
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Deoxygenative dual CO2 conversions: methylenation and switchable N-formylation/N-methylation of tryptamines†
Kazuto
Takaishi
 *,
Hajime
Morishita
,
Kosuke
Iwaki
and
Tadashi
Ema
*
*,
Hajime
Morishita
,
Kosuke
Iwaki
and
Tadashi
Ema
*
Division of Applied Chemistry, Graduate School of Environmental, Life, Natural Science and Technology, Okayama University, Tsushima, Okayama 700-8530, Japan. E-mail: takaishi@okayama-u.ac.jp; ema@cc.okayama-u.ac.jp
First published on 8th April 2025
Abstract
The unprecedented one-pot synthesis of N-formyl/N-methyltryptolines from tryptamines was achieved via phenylsilane-assisted deoxygenative dual CO2 conversions. Two CO2 molecules acted as different synthons and were converted into methylene and N-formyl/N-methyl groups. The CO2 reduction step was catalyzed by a pentanuclear zinc complex at atmospheric pressure under solvent-free conditions. The N-formyl/N-methyl products could be switched by changing the amount of phenylsilane, and the amounts of in situ generated bis(silyl)acetals and silyl formates were key to the chemoselectivity. Methylenation, N-formylation, and N-methylation proceeded via the Pictet–Spengler reaction, amine–acid condensation, and the Eschweiler–Clarke reaction, respectively. The CO2 reduction with phenylsilane could also be applied to the one-pot three-step synthesis of spiro[oxindole-pyrrolidine]s.
Green foundation1. We have developed new types of deoxygenative dual CO2 conversions in which two CO2 molecules act as different synthons and are converted into methylene and N-formyl/N-methyl groups of tryptolines, known as important compounds in biology. These CO2 fixation methods will expand the usefulness of CO2.2. The CO2 conversions have been achieved under solvent-free conditions at atmospheric pressure. The CO2 reduction step was catalyzed by a small amount (0.07 mol%) of zinc complex, which could be prepared easily, and therefore noble and toxic metals are not required. Moreover, H2O could be used as an additive. 3. Our methods could be made greener by catalyst reuse strategies in flow chemistry. In addition, methods that allow for the application of a variety of sustainable reductants are recommended. |
Introduction
CO2 is a renewable C1 building block and is expected to serve as an alternative to petroleum-based chemicals. Therefore, the development of fine organic synthetic methods with CO2 for value-added chemicals has become increasingly important.1 Among CO2 fixations, deoxygenative conversions involving C–H/C–C bond formation, such as conversion to methyl (–CH3)2 or methylene (–CH2–)3 groups, are still rare despite their importance. Deoxygenative CO2 conversions require reductants, and H2,4 hydroboranes,5,6 and hydrosilanes6,7 are often used in the presence of catalysts. In the case of hydrosilanes, the reduction of CO2 generates reactive species: silyl formates (HCO2Si), bis(silyl)acetals (SiOCH2OSi), and methoxysilanes (CH3OSi), which are easily hydrolyzed to formic acid, formaldehyde, and methanol, respectively (Scheme 1a). Among the silyl species, bis(silyl)acetals are potentially useful for C–H/C–C bond-forming reactions, but there are few reports.6 | ||
| Scheme 1 CO2 fixation with ZnII complex 1. | ||
Double CO2 conversions, where two CO2 molecules are captured by one substrate molecule, are attractive molecular conversions. Although several double carboxylations have been reported,8 the number of double deoxygenative CO2 conversions reported is severely limited. For example, the Yan–You–Jiang group reported the construction of a tetrahydropyrimidine ring in which two CO2 molecules were converted into two methylene groups,9 and our group reported the construction of a fused benzene ring by conversion into two aromatic CH groups.10 In contrast, the Qi–Jiang group reported the synthesis of α-methyl-β-diketones from aryl iodides, alkynes, and CO2, where two CO2 molecules were transformed into two different moieties, carbonyl and methyl groups.11 The Nan group has recently developed the synthesis of methylindoloquinoxalines from indolylanilines with CO2via double N-formylation in which two CO2 molecules were converted into an aromatic carbon and a methyl group.12 Such dual CO2 conversions will expand the usefulness of CO2, and the development of new types of conversions would be a highly valuable achievement.
We have previously reported a macrocyclic pentanuclear ZnII complex 1, which can be easily prepared via the self-assembly of the binaphthyl–bipyridyl ligand H2L and Zn(OAc)2·2H2O (Scheme 1b).13 Complex 1 exhibited catalytic activity toward deoxygenative CO2 conversions via the hydrosilylation of CO2:14 temperature-switched N-formylation/N-methylation of amines (Scheme 1c),13,15C-methylenation of arenes (Scheme 1d),14a and the synthesis of 3,4-dihydropyrans from β-dicarbonyl compounds and styrenes (Scheme 1e).14b
More recently, during the investigation of the scope of 1-catalyzed CO2 fixation, we unexpectedly noticed that tryptamine (2a) was converted into 2-formyltryptoline (3a) (tryptoline is also known as 1,2,3,4-tetrahydro-β-carboline), a novel compound (Scheme 1f). In this synthesis, two CO2 molecules were incorporated into two different moieties, methylene and N-formyl groups, and this type of CO2 conversion is unprecedented. The Pictet–Spengler cyclization involving formaldehyde and amine–formic acid condensation seems to proceed. Tryptoline derivatives often exhibit pharmacological activities in the treatment of serious diseases such as mitochondrial disease, Alzheimer's disease, and malaria.16 Therefore, expanding the synthetic methods of tryptolines would be a significant contribution, and we decided to investigate this synthesis. As a result, we established the synthetic conditions leading to 3a and found that the product could be switched to 2-methyltryptoline (4a). In addition, one-pot three-step reactions, Br-assisted semi-pinacol rearrangement17 after the formation of 3a or 4a, proceeded to form spiro[oxindole-pyrrolidine]s 5a and 6a (Scheme 1g). Some derivatives with this spiro-skeleton also exhibit pharmaceutical activity,18 and the method matches the pot-economy concept.19 Therefore, this strategy is promising.
Results and discussion
Optimizing the reaction conditions
We first optimized the reaction conditions for the synthesis of formyltryptoline 3a from 2a (Table 1). After the reaction of CO2 (1 atm) with PhSiH3 (8 equiv.) in the presence of catalyst 1 (0.07 mol%) under solvent-free conditions, 2a was added along with additives AcOH and H2O (entries 1–5). The yield was sensitive to temperature, and the optimal temperatures were 55 and 120 °C for the first and second steps, respectively (83%, entry 4).|
|
|||||
|---|---|---|---|---|---|
| Entry | X (equiv.) | T 1 (°C) | T 2 (°C) | Yieldb (%) | |
| 3a | 4a | ||||
| a Conditions: CO2 (1 atm, balloon, 3.7 L), PhSiH3 (6–14 equiv.), cat. 1 (0.07 mol% based on PhSiH3), 2a (0.25 mmol), AcOH (200 μL), H2O (100 μL). b Determined by 1H NMR using styrene as an internal standard. c Isolated yield. d Absence of AcOH. e Absence of H2O. f DMSO (100 μL) was added. | |||||
| 1 | 8 | 45 | 100 | 38 | 5 |
| 2 | 8 | 55 | 100 | 62 | 8 |
| 3 | 8 | 65 | 100 | 49 | 12 |
| 4 | 8 | 55 | 120 | 83 (80)c | 6 |
| 5 | 8 | 55 | 140 | 29 | 4 |
| 6d | 8 | 55 | 120 | 27 | 21 |
| 7e | 8 | 55 | 120 | 60 | 19 |
| 8 | 6 | 55 | 120 | 43 | 3 |
| 9 | 10 | 55 | 120 | 64 | 34 |
| 10 | 12 | 55 | 120 | 14 | 82 (83)c |
| 11f | 14 | 55 | 120 | 4 | 82 |
| 12 | 14 | 55 | 120 | Trace | 76 |

In the first step, key C1 intermediates, bis(silyl)acetals and silyl formates, are considered to accumulate. When AcOH or H2O was absent, the yields decreased (27–60%, entries 6 and 7). We also screened hydrosilanes and additives, which resulted in lower yields (Tables S1–S4†). We noticed that methyltryptoline 4a was generated as a byproduct (4–21%, entries 1–7) and changed the amount of PhSiH3 to increase the yield of 4a (entries 8–12). Compound 4a was formed in the highest yield (82%, entry 10) when 12 equiv. of PhSiH3 were used. The addition of DMSO did not improve the yield (entry 11). On the basis of the above results, entries 4 and 10 were found to be the best conditions for the synthesis of 3a and 4a, respectively. The structures of 3a and 4a were unambiguously confirmed by X-ray crystallography (Fig. 1 and S2†).20
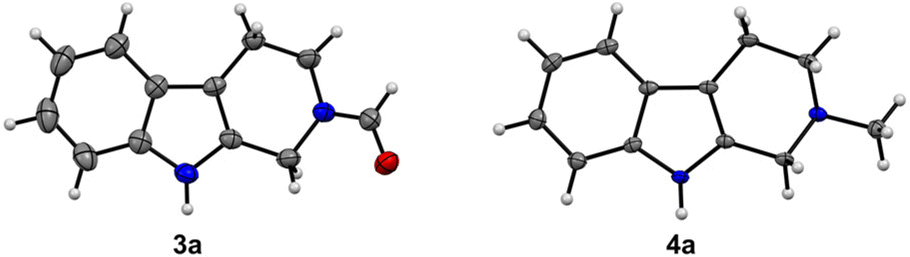 | ||
| Fig. 1 ORTEP drawings of X-ray crystal structures of 3a and 4a. The thermal ellipsoids are scaled to the 50% probability level. | ||
Substrate scope
We next investigated the substrate scope for the chemoselective dual CO2 conversions (Scheme 2). When 5-methyl-, 5-methoxy-, 5-bromo-, 7-methyl-, 1-methyl-, and 1-ethyl-substituted tryptamines were used as substrates, the corresponding formyltryptolines 3b–3g (51–78%) and methyltryptolines 4b–4g (46–74%) were obtained selectively in moderate to high yields, depending on the amount of phenylsilane. Moreover, 1-benzyl-, 1-(4-methoxybenzyl)-, and 1-(4-chlorobenzyl)-substituted tryptamines were also converted into formyltryptolines 3h–3j (45–84%) or methyltryptolines 4h–4j (55–89%) in gratifying yields despite the large steric hindrance. The fact that even 1-substituted tryptamines underwent the reactions demonstrates the usefulness of this synthetic method. In addition, (S)-tryptophan methyl ester could be transformed into (S)-3k (26%, 78% ee at T2 of 130 °C) or (S)-4k (38%, 74% ee at T2 of 80 °C) although partial racemization proceeded. The racemization could be suppressed to a minimum by lowering the reaction temperature ((S)-3k: 17%, 97% ee at T2 of 90 °C; (S)-4k: 18%, 98% ee at T2 of 45 °C). For the scope of substrates beyond tryptamines, benzofuran and dimethoxybenzene analogs were converted into methylated tetrahydrobenzofuropyridine 4l (60%) and tetrahydroisoquinoline 8 (70%), respectively, in gratifying yields, although the yields of formylated products were low. The aforementioned results indicate that the substrate scope is relatively broad.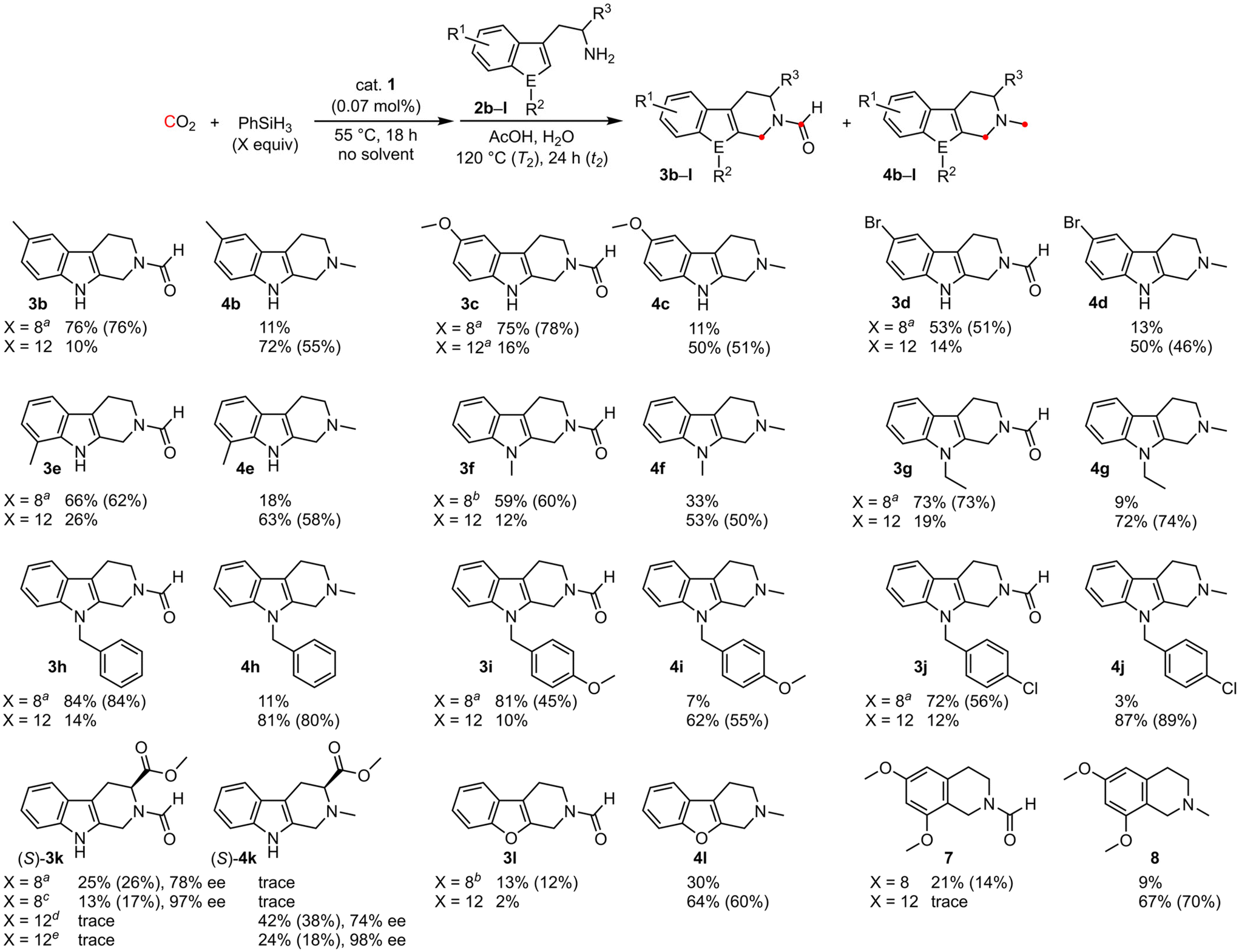 | ||
Scheme 2 Substrate scope. Yields were determined by 1H NMR using styrene as an internal standard. Isolated yields are shown in parentheses. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) T2 = 130 °C. bT2 = 110 °C. cT2 = 90 °C. dT2 = 80 °C, t2 = 3 h. eT2 = 45 °C. T2 = 130 °C. bT2 = 110 °C. cT2 = 90 °C. dT2 = 80 °C, t2 = 3 h. eT2 = 45 °C. | ||
Isotope-labeling and control experiments
To elucidate the carbon and hydrogen sources for the construction of 3a and 4a, isotope-labeling experiments were conducted. When the reaction was performed with 13CO2, 3a′ possessing one each of 13C-methylene and 13C-formyl groups and 4a′ possessing one each of 13C-methylene and 13C-methyl groups were obtained without the formation of 3a and 4a (Scheme 3a).21 When the reaction was performed with PhSiD3, 3a′′ possessing deuterated methylene and formyl groups and 4a′′ possessing deuterated methylene and methyl groups were obtained without the formation of 3a and 4a (Scheme 3b). These results clearly demonstrate that the incorporated carbon and hydrogen atoms originated from carbon dioxide and phenylsilane, respectively.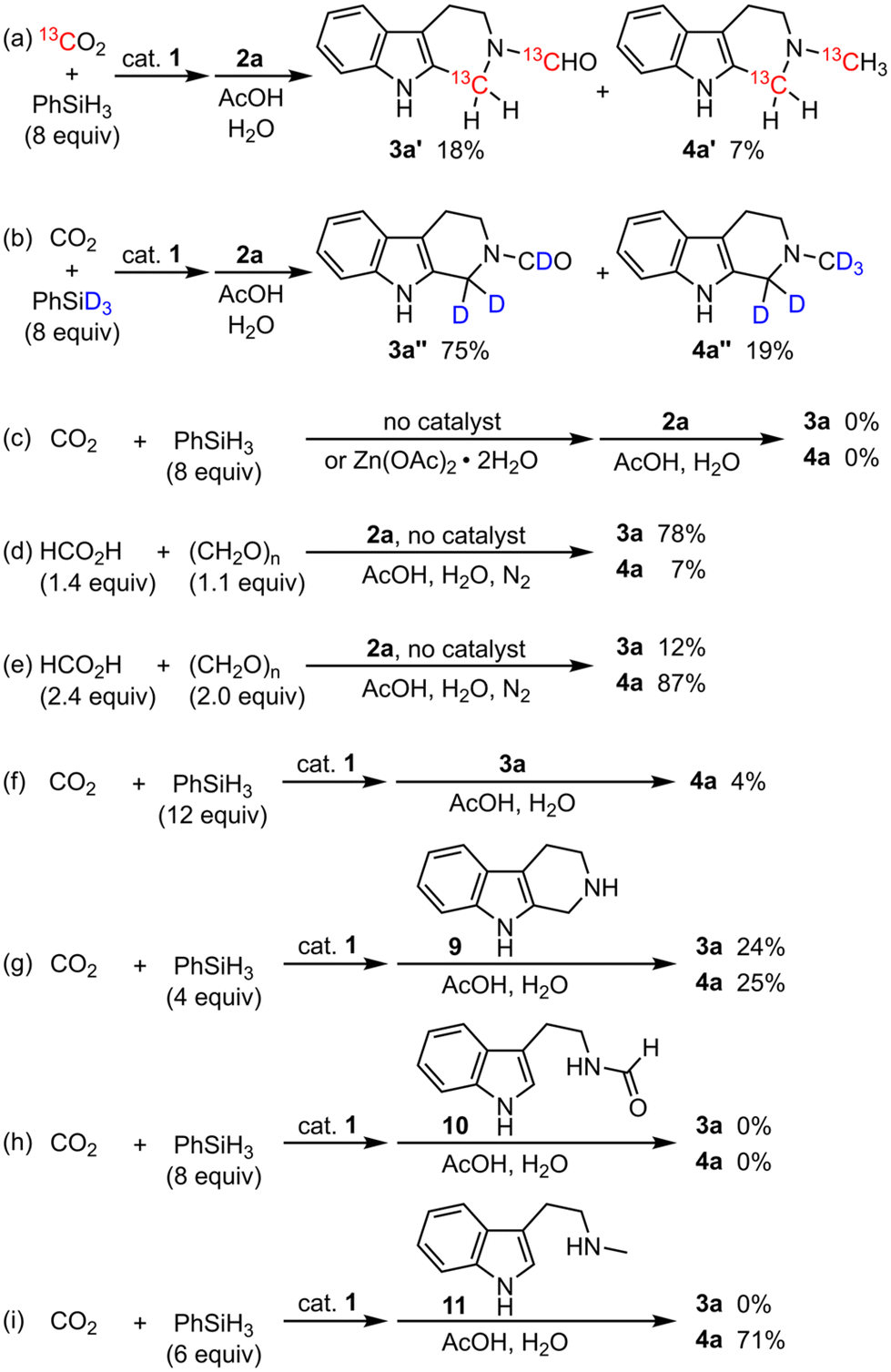 | ||
| Scheme 3 Isotope-labeling and control experiments. | ||
We performed control experiments to predict the reaction mechanisms. An experiment without catalyst 1 resulted in no formation of 3a or 4a, and Zn(OAc)2·2H2O showed no catalytic activity, both indicating that 1 is an essential catalyst (Scheme 3c). When formic acid and paraformaldehyde were used instead of CO2 and PhSiH3, 3a or 4a was obtained selectively according to the amounts of reagents (Scheme 3d and e).22 In contrast, the reactions did not proceed with paraformaldehyde or formic acid alone (not shown), which strongly suggests that the system requires both paraformaldehyde and formic acid (or their equivalents) as intermediates. When 3a was used as a substrate, 4a was hardly obtained, which indicates that 3a is not a major intermediate leading to 4a (Scheme 3f). When tryptoline (9) was used as a substrate, both 3a and 4a were obtained, indicating that 9 is a common intermediate (Scheme 3g). In contrast, formyltryptamine 10 did not give 3a, suggesting that 10 is not an intermediate and that the electron-withdrawing formyl group prevents the Pictet–Spengler cyclization (Scheme 3h). On the other hand, methyltryptamine 11 afforded 4a, which indicates that 11 as well as 9 is an intermediate for 4a (Scheme 3i).
Proposed pathways
The above isotope-labeling and control experiments enabled us to propose the following pathways (Scheme 4). First, (i–ii) silyl formates and bis(silyl)acetals are generated from CO2 and PhSiH3via the formation of a Zn–hydride complex, and (iii) subsequent hydrolysis gives formic acid and formaldehyde (Scheme 4a).13 Next, (iv) the Pictet–Spengler reaction3d,e of 2a with formaldehyde affords 9, and (v) the formylation with formic acid gives 3a (Scheme 4b). Instead of this formylation, (vi) the Eschweiler–Clarke reaction with formaldehyde and formic acid gives 4a. As for the synthesis of 4a, another route is possible: (vii) the Eschweiler–Clarke reaction of 2a to form 11 and (viii) the subsequent Pictet–Spengler reaction. Because 3a could be generated enough experimentally, it is supposed that reaction (iv) is faster than reaction (vii) and that the route via9 is the main pathway for the synthesis of 4a. Here, 9 was not detected experimentally even when the reaction time was shortened, which indicates that reaction (iv) includes the rate-determining step. | ||
| Scheme 4 Proposed pathways for the synthesis of 3a and 4a. | ||
Chemoselectivity in the formation of 3a or 4a
We next turned our attention to the origin of the chemoselectivity in the formation of 3a or 4a. The amounts and/or molar ratio of C1 intermediates generated in the first step (CO2 reduction step) were strongly expected to depend on the amount of PhSiH3, which could be considered a determinant of the selectivity. Therefore, we measured their amounts in the reaction mixtures from the first step under the standard conditions (Table 2 and Fig. S6†).23 When 2.00 mmol of PhSiH3 (for the synthesis of 3a) was used, 0.46 mmol of silyl formates and 0.35 mmol of bis(silyl)acetals were detected together with methoxysilanes. When 3.00 mmol of PhSiH3 (for the synthesis of 4a) was used, 0.73 mmol of silyl formates and 0.60 mmol of bis(silyl)acetals were detected. Although the quantities changed, the proportions were remarkably similar: 30% for silyl formates, 25% for bis(silyl)acetals, and 45% for methoxysilanes. In other words, the amounts, not the ratio, of C1 intermediates determine the main product. In the case that an amount of bis(silyl)acetals is small, generated formaldehyde is depleted in the Pictet–Spengler reaction of 2a and cannot participate in the subsequent Eschweiler–Clarke reaction; thus, N-formylation proceeds selectively to give 3a. In the case that a sufficient amount of bis(silyl)acetals is accumulated, 4a forms selectively because the Eschweiler–Clarke reaction of 9 is faster than the N-formylation.a
| PhSiH3 (mmol) | Production amountb (mmol) | ||
|---|---|---|---|
| HCO2Si | SiOCH2OSi | CH3OSi | |
| a Conditions: CO2 (1 atm), PhSiH3 (2.00 or 3.00 mmol), cat. 1 (0.07 mol%), 55 °C, 18 h. b Determined by proton-coupled 13C NMR using mesitylene as an internal standard. Product selectivity is shown in parentheses. | |||
| 2.00 | 0.46 (31%) | 0.35 (24%) | 0.65 (45%) |
| 3.00 | 0.73 (31%) | 0.60 (25%) | 1.05 (44%) |
To more reliably elucidate the pathways and chemoselectivity, we carried out DFT calculations. Selected elementary reactions and overall pathways with energy profiles are shown in Scheme 5 and S2–S4,† respectively. As a result, the pathways shown in Scheme 4b are acceptable, and some highlights are as follows. Iminium salt I2 is formed from 2a as a common intermediate via a rate-determining dehydration step with a ΔG‡ value of 24.1 kcal mol−1 (Scheme 5a). Then, I2 branches into the reactions to I3 (for 9) and 11, and the ΔG‡ value of the transition state (TS) to I3 (4.3 kcal mol−1) is slightly greater than that to 11 (2.3 kcal mol−1). Here, it should be noted that the counterion in I2 for I3 can be either a formate or an acetate ion, whereas that for 11 must be a formate ion. In the actual experiments, the amount of added AcOH (3.5 mmol, 200 μL) is much larger than that of the generated formic acid (up to 0.73 mmol), which likely leads to the preferential production of 9 rather than 11. The ΔG‡ value of the TS from 9 to 3a (10.7 kcal mol−1) is larger than that from 9 to 4a (6.4 kcal mol−1), which suggests that 4a is certainly formed preferentially when the reactive C1 intermediates are present in sufficient concentration (Scheme 5b). Several TSs are stabilized by water-involved hydrogen bonds, and the proton transfer is mediated by water; an example is P1_TS, which involves dehydration.
 | ||
| Scheme 5 Selected theoretical elementary reactions and TS structures for the formation of 3a and 4a at the B3LYP/6-31+G(d,p) level with the self-consistent reaction field method (H2O) at 393.15 K. ΔG‡ values and distances are shown in kcal mol−1 and Å, respectively. ΔG‡ values are based on each elementary reaction. | ||
One-pot three-step reactions
Finally, we explored one-pot three-step reactions of 2via3 or 4 to demonstrate the potential of the aforementioned CO2 conversions (Scheme 6). After the two-step reaction, hydrolysis of 3a with aqueous HCl gave tryptoline (9) in good yield (72%). When N-bromosuccinimide (NBS) was added in the third step, formylated spiro[oxindole-pyrrolidine] 5a was obtained in a high yield (76%). This method could also be applied to 4a, and methylated spiro-compound 6a, which is known as coerulescine, was obtained. In addition, the spiro-ring formation from the 1-benzyl analog 2h to 5h and 6h was also achieved. These results further demonstrated the substantial potential of the one-pot synthesis strategy using mixtures of 1-catalyzed CO2 reduction products. | ||
| Scheme 6 One-pot three-step reactions for the synthesis of 5, 6, and 9. | ||
Conclusions
We have developed new deoxygenative dual CO2 conversions in which CO2 molecules were converted into different groups, methylene and N-formyl/N-methyl groups. Phenylsilane and a pentanuclear Zn complex acted as a highly efficient reductant and catalyst, respectively, and the reactions proceeded at atmospheric pressure without rare and harmful metals and solvents. The chemoselectivity of N-functionalization was switched by modifying only the amount of phenylsilane. These new CO2 conversions will contribute greatly to the further development of CO2 fixation.Author contributions
K. T. and T. E. conceived the project. H. M. and K. I. synthesized and characterized the compounds. K. T. conducted the DFT calculations. K. T. and T. E. wrote the initial draft of the manuscript, and all authors discussed the results and commented on the manuscript.Data availability
The data supporting the findings of this study are available in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Takahashi Industrial and Economic Research Foundation, the Yakumo Foundation for Environmental Science, and a Grant for the Promotion of Science and Technology in Okayama Prefecture by MEXT. We thank Dr Shigeki Mori (Ehime University) for the X-ray analyses. The computations were performed using RCCS, Okazaki, Japan (project: 24-IMS-C097).References
- For selected reviews, see: (a) A. Tortajada, F. Juliá-Hernández, M. Börjesson, T. Moragas and R. Martin, Angew. Chem., Int. Ed., 2018, 57, 15948 CrossRef CAS PubMed; (b) X. Jiang, X. Nie, X. Guo, C. Song and J. G. Chen, Chem. Rev., 2020, 120, 7984 CrossRef CAS PubMed; (c) Y. Zhang, T. Zhang and S. Das, Green Chem., 2020, 22, 1800 RSC; (d) A. Modak, P. Bhanja, S. Dutta, B. Chowdhury and A. Bhaumik, Green Chem., 2020, 22, 4002 RSC; (e) D. A. Sable, K. S. Vadagaonkar, A. R. Kapdi and B. M. Bhanage, Org. Biomol. Chem., 2021, 19, 5725 RSC.
- (a) M. Khandelwal and R. J. Wehmschulte, Angew. Chem., Int. Ed., 2012, 51, 7323 CrossRef CAS PubMed; (b) R. J. Wehmschulte, M. Saleh and D. R. Powell, Organometallics, 2013, 32, 6812 CrossRef CAS; (c) Y. Li, T. Yan, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2014, 53, 10476 CrossRef CAS PubMed; (d) X. Zhang, S. Wang and C. Xi, J. Org. Chem., 2019, 84, 9744 CrossRef CAS PubMed; (e) Q. Shi, H. Hu, M. Du, Y. Sun, Y. Li and Y. Li, Org. Lett., 2023, 25, 7100 CrossRef CAS PubMed.
- (a) X. Frogneux, E. Blondiaux, P. Thuéry and T. Cantat, ACS Catal., 2015, 5, 3983 CrossRef CAS; (b) D.-Y. Zhu, L. Fang, H. Han, Y. Wang and J.-B. Xia, Org. Lett., 2017, 19, 4259 CrossRef CAS PubMed; (c) C. Zhang, Y. Lu, R. Zhao, W. Menberu, J. Guo and Z.-X. Wang, Chem. Commun., 2018, 54, 10870 RSC; (d) M. Rauch, Z. Strater and G. Parkin, J. Am. Chem. Soc., 2019, 141, 17754 CrossRef CAS PubMed; (e) W.-D. Li, J. Chen, D.-Y. Zhu and J.-B. Xia, Chin. J. Chem., 2021, 39, 614 CrossRef CAS; (f) T. Murata, M. Hiyoshi, S. Maekawa, Y. Saiki, M. Ratanasak, J. Hasegawa and T. Ema, Green Chem., 2022, 24, 2385 RSC; (g) Z. Guo, J. Wu, X. Wei and C. Xi, ChemSusChem, 2025, 18, e202401491 CrossRef CAS PubMed; (h) A. Kumar, R. Gupta, V. Subramaniyan and G. Mani, Catal. Sci. Technol., 2025, 15, 678 RSC.
- W.-H. Wang, Y. Himeda, J. T. Muckerman, G. F. Manbeck and E. Fujita, Chem. Rev., 2015, 115, 12936 CrossRef CAS PubMed.
- S. Bontemps, Coord. Chem. Rev., 2016, 308, 117 CrossRef CAS.
- S. Desmons, J. Bonin, M. Robert and S. Bontemps, Chem. Sci., 2024, 15, 15023 RSC.
- (a) F. J. Fernández-Alvarez and L. A. Oro, ChemCatChem, 2018, 10, 4783 CrossRef; (b) Y. Zhang, T. Zhang and S. Das, Green Chem., 2020, 22, 1800 RSC; (c) R. A. Pramudita and K. Motokura, ChemSusChem, 2021, 14, 281 CrossRef CAS PubMed.
- For recent examples of double carboxylations and related reactions, see: (a) Y. You, W. Kanna, H. Takano, H. Hayashi, S. Maeda and T. Mita, J. Am. Chem. Soc., 2022, 144, 3685 CrossRef CAS PubMed; (b) M. Shigeno, I. Tohara, K. Sasaki, K. Nozawa-Kumada and Y. Kondo, Org. Lett., 2022, 24, 4825 CrossRef CAS PubMed; (c) R. Giovanelli, L. Lombardi, R. Pedrazzani, M. Monari, M. Castiñeira, R. Carlos, S. López, G. Bertuzzi and M. Bandini, Org. Lett., 2023, 25, 6969 CrossRef CAS PubMed; (d) C. Maeda, T. Cho, R. Kumemoto and T. Ema, Org. Biomol. Chem., 2023, 21, 6565 RSC; (e) F. Zhang, X.-Y. Wu, P.-P. Gao, H. Zhang, Z. Li, S. Ai and G. Li, Chem. Sci., 2024, 15, 6178 RSC; (f) Y.-Y. Gui, X.-W. Chen, X.-Y. Mo, J.-P. Yue, R. Yuan, Y. Liu, L.-L. Liao, J.-H. Ye and D.-G. Yu, J. Am. Chem. Soc., 2024, 146, 2919 CrossRef CAS PubMed; (g) A. Brunetti, M. Garbini, G. Autuori, C. Zanardi, G. Bertuzzi and M. Bandini, Chem. – Eur. J., 2024, 30, e202401754 CrossRef CAS PubMed; (h) H. Liu, M. Guo, M. Jia, J. Zhang and X. Xu, Org. Lett., 2025, 27, 778 CrossRef CAS PubMed.
- Y. Zhao, X. Liu, L. Zheng, Y. Du, X. Shi, Y. Liu, Z. Yan, J. You and Y. Jiang, J. Org. Chem., 2020, 85, 912 CrossRef CAS PubMed.
- S. Li, S. Nakahara, T. Adachi, T. Murata, K. Takaishi and T. Ema, J. Am. Chem. Soc., 2024, 146, 14935 CrossRef CAS PubMed.
- W. Xiong, X. Tan, H. Liu, B. Zhu, J. Zhao, J. Li, C. Qi and H. Jiang, Sci. China: Chem., 2024, 67, 841 CrossRef CAS.
- Q. Yan, J. Nan, R. Cao, L. Zhu, S. Liu, C. Liang and C. Zhang, Org. Lett., 2025, 27, 510 CrossRef CAS PubMed.
- K. Takaishi, B. D. Nath, Y. Yamada, H. Kosugi and T. Ema, Angew. Chem., Int. Ed., 2019, 58, 9984 CrossRef CAS PubMed.
- (a) K. Takaishi, H. Kosugi, R. Nishimura, Y. Yamada and T. Ema, Chem. Commun., 2021, 57, 8083 RSC; (b) K. Takaishi, R. Nishimura, Y. Toda, H. Morishita and T. Ema, Org. Lett., 2023, 25, 1370 CrossRef CAS PubMed; (c) T. Ema, Bull. Chem. Soc. Jpn., 2023, 96, 693 CrossRef CAS.
- For recent reviews on N-formylation and N-methylation, see: (a) A. Tlili, E. Blondiaux, X. Frogneux and T. Cantat, Green Chem., 2015, 17, 157 RSC; (b) J.-Y. Li, Q.-W. Song, K. Zhang and P. Liu, Molecules, 2019, 24, 182 CrossRef PubMed; (c) J. R. Cabrero-Antonino, R. Adam and M. Beller, Angew. Chem., Int. Ed., 2019, 58, 12820 CrossRef CAS PubMed; (d) M. Hulla and P. J. Dyson, Angew. Chem., Int. Ed., 2020, 59, 1002 CrossRef CAS PubMed; (e) Z. Li, Z. Yu, X. Luo, C. Li, H. Wu, W. Zhao, H. Li and S. Yang, RSC Adv., 2020, 10, 33972 RSC; (f) G. Naik, N. Sarki, V. Goyal, A. Narani and K. Natte, Asian J. Org. Chem., 2022, 11, e202200270 CrossRef CAS.
- For recent examples, see: (a) H. Kobayashi, H. Hatakeyama, H. Nishimura, M. Yokota, S. Suzuki, Y. Tomabechi, M. Shirouzu, H. Osada, M. Mimaki, Y. Goto and M. Yoshida, Nat. Chem. Biol., 2021, 17, 335 CrossRef CAS PubMed; (b) L. Ting, L. Shiru, D. Baiyun, L. Xiaofa, G. Yifan, R. R. Raphael, H. Jiadong, L. Long, Y. Peiyu, W. Ruotian, Z. Meng, G. Jinming, Y. Xia and C. Xin, Eur. J. Med. Chem., 2024, 275, 116624 CrossRef PubMed; (c) S. Eagon, J. T. Hammill, J. Bach, N. Everson, T. A. Sisley, M. J. Walls, D. Sierra, D. R. Pillai, M. O. Falade, A. L. Rice, J. J. Kimball, H. Lazaro, C. DiBernardo and R. K. Guy, Bioorg. Med. Chem. Lett., 2024, 30, 127502 CrossRef PubMed.
- (a) C. Pellegrini, C. Strässler, M. Weber and H.-J. Borschberg, Tetrahedron:Asymmetry, 1994, 5, 1979 CrossRef CAS; (b) S. Edmondson, S. J. Danishefsky, L. Sepp-Lorenzino and N. Rosen, J. Am. Chem. Soc., 1999, 121, 2147 CrossRef CAS.
- (a) I. V. Efremov, F. F. Vajdos, K. A. Borzilleri, S. Capetta, H. Chen, P. H. Dorff, J. K. Dutra, S. W. Goldstein, M. Mansour, A. McColl, S. Noell, C. E. Oborski, T. N. O'Connell, T. J. O'Sullivan, J. Pandit, H. Wang, B. Wei and J. M. Withka, J. Med. Chem., 2012, 55, 9069 CrossRef CAS PubMed; (b) H. Chen, P. Hua, D. Huang, Y. Zhang, H. Zhou, J. Xu and Q. Gu, J. Med. Chem., 2023, 66, 752 CrossRef CAS PubMed.
- Y. Hayashi, Chem. Sci., 2016, 7, 866 RSC.
- Conformational analyses of 3a and 4a in solution were also carried out, see section 3 in the ESI.†.
- The volume of the 13CO2 balloon (0.3 L) was much smaller than that of the 12CO2 balloon (3.7 L), which probably led to the low yields of 3a′ and 4a′.
- The equivalents of formic acid and paraformaldehyde were roughly optimized on the basis of amounts of silyl formates and bis(silyl)acetals generated by 1-catalyzed CO2 reduction, as discussed later (Table 2).
- H. H. Cramer, B. Chatterjee, T. Weyhermüller, C. Werlé and W. Leitner, Angew. Chem., Int. Ed., 2020, 59, 15674 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2413450 and 2413452. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5gc00942a |
| This journal is © The Royal Society of Chemistry 2025 |