
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Amino and hydroxyl functionalization of nucleosides via resonant acoustic mixing†
Julian
Marlyn
,
Olivia
Del Carlo
,
James D.
Thorpe
and
Masad J.
Damha
 *
*
Department of Chemistry, McGill University, Montreal, QC, Canada. E-mail: masad.damha@mcgill.ca
First published on 17th June 2025
Abstract
The synthesis of oligonucleotides for therapeutics faces significant challenges such as cost, waste generation, and energy-intensive processes. While synthesis of oligonucleotide strands has received a great deal of attention, sustainable synthesis of the crucial nucleoside monomers has been largely disregarded. Herein, we present an application of resonant acoustic mixing (RAM), a novel mechanochemical technique, to common functionalization reactions on nucleosides. Each stage of oligonucleotide synthesis was addressed and various protected nucleosides, as well as phosphitylated and succinylated nucleosides were synthesized with significant reduction in solvent and reagent consumption. Generally, RAM reactions proceeded faster compared to reactions carried out in solution, with significant reduction in solvent use, and could be easily scaled up while maintaining yields.
Green foundation1. This work is among the first in the use of resonant acoustic mixing (RAM) application to organic synthesis.2. This work describes sustainable methods of synthesis, reducing or eliminating solvent use. PMI reduction of ca. 50% (reaction/purification) was achieved in a large-scale reaction when compared to the corresponding solution phase process. 3. Reaction rate increases of one to two orders of magnitude were also achieved, highlighting further the advantages of RAM in the reactions studied. |
Introduction
Solid-phase synthesis, pioneered by Merrifield and Letsinger in the 1960s, has been the dominant mode of synthesis for oligonucleotides owing to the ease of purification, rapid and high-yield generation of DNA and RNA strands, and the discovery and development of oligonucleotides as therapeutics.1–3 To this point, small (micromole) quantities of oligonucleotides have largely been sufficient for preliminary studies. However, the growth of oligonucleotide drugs demands large (mole) quantities of drug substance.4 With the oligonucleotide therapeutics market projected to grow at an annual rate of 17.5% through 2030, the industry faces significant challenges such as high production costs, waste generation, and energy-expenditure.5 The American Chemical Society Green Chemistry Industrial Pharmaceutical Roundtable (ACS GCIPR), a forum of global pharmaceutical and allied industries focused on the sustainability of manufacturing medicines, has highlighted the need for sustainable practices to help reduce the environmental impact of oligonucleotide synthesis. Their 2020 study outlined sustainability challenges and opportunities for oligonucleotide synthesis and laid out long and short-term goals in developing greener methodology for the field.6 Particularly concerning is the finding that scale had no impact on the process mass intensity (PMI), a feature unheard of in small molecule synthesis. As a result, industry and academia have presented a variety of solutions to ameliorate this problem.7–10While there has been a great deal of focus in the field regarding the greenness of oligonucleotide synthesis, the field has largely overlooked the upstream production of nucleosides. Current studies reporting synthesis PMI of oligonucleotides fail to account for the production of building blocks (nucleoside phosphoramidites), and therefore dramatically underestimate the overall PMI for the process.6,11 All current forms of oligonucleotide synthesis use highly protected or modified nucleosides adding to the synthetic challenge and waste. The use of environmentally damaging solvents and reagents throughout the process of nucleoside production further increases cost and environmental impact.12 Hence, implementing green chemistry principles to nucleoside synthesis would significantly enhance the sustainability of oligonucleotide production by promoting safer, more efficient, and environmentally friendly processes.
Resonant acoustic mixing (RAM)
Resonant acoustic mixing (RAM) technology was initially studied for its remarkable ability to efficiently produce pharmaceutical co-crystals, safely mix energetic materials, and generate metal–organic frameworks.13–15 While other mechanochemical methods (ball-milling, extrusion, etc.) have been used in the context of organic synthesis for some time, the possibility of RAM as a tool in organic-synthetic chemistry has only recently received significant attention. Mechanochemistry has been an important tool in organic synthesis, allowing chemists to significantly reduce quantities of solvent while improving rates and regio/stereoselectivity of reactions.7,13,14RAM as a mechanochemical tool is characterized by the lack of milling media. Rapid mixing and mechanochemical action are induced by linear vibration along the vertical axis with moderate displacement (∼1 cm) and moderate frequency (60 Hz). The result is a system in which the reaction vessel contents experience high accelerations of 30–100 times the force of gravity on earth (expressed in units of g), resulting in highly efficient, yet low-impact mixing of substrates.15,16 There are several advantages to RAM as a media-free mechanochemical method. Chiefly, it eliminates the constant impact forces of media on the vessel, which greatly reduces both wear on vessels and prevents contamination as seen with other mechanochemical techniques. Additionally, vessels can be constructed in a variety of more fragile materials including glass and conventional plastics.17 Relevant to this work, our group has recently demonstrated the utility of mechanochemistry in oligonucleotide chemistry with encouraging results.18,19
In this work, we report the first application of RAM in the functionalization of nucleosides, culminating in a method for the protection and preparation of nucleoside phosphoramidites. We found RAM to be effective in significantly reducing solvent waste, and in increasing the rate of nucleoside protection and modification reactions.
Results and discussion
Nucleoside 3′-O-phosphoramidites were first described in 1981![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 20 and remain the standard building blocks used in modern DNA and RNA synthesis. These molecules permit the sequential addition nucleosides to the oligonucleotide chain in a simple and exceptionally efficient cyclic process. They are currently prepared in solution on the metric ton scale, requiring a few basic steps starting from the free nucleosides, as demonstrated in Scheme 1.21 It is important to note that reactions were appropriately quenched prior to NMR analysis and purification to ensure that transformations were a result of RAM reaction conditions.
20 and remain the standard building blocks used in modern DNA and RNA synthesis. These molecules permit the sequential addition nucleosides to the oligonucleotide chain in a simple and exceptionally efficient cyclic process. They are currently prepared in solution on the metric ton scale, requiring a few basic steps starting from the free nucleosides, as demonstrated in Scheme 1.21 It is important to note that reactions were appropriately quenched prior to NMR analysis and purification to ensure that transformations were a result of RAM reaction conditions.
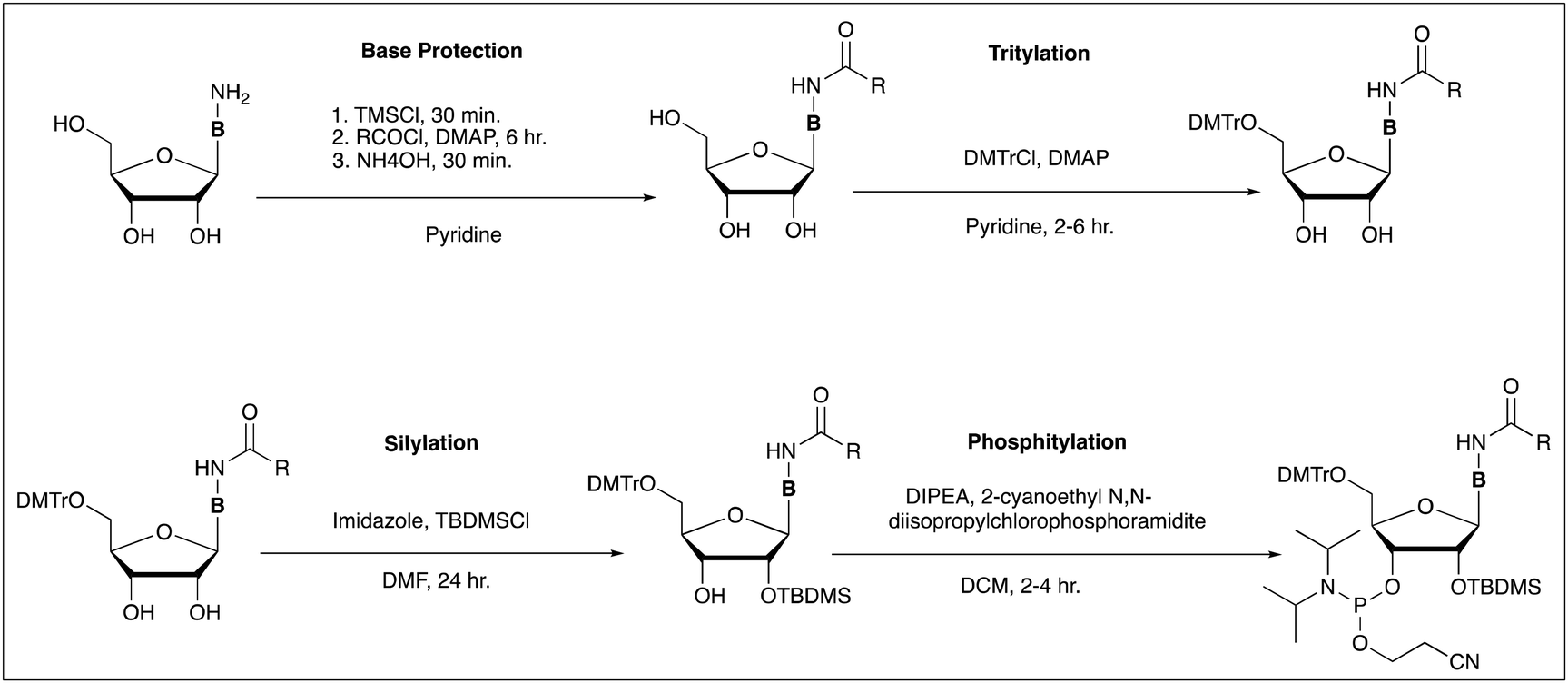 | ||
| Scheme 1 Generalized scheme for the preparation of nucleoside phosphoramidites suitable for oligonucleotide synthesis. | ||
Base protection
Base protection describes a variety of acylation reactions which protect the nucleobase exocyclic amines during oligonucleotide synthesis and is typically the first step in the preparation of nucleoside phosphoramidites. A variety of acyl groups are utilized and installed under similar conditions, most commonly using pyridine or dimethylformamide (DMF) as solvent.We began by attempting the selective N-acetylation of cytidine following the method of Bhat and coworkers.21 Cytidine was combined with equivalent of liquid acetic anhydride in the absence of DMF, and subjected to RAM (Scheme 2). Product 2 was obtained in near quantitative yield in considerably less time when compared to a reaction carried out in a flask using DMF as the solvent (30 min by RAM at 60g vs. 4 h in solution).
 | ||
| Scheme 2 Preparation of acetyl protected cytidine via RAM. | ||
Based on our past work,18 we found that 60g was a suitable acceleration to achieve good reactivity, while balancing power demands and wear on the equipment. Little improvement in apparent mixing efficiency and rate was observed at higher accelerations (e.g., 90g), and 60g acceleration used throughout our syntheses.
Nucleobase acetylation of adenosine and guanosine require transient trimethylsilyl (TMS) protection with excess pyridine employed as a solvent and to quench the resulting hydrochloric acid byproduct. However, we hypothesized that with the application of RAM, pyridine may only be required in equivalent quantities. Indeed, we found that only a slight excess of pyridine was sufficient to accomplish the silylation and subsequent benzoylation of both nucleosides (Scheme 3). Benzoyl and isobutyryl protection were achieved in similar yields to their solution phase counterparts, but with significantly reduced reaction times. To better describe reaction conditions, we calculated the η value for this reaction. η, or eta, represents the ratio of liquid to solid components in a chemical system, and is utilized in mechanochemistry as a point of objective comparison between systems. Notably, the above reactions remained viscous slurries throughout the reaction with η values in the range of 1 μL mg−1 suggesting mechanochemical action. This result encouraged us to investigate further nucleoside functionalization.
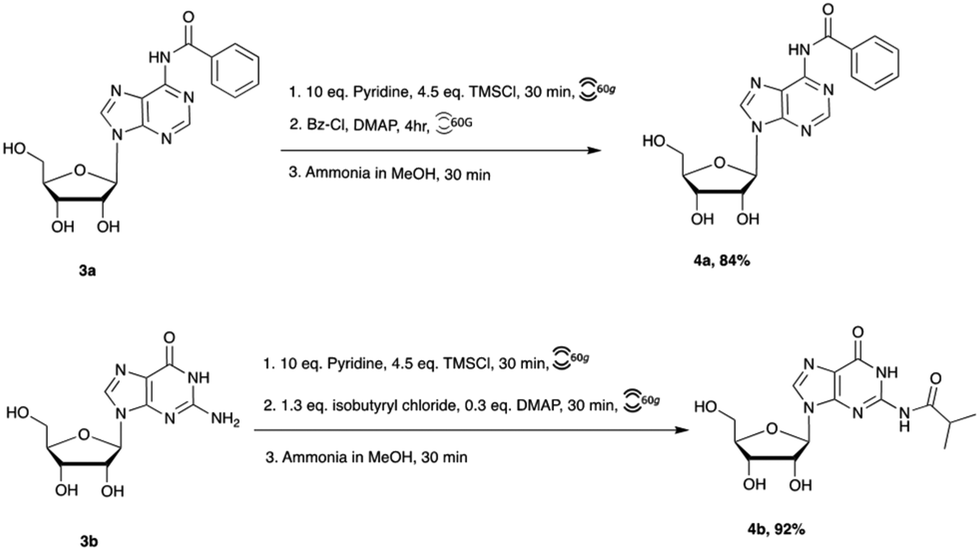 | ||
| Scheme 3 Preparation of acyl-protected pyrimidine nucleosides via RAM. | ||
5′-OH tritylation
Building from our results in nucleobase protection, we next investigated 5′-OH protection with 4,4-dimethoxytrityl (DMTr) chloride, ubiquitous in nucleoside chemistry. From a green chemistry perspective, both the use and installation of DMTr have major issues. Protection utilizes neat pyridine as a solvent, is often slow, and suffers from relatively moderate yields due to the formation of 2′,5′- and 3′,5′-ditritylated byproducts.22 Standard reaction conditions for DMTr installation call for 1.5–2.0 equivalents of DMTrCl, ca. 10 mL pyridine per gram of nucleoside, and purification by column chromatography to isolate the desired 5′-DMTr nucleoside.23,24Our optimization began by reducing the quantity of pyridine from a standard solvent (25 eq.) to the minimal quantities (1–2 eq.) required to quench the hydrochloric acid produced. While the starting material formed a smooth slurry in pyridine, addition of DMTrCl caused a visible increase in viscosity, with large aggregates of DMTrCl remaining. Purification and analysis showed significant quantities of both starting material and di-tritylated nucleoside. We hypothesize this to be a result of high concentration pockets of DMTrCl at the surface of aggregated material, resulting in increased di-tritylation. Addition of larger quantities of pyridine (5.0–6.0 equivalents) was effective in eliminating aggregated material. Further reduction of pyridine to 2.0 equivalents was possible by the addition of 2.0 equivalents of ethyl acetate (Scheme 4). Ethyl acetate was selected as our preferred substitute in this, and other reactions due to its favourable green chemistry profile and compatibility with nucleoside substrates.25
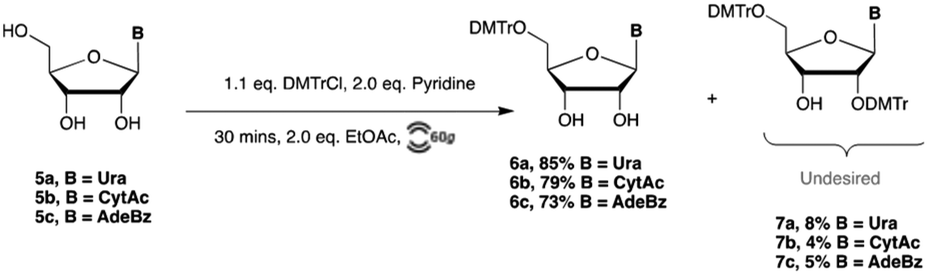 | ||
| Scheme 4 Tritylation of ribonucleosides via RAM. | ||
Gratifyingly, under these conditions reactions also proceeded significantly more rapidly (30 min vs. 3–5 h in solution) and with excellent regioselectivity providing the desired 5′-O-DMTr protected nucleosides in 73–85% yields. Interestingly, the dramatic increase in overall concentration of the reported tritylation reactions did not increase the quantity of di-tritylated nucleoside by-products. So long as DMTrCl was added in increments and a free-flowing slurry phase was maintained, nearly exclusive 5′-tritylation was observed.
To demonstrate scalability and assess the impact of our methodology on process mass intensity (PMI), 5′-OH tritylation of uridine was scaled from 2 mmol (488 mg) to 40 mmol (9.77 g) without the need for further optimization, affording 5′-DMTr- in 74% yield (18.42 g) and high purity (1H NMR; ∼95%) after recrystallization from toluene and hexanes. This method resulted in a PMI reduction of ∼48% when compared to our control solution phase method (Fig. 1 and S4†).
 | ||
| Fig. 1 PMI comparison between RAM and solution phase tritylation of uridine. | ||
2′-OH protection
Protection of the ribose 2′-OH group is the basis for all RNA monomers which are currently commercially available. The tert-butyldimethylsilyl (TBDMS) group26 has become the standard and most widely used 2′-OH protecting group in both solution and solid-phase oligomerization of ribonucleotides.27 TBDMS remains stable through both acidic and basic intermediate deprotection of DMTr and acyl groups, and is removed under mild fluoride-based conditions.The TBDMS group is typically installed using TBDMSCl in the presence of silver nitrate and pyridine.28 We found this method to be ineffective when applying RAM, as little to no silylated products were observed. Following this, we substituted silver nitrate and pyridine with dimethylformamide (DMF) and imidazole. Initial trials of RAM silylation under these conditions were monitored by thin-layer chromatography (TLC) at one-hour intervals. To our surprise, after the first hour, TLC showed only degradation products, including detritylated material.26 Gratifyingly, reducing the reaction time to 10 min provided the desired 2′ and 3′-O-TBDMS products in 56% and 34% yield, respectively (Scheme 5). This was an encouraging result, as a similar control reaction carried out in solution took considerably longer, reaching completion at 18 hours.
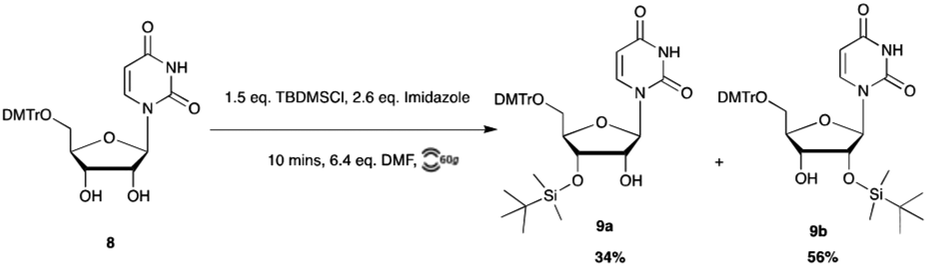 | ||
| Scheme 5 TBDMS protection of uridine via RAM. | ||
3′-OH phosphitylation and succinylation
The nucleoside 3′-hydroxyl group is a target of several functionalization reactions, most commonly the introduction of an active phosphoramidite moiety as a coupling point during solid-phase oligonucleotide synthesis. The 3′-OH also functions as an attachment point to the solid support such as glass or polystyrene based materials. Functionalization for this purpose include succinylation,27 or esterification with an lipophilic or ionic support.4,10,29 To prepare the 3′-O-phosphoramidite derivatives of thymidine, 2′-deoxycytidine, and 2′-deoxyadenosine (Scheme 6), we first reduced the quantity of dichloromethane (DCM) to five equivalents.27 Under these conditions, the reaction reached completion within 5–10 minutes affording the desired products in excellent yields (>80%). | ||
| Scheme 6 3′-OH phosphitylation of protected ribonucleosides via RAM. | ||
Gratifyingly, removing DCM entirely, but maintaining the amount of base resulted in functionally identical results (Scheme 6). The reaction mixture maintained a slurry regime before and after RAM was applied, however it was observed that the quantity of precipitate increased upon completion of the reaction. Attempts to filter the reaction mixture were of mixed success and ultimately short column purification was required to obtain spectroscopically pure material. In the stirred reaction control, formation of H-phosphonate was observed, likely due to protracted exposure to small quantities of water. Beneficially, RAM showed no such degradation by TLC.
Succinylation of nucleosides has previously been reported via ball milling.30 Reported conditions showed complete conversion of starting material to the desired product in crude mixtures by 1H-NMR analysis. However, we wanted to investigate alternative bases and solvents as both 4-dimethylaminopyridine (DMAP) and DCM, used in the reported procedure, pose significant health and environmental risks.25
Substitution of base with other amine bases (triethylamine, pyridine, DIPEA) showed marked decreases in yield. However, reduction in the quantity of DMAP to half an equivalent showed only a slight decrease in yield, remedied by a small increase in reaction time. In addition, substitution of DCM with ethyl acetate posed no consequence. The resulting optimised condition is shown in Scheme 7.
 | ||
| Scheme 7 Succinylation of thymidine. aNMR yield. | ||
Ionic tag installation at 3′-OH
Previously our lab has investigated the utility of phosphonium and imidazolium tags to induce precipitation as an alternative to column chromatography in solution-phase oligonucleotide synthesis.29 We looked to apply our succinylation chemistry towards functionalization of 3′-hydroxyl groups with an ionic tag in one-pot. To the crude succinylated material 13 produced with our optimized method, was directly added a coupling reagent (dicyclohexylcarbodiimide, DCC) and ionic liquid 14 (Scheme 8), and subjected to RAM at 60g for an additional 30 minutes. Compound 15 was isolated in 57% over the two steps. Precipitation afforded product with slight contamination of starting materials, and further column chromatography (1–6% MeOH in DCM) provided pure product. | ||
| Scheme 8 One pot synthesis of ionic liquid tagged nucleoside. | ||
Comparison of RAM reaction conditions with solution-phase conditions
Table 1 summarizes the scope of reactions investigated. Without exception, all transformations occurred exclusively under slurry type regimes. Optimal reaction conditions. consistently fell in a narrow range of η = 0.5–1.0 μL mg−1, while solution-phase counterparts had an average value of η = 12 μL mg−1. As such, RAM provided a minimum of 80% reduction in solvent consumption compared to solution-phase counterparts.| Reaction | Solvent reduction | Solution phase solvent | RAM solvent | RAM η (μL mg−1) | RAM regime |
|---|---|---|---|---|---|
| 5′-DMTr | 93% | Pyridine | EtOAc | 0.72 | Slurry |
| 2′-TBDMS | 89% | DMF | DMF | 0.51 | Slurry |
| 3′-Phosphitylation | 100% | DCM | None | 0.51 | Slurry |
| N-Acylation | 100%–88% | Pyridine | Pyridine | 0.84 | Slurry |
| 3′-Succinylation | 81% | DCM | EtOAc | 0.71 | Slurry |
Considering our findings above, along with observations of the systems studied, we assert that for RAM reactions on nucleosides, optimization trends towards slurries, and a narrow η window. Ratios below the window (low η) result in sticky reaction mixtures leading to adhesion of materials to the reaction vessel surface. In addition, lower η values encourage heterogeneous mixtures and generally poor mass transfer, negatively impacting outcomes. On the other hand, high η regimens dilute the mixture to the point of solubilization, inhibiting mechanochemical action despite beneficial mass transfer. RAM consistently and significantly accelerated reactions one to two orders of magnitude relative to control reactions carried in parallel in solution (Table 2, Fig. S4†).
Conclusions
Herein, we demonstrate the utility of RAM in sustainable functionalization and protection of nucleosides. RAM allowed for dramatic reduction in solvent quantity in diverse chemistries, and in some cases favoured total elimination of reaction solvent. Additionally, we found that RAM substantially increased the rate of nucleoside functionalization reactions compared to solution phase counterparts. The ease of optimization provided by RAM was also notable. Screening of ten or more reactions simultaneously allowed for rapid parameter optimization while ensuring batch consistency. RAM's compatibility with inexpensive, diverse, and commercially available reaction vessels allowed us work far faster and more efficiently than our experiments in ball milling, and in solution phase.19 The ability to rapidly develop and monitor reactions is a distinct advantage of RAM over other mechanochemical techniques.Crucially from a green chemistry standpoint, we had a great deal of success in eliminating, reducing, and replacing detrimental solvents, with ethyl acetate serving as an excellent functional replacement for pyridine, dimethylformamide, and dichloromethane.
The characteristics of a reaction which allow effective translation to RAM are still yet to be fully determined. Further study into factors contributing to the effectiveness and limitations of RAM as a synthetic tool is of great importance in the advancement of the field. We plan in future studies to better identify mechanisms by which rate enhancement occurs. Additionally, we identify the need for investigation of a diversity of reactions under RAM conditions, with the aim of working towards an understanding of reactivity akin to that of solution phase chemistry. Of further interest is the comparison of RAM to other mechanochemical methods at process scale, considering factors such as energy consumption, PMI, and safety considerations.
Conflicts of interest
There are no conflicts of interest to declare.Data availability
The data supporting this article have been included as part of the ESI.†Acknowledgements
This investigation was supported by research grants from the National Science and Engineering Council of Canada (Discovery Grant and Alliance Grant to MJD, and an Undergraduate Student Research Award to ODC).References
- R. L. Letsinger and M. J. Kornet, J. Am. Chem. Soc., 1963, 85, 3045–3046 CrossRef CAS.
- R. B. Merrifield, J. Am. Chem. Soc., 1963, 85, 2149–2154 CrossRef CAS.
- R. T. Pon, Curr. Protoc. Nucleic Acid Chem., 2000, 3.1.1–3.1.28 Search PubMed.
- R. Obexer, M. Nassir, E. R. Moody, P. S. Baran and S. L. Lovelock, Science, 2024, 384, eadl4015 CrossRef CAS PubMed.
- P. Markolin, European Pharmaceutical Manufacturer, 2024 Search PubMed.
- B. I. Andrews, F. D. Antia, S. B. Brueggemeier, L. J. Diorazio, S. G. Koenig, M. E. Kopach, H. Lee, M. Olbrich and A. L. Watson, J. Org. Chem., 2021, 86, 49–61 CrossRef CAS PubMed.
- J. F. Kim, P. R. J. Gaffney, I. B. Valtcheva, G. Williams, A. M. Buswell, M. S. Anson and A. G. Livingston, Org. Process Res. Dev., 2016, 20, 1439–1452 Search PubMed.
- World Intellectual Property Organization, WO/2022/103802A1, 2022.
- A. G. Molina and Y. S. Sanghvi, Curr. Protoc., 2019, 77, e82 Search PubMed.
- X. Zhou, W. F. Kiesman, W. Yan, H. Jiang, F. D. Antia, J. Yang, Y. A. Fillon, L. Xiao and X. Shi, J. Org. Chem., 2022, 87, 2087–2110 CrossRef CAS PubMed.
- E. R. Moody, R. Obexer, F. Nickl, R. Spiess and S. L. Lovelock, Science, 2023, 380, 1150–1154 CrossRef CAS PubMed.
- L. K. McKenzie, R. El-Khoury, J. D. Thorpe, M. J. Damha and M. Hollenstein, Chem. Soc. Rev., 2021, 50, 5126–5164 RSC.
- J.-L. Do and T. Friščić, ACS Cent. Sci., 2017, 3, 13–19 CrossRef CAS PubMed.
- J. G. Hernández and C. Bolm, J. Org. Chem., 2017, 82, 4007–4019 CrossRef PubMed.
- H. M. Titi, J.-L. Do, A. J. Howarth, K. Nagapudi and T. Friščić, Chem. Sci., 2020, 11, 7578–7584 RSC.
- F. Effaty, L. Gonnet, S. G. Koenig, K. Nagapudi, X. Ottenwaelder and T. Friščić, Chem. Commun., 2023, 59, 1010–1013 RSC.
- A. A. L. Michalchuk and F. Emmerling, Angew. Chem., Int. Ed., 2022, 61, e202117270 CrossRef CAS PubMed.
- J. D. Thorpe, J. Marlyn, S. G. Koenig and M. J. Damha, RSC Mechanochem., 2024, 1, 244–245 RSC.
- J. D. Thorpe, D. O'Reilly, T. Friščić and M. J. Damha, Chem. – Eur. J., 2020, 26, 8857–8861 CrossRef CAS PubMed.
- S. L. Beaucage and M. H. Caruthers, Tetrahedron Lett., 1981, 22, 1859–1862 CrossRef CAS.
- V. Bhat, B. G. Ugarkar, V. A. Sayeed, K. Grimm, N. Kosora, P. A. Domenico and E. Stocker, Nucleosides Nucleotides, 1989, 8, 179–183 CrossRef CAS.
- H. Seliger and Y. S. Sanghvi, Current Protocols, 2024, 4, e999 CrossRef CAS PubMed.
- M. Smith, D. H. Rammler, I. H. Goldberg and H. G. Khorana, J. Am. Chem. Soc., 1962, 84, 430–440 CrossRef CAS.
- A. K. McPherson, D. Capaldi, L. Chen and P. Olsen, Org. Process Res. Dev., 2020, 24, 2583–2590 CrossRef CAS.
- D. Prat, J. Hayler and A. Wells, Green Chem., 2014, 16, 4546–4551 RSC.
- E. J. Corey and A. Venkateswarlu, J. Am. Chem. Soc., 1972, 94, 6190–6191 CrossRef CAS.
- M. J. Damha and K. K. Ogilvie, in Protocols for Oligonucleotides and Analogs, Humana Press, New Jersey, 1993, vol. 20, pp. 81–114 Search PubMed.
- G. H. Hakimelahi, Z. A. Proba and K. K. Ogilvie, Can. J. Chem., 1982, 60, 1106–1113 CrossRef CAS.
- R. A. Donga, S. M. Khaliq-Uz-Zaman, T.-H. Chan and M. J. Damha, J. Org. Chem., 2006, 71, 7907–7910 CrossRef CAS PubMed.
- C. Johnston, C. Hardacre and M. E. Migaud, Chem.:Methods, 2021, 1, 382–388 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5gc01768h |
| This journal is © The Royal Society of Chemistry 2025 |