
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTwo-step hybrid photo-thermochemical looping process, using metallic clusters on metal oxide carriers, for very efficient green hydrogen production†
Anh Dung
Nguyen‡
a,
David
Buceta‡
b,
Qingqing
Wu‡
 c,
Moteb
Alotaibi
d,
Julian T.
Müller
e,
Iria R.
Arias
b,
Albert
Gili
f,
Maged F.
Bekheet
e,
Martin
Dieste
g,
Nerea
Davila-Ferreira
g,
Fatimah
Alhawiti
h,
Colin
Lambert
*c,
M. Arturo
López-Quintela
*b and
Reinhard
Schomäcker
*a
c,
Moteb
Alotaibi
d,
Julian T.
Müller
e,
Iria R.
Arias
b,
Albert
Gili
f,
Maged F.
Bekheet
e,
Martin
Dieste
g,
Nerea
Davila-Ferreira
g,
Fatimah
Alhawiti
h,
Colin
Lambert
*c,
M. Arturo
López-Quintela
*b and
Reinhard
Schomäcker
*a
aTechnische Universität Berlin, Department of Chemistry, Straße des 17. Juni 124, 10623, Berlin, Germany
bLaboratory of Magnetism and Nanotechnology (NANOMAG), Department of Physical Chemistry, Institute of Materials (iMATUS), Universidade de Santiago de Compostela, 15782 Santiago de Compostela, Spain
cPhysics Department, Lancaster University, Lancaster LA1 4YB, UK
dDepartment of Physics, College of Science and Humanities in Al-Kharj, Prince Sattam Bin Abdulaziz University, Al-Kharj 11942, Saudi Arabia
eTechnische Universität Berlin, Faculty III Process Sciences, Institute of Materials Science and Technology, Chair of Advanced Ceramic Materials, Straße des 17. Juni 135, 10623 Berlin, Germany
fHelmholt-Zentrum Berlin für Materialien und Energie, 14109, Berlin, Germany
gNanogap, Polig.Ind. Novo Milladoiro, c/Xesta 74, 15895 Milladoiro, A Coruña, Spain
hDepartment of Physics, University College of Taraba, Taif University, Taraba 21944, Saudi Arabia. E-mail: frhawiti@tu.edu.sa
First published on 26th June 2025
Abstract
In this work, we demonstrate a sustainable method for producing high-purity hydrogen through a two-step water-splitting process that leverages reducible oxides to store and release oxygen independently of hydrogen. Unlike conventional solar thermochemical (STCH) water-splitting techniques, which require extremely high temperatures exceeding 1000 °C, our approach operates at significantly lower temperatures—below ≈600 °C—thanks to a sunlight-driven photocatalyst composed of silver metal clusters (Ag5) supported on ceria and Ce–Zr oxygen storage materials. This lower-temperature operation not only reduces the demand for high-performance materials for the design of the process but also enhances safety, simplifies system design, and improves the long-term stability of both materials and equipment. Overall, this green technology offers an energy-efficient and environmentally responsible pathway for clean hydrogen production. Density-functional-theory calculations show that Ag5 clusters (1) enhance the photo-absorption, especially in the visible range, by increasing the gap states of the CeO2 surface, and decrease the oxygen vacancy formation energy (EVO) in certain positions around the clusters dramatically, and (2) create active sites in the Ag-CeO2 interface possessing lower reaction energy and activation barrier for the hydrogen evolution reaction. Guided by these studies, we demonstrate a cleaner and more energy-efficient hydrogen production process, achieving an average output of ≈55 mL per cycle (≈435 μmol g−1 h−1). This corresponds to an oxygen vacancy parameter δ ≈ 0.26 per cycle—about 51% of the theoretical maximum—significantly surpassing the performance of traditional high-temperature STCH methods. Notably, our process reaches a solar-to-hydrogen (STH) conversion efficiency of ≈9.7%, placing it at the upper end of the typical STCH range (1–10%) while operating at much lower, intermediate temperatures. These results highlight the strong potential of this greener approach to hydrogen production, offering high efficiency due to the utilization of a broader wavelength range of the solar light and a smaller environmental footprint. Additionally, the use of methane in the reduction cycle promotes the formation of oxygen vacancies, while selectively generating carbon monoxide. The two-step concept has the potential to convert biomethane into a higher-value syngas product, with the added benefit of producing extra hydrogen. This allows for the adjustment of the CO/H2 ratio, enabling subsequent Fischer–Tropsch processing for liquid fuel production.
Green foundation1. Advancement in green chemistry: our work reduces the temperature for water splitting, enhancing hydrogen production efficiency while lowering energy consumption compared to traditional high-temperature SCTH methods, making the process more sustainable.2. Green chemistry achievement: we achieved an hydrogen production at 600 °C using a photo-catalyst (silver pentamers on ceria based oxides), improving light absorption and reaction efficiency. This process yields high-purity hydrogen with a solar-to-hydrogen conversion rate of 9.7%. 3. Further research for greener impact: to make this process even greener, future research could focus on improving efficiencies using other supporting metal oxides and optimizing it using renewable methane. This could further minimize the environmental impact. |
1. Introduction
Hydrogen is one of the most promising green energy carriers for today's energy transition. Hydrogen that is produced from renewable energies and without the use of fossil raw materials is generally referred to as green hydrogen, as these production methods are not associated with the emission of greenhouse gases, especially CO2.1 In the established processes that use fossil raw materials, CO2 is inevitably produced and emitted in a stoichiometric ratio to the hydrogen produced, which is then referred to as grey hydrogen. However, a detailed analysis of the processes for the production of green hydrogen by LCA (life-cycle assessment) also shows significant contributions to eco-indicators such as GWP (global warming potential), consumption of scarce resources or land use for these processes.2 These contributions arise primarily from the manufacture of equipment for the generation of renewable energy, e.g. PV (Photovoltaic) panels, or their conversion into hydrogen in electrolysers with precious metal electrodes. For photocatalysis, such LCAs show great potential for more efficient use of resources, as readily available materials are essentially used.3,4 In the Ag clusters investigated in this work, for example, significantly lower quantities of metals are used than in the nanoparticles of the catalysts in the electrolyzers.Photocatalytic processes utilize the energy provided by the sun to split water into its hydrogen and oxygen constituents. Since hydrogen and oxygen are the main products, the near-zero global and air emissions from these processes are a big advantage.5–7 However, currently, their low efficiencies and the lack of suitable separation of hydrogen and oxygen with sufficient purity make this process unsuitable for application at an industrial scale.8
To separate the release of hydrogen and oxygen in the same device photocatalytic hydrogen evolution is often operated in the presence of a sacrificial agent that is oxidized instead of the water, avoiding the oxygen evolution.9 Although this mode of hydrogen production is possible at ambient conditions it is economically not as attractive as it appears, due to the cost and energy demand of the sacrificial agents. The same holds for thermochemical water splitting at room temperature where a metal-water reaction can be operated in apparently catalytic mode, but also only at the expense of another sacrificial agent. For example, in ref. 10 this role is taken by tungsten carbide that is oxidized to tungsten oxide. For economic reasons, these systems will only be interesting on an academic level.
Electrochemical methods, which are the most advanced ones to provide alternatives to the currently used industrial chemical methods, can provide hydrogen and oxygen separately at the cathode and anode, respectively.11–13 However, they suffer from several disadvantages, including dependence on the electric grid, low durability, usage of noble metals as catalysts, and subsequently high capex costs.14 Although one can increase the efficiencies of these processes, using electricity coupled with light in the photoelectrochemical methods, the drawbacks remain.15
Another well-established method to separately produce hydrogen and oxygen is the thermal looping process or solar thermochemical (STCH) water-splitting.16–18 This process uses stable metal oxides from which oxygen can be released in the first step of the loop and a subsequent step in which water is split using an appropriate catalyst attached to the oxides. In the latter step, pure hydrogen is generated, and the oxygen replaces the oxygen vacancies (VO) created in the oxide during the first step. The main drawback of this method is the high temperatures needed for the process, which are ≈1800 K for the creation of oxygen vacancies (VO) in the first step and ≈1120 K for the water splitting.19,20 Such high temperatures result in handling issues of the catalysts, because of rapid quenching rates for volatile materials. This fact and the high temperatures needed are the biggest impediments to this technology being industrially viable. Although the high temperatures needed can be provided using solar concentrators, the handling of the catalysts makes it industrially not viable.21 Therefore, green efficient alternatives to the above processes are urgently needed.
Metal clusters of small atomicity, also known as atomic quantum clusters (AQCs), display unique properties due to their strong quantum confinement effects, which drive their catalytic activities for different reactions.22 Furthermore, because they possess several atoms to interact with the reactants, they provide many more degrees of freedom than single-atom catalysts. This allows fine-tuning of their catalytic properties, which can be further expanded by selecting the appropriate supports.23 At the same time, the excitonic behavior of AQCs, can be further exploited for photocatalytic processes,24 alongside the above-mentioned thermo-catalytic applications.
In this work, our density functional theory (DFT) calculations show that Ag5 clusters: (1) enhance photoabsorption—particularly in the visible range—by introducing gap states in the CeO2 surface and significantly lowers the oxygen vacancy formation energy (EVO) at certain positions around the clusters, and (2) create interfacial sites with lower reaction energy and activation barriers for the hydrogen evolution reaction. Driven by these studies and a commitment to advancing sustainable hydrogen production, we present an environmentally friendly and energy-efficient solution based on a hydrogen evolution looping (HEL) two-step water-splitting process. This green approach achieves efficiencies comparable to—or better than—the conventional solar thermochemical hydrogen (STCH) methods, while operating at significantly lower temperatures. Our method utilizes silver pentamers (Ag5 AQCs) supported on metal oxides, serving as both thermal and photocatalysts in the initial low-temperature (≈773 K) generation of oxygen vacancies (VO), and as thermal catalysts in the subsequent hydrogen production step. The entire process is designed to minimize energy input and maximize sustainability, making it a promising pathway for green hydrogen production, as shown schematically in Fig. 1.
 | ||
| Fig. 1 Looping scheme of the HEL water-splitting process. | ||
Overall, by operation of this photocatalytic reaction in a chemical looping mode, no other resources than water and solar light are consumed for the production of hydrogen. This justifies considering it as Green Hydrogen according to the International Energy Agency's definition.25
This process concept can also be considered as a contribution to Green Chemistry because it avoids fossil raw materials, emission of greenhouse gases, and hazardous chemicals by using catalysts for a by-product-free production of hydrogen in a safe and well-controllable process according to the principles of Green Chemistry.26
We will also show that this HEL concept can be extended by using CH4 in the reduction cycle to facilitate the formation of VO. Through this extension, the amount of H2 per cycle was increased by a factor of ≈60, thereby allowing the production of syngas, with an adjustable CO/H2 ratio, which could be used for the production of liquid fuels through a Fischer–Tropsch process.
2. Results and discussion
2.1. Preliminary results
First, photocatalytic water splitting was carried out with TiO2/Cu5 AQCs deposited onto TiO2 nanoparticle supports. It was observed that H2 evolved, but O2 was missing as a product (Fig. S1†). The amount of evolved H2 decreased over time until it disappeared, and the colour of the catalyst changed. Since the TiO2 nanoparticles possess oxygen vacancies and O2 could not be detected as a significant product, we speculate that VO present in the TiO2 nanoparticles must play a crucial role in the observed reaction, which resembles thermochemical water splitting, but at much lower temperatures. We could then assume that Ag5 AQCs also catalyse the H2O splitting, generating H2, and the O from H2O eliminates the VO of the TiO2 nanoparticles. Therefore, oxygen is trapped within the TiO2 and could not be detected as a product after the reaction. To continue the H2 production, the metal oxide needs to be regenerated, creating new VO in the material. We further observed that the Ag5 AQCs could also catalyse the regeneration of the VO at 773 K (increasing the amount using light irradiation – see later), confirming the dual catalytic/photocatalytic activity of such clusters. To carry out further studies, we choose CeO2, which is an oxide with higher oxygen storage capacity than TiO2.Fig. S2† shows the feasibility of the hydrogen evolution looping (HEL) process as a photocatalytic modified thermochemical approach. For this purpose, the production of H2, where H2O was introduced as steam during the second step of the HEL, was carried out at the same temperature (773 K) used for the VO formation in the first step of the HEL. Although the hydrogen production in this preliminary experiments was small (≈ at 1.5 μmol per cycle or ≈10 μmol g−1 h−1) it indicates the potentialities of the process. To get inside into the HEL looping process and enhance the understanding of the whole process, and with the aim of optimizing the whole process we first carried out more experiments and in-depth simulations based on DFT for the two thermal/photocatalytic steps.
2.2. Step 1: oxygen vacancies’ formation
 | ||
| Fig. 2 Results of DFT-calculation of trapezoidal (2d) and bipyramidal (3d) Ag5 on CeO2 in the absence and presence of VO. (A) Energetical stability of a 2d and 3d Ag5 deposited on pristine CeO2 surface. (B) Geometries and oxygen vacancy positions of a trapezoidal Ag5 AQC adsorbed on CeO2. (C) Geometries and oxygen vacancy positions of a bipyramidal Ag5 AQC adsorbed on CeO2. (D) Related oxygen vacancy formation energy (EVO) of the different positions around AQCs. (E) The density of states and wavefunctions of the polaron of the 3d Ag5 on a pristine CeO2 surface. (F) Density of states of the 3d Ag5 on reduced CeO2 surface with VO on site 1. (G) Density of states of the 3d Ag5 on reduced CeO2 surface with VO on site 6. | ||
First, we find that the AQCs could enhance the photon absorption, especially in the visible range, by increasing the gap states of the CeO2 surface, which is in line with previous results.28,29 Compared to the bare surface (Fig. S3†), Fig. 2E shows some electronic states (blue peaks) originating from the Ag atoms and the polaronic state (green peak) stemming from charge transfer from Ag5 to the substrate in the ∼3.1 eV band gap. The inset picture displays the polaronic state localized on the surface Ce near the Ag5 at an energy of 1 eV below the conduction band, due to the 0.6 electron transfer from the AQC. This excess electron reduces the particular Ce4+ to Ce3+. More polaronic states are observed in the presence of VO. Fig. 2F and G (VO on site 1 and 6 of 3d geometry) each show four polarons created by the two excess electrons due to the one VO and the 1.0 electron transfers (forming two excess electrons) from the Ag5 depicted by the green peaks in the band gap. Similar phenomena can be found for 2d isomers in Fig. S4.† The detailed localization of Ce atoms can be found in Fig. S5.† In addition, based on the case of 3d Ag5 on the pristine surface in Fig. 3E, one further electron is transferred from the AQC to the ceria substrate. Therefore, we conclude that VO and Ag5 AQCs reinforce each other since the polaronic states caused by Ag5 could not only coexist, but also be enhanced by those generated by VO (the charge transfer from AQCs is enhanced by VO). Also, two spin-polarized states (blue peaks in the band gap) are observed and attributed to the states sitting on the Ag5 cluster. Since the unoccupied state above the Fermi energy (0 eV) could accept an excited electron from the valence band, while the electron on the other occupied state could be excited to the conduction band, the AQCs are expected to enhance the separation of electron–hole pairs by absorbing the photons in the visible range.30,31
 | ||
Fig. 3
In situ XRD of (A) CeO2 and (B) CeO2/Ag5 AQCs, with the insights showing a magnification of the CeO2(202) reflection. For both experiments, the initial black patterns were obtained in (Step-1), rt-723 K under Ar![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2 = 2:2 mL min−1; alternating red and blue patterns were obtained at the end of the respective Ar:H2 = 2:2 mL min−1 (red) or Ar:O2 = 2:2 mL min−1 (blue) segments of 2 min. :H2 = 2:2 mL min−1; alternating red and blue patterns were obtained at the end of the respective Ar:H2 = 2:2 mL min−1 (red) or Ar:O2 = 2:2 mL min−1 (blue) segments of 2 min. | ||
Secondly, we predict a decrease in the EVO by depositing Ag5 AQCs on the surface of CeO2. Table D in Fig. 2 demonstrates that the EVO is dramatically decreased in some positions around the AQCs (e.g., 0.14 eV, 0.59 eV) compared to the bare substrate 1.85 eV (more details could be found in Fig. S6†). It should be noted that these values depend on the parameters chosen in the DFT calculations, especially the value of Hubbard U.32 The dependence is further confirmed and demonstrated in Fig. S7:† 2.6 eV is predicted at Hubbard U = 4.5 eV for the bare substrate which agrees well with the cited literature. Some lattice distortions around the AQCs are observed, which are speculated to be the main factor leading to a decreasing oxygen vacancy formation energy together with the injection of electrons to the substrate from the AQCs destabilizing the lattice. This means that the bond energy between the Ce atoms and the lattice oxygen atom on the surface is decreased, thereby facilitating oxygen vacancy formation.
Time-resolved in situ XRD measurements also confirm the formation of a higher amount of oxygen vacancies VO in the CeO2 lattice when Ag5 AQCs are deposited on CeO2 compared to pure commercial CeO2, meaning Ag5 AQCs catalyze oxygen vacancy formation. Fig. 3 shows in situ XRD analysis of both the CeO2 commercial material (A, left) and the CeO2/Ag5 AQCs (B, right) (see ESI, Note 1†). An inset in each graph displays a magnified view of the CeO2(202) reflection at 2Θ angles of approximately 14.8°. The patterns are stacked and progress with TOS (time on stream) from bottom to top. All along the experiment shown, both materials maintain the fluorite Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m structure, without the appearance of side phases, which can be better fitted to a PDF 96-434-3162. Initially, during heating under Ar:H2=1:1 (black patterns), the position of the reflections shifts to lower 2Θ angles. This shift is caused by the expansion of the unit cell volume, due to thermal expansion and potentially, a contribution caused by the reduction of Ce4+ to Ce3+ and the associated formation of VO.33,34 To disentangle these contributions, alternating cycles of 2 min of H2 and O2 in Ar are applied: the graphs display the last acquired pattern of the H2 (red) and O2 (blue) cycles. On the one hand, the commercial CeO2 material reflections position barely shifts during the alternating cycles, as shown in the magnification insight of panel 3.A.1. On the other hand, the CeO2/Ag5 material reflections position significantly shift during the cycles (see Fig. 3B.2). At the end of the H2-rich periods, the unit cell has expanded (decrease of the 2Θ angle). This expansion is caused by the generation of VO,34,35 which reaches the bulk of the material (powder XRD is inherently a bulk characterization technique). At the end of the O2-rich periods, the peak maximum position is recovered, meaning that the VO is reoccupied. We note that such processes are relatively fast and occur within 2 minutes of exposure to the gas phase. Upon cooling, the peak position shifts back to higher angles due to unit cell thermal contraction.
m structure, without the appearance of side phases, which can be better fitted to a PDF 96-434-3162. Initially, during heating under Ar:H2=1:1 (black patterns), the position of the reflections shifts to lower 2Θ angles. This shift is caused by the expansion of the unit cell volume, due to thermal expansion and potentially, a contribution caused by the reduction of Ce4+ to Ce3+ and the associated formation of VO.33,34 To disentangle these contributions, alternating cycles of 2 min of H2 and O2 in Ar are applied: the graphs display the last acquired pattern of the H2 (red) and O2 (blue) cycles. On the one hand, the commercial CeO2 material reflections position barely shifts during the alternating cycles, as shown in the magnification insight of panel 3.A.1. On the other hand, the CeO2/Ag5 material reflections position significantly shift during the cycles (see Fig. 3B.2). At the end of the H2-rich periods, the unit cell has expanded (decrease of the 2Θ angle). This expansion is caused by the generation of VO,34,35 which reaches the bulk of the material (powder XRD is inherently a bulk characterization technique). At the end of the O2-rich periods, the peak maximum position is recovered, meaning that the VO is reoccupied. We note that such processes are relatively fast and occur within 2 minutes of exposure to the gas phase. Upon cooling, the peak position shifts back to higher angles due to unit cell thermal contraction.
We conducted a Rietveld refinement analysis on the XRD data to make a quantitative analysis of the oxygen vacancies. The results indicate that the lattice parameter of fluorite-type CeO2 in the initial CeO2/Ag5 sample (a = 5.4129 ± 0.0001 Å) is larger than that of bare CeO2 (a = 5.4107 ± 0.0001 Å). The higher lattice parameter of CeO2 in the CeO2/Ag5 is attributed to the partial reduction of smaller Ce4+ cations to larger Ce3+ in the fluorite structure [r(Ce3+) = 1.143 Å, r(Ce4+) = 0.970 Å; all cations are 8-fold coordinated]36 and the associated formation of VO. Fig. 4A shows the lattice parameter of CeO2 in both samples as a function of time and gas atmosphere at an isothermal temperature of 723 K. The reduction and oxidation periods are marked in green and red, respectively. The lattice parameter of the fluorite structure in the CeO2 sample remains constant, regardless of the atmosphere. In contrast, the CeO2/Ag5 sample exhibits a significant increase in its lattice parameter over time when the atmosphere is switched to H2. This increase is followed by a return to the initial value during the subsequent oxidation cycle. The mole fraction x of Ce3+ in the reduced fluorite phases, described as CeIIIx CeIV1−xO2−δ and the oxygen vacancies parameter δ (i.e. x/2) were also determed using the following relationship:33

 | ||
| Fig. 4 (A) Lattice and (B) oxygen vacancy parameters (δ) for pure CeO2 and CeO2/Ag5 AQCs as a function of time and gas atmosphere at 723 K. Reduction (green) and oxidation (red) periods are highlighted. | ||
Fig. 4B shows δ as a function of time and gas atmosphere at an isothermal temperature of 450 °C. Reduction and oxidation periods are represented in green and red, respectively. As shown, the VO increases from about 0.004 to 0.02 over time when the atmosphere is switched to H2 and decreases back to the initial value when the atmosphere is switched to O2. Notably, the maximum level of VO during the reduction step decreases with each cycle, dropping from 0.02 in the first cycle to ≈0.016 in the last reduction cycle (this fact could be related to the increase in crystallite size of CeO2 observed in the CeO2/Ag5 sample see ESI, Note 2†). In contrast, the lattice parameter, the VO and the crystallite size in the bare CeO2 sample remain constant. These results demonstrate that the presence of Ag5 on the surface of CeO2 enables the formation of VO in the bulk of the support at the operating conditions tested, as theoretically predicted in the previous section, whereas the lack of Ag5 results in a material showing no significant bulk appearance of vacancies.
Finally, to analyze the influence of light, we show in Fig. 5 the XPS results of the used catalyst before and after irradiation. The comparison of the XPS Ce 3d spectra clearly shows that there is an increase in the relative ratio of the Ce3+ features u0 and v0 at ∼903.0 and 885 eV, respectively.37,38 The deconvolution of both before and after irradiation samples is included in Fig. 5, with quantitative results displayed in Table S1† and indicating an increase of the surface atomic concentration of %Ce3+ from 26.0 to 40.4%. Following XRD, the XPS results further prove that Ag5 clusters photocatalyze the reduction of Ce4+ and the formation of oxygen vacancies, confirming the dual catalytic/photo-catalytic role played by the clusters.
 | ||
| Fig. 5 XPS Ce3d from CeO2/Ag5 AQCs system before and after irradiation (top) and deconvoluted spectra. | ||
To further explain the experimental results, we can use the Gibbs free energy change for the thermal transformation 2CeO2 → Ce2O3 + 0.5 O2, represented in Fig. 6 (we also include in the figure the transformation in H2 and CH4 atmospheres, corresponding to the reactions 2CeO2 + H2 → Ce2O3 + H2O and 2CeO2 + CH4 → Ce2O3 + CO + H2, respectively). From this figure, the temperature at which this is a spontaneous reaction can be estimated. It is observed that the thermal reduction needs very high temperatures, but the reduction of CeO2 by H2 (red line) has a more negative value of ΔG compared to the thermal reduction of CeO2 (black continuous line), indicating that the reduction is facilitated in a H2 atmosphere. At 773 K, although there is a significant reduction of ΔG° (≈205 kJ mol−1), H2 alone cannot transform CeO2 into Ce2O3, suggesting that it could not produce VO at 773 K (ΔG° ≈ 75 kJ mol−1 > 0). However, one has to consider that the energy needed for vacancy formation is smaller than for transforming CeO2 into a new thermodynamic phase (Ce2O3). Indeed, it is reported that at 0 K bulk CeO2 can be transformed into CeO2−x with EVO ≈ 251 kJ mol−1 (2.6 eV at Hubbard U = 4.5 eV shown in Fig. S7†), which is smaller than the value observed in Fig. 6 for the transformation of CeO2 into Ce2O3 (EVO at 0 K ≈ 380 kJ mol−1) implying a reduction of ≈130 kJ mol−1. Assuming a similar thermodynamic dependence of EVO with temperature for values of x < 0.5 than for x = 0.5 (a similar behavior was calculated by Wang et al.40 for SrFeO3), then the calculated EVO at T = 773 K in a H2 atmosphere should be reduced to ≈75 kJ mol−1–130 kJ mol−1 ≈ −55 kJ mol−1, indicating that H2 alone could produce VO at 773 K. On the other hand, the experimental results show that without clusters, the formation of vacancies is negligible, probably because of the large activation energy involved in the process, requiring the use of a catalyst to overcome the energy barrier.
 | ||
| Fig. 6 Gibbs free energy change for CeO2 and CeO2 reacting with CH4 and H2 to Ce2O3 (entropy and enthalpy taken from ref. 39). | ||
As it was seen above, Ag5 AQCs can also reduce the energy of vacancy formation. According to DFT, there is a reduction in ΔG° (at 0 K) for the vacancy formation of about 170 kJ mol−1 (≈1.76 eV) in some positions41 (see Fig. 2). Considering the reduction noted above of EVO for x < 0.5, ΔG° at 773 K for pure CeO2 ≈ 150 kJ mol−1 (280 kJ mol−1–130 kJ mol−1). Then, for CeO2/Ag5 AQCs, the predicted value of ΔG° at 773 K ≈ 150 kJ mol−1–170 kJ mol−1 ≈ −20 kJ mol−1, indicating that the reaction is thermodynamically possible and at the same time, Ag5 AQCs can catalyze the reaction, which explains the experimental results that the formation of VO is spontaneous at this temperature. Of course, the use of a H2 atmosphere with CeO2/Ag5 AQCs should imply a much larger driving force for the production of vacancies, so that the process of vacancy formation takes place in only 2 minutes in a H2 atmosphere, as it was noted above (results shown in Fig. 3).
We now proceed to look into the second step of the HEL looping process in more detail. For this purpose, we carried out DFT calculations and more experiments for the water-splitting step.
2.3. Step 2: water splitting
In practice, there are VO on the surface and even in the bulk of ceria, and water splitting occurs on these VO31,32. Therefore, the effect of an oxygen vacancy, but in different positions is considered (Fig. 7). For the reduced surface in the absence of Ag5, it is found the water prefers the VO site, and no energy activation barrier is found for the water dissociation process, which is consistent with the reports of Wu, et al. and Li, et al. mentioned above. The dissociated [OH] group fills the VO and the [H] binds to the neighbor O, forming two –OH groups. However, a huge energy barrier of around 4.3 eV is calculated for the breaking of two O–H bonds and formation of the H2 molecule, which is the rate-determining step of HER. This feature agrees well with the above literature. The configuration of the transition state and the reaction path for H2 forming from separate H are shown in Fig. S9A.† This shows that one O–H bond breaks first and the dissociated H moves towards the other activated O–H, leading to a H⋯H distance of 0.85 Å. The next step involves the desorption of H2 molecules from the ceria surface, which is an exothermic process. In contrast, two reaction paths corresponding to VO positions 1 and 6 in Fig. 2C are shown as pink and blue plots in Fig. 7 and reveal that the Ag5 not only provides active sites on the surface of the AQC, but also the deposition of Ag5 induces some active sites in the interface of AQC and substrate. The corresponding reaction free energy diagrams for hydrogen evolution reaction at 773 K are shown in Fig. 7. In the case of vacancy 6 (blue path), we find a spontaneous water dissociation process which means no intact adsorbed water around AQC on the substrate can be found above vacancy site 6. For the following H2 formation process, it is found the reaction energy decreases by 0.64 eV and the activation energy decreases by 1.32 eV compared to the bare reduced substrate (black reaction path; see the definition of activation and reaction energies in Fig. S9C†). As a consequence, the 3d Ag5 AQC creates at least two positions at the interface (the symmetric position on the right side of AQC) possessing lower reaction and activation energies which improve the catalytic activity for HER. In the case of vacancy 1 (pink path), the adsorption free energy of a water molecule is 0.35 eV. For the subsequent step, a 1.2 eV activation energy is found for the water dissociation, which is an exothermic reaction where one H binds to AQC and the remaining [OH] fills the O vacancy site 1. However, a zero-activation energy is obtained for H2 formation from the intermediate where one [H] binds to Ag5 and the other binds to the oen oxygen on CeO2 surface. And the energy barrier of this H2 formation step (reaction energy of 1.81 eV) is suppressed by around 1 eV compared to that of the blue path (activation energy of 2.83 eV; site 6: reduced surface with Ag5) and by 2.34 eV compared to that of the black path (activation energy of 4.15 eV; reduced bare surface). Therefore, the path corresponding to vacancy 1 (pink) shows further improvement for HER. More details about transition states for water splitting and H2 formation can be found in Fig. S9 and S10.† In the final desorption step, one H2 desorption process remains exothermic but has a low free energy of less than 0.1 eV (blue path), while the other becomes endothermic with a free energy of 0.3 eV (pink path) in the presence of AQCs. This represents a significant deviation from the free energy of 0.77 eV on the bare surface, which is attributed to the extra coupling from Ag5 compared to the bare substrate. In conclusion, the AQCs could provide active sites in the H2 formation step, which means the deposition of Ag5 AQC increases the catalytic activity in this thermochemical process. Overall, the doping or deposition of Ag5 AQCs works better than the metal atom doping in the ceria system reported by Li et al., where the HER process is inhibited.31
 | ||
| Fig. 7 Reaction free energy diagrams for H2 generation on the reduced CeO2 surface in the absence (black path) and presence (site 1: pink path and site 6: blue path) of pyramidal Ag5 AQC at 773 K. | ||
 | ||
| Fig. 8 Net H2 evolution per cycle with the optimized HEL looping method with Ce0.5Zr0.5O2/Ag5 AQCs (0.5%). Reaction conditions: T = 873 K, m Ce0.5Zr0.5O2 = 2.727 g. | ||
It is noteworthty that the results obtained by the HEL procedure surpass all previously reported results for ceria-based compounds in high temperature STCH, as shown in Table S2.† As indicated in the same table, our results are comparable even to the STCH outcomes obtained with other Ce-based oxides, perovskites and high entropy oxides at high temperatures, showing that a much greener procedure for the H2 production can be achieved by this procedure.
These results clearly indicate that the lower temperatures used in our looping process are due to the photo-catalytic activity of clusters overcoming the thermal instabilities observed in different materials, with better performances than the Ce-based compounds (such as single and double-oxide substituted ABO3 perovskites, iron aluminate-based spinels or high entropy oxides16), which is a major drawback for their application in STCH.
2.4. Further experiments using the HEL looping concept: oxygen vacancy regeneration with CH4
To further show the versatility of the HEL looping concept, we studied the introduction of methane in the first step of oxygen vacancy formation. We can observe in Fig. 6 that at 773 K, the difference between the Gibbs free energy change in H2 as a reducing agent and CH4 as a reducing agent is 40 kJ mol−1. This highlights the possibility that CH4 could act as a reducing agent at 773 K to facilitate the formation of VO. It is known that doping can increase oxygen ion conductivity. With increased oxygen ion conductivity, the number of VO formed by Ag5 AQCs could be increased, resulting in a higher entropy and thus, a larger gradient/slope. Since vacancies affect the catalyst's band gap, a reversible color change already points to the formation of VO (Fig. S8†). Fig. 9 shows the DRS spectra of CeO2/Ag AQCs after reduction in CH4/light, after impregnation (original state), and after oxidation in air. It has been observed that the optical properties depend on the catalyst's oxygen vacancy density. A higher amount of VO inside the material results in enhanced optical properties in the light's visible spectrum.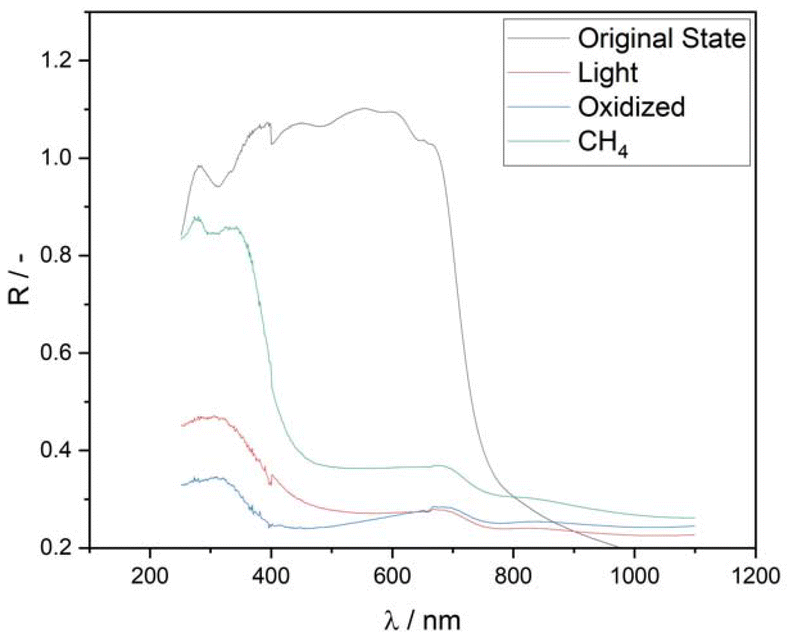 | ||
| Fig. 9 DRS spectra of CeO2/Ag5 AQCs after impregnation, reduction with light/CH4 and its oxidized state. | ||
Fig. 10 shows the XRD of CeO2/Ag5 AQCs after different catalyst treatments. As can be seen, shifts in the characteristic peaks occurred compared to the catalyst's oxidized state. When the catalyst is irradiated, reduced by methane, or after impregnation (so that the support material already has VO), the characteristic peaks are shifted to lower 2θ angles compared to the catalyst's oxidized state. Therefore, it can be concluded that with Ag AQCs deposited on CeO2 the oxygen vacancy formation with CH4 or light is no longer a surface effect, since a change in the bulk phase can be observed. This can be explained by the enhanced interaction between Ag and Ce species, as the oxygen mobility and activation are facilitated, similar to Ag-supported manganese catalysts for the oxidation of toluene42. The peaks shift to lower 2θ angles and are associated with an expansion of the unit cell caused by the oxygen extraction from the lattice.35
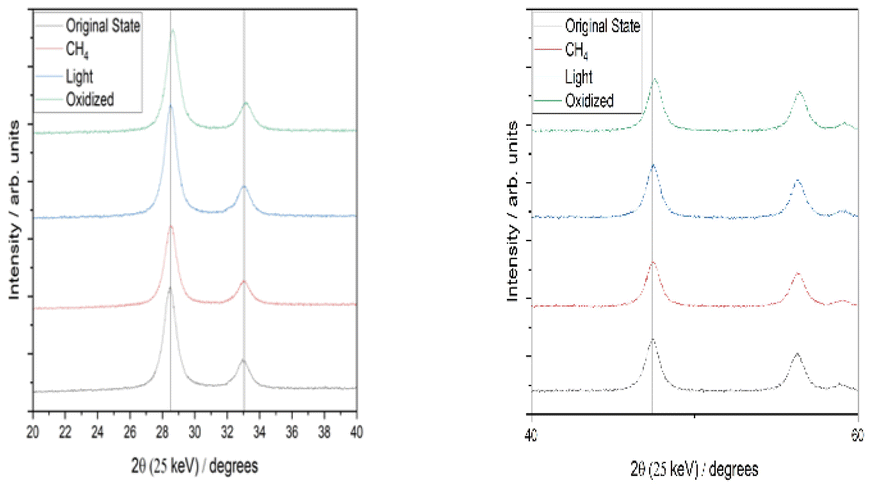 | ||
| Fig. 10 XRD spectra of CeO2/Ag5 AQCs with different treatments. | ||
By looking at the XPS spectra of the Ce 3d (Fig. 11A), it can be observed that reducing the catalyst with CH4/light results in an increase of the typical features associated to Ce3+ at ≈885 eV and ∼903 eV. Deconvolution leads to a surface atomic concentration of Ce3+ of 42.6% (see Table S1†). Fig. 11B depicts the Ag 3d XPS spectra of the sample after reduction with CH4/light and the oxidized state of the catalyst. There is no shift of the characteristic peak for the sample exposed to light, hinting that the silver clusters remain in the same oxidation state in both steps of the looping process. Using CH4 as a reducing agent, CeO2 with Ag5 AQCs deposited on the surface can be recovered at 773 K for the following oxidation cycle, even without light. As light can induce VO, exposing the catalyst to light in a CH4 environment can accelerate the oxide regeneration. The following reaction is proposed since H2 and CO were identified as products with VO, while formed VO caused the catalyst's color change:
| 2Ce4+ + O2− + CH4 → 2H2 + CO + Ce3+ + VO |
 | ||
| Fig. 11 XPS spectra of CeO2/Ag5 AQCs. Ce3d (left) Ag3d (right) after reduction with CH4/light and in the oxidized state. | ||
If CH4 can be used as a reducing agent, then CeO2/Ag5 AQCs should be able to be reoxidized by H2O to produce H2, as we have already seen before. By curing the CH4/light-induced vacancies, the final gas should only contain H2 after reoxidation. Since the oxidation and reduction of the catalyst are reversible, the amount of H2 per cycle should not decrease after several cycles. Fig. 12 shows the amount of H2 evolved per cycle during 16 cycles. Each experiment is performed under the same conditions with the same catalyst, only with a different irradiation source. The average amount of H2 produced per cycle is shown on the right.
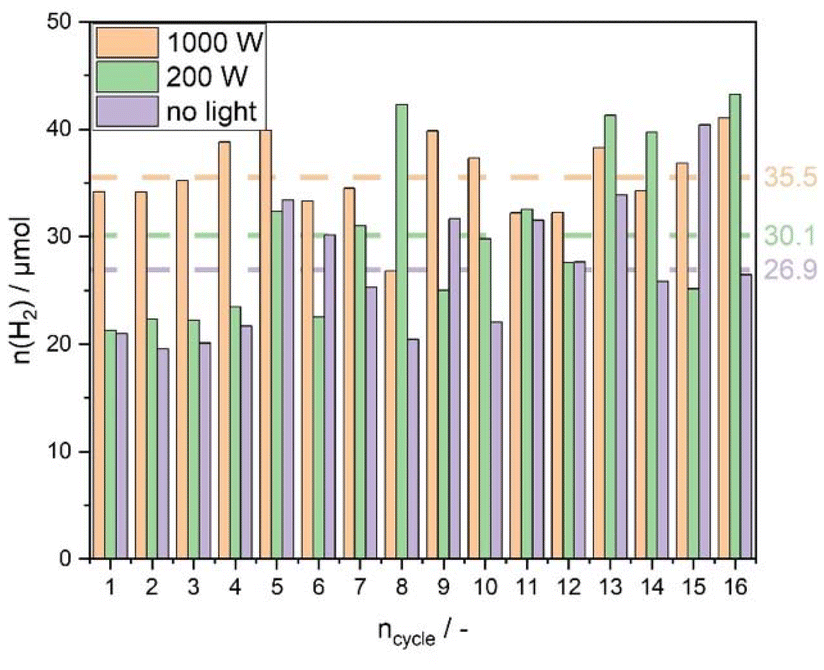 | ||
| Fig. 12 HEL loop with CeO2/Ag5 AQCs (0.5 wt% Ag5) and CH4 as reducing agent, reaction conditions: m CeO2 = 50 mg, Airradiation = 19.6 cm2, dV/dt = 15 ml min−1 with 2:1 Ar:CH4 for the reduction cycle, dV/dt = 13 ml min−1 with 10:3 Ar:H2O, T = 773 K, irradiation source: no light, 200 W Hg (Xe) lamp/1000 W Xe lamp, λ < 1450 nm, toxidation = 0.5 h, treduction = 0.5 h. | ||
It is observed that with light, the average amount of H2 evolved per cycle is higher compared to the experiment in which the catalyst was not exposed to light during the reduction cycle. With higher intensity, the amount per cycle even increased to ≈35% with 1000 W intensity. Looking at the m/z = 2 signal from the mass spectrometer (Fig. S14†) it can be concluded that the oxidation cycle time is too short to consume all the vacancies formed during the reduction cycle. This means that the reaction rate for the oxidation cycle at 773 K is not high enough to oxidize the catalyst completely. Since the amount of H2 generated per cycle depends on the light source used, the reaction rate for the oxidation cycle does not follow a zero-order kinetic. With a higher amount of available VO formed in the reduction cycle, the reaction rate of the oxidation cycle is increased, resulting in an amount of evolved H2 of 600 μmol g−1 h−1, which corresponds to a δ ≈ 0.10, and represents ≈20% of the theoretical maximun number of vacancies. The proposed reaction for the HEL with CH4 can be described similarly to that proposed by Wang et al. for the Rh-CeO2/Al2O3 catalyst43 with roxidation < rreduction.

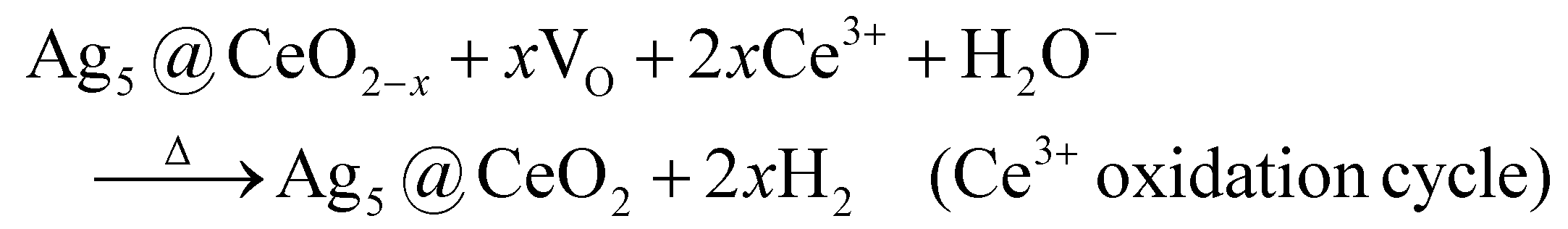
These results clearly show the possibility of extending our HEL concept to transform biomethane into a more added-value syngas product, with the possibility to obtain separately more hydrogen, so that the CO/H2 ratio could be adjusted to carry out subsequently a Fischer–Tropsch process to produce liquid fuels.
The comparison of different hydrogen technologies is very challenging, due to the utilization of different reaction feedstocks (water, methane, biomass) and auxiliaries (catalysts, microorganisms, electrodes).44 Due to the huge research activities in this field, there are many new developments on very different technology readiness levels.45 A comparison of technologies is only possible if performance or economic indicators can be determined within the same system boundaries.46 Therefore, often the same technology is assessed with some variations, for example, the combination of photovoltaic electricity production and electrolysis for hydrogen production.47 But even here, different boundary conditions cause a huge variation, e.g. in the production cost of a kg of hydrogen for €6 to €15 per kg.48 Another interesting case for comparison is thermochemical water splitting, using the thermal energy of the sun to drive reduction-oxidation cycles of redox systems, especially metal oxides at high temperatures.34 Due to the very different TRLs (technology readiness levels) and integration in value chains, a direct comparison is only possible using key indicators like solar-to-hydrogen efficiency, land use expressed in the hydrogen yield per m2 and year, and the production costs. Even without being able to make a quantitative comparison between STCH and HEL technology, there are clear advantages to the HEL concept. (1) Through photocatalysis, HEL utilizes a significantly larger proportion of the spectrum of sunlight, as the UV/VIS range is used in addition to infrared radiation. (2) Due to the lower process temperature of approx. 600 °C, the pressure drop in the product gas stream is lower than at the temperature of the STCH process at about 1200 °C and thus requires less compression energy for feeding the hydrogen into the downstream processes. Together with point #1 this results in a more efficient utilization of the available solar energy at a given area. (3) Evaluations of technologies for the use of solar energy that performed TEAs (Techno-economic analysis) and LCAs for high-temperature processes such as STCH, that requires so-called solar towers or heliostats, and technologies that use parabolic mirrors to collect sunlight have repeatedly come to the conclusion that the investment costs for the parabolic mirror systems are significantly lower.49–51 The main reason for this is the lower demands on the materials used, which only have to withstand temperatures of up to 600 °C.29,31 For PV/electrolysis, the STH efficiency is about 15%, which yields 5 to 7 kg H2 per m2 and year resulting in production costs in a range of €5 to €10 per kg.45 The same indicator of STCH is STH = 2 to 10%, hydrogen yield of 0.3 to 0.75 kg m−2 years, (which represents the target value of the EU Green Deal), and production costs of €10 to €15 per kg H2.52 For a comparison of the presented results, the obtained experimental data were converted into macroscopic units and a scale-up scenario was used. A solar concentrator is assumed to focus the sunlight with 10-fold intensity on a receiver tube. This is coated with 100 g m−2 of AQC/CeO2. Assuming the same utilization of 50% of the oxygen of the oxide, as in the optimized procedure, in each redox cycle would produce ≈ 87 × 2 (assuming only a moderate increase with the light intensity as we saw above, Fig. 12) = 174 mg per m2 of reactor surface and cycle. With 4 cycles per hour and 2000 sun hours per year, this would yield 1.39 kg H2 m−2 of reactor surface and year, with an STH efficiency of ≈10% which clearly competes with the other mentioned methods. Due to the high purity of the hydrogen, no purification is required. For light collection parabolic mirror should be sufficient for providing the intensity and temperature for the proposed process, keeping the design rather simple and inexpensive.
3. Conclusions
Building huge photoreactors to harvest the energy for H2 would explode the CAPEX costs (photoreactor53 €1000 per m2vs. parabolic solar collector54 €220 per m2), resulting in uneconomically high H2 production costs. One approach is to concentrate the light using parabolic mirror systems as an example. Concentrating the light increases the photon density and the operating temperature. Photocatalysts with a core–shell structure have been studied in the literature, which can split water with a total quantum efficiency of close to 100%.55 A disadvantage that occurs is that H2 and O2 are formed in a ratio of 2:1 without any spatial separation.
Here we demonstrated an alternative approach for producing H2 is a two-step hybrid thermochemical cycle called Hydrogen Evolution Looping (HEL). DFT calculations were performed, predicting that the Vo formation energy is reduced by depositing Ag5 AQCs on CeO2. The operating temperature of the Vo reduction cycle was lowered by exposing the catalytic system (CeO2/Ag AQCs) to light compared to pure CeO2. Pure CeO2 required 1670 K–1870 K for its reduction.56 With CeO2/Ag AQCs at 773 K, VO were already formed on the catalyst's surface. DFT calculations also show that Ag5 AQCs create active sites in the interface possessing lower reaction energy and an activation barrier for the hydrogen evolution reaction. H2 production via HEL was stopped by preventing the exposure to light of the catalyst. Light-induced oxygen vacancy formation was identified as a surface effect under the investigated reaction conditions, which confirms the latest results from the literature.31 By creating a metal–ceria interface, the oxygen ion conductivity can be increased, facilitating the transfer of bulk oxygen to the surface.57 Using the H2-assisted method under optimized conditions, with Ce0.5Zr0.5O2/Ag AQCs (under 2.5 suns and at 873 K), we can generate approximately half of the theoretical maximun number of vacancies per cycle. This results in a production rate of 435 μmol g−1 h−1 (≈10 mL g−1 h−1), which surpasses all previously reported results for ceria-based compounds in high temperature STCH, as shown in ESI, Table 1.† As indicated in the same table, our results are comparable to the STCH outcomes obtained with other Ce-based oxides, perovskites and high entropy oxides at high temperatures. This suggest the potential to further enhance the efficiency of our method by using such compounds while also reducing the risk of the oxide deterioration by lowering the operating temperature. CH4 can be used at 773 K as an educt to create vacancies in CeO2/Ag AQCs for the reduction cycle. In this case, this is no longer a surface effect as the vacancies are formed throughout the bulk phase. The reduction can be accelerated by exposing the catalyst to light. Higher intensity leads to higher reaction rates, but the dependence is non-linear. By using CH4 in the reduction cycle to facilitate the formation of VO, the amount of H2 per cycle is increased, resulting in an amount of evolved H2 of 600 μmol g−1 h−1 (10% of the maximum number of vacancies per cycle) and allowed the production of syngas with an adjustable CO/H2 ratio, which could be used for the production of liquid fuels through a Fischer–Tropsch process.
4. Experimental
4.1. Chemicals
The following chemicals/materials were used as purchased: silver foils (99.95% purity) were purchased from Goodfellow, UK. Sandpaper (1000 grit) was supplied by Wolfcraft España S.L, Madrid, Spain. All aqueous solutions were prepared with MilliQ-grade water using a Direct-Q8UV system from Millipore (Millipore Ibérica S.A., Madrid, Spain). Commercially available Titanium oxide (P25, Evonic), Cerium oxide (>99.5%, Alfa Aesar) and Ce0.5Zr0.5O2 (Sigma Aldrich) were used as supports. Argon (99.999%, Air Liquide Deutschland GmbH & Sigma Aldrich), Methane (99.5%, Linde Gas GmbH), Hydrogen (99.999%, Air Liquide Deutschland GmbH & Sigma Aldrich).4.2. Catalyst preparation
There is a well-established correlation between the emission energy (Eem) of clusters and the number of atoms (N) they contain, as predicted by the spherical jellium model: Eem ≈ EHOMO–LUMO = EF/N1/3, where EHOMO–LUMO gap is the energy gap between the highest occupied and lowest unoccupied molecular orbitals, and EF is the Fermi energy of the bulk metal.59 The fluorescence spectra can therefore be used to determine the size distribution of clusters, which is consistent with other characterization techniques, such as Mass Spectrometry (see ref. 58 for further details). Fig. S15† shows the emission spectra of the cluster samples used in this study, featuring a single peak at λ ≈ 409 nm (3.1 eV). Using the jellium model and assuming EF ≈ 5.3 eV for Ag, it can be inferred that the clusters primarily consist of five atoms [Njellium ≈ 5.3], which aligns with the sizes previously reported for clusters with a similar emission peak.56
X-ray Photelectron Spectroscopy (XPS) measurements were carried out using a K-Alpha system (Thermo Fisher Scientific, USA) equipped with a monochromatic Al Kα source. Samples were prepared on carbon pads. Spectra were acquired in constant analyzer energy mode with a pass energy of 50 eV, a step size of 0.1 eV, and a spot size of 400 μm. All XPS spectra were calibrated using the C 1s core level at a binding energy of 284.8 eV. The spectra were deconvoluted using Fityk software,60 with assigned peaks according to literature37,38 : Ce+4 (v = ∼882.0 eV, v′′ = ∼888.2 eV, v′′′ = ∼898.0 eV, u = ∼900.8 eV, u′′ = ∼907.3 eV, u′′′ = ∼916.6 eV) and Ce3+ (v0 = ∼885.0 eV, v′ = ∼880.0 eV, u0 = ∼903.0 eV, u′ = ∼898.3 eV). The background signal was removed using the Shirley method61 and the different peaks were fitted using Pseudovoigt functions. The FWHM (Full width at half maximum) for the Ce3+ and Ce+4 groups was fixed, separately. The atomic surface concentrations of Ce3+, Ce4+ were calculated from peak areas according to eqn (X), where ACe3+ is the sum of the areas of the features described above.
 | (X) |
The reactor is connected to a 3-way valve, which can introduce dry argon or water-saturated argon into the reactor. Fig. S16a† (reduction cycle) and Fig. S16b† (oxidation cycle) show the flow charts of the used setup to perform HEL experiments. First, the catalyst (mcat = 50–400 mg) was added to 10–40 mL of pure water followed by an ultrasonication for 5 min. Afterward, the suspension was added on top of an Erlenmeyer flask containing a stainless-steel sinter plate surrounded by a hollow cylinder of Teflon. The flask is connected to a vacuum pump and by evacuating, the catalyst is deposited on the top of the sinter plate. The deposited catalyst is then dried overnight in an oven at 333 K. After putting the sintered plate into the reactor, the pipes and the reactor are evacuated and purged with Ar four times, followed by purging the water saturator with Ar for 30 min with a volume flow of 200 mL min−1 to remove air in the pipes. After heating up the reactor to the required temperature with a ramp of 3.3 K min−1. For some experiments, the sample was pretreated with H2 or CH4. The cyclic experiment contains 2 phases characterized by their duration time: an irradiation phase (tirradation) for the creation of VO and an oxidation phase (toxidation) for the formation of hydrogen. While irradiating, dry Ar passes the reactor. By introducing water vapor in the oxidation phase, H2 is evolving. Switching between dry Ar and with water-saturated Ar is conducted by a 3-way valve controlled by a servo motor. The outgoing gas was analyzed once every 30 min by gas chromatography. (7890A, Agilent Technologies, Porapak-N column). The reaction was performed at 673 K–773 K with an intensity of 4000 W m−2 supplied by a 200 W Hg lamp/40000 W m−2 supplied by a 1000 W Xe lamp.
The optimized HEL experiments were performed in a quartz reactor with an internal diameter of 9 mm and length of 50 cm
The sample is supported by quartz wool in the irradiation zone of the reactor, heated with an oven (Hobersal HTF), irradiated with an intensity of 2500 W m−2 supplied by a 1000 W Xe lamp (Quantum Design). Set up and measure conditions were similar to ones described in 5.1. The outgoing gas was analyzed once every 15 min by gas chromatography (Clarus 580, PerkinElmer) and every second by a MS spectrometer (OmniStar GSD 350, Pfeiffer vacuum).
4.3. Theoretical methods
The periodic slab model CeO2(111) (Ce atoms: 48; O atoms: 96) is composed of 15.58 Å × 15.58 Å unit cells arranged in three O–Ce–O trilayers with a 15 Å vacuum layer above the surface. The geometrical and electrical properties for pristine or reduced CeO2(111) surface and Ag5 deposited on pristine or reduced CeO2(111) were calculated by density functional theory (DFT) based simulation package Vienna Ab initio simulation package (VASP). The periodic boundary condition method and spin-polarized DFT were adopted. The projector augmented wave (PAW) method was performed to describe the interaction between valence electrons and the ion cores and the generalized-gradient approximation (GGA) of Perdew, Burke, and Ernzerhof (PBE)67 was utilized for the exchange–correlation functionals. A plane-wave basis set with a 500 eV cut-off energy was used to expand the Kohn–Sham wave functions. Hubbard U term68U = 6.3 eV to correct on-site coulombic interactions of cerium 4f electrons was adopted for the lattice relaxation.69,70 More specifically, the results presented in Fig. 3 and 7 were obtained by using U = 6.3 eV while U = 4.0, 4.5, 5, 5.5, 6, 6.5 were utilized to optimize the unit cell of CeO2 to build each surface slab respectively and the dependence of Hubbard U for the VO formation energy was finally characterized and shown in Fig. S7.† In all cases, the Brillouin zone was sampled at the Γ point due to the large unit cell (15.576 Å × 15.576 Å) in the two directions with periodic boundary conditions (x and y directions). The Becke-Jonson (BJ) damping function in Grimme's D3 method71 was adopted to include van der Waals corrections due to the accuracy for reproducing the binding or adsorption energies on metal oxide surfaces.The electronic structures (wavefunctions and density of states) of the CeO2(111) substrate, and Ag5 AQCs deposited on CeO2(111) in the absence and presence of O vacancy were performed with spin-polarized HSE06 calculations based on the optimized configurations in the earlier DFT+U with D3 (BJ) performance since HSE06 is really time-consuming.72–74 In the HSE06 hybrid exchange–correlation functional, the exchange potential consists of a short-range PBE part mixed with an HF exchange and a long-range PBE part. Bader analysis method75 is applied to the results of spin-polarized HSE06 calculation for atomic charge distributions. The VO formation energy is defined below:
| EVO = 1/xE(CeO2−x) + 1/2E(O2) − 1/xE(CeO2) |
To find transition states (TS), a damped molecular dynamics algorithm was combined with the climbing-image nudged elastic band (CI-NEB) method.76,77 During the CI-NEB calculation, the structural optimization would be completed when the maximum forces on all atoms were smaller than 0.04 eV Å−1. Finally, a check was made to confirm that only one imaginary frequency is present for the transition state. All the optimized structures were visualized by VESTA.78
Free energy diagrams display the free energy variation (ΔG) along the hydrogen evolution reaction from the reactants to the final products through various intermediate states. The Free energy was calculated by
| G = EDFT + EZPE − TS |
Author contributions
Anh Dung Nguyen designed and conducted the photocatalytic experiments and contributed to writing the manuscript. David Buceta designed and supervised the synthesis and deposition of the Ag5 AQCs, and collaborates in the overall conception and discussions of the photocatalytic experiments. Qingqing Wu and Moteb Alotaibi conducted the theoretical calculations and wrote the corresponding part of the manuscript. Iria R. Arias synthesised and purified the Ag5 AQCs, prepared the deposited AQCs in the substrates and collaborates in the discussion and analysis of the optimized HEL experiments. Julian Müller and Albert Gili contributed with the in situ XRD investigation of the catalysts. Maged F. Bekheet performed Rietveld refinement analysis on the obtained in situ XRD data, and contributed to the discussion of the results. Martin Dieste and Nerea Davila-Ferreira conducted the optimized HEL experiments and collaborate in their overal conception, discussion and analyis. Fatimah Alhawiti contributed to the discussion and examination of the VO calculations. Colin Lambert designed and supervised the theoretical results, and collaborated in the overall conception and discussion of the manuscript. M. Arturo López-Quintela, supervised the results and contributed to the overall conception, discussion, and writing the manuscript. Reinhard Schomäcker supervised the results and contributed to the overall conception, discussion, and writing the manuscript. Review and editing were done by all co-authors.Conflicts of interest
Anh Dung Nguyen, David Buceta, Qingqing Wu, Colin Lambert, M. Arturo López Quintela and Reinhard Schomäcker are co-inventors of a patent filed, which is related to the looping process described in the manuscript.Data availability
Data for this article are available at: (1) Experimental data can be uploaded from the following link: https://doi.org/10.14279/depositonce-19490. (2) Theoretical data can be uploaded from the following link: https://www.dropbox.com/scl/fo/t65j2xo9njcrm1e69jvh2/AJrSMnzyd9epuEWaV-MyZyM?rlkey=zxi0i7tlukzc7dkbetf2nz03o&dl=0. (3) HEL optimized results can be uploaded from the following link: data optimized HEL (https://nubeusc-my.sharepoint.com/:f:/g/personal/malopez_quintela_usc_es/Ek4qcGJoQNVGkfgh_DzyLEoBKgeqj03RnPkNGOsQXhcD9A?e=WtVXei).Acknowledgements
This research was partially supported by the Consellería de Educación (Xunta de Galicia), Grants No. Grupos Ref. Comp. ED431C-2021/16; Spanish Ministerio de Ciencia e Innovación (MICIU) PID2022-142334OB-I0, financed by MICIU/AEI/10.13039/501100011033 and European Union (ERDF/EU); and TED2021-131899B-I00, financed by MICIU/AEI /10.13039/501100011033 and EU Next Generation (EU/PRTR). CJL and QW acknowledge support from the EPSRC Program grant “Quantum Engineering of Energy-Efficient Molecular Materials”. A. Gili acknowledges support by the German Federal Ministry of Education and Research in the framework of the project Catlab (03EW0015A). JTM and MFB thank the Advanced Light Source, which is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the US Department of Energy under Contract No. DE-AC02-05CH11231, and where the in situ XRD measurements where conducted at beamline 12.2.2 in the framework of the AP proposals (ALS-11921).References
- J. Incer-Valverde, A. Korayem, G. Tsatsaronis and T. Morosuk, Energy Convers. Manage., 2023, 291, 117294 CrossRef CAS.
- D. Alghool, M. Haouari and P. Trucco, Int. J. Hydrogen Energy, 2024, 81, 214–224 CrossRef CAS.
- J. Maurya, E. Gemechu and A. Kumar, Int. J. Hydrogen Energy, 2023, 48, 20077–20095 CrossRef CAS.
- C. Nelson, M. M. Gui and P. K. J. Robertson, Adv. Energy Sustainability Res., 2025, 6, 2400337 CrossRef CAS.
- H. Nishiyama, T. Yamada, M. Nakabayashi, Y. Maehara, M. Yamaguchi, Y. Kuromiya, Y. Nagatsum, H. Tokudome, S. Akiyama, T. Watanabe, R. Narushima, S. Okunaka, N. Shibata, T. Takata, T. Hisatomi and K. Domen, Nature, 2021, 598, 304–318 CrossRef CAS.
- A. Gupta, B. Likozar, R. Jana, W. C. Chanu and M. K. Singh, Int. J. Hydrogen Energy, 2022, 47, 33282–33307 CrossRef CAS.
- M. J. Molaei, Fuel, 2024, 365, 131159 CrossRef CAS.
- S. Alsayegh, J. R. Johnson, B. Ohs and M. Wessling, J. Cleaner Prod., 2019, 208, 1446e1458 CrossRef.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS.
- T. Yamamoto, S. Ashida, N. Inubuse, S. Shimizu, Y. Miura, T. Mizutani and K.-ichi Saitow, J. Mater. Chem., 2024, 12, 30906–30918 RSC.
- B. You and Y. Sun, Acc. Chem. Res., 2018, 51, 1571–1580 CrossRef CAS.
- X. Xiao, L. Yang, W. Sun, Yu Chen, H. Yu, K. Li, B. Jia, L. Zhang and T. Ma, Small, 2022, 18, 2105830 CrossRef CAS.
- D. T. Tran, P. K. L. Tran, D. Malhotra, T. H. Nguyen, T. T. An Nguyen, N. T. A. Duong, N. H. Kim and J. H. Lee, Nano Convergence, 2025, 12, 9 CrossRef CAS.
- Md M. Rashid, M. K. Al Mesfer, H. Naseem and M. Danishshould, Int. J. Eng. Adv. Technol., 2015, 4(3), 2249–8958 Search PubMed.
- Yi Yang, S. Niu, D. Han, T. Liu, G. Wang and Y. Li, Adv. Energy Mater., 2017, 7, 1700555 CrossRef.
- J. T. Tran, K. J. Warren, S. A. Wilson, C. L. Muhich, C. B. Musgrave and A. W. Weimer, Wiley Interdiscip. Rev.: Energy Environ., 2024, 13, e528 CAS.
- S. Abanades, Chem. Eng., 2019, 3, 63 CAS.
- D. Oudejans, M. Offidani, A. Constantinou, S. Albonetti, N. Dimitratos and A. Bansode, Energies, 2022, 15, 3044 CrossRef CAS.
- F. Safari and I. Dincer, Energy Convers. Manage., 2020, 205, 112182 CrossRef CAS.
- Y. Mao, Y. Gao, W. Dong, H. Wu, Z. Song, X. Zhao, J. Sun and W. Wang, Appl. Energy, 2020, 267, 114860 CrossRef CAS.
- C. L. Muhich, B. D. Ehrhart, I. Al-Shankiti, B. J. Ward, C. B. Musgrave and A. W. Weimer, Wiley Interdiscip. Rev.: Energy Environ., 2016, 5, 261–287 CAS.
- H. Rong, S. Ji, J. Zhang, D. Wang and Y. Li, Nat. Commun., 2020, 11, 5884 CrossRef CAS.
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS.
- See e.g. Y. A. Attia, D. Buceta, C. Blanco-Varela, M. B. Mohamed, G. Barone and M. A. López-Quintela, J. Am. Chem. Soc., 2014, 136, 1182–1185 CrossRef CAS; N. Vilar-Vidal, J. R. Rey and M. A. López-Quintela, Small, 2014, 10, 3632–3636 CrossRef; Ye Liu, Yu Wang and N. Pinna, ACS Mater. Lett., 2024, 6, 2995–3006 CrossRef.
- Global Hydrogen Review 2022-Analysis, International Energy Agency, 2022, p 71, Available from: https://www.iea.org Search PubMed.
- P. T. Anastas and J. C. Warner. Green chemistry: theory and practice, Oxford University Press, Oxford [England]; New York, 1998. ISBN 9780198502340 Search PubMed.
- M. Alotaibi, Q. Wu and C. Lambert, Appl. Surf. Sci., 2013, 613, 156054 CrossRef.
- M. P. de Lara-Castells, A. W. Hauser, J. M. Ramallo-López, D. Buceta, L. J. Giovanetti, M. A. López-Quintela and F. G. Requejo, J. Mater. Chem. A, 2019, 7, 7489–7500 RSC.
- P. López-Caballero, J. M. Ramallo-López, L. J. Giovanetti, D. Buceta, S. Miret-Artés, M. A. López-Quintela, F. Requejo and M. P. de Lara-Castells, J. Mater. Chem. A, 2020, 8, 6842–6853 RSC.
- Yu Zhang, D. T. Payne, C. L. Pang, C. Cacho, R. T. Chapman, E. Springate, H. H. Fielding and G. Thornton, J. Phys. Chem. Lett., 2019, 10, 5265–5270 CrossRef CAS.
- R. Li, C. Wen, K. Yan, T. Liu, B. Zhang, M. Xub and Z. Zhouc, J. Mater. Chem. A, 2023, 11, 7128–7141 RSC.
- T. Wu, Q. Deng, H. A. Hansen and T. Vegge, J. Phys. Chem. C, 2019, 123, 5507–5517 CrossRef CAS.
- M. F. Bekheet, M. Grünbacher, L. Schlicker, A. Gili, A. Doran, J. D. Epping, A. Gurlo, B. Klötzerb and S. Penner, CrystEngComm, 2019, 21, 145–154 RSC.
- A. Gili, M. F. Bekheet, F. Thimm, B. Bischoff, M. Geske, M. Konrad, S. Praetz, C. Schlesiger, S. Selve, A. Gurlo, F. Rosowskidg and R. Schomäcker, Catal. Sci. Technol., 2024, 14, 4174 RSC.
- M. F. Bekheet, M. Grünbacher, L. Schlicker, A. Gili, A. Doran, J. D. Epping, A. Gurlo, B. Klötzerb and S. Penner, CrystEngComm, 2019, 21, 145 RSC.
- R. D. Shannon and C. T. Prewitt, Acta Crystallogr., Sect. A, 1969, 25, 925 CrossRef CAS.
- P. Burroughs, A. Hamnett, A. F. Orchard and G. Thornton, J. Chem. Soc., Dalton Trans., 1976, 17, 1686–1698 RSC.
- E. Bêche, P. Charvin, D. Perarnau, S. Abanades and G. Flamant, Surf. Interface Anal., 2008, 40, 264–267 CrossRef.
- CRC Handbook of Chemistry and Physics, ed. D. R. Lide, 76th edn, 2003, vol. 84, p. 2474 Search PubMed.
- X. Wang, Y. Gao, E. Krzystowczyk, S. Iftikhar, J. Dou, R. Cai, H. Wang, C. Ruan, S. Ye and F. Li, Energy Environ. Sci., 2022, 15, 1512–1528 RSC.
- Note: 1.76 eV is obtained by the energy reduction from the oxygen vacancy formation energy 2.6 eV of CeO2 in the absent of Ag5 to 0.84 eV of Ag5/CeO2. 0.84 eV is calculated by 0.14 eV + 0.7 eV since the oxygen vacancy formation energy elevates with the decrease of Hubbard U, where 0.84 eV is a linear prediction (0.14 eV for U = 6.3; then 0.84 eV for U = 4.5) from the dependence plotting on Hubbard U of the oxygen formation energy.
- Z. Qua, Y. Bua, Y. Qina, Yi Wanga and Q. Fub, Appl. Catal., B, 2013, 132–133, 353–362 CrossRef.
- R. Wang, H. Xu, X. Liu, Q. Ge and W. Li, Appl. Catal., A, 2006, 305, 204–210 CrossRef CAS.
- I. Dincer and C. Acar, Int. J. Hydrogen Energy, 2014, 40, 11094–11111 CrossRef.
- I. Dincer and C. Acar, Int. J. Hydrogen Energy, 2015, 42, 14843–14864 CrossRef.
- A. J. Carrillo, J. González-Aguilar, M. Romero and J. M. Coronado, Chem. Rev., 2019, 119, 4777–4816 CrossRef CAS.
- M. Chatenet, B. G. Pollet, D. R. Dekel, F. Dionigi, J. Deseure, P. Millet, R. D. Braatz, M. Z. Bazant, M. Eikerling, I. Staffell, P. Balcombe, Y. Shao-Horn and H. Schäfer, Chem. Soc. Rev., 2022, 51, 4583–4762 RSC.
- J. Collis and R. Schomäcker, Front. Energy Res., 2022, 10, 909298 CrossRef.
- N. A. Jailani, A. Ahmad and N. Norazahar, J. Energy Saf. Technol., 2021, 4, 13–28 Search PubMed.
- F. Hosseinifard, G. R. Aghdami, M. Salimi and M. Amidpour, Fuel Process. Technol., 2025, 271, 108207 CrossRef CAS.
- L. B. Gobio-Thomas, M. Darwish and V. Stojceska, Therm. Sci. Eng. Prog., 2023, 46, 102224 CrossRef.
- F. Safari and I. Dincer, Energy Convers. Manage., 2020, 205, 112182 CrossRef CAS.
- A. Cabrera Reina, S. Miralles-Cuevas, L. Cornejo, L. Pomares, J. Polo, I. Oller and S. Malato, Renewable Energy, 2020, 145, 1890–1900 CrossRef CAS.
- A. Giostri, M. Binotti, P. Silva, E. Macchi and G. Manzolini, J. Sol. Energy Eng., 2013, 135, 011001 CrossRef.
- T. Takata, J. Jiang, Y. Sakata, M. Nakabayashi, N. Shibata, V. Nandal, K. Seki, T. Hisatomi and K. Domen, Nature, 2020, 581, 411–414 CrossRef CAS.
- H. S. Cho, T. Myojin, S. Kawakami, N. Gokon, T. Kodama, Y. H. Kang, S. N. Lee, K. K. Chai, H. K. Yoon and H. J. Lee, Energy Procedia, 2014, 49, 1922–1931 CrossRef CAS.
- R. J. Gorte, AIChE J., 2010, 56, 1226–1135 CrossRef.
- V. Porto, D. Buceta, B. Domínguez, C. Carneiro, E. Borrajo, M. Fraile, N. Davila-Ferreira, I. R. Arias, J. M. Blanco, M. C. Blanco, J. M. Devida, L. J. Giovanetti, F. G. Requejo, J. C. Hernández-Garrido, J. J. Calvino, M. López-Haro, G. Barone, A. M. James, T. García-Caballero, D. M. González-Castaño, M. Treder, W. Huber, A. Vidal, M. P. Murphy, M. A. López-Quintela and F. Domínguez, Adv. Funct. Mater., 2022, 32, 2113028 CrossRef CAS.
- see e.g. J. Zheng, C. Zhang and R. M. Dickson, Phys. Rev. Lett., 2004, 93, 077402 CrossRef; B. Santiago-González and M. A. López-Quintela, in Functional Nanometer-Sized Clusters of Transition Metals: Synthesis, Properties, and Applications, ed. W. Chen and S. Chen, The Royal Society of Chemistry, 2014, ch. 2, pp. 25–50 Search PubMed.
- M. Wojdyr, J. Appl. Crystallogr., 2010, 43, 1126–1128 CrossRef CAS.
- D. A. Shirley, Phys. Rev. B, 1972, 5, 4709 CrossRef.
- A. Gili, et al. , ACS Catal., 2019, 9(8), 6999–7011 CrossRef CAS.
- L. Schlicker, et al. , Rev. Sci. Instrum., 2018, 89, 033904 CrossRef.
- C. Prescher and V. P. Prakapenka, High Pressure Res., 2015, 35, 223–230 CrossRef CAS.
- J. Rodriguez-Carvajal, J. Phys. B, 1993, 192, 5 Search PubMed.
- L. W. Finger, D. E. Cox and A. P. Jephcoat, J. Appl. Crystallogr., 1994, 27, 892–900 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. J. Humphreys and A. P. Sutton, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 1505–1509 CrossRef CAS.
- R.K Singha, Y. Tsuji, M.H Mahyuddin and K. Yoshizawa, J. Phys. Chem. C, 2019, 123, 9788–9798 CrossRef CAS.
- Y. Jiang, J. B. Adams and M. van Schilfgaarde, J. Chem. Phys., 2005, 123, 064701 CrossRef.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef.
- P López-Caballero, A W. Hauser and M P de Lara-Castells, J. Phys. Chem. C, 2019, 123, 23064–23074 CrossRef.
- S. Dong, B. Li, X. Cui, S. Tan and B. Wang, J. Phys. Chem. Lett., 2019, 10, 4683–4691 CrossRef CAS.
- F. Alhawiti, et al. , Phys. Chem. Chem. Phys., 2024, 26, 27088–27097 RSC.
- E. Sanville, S. D. Kenny, R. Smith and G. Henkelman, J. Comput. Chem., 2007, 28, 899–908 CrossRef CAS.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- G. Henkelman and H. Jónsson, J. Chem. Phys., 2000, 113, 9978–9985 CrossRef CAS.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5gc01425e |
| ‡ These authors are considered as co-first authors. |
| This journal is © The Royal Society of Chemistry 2025 |