 (1:1:1) for 400 min at 500 °C; (c) the used catalyst exposed to H2/CO2/N2 (1:1:1) with 0.5% SO2 for 400 min at 500 °C. | ||
:1:1 ratio, was then introduced to interact with the reduced catalyst at 500 °C. The bands observed at 1315 and 1455 cm−1 were attributed to carbonate species on the Cu–ZnOx interface,36–38 while no bands in the 3000–2400 cm−1 range were detected, indicating no formate species exist (Fig. 5a and S5†). Gas-phase CO was also identified with bands at 2177 and 2114 cm−1.37,39 During this process, the CO2 conversion maintains at 27.5%, with the 100% CO selectivity (Fig. S5†).
 | ||
| Fig. 5
Operando DRIFT spectra over the Cu–ZnO–Al2O3 catalyst under different feed gas compositions. The catalyst was first exposed to (a) CO2/H2/N2 (1:1:1) for 60 min, followed by (b) switching the gas flow to CO2/H2/N2 (1:1:1) with 0.5% SO2. Reaction condition: 500 °C, 1 bar, and 30 mL min−1, 0.5% SO2. | ||
Subsequently, the gas flow was switched to a 1:1 mixture of H2 and CO2 with 0.5% SO2, and the reaction was monitored for 180 min (Fig. 5b). After 180 min, CO2 conversion dropped from 27.5% to lower than 0.1%, indicating complete catalyst deactivation (Fig. S5†). At this point, the gas-phase CO bands (2177 and 2114 cm−1) disappeared, suggesting a significant reduction in CO formation. Additionally, a new band at 2077 cm−1 emerged, which is characteristic of carbonyl species,40 indicating these species are not desorbing into the gas phase as CO. These carbonyl species are strongly adsorbed on the catalyst surface, likely due to strong interaction with active sites, making them difficult to desorb.
Moreover, the DRIFT spectra revealed the formation of new bands at 1401 and 1636 cm−1, which are assigned to sulfate species, while the band at 1501 cm−1 is characteristic of sulfite species, respectively.41–43 These findings indicate that SO2 is strongly adsorbed onto the catalyst surface, predominantly in the form of sulfate and sulfite. The strong adsorption of these species leads to the occupation and blockage of the active sites, preventing the adsorption of reactants and ultimately inhibiting catalytic activity.11,40,41 Consequently, the accumulation of these sulfur compounds accelerates catalyst deactivation, further exacerbating the decline in catalytic performance over time. Notably, the absence of characteristic bands in the 1211–1119 cm−1 region, which are typically associated with bulk sulfate species,43–46 suggests that the sulfates formed during SO2 exposure remain confined to the catalyst surface rather than incorporating into the bulk structure.
 | ||
| Fig. 6 XRD of the fresh and used catalysts with or without 0.5% SO2. (a) XRD in the 2θ range of 25–80°; (b) XRD in the 2θ range of 46–49°; (c) XRD in the 2θ range of 55–58°. | ||
For the used catalyst under 0.5% SO2 conditions, the peaks corresponding to metallic Cu disappeared, and new peaks at 26.6°, 30.8°, 44.1°, and 52.3° emerged, which were attributed to Cu2S (JCPDS 75-2241).53,54 New peaks at 27.9°, 47.5°, and 56.4° emerged, indicating the formation of either ZnS or CuS (JCPDS 01-0792 and 89-2073).55,56 These peaks were located at similar positions, making it difficult to distinguish between CuS and ZnS. Moreover, it was not possible to unequivocally assign the peaks at 47.5° and 56.4° to either newly formed metal sulfides or to ZnO (Fig. 6b and c). To clarify these ambiguities, operando XRD measurements were conducted. Moreover, the crystallite size for Cu2S was 49.5 nm, overlapping peaks for CuS and ZnS suggest a composite crystallite size of 67.9 nm, though this value should be interpreted cautiously due to peak overlap. The growth of the particle sizes indicates that SO2 exposure induces moderate crystallite growth in Cu-derived sulfides (Cu2S/CuS) and ZnS, likely driven by phase restructuring during sulfidation.
The catalytic behavior of Cu–ZnO–Al2O3 catalysts during CO2 hydrogenation was investigated using operando XRD analysis under both SO2-free and 0.5% SO2 conditions (Fig. 7). Under SO2-free conditions, characteristic peaks corresponding to Cu (42.5° and 49.4°) and ZnO (31.1°, 34.0°, and 35.6°) were observed (Fig. 7a). These peaks exhibited slight shifts to lower 2θ values compared to the ex situ XRD at room temperature, which can be attributed to the lattice expansion at elevated temperatures. Over a time-on-stream (TOS) of 400 min, the catalyst retained its original structure, with XRD patterns displaying consistent peaks for Cu, ZnO, and Al2O3, without significant shifts in peak positions or intensities. It is worth noting that the ZnO peaks (47.0° and 56.0°) were not as pronounced, likely due to the high intensity of the Cu peaks. Throughout the reaction, CO2 conversion and CO selectivity remained stable at 26.3% and 88.9%, respectively (Fig. 7b).
 | ||
| Fig. 7
Operando XRD over the Cu–ZnO–Al2O3 catalyst. (a) Operando XRD without SO2 in the 2θ range of 25° to 65°; (b) CO2 conversion and CO selectivity over 400 min of TOS under SO2 free condition (reaction conditions: 500 °C, 1 bar, 150 mg catalyst loaded, H2:CO2:N2 = 1:1:1, with a flow rate of 100 mL min−1); (c) operando XRD under 0.5% SO2 in the 2θ range of 25° to 65°; (d) CO2 conversion and CO selectivity over 300 min of TOS under 0.5% SO2 condition; (e) operando XRD under 0.5% SO2 in the 2θ range of 45° to 48°; (f) operando XRD under 0.5% SO2 in the 2θ range of 53° to 58° (reaction condition: 1 bar, 500 °C, 150 mg catalyst loaded, CO2/H2/N2 (1:1:1) with 0.5% SO2, with a flow rate of 100 mL min−1). | ||
Upon introducing SO2 into the reaction environment, significant changes in the structure and catalytic properties were observed (Fig. 7c). Catalytic performance was continuously monitored using a gas analyzer (Fig. 7d). X-ray diffraction (XRD) analysis revealed a gradual decrease in the intensity of the ZnO peaks at 31.1°, 34.0°, and 35.6°, as well as a decrease in the intensity of the Cu peaks at 42.5° and 49.5°. Meanwhile, new peaks corresponding to metal sulfides appeared and became more pronounced at 27.9°, 46.8°,47.0 55.8° and 56.0.
Both ZnS and CuS exhibit peaks at similar positions in the XRD patterns. According to the standard PDF cards, CuS has characteristic peaks at 28.7°, 47.7°, and 56.6°, whereas ZnS displays peaks at 28.6°, 47.5°, and 56.4°. Based on these values, the peaks observed at 46.8° and 47.0° are assigned to CuS and ZnS, respectively, while the peaks at 55.8° and 56.0° are attributed to CuS and ZnS, respectively (Fig. 7e and f). Notably, prior to the introduction of SO2, the peaks at 47.0° and 56.0° were weak or absent, suggesting that the newly intensified peaks at these positions after SO2 exposure do not correspond to ZnO. Instead, the weakening of other ZnO peaks supports the assignment of these new peaks to metal sulfides, indicating the formation of CuS and ZnS as a result of SO2-induced catalyst deactivation. Additionally, the peak at 27.9° is attributed to a mixture of CuS and ZnS.
The formation of CuS and ZnS was accompanied by a significant decrease in CO2 conversion. Combining the phase transitions with the catalytic performance, we observed that after the introduction of SO2 into the flow for 10 min, metal sulfides began to form, and CO2 conversion started to decline. Meanwhile, CO selectivity increased due to the rapid decrease in CH4 formation, exhibiting trends similar to those observed in the fixed-bed reactor (Fig. 7d).
Subsequently, XRD analysis showed the disappearance of metallic Cu at point A, accompanied by the appearance of three new peaks at 30.8°, 44.5°, and 52.6°, which were attributed to Cu2S. At this point, the catalyst exhibited complete deactivation. This indicates that the gradual decrease in CO2 conversion was linked to the formation of metal sulfides and the disappearance of metallic Cu. The full conversion of ZnO and Cu into metal sulfides led to catalyst deactivation. Notably, no sulfates or sulfites were detected during the process.57–59 It is important to note that in the operando XRD setup, the contact time is 0.09 s g mL−1, whereas in the quartz tube reactor, the contact time is shorter at 0.05 s g mL−1. The longer contact time in the operando XRD experiment leads to a more prolonged interaction between the catalyst and reactants or poisons, which accelerates the accumulation of these species on the catalyst surface. This may result in faster catalyst deactivation. Therefore, although the catalyst in the quartz tube reactor eventually reaches full deactivation, some residual zero-valent copper remains detectable (shown in the XPS result, Fig. 3a) due to the shorter contact time and lower accumulation of reaction products and poisons.
We then performed a detailed kinetic analysis of the time-dependent XRD data. Due to the overlapping characteristic peaks of ZnS and CuS in the low-angle region (2θ ≤ 0.2°), it is challenging to unambiguously distinguish these sulfide phases solely via XRD. To address this limitation, our kinetic analysis focuses on the disappearance of the metallic Cu and ZnO phases, which exhibit distinct and non-overlapping diffraction signatures. This semi-quantitative approach tracks the decay of the reactant phases (Cu and ZnO) rather than directly quantifying the sulfide products (CuS/Cu2S and ZnS), providing robust insights into the sulfidation kinetics while mitigating phase deconvolution uncertainties.
For Cu-to-CuS/Cu2S transformation (Fig. S6†), a power-law logistic function (eqn (1)) captures a three-stage diffusion-controlled mechanism: (1) a lag phase (0–15 min) with slow nucleation due to sulfur incorporation barriers, (2) a growth phase (15–60 min) dominated by accelerated 3D sulfur diffusion into the Cu lattice, and (3) a saturation phase (>60 min) where unreacted Cu is kinetically trapped within a core-shell structure. In contrast, ZnO-to-ZnS conversion (Fig. S7†) follows a single-exponential decay model (eqn (2)), revealing a two-phase process: (1) an initial fast phase (0–30 min) driven by rapid surface reactions between ZnO and sulfur sources, and (2) a slow phase (>30 min) limited by ZnS passivation layer formation, which impedes bulk diffusion. These analyses collectively highlight how material-specific properties—Cu's metallic lattice favoring diffusion-limited nucleation-growth versus ZnO's semiconductor surface reactivity—govern sulfidation kinetics.
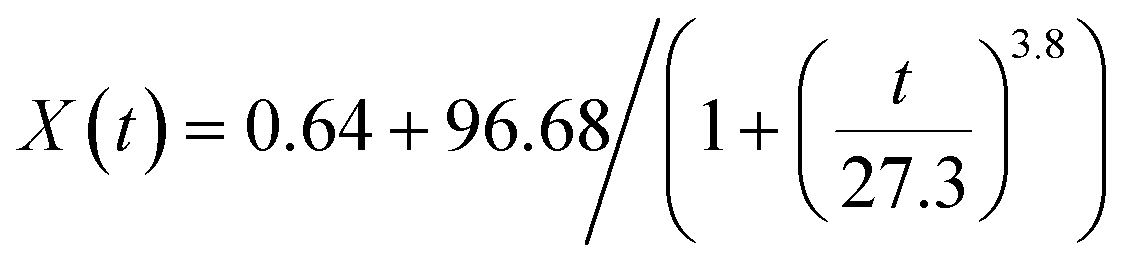 | (1) |
 | (2) |
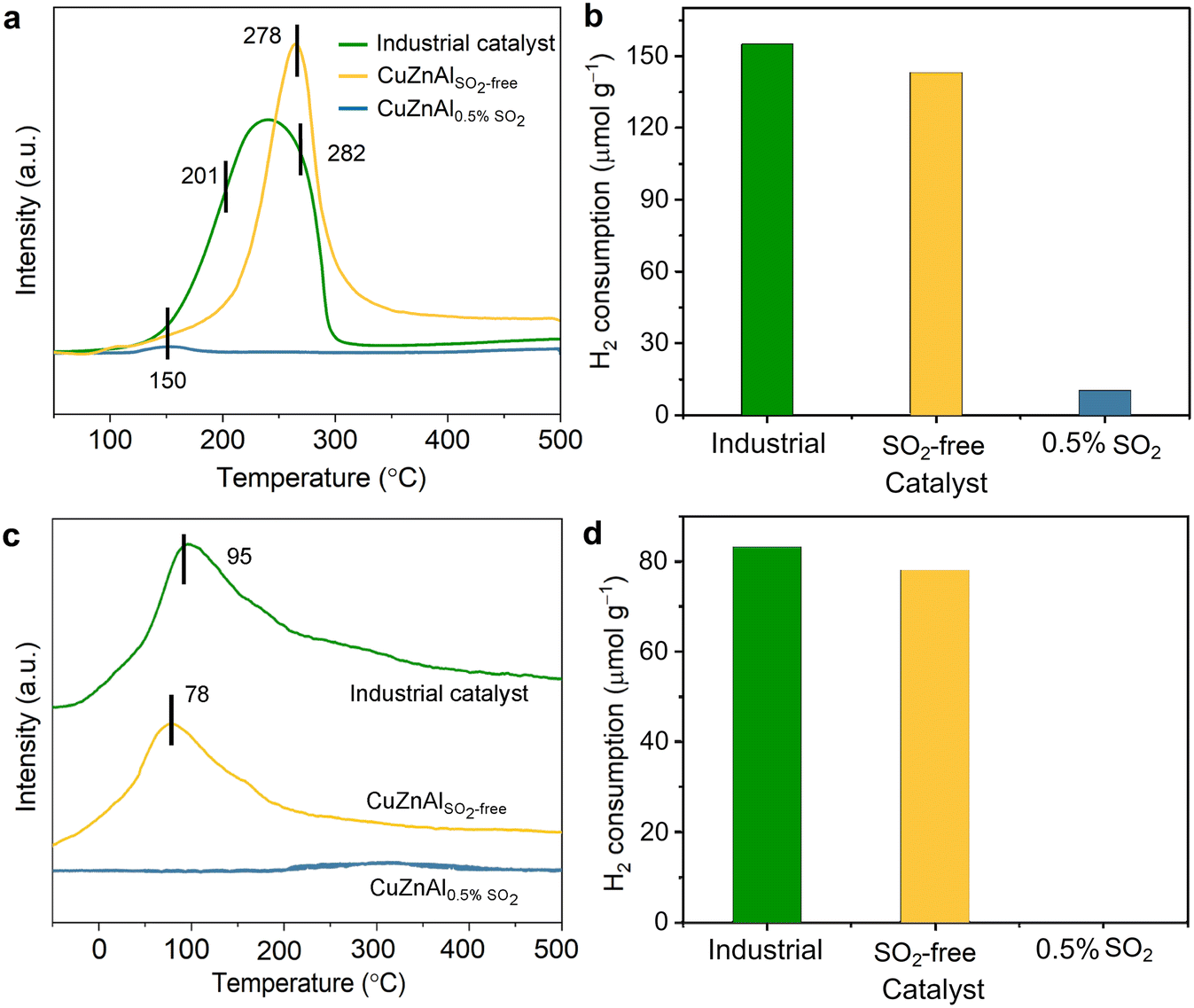 | ||
| Fig. 8 H2-TPR (a and b) and H2-TPD (c and d) of the fresh catalyst and the used catalysts under CO2 hydrogenation condition with or without 0.5% SO2. The used catalysts were treated using H2/CO2/N2 (1:1:1) with or without 0.5% SO2 for 400 min at 500 °C. | ||
To probe the spatial distribution of deactivated catalytic sites, H2-TPD analysis was conducted (Fig. 8c). H2-TPD reveals that the number of H2-adsorbing active sites (attributed to metallic Cu) sharply decreases in the presence of SO2. Specifically, the H2 uptake drops from 83 μmol g−1 (fresh catalyst) and 78 μmol g−1 (post-reaction without SO2) to nearly zero under SO2-containing conditions (Fig. 8d). This aligns with the complete loss of catalytic activity, confirming that SO2-induced sulfidation fully converts surface Cu sites into inactive sulfur-containing species (whether as sulfides, sulfates, or sulfites), thereby irreversibly deactivating the catalyst.
Inductively coupled plasma optical emission spectrometer (ICP-OES) was used to identify the sulfur content in the used catalyst (Table S3†). The experimental sulfur content of 235.3 mg g−1 (∼33.5 wt%), lying between the theoretical thresholds for complete Cu→Cu2S (26.3 wt%) and Cu→CuS (41.2 wt%) conversions, strongly indicates a mixed sulfide phase composition. This intermediate sulfur uptake demonstrates that copper species undergo partial sulfidation to both Cu2S and CuS, while zinc predominantly forms ZnS as evidenced by its stoichiometric sulfur demand. The quantitative alignment with sulfide-phase predictions, coupled with XRD identification of crystalline Cu2S/CuS and ZnS phases, confirms that irreversible deactivation originates from bulk sulfide formation rather than surface adsorption. This mechanistic interpretation is further corroborated by H2-TPD results showing near-complete loss of hydrogen adsorption capacity, consistent with sulfide-induced active site blockage at both surface and subsurface levels.
2.3 Discussion
XPS analysis showed that Cu predominantly exists as Cu+ ions, along with the presence of metal sulfides, sulfate, and sulfite species. Operando DRIFTS further confirmed the presence of these sulfur species by showing characteristic peaks for sulfates and sulfites. However, no sulfate or sulfite signals were detected in the bulk phase, suggesting that these species are exclusively formed on the surface.Referring to the previous studies on the effect of SO2 on NH3-SCR response and ICCU,23,25,26,60–62 the deactivation mechanisms induced by SO2 in NH3-SCR systems and CO2 hydrogenation processes exhibit fundamental differences due to their distinct reaction environments. In NH3-SCR systems operating under oxidizing conditions (with flue gas containing O2), SO2 oxidation is strongly promoted, leading to the predominant formation of ammonium sulfate and surface-adsorbed sulfates/sulfites.25,60–62 These species progressively block active sites and deplete catalytically active Cu species through bulk sulfate formation. In contrast, CO2 hydrogenation process contains the weakly oxidizing species CO2 and strongly reducing sepcies H2. Thermodynamic calculations (ΔG < 0 at elevated temperatures, Fig. S8†) and experimental evidence demonstrate that H2 drives the reduction of SO2 to H2S (Fig. S9†), which subsequently reacts with catalyst components to form bulk metal sulfides (CuS, Cu2S, ZnS). Furthermore, parallels emerge when comparing SO2 interactions in CO2 capture systems due to the presence of O2 5,6. For instance, in ICCU processes,23,26 SO2 exposure under oxidative conditions rapidly converts CaO to thermally stable CaSO4, significantly degrading CO2 capture capacity due to pore blockage and mass transfer limitations. This underscores the universal challenge of sulfur poisoning across systems, while emphasizing that the dominant sulfur species (sulfates vs. sulfides) are dictated by the local chemical environment—oxidizing conditions favor sulfation, whereas reducing conditions drive sulfidation.
The surface sulfur speciation analysis by XPS reveals an intriguing distribution pattern: sulfates and sulfites collectively account for only 33% of surface sulfur species, with sulfides constituting the majority. This observation can be rationalized through three key factors: (1) the absence of gaseous O2 restricts sulfite formation to ZnO vicinal sites through Zn–O–SO2 interactions; (2) sulfate generation requires dual participation of metal oxides and reactive oxygen species derived from CO2 activation, imposing strict spatial constraints; (3) dynamic interconversion between sulfur species under reaction conditions. Literature reports indicate sulfites exhibit thermal lability (decomposition >200 °C), while sulfates demonstrate higher stability (decomposition >650 °C).63–65 Crucially, in the H2-rich environment, both sulfites and sulfates undergo progressive reduction to sulfides, establishing a cyclic transformation pathway (sulfites ↔ sulfates → sulfides).
Exposure to SO2 leads to a gradual decline in catalytic activity. Partial recovery is observed in the short term, but prolonged exposure results in irreversible deactivation, with no recovery after complete deactivation. The partial reversibility observed during transient H2S exposure likely arises from incomplete poisoning due to insufficient sulfidation time, allowing the decomposition of sulfites/sulfites to release the active sites. However, once full bulk sulfidation is achieved (confirmed by XRD), the deactivation becomes irreversible, with no significant activity recovery. XRD results indicate that complete deactivation occurs after Cu and ZnO are transformed into CuS, Cu2S, and ZnS phases, confirming the catalytic inactivity of these bulk sulfides. The short-term recovery of activity is attributed to the residual catalytic activity of un-sulfidized material. However, once a complete phase transformation occurs, catalytic activity cannot be restored. Together, these findings highlight the detrimental effects of SO2 on the catalyst, emphasizing the need for developing sulfur-resistant catalysts or advanced sulfur-removal methods. These insights are critical for improving the durability of CO2 hydrogenation processes under industrial conditions.
3 Conclusions
This study systematically investigated the impact of SO2 on the structural and catalytic properties of Cu–ZnO–Al2O3 catalysts. The results revealed that SO2 exposure induces significant changes in the catalyst composition, leading to substantial deactivation. Specifically, sulfates and sulfites are adsorbed on the catalyst surface blocking active sites and impairing catalytic performance. This is further corroborated by the formation of metal carbonyl species, indicating that sulfur species hinder the desorption of CO. In addition, SO2 exposure caused the progressive transformation of Cu and Zn into catalytically inactive metal sulfides (CuS, Cu2S, and ZnS). Overall, the combined effects of metal sulfide formation and surface sulfate/sulfite accumulation significantly reduce catalytic activity, underscoring the detrimental role of SO2 in catalyst deactivation. These findings provide critical insights into the mechanisms of SO2-induced catalyst poisoning, which are essential for developing sulfur-tolerant catalysts for CO2 hydrogenation under industrial conditions.4 Experimental section
4.1 Materials
The industrial Cu–ZnO–Al2O3 catalyst used in this study was purchased from Tianjin Kaite New Material Technology Co., Ltd. The concentrations of Cu, ZnO, and Al2O3 in the catalysts were 59.1%, 29.0%, and 11.9%, respectively, as determined by ICP-OES (iCAP 7400, Thermo Fisher, Table S3†).4.2 CO2 hydrogenation
The CO2 hydrogenation reactions were carried out in a fixed-bed quartz reactor with an inner diameter of 6 mm. The catalyst (0.05 g) was loaded into the reactor and held in place with quartz wool. The reactor was connected to a gas feed system that allowed precise control over the flow rates of CO2, H2, and SO2. Before the catalytic tests, the Cu–ZnO–Al2O3 catalyst was reduced in situ at 500 °C in a 5% H2/N2 flow for 2 h to generate the active metallic Cu phase. The reaction gas mixture was passed through the reactor at a total flow rate of 60 mL min−1, and the pressure inside the reactor was maintained at 1 bar. The reaction was conducted at a temperature range from 300–700 °C, and the products were analyzed using a Gasboard 3100 Gas Analyzer (Hubei Ruiyi Co., Ltd). The catalyst was exposed to different concentrations of SO2 (ranging from 0 to 2%) in the CO2/H2 (1:1) gas mixture to evaluate the effect of SO2 on the catalytic performance. For each condition, the experiment was repeated three times to ensure reproducibility.
4.3 Catalyst characterization
STEM with energy-dispersive X-ray (EDX) spectroscopy was carried out using a high-resolution field emission TEM (JEM-2100F), which features advanced drift correction technology. The instrument was operated at an electron acceleration voltage of 200 kV, offering a point resolution of 0.23 nm and a scanning transmission electron microscopy (STEM) resolution of 0.20 nm. The lattice resolution achieved was 0.10 nm, enabling detailed elemental analysis across a broad spectrum, from beryllium (Be, atomic number 4) to uranium (U, atomic number 92). Molybdenum grids were employed to prevent interference from copper scattering.Powder XRD analysis was performed using a PANalytical Empyrean series diffractometer equipped with a high-definition Bragg–Brentano mirror. The instrument operated at 45 kV and 40 mA, utilizing Cu Kα radiation (λ = 1.5406 Å). Data were collected within a 2θ range of 5–90° with scanning speed of 0.013° per step. Operando XRD experiments were performed in PANalytical Empyrean series diffractometer equipped with reactor chamber XRK 900 from Anton Paar. The collected 2 theta range is 25–65° with scanning speed of 0.013° and 42 second per step. The sample was pre-reduced in 5% H2/N2 stream (100 mL min−1) at 500 °C for 2 h. After reduction, the sample were in the different flow by 100 mL min−1 CO2/H2 (1:1) or CO2/H2 with 0.5% SO2 under 500 °C for different min. The XRD patterns were recorded continuously for all stages.
XPS was carried out with an ESCALAB Xi+ instrument (Thermo Fisher Scientific) in an ultra-high vacuum environment (less than 1.0 × 10−9 mbar). A monochromatic Al target and magnetic lens mode were used, with a 500 μm X-ray beam spot. Survey spectra were recorded with a pass energy of 160 eV and a step size of 1.0 eV, while narrow scan spectra were recorded at a pass energy of 20 eV and a step size of 0.05 eV. Samples, supported on carbon tapes, were transferred from the glovebox to the XPS chamber via an air-tight cell, ensuring no exposure to air.
H2-TPR was carried out using a Micromeritics AutoChem 2920 instrument equipped with a thermal conductivity detector (TCD). Approximately 100 mg of sample was placed in a U-shaped quartz reactor. Prior to measurements, the sample was dried in an argon flow at 300 °C for 30 min with a heating rate of 10 °C min−1. H2-TPR was conducted under a 5% H2/Ar mixture, heating from room temperature to 500 °C at a rate of 10 °C min−1.
DRIFTS measurements were carried out using a Nicolet 6700 FTIR spectrometer, equipped with a mercury cadmium telluride detector. Operando DRIFT spectra were recorded with 32 scans at a resolution of 4 cm−1. Prior to measurements, the catalysts were pretreated in situ with a 30 ml min−1 flow of 5% H2 at 500 °C for 2 h. Background spectra were recorded at the same temperature under a nitrogen flow. Following this, the catalysts were exposed to a gas flow with the desired composition, maintained at a constant flow rate of 30 ml min−1 at the required temperature.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Author contributions
H. Z. and X. B. conceived the research project. X. B. designed the experimental work. R. W. performed the CO2 hydrogenation experiments. B. Y. contributed to the operando XRD experiments. S. Z. and X. Q. assisted with the catalyst characterization. Q. L. and Y. Z. supported the CO2 hydrogenation reactions. Data analysis and interpretation were discussed among all coauthors. X. B. and H. Z. wrote the manuscript, with contributions from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Beijing Natural Science Foundation (JQ24053), National Natural Science Foundation of China (52276202), International Joint Mission on Climate Change and Carbon Neutrality, Natural Science Foundation of Shanxi Province (202403021211024, 202403021212148), and Tsinghua University Initiative Scientific Research Program.References
- L. J. R. Nunes, The rising threat of atmospheric CO2: A review on the causes, impacts, and mitigation strategies, Environments, 2023, 10, 1–22 CrossRef.
- M. Ramonet, A. Chatterjee, P. Ciais, I. Levin, M. K. Sha, M. Steinbacher and C. Sweeney, CO2 in the atmosphere: Growth and trends since 1850, Oxford University Press, 2023 Search PubMed.
- Z. Yang, R. Chen, L. Zhang, Y. Li and C. Li, Recent progress in nickel single-atom catalysts for the electroreduction of CO2 to CO, Ind. Chem. Mater., 2024, 2, 533–555 RSC.
- J. Hu, L. Yu, J. Deng, Y. Wang, K. Cheng, C. Ma, Q. Zhang, W. Wen, S. Yu, Y. Pan, J. Yang, H. Ma, F. Qi, Y. Wang, Y. Zheng, M. Chen, R. Huang, S. Zhang, Z. Zhao, J. Mao, X. Meng, Q. Ji, G. Hou, X. Han, X. Bao, Y. Wang and D. Deng, Sulfur vacancy-rich MoS2 as a catalyst for the hydrogenation of CO2 to methanol, Nat. Catal., 2021, 4, 242–250 CrossRef CAS.
- M. D. Porosoff, B. Yan and J. G. Chen, Catalytic reduction of CO2 by H2 for synthesis of CO, methanol and hydrocarbons: Challenges and opportunities, Energy Environ. Sci., 2016, 9, 62–73 RSC.
- S. Wang, L. Zhang, P. Wang, W. Jiao, Z. Qin, M. Dong, J. Wang, U. Olsbye and W. Fan, Highly selective hydrogenation of CO2 to propane over GaZrOx/H-SSZ-13 composite, Nat. Catal., 2022, 5, 1038–1050 CrossRef CAS.
- H. Zhou, Z. Chen, A. V. López, E. D. López, E. Lam, A. Tsoukalou, E. Willinger, D. A. Kuznetsov, D. Mance, A. Kierzkowska, F. Donat, P. M. Abdala, A. Comas-Vives, C. Copéret, A. Fedorov and C. R. Müller, Engineering the Cu/Mo2CTx (MXene) interface to drive CO2 hydrogenation to methanol, Nat. Catal., 2021, 4, 860–871 CrossRef CAS.
- H. Zhou, Z. Chen, E. Kountoupi, A. Tsoukalou, P. M. Abdala, P. Florian, A. Fedorov and C. R. Müller, Two-dimensional molybdenum carbide 2D-Mo2C as a superior catalyst for CO2 hydrogenation, Nat. Commun., 2021, 12, 5510 Search PubMed.
- S. Saeidi, S. Najari, V. Hessel, K. Wilson, F. J. Keil, P. Concepción, S. L. Suib and A. E. Rodrigues, Recent advances in CO2 hydrogenation to value-added products—Current challenges and future directions, Prog. Energy Combust. Sci., 2021, 85, 100905 CrossRef.
- S. Sun, Y. Wang, Y. Xu, H. Sun, X. Zhao, Y. Zhang, X. Yang, X. Bie, M. Wu, C. Zhang, Y. Zhu, Y. Xu, H. Zhou and C. Wu, Ni-functionalized Ca@Si yolk-shell nanoreactors for enhanced integrated CO2 capture and dry reforming of methane via confined catalysis, Appl. Catal., B, 2024, 348, 123838 Search PubMed.
- M. Zhang, X. Lu, K. Luo, J. Ye, J. L. Dong, N. Lu, X. Wang, Q. Niu, P. Zhang and S. Dai, High entropy oxide catalysts with SO2 resistance in RWGS reaction, Appl. Catal., B, 2024, 349, 123845 CrossRef CAS.
- N. Sadokhina, G. Smedler, U. Nylén, M. Olofsson and L. Olsson, Deceleration of SO2 poisoning on PtPd/Al2O3 catalyst during complete methane oxidation, Appl. Catal., B, 2018, 236, 384–395 CrossRef CAS.
- L. Kang, L. Han, J. He, H. Li, T. Yan, G. Chen, J. Zhang, L. Shi and D. Zhang, Improved NOx reduction in the presence of SO2 by using Fe2O3-promoted halloysite-supported CeO2–WO3 catalysts, Environ. Sci. Technol., 2019, 53, 938–945 CrossRef CAS PubMed.
- M. Pasichnyk, P. Stanovsky, P. Polezhaev, B. Zach, M. Šyc, M. Bobák, J. C. Jansen, M. Přibyl, J. E. Bara, K. Friess, J. Havlica, D. L. Gin, R. D. Noble and P. Izák, Membrane technology for challenging separations: Removal of CO2, SO2 and NOx from flue and waste gases, Sep. Purif. Technol., 2023, 323, 124436 CrossRef CAS.
- J. Xie, D. Wang, L. Liu, T. Shao, H. Zhou and D. Zhang, An overview of flue gas so2 capture technology based on absorbent evaluation and process intensification, Ind. Eng. Chem. Res., 2024, 63, 6066–6086 CrossRef CAS.
- Z. Zhu and B. Xu, Purification technologies for NOx removal from flue gas: A review, Separations, 2022, 9, 307 CrossRef CAS.
- R. T. J. Porter, M. Fairweather, M. Pourkashanian and R. M. Woolley, The range and level of impurities in CO2 streams from different carbon capture sources, Int. J. Greenhouse Gas Control, 2015, 36, 161–174 CrossRef CAS.
- A. Wang and L. Olsson, Insight into the SO2 poisoning mechanism for NOx removal by NH3-SCR over Cu/LTA and Cu/SSZ-13, Chem. Eng. J., 2020, 395, 125048 CrossRef CAS.
- L. Liu, S. Su, D. Chen, T. Shu, X. Zheng, J. Yu, Y. Feng, Y. Wang, S. Hu and J. Xiang, Highly efficient NH3-SCR of NO over MnFeW/Ti catalyst at low temperature: SO2 tolerance and reaction mechanism, Fuel, 2022, 307, 121805 CrossRef CAS.
- J. Chen, Y. Xu, P. Liao, H. Wang and H. Zhou, Recent progress in integrated CO2 capture and conversion process using dual function materials: A state-of-the-art review, Carbon Capture Sci. Technol., 2022, 4, 100052 CrossRef CAS.
- L. Zhu, J. Yao, G. Ma, P. Cao, S. Wu and Z. Li, NH3-SCR performance and SO2 resistance comparison of CeO2 based catalysts with Fe/Mo additive surface decoration, Chem. Eng. J., 2022, 428, 131372 CrossRef CAS.
- R. Purbia, S. Y. Choi, H. J. Kim, B. Ye, B. Jeong, D. H. Lee, H. Park, H.-D. Kim and J. M. Baik, Cu- and Ce-promoted nano-heterostructures on vanadate catalysts for low-temperature NH3–SCR activity with improved SO2 and water resistance, Chem. Eng. J., 2022, 437, 135427 CrossRef CAS.
- X. Xie, L. Liu, H. Liu and Z. Sun, Mechanistic insights into SO2-induced deactivation of Ni-based materials for integrated CO2 capture and methanation, Fuel, 2025, 382, 133755 CrossRef CAS.
- H. Zhou, S. Sun, Y. Xu, Y. Zhang, S. Yi and C. Wu, Upcycling municipal solid waste to sustainable hydrogen via two-stage gasification-reforming, J. Energy Chem., 2024, 96, 611–624 CrossRef CAS.
- K. Wijayanti, K. Xie, A. Kumar, K. Kamasamudram and L. Olsson, Effect of gas compositions on SO2 poisoning over Cu/SSZ-13 used for NH3-SCR, Appl. Catal., B, 2017, 219, 142–154 CrossRef CAS.
- S. Cimino, E. M. Cepollaro and L. Lisi, Ageing study of Li-Ru/Al2O3 dual function material during the integrated CO2 capture and methanation with SO2-containing flue gas, Carbon Capture Sci. Technol., 2023, 6, 100096 CrossRef CAS.
- H. Zhou, S. R. Docherty, N. Phongprueksathat, Z. Chen, A. V. Bukhtiyarov, I. P. Prosvirin, O. V. Safonova, A. Urakawa, C. Copéret, C. R. Müller and A. Fedorov, Combining atomic layer deposition with surface organometallic chemistry to enhance atomic-scale interactions and improve the activity and selectivity of Cu–Zn/SiO2 catalysts for the hydrogenation of CO2 to methanol, JACS Au, 2023, 3, 2536–2549 CrossRef CAS PubMed.
- R. Ye, L. Ma, J. Mao, X. Wang, X. Hong, A. Gallo, Y. Ma, W. Luo, B. Wang, R. Zhang, M. S. Duyar, Z. Jiang and J. Liu, A Ce-CuZn catalyst with abundant Cu/Zn-OV-Ce active sites for CO2 hydrogenation to methanol, Nat. Commun., 2024, 15, 2159 CrossRef CAS PubMed.
- S. Kuld, C. Conradsen, P. G. Moses, I. Chorkendorff and J. Sehested, Quantification of zinc atoms in a surface alloy on copper in an industrial-type methanol synthesis catalyst, Angew. Chem., Int. Ed., 2014, 53, 5941–5945 CrossRef CAS PubMed.
- D. Li, F. Xu, X. Tang, S. Dai, T. Pu, X. Liu, P. Tian, F. Xuan, Z. Xu, I. E. Wachs and M. Zhu, Induced activation of the commercial Cu/ZnO/Al2O3 catalyst for the steam reforming of methanol, Nat. Catal., 2022, 5, 99–108 CrossRef CAS.
- V. V. Mesilov, S. L. Bergman, S. Dahlin, Y. Xiao, S. Xi, M. Zhirui, L. Xu, W. Chen, L. J. Pettersson and S. L. Bernasek, Differences in oxidation-reduction kinetics and mobility of Cu species in fresh and SO2-poisoned Cu-SSZ-13 catalysts, Appl. Catal., B, 2021, 284, 119756 CrossRef CAS.
- X. Pang, W. Liu, H. Xu, Q. Hong, P. Cui, W. Huang, Z. Qu and N. Yan, Selective uptake of gaseous sulfur trioxide and mercury in ZnO-CuS composite at elevated temperatures from SO2-rich flue gas, Chem. Eng. J., 2022, 427, 132035 Search PubMed.
- J. Li, M. Li, C. Zhang, C. L. Liu, R. Z. Yang and W. S. Dong, Construction of mesoporous Cu/ZrO2-Al2O3 as a ternary catalyst for efficient synthesis of γ-valerolactone from levulinic acid at low temperature, J. Catal., 2020, 381, 163–174 CrossRef CAS.
- A. Venugopal, J. Palgunadi, J. K. Deog, O. S. Joo and C. H. Shin, Dimethyl ether synthesis on the admixed catalysts of Cu-Zn-Al-M (M = Ga, La, Y, Zr) and γ-Al2O3: The role of modifier, J. Mol. Catal. A:Chem., 2009, 302, 20–27 CrossRef CAS.
- R. Ahmad, M. Hellinger, M. Buchholz, H. Sezen, L. Gharnati, C. Wöll, J. Sauer, M. Döring, J.-D. Grunwaldt and U. Arnold, Flame-made Cu/ZnO/Al2O3 catalyst for dimethyl ether production, Catal. Commun., 2014, 43, 52–56 CrossRef CAS.
- A. Beck, M. Zabilskiy, M. A. Newton, O. Safonova, M. G. Willinger and J. A. Van Bokhoven, Following the structure of copper-zinc-alumina across the pressure gap in carbon dioxide hydrogenation, Nat. Catal., 2021, 4, 488–497 Search PubMed.
- H. Zhang, J. Chen, X. Han, Y. Pan, Z. Hao, S. Tang, X. Zi, Z. Zhang, P. Gao, M. Li, J. Lv and X. Ma, High-performance Cu/ZnO/Al2O3 catalysts for CO2 hydrogenation to methanol, Ind. Eng. Chem. Res., 2024, 63, 6210–6221 Search PubMed.
- M. Behrens, F. Studt, I. Kasatkin, S. Kühl, M. Hävecker, F. Abild-Pedersen, S. Zander, F. Girgsdies, P. Kurr, B.-L. Kniep, M. Tovar, R. W. Fischer, J. K. Nørskov and R. Schlögl, The active site of methanol synthesis over Cu/ZnO/Al2O3 industrial catalysts, Science, 2012, 336, 893–897 CrossRef CAS PubMed.
- X. Bie, Y. Pan, X. Wang, S. Zhang, J. Hu, X. Yang, Q. Li, Y. Zhang, R. E. Przekop, Y. Zhang and H. Zhou, NH3-induced challenges in CO2 hydrogenation over the Cu/ZnO/Al2O3 catalyst, JACS Au, 2025, 5, 1243–1257 Search PubMed.
- J. Schumann, J. Kröhnert, E. Frei, R. Schlögl and A. Trunschke, IR-spectroscopic study on the interface of Cu-based methanol synthesis catalysts: Evidence for the formation of a ZnO overlayer, Top. Catal., 2017, 60, 1735–1743 Search PubMed.
- P. A. Kumar, Y. E. Jeong, S. Gautam, H. P. Ha, K. J. Lee and K. H. Chae, XANES and DRIFTS study of sulfated Sb/V/Ce/TiO2 catalysts for NH3-SCR, Chem. Eng. J., 2015, 275, 142–151 Search PubMed.
- Q. Wu, H. Gao and H. He, Conformational analysis of sulfate species on Ag/Al2O3 by means of theoretical and experimental vibration spectra, J. Phys. Chem. B, 2006, 110, 8320–8324 Search PubMed.
- Y. Cai, B. Zhang, H. Yu, X. Ji, J. Sun, X. Wang, Q. Qian, L. Li, A. Liu, W. Tan, F. Gao and L. Dong, Shielding ceria based catalysts from SO2 poisoning in NH3-SCR reaction: Modification effect of acid metal oxides, Appl. Catal., B, 2024, 342, 123424 CrossRef CAS.
- R. Chen, X. Fang, J. Li, Y. Zhang and Z. Liu, Mechanistic investigation of the enhanced SO2 resistance of Co-modified MnOx catalyst for the selective catalytic reduction of NOx by NH3, Chem. Eng. J., 2023, 452, 139207 CrossRef CAS.
- Z. Zhang, R. Li, M. Wang, Y. Li, Y. Tong, P. Yang and Y. Zhu, Two steps synthesis of CeTiOx oxides nanotube catalyst: Enhanced activity, resistance of SO2 and H2O for low temperature NH3-SCR of NOx, Appl. Catal., B, 2021, 282, 119542 CrossRef CAS.
- R. Chen, X. Fang, J. Li, Y. Zhang and Z. Liu, Mechanistic investigation of the enhanced SO2 resistance of Co-modified MnOx catalyst for the selective catalytic reduction of NOx by NH3, Chem. Eng. J., 2023, 452, 139207 CrossRef CAS.
- B. Liang, J. Ma, X. Su, C. Yang, H. Duan, H. Zhou, S. Deng, L. Li and Y. Huang, Investigation on deactivation of Cu/ZnO/Al2O3 catalyst for CO2 hydrogenation to methanol, Ind. Eng. Chem. Res., 2019, 58, 9030–9037 Search PubMed.
- M. Sahibzada, D. Chadwick and I. S. Metcalfe, Hydrogenation of carbon dioxide to methanol over palladium-promoted Cu/ZnO/Al2O3 catalysts, Catal. Today, 1996, 29, 367–372 CrossRef CAS.
- T. W. Hansen, A. T. Delariva, S. R. Challa and A. K. Datye, Sintering of catalytic nanoparticles: Particle migration or ostwald ripening?, Acc. Chem. Res., 2013, 46, 1720–1730 CAS.
- M. Salavati-Niasari and F. Davar, Synthesis of copper and copper(I) oxide nanoparticles by thermal decomposition of a new precursor, Mater. Lett., 2009, 63, 441–443 CAS.
- Y. Zhang, Y. Yang, H. Han, M. Yang, L. Wang, Y. Zhang, Z. Jiang and C. Li, Ultra-deep desulfurization via reactive adsorption on Ni/ZnO: The effect of ZnO particle size on the adsorption performance, Appl. Catal., B, 2012, 119–120, 13–19 CrossRef CAS.
- J. Wu, M. Saito, M. Takeuchi and T. Watanabe, The stability of Cu/ZnO-based catalysts in methanol synthesis from a CO2-rich feed and from a CO-rich feed, Appl. Catal., A, 2001, 218, 235–240 Search PubMed.
- M. Mousavi-Kamazani, Z. Zarghami and M. Salavati-Niasari, Facile and novel chemical synthesis, characterization, and formation mechanism of copper sulfide (Cu2S, Cu2S/CuS, CuS) nanostructures for increasing the efficiency of solar cells, J. Phys. Chem. C, 2016, 120, 2096–2108 Search PubMed.
- R. K. Sithole, L. F. E. Machogo, M. J. Moloto, S. S. Gqoba, K. P. Mubiayi, J. Van Wyk and N. Moloto, One-step synthesis of Cu3N, Cu2S and Cu9S5 and photocatalytic degradation of methyl orange and methylene blue, J. Photochem. Photobiol., A, 2020, 397, 112577 CAS.
- M. Sookhakian, Y. M. Amin, W. J. Basirun, M. T. Tajabadi and N. Kamarulzaman, Synthesis, structural, and optical properties of type-II ZnO–ZnS core–shell nanostructure, J. Lumin., 2014, 145, 244–252 CAS.
- C. Ramamoorthy and V. Rajendran, Synthesis and characterization of CuS nanostructures: Structural, optical, electrochemical and photocatalytic activity by the hydro/solvothermal process, Int. J. Hydrogen Energy, 2017, 42, 26454–26463 CAS.
- R. Ma, J. Stegemeier, C. Levard, J. G. Dale, C. W. Noack, T. Yang, G. E. Brown and G. V. Lowry, Sulfidation of copper oxide nanoparticles and properties of resulting copper sulfide, Environ. Sci.:Nano, 2014, 1, 347–357 Search PubMed.
- Y.-C. Li, S.-N. Zhuo, B. Peng, X.-B. Min, H. Liu and Y. Ke, Comprehensive recycling of zinc and iron from smelting waste containing zinc ferrite by oriented transformation with SO2, J. Cleaner Prod., 2020, 263, 121468 CAS.
- H. Chen, C. Dai, F. Xiao, Q. Yang, S. Cai, M. Xu, H. J. Fan and S. Bao, Reunderstanding the reaction mechanism of aqueous Zn–Mn batteries with sulfate electrolytes: Role of the zinc sulfate hydroxide, Adv. Mater., 2022, 34, 2109092 CAS.
- L. Zhang, D. Wang, Y. Liu, K. Kamasamudram, J. H. Li and W. Epling, SO2 poisoning impact on the NH3-SCR reaction over a commercial Cu-SAPO-34 SCR catalyst, Appl. Catal., B, 2014, 156–157, 371–377 CAS.
- J. D. Bjerregaard, M. Votsmeier and H. Grönbeck, Mechanism for SO2 poisoning of Cu-CHA during low temperature NH3-SCR, J. Catal., 2023, 417, 497–506 CrossRef CAS.
- Y. Jangjou, D. Wang, A. Kumar, J. H. Li and W. S. Epling, SO2 poisoning of the NH3-SCR reaction over Cu-SAPO-34: Effect of ammonium sulfate versus other S-containing species, ACS Catal., 2016, 6, 6612–6622 CrossRef CAS.
- W. K. Su, Z. G. Li, Y. N. Zhang, C. C. Meng and J. H. Li, Identification of sulfate species and their influence on SCR performance of Cu/CHA catalyst, Catal. Sci. Technol., 2017, 7, 1523–1528 RSC.
- M. Happel, Y. Lykhach, N. Tsud, T. Skála, V. Johánek, K. C. Prince, V. Matolín and J. Libuda, SO2 decomposition on Pt/CeO2(111) model catalysts: On the reaction mechanism and the influence of H2 and CO, J. Phys. Chem. C, 2012, 116, 10959–10967 CrossRef CAS.
- M. S. Wilburn and W. S. Epling, Formation and decomposition of sulfite and sulfate species on Pt/Pd catalysts: An SO2 oxidation and sulfur exposure study, ACS Catal., 2019, 9, 640–648 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S9, Tables S1–S3. See DOI: https://doi.org/10.1039/d5im00025d |
| This journal is © Institute of Process Engineering of CAS 2025 |