
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInfluence of functional additives, fillers, and pigments on thermal and catalytic pyrolysis of polyethylene for waste plastic upcycling†
Harish
Radhakrishnan
 a,
Abdulrahman A. B. A.
Mohammed
a,
Isabel
Coffman
a and
Xianglan
Bai
*ab
a,
Abdulrahman A. B. A.
Mohammed
a,
Isabel
Coffman
a and
Xianglan
Bai
*ab
aDepartment of Mechanical Engineering, Iowa State University, Ames, IA, USA. E-mail: bxl9801@iastate.edu
bDepartment of Chemical & Biological Engineering, Iowa State University, Ames, IA, USA
First published on 16th April 2025
Abstract
Pyrolysis offers a relatively green and economical method to convert waste plastics into valuable chemicals and fuels without the need for harmful solvents, toxic chemicals, or costly high-pressure reactors. Despite its popularity among chemical upcycling technologies, industrial adoption suffers from feedstock heterogeneity, low-quality products, and catalyst deactivation. Most plastics in our daily lives are formulated with functional additives, fillers, and colorants. These additives remaining in end-of-life waste streams increase feedstock heterogeneity, creating a challenging issue in recycling plastics. Still, the potential impacts of additives on the chemical upcycling of plastics have been poorly understood. In this study, polyethylene compounded with a range of widely used additives (antioxidants, stabilizers, pigments, fillers, slip agents, and flame retardants) was subjected to both thermal pyrolysis and catalytic pyrolysis in different catalyst-to-feedstock contact modes. It showed that many inorganic additives, such as talc, kaolin, CaCO3, TiO2, carbon black, and zinc stearate, facilitated polymer decomposition during pyrolysis, increasing light hydrocarbons while also promoting aromatic and carbon residue formation. Conversely, antioxidants and stabilizers inhibited depolymerization, favoring heavier hydrocarbons. During catalytic pyrolysis with HZSM-5 zeolite, additives strongly enhanced aromatic and catalytic coke formation, especially when there was direct contact between plastics and catalysts. Although certain additives seem beneficial in the short term by promoting polymer cracking and improving the selectivity of aromatics, the transport of the additives and their degradation products and increased carbon coking can contaminate products, deactivate or modify catalysts, and foul reactors. These findings address a critical knowledge gap in effectively converting waste plastics via a greener route.
Green foundation1. This work addresses a long-overlooked yet key issue in waste plastic upcycling—the role of embedded additives, fillers, and pigments in the thermal and catalytic pyrolysis of plastics. By linking the effect of a wide range of additives to changes in product distribution and coking, we bridge the gap between virgin polymer studies and real, heterogeneous feedstocks, enabling more resource-efficient upcycling.2. Most inorganic additives served as catalysts to facilitate polymer cracking while increasing aromatic and carbon residue formation. Conversely, antioxidants and stabilizers inhibited depolymerization, favoring heavier molecules. During catalytic pyrolysis, additives promoted aromatic and catalytic coke formation, and these effects were further amplified by direct contact between plastics and catalysts. Furthermore, additive degradation and their transport contaminated pyrolysis products. 3. Improved feedstock characterization methods and long-term implications of additives on the reactor and catalyst integrity should be studied in the future. |
1. Introduction
Plastics are utilized in a wide array of applications, ranging from packaging and construction materials to consumer goods and automotive components.1–3 This widespread use of plastics has led to significant environmental challenges, particularly concerning the disposal of end-of-life plastics.4–6 Notably, global annual plastic waste generation has increased from 2 Mt in 1950 to 367 Mt in 2020 and is forecasted to be up to 1100 Mt by 2050.1,7 Plastic waste accumulated in landfills and natural environments poses serious threats to ecosystems and human health.4,8,9 Polyolefins, such as polyethylene (PE) and polypropylene (PP), represent a major fraction of waste plastic due to their extensive use in packaging and other single-use applications.1,2 In general, these thermoplastics can be mechanically recycled and reshaped into new products. However, re-melting and extrusion processes of recycled plastics downgrade the product quality, limiting their circularity.Historically, many countries have employed incineration to reduce the volume of various landfill-bound wastes, especially plastics, and promote energy recovery.10 However, incineration has low energy efficiency, and it also releases pollutants and toxic chemicals.11 Chemical recycling and upcycling offer an attractive avenue for transforming waste plastics into platform chemicals and other higher-value molecules.2,12 Recently, several promising thermochemical recycling and upcycling strategies, such as dehydrogenation, metathesis, ethenolysis, oxidation, and hydrogenation, have been proposed in the literature to improve process efficiency and enhance value-addition.13–19 Additionally, other emerging upcycling strategies involve photothermal, non-thermal plasma, and electrocatalytic conversion.20–22 However, pyrolysis continues to be the most studied technology for waste plastic conversion, largely due to its process that eliminates the need for solvents or harsh chemicals, simpler reactor design, and scalability for commercial operations.2,23–26 Regardless of the technology employed, waste plastics and their inherent heterogeneity remain a key issue. During pyrolysis, plastics are depolymerized under atmospheric pressure in the absence of oxygen to produce smaller molecules, which can be directly used as low-quality fuels or upgraded to chemicals and advanced fuels. Other than thermally decomposing, catalysts can also be introduced to upgrade pyrolysis vapors and improve product selectivity.25,27–30 Despite these advantages, the inherent heterogeneity of waste plastics and the complexity of the decomposition products pose significant challenges for pyrolytic conversion.31 Pyrolysis of polyolefins usually produces aliphatic and aromatic hydrocarbons with a wide range of carbon numbers and boiling points. Additionally, pyrolysis oils of waste plastics frequently contain various contaminant elements such as nitrogen, oxygen, chlorine, iron, sodium, calcium, and other inorganics, exceeding the acceptable levels for downstream upgrading processes like steam cracking.32 Previous studies suggest that waste plastic conversion can differ considerably from virgin polymers. For instance, Yan et al. observed that waste polyolefins exhibit lower activation energies than virgin polymers while attributing these differences to unknown additives and contaminants.33 In their study, Abbas-Abadi et al. noted that lower liquid products and higher gas yields observed from waste plastics compared to virgin polymers could be associated with organic and inorganic contaminants.34 In a recent study, Wu et al. evaluated pyrolysis oil derived from post-consumer polyolefins, observing greater hydrocarbon isomerization compared to the pyrolysis oils from virgin polymers.35 These authors also speculated that the variations in waste plastic-derived oils may be connected to certain additives and contaminants.
The heterogeneity of waste plastics originates from two major sources: physically removable impurities and embedded impurities. The first type includes different constituent plastic polymers, organic impurities, or other environmental contamination. These materials can be removed by mechanical sorting and washing.2 The embedded impurities are the additives incorporated during the plastic manufacturing process. In fact, day-to-day plastics are polymers formulated with various additives tailored to specific applications.36–39 Incorporating these additives can provide functionality such as mechanical property improvement, processability enhancement, and thermal stability, among other functions.9,36,37 Typical plastic additives include antioxidants, stabilizers, fillers, pigments, flame retardants, plasticizers, and slip agents, with concentrations ranging from less than 1% to more than 50%, depending on their purposes.9 These embedded impurities participate in the chemical upcycling of plastics because they cannot be removed before plastic conversion. Evidently, the heterogeneous elements found in pyrolysis oils are often associated with the migration of organic and inorganic additives during pyrolysis.32,40 Furthermore, additives can influence the decomposition of polymers through their physical and chemical interactions with the polymer. For instance, antioxidants and stabilizers are commonly added to polymers to prevent oxidative degradation of polymers via coupling reactions with polymer-derived radicals.36,38 Titanium oxide (TiO2), a pigment and a filler widely used in plastics, is a photocatalyst. Kaolin, a reinforcing filler, is also known as a catalyst. While the above are only a few examples, plastics contain many different types of additives.9,37 Since waste plastics, especially post-consumer plastics, are derived from multiple sources, a diverse mixture of additives inevitably appears within the waste stream.
Although additives are inherently present in waste plastics, their effects on plastic conversion remain poorly studied. Previous studies have predominantly employed virgin polymers or waste plastics with minimal attention to additive effects.2,29,41,42 Although recent investigations speculate that variations in pyrolysis oil composition may stem from additives in post-consumer wastes,33–35,43 the effects of individual additives remain largely unknown. Apart from the limitations in feedstock choice and characterization, different reactor types and pyrolysis conditions used in individual studies also led to variability in conversion results, making it even more difficult to assess additive effects quantitatively. Notably, one study reported the thermal degradation of PE containing a few functional additives using a thermogravimetric analyzer and claimed no measurable impact without investigating pyrolysis products.44 Such shortfalls in understanding, coupled with the unknown effects of additives, especially on product composition and carbon distribution, have led to discrepancies in the reported results,2,29 representing a significant knowledge gap in the literature. Unknown additive effects also have substantial technical implications during industrial operations, such as the batch-to-batch variation in product quality and selectivity, the issues with maintaining the reactor and catalyst integrity, and proper disposal of residual char.
The present study aimed to fill this important knowledge gap by investigating the effects of common polymer additives on polyolefin pyrolysis. PE was co-extruded or compounded with 12 common additives or their mixtures, including antioxidants, stabilizers, fillers, pigments, flame retardants, and slip agents. PE was selected as the polymer matrix due to its prevalence in plastic production and single-use plastic waste streams. The additives were selected based on the volume produced to reflect those frequently identified in post-consumer plastic waste. By performing pyrolysis or catalytic pyrolysis of virgin polymer and plastics containing known concentrations of additives in addition to pyrolyzing individual additives using the same reactors and conditions, we systematically evaluated their influence on plastic deconstruction and product composition. Given a large number of experiments involving 12 different additives and three different pyrolytic conversion scenarios for each additive (thermal pyrolysis, in situ catalytic pyrolysis, and ex situ catalytic pyrolysis), a micropyrolyzer was used to convert plastics and individual additives in this work, in addition to employing a thermogravimetric analyzer. A micropyrolyzer is a well-known and reliable tool, frequently used to study pyrolysis and catalytic pyrolysis of both waste plastics and biomass feedstocks.45–50 While it can provide high heating rates and short vapor residence times similar to a fluidized-bed setup, it has multiple advantages over larger reactors, such as a short turnaround time, smaller samples, reduced heat transfer limitation and secondary reactions, precise control of the reactor conditions, online product analysis (thus, higher product mass closure), and reproducible results. Despite its small scale, the trends observed from micropyrolyzer studies are transferable across reactor scales. In this study, the implications of plastic additives on industrial upcycling processes are also described in detail.
2. Materials and methods
2.1 Materials
Virgin high-density polyethylene (HDPE) was sourced from DOW Chemicals. As detailed in Table 1, functional additives and fillers were procured in powder form, except the pigments, which were sourced as polyethylene masterbatches. Calcium carbonate (CaCO3), talc, kaolin, and barium sulfate (BaSO4) were purchased from Fisher Scientific. A hindered phosphite antioxidant (HPSAO), a hindered phenol antioxidant (HPAO), and a hindered amine light stabilizer (HALS) were procured, and an equal mixture of the three additives was also compounded in-house. Their chemical names are tris(2,4-di-tert-butylphenyl)phosphite, pentaerythrityl tetrakis(3-(3,5-di-tert-butyl-4-hydroxyphenyl)propionate), and poly(4-hydroxy-2,2,6,6-tetramethyl-1-piperidine ethanol-alt-1,4-butanedioic acid), respectively. These additives are also commonly called Irgafos 168, Irganox 1010, and Tinuvin 622 LD, respectively. Additionally, zinc stearate and aluminum trihydrate (ATH) were also purchased from Fisher Scientific, while titanium dioxide (TiO2) and carbon black were sourced as linear low-density polyethylene (LLDPE) masterbatches (70%/30% of TiO2/LLDPE and 40%/60% of carbon black/LLDPE) from a global masterbatch and additive producer. To prepare PE plastics containing additives, virgin PE and known amounts of additives were co-extruded using a twin-screw compounding extruder at 150–180 °C. Virgin PE and additive-added PE samples were size reduced to a 125–250 μm particle distribution using a cryogenic mill before plastic characterization and conversion. Aliphatic hydrocarbon standards were obtained from Sigma Aldrich. Mixed gas standards (He, CO, CO2, CH4, C2H6, C3H8, C4H10, C5H12, C2H4, C3H6, and C4H8) were purchased from Praxair, USA. The HZSM-5 zeolite catalyst with a SiO2/Al2O3 ratio of 23![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and a surface area of 425 m2 g–1 (CBV 2314, Zeolyst International) was calcined at 550 °C for 5 hours in a muffle furnace with a constant air supply. The activated zeolite catalyst was pelletized, pulverized, and sieved to a 50–70 mesh size. The purity and detailed supplier information are provided in the ESI.†
:1 and a surface area of 425 m2 g–1 (CBV 2314, Zeolyst International) was calcined at 550 °C for 5 hours in a muffle furnace with a constant air supply. The activated zeolite catalyst was pelletized, pulverized, and sieved to a 50–70 mesh size. The purity and detailed supplier information are provided in the ESI.†
| Class | Plastic compounded with additives | Mass loading | Ultimate analysis | Functions29 | Typical amount9 | ||||
|---|---|---|---|---|---|---|---|---|---|
| Additive | Polymer | N% | C% | H% | S% | ||||
| a Added to the LLDPE masterbatch. | |||||||||
| Virgin polymer | HDPE, CnH2n | 0 | 100 | — | 85.6 | 14.4 | — | — | — |
| w/fillers | Kaolin, Al2Si2O5(OH)4 | 20 | 80 | — | 68.5 | 11.8 | — | Modifies mechanical properties, diluent, and extender (cost reduction) | Up to 50% |
| Talc, Mg3Si4O10(OH)2 | 20 | 80 | — | 68.5 | 11.6 | — | |||
| Calcium carbonate, CaCO3 | 20 | 80 | — | 70.1 | 11.5 | — | |||
| Barium sulfate, BaSO4 | 20 | 80 | — | 66.7 | 11.2 | 2.7 | |||
| w/slip agent | Zinc stearate, Zn(C18H35O2)2 | 3 | 97 | — | 85.1 | 14.3 | — | Improve manufacturing process: anti-friction, anti-slip, and anti-sticking agent | 0.1–3% |
| w/flame retardant | Aluminum trihydrate, AlH6O3 | 25 | 75 | — | 64.2 | 10.8 | — | Curtails flames by disrupting combustion | Up to 25% |
| w/pigmentsa | Titanium dioxide, TiO2 + CnH2n | 7.5 | 92.5 | — | 79.1 | 13.3 | — | Various colors and aesthetic looks | 0.001–10% |
| Carbon black, C + CnH2n | 7.5 | 92.5 | — | 86.4 | 13.6 | — | |||
| w/antioxidants (AO) and light stabilizer (LS) | Hindered phosphite AO, C42H63O3P | 3 | 97 | — | 85.3 | 14.2 | — | Delays oxidative degradation | 0.05–3% |
| Hindered phenol AO, C73H108O12 | 3 | 97 | — | 85.2 | 14.2 | — | |||
| Hindered amine LS, C17H33O6N | 3 | 97 | 0.1 | 84.8 | 14.2 | — | Delays UV-based degradation | 0.05–3% | |
| Hindered phosphite & phenol AOs and hindered amine LS, equal mixture | 3 | 97 | — | 85.3 | 14.2 | — | Delays different modes of oxidative and photodegradation | 0.05–3% | |
2.2 Characterization methods
The elemental composition of plastic feedstocks was analyzed using an elemental analyzer (vario MICRO Cube, Elementar Americas, USA). A standard operating procedure involving the acetanilide standard and 5 ± 0.25 mg of samples was used. Each test was performed three times to ensure reproducibility, and the average was reported.Inductively-coupled plasma-optical emission spectroscopy (ICP-OES) analysis was performed after microwave digesting (Multiwave Pro, Anton Paar USA Inc.) virgin PE (500 ± 5 mg) in concentrated trace metal-grade nitric acid, followed by dilution of the dissolved metals into a 2% nitric acid matrix. ICP-OES analysis (Optima™ 8000, PerkinElmer, USA) was conducted to quantify the concentration of the inorganic impurities such as Sb, Ba, Ca, Cd, Cr, Cu, Pb, Mn, Ni, Fe, Zn, Co, Ti, V, Mo, Al, Mg, K and Na. High-accuracy (>0.999) standard calibration was performed for all the tested inorganic elements. Duplicate measurements were performed with blank subtractions to ensure proper baseline and reproducibility.
Thermal stability was measured using a thermogravimetric analyzer (TGA/DSC 1 STARe system, Mettler Toledo, USA) at a 10 °C min−1 heating rate to evaluate PE with and without additives. Plastic or individual additives of about 20 ± 0.25 mg were placed in a crucible and heated from room temperature to 900 °C. Nitrogen sweep and cell gas flow rates were 100 and 20 mL min−1, respectively. For proximate analysis, air was introduced at 900 °C for 30 min to combust the organic residue.
2.3 Pyrolysis and product analysis
Thermal pyrolysis and catalytic pyrolysis of PE with and without additives were conducted using a tandem micro-pyrolyzer system (Rx-3050 TR, Frontier Laboratory, Japan) where two furnaces are sequentially connected with the ability to heat at approximately 250 °C s−1.51 Individual additives were also thermally pyrolyzed. For thermal pyrolysis, deactivated stainless sample cups containing samples were dropped into the first furnace, preheated to 600 °C, and maintained inert using helium gas (<1 s vapor-phase residence time). The mixture of plastics and catalysts was converted in the first furnace for in situ catalytic pyrolysis. For ex situ pyrolysis, plastics were pyrolyzed in the first furnace, and a quartz tube with catalysts was inserted into the second furnace to upgrade the pyrolysis vapor. The second furnace temperature was 600 °C, and the catalyst test bed had a residence time of about 10 ms. Schematics of the pyrolysis reactor are given in our previous work.52 For individual tests, 0.1–0.5 mg of samples were pyrolyzed, and a plastic-to-catalyst ratio of 1:20 was used.
For online product analysis, the pyrolyzer was connected to a GC/MS-Polyarc-FID-TCD multi-dimensional chromatography system (GC 7890B, Agilent, USA). Helium was used as both pyrolysis gas and carrier gas with a GC split ratio of 1:50 and a septum purge of 3 mL min−1, translating to a total flow of 156 mL min−1. Two Phenomenex ZB-1701 (60 m × 250 μm × 0.25 μm) capillary columns were connected to a mass spectrometer (MS 5977A, Agilent, USA) and a flame ionization detector (FID), respectively. A porous layer open tubular (PLOT) column (60 m × 0.320 mm) (GS-GasPro, Agilent, USA) was connected to a thermal conductivity detector (TCD). A guard column (part# 160-2255-5, Agilent, USA) was installed between the GC inlet and the multidimensional splitter to protect the analytical columns from heavy condensates. The guard column was set to a flow of 3 mL min−1 feeding into the splitter, where the MS, FID, and TCD columns are set to flows of 1, 1, and 2.7 mL min−1, respectively, with auxiliary gas and pressure control from an electronic pressure controller connected to the splitter. The front-injector temperature of the GC was set at 280 °C to prevent product condensation. The GC oven temperature was held at 40 °C for 3 minutes, increased to 280 °C at 6 °C min−1, and held at 280 °C for another 3 minutes. The electron ionization (EI) mode mass spectrometer was operated at 70 eV, with source and quadrupole temperatures of 230 and 150 °C, respectively, with a scanning range of m/z 40 to 550 m/z. The FID and TCD were operated at 300 °C and 250 °C. The FID H2, air, and makeup flows were set to 1, 350, and 20 mL min−1, respectively. A serial Polyarc reactor operated at 450 °C with externally controlled H2 and air flows of 35 and 2.5 mL min−1 was added in between the column and the FID to convert a variety of analytes into methane and provide a uniform FID response. The reference and makeup flow for the TCD were set at 10 and 5 mL min−1, respectively. Data acquisition and processing were performed using Agilent MassHunter software. Condensable products were identified by MS using the NIST compound database and quantified using Polyarc-FID. Five concentrations of C6–C30 alkane standards were injected into the GC to create calibration curves. Due to the uniform carbon response of Polyarc-FID, calibration for an alkane with a specific number of carbon atoms can be applied to other compounds with the same carbon count. Gaseous compounds were quantified by TCD by injecting standard gas mixtures with known concentrations and creating calibration curves with a regression coefficient of >0.99. Each pyrolysis condition was triplicated to ensure reproducibility, and average product yields were reported. After reactions, solid residues remaining in the sample cups were measured using a microbalance with an accuracy of 1 μg. Spent catalytic beds were also measured to determine the mass of the spent catalyst. Carbon contents in the solid residue and the catalytic coke in the spent catalyst were analyzed using an elemental analyzer. Due to their high boiling points, long carbon chain hydrocarbons are usually difficult to analyze using regular GC/MS. In the present study, hydrocarbons larger than C20 were quantified by mass difference.
The carbon yield of an individual product per plastic mass was calculated using the following equation:
 | (1) |
Carbon selectivity for a specific group of hydrocarbons among the C1–C20 products was calculated based on the following equation:
 | (2) |
3. Results and discussion
3.1 Thermal stability of PE with additives
Twelve different types of major plastic additives and their combination were studied based on volumes reported in the literature.37 Their functions in polymers, their typical concentrations, the concentrations used in this study, and the elemental analysis of virgin PE and the additive-compounded plastics are given in Table 1. The concentrations of the additives are 3–20% for different types of additives. Although these concentrations fall in typical ranges of additives in industrial applications,9 higher-end or above-average concentrations within the ranges were used to capture the additive effects. The virgin PE used in this study is an unstabilized polymer without additives. The inorganic compositional analysis shown in Table S1† indicates that it contained ppm levels of Ti and Al that originate from Ziegler–Natta catalysts used to synthesize PE. Their molecular structures are shown in Fig. 1. Fillers like kaolin, talc, calcium carbonate (CaCO3), and barium sulfate are incorporated into polymers to improve thermal stability, assign mechanical and electrical properties, and reduce costs.9 Zinc stearate is a slip agent, acting as a lubricant to enhance plastic processability.39 Aluminum trihydrate is the most commonly used non-halogen fire retardant.39 It is usually added at high concentrations to overcome the intrinsic flammability of polymers. Titanium dioxide (TiO2) is a widely used white pigment.39 It can also improve plastic durability and resistance to weather. Carbon black is a black pigment that can also improve the electrical conductivity of polymers.39 In industrial practice, pigments for PE are usually processed as polyethylene masterbatches.39 Hindered phenol and hindered phosphite are antioxidants (AOs), while hindered amines are predominantly used as light stabilizers (LS). They are commonly added to polymers to prevent oxidative degradation during plastic melt processing and delay the degradation of plastics.9,37,39 Most additives were found to be used in more than one application or serve more than one purpose. For instance, some fillers were also used as reinforcements and pigments, and vice versa.39 Also, all the hindered additives can be used separately or together for antioxidant and UV/light stabilizer applications.39 | ||
| Fig. 1 Molecular structures of organic additives tested. | ||
The thermal stabilities of virgin PE, individual additives, and PE with the compounded additives are analyzed based on TGA results given in Fig. 2. The thermal decomposition temperatures of the PE with the different additives are listed in Table 2. During the TGA tests, virgin PE decomposed at a temperature range of around 400–500 °C with a single mass loss curve peak at 475 °C. Kaolin was stable at below 450 °C. The mass loss at temperatures above 450 °C was due to its dehydroxylation to form metakaolin and water. Other inorganic fillers, such as talc, calcium carbonate, and barium sulfate, are thermally stable at the typical pyrolysis temperature of PE. There was a minor mass loss at temperatures above 600 °C for barium sulfate. At higher temperatures, barium sulfate can decompose to release SO2 and O2. The volatility varied widely for specific functional additives like aluminum trihydrate, TiO2, and carbon black. Aluminum trihydrate showed a mass loss at 200–300 °C, related to water released from aluminum trihydrate decomposition. This endothermic reaction, leading to water formation, is the working principle of aluminum trihydrate serving as a flame retardant. TiO2 and carbon black in LLDPE masterbatches showed mass losses at 400–500 °C. However, the mass losses were due to the depolymerization of the LLDPE fraction since TiO2 and carbon black are thermally stable. In comparison, zinc stearate, hindered phenol, hindered phosphite, hindered amine, and the mixture of hindered AOs and LS exhibited high volatility at temperatures below 500 °C. Zinc stearate showed two mass loss peaks centered at 342 and 431 °C, which are associated with the volatilization of organic decomposition products of zinc stearate. Hindered phosphite exhibited mass loss in the range of 200–350 °C, whereas devolatilization of hindered phenol occurs in the broader range between 300 and 500 °C. Hindered amine showed a narrower and sharper mass loss range between 280 and 380 °C. As expected, the mixed hindered additives showed a broad mass loss between 200 and 500 °C with two mass loss curves with each centered at 321 and 385 °C, respectively. Hindered additives can undergo extensive decomposition during PE pyrolysis.53,54
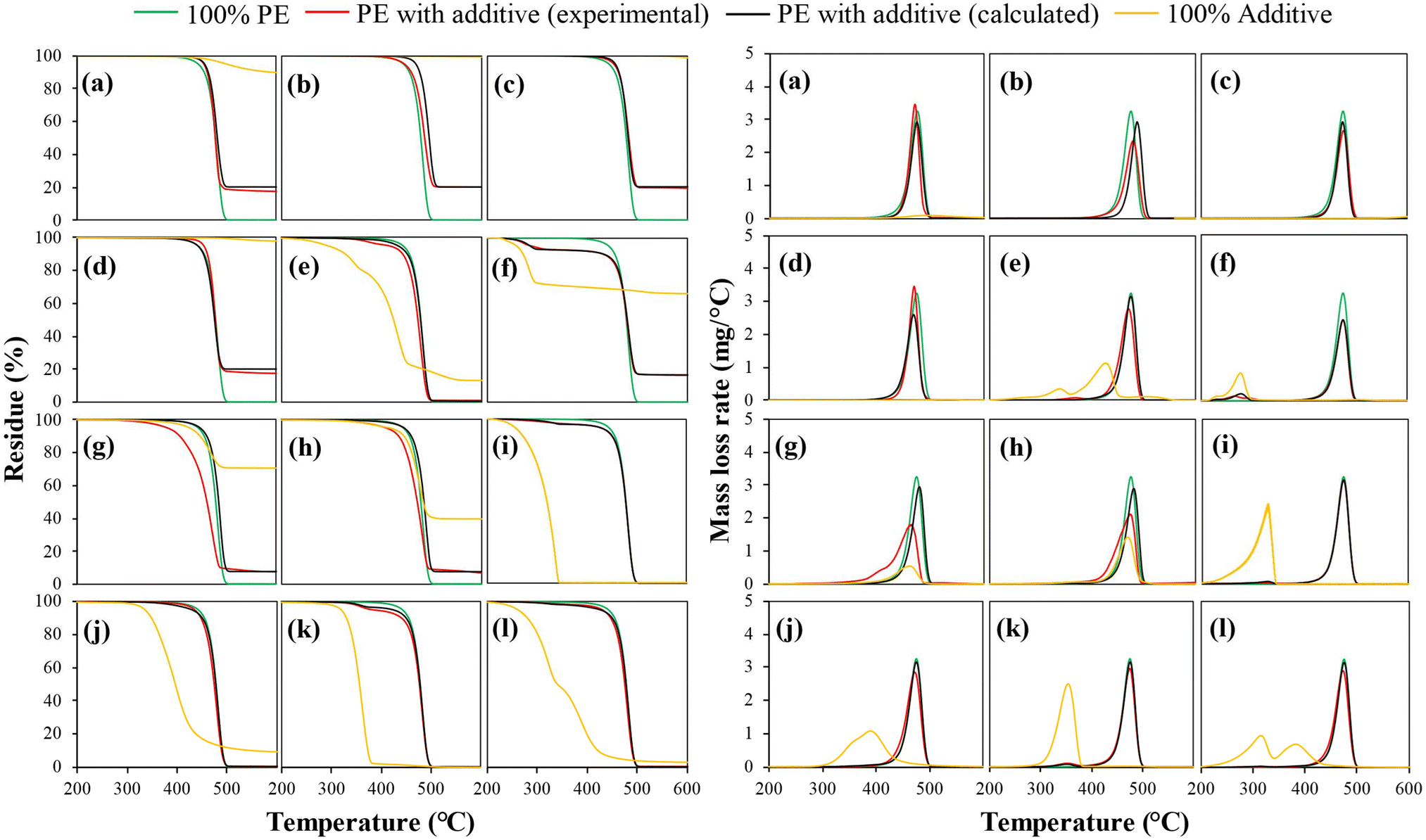 | ||
| Fig. 2 Experimental versus calculated TGA mass loss and mass loss rate profiles of PE with additives: (a) kaolin, (b) talc, (c) calcium carbonate, (d) barium sulfate, (e) zinc stearate, (f) aluminum trihydrate, (g) titanium dioxide, (h) carbon black, (i) hindered phosphite AO, (j) hindered phenol AO, (k) hindered amine LS, and (l) AOs & LS mixture. | ||
| Additive type |
T
da (°C) |
T
maxa (°C) |
||
|---|---|---|---|---|
| Experimental | Calculated | Experimental | Calculated | |
| a T d and Tmax are 5% mass loss and maximum degradation temperatures, respectively. | ||||
| 0% additive | 443 | 475 | ||
| 20% kaolin | 455 | 456 | 471 | 474 |
| 20% talc | 455 | 456 | 471 | 474 |
| 20% calcium carbonate | 453 | 456 | 476 | 474 |
| 20% barium sulfate | 455 | 455 | 478 | 474 |
| 3% zinc stearate | 415 | 433 | 470 | 475 |
| 25% aluminum trihydrate | 289 | 286 | 475 | 475 |
| 7.5% titanium dioxide | 381 | 447 | 465 | 480 |
| 7.5% carbon black | 409 | 445 | 474 | 481 |
| 3% hindered phosphite AO | 423 | 424 | 475 | 475 |
| 3% hindered phenol AO | 436 | 430 | 472 | 475 |
| 3% hindered amine LS | 385 | 425 | 475 | 475 |
| 3% AOs & LS mixture | 431 | 427 | 473 | 475 |
The TGA curves of the additive-compounded PE were somewhere between that of virgin PE and additives only. To distinguish the additive effects on the thermal decomposition of PE, the “calculated” TGA profiles and “calculated” Td and Tmax values are also included in Fig. 2 and Table 2. The “calculated” values are obtained using the TGA results of 100% virgin PE and 100% individual additives and calculated based on their respective mass fractions in the PE compounded with additives. Since the “calculated” results assume no interaction between PE and additives, a discrepancy between the measured and calculated values could represent a positive or negative effect on PE thermal decomposition caused by additives. From the figure, the shifts in the TGA and DTG curves to the lower temperature regions compared to their “calculated” profiles were noticeable for inorganic additives like talc, kaolin, TiO2, carbon black, and zinc stearate. In the table, the thermal decomposition temperatures (Td and Tmax) of these additive-compounded plastics were lower than their calculated values. For example, the Tmax value of PE with TiO2 was 465 °C, 15 °C lower than its calculated value. The results suggest that the incorporation of these additives reduced the thermal stability of PE and facilitated its devolatilization at lower temperatures. The increased instability is mainly attributed to the catalytic effects of the additives,32,33 which will be discussed in detail later. The Tmax value of PE with CaCO3 was slightly higher than the calculated value, suggesting a slightly increased thermal stability. Barium sulfate or aluminum trihydride had minimal effects on the mass loss rate of PE, as their measured and calculated temperatures were nearly identical. Among the hindered additives, hindered phosphite and hindered amine had no noticeable impact, whereas hindered phenol slightly lowered the thermal stability.
3.2 Thermal pyrolysis of PE with additives
Fast pyrolysis of both virgin PE and PE with additives produced a broad range of saturated and unsaturated hydrocarbons. The pyrolysis vapor products are grouped in Fig. 3 based on the physical appearance of the compounds at room temperature, which are light gases (C1–C4), oils (C5–C20), and waxes (C20+). The yields of different fractions obtained from virgin PE pyrolysis were 5.7% for gases, 52.2% for oils, and 41.7% for waxes. The high wax yield is typically observed when polyolefins are fast pyrolyzed with short vapor residence times.19 While random chain scissions primarily depolymerize PE into a broad hydrocarbon distribution,42 the short residence time in the micropyrolyzer limits the secondary cracking of long carbon chain hydrocarbons.55,56 A strong shift of the products to lighter hydrocarbons was observed with several inorganic fillers. At equal concentrations in PE (i.e., 20%), the wax yield was reduced to 5.9% for PE with talc, 8.7% for kaolin, and 18.2% for CaCO3. While oil and gas fractions increased with these fillers, the highest oil yield of 74.3% was obtained with PE with talc. The effect of kaolin on enhancing polymer cracking was comparable to talc and higher than that of CaCO3. The increased polymer cracking by the fillers was accompanied by increased carbon residue formation. Solid carbon residue was negligible for virgin PE, whereas the highest yield of 3.6% was observed for the PE compounded with talc. Importantly, solid residue yields reported in this study are based on feedstock carbon and thus do not include the non-volatile fillers remaining after pyrolysis.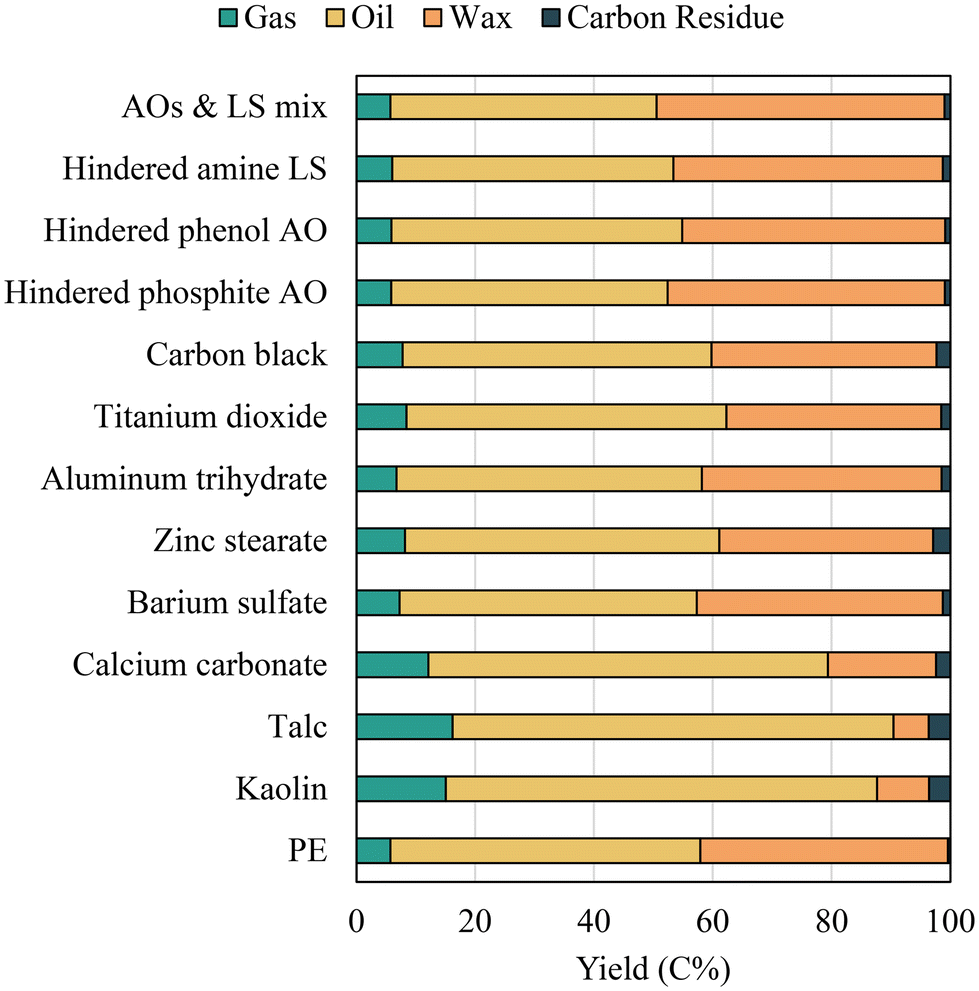 | ||
| Fig. 3 Pyrolysis product distribution of PE with additives. | ||
The wax yield was reduced to 36%, whereas the carbon residue yield increased to 2.9% for PE with zinc stearate. Considering its concentration was only 3%, zinc stearate had a pronounced effect on the polymer cracking. The shift to shorter-chain hydrocarbons was also observed with PE containing TiO2 and carbon black. The wax yield was slightly lower upon adding TiO2 compared to carbon black (36.2% vs. 37.9%), whereas adding carbon black produced slightly more carbon residue than TiO2. Among the inorganic additives, aluminum trihydrate and barium sulfate showed minor effects on the product distribution.
A contrasting effect was observed with AOs and LS compared to the above-described additives. The wax yield increased by pyrolyzing PE with AOs and LS, indicating that these additives suppress polymer cracking. The effect of hindered amine was similar to hindered phosphite, whereas hindered phenol had a slightly less effect on the wax yield. On the other hand, incorporating the AOs and LS mixture resulted in the highest wax yield of 48.5%, which was higher than the wax yields obtained with PE with individual hindered additives. Possible synergy among different AOs and LS strongly hindered the polymer chain cleavage.
The selectivity of different product functional groups (for C1–C20 products) is shown in Fig. 4, and the yield distributions of saturated and unsaturated hydrocarbon products per their carbon numbers are plotted in Fig. 5. During PE pyrolysis, the β-scission of the polymer chain to form free radicals primarily produces alkenes.41,57 Meanwhile, intra- and intermolecular hydrogen transfers during pyrolysis lead to alkanes and other unsaturated hydrocarbons, such as dienes, and cyclic and aromatic hydrocarbons.56,58 Deeply unsaturated hydrocarbons, such as aromatics, are usually associated with secondary reactions of primary products involving dehydrogenation, cyclization, and aromatization.56 Aromatic hydrocarbons were not found during virgin PE pyrolysis due to the short gas residence time in the pyrolyzer suppressing secondary reactions. However, aromatics, such as benzene, toluene, ethylbenzene, and xylene, were among the products of PE with all types of additives, with kaolin, talc, CaCO3, and zinc stearate, profoundly promoting aromatic formation. The highest aromatic selectivity of 11.8% was observed for PE with kaolin, which was accompanied by a decreased alkene selectivity of 52% from 62% for virgin PE. The aromatic selectivity followed the order of kaolin > talc > CaCO3 > zinc stearate > TiO2 > carbon black > mixed AOs and LS > hindered phosphite > hindered amine ≈ hindered phenol. In comparison, barium sulfate and aluminum trihydrate resulted in the least aromatic formation. The aromatic formation in the PE with additives explains the increased carbon residue yields since polyaromatics are the precursor for carbonaceous residues.
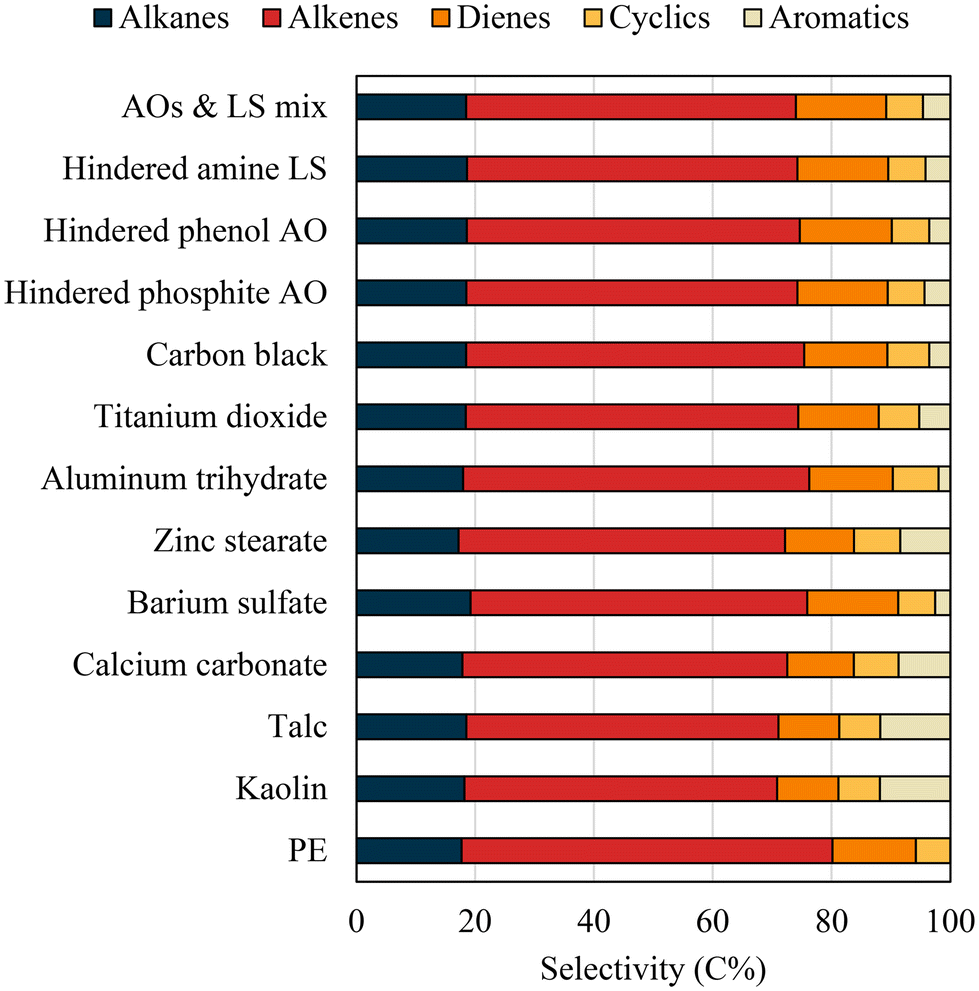 | ||
| Fig. 4 Functional group selectivity of pyrolysis products of PE with additives. | ||
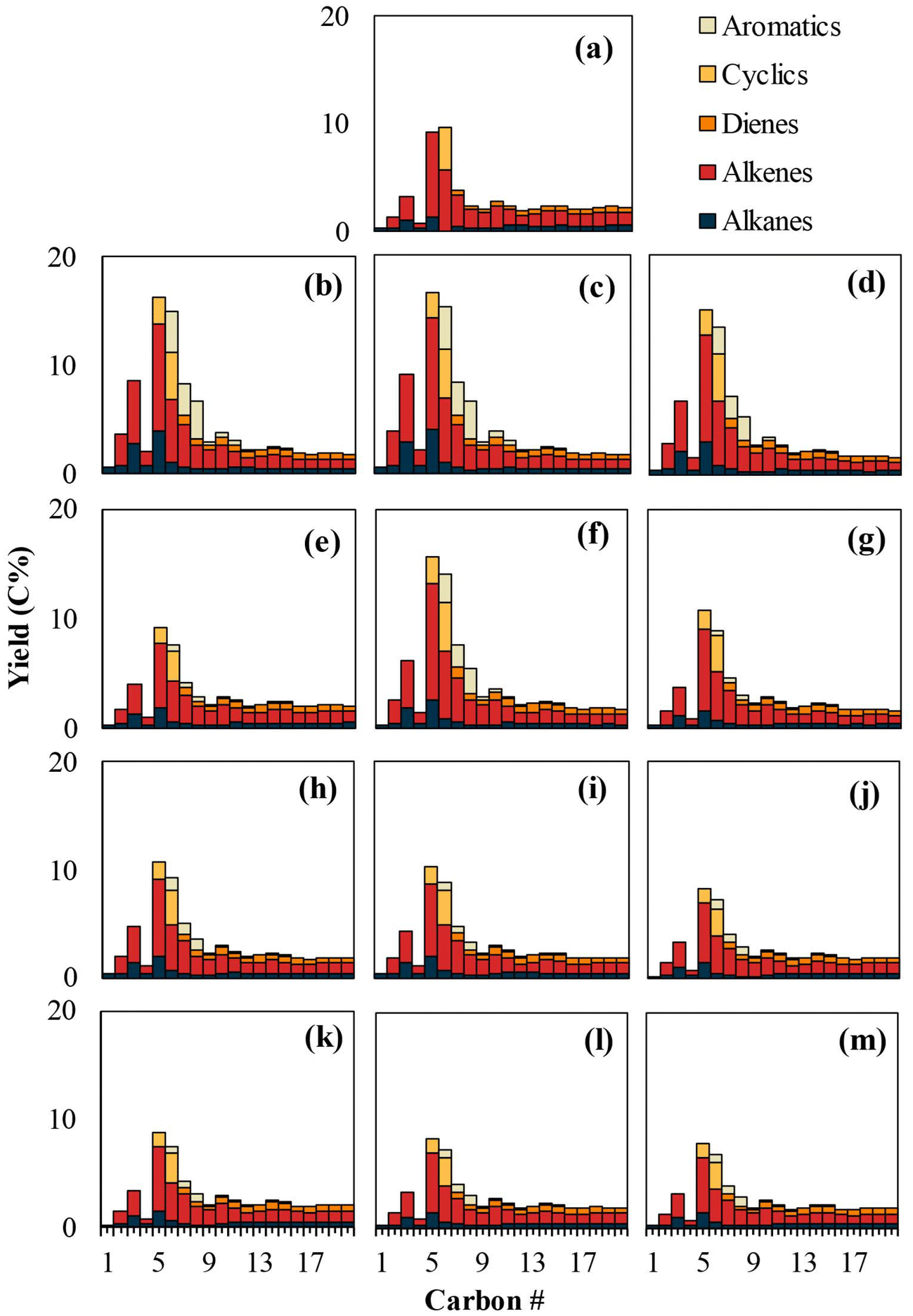 | ||
| Fig. 5 The carbon number distribution of PE pyrolysis products (C1–C20): (a) virgin PE, and PE with (b) kaolin, (c) talc, (d) calcium carbonate, (e) barium sulfate, (f) zinc stearate, (g) aluminum trihydrate, (h) titanium dioxide, (i) carbon black, (j) hindered phosphite AO, (k) hindered phenol AO, (l) hindered amine LS, and (m) AOs & LS mixture. | ||
In addition to the additive-compound PE, additives were independently pyrolyzed in the same reactor and under the same conditions to determine their decomposition products. The solid recovery and the GC/MS detected compound lists are shown in Tables S2–S7.† Upon pyrolysis, 89.7% solid was recovered from kaolin. The mass loss was due to kaolin dehydroxylation and water evaporation, as discussed above. Due to their high stability, the solid recovery was 97.9%, 98.7%, and 99.4%, respectively, from talc, CaCO3, and barium sulfate. Despite their high thermal stability, the missing mass balance is an indication that small amounts of additive particles can be entrained in the sweep gas and transported out of the sample cup. The solid remaining from zinc stearate pyrolysis was 13.3%. It also produced various short-chain hydrocarbons as the decomposition products. The solid recovery was 65.7% from aluminum trihydrate due to water release. When the polyethylene content in their masterbatches is considered, the solid yields from TiO2 and carbon black were 99.9% and 99.5%. Hindered phosphite and hindered amine were nearly completely devolatilized, whereas there was 9.3% solid after hindered phenol was pyrolyzed. In addition, pyrolysis products from AOs and LS also included significant amounts of light gases and hydrocarbon liquids. However, a majority of the volatile decomposition products contained compounds with heterogeneous atoms (P, N, S, and O), which will be discussed further in the following section.
3.3 The role of additives during thermal pyrolysis
In general, thermal pyrolysis of PE is initiated by random homolytic chain cleavages for forming primary radicals.19,41 The β–β scission of the primary hydrocarbon radicals can form a shorter chain primary radical and ethylene. The primary radicals are also converted to a more stable secondary radical through intra- or intermolecular hydrogen transfer, which can further undergo β-scission to form an alkene and another shorter chain primary radical. The alkene can also undergo hydrogen abstraction and β-scission to form a diene. Meanwhile, an alkane is formed when the primary radical is saturated by free hydrogen or when two primary radicals combine. Cyclization and aromatization are less dominant during thermal pyrolysis because they require more steps and deeper dehydrogenation.56 Aromatics are usually the result of Diels–Alder reactions between small alkenes or dienes to form cyclic intermediates, followed by sequential dehydrogenation. Monoaromatic products like benzene, toluene, and xylene can also further undergo alkylation and condensation to form longer alkylated aromatics and polyaromatics. The presented results suggest that the thermal decomposition mechanism of PE is strongly affected by plastic additives. The inorganic fillers, such as talc, kaolin, and CaCO3, strongly promoted both C–C bond scissions to lighter compounds and dehydrogenation, which subsequently promoted Diels–Alder cyclization, followed by further dehydrogenation to form aromatics.42,59 These fillers are thermally stable at the pyrolysis temperatures, so they affect PE decomposition by serving as catalysts. While kaolin clay is present in plastics as a reinforcing filler, it can act as an acid catalyst. In a previous study, employing a kaolin catalyst in low-density polyethylene (LDPE) pyrolysis using a fixed bed was found to increase the gas yield.60 Kaolin was also added to a fluidized bed for pyrolyzing LDPE, resulting in increased oil yield and higher aromatic compounds than without kaolin.61 Kaolin is a layered phyllosilicate mineral with macropores. As an aluminosilicate mineral, it exhibits inherent acidity due to the presence of Brønsted and Lewis acid sites.62 Attributed to its acidity and porous structure, kaolin can promote C–C and C–H bond cleavages. The proton transfer to PE at Brønsted acid sites of kaolin can form a carbonium ion intermediate, which can subsequently undergo β-scission to form alkenes and a shorter chain carbonium ion. On the other hand, Lewis acid can enhance the PE dehydrogenation to form alkene intermediates, promoting the formation of carbonium and a subsequent catalytic cracking reaction.27 The hydrogen transfer catalyzed by kaolin can increase unsaturated hydrocarbons, especially aromatics.60 Talc is less known as a catalyst. However, a few studies reported its ability to catalyze bond cleavages. Talc was found to catalyze the dehydrogenation of ethanol to 1,3-butadiene.63 Zhou et al. reported that PE with a talc filler produced higher amounts of lighter hydrocarbons than neat PE during microwave pyrolysis in the presence of zeolite.64 In another study, PE composites with a talc nanofiller were found to increase PE degradation.65 In talc, a trioctahedral sheet of Mg and OH ions is sandwiched between two tetrahedral sheets of Si–O units. Mg–O is a base, whereas Si–O is acidic. Since the molar ratio of Mg–O and Si–O in talc is 1:2, the imbalanced charges of the acid and base-paired structure (Mg–O–Si bonds) can make talc a weak acid catalyst.63,66 The drastic reduction in wax yields and the formation of aromatics in this study indicate that the acidity of the talc filler can effectively facilitate C–C scission and dehydrogenation.
The CaCO3 filler also had a pronounced effect on C–C bond cleavages and aromatization during PE pyrolysis. CaCO3 is a Brønsted base catalyst due to its strong base anion.67 In a literature study, utilizing CaCO3 as a base catalyst for PE conversion increased lighter oil yield and reduced the reaction time.68 Another study reported that a carbonate base catalyst can reduce the wax formation from PE conversion.69 Base-catalyzed depolymerization is likely initiated when CaCO3 abstracts hydrogen from PE to form polymer carbanions because the abstracted H+ is stabilized on the CaCO3 surface. Subsequent β-scission of the carbanion forms an alkene and a shorter carbanion until the combination of the carbanion and the hydrogen metal catalyst anion terminates the reaction. Simultaneously, hydrogen abstraction by CaCO3 promotes aromatic hydrocarbons. The observation of naphthalene in the pyrolysis products suggests that extensive hydrogen transfer and dehydrogenation promoted the growth of mono-aromatics into large polyaromatics, which served as the precursor for carbon residue. While the fillers affect PE decomposition through their catalytic activities, the high percentage of fillers added to the polymer matrix may also act as a physical barrier to limit the diffusion of the volatile products out of the molten polymer matrix, increasing their secondary cracking and dehydrogenation reactions. Although the catalytic effect of the fillers for enhancing polymer cracking is expected to lower the temperature required for volatilizing the hydrocarbon products, the increased mass transfer limitation is having the opposite impact. The counterbalance between the two contradictory effects can diminish the effects of fillers for promoting early volatilization or even delay the volatilization, which was observed in the TGA results of PE with CaCO3 described above. Noteworthily, the catalytic effects of CaCO3 presented in this study do not agree with the results reported by Ishimura and co-workers.70 They pyrolyzed a physical mixture of microplastics and CaCO3 using a micro-pyrolyzer and found no changes in PE decomposition products upon adding CaCO3. However, their study also reported that CaCO3 affected non-PE plastics. The discrepancy between the present study and the literature results regarding the CaCO3 effect could be associated with how it was introduced during pyrolysis. When CaCO3 is compounded into the polymer as a filler, as shown in this study, a much intensive physical and chemical interaction between the polymer and filler occurs.
Among the inorganic additives, barium sulfate and aluminum trihydride had a much subtler impact than others. Barium sulfate is a stable and neutral salt and remains chemically inert when PE with this additive is pyrolyzed. As a fire retardant, aluminum trihydrate decomposes at elevated temperatures (∼220 °C) to release water vapor and form Al2O3.39 Al2O3 can act as both an acid and a base due to the presence of Lewis acidic sites and basic sites.71 Additionally, Al2O3 has a high surface area for adsorbing reactants, and is used as a catalyst support known to enhance activity for dehydrogenation and aromatization reactions.72 However, the aluminum trihydrate effect was minor during PE pyrolysis, likely because its endothermic decomposition reaction and water evaporation consumed the supplied thermal energy, resulting in lower reaction rates for PE. Thus, the catalytic effect of Al2O3 may be countered by aluminum trihydrate decomposition.
Zinc stearate exhibited a stronger tendency to increase light hydrocarbons and aromatics. This present result also corresponds with the previous finding by Gönen et al., as they used zinc stearate to increase paraffin decomposition.73 Zinc stearate can decompose at temperatures above 200 °C to form ZnO, ZnCO3, and short-chain hydrocarbons as its decomposition products (given in Table S3†). ZnO is an amphoteric oxide with both Lewis acid and base sites,74 while ZnCO3 is basic. When PE with zinc stearate is pyrolyzed, the zinc oxide and zinc salt derived from zinc stearate can serve as catalysts to facilitate polymer cracking and dehydrogenation.75 Additionally, the light hydrocarbons produced from zinc stearate decomposition can also contribute to the shift in the product distributions to shorter carbon chain lengths.
PE cracking and aromatic hydrocarbons were also promoted with TiO2 and carbon black. TiO2 can enhance bond cleavages and cyclization due to its Lewis acid activity.76 While TiO2 is known for its catalytic activity for photocatalysis, it was also previously used as a catalyst in plastic pyrolysis.77 Carbon black has a graphite-like structure with high structural defects. Its high surface area, oxygen functional groups (COOH, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), and defective structure help to establish its reactivity.78 Previously, carbon black has been used as an effective dehydration catalyst for producing hydrogen and carbon from methane.79 It has been postulated in the literature that the edges or defects of the graphene layers in carbon black serve as active sites.80 The increased light hydrocarbons from PE with TiO2 or carbon black are also partially attributed to the LLDPE masterbatch used to disperse pigments. LLDPE is a hydrocarbon polymer with tertiary carbons and a shorter carbon chain length than HDPE, requiring lower pyrolysis activation energy to promote lighter hydrocarbons.41,81
O), and defective structure help to establish its reactivity.78 Previously, carbon black has been used as an effective dehydration catalyst for producing hydrogen and carbon from methane.79 It has been postulated in the literature that the edges or defects of the graphene layers in carbon black serve as active sites.80 The increased light hydrocarbons from PE with TiO2 or carbon black are also partially attributed to the LLDPE masterbatch used to disperse pigments. LLDPE is a hydrocarbon polymer with tertiary carbons and a shorter carbon chain length than HDPE, requiring lower pyrolysis activation energy to promote lighter hydrocarbons.41,81
Contrary to the inorganic additives, AOs and LS increased heavier hydrocarbon yields. Hindered amines and phenols are primary antioxidants and stabilizers. In an oxidative environment, they can donate hydrogen, enabling peroxyl radicals to form polymer hydroperoxide complexes, terminating the autocatalytic oxidation of plastics. Hindered phosphite is a secondary AO usually used in combination with primary AOs. It can further react with hydroperoxide to prevent oxidative branching or substitute with the polymer alkoxy radicals.82 Due to the inert environment, oxygen-centered radical scavenging does not occur during PE pyrolysis. However, AOs and LS are extremely unstable and can decompose at much lower temperatures than the pyrolysis temperature of PE (as shown in Fig. 2). Thus, the AOs and LS would undergo extensive decomposition during pyrolysis (see Tables S4–S7† for the decomposition products). The primary bond cleavage occurs at the phosphite and ester linkages for the hindered phosphite and phenol.83,84 In the hindered phosphite, the O–P bond cleavages led to the formation of phosphorus oxides and butylated phenols, with 2,4-di-tert-butylphenol being the major product. The secondary depolymerization of the phenolics also resulted in various mono- and di-aromatic hydrocarbons as well as aromatics with oxygenated side chains. In hindered phenol, the ester bonds connecting the antioxidant moieties to the central core are cleaved, releasing the antioxidant moieties and potentially leaving behind pentaerythritol, which can further decompose to form phenolics and alkylated aromatics. On the other hand, hindered amines were decomposed to produce various aliphatic hydrocarbons and N- or O-containing compounds. The ester bond cleavage in hindered amines84,85 leads to the formation of succinates and potential ring-opening followed by rearrangement reactions in the piperidine structure to form 1-methyl-cyclopentene, p-xylene, 2,3-dimethyl pyridine, and succinic anhydride as major products. When PE with AOs and LS were pyrolyzed, the AOs or LS-derived radicals and molecules could stabilize the PE-derived radicals through radical coupling or radical substitution. For example, aromatic alkoxy radicals derived from hindered phosphite can combine with the primary or secondary hydrocarbon radicals produced from PE pyrolysis. Amine groups in the hindered amine decomposition products can stabilize radical intermediates of PE through resonance and hydrogen donation.84 Scavenging PE radicals by the AOs and LS would terminate the propagation of β-scission, thus hindering effective depolymerization of PE to increase the heavy hydrocarbon fraction in the pyrolysis products. Aromatic hydrocarbons were also found in the pyrolysis of PE with AOs and LS, which could be attributed to the decomposition of the aromatic ring-containing AOs (hindered phenol or hindered phosphite). Aromatic hydrocarbons can also be produced via the cyclization reactions between the PE-derived hydrocarbons and AOs or LS decomposition products, with benzene, toluene, and xylene being the major aromatics. PE containing all three hindered additives further suppressed PE depolymerization to produce a higher wax yield than PE with individual additives. Notably, a previous study by Almeida et al. suggests that a combination of primary and secondary antioxidants can result in synergistic radical scavenging activities during melt processing.86
The PE pyrolysis mechanism and the summarized effect of additives are illustrated in Fig. 6, where the intensity of the heat map is derived from the deviation in pyrolysis product distribution for PE with additives compared to the virgin polymer.
 | ||
| Fig. 6 Degradation mechanisms for the pyrolysis of polyolefins containing common additives. | ||
3.4 Catalytic pyrolysis of PE with additives
The effect of additives on the catalytic pyrolysis of PE was investigated by employing HZSM-5 zeolite as the catalyst. Zeolite is a commercial solid acid catalyst frequently used to convert biomass and waste plastics.25,27,30 In this study, two different catalyst-to-plastic contact modes were studied. Ex situ catalytic pyrolysis consists of two stages, which includes plastic pyrolysis and subsequent catalytic upgrading of pyrolysis vapors. In situ catalytic pyrolysis is a single-stage conversion in which plastics and catalyst mixtures are pyrolyzed. In a scaled process, ex situ catalytic pyrolysis can mimic pyrolysis using a fluidized bed with a downstream catalytic bed configuration, whereas in situ catalytic pyrolysis can be related to configurations where catalysts are placed inside a fluidized bed reactor along with heat carriers.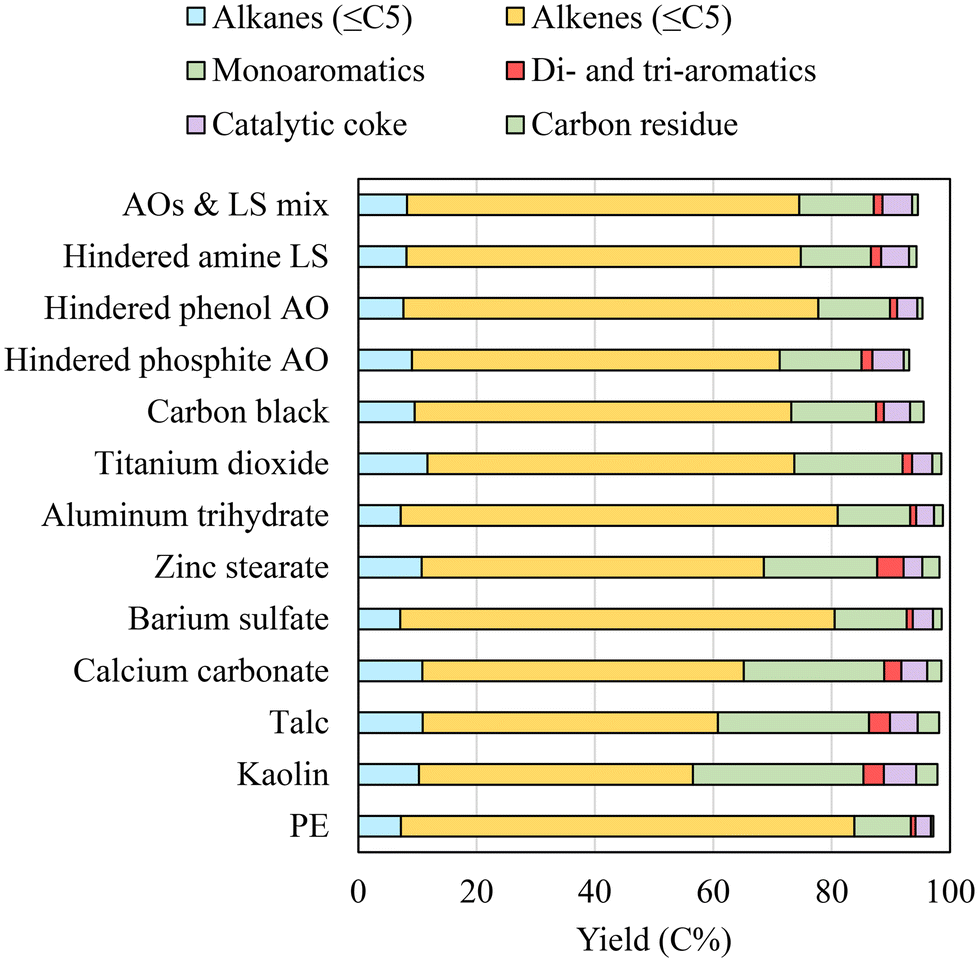 | ||
| Fig. 7 The product distribution during ex situ catalytic pyrolysis of PE with additives. | ||
| Compounds | A | B | C | D | E | F | G | H | I | J | K | L | M |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Aliphatic monomer yield & selectivity (C%) | |||||||||||||
| Methane | 0.95 (1.13) | 1.26 (2.23) | 1.03 (1.69) | 0.91 (1.4) | 0.91 (1.13) | 1.33 (1.95) | 0.93 (1.14) | 0.97 (1.32) | 0.76 (1.04) | 1.45 (2.04) | 1.2 (1.54) | 0.9 (1.2) | 0.93 (1.25) |
| Ethane | 1.58 (1.89) | 0.6 (1.06) | 0.55 (0.9) | 0.48 (0.73) | 1.53 (1.9) | 0.63 (0.92) | 1.53 (1.89) | 0.39 (0.53) | 0.33 (0.44) | 0.71 (1) | 1.52 (1.96) | 1.76 (2.35) | 0.43 (0.58) |
| Propane | 2.98 (3.56) | 3.86 (6.83) | 4.07 (6.69) | 4.07 (6.24) | 3.02 (3.75) | 4.08 (5.94) | 3.04 (3.75) | 4.85 (6.58) | 3.84 (5.25) | 3.94 (5.53) | 3.17 (4.07) | 3.61 (4.83) | 3.77 (5.06) |
| Butane | 1.24 (1.48) | 2.6 (4.6) | 2.87 (4.72) | 2.97 (4.56) | 1.18 (1.47) | 2.73 (3.98) | 1.2 (1.49) | 3.53 (4.79) | 2.96 (4.04) | 2.36 (3.32) | 1.28 (1.65) | 1.44 (1.92) | 2.39 (3.2) |
| Ethylene | 17.13 (20.43) | 12.46 (22.03) | 12.69 (20.87) | 11.92 (18.29) | 12.76 (15.85) | 16.22 (23.65) | 13.02 (16.07) | 13.53 (18.36) | 13.32 (18.2) | 15.19 (21.32) | 13.08 (16.83) | 14.98 (20.03) | 13.89 (18.64) |
| Propylene | 44.18 (52.7) | 25.69 (45.42) | 27.41 (45.09) | 31.69 (48.64) | 45.53 (56.54) | 31.37 (45.76) | 45.62 (56.3) | 36.4 (49.39) | 37.91 (51.8) | 35.75 (50.2) | 43.06 (55.38) | 40.42 (54.04) | 38.49 (51.64) |
| Butene | 12.78 (15.24) | 6.57 (11.62) | 7.94 (13.07) | 8.56 (13.14) | 12.79 (15.89) | 8.31 (12.12) | 12.82 (15.82) | 9.6 (13.03) | 10.05 (13.73) | 9.79 (13.75) | 12.21 (15.71) | 9.78 (13.08) | 11.36 (15.24) |
| Pentane | 0.47 (0.56) | 1.94 (3.44) | 2.38 (3.92) | 2.41 (3.69) | 0.47 (0.59) | 1.96 (2.86) | 0.48 (0.59) | 1.97 (2.68) | 1.65 (2.25) | 0.62 (0.87) | 0.47 (0.61) | 0.45 (0.6) | 0.72 (0.96) |
| Pentene | 2.53 (3.02) | 1.56 (2.76) | 1.85 (3.04) | 2.14 (3.29) | 2.32 (2.89) | 1.93 (2.82) | 2.39 (2.95) | 2.45 (3.33) | 2.37 (3.24) | 1.41 (1.98) | 1.76 (2.26) | 1.46 (1.95) | 2.56 (3.43) |
| Aliphatics (≤C 5 ) | 83.84 | 56.55 | 60.78 | 65.15 | 80.51 | 68.56 | 81.02 | 73.69 | 73.19 | 71.22 | 77.75 | 74.80 | 74.54 |
| Aromatic monomer yield & selectivity (C%) | |||||||||||||
| Benzene | 1.93 (18.62) | 6.44 (19.93) | 5.11 (17.56) | 4.73 (17.74) | 2.43 (18.46) | 4.08 (17.29) | 2.46 (18.59) | 3.71 (18.65) | 2.86 (18.3) | 4.74 (30.25) | 2.94 (22.07) | 2.25 (16.58) | 2.98 (21.19) |
| Toluene | 4.38 (42.35) | 12.04 (37.31) | 11.07 (38.05) | 10.27 (38.56) | 5.62 (42.64) | 7.54 (31.96) | 5.64 (42.59) | 8.08 (40.58) | 6.31 (40.38) | 5.83 (37.15) | 5.18 (38.93) | 4.9 (36.16) | 5.43 (38.61) |
| Ethylbenzene | 0.36 (3.49) | 0.75 (2.32) | 0.78 (2.67) | 0.77 (2.88) | 0.46 (3.49) | 0.48 (2.03) | 0.47 (3.53) | 0.61 (3.07) | 0.47 (3.03) | 0.25 (1.57) | 0.37 (2.77) | 0.22 (1.62) | 0.44 (3.15) |
| p-Xylene | 2.1 (20.31) | 5.76 (17.83) | 5.11 (17.58) | 4.89 (18.36) | 2.64 (19.99) | 3.61 (15.31) | 2.65 (19.98) | 4.04 (20.3) | 3.11 (19.9) | 2.2 (14.02) | 2.35 (17.68) | 2.49 (18.35) | 2.61 (18.55) |
| o-Xylene | 0.28 (2.67) | 1.03 (3.19) | 0.86 (2.96) | 0.79 (2.97) | 0.36 (2.74) | 0.65 (2.77) | 0.37 (2.76) | 0.7 (3.54) | 0.56 (3.59) | 0.4 (2.56) | 0.3 (2.24) | 0.39 (2.88) | 0.44 (3.12) |
| Styrene | 0.13 (1.27) | 0.16 (0.48) | 0.13 (0.44) | 0.1 (0.39) | 0.17 (1.28) | 0.1 (0.42) | 0.17 (1.28) | 0.3 (1.49) | 0.23 (1.49) | 0.08 (0.48) | 0.51 (3.87) | 0.11 (0.8) | 0.09 (0.62) |
| 1-Ethyl-3-methylbenzene | 0.21 (2.07) | 1.12 (3.46) | 1.12 (3.86) | 1.04 (3.92) | 0.26 (1.99) | 1.12 (4.75) | 0.27 (2.02) | 0.42 (2.13) | 0.37 (2.39) | 0.18 (1.13) | 0.25 (1.87) | 0.61 (4.47) | 0.35 (2.52) |
| 1,2,3-Trimethylbenzene | 0.18 (1.75) | 1.52 (4.72) | 1.37 (4.69) | 1.12 (4.22) | 0.22 (1.64) | 1.55 (6.58) | 0.22 (1.68) | 0.42 (2.1) | 0.37 (2.37) | 0.16 (1.05) | 0.22 (1.64) | 0.88 (6.46) | 0.25 (1.78) |
| Indane | 0.18 (1.74) | 0.47 (1.47) | 0.54 (1.86) | 0.65 (2.43) | 0.23 (1.73) | 0.48 (2.05) | 0.23 (1.76) | 0.13 (0.66) | 0.12 (0.74) | 0.29 (1.88) | 0.35 (2.6) | 0.51 (3.79) | 0.08 (0.54) |
| Indene | 0.2 (1.97) | 0.59 (1.84) | 0.39 (1.34) | 0.32 (1.21) | 0.25 (1.92) | 0.59 (2.52) | 0.26 (1.96) | 0.11 (0.55) | 0.09 (0.6) | 0.19 (1.19) | 0.26 (1.95) | 0.3 (2.2) | 0.13 (0.91) |
| 1-Methyl-1H-indene | 0.17 (1.63) | 0.5 (1.56) | 0.37 (1.28) | 0.28 (1.07) | 0.22 (1.68) | 0.5 (2.11) | 0.22 (1.69) | 0.09 (0.47) | 0.08 (0.51) | 0.15 (0.98) | 0.21 (1.6) | 0.26 (1.89) | 0.11 (0.77) |
| Naphthalene | 0.11 (1.08) | 0.77 (2.39) | 0.6 (2.06) | 0.53 (2) | 0.14 (1.09) | 0.78 (3.31) | 0.14 (1.09) | 0.29 (1.47) | 0.26 (1.67) | 0.45 (2.87) | 0.27 (2) | 0.36 (2.67) | 0.44 (3.1) |
| 2-Methylnaphthalene | 0.06 (0.62) | 0.64 (1.99) | 0.71 (2.44) | 0.8 (3) | 0.08 (0.62) | 0.65 (2.75) | 0.08 (0.64) | 0.45 (2.26) | 0.39 (2.51) | 0.49 (3.15) | 0.11 (0.79) | 0.29 (2.13) | 0.43 (3.07) |
| 1,2-Dimethyl-naphthalene | 0.05 (0.45) | 0.49 (1.51) | 0.38 (1.3) | 0.34 (1.27) | 0.09 (0.72) | 0.49 (2.09) | 0.06 (0.44) | 0.34 (1.7) | 0.3 (1.92) | 0.17 (1.11) | — | — | 0.13 (0.93) |
| Anthracene | — | — | 0.56 (1.91) | — | — | 0.64 (2.72) | — | 0.11 (0.55) | 0.1 (0.62) | 0.1 (0.61) | — | — | 0.09 (0.61) |
| 2-Methylanthracene | — | — | — | — | — | 0.32 (1.34) | — | 0.1 (0.49) | — | — | — | — | 0.07 (0.51) |
| Aromatics | 10.34 | 32.29 | 29.09 | 26.65 | 13.19 | 23.59 | 13.25 | 19.90 | 15.64 | 15.68 | 13.30 | 13.56 | 14.05 |
The aromatic yield was 19.9% for PE with TiO2 and 15.6% for PE with carbon black, both considerably higher than that obtained with virgin PE. On the other hand, aluminum trihydrate and barium sulfate had minimal impacts on the ex situ catalytic pyrolysis of PE. This result was expected because the composition of the upstream pyrolysis vapors was not altered much for PE with these additives, as shown in Fig. 3 and 4 above.
The aromatic yields from ex situ pyrolysis of PE with AOs and LS were between 13.3 and 15.7%. The effect of hindered phosphite was more profound than those of hindered phenol and hindered amine. The alkene (<C5) yield was decreased to 62.1% for PE with hindered phosphite compared to 70.1% with hindered phenol and 66.6% with hindered amine. The presence of AOs and LS also significantly promoted catalytic coke formation. The coke yield from PE with hindered phosphite was 5.3%, comparable to that produced from PE with kaolin. As shown in Fig. 7, the ex situ pyrolysis of PE with AOs and LS had relatively lower carbon mass closures for the quantified products (93.1–95.3%) than that of PE with other additives. This is because ex situ pyrolysis of PE with AOs and LS also produced >C5 aliphatic and carbon oxide gases that were not accounted for.
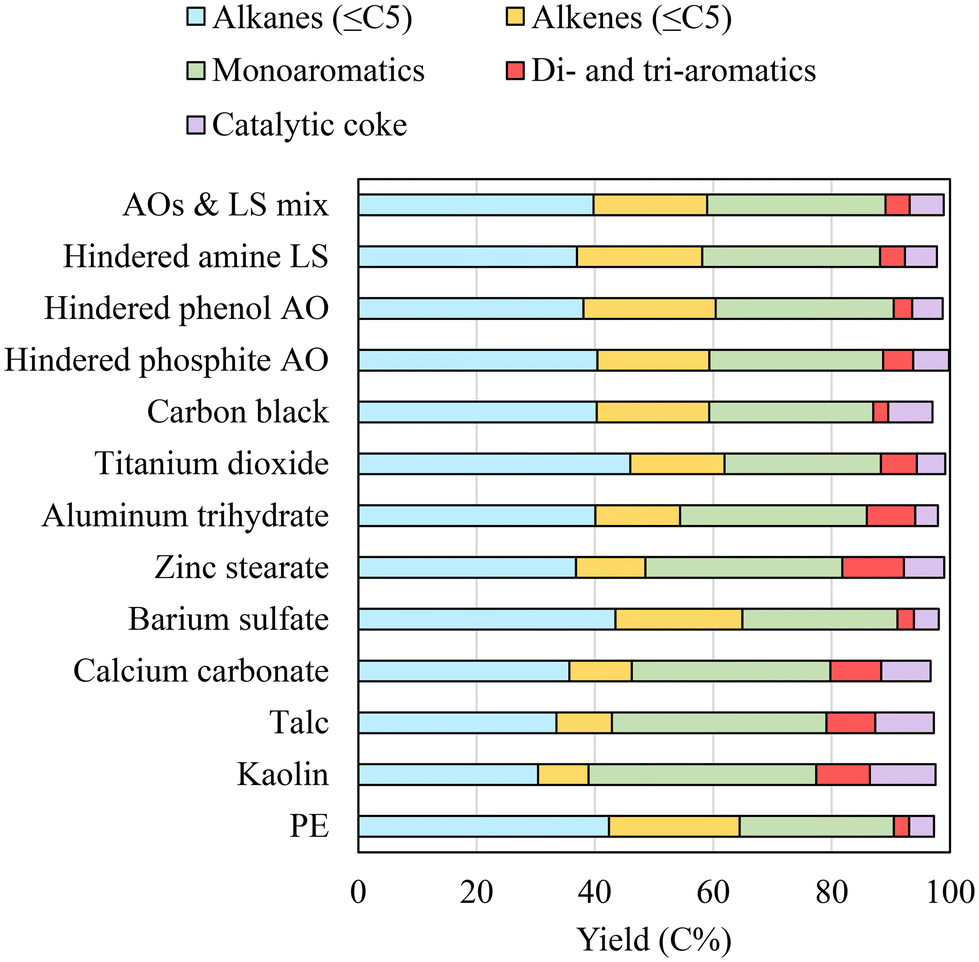 | ||
| Fig. 8 The product distribution during in situ catalytic pyrolysis of PE with additives. | ||
| Compounds | A | B | C | D | E | F | G | H | I | J | K | L | M |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Aliphatic monomer yield/selectivity (C%) | |||||||||||||
| Methane | 1.3 (2.02) | 0.81 (2.09) | 0.69 (1.61) | 0.63 (1.36) | 1.29 (1.99) | 0.99 (2.05) | 0.61 (1.13) | 0.73 (1.18) | 1.24 (2.09) | 1.37 (2.32) | 1.44 (2.38) | 0.95 (1.63) | 0.89 (1.51) |
| Ethane | 2.15 (3.34) | 0.38 (0.98) | 0.36 (0.84) | 0.32 (0.69) | 2.17 (3.34) | 0.46 (0.94) | 0.26 (0.48) | 0.28 (0.46) | 2.05 (3.45) | 0.67 (1.13) | 1.76 (2.91) | 1.81 (3.11) | 0.4 (0.68) |
| Propane | 21.55 (33.43) | 13.12 (33.71) | 13.85 (32.29) | 14.39 (31.13) | 22.79 (35.1) | 15.55 (32.04) | 16.06 (29.52) | 19.01 (30.72) | 21.18 (35.7) | 18.95 (31.94) | 19.51 (32.29) | 19.52 (33.59) | 18.67 (31.66) |
| Butane | 14.04 (21.78) | 13.82 (35.5) | 15.47 (36.08) | 16.75 (36.23) | 13.8 (21.26) | 16.71 (34.43) | 19.64 (36.1) | 21.95 (35.46) | 13.29 (22.39) | 16.29 (27.45) | 12.48 (20.66) | 12.24 (21.06) | 16.3 (27.65) |
| Ethylene | 6.07 (9.42) | 2.84 (7.29) | 2.9 (6.77) | 2.84 (6.14) | 4.74 (7.3) | 3.99 (8.22) | 3.78 (6.94) | 4.37 (7.05) | 4.21 (7.1) | 5.64 (9.51) | 5.16 (8.54) | 5.83 (10.03) | 4.99 (8.46) |
| Propylene | 9.49 (14.72) | 3.51 (9.01) | 3.78 (8.81) | 4.64 (10.04) | 10.08 (15.53) | 4.75 (9.78) | 6.5 (11.94) | 7.05 (11.39) | 8.89 (14.98) | 8.13 (13.7) | 10.39 (17.19) | 9.82 (16.9) | 8.33 (14.13) |
| Butene | 6 (9.31) | 1.98 (5.09) | 2.45 (5.7) | 2.75 (5.96) | 6.14 (9.46) | 2.72 (5.6) | 3.69 (6.78) | 4.06 (6.56) | 5.47 (9.22) | 4.84 (8.15) | 6.39 (10.57) | 5.17 (8.9) | 5.35 (9.07) |
| Pentane | 3.34 (5.18) | 2.26 (5.8) | 3.14 (7.32) | 3.61 (7.8) | 3.42 (5.27) | 3.1 (6.39) | 3.48 (6.4) | 3.98 (6.44) | 2.57 (4.33) | 3.13 (5.28) | 2.89 (4.78) | 2.45 (4.22) | 3.5 (5.94) |
| Pentene | 0.52 (0.81) | 0.21 (0.53) | 0.25 (0.57) | 0.3 (0.65) | 0.48 (0.75) | 0.27 (0.56) | 0.38 (0.71) | 0.47 (0.75) | 0.44 (0.74) | 0.31 (0.52) | 0.41 (0.68) | 0.34 (0.58) | 0.53 (0.9) |
| Aliphatics (≤C 5 ) | 64.46 | 38.92 | 42.87 | 46.22 | 64.92 | 48.54 | 54.41 | 61.90 | 59.33 | 59.34 | 60.42 | 58.13 | 58.97 |
| Aromatic monomer yield/selectivity (C%) | |||||||||||||
| Benzene | 5.11 (17.83) | 8.05 (16.94) | 6.75 (15.18) | 6.19 (14.68) | 5.04 (17.37) | 6.71 (15.37) | 5.98 (15.04) | 5.15 (15.86) | 5.41 (17.88) | 9.98 (28.97) | 7.53 (22.68) | 5.68 (16.57) | 7.03 (20.52) |
| Toluene | 10.97 (38.31) | 14.09 (29.65) | 13.91 (31.26) | 12.83 (30.42) | 11.31 (38.99) | 11.42 (26.15) | 12.53 (31.55) | 10.54 (32.43) | 11.87 (39.26) | 11.56 (33.55) | 12.68 (38.2) | 11.8 (34.45) | 12.18 (35.56) |
| Ethylbenzene | 0.71 (2.47) | 0.69 (1.46) | 0.76 (1.72) | 0.75 (1.79) | 0.72 (2.48) | 0.58 (1.33) | 0.76 (1.9) | 0.62 (1.91) | 0.76 (2.5) | 0.38 (1.11) | 0.72 (2.17) | 0.42 (1.22) | 0.8 (2.32) |
| p-Xylene | 5.7 (19.91) | 7.3 (15.36) | 7.03 (15.81) | 6.7 (15.89) | 5.53 (19.07) | 5.99 (13.72) | 6.66 (16.76) | 5.71 (17.56) | 5.82 (19.24) | 4.6 (13.37) | 6.23 (18.75) | 6.51 (18.99) | 6.25 (18.25) |
| o-Xylene | 1.76 (6.13) | 3.15 (6.64) | 2.81 (6.32) | 2.53 (6.01) | 1.82 (6.26) | 2.59 (5.94) | 2.86 (7.2) | 2.35 (7.22) | 1.95 (6.44) | 2.02 (5.87) | 1.89 (5.7) | 2.37 (6.92) | 2.55 (7.43) |
| Styrene | 0.16 (0.57) | 0.11 (0.23) | 0.09 (0.21) | 0.08 (0.18) | 0.2 (0.69) | 0.08 (0.19) | 0.28 (0.7) | 0.23 (0.71) | 0.21 (0.69) | 0.06 (0.18) | 0.3 (0.89) | 0.09 (0.26) | 0.07 (0.19) |
| 1-Ethyl-3-methylbenzene | 1.05 (3.65) | 2.6 (5.47) | 2.64 (5.94) | 2.55 (6.04) | 1.02 (3.51) | 3.01 (6.89) | 1.46 (3.67) | 1.07 (3.31) | 1.09 (3.61) | 0.46 (1.35) | 0.45 (1.36) | 1.59 (4.65) | 0.87 (2.54) |
| 1,2,3-Trimethylbenzene | 0.62 (2.17) | 2.49 (5.23) | 2.24 (5.04) | 1.95 (4.63) | 0.58 (2.01) | 2.87 (6.57) | 1.02 (2.56) | 0.76 (2.33) | 0.63 (2.08) | 0.3 (0.86) | 0.29 (0.86) | 1.61 (4.69) | 0.42 (1.23) |
| Indane | 0.19 (0.65) | 0.23 (0.48) | 0.26 (0.6) | 0.34 (0.8) | 0.18 (0.64) | 0.27 (0.61) | 0.09 (0.24) | 0.07 (0.22) | 0.2 (0.66) | 0.17 (0.48) | 0.14 (0.41) | 0.28 (0.81) | 0.04 (0.11) |
| Indene | 0.11 (0.38) | 0.15 (0.31) | 0.1 (0.22) | 0.08 (0.2) | 0.1 (0.36) | 0.17 (0.38) | 0.04 (0.1) | 0.03 (0.09) | 0.11 (0.38) | 0.05 (0.15) | 0.05 (0.16) | 0.08 (0.24) | 0.03 (0.1) |
| 1-Methyl-1H-indene | 0.1 (0.35) | 0.14 (0.3) | 0.11 (0.24) | 0.08 (0.2) | 0.1 (0.36) | 0.16 (0.36) | 0.04 (0.09) | 0.03 (0.09) | 0.11 (0.36) | 0.05 (0.14) | 0.05 (0.14) | 0.08 (0.24) | 0.03 (0.09) |
| Naphthalene | 0.54 (1.9) | 1.76 (3.7) | 1.39 (3.13) | 1.3 (3.08) | 0.54 (1.86) | 2.07 (4.74) | 1.02 (2.57) | 0.74 (2.28) | 0.58 (1.92) | 1.14 (3.31) | 0.49 (1.48) | 0.95 (2.78) | 1.05 (3.07) |
| 2-Methyl-naphthalene | 0.67 (2.33) | 3.18 (6.69) | 3.58 (8.03) | 4.13 (9.81) | 0.68 (2.34) | 3.65 (8.37) | 3.27 (8.23) | 2.45 (7.54) | 0.74 (2.46) | 2.71 (7.86) | 0.42 (1.26) | 1.64 (4.79) | 2.26 (6.59) |
| 1,2-Dimethyl-naphthalene | 0.72 (2.5) | 3.58 (7.54) | 2.81 (6.32) | 2.65 (6.28) | 1.17 (4.05) | 4.1 (9.39) | 3.73 (9.38) | 2.75 (8.46) | 0.76 (2.52) | 0.97 (2.81) | 1.97 (5.93) | 1.17 (3.4) | 0.68 (1.99) |
| Anthracene | 0.17 (0.59) | — | — | — | — | — | — | — | — | — | — | — | — |
| 2-Methylanthracene | 0.07 (0.26) | — | — | — | — | — | — | — | — | — | — | — | — |
| Aromatics | 28.64 | 47.53 | 44.50 | 42.16 | 29.00 | 43.67 | 39.73 | 32.51 | 30.23 | 34.45 | 33.20 | 34.26 | 34.24 |
While it had minor effects during thermal pyrolysis and ex situ pyrolysis, aluminum trihydrate noticeably increased aromatic yield during in situ pyrolysis. It was also noted that the increased aromatics (39.7%) were mainly balanced by the decreased alkenes, since alkane and coke yields only changed slightly. Barium sulfate was inert and thus had no significant impact during in situ pyrolysis.
TiO2 and carbon black affected ex situ pyrolysis of PE to a similar extent. However, they showed noticeable differences during in situ pyrolysis. The aromatic yield was 32.5% for PE with TiO2, higher than 30.2% for PE with carbon black. The higher aromatic yield with TiO2 was mostly attributed to the increase in polyaromatics. On the other hand, carbon black caused a higher coke yield of 7.5% compared to 4.8% with TiO2. The presence of carbon black also reduced the alkane yield more than TiO2 did.
The presence of AOs and LS also promoted higher aromatic and coke yields during in situ pyrolysis. Hindered phosphite led to slightly more aromatics (34.5%) than hindered phenol (33.2%) and hindered amine (34.3%). The coke yield produced from PE with hindered phosphite was 6%, comparable to the 6.8% produced from PE with zinc stearate.
3.5 The impact of additives during catalytic pyrolysis of PE
In zeolite, tetrahedral networks of alumina and silica form a microporous structure with both Brønsted and Lewis acid sites. While most active sites are inside the pores, the porous structures provide large surface areas for catalyzing various reactions, such as cracking, deoxygenation, isomerization, cyclization, oligomerization, and dehydrogenation.27,87 The mechanism of PE decomposition changes from the free radical-initiated chain scission during thermal pyrolysis to carbocationic-intermediate chain scission during catalytic pyrolysis.29 Hydrogen transfer reactions of alkenes can yield alkanes and highly unsaturated compounds like aromatics. The increasing number of acid sites could enhance the transformation of alkenes into carbonium ions, which provide intermediates for secondary reactions, including hydrogen transfer reactions. For hydrocarbon polymer conversion, Brønsted acid primarily promotes β-scission, while the Lewis acid site plays a role in transforming primary products from chain scissions into cyclics and aromatics via carbonium ion formation.87 There are several different pathways to form aromatics over zeolite catalysts, such as Diels–Alder reactions between C2–C5 dienes and alkenes, cyclization between unsaturated carbonium ions and alkanes, followed by dehydrogenation of cycloalkanes.87 Another pathway is a hydrocarbon pool mechanism in which alkenes form a pool of (CH2)n species inside the pores and further convert to aromatics.88,89 Several factors, such as acidity, pore size, and the configuration of pore channels, can impact the effects of a zeolite catalyst.87 Strong acidity of zeolite enhances cracking and aromatization, but it can also increase coke formation due to the growth of polyaromatics.87,90 Pore size affects the accessibility of the reactants for the active sites since the bulky molecules cannot enter micropores. HZSM-5 is a catalyst with medium-sized pores and moderately high acidity. In general, coke formation in zeolite can affect the catalytic activity by decreasing the density of Brønsted acid sites, limiting the diffusion of bulkier products, and intensifying the intrinsic shape selectivity of the zeolite to aromatics.91During ex situ pyrolysis of PE, thermal pyrolysis of PE is followed by catalytic upgrading of the pyrolysis vapor. Due to the short vapor retention time over the catalyst bed, cracking was the dominant reaction at the catalyst sites. Although the solid recovery results described in section 3.2 indicate that small amounts of additive particles can still be transported by sweeping gas, mineral additives like kaolin, talc, CaCO3, carbon black, and TiO2 are mostly non-volatile. Thus, these additives mostly affect the ex situ catalytic pyrolysis by altering the composition of the pyrolysis vapors. Pyrolysis vapors derived from the PE containing these additives consist of shorter chain hydrocarbons than the virgin PE-derived vapor, requiring fewer C–C scissions in the subsequent stage for catalytic conversion. Smaller molecules also have improved accessibility to the zeolite pores where most acid sites are present, thereby promoting zeolite activities for cracking and aromatization. Moreover, the pyrolysis vapors reaching the catalyst already contain aromatics as well as higher concentrations of dienes and cyclic compounds that are the intermediates of aromatics. These changes in the pyrolysis vapor composition contributed to the higher aromatic yield obtained during ex situ catalytic pyrolysis. The increased aromatization also promoted di- and tri-aromatic hydrocarbons, enhancing coke formation.
Some additives decompose during pyrolysis, entraining their decomposition products into the pyrolysis vapors. In such cases, the additives can affect the catalytic conversion of PE both indirectly and directly. As mentioned earlier, zinc stearate decomposition produces light aliphatic hydrocarbons in addition to non-volatile ZnO and ZnCO3. While ZnO and ZnCO3 promote PE cracking during the pyrolysis step to increase the shorter chain hydrocarbons, the additive-derived hydrocarbons can be converted by zeolite to increase the aromatic content. AOs and LS also completely decompose during pyrolysis. As described earlier, pyrolysis vapors of PE with AOs and LS contained increased amounts of heavy hydrocarbons due to the radical scavenging effects of AOs and LS for inhibiting PE chain cracking. These heavier hydrocarbons require more intensive cracking at the catalytic sites, whereas their larger molecular sizes hinder their access to the active sites inside the zeolite pores. Instead, these large molecules with unsaturated functional groups would be adsorbed onto the catalyst surface, where the active sites are scarce, to dehydrogenate into aromatics and coke. Hindered phosphite and hindered phenol decomposition also produce various phenolic compounds (Tables S4–S7†). Due to their strong tendency for adsorption onto zeolite surfaces, these phenolic decomposition products can compete with PE-derived hydrocarbons for the active sites. Hindered amine, characterized by multiple nitrogen-containing functional groups, has a pronounced ability to neutralize the acidic sites of zeolite catalysts.91 The basic nature of the nitrogen atoms in the volatile decomposition products of hindered amine allows them to titrate the Brønsted acid sites within the zeolite framework.44 As such, the catalyst efficiency of zeolite can be decreased when the pyrolysis vapors of PE with AOs and LS are converted. This speculation is supported by the observation of >C5 aliphatic hydrocarbons among the ex situ conversion products of PE with AOs and LS, which was not found with other additives. The increased adsorptions of reactants and decreased catalytic activity also strongly promoted catalytic coke formation.
Unlike ex situ pyrolysis, PE and additives can directly interact with zeolite during in situ pyrolysis. Since the macromolecule of the polymer cannot enter zeolite pores, the molten polymer is first cracked on the catalyst surface before the fragmented hydrocarbons diffuse into the catalyst pores. Zeolite covered with a molten phase polymer effectively increased the physical contact between PE and the catalyst, significantly promoting aromatization. Increased aromatization also led to much higher coke formation during in situ pyrolysis than during ex situ pyrolysis. Alkanes also increased, attributed to the increased hydrogen available as a result of aromatization. The additives can affect in situ pyrolysis of PE in several different ways. As seen during pyrolysis, the acidity or basicity of the additives can assist PE cracking to increase shorter chain hydrocarbons, thereby improving their diffusion into the zeolite pores. On the other hand, the deposition of the additives on zeolite can reduce the surface area of the catalyst and clog pores, especially when the additive concentration is high. In a previous study, HZSM-5 zeolite with wet-impregnated CaCO3 had a poor HDPE conversion compared to the untreated HZSM-5 catalyst due to blocked zeolite pores by CaCO3.92 In this work, the presence of kaolin, talc, and CaCO3 strongly enhanced aromatic formation from PE. Nonetheless, catalytic coke also increased due to the presence of additives. High catalytic coke indicates a pronounced loss of feedstock carbon to a less desired product. Catalytic coke also promotes zeolite deactivation by clogging pores and blocking active sites.
Metal-based additives like zinc stearate may also modify the aromatization pathway of zeolite by activating C–H bonds in PE. Its metal centers provide additional Lewis acid sites for dehydrogenation and thus can directly convert light hydrocarbons into aromatics.93 Direct contact between zinc stearate and zeolite can also cause metal-ion exchange at the catalyst. It was reported that after PE with zinc stearate was converted by zeolite, zinc remaining on the used catalyst can permanently change the catalyst structure by forming new Brønsted acid sites (Zn(OH)+) during the regeneration of the used catalyst.44 In the literature, zinc-modified zeolite exhibited higher activity in activating alkanes compared to fresh zeolite for achieving significantly higher conversion rates and altering product distributions to favor the formation of aromatic compounds like benzene.93,94 The introduction of zinc sites accelerates reaction rates and promotes aromatization during alkane cracking.
Aluminum trihydrate had a minimal effect during ex situ pyrolysis of PE, whereas it notably increased aromatic yield during in situ pyrolysis. During in situ pyrolysis, Al2O3, the decomposition product of aluminum trihydrate, combined with zeolite, may increase the acidity of the catalyst. Additionally, water produced during the decomposition of aluminum trihydrate can adsorb onto the Brønsted acid sites of zeolite to form hydronium ions.95 The presence of hydronium ions can further increase the acidity of the catalyst. As mentioned earlier, the higher acidity of zeolite is known to increase aromatization. On the other hand, barium sulfate had a minimal effect during both ex situ catalytic pyrolysis and in situ catalytic pyrolysis due to its relatively inert structure.
The role of AOs and LS in in situ pyrolysis can differ from ex situ pyrolysis. The radical scavenging effects of AOs and LS were diminished during in situ pyrolysis because the radical-based mechanism of thermal pyrolysis was replaced by a carbonium ion-based mechanism when PE was converted by zeolite. On the other hand, AOs and LS can affect the conversion through direct interaction with zeolite. Among them, hindered phosphite exhibited a much more pronounced effect on increasing aromatization. It has been suggested that phosphorus atoms can exchange with Brønsted acid sites, decreasing the overall acidity of the zeolite.96,97 In industrial applications, phosphorus is intentionally incorporated into zeolite catalysts to enhance long-term stability and minimize coke formation by modifying stronger acid sites while preserving weaker ones.96–98 The decreased acidity is supposed to reduce the efficiency of catalytic reactions. However, aromatization and coke formation increased in this study, suggesting phosphorus ion exchange was less significant. On the other hand, the aromatic rings and branched alkyl groups of phosphite can directly produce aromatics or contribute to the hydrocarbon pool mechanism to form aromatics when they interact with zeolite. The naphthalene and alkylated naphthalene noticeably increased, suggesting the aromatic ring of the hindered phosphite served as a precursor for further dehydrocyclization and alkylated aromatics. For hindered amine, its decomposition products can also contribute to the hydrocarbon pool mechanism. Additionally, its nitrogen atoms can strongly adsorb and neutralize some acid sites of zeolite to increase coke formation.44,91 Hindered phenol exerted a slightly lesser effect on zeolite activity compared to other hindered additives. Lacking inorganic components or strongly binding basic functional groups, they are less likely to engage in ion exchange or acid site neutralization. Its phenolic structure can contribute to aromatic formation while also potentially blocking active sites to increase coke. On the other hand, the hindered phenol molecule has a highly branched structure with non-oxygenated and oxygenated aliphatic branches. The cracking and deoxygenation of its abundant branches over zeolite can produce aliphatic hydrocarbons, although they can also further form aromatics. The increase in coke formation was more noticeable during in situ pyrolysis, likely attributed to the longer contact times between zeolite and heterogeneous atom-containing decomposition products of AOs and LS.
The catalytic pyrolysis mechanism of PE and the summarized effects of plastic additives are shown in Fig. 9.
 | ||
| Fig. 9 Mechanism for catalytic pyrolysis of polyolefins containing common additives. | ||
3.6 Implications of plastic additives for industrial chemical upcycling
Although polymer decompositions are well documented in the literature, converting real-world post-consumer plastics suffers from great technical challenges for commercial operations. Plastics are more than just polymers. The present study shows that functional additives, colorants, and fillers added to polymers during plastic manufacturing processes can significantly influence both thermal decomposition and catalytic upgrading of plastics, ultimately affecting product distribution and selectivity. During thermal pyrolysis, most inorganic additives serve as catalysts to promote polymer cracking, although AOs and LS suppressed the decomposition of PE. The impact of the additives was more noticeable during catalytic depolymerization, where additives altered catalyst activity likely through mechanisms such as supplemental catalyst sites, ion exchange, acid site neutralization, or competitive adsorption.44 Some additives like kaolin, talc, CaCO3, and zinc stearate showed potentially beneficial effects, such as reducing heavy hydrocarbons during pyrolysis and increasing aromatic production during catalytic conversion. Minimizing heavy hydrocarbon formation during plastic pyrolysis is advantageous, as their high viscosity and high boiling points can lead to more energy-intensive separation and downstream processing. The increasingly produced aromatic monomers are valuable platform chemicals, especially benzene, toluene, and xylene. However, the increase in aromatic monomers is accompanied by lower olefinic monomer yields. Other than understanding the product selectivity affected by different additives, close attention should also be paid to the fate of the additives and their long-term impact on the reactor operation and catalysts in the industrial process of advanced plastic upcycling.Besides increasing aromatic yields, a common effect of additives observed in this study was increased charring or coking, which can increase reactor fouling and reduce the overall carbon efficiency of plastic conversion. Coke deposits on catalysts will also deactivate catalyst sites, reducing the long-term efficiency of catalysts. Some additives, such as aminic antioxidants, can block catalyst sites by strong adsorption and further neutralize acidic sites in zeolite, accelerating catalyst deactivation.44 The fast deactivation of catalysts and frequent regeneration of used catalysts will increase operation costs and potentially reduce catalyst lifetime. Furthermore, inorganic additives containing zinc, phosphorus, calcium, magnesium, titanium, aluminosilicates, and aluminum remaining on the used catalysts can irreversibly alter the catalyst pore structure and acid sites, which have been discussed in previous studies about the effect of biomass feedstock impurities or intentional metal-impregnation on zeolite activity.44,92,93,96,99–102 These changes can profoundly influence future reactivity and selectivity, potentially necessitating catalyst replacement or reconditioning to maintain optimal performance. Such effects of additives on used catalysts will be much more profound with in situ pyrolysis because of their direct contact and higher coke formation.
The transport of additives and their derivatives during conversion may also contaminate the product streams and reactors, increasing recycling costs.31,32 The present study shows that antioxidants, stabilizers, and slip agents can easily decompose under pyrolysis, yielding compounds containing heterogeneous atoms (i.e., P, N, S, and O). These heterogeneous elements containing molecules entrained in the pyrolysis vapors can contaminate the hydrocarbon products. Metal-based additives can accumulate in char and deposit on catalysts and reactor parts. Although less volatile, some can still migrate into the product streams by their nanoparticles riding on the sweep gas, as shown in this study. The presence of heterogeneous atoms in the product streams will lower the product quality and create challenges for further processing.32,40 Depending on the plastic applications, some commercial plastics can contain lower concentrations of additives than those tested here. While immediate effects on catalyst activity may be less noticeable in those cases, accumulation over repeated operation cycles can substantially impact catalyst performance. Additionally, from the perspective of upgrading pyrolysis oils, downstream catalytic naphtha-cracking processes such as fluid catalytic cracking (FCC) and steam cracking could also face significant challenges due to the presence of inorganics and heteroatoms in the pyrolysis oils. Typically, steam crackers lay out strict specifications for feeds and do not allow over 100 ppm for elements such as N and S, while sulfur has <500 ppm, metals have <1 ppm, and halogens have <3 ppm limits.32 There are also specifications for the maximum wax content that can be fed into naphtha crackers. Extensive pretreatment, such as hydrocracking and hydrotreatment, may be required before waste plastic-derived pyrolysis oil can be used for commercial monomer or fuel production.103 Again, since hydrotreatment is also a catalytic process, contaminants may inhibit catalytic activity, since most waste plastic feedstocks contain multiple additives and impurities co-existing in them. Until recently, the global outlook for waste plastic pyrolysis oils as co-feeds in commercial crackers was promising due to the existing infrastructure and the possibility of carbon penalties associated with refineries all over the world.32 However, the costly upgrading, catalyst optimization, and frequent regeneration necessitated by additives and impurities may lower the economic feasibility.
The plastic recycling industry can utilize the present research results to help their operation in several ways. Based on the additive effects described above and feedstock characterization, one can predict pyrolysis outcomes, such as product composition, reactor fouling, and catalytic activity. The results provided from this study can also guide process modelers in selecting optimal reactor configurations (for example, in situ vs. ex situ) and process conditions tailored to their products. Knowing the additive effects can also assist in the selection of optimal waste feedstock for their reactors, contributing to better process economics. Meanwhile, the effect of additives on catalysts described here can also inform catalyst selection and design. Furthermore, by clarifying the transport of certain additive components through the conversion pathway, this work facilitates the development of targeted recovery methods for different product streams to minimize secondary contamination. The insights gained in this study also provide information for next-generation additive formulations, guiding manufacturers toward additives that are inherently more amenable to plastic circularity.
While known additives with known concentrations are used to elucidate their effects, actual waste streams originate from diverse sources and inherently contain various additives of unknown amounts.31,104 Consequently, thorough feedstock characterization is paramount for developing optimal strategies that yield consistent, high-quality products in real-world upcycling scenarios. Yet, identifying and quantifying these additives is notoriously difficult, as commercial polymer manufacturers and compounders rarely disclose detailed formulations.6,31 Potential methods for identifying incorporated additives are analyzing the compositions of solvent-extracted plastics or analyzing the pyrolysates of individual plastics to track the fragments of decomposed additives. However, these techniques are mainly applied to volatile additives. Developing improved feedstock characterization methods or establishing a public database for additive composition in commercial plastics would enable more predictable process design and operational control. Such concerted efforts will accelerate the adoption of truly sustainable plastic upcycling practices and bring the field closer to achieving green, closed-loop waste management.
4. Conclusion
This study has systematically explored the impact of commonly used polymer additives on the thermal and catalytic pyrolysis of PE. The findings reveal that additives such as antioxidants, stabilizers, colorants, and fillers distinctly modify both the thermal and catalytic pyrolysis behaviors of PE, affecting both the degradation rates and the composition of the resulting products. A significant interaction between additives and catalysts was also discovered. The main conclusions from this work include the following:(1) Additives significantly influence both thermal stability and pyrolysis of PE.
(2) Inorganic additives, such as kaolin, talc, CaCO3, zinc stearate, TiO2, and carbon black, promoted cracking reactions during plastic pyrolysis due to their catalytic effects. Zinc stearate, which was depolymerized during pyrolysis, affected plastic conversion at a much lower concentration than other additives. Barium sulfate and aluminum trihydride had no noticeable impact on the thermal pyrolysis of plastics.
(3) Antioxidants and stabilizers, such as hindered phosphites, phenols, and amines, can decompose during pyrolysis. Their radical scavenging effects led to a nominal increase in wax yields from pyrolysis, hindering the effective depolymerization of polymers.
(4) Most tested additives promoted aromatic products and carbon residue or coke formation from PE during thermal and catalytic pyrolysis. The additive effects were more pronounced under in situ catalytic pyrolysis than under ex situ catalytic pyrolysis. Aluminum trihydride had a minor effect on ex situ pyrolysis but significantly impacted in situ pyrolysis.
(5) While additives could enhance polymer cracking and aromatic production, high coke formation can be problematic to catalyst stability and lower carbon efficiency. The transport and deposit of metal additives can also affect the catalyst. Moreover, the heterogeneous atom-containing decomposition compounds of the additives and the physical transport of inorganic additive particles can contaminate the product stream and lower its quality.
Overall, this article investigated the key functional additives, colorants, and fillers present in waste plastics and quantified their impacts on both thermal and catalytic pyrolytic conversions of plastics. Such information is highly desired for developing recycling technologies that can effectively handle heterogeneous plastic wastes and support circular economy initiatives.
Data availability
The data supporting this article have been included in the main article and the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the U.S. Department of Energy, Office of Energy Efficiency and Renewable Energy under award numbers DE-EE0009285, DE-EE0009943, and DE-EE0010296.References
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef.
- H. Li, H. A. Aguirre-Villegas, R. D. Allen, X. Bai, C. H. Benson, G. T. Beckham, S. L. Bradshaw, J. L. Brown, R. C. Brown, V. S. Cecon, J. B. Curley, G. W. Curtzwiler, S. Dong, S. Gaddameedi, J. E. García, I. Hermans, M. S. Kim, J. Ma, L. O. Mark, M. Mavrikakis, O. O. Olafasakin, T. A. Osswald, K. G. Papanikolaou, H. Radhakrishnan, M. A. Sanchez Castillo, K. L. Sánchez-Rivera, K. N. Tumu, R. C. Van Lehn, K. L. Vorst, M. M. Wright, J. Wu, V. M. Zavala, P. Zhou and G. W. Huber, Green Chem., 2022, 24, 8899–9002 RSC.
- A. L. Andrady and M. A. Neal, Philos. Trans. R. Soc., B, 2009, 364, 1977–1984 CrossRef CAS.
- M. Cherif Lahimer, N. Ayed, J. Horriche and S. Belgaied, Arabian J. Chem., 2017, 10, S1938–S1954 CrossRef CAS.
- E. S. Beach, B. R. Weeks, R. Stern and P. T. Anastas, Pure Appl. Chem., 2013, 85, 1611–1624 CrossRef CAS.
- A. A. Cuthbertson, C. Lincoln, J. Miscall, L. M. Stanley, A. K. Maurya, A. S. Asundi, C. J. Tassone, N. A. Rorrer and G. T. Beckham, Green Chem., 2024, 26, 7067–7090 RSC.
- M. Dokl, A. Copot, D. Krajnc, Y. Van Fan, A. Vujanović, K. B. Aviso, R. R. Tan, Z. Kravanja and L. Čuček, Sustainable Prod. Consumption, 2024, 51, 498–518 CrossRef.
- E. Hansen, N. Nillson, D. Lithner and C. Lassen, Hazardous substances in plastic materials, COWI in cooperation with Danish Technological Institute, 2013 Search PubMed.
- J. N. Hahladakis, C. A. Velis, R. Weber, E. Iacovidou and P. Purnell, J. Hazard. Mater., 2018, 344, 179–199 CrossRef CAS.
- J. Scheirs and W. Kaminsky, Feedstock recycling and pyrolysis of waste plastics:converting waste plastics into diesel and other fuels, J. Wiley & Sons, 2006 Search PubMed.
- R. Hermanns, A. Kraft, P. Hartmann and R. Meys, Chem. Ing. Tech., 2023, 95, 1259–1267 CrossRef CAS.
- J. Sun, J. Dong, L. Gao, Y.-Q. Zhao, H. Moon and S. L. Scott, Chem. Rev., 2024, 124, 9457–9579 CrossRef CAS PubMed.
- K. V. Khopade, N. S. Rajput, R. Rangappa, N. Barsu and S. H. Chikkali, Green Chem., 2024, 26, 10558–10566 RSC.
- N. M. Wang, G. Strong, V. DaSilva, L. Gao, R. Huacuja, I. A. Konstantinov, M. S. Rosen, A. J. Nett, S. Ewart, R. Geyer, S. L. Scott and D. Guironnet, J. Am. Chem. Soc., 2022, 144, 18526–18531 CrossRef CAS PubMed.
- R. J. Conk, S. Hanna, J. X. Shi, J. Yang, N. R. Ciccia, L. Qi, B. J. Bloomer, S. Heuvel, T. Wills, J. Su, A. T. Bell and J. F. Hartwig, Science, 2022, 377, 1561–1566 CrossRef CAS PubMed.
- R. J. Conk, J. F. Stahler, J. X. Shi, J. Yang, N. G. Lefton, J. N. Brunn, A. T. Bell and J. F. Hartwig, Science, 2024, 385, 1322–1327 CrossRef CAS.
- H. Li, J. Wu, Z. Jiang, J. Ma, V. M. Zavala, C. R. Landis, M. Mavrikakis and G. W. Huber, Science, 2023, 381, 660–666 CrossRef CAS PubMed.
- Z. Xu, N. E. Munyaneza, Q. Zhang, M. Sun, C. Posada, P. Venturo, N. A. Rorrer, J. Miscall, B. G. Sumpter and G. Liu, Science, 2023, 381, 666–671 CrossRef CAS PubMed.
- H. Radhakrishnan, V. S. Cecon, A. Lusi, M. K. Islam, I. Coffman, K. Vorst and X. Bai, Energy Fuels, 2024, 38, 16692–16704 CrossRef CAS.
- H. Jiang, Y.-J. Jang, C. Tagnon, E. A. Medina and E. E. Stache, ACS Appl. Polym. Mater., 2025, 7, 1163–1174 CrossRef CAS.
- H. Radhakrishnan, S. Gnangbe, A. Duereh, S. U. Iffat Uday, A. Lusi, H. Hu, H. Hu, M. M. Wright and X. Bai, Green Chem., 2024, 26, 9156–9175 RSC.
- G. Wang, Z. Chen, W. Wei and B. Ni, Electron, 2024, 2, e34 CrossRef CAS.
- M. H. Rahman, P. R. Bhoi and P. L. Menezes, Renewable Sustainable Energy Rev., 2023, 188, 113799 CrossRef CAS.
- S. H. Chang, Sci. Total Environ., 2023, 877, 162719 CrossRef CAS PubMed.
- S. M. Al-Salem, A. Antelava, A. Constantinou, G. Manos and A. Dutta, J. Environ. Manage., 2017, 197, 177–198 CrossRef CAS PubMed.
- H. Li, A. A. Cuthbertson, A. A. Alamer, V. S. Cecon, H. Radhakrishnan, J. Wu, G. W. Curtzwiler, K. L. Vorst, X. Bai, C. R. Landis, G. T. Beckham and G. W. Huber, Green Chem., 2024, 26, 8718–8727 RSC.
- R. Miandad, M. A. Barakat, A. S. Aburiazaiza, M. Rehan and A. S. Nizami, Process Saf. Environ. Prot., 2016, 102, 822–838 CrossRef CAS.
- W.-H. Chen, P. Pratim Biswas, E. E. Kwon, Y.-K. Park, S. Rajendran, L. Gnanasekaran and J.-S. Chang, Chem. Eng. J., 2023, 471, 144695 CrossRef CAS.
- M. S. Abbas-Abadi, Y. Ureel, A. Eschenbacher, F. H. Vermeire, R. J. Varghese, J. Oenema, G. D. Stefanidis and K. M. Van Geem, Prog. Energy Combust. Sci., 2023, 96, 101046 CrossRef.
- G. Lopez, M. Artetxe, M. Amutio, J. Bilbao and M. Olazar, Renewable Sustainable Energy Rev., 2017, 73, 346–368 CrossRef CAS.
- M. Roosen, N. Mys, M. Kusenberg, P. Billen, A. Dumoulin, J. Dewulf, K. M. Van Geem, K. Ragaert and S. De Meester, Environ. Sci. Technol., 2020, 54, 13282–13293 CrossRef CAS PubMed.
- M. Kusenberg, A. Eschenbacher, M. R. Djokic, A. Zayoud, K. Ragaert, S. De Meester and K. M. Van Geem, Waste Manage., 2022, 138, 83–115 CrossRef CAS PubMed.
- G. Yan, X. Jing, H. Wen and S. Xiang, Energy Fuels, 2015, 29, 2289–2298 CrossRef CAS.
- M. S. Abbas-Abadi, M. Kusenberg, A. Zayoud, M. Roosen, F. Vermeire, S. Madanikashani, M. Kuzmanović, B. Parvizi, U. Kresovic, S. De Meester and K. M. Van Geem, Waste Manage., 2023, 165, 108–118 CrossRef CAS PubMed.
- J. Wu, Z. Jiang, V. S. Cecon, G. Curtzwiler, K. Vorst, M. Mavrikakis and G. W. Huber, Green Chem., 2024, 26, 11908–11923 RSC.
- J. C. J. Bart, Additives in Polymers, Wiley, 2005 Search PubMed.
- H. Wiesinger, Z. Wang and S. Hellweg, Environ. Sci. Technol., 2021, 55, 9339–9351 CrossRef CAS PubMed.
- V. Marturano, P. Cerruti and V. Ambrogi, Phys. Sci. Rev., 2017, 2(6), 20160130 Search PubMed.
- Handbook of Industrial Polyethylene and Technology, ed. M. A. Spalding and A. M. Chatterjee, Wiley, 2017 Search PubMed.
- W. Zeb, M. Roosen, P. Knockaert, S. Janssens, D. Withoeck, M. Kusenberg, J. Hogie, P. Billen, S. Tavernier, K. M. Van Geem and S. De Meester, J. Cleaner Prod., 2023, 416, 137881 CrossRef CAS.
- Y. Zhang, Z. Fu, W. Wang, G. Ji, M. Zhao and A. Li, ACS Sustainable Chem. Eng., 2022, 10, 91–103 CrossRef CAS.
- D. Zhao, X. Wang, J. B. Miller and G. W. Huber, ChemSusChem, 2020, 13, 1764–1774 CrossRef CAS PubMed.
- M. Kusenberg, A. Zayoud, M. Roosen, H. D. Thi, M. S. Abbas-Abadi, A. Eschenbacher, U. Kresovic, S. De Meester and K. M. Van Geem, Fuel Process. Technol., 2022, 227, 107090 CrossRef CAS.
- A. C. Jerdy, T. Pham, M. Á. González-Borja, P. Atallah, D. Soules, R. Abbott, L. Lobban and S. Crossley, Appl. Catal., B, 2023, 325, 122348 CrossRef CAS.
- C. Mukarakate, M. J. Watson, J. ten Dam, X. Baucherel, S. Budhi, M. M. Yung, H. Ben, K. Iisa, R. M. Baldwin and M. R. Nimlos, Green Chem., 2014, 16, 4891–4905 RSC.
- Y. Xue, A. Kelkar and X. Bai, Fuel, 2015, 166, 227–236 CrossRef.
- J. V. Jayarama Krishna, B. A. Perez and H. E. Toraman, ACS Sustainable Chem. Eng., 2024, 12, 7508–7518 CrossRef CAS.
- A. Eschenbacher, R. J. Varghese, M. S. Abbas-Abadi and K. M. Van Geem, Chem. Eng. J., 2022, 428, 132087 CrossRef CAS.
- J. Wang, J. Jiang, X. Wang, S. Liu, X. Shen, X. Cao, Y. Sun, L. Dong, X. Meng, A. J. Ragauskas and Y. Wang, Chem. Eng. J., 2022, 444, 136360 CrossRef CAS.
- J. Wang, J. Jiang, Y. Zhang, X. Meng and A. J. Ragauskas, Energy, 2023, 263, 125843 CrossRef CAS.
- J. Proano-Aviles, J. K. Lindstrom, P. A. Johnston and R. C. Brown, Energy Technol., 2017, 5, 189–195 CrossRef CAS.
- Y. Xue, P. Johnston and X. Bai, Energy Convers. Manage., 2017, 142, 441–451 CrossRef CAS.
- F. Akoueson, C. Chbib, S. Monchy, I. Paul-Pont, P. Doyen, A. Dehaut and G. Duflos, Sci. Total Environ., 2021, 773, 145073 CrossRef CAS PubMed.
- K. D. Jansson, C. P. Zawodny and T. P. Wampler, J. Anal. Appl. Pyrolysis, 2007, 79, 353–361 CrossRef CAS.
- D. G. Kulas, A. Zolghadr and D. Shonnard, ACS Sustainable Chem. Eng., 2021, 9, 14443–14450 CrossRef CAS.
- U. R. Gracida-Alvarez, M. K. Mitchell, J. C. Sacramento-Rivero and D. R. Shonnard, Ind. Eng. Chem. Res., 2018, 57, 1912–1923 CrossRef CAS.
- Supriyanto, P. Ylitervo and T. Richards, J. Anal. Appl. Pyrolysis, 2021, 158, 105248 CrossRef CAS.
- M. del R. Hernández, A. Gómez, Á. N. García, J. Agulló and A. Marcilla, Appl. Catal., A, 2007, 317, 183–194 CrossRef.
- R. Cypres, Fuel Process. Technol., 1987, 15, 1–15 CrossRef CAS.
- W. Luo, Z. Fan, J. Wan, Q. Hu, H. Dong, X. Zhang and Z. Zhou, Fuel, 2021, 302, 121164 CrossRef CAS.
- Y. Choi, Y. Min Yoon, J. Jun Jang, D. Kim, H. J. Ryu, D. Lee, Y. Won, H. Nam and B. Hwang, Chem. Eng. J., 2024, 490, 151503 CrossRef CAS.
- A. C. Zettlemoyer and J. J. Chessick, J. Phys. Chem., 1960, 64, 1131–1134 CrossRef CAS.
- Y. Sekiguchi, S. Akiyama, W. Urakawa, T. R. Koyama, A. Miyaji, K. Motokura and T. Baba, Catal. Commun., 2015, 68, 20–24 CrossRef CAS.
- N. Zhou, L. Dai, Y. Lyu, H. Li, W. Deng, F. Guo, P. Chen, H. Lei and R. Ruan, Chem. Eng. J., 2021, 418, 129412 CrossRef CAS.
- P. B. Linares, L. A. Castillo and S. E. Barbosa, J. Polym. Environ., 2019, 27, 1666–1676 CrossRef CAS.
- B. Szabó, G. Novodárszki, Z. Pászti, A. Domján, J. Valyon, J. Hancsók and R. Barthos, ChemCatChem, 2020, 12, 5686–5696 CrossRef.
- G. J. Suppes, K. Bockwinkel, S. Lucas, J. B. Botts, M. H. Mason and J. A. Heppert, J. Am. Oil Chem. Soc., 2001, 78, 139–145 CrossRef CAS.
- M. Rasul Jan, J. Shah and H. Gulab, Fuel, 2013, 105, 595–602 CrossRef CAS.
- M. R. Jan, J. Shah and H. Gulab, Fuel Process. Technol., 2010, 91, 1428–1437 CrossRef CAS.
- T. Ishimura, I. Iwai, K. Matsui, M. Mattonai, A. Watanabe, W. Robberson, A. M. Cook, H. L. Allen, W. Pipkin, N. Teramae, H. Ohtani and C. Watanabe, J. Anal. Appl. Pyrolysis, 2021, 157, 105188 CrossRef CAS.
- P. Zhang, L. Bai, Y. Wang, Z. Sun, Y.-Y. Liu and A. Wang, Catal. Sci. Technol., 2024, 14, 2441–2451 RSC.
- H. Dong, W. Luo, X. Yan, B. Li, J. Hu, S. Huang, M. Xia, M. Zhong, Q. Tang, Z. Zhou and N. Zhou, Fuel, 2022, 325, 124812 CrossRef CAS.
- M. Gönen, D. Balköse, F. İnal and S. Ülkü, J. Therm. Anal. Calorim., 2008, 94, 737–742 CrossRef.
- H. Xu, X. Ye, X. Shi, H. Zhong, D. He, B. Jin and F. Jin, Mol. Catal., 2022, 522, 112241 CrossRef CAS.
- S. P. Tekade, P. P. Gugale, M. L. Gohil, S. H. Gharat, T. Patil, P. K. Chaudhari, D. S. Patle and A. N. Sawarkar, Energy Sources, Part A, 2020, 47(1), 3597–3610 CrossRef.
- K. Nakajima, R. Noma, M. Kitano and M. Hara, J. Phys. Chem. C, 2013, 117, 16028–16033 CrossRef CAS.
- E. Obuchi, M. Suyama and K. Nakano, J. Mater. Cycles Waste Manage., 2001, 3, 88–92 CAS.
- S. Khodabakhshi, P. F. Fulvio and E. Andreoli, Carbon, 2020, 162, 604–649 CrossRef CAS.
- N. Zhou, D. Zhao, Q. Su, Q. Li, W. Zha, S. Feng and S. Feng, RSC Adv., 2024, 14, 15656–15663 RSC.
- E. Jakab and M. Blazsó, J. Anal. Appl. Pyrolysis, 2002, 64, 263–277 CrossRef CAS.
- R. E. Harmon, G. Sribala, L. J. Broadbelt and A. K. Burnham, Energy Fuels, 2021, 35, 6765–6775 CrossRef CAS.
- S. Dorey, F. Gaston, N. Girard-Perier, N. Dupuy, S. R. A. Marque, M. Barbaroux and G. Audran, J. Appl. Polym. Sci., 2020, 137, 49336 CrossRef CAS.
- F. C.-Y. Wang, J. Chromatogr. A, 2000, 891, 325–336 CrossRef CAS PubMed.
- N. Bartsch, M. Girard, A. Wilde, T. Bruhn, O. Kappenstein, B. Vieth, C. Hutzler and A. Luch, J. Vinyl Addit. Technol., 2019, 25, E12–E27 CAS.
- A. I. Balabanovich, I. A. Klimovtsova, V. P. Prokopovich and N. R. Prokopchuk, Thermochim. Acta, 2007, 459, 1–8 CrossRef CAS.
- S. Almeida, S. Ozkan, D. Gonçalves, I. Paulo, C. S. G. P. Queirós, O. Ferreira, J. Bordado and R. Galhano Dos Santos, Polymers, 2022, 15, 6 CrossRef PubMed.
- Z. Dong, W. Chen, K. Xu, Y. Liu, J. Wu and F. Zhang, ACS Catal., 2022, 12, 14882–14901 CrossRef CAS.
- Md. M. Rahman, R. Liu and J. Cai, Fuel Process. Technol., 2018, 180, 32–46 CrossRef CAS.
- J. Gancedo, H. Li, J. S. Walz, L. Faba, S. Ordoñez and G. W. Huber, Appl. Catal., A, 2024, 669, 119484 CrossRef CAS.
- M. Artetxe, G. Lopez, M. Amutio, G. Elordi, J. Bilbao and M. Olazar, Ind. Eng. Chem. Res., 2013, 52, 10637–10645 CrossRef CAS.
- G. Caeiro, P. Magnoux, P. Ayrault, J. M. Lopes and F. R. Ribeiro, Chem. Eng. J., 2006, 120, 43–54 CrossRef CAS.
- S. Fathi, M. Sohrabi and C. Falamaki, Fuel, 2014, 116, 529–537 CrossRef CAS.
- S. Dong, T. Ryu, C. Oi, J. Wu, N. R. Altvater, R. Hagmann, Z. Alikhani, E. A. Lebrón-Rodríguez, J. H. Jansen, V. S. Cecon, G. W. Curtzwiler, K. L. Vorst, G. W. Huber and I. Hermans, Chem. Eng. J., 2024, 482, 148889 CrossRef CAS.
- S. Tamiyakul, T. Sooknoi, L. L. Lobban and S. Jongpatiwut, Appl. Catal., A, 2016, 525, 190–196 CrossRef CAS.
- K. Stanciakova and B. M. Weckhuysen, Trends Chem., 2021, 3, 456–468 CrossRef CAS.
- H. E. Van Der Bij and B. M. Weckhuysen, Chem. Soc. Rev., 2015, 44, 7406–7428 RSC.
- A. Rahman, G. Lemay, A. Adnot and S. Kaliaguine, J. Catal., 1988, 112, 453–463 CrossRef CAS.
- T. Blasco, A. Corma and J. Martínez-Triguero, J. Catal., 2006, 237, 267–277 CrossRef CAS.
- R. Mahadevan, S. Adhikari, R. Shakya and O. Fasina, Catalysts, 2021, 11, 124 CrossRef CAS.
- N. Nikolopoulos, R. G. Geitenbeek, G. T. Whiting and B. M. Weckhuysen, J. Catal., 2021, 396, 136–147 CrossRef CAS.
- J. Cen, N. Zhang, H. Hu, N. Yao, Z. Li, L. Yang, F. Feng, C. Lu and X. Li, Catalysts, 2020, 10, 693 CrossRef CAS.
- N. Miskolczi, T. Juzsakova and J. Sója, J. Energy Inst., 2019, 92, 118–127 CrossRef CAS.
- B. S. Yoon, C. Kim, G. J. Park, S. G. Jeon and C. H. Ko, Fuel, 2024, 369, 131688 CrossRef CAS.
- P. Hennebert, Detritus, 2023, 23, 83–93 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5gc00688k |
| This journal is © The Royal Society of Chemistry 2025 |