
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Highly efficient benzyl alcohol valorisation via the in situ synthesis of H2O2 and associated reactive oxygen species†
Gregory
Sharp‡
a,
Richard J.
Lewis‡
 *a,
G.
Magri
b,
J.
Liu
c,
David J.
Morgan
ad,
Thomas E.
Davies
a,
Ángeles
López-Martín
a,
Damien M.
Murphy
b,
Andrea
Folli
e,
Liwei
Chen
c,
Xi
Liu
*cf and
Graham J.
Hutchings
*a
*a,
G.
Magri
b,
J.
Liu
c,
David J.
Morgan
ad,
Thomas E.
Davies
a,
Ángeles
López-Martín
a,
Damien M.
Murphy
b,
Andrea
Folli
e,
Liwei
Chen
c,
Xi
Liu
*cf and
Graham J.
Hutchings
*a
aMax Planck-Cardiff Centre on the Fundamentals of Heterogeneous Catalysis FUNCAT, Cardiff Catalysis Institute, School of Chemistry, Cardiff University, Translational Research Hub, Maindy Road, Cardiff, CF24 4HQ, UK. E-mail: LewisR27@Cardiff.ac.uk; Hutch@Cardiff.ac.uk
bSchool of Chemistry, Cardiff University, Main Building, Park Place, Cardiff, CF10 3AT, UK
cIn situ Centre for Physical Sciences, School of Chemistry and Chemical, Frontiers Science Centre for Transformative Molecules, Shanghai 200240, P. R. China. E-mail: LiuXi@stju.edu.cn
dHarwellXPS, Research Complex at Harwell (RCaH), Didcot, OX11 OFA, UK
eNet Zero Innovation Institute, Cardiff Catalysis Institute, School of Chemistry, Cardiff University, Translational Research Hub, Maindy Road, Cardiff, CF24 4HF, UK
fSchool of Chemistry and Chemical Engineering, Ningxia, University, Yinchuan 750021, P. R. China
First published on 25th April 2025
Abstract
The selective oxidation of chemical feedstocks via in situ production of reactive oxygen species (H2O2, ˙OOH, ˙OH, ˙O2−), represents an attractive, environmentally friendly alternative to the use of stoichiometric oxidants. Within this contribution, we demonstrate the efficacy of the in situ approach to the selective oxidation of benzyl alcohol to the commodity chemical benzaldehyde, with the alloying of Au with Pd shown to be key in significantly promoting catalytic performance. The immobilisation of AuPd nanoalloys, particularly on to a γ-Al2O3 carrier, is demonstrated to result in high selective utilisation of H2 (ca. 80%), overcoming a major hurdle that has often precluded the adoption of the in situ approach to chemical synthesis on a commercial scale, while also achieving yields of benzaldehyde in excess of 60%, over successive experiments, representing a significant step towards competitiveness with traditional oxidative processes reliant on stoichiometric oxidants. Evaluation of catalyst performance towards individual reaction pathways (i.e. H2O2 direct synthesis and benzyl alcohol oxidation in the presence of preformed H2O2), analysis by EPR spectroscopy and radical quenching experiments, indicates that reactive oxygen-based species (ROS), rather than H2O2, are primarily responsible for the observed catalysis. While the origin of these oxygen-based radicals is not fully understood, we consider that they are generated primarily as reaction intermediates formed during H2O2 synthesis over active metal surfaces.
Green foundation1. Currently the oxidative valorisation of alcohols is reliant on costly and atom-inefficient stoichiometric oxidants, which generate large quantities of unwanted by-products. However, the in situ production of hydrogen peroxide and related oxidative species avoids these concerns and allows for significant process intensification.2. To date, low yields and poor H2 utilisation have prevented the adoption of an in situ approach to feedstock upgrading. This work demonstrates that through rational catalyst design it is possible to overcome these drawbacks, with our optimised AuPd catalyst offering H2 selectivity approaching 80%, and product yields in excess of 60% through successive reactions. 3. Further catalyst design is required in order to improve reactivity and H2 utilisation rates. |
Introduction
Primary alcohol valorisation to the corresponding aldehyde is of significant industrial importance, representing key feedstocks in sectors ranging from perfumery to dyestuff and agrochemicals.1 Typically, the selective oxidation of these platform chemicals on an industrial scale is reliant on the utilisation of energy- and atom-inefficient stoichiometric oxidants, resulting in the production of undesirable and complex product streams.2 Such concerns have led to an interest in alternative, aerobic approaches, with numerous reports demonstrating that is possible to achieve high aldehyde yields when using molecular oxygen.3 However, while the green credentials associated with aerobic oxidation are clear, such routes typically require elevated reaction temperatures to be effective.3–6 Thus, while the use of O2 as the terminal oxidant can overcome the atom-inefficiency concerns of current industrial routes, reliant on stoichiometric oxidants, the utilisation of aerobic routes may still be hampered by poor energy usage. Alternatively, unlike O2-mediated pathways, the utilisation of preformed H2O2 allows for relatively low reaction temperatures to be exploited,7–10 but suffers from (i) high production costs of the oxidant, (ii) the continual dilution of product streams (iii) the presence of proprietary stabilising agents, which can lead to reactor corrosion, and (iv) the need for constant monitoring to avoid the development of H2O2 hot spots.Offering the potential for improved atom efficiency compared to the use of alternative stoichiometric oxidants (and preformed H2O2) and lower operating temperatures than aerobic pathways, the in situ synthesis and subsequent utilisation of H2O2 and related reactive oxygen species (ROS; ˙OOH, ˙OH, ˙O2−), in chemical feedstock valorisation is an area of significant and growing research interest.11–13 However, while considerable efforts have been made in this field, particularly around propylene epoxidation,14–16 very few examples of an in situ approach to oxidative upgrading which rival the performance metrics of mature industrial processes have been reported. In many cases, a combination of poor selective H2 utilisation, rapid catalyst deactivation, and the formation of complex product streams, necessitating energy-intensive purification steps, have prevented development beyond the laboratory scale. Indeed, the presence of H2, necessary for H2O2 and ROS production, in conjunction with effective hydrogenation catalysts, such as Pd and Pt, which are typically used for H2O2 synthesis,17–22 are responsible for many of these challenges.23–25
The in situ selective oxidation of benzyl alcohol to benzaldehyde (Scheme 1), is one such chemical transformation that has received considerable research interest.26–28 In part, this is due to the limited number of reaction products and the relatively well-known pathways to their formation. However, benzaldehyde is a valuable feedstock in its own right, finding use in the cosmetics and pharmaceutical sectors. Indeed, the benzaldehyde market was valued at approximately $580 million per annum in 2024,29 so interest in the oxidative upgrading of this feedstock chemical is not purely academic.
 | ||
| Scheme 1 General reaction scheme for the oxidation of benzyl alcohol. | ||
Supported AuPd nanoalloys have been reported to offer exceptional activity towards the aerobic valorisation of several platform chemicals30,31 and are also considered among the state-of-the-art for the direct synthesis of H2O2.32 With regard to the latter transformation, the alloying of Pd with Au has been demonstrated to significantly enhance catalytic performance (compared to the monometallic analogues), due to a combination of electronic and isolation effects, resulting in improved activity and selectivity towards H2O2.33–36 In part, this has been attributed to the weaker interaction between the metal surface and the synthesised H2O2, when compared to Pd-only surfaces.37 Importantly, the formation of these mixed metal surfaces has recently been shown to favour the release of highly reactive oxygen species, which are formed as intermediates during H2O2 synthesis.38 The potential for such ROS to be utilised in chemical synthesis, particularly in processes where proton-abstraction is key, is particularly intriguing and in this contribution, we investigate the performance of supported AuPd nanoparticles to effectively generate H2O2 (and related ROS), and the subsequent efficacy of these species towards the in situ selective oxidation of benzyl alcohol to benzaldehyde.
Experimental
Catalyst synthesis
Mono- and bi-metallic 1%Au–Pd/X catalysts were prepared (on a weight basis) by a wet co-impregnation of chloride metal precursors onto a range of common oxide supports (TiO2 (P25), γ-Al2O3, CeO2, Nb2O5, ZrO2), based on a methodology previously reported in the literature.39 The procedure to produce 0.5% Au–0.5%Pd/Al2O3 (2 g) is outlined below, with a similar methodology being utilised for all catalysts.Aqueous PdCl2 solution (1.667 mL, [Pd] = 6 mg mL−1, Merck) and aqueous HAuCl4·3H2O solution (0.8263 mL, [Au] = 12.25 mg mL−1, Strem Chemicals) were mixed in a 50 mL round-bottom flask and heated to 65 °C with stirring (1000 rpm) in a thermostatically controlled oil bath, with the total volume fixed to 16 mL using H2O (HPLC grade, Fisher Scientific). When a temperature of 65 °C was reached, γ-Al2O3 (1.98 g, Fischer Scientific) was added over the course of 5 min with constant stirring. The resulting slurry was stirred at 65 °C for a further 15 min, after which the temperature was raised to 95 °C for 16 h to allow for the complete evaporation of water. The resulting solid was mechanically ground prior to heat treatment under a reductive atmosphere (flowing 5% H2/Ar, 500 °C, 4 h, and ramp rate of 10 °C min−1).
Catalyst testing
Note 1: The reaction conditions used within this study operate below the flammability limits of gaseous mixtures of H2 and O2.Note 2: The conditions used within this work for H2O2 synthesis and degradation have previously been investigated, where the presence of CO2 as a diluent for reactant gases and methanol as a co-solvent has been identified as key to maintaining high catalytic efficacy towards H2O2 production.40 In particular, the CO2 gaseous diluent has been found to act as an in situ promoter of H2O2 stability through dissolution in the reaction solution and the formation of carbonic acid. We have previously reported that the use of the CO2 diluent has a comparable promotive effect to that observed when acidifying the reaction solution to a pH of 4 using HNO3.41
Direct synthesis of H2O2
Hydrogen peroxide synthesis activity was evaluated using a Parr Instruments stainless steel autoclave with a nominal volume of 50 mL and a maximum working pressure of 2000 psi, equipped with a PTFE liner. To test each catalyst for H2O2 synthesis, the autoclave liner was charged with catalyst (0.01 g), solvent (5.6 g methanol, HPLC grade, Fisher Scientific) and H2O (2.9 g, HPLC grade, Fisher Scientific). The charged autoclave was then purged three times with 5% H2/CO2 (100 psi) before filling with 5%H2/CO2 to a pressure of 420 psi, followed by the addition of 25%O2/CO2 (160 psi). The pressures of 5%H2/CO2 and 25%O2/CO2 were taken as gauge pressures. The reaction mixture was stirred (1200 rpm) for 0.5 h, with the temperature being maintained at 20 °C. Reactor temperature control was achieved using a HAAKE K50 bath/circulator using an appropriate coolant. The reactor was not continuously supplied with gas. H2O2 productivity was determined by titrating aliquots of the final solution after reaction with acidified Ce(SO4)2 (0.01 M) in the presence of ferroin indicator. Catalyst productivities are reported as molH2O2 kgcat−1 h−1.In all cases, reactions were run multiple times, over multiple batches of catalyst, with the data presented an average of these experiments. Catalytic activity towards H2O2 synthesis was found to be consistent to within ±2% based on multiple reactions.
Degradation of H2O2
Catalytic activity towards H2O2 degradation (via hydrogenation and decomposition pathways) was determined in a manner similar to that used for measuring the H2O2 direct synthesis activity of a catalyst. The autoclave liner was charged with methanol (5.6 g, HPLC grade, Fisher Scientific), H2O2 (50 wt% 0.69 g, Merck), H2O (2.21 g, HPLC grade, Fisher Scientific) and catalyst (0.01 g), with the solvent composition equivalent to a 4 wt% H2O2 solution. From the solution, two aliquots of 0.05 g were removed and titrated with acidified Ce(SO4)2 solution using ferroin as an indicator to determine an accurate concentration of H2O2 at the start of the reaction. The charged autoclave was then purged three times with 5%H2/CO2 (100 psi) before filling with 5%H2/CO2 (420 psi). The pressures of 5%H2/CO2 was taken as gauge pressure. The reaction mixture was stirred (1200 rpm) for 0.5 h, with the reaction temperature maintained at 20 °C. After the reaction was complete, the catalyst was removed from the reaction solvents via filtration and as described previously, two aliquots of 0.05 g were titrated against the acidified Ce(SO4)2 solution using ferroin as an indicator. Catalyst degradation activity is reported as molH2O2 kgcat−1 h−1.In all cases, reactions were run multiple times, over multiple batches of catalyst, with the data presented an average of these experiments. Catalytic activity towards H2O2 degradation was found to be consistent to within ±5% based on multiple reactions.
Benzyl alcohol oxidation via in situ production of H2O2
Note 3: Reaction conditions utilised within this study are based on those previously identified by our laboratory for the in situ oxidation of benzyl alcohol, they have been maintained in this work to allow for ease of comparison to the established literature.The oxidation of benzyl alcohol has been investigated in a 50 mL Parr Instruments stainless steel autoclave, equipped with PTFE liner. The autoclave liner was charged with catalyst (0.01 g), methanol (7.13 g, HPLC grade, Fisher Scientific) and benzyl alcohol (1.04 g, 9.62 mmol, Merck) along with 0.5 mL of the internal standard mesitylene (0.43 g, 3.58 mmol, Merck). The charged autoclave was then purged three times with 5% H2/CO2 (100 psi) before filling with 5% H2/CO2 to a pressure of 420 psi, followed by the addition of 25%O2/CO2 (160 psi). The pressures of 5%H2/CO2 and 25%O2/CO2 were taken as gauge pressures. The reactor was subsequently heated to 50 °C, followed by stirring at 1200 rpm for 0.5 h, unless otherwise stated. The reactor was not continuously supplied with gas. After the reaction was complete, the reactor was cooled in ice water to a temperature of 15 °C, after which a gas sample was taken for analysis by gas chromatography using a Varian CP-3380 equipped with a TCD detector and a Porapak Q column, to allow for the determination of H2 conversion. Once cooled to the desired temperature, the catalyst was removed from the reaction solvents via filtration and the liquid product yield was determined by gas chromatography using a Varian 3200 GC, equipped with a CP Wax 42 column and FID. The concentration of residual H2O2 was determined by titrating aliquots of the final solution after reaction with acidified Ce(SO4)2 (0.01 M) in the presence of ferroin indicator.
Further studies were conducted in the presence of radical quenching agents (Na2SO3 or NaNO2) at a concentration of 0.05 M.
The total capacity of the autoclave was determined via water displacement to allow for accurate determination of H2 conversion and H2 selectivity. When equipped with the PTFE liner and liquid reagents the total available gaseous space within the autoclave and is equivalent to 2.8 mmol of H2.
In all cases, reactions were run multiple times, over multiple batches of catalyst, with the data presented an average of these experiments. For benzyl alcohol oxidation total product yield was observed to be consistent to within ±4% based on multiple reactions.
H2 conversion (eqn (1)), benzyl alcohol conversion (eqn (2)), product yield (eqn (3)), product selectivity (eqn (4)) and H2 selectivity (eqn (5)) are defined as follows:
 | (1) |
 | (2) |
 | (3) |
 | (4) |
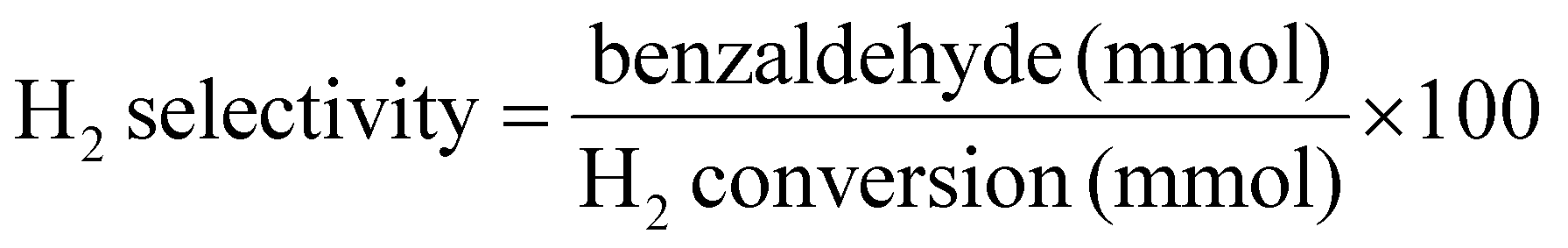 | (5) |
Catalyst re-useability in benzyl alcohol oxidation via in situ production of H2O2
In order to determine catalyst reusability, a similar procedure to that outlined above for benzyl alcohol oxidation via the in situ production of H2O2 was followed utilizing 0.3 g of catalyst. Following the initial test, the catalyst was recovered by filtration, washed with MeOH (5 g, HPLC grade, Fischer Scientific) and dried (30 °C, 16 h, under vacuum). Next, 0.01 g of material from the recovered catalyst sample was used to conduct a standard oxidation experiment.Gas replacement experiments for the oxidation of benzyl alcohol via the in situ production of H2O2
An identical procedure to that outlined above for the oxidation of benzyl alcohol was followed for a reaction time of 0.5 h. After this, stirring was stopped and the reactant gas mixture was vented prior to replacement with the standard pressures of 5%H2/CO2 (420 psi) and 25% O2/CO2 (160 psi). The reaction mixture was then stirred (1200 rpm) for a further 0.5 h. To collect a series of data points, as in the case of Fig. 4, it should be noted that individual experiments were carried out and the reactant mixture was not sampled online.Benzyl alcohol upgrading using individual gaseous reagents (H2 or O2) or commercial H2O2
An identical procedure to that outlined above for the in situ oxidation of benzyl alcohol was followed for a reaction time of 0.5 h, with the reactor charged with either 5%H2/CO2 (420 psi) or 25% O2/CO2 (160 psi), with total reactor pressure maintained at 580 psi with CO2. As above, the reactor was not continually supplied with gas. Additional experiments were conducted using commercial H2O2 (50 wt%, Merck), at a concentration equal to that which may be synthesised during the in situ oxidation of benzyl alcohol, assuming total H2 conversion to H2O2. In this case, the preformed H2O2 was added to the reactor prior to the reaction commencing (i.e. not continually introduced) and total pressure was maintained at 580 psi using CO2.Radical trapping experiments with electron paramagnetic resonance spectroscopy
The catalyst (0.01 g), methanol (7.13 g, HPLC grade, Fisher Scientific) and benzyl alcohol (1.04 g, 9.6 mmol, Merck) were added to the reactor along with 0.5 mL of the internal standard mesitylene (0.43 g, 3.58 mmol, Merck) and 5,5-dimethyl-1-pyrroline N-oxide (12 μL, Merck). The reactor was purged three times with 5%H2/CO2 (100 psi) and then filled with 5%H2/CO2 (420 psi) and 25%O2/CO2 (160 psi). The reactor was then heated to 50 °C and once the temperature was reached, stirring (1200 rpm) was commenced for 0.5 h. Once the reaction was complete, the reactor was purged with 20 bar N2 for 20 min before the catalyst was separated by filtration and the filtered solution loaded into a 1.1 mm quartz tube for analysis by EPR spectroscopy.Various blank reactions were also analysed by EPR spectroscopy to determine any background activity.
Continuous wave X-band EPR spectra were recorded at 298 K using a Bruker EMX Micro spectrometer equipped with a Bruker ER 4123d dielectric resonator. Spectra were recorded at ca. 9.75 GHz and 2 mW microwave power, with 100 kHz field modulation frequency, 1 G field modulation amplitude, 5 × 104 receiver gain, 10.00 ms conversion time and 5.02 ms time constant. EPR spectra were simulated using the EasySpin toolbox42 running within the MathWorks Matlab environment.
Catalyst characterisation
A Kratos Axis Ultra DLD spectrometer was used to collect X-ray photoelectron spectra utilising a monochromatic Al Kα X-ray source operating at 144 W (12 mA × 12 kV). Samples for analysis were mounted by pressing into double-sided adhesive tape attached to a microscope slide attached to a standard Kratos sample bar assembly. Data was collected using the Hybrid mode with a Slot aperture, yielding an analysis area of ca. 700 × 300 μm2. The pass energies for acquisition were 40 (step size 0.1 eV) and 160 eV (step size 1 eV) for high-resolution and survey spectra, respectively. Sample charging effects were minimised through magnetically confined low-energy electrons using the Kratos immersion lens system and the resulting spectra were calibrated to the C(1s) line at 284.8 eV for all samples, a secondary charge reference of the Al(2p) peak was also used to confirm the suitability of the C(1s) line and found to be 74.5 eV, typical for Al2O3. The uncertainty in binding energies is ±0.2 eV. All data was processed using CasaXPS v2.3.27. The data was analysed typically using a Shirley background, although a linear background was used where the signal was low or negative. Peak areas were corrected using modified Wagner sensitivity factors. Peak fitting where required was achieved using line shape models derived from bulk compounds (Pd and Au metal foils and PdO) using Voigt-like functions.Brunauer Emmett Teller (BET) surface area measurements were conducted using a Quadrasorb surface area analyser. A 5-point isotherm of each material was measured using N2 as the adsorbate gas. Samples were degassed at 250 °C for 2 h prior to the surface area being determined by 5-point N2 adsorption at −196 °C, and data analysed using the BET method. Surface area measurements of key samples (and corresponding bare supports) are reported in Table S1,† with a minor loss in surface area found to result from metal deposition and thermal treatment of the catalytic samples.
The bulk structure of the catalysts was determined by powder X-ray diffraction using a (θ–θ) PANalytical X′pert Pro powder diffractometer using a Cu Kα radiation source, operating at 40 keV and 40 mA. Standard analysis was carried out using a backfilled sample, between 2θ values of 10–80°. Phase identification was carried out using the International Centre for Diffraction Data (ICDD). XRD analysis of supported AuPd catalysts prepared on a range of common oxide supports is reported in Figure S1A–E,† with no clear reflections associated with either precious metal, which may be attributed to the low total metal loading.
Total metal loading of key catalytic samples was quantified by digestion of as-prepared (dried only) samples via microwave-assisted aqua regia digestion. Digested samples were analysed via inductively coupled plasma mass spectrometry (ICP-MS). All calibrants were matrix-matched and measured against a five-point calibration using certified reference materials purchased from PerkinElmer and certified internal standards acquired from Agilent. Actual metal loadings of key catalytic samples are provided in Table S2A and B.†
Total metal leaching from the supported catalyst was quantified via inductively coupled plasma mass spectrometry (ICP-MS). Post-reaction solutions were analysed using an Agilent 7900 ICP-MS equipped with I-AS auto-sampler. All samples were diluted by a factor of 10 using HPLC grade H2O (1%HNO3 and 0.5% HCl matrix). All calibrants were matrix-matched and measured against a five-point calibration using certified reference materials purchased from PerkinElmer and certified internal standards acquired from Agilent.
DRIFTS measurements were taken on a Bruker Tensor 27 spectrometer fitted with a mercury cadmium telluride (MCT) detector. A sample was loaded into the Praying Mantis high temperature (HVC-DRP-4) in situ cell before exposure to N2 and then 1% CO/N2 at a flow rate of 50 cm3 min−1. A background spectrum was obtained using KBr, and measurements were recorded every 1 min at room temperature. Once the CO adsorption bands in the DRIFT spectra ceased to increase in size, the gas feed was changed back to N2 and measurements were repeated until no change in subsequent spectra was observed.
Aberration-corrected scanning transmission electron microscopy (AC-STEM) was performed using a probe-corrected S/TEM instrument (Thermo Fisher, Thermis Z), operating at 300 kV. The latter instrument was equipped with a Super-X EDS detector for high-spatial XEDS characterization.
Results and discussion
Our initial studies identified the crucial role of the support in dictating catalytic activity towards the direct synthesis and subsequent degradation of H2O2 (Table 1). In keeping with earlier investigations43,44 into the performance of AuPd catalysts prepared by a co-impregnation procedure, a strong correlation between reactivity and the choice of nanoparticle carrier was observed, with the high activity of the 0.5%Au–0.5%Pd/TiO2 (75 molH2O2 kgcat−1 h−1), and 0.5%Au–0.5%Pd/Al2O3 (62 molH2O2 kgcat−1 h−1), catalysts towards H2O2 direct synthesis, in comparison to alternative oxide supported analogues, clear. Numerous studies have identified the role of catalyst support in dictating both reactivity and selectivity in the direct synthesis of H2O2, through the control of alloy formation and particle dispersion, amongst other factors. Indeed, we have recently demonstrated the role of the particle carrier in dictating metal speciation, particularly that of Pd, a key factor in the production of the oxidant, and direct the reader to these earlier works for an in-depth analysis of many of the formulations reported in Table 1.44| Catalyst | Productivity/molH2O2 kgcat−1 h−1 | H2O2 Conc./wt% | H2 Conv./% | H2O2 Sel./% | Degradation/molH2O2 kgcat−1 h−1 |
|---|---|---|---|---|---|
| H2O2 direct synthesis reaction conditions: mass of catalyst (0.01 g), H2O (2.9 g), MeOH (5.6 g), 5%H2/CO2 (420 psi), 25%O2/CO2 (160 psi), 0.5 h, 20 °C, 1200 rpm. H2O2 degradation reaction conditions: catalyst (0.01 g), H2O2 (50 wt% 0.68 g) H2O (2.22 g), MeOH (5.6 g), 5%H2/CO2 (420 psi), 0.5 h, 20 °C, 1200 rpm. | |||||
| 0.5%Au–0.5%Pd/TiO2 | 75 | 0.15 | 38 | 36 | 390 |
| 0.5%Au–0.5%Pd /Al2O3 | 62 | 0.12 | 33 | 34 | 366 |
| 0.5%Au–0.5%Pd /CeO2 | 30 | 0.06 | 14 | 40 | 289 |
| 0.5%Au–0.5%Pd /Nb2O5 | 25 | 0.06 | 11 | 43 | 255 |
| 0.5%Au–0.5%Pd /ZrO2 | 30 | 0.04 | 12 | 47 | 270 |
The performance of the TiO2(P25) and γ-Al2O3-based formulations is particularly noteworthy given the relatively challenging reaction temperatures utilised within this work and the poor stability of H2O2 under such conditions, with sub-ambient temperatures widely applied to inhibit competitive H2O formation via H2O2 hydrogenation and decomposition pathways.40
Subsequent investigation of catalytic performance towards the in situ oxidation of benzyl alcohol (Fig. 1, with additional data reported in Table S3†) revealed no clear correlation between individual reaction pathways (i.e. H2O2 synthesis and benzyl alcohol valorisation), with almost all formulations offering similar reactivity (<3.0% product yield, 100% benzaldehyde selectivity), despite the relatively varied activity towards H2O2 synthesis (Table 1). The limited reactivity of the 0.5%Au–0.5%Pd/TiO2 catalyst towards the in situ oxidation of benzyl alcohol is particularly noteworthy given the high H2O2 synthesis activity observed over this formulation. While it is important to highlight the variation in reaction conditions used to probe individual reaction pathways, such observations, particularly when considered alongside recent studies38 which report the ability of Au to promote the desorption of oxygen-based radical species (˙OOH, ˙OH, ˙O2−) from AuPd surfaces, may indicate that H2O2 is not the primary species responsible for the observed catalysis. Indeed, we have recently proposed the key role of such radical species for the selective oxidation of alternative chemical feedstocks.45,46
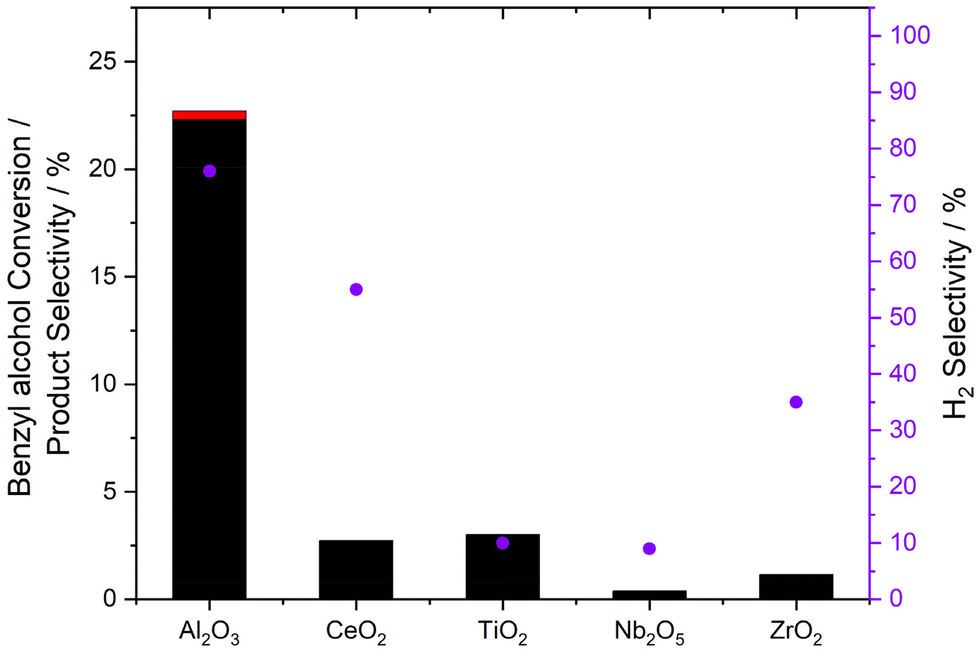 | ||
| Fig. 1 Catalytic activity of bimetallic 0.5%Au–0.5%Pd/Al2O3 catalysts towards the selective oxidation of benzyl alcohol via in situ H2O2 synthesis, as a function of the catalyst support. Key: benzaldehyde (black bars), benzoic acid (red bars); H2 selectivity towards benzaldehyde (purple circles). Reaction conditions: mass of catalyst (0.01 g), benzyl alcohol (1.04 g, 9.62 mmol), MeOH (7.1 g), 5%H2/CO2 (420 psi), 25%O2/CO2 (160 psi), 50 °C, 0.5 h, 1200 rpm. Note: H2 selectivity was determined, based on the mol of H2 utilised in the formation of benzaldehyde. | ||
The 0.5%Au–0.5%Pd/Al2O3 catalyst was found to offer exceptional activity towards benzyl alcohol oxidation (22.7% product yield, 98% benzaldehyde selectivity). Indeed, the performance of this formulation is particularly noteworthy given the high selective utilisation of H2 in the valorisation of benzyl alcohol (76%), with poor H2 efficiency a longstanding hurdle for numerous in situ approaches to feedstock valorisation. In particular, catalytic hydrogenation of both reagents and products, as well as the degradation of H2O2 to H2O via hydrogenation pathways, have been identified as major contributors to process inefficiency.23 Although here we highlight our recent contributions centred around the in situ ammoximation of cyclic ketones to the corresponding oxime, which has demonstrated that such concerns may be overcome through rational catalyst and process design.47
Together with other laboratories, we have previously identified a strong dependence between the catalytic performance of bimetallic AuPd formulations towards the direct synthesis of H2O2,35,40,48,49 as well as the aerobic valorisation of a range of chemical feedstocks (including benzyl alcohol), and the elemental composition of active sites.50–53 In keeping with these earlier studies, and with a focus on γ-Al2O3 supported formulations, we subsequently established the synergistic enhancement that results from the formation of AuPd nanoalloys (Table 2 and Fig. 2, with additional data reported in Table S4†). This enhancement can, at least in part be attributed to the electronic modification of Pd species through alloying with Au, as evidenced by CO-DRIFTS (Figure S2 and ESI Note 1†), and XPS analysis (Figure S3†), with further investigation of the catalytic series by TEM (Table 3, with representative micrographs reported in Figure S4†), ruling out variation in mean particle size, another key parameter known to dictate catalytic activity, especially to H2O2 synthesis,54 as a source for the underlying promotive effect observed upon the formation of AuPd alloys.
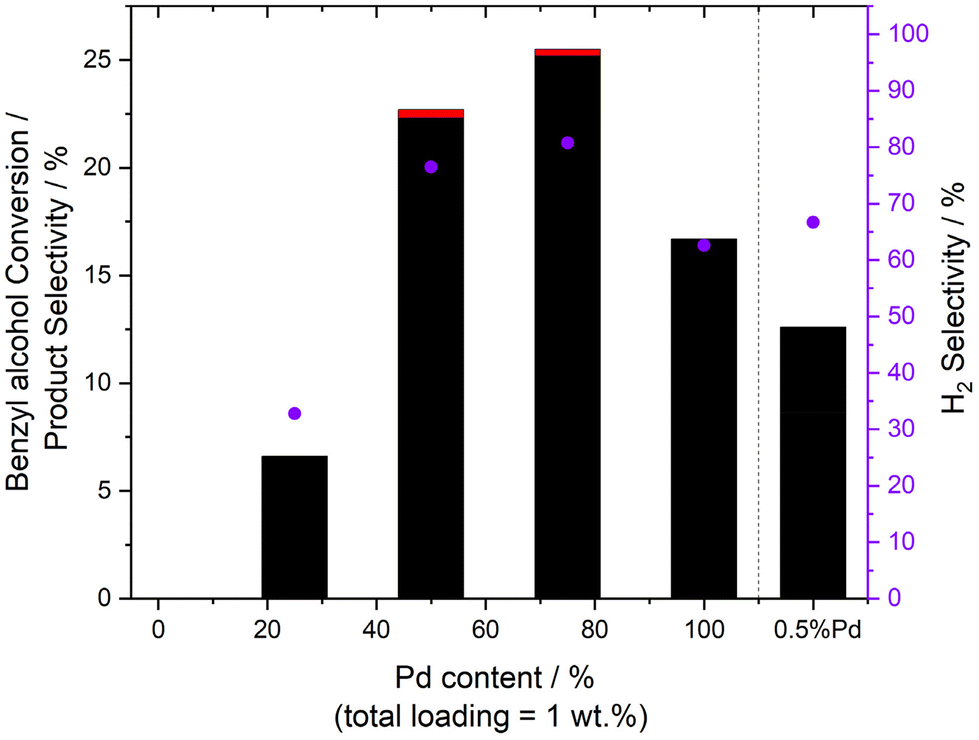 | ||
Fig. 2 Catalytic activity of bimetallic 1%AuPd/Al2O3 catalysts towards the selective oxidation of benzyl alcohol via in situ H2O2 synthesis, as a function of Au![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Pd ratio. Key: benzaldehyde (black bars), benzoic acid (red bars), H2 selectivity (purple circles). Reaction conditions: mass of catalyst (0.01 g), benzyl alcohol (1.04 g, 9.62 mmol), MeOH (7.1 g), 5%H2/CO2 (420 psi), 25%O2/CO2 (160 psi), 50 °C, 0.5 h, 1200 rpm. Note: H2 selectivity is determined based on the mol of H2 utilised in the formation of benzaldehyde. The 1%Au/Al2O3 offered no measurable activity towards benzyl alcohol oxidation. :Pd ratio. Key: benzaldehyde (black bars), benzoic acid (red bars), H2 selectivity (purple circles). Reaction conditions: mass of catalyst (0.01 g), benzyl alcohol (1.04 g, 9.62 mmol), MeOH (7.1 g), 5%H2/CO2 (420 psi), 25%O2/CO2 (160 psi), 50 °C, 0.5 h, 1200 rpm. Note: H2 selectivity is determined based on the mol of H2 utilised in the formation of benzaldehyde. The 1%Au/Al2O3 offered no measurable activity towards benzyl alcohol oxidation. | ||
| Catalyst | Productivity/molH2O2 kgcat−1 h−1 | H2O2 Conc./wt% | H2 Conv./% | H2O2 Sel./% | Degradation/molH2O2 kgcat−1 h−1 |
|---|---|---|---|---|---|
| H2O2 direct synthesis reaction conditions: Mass of catalyst (0.01 g), H2O (2.9 g), MeOH (5.6 g), 5%H2/CO2 (420 psi), 25%O2/CO2 (160 psi), 0.5 h, 20 °C, 1200 rpm. H2O2 degradation reaction conditions: mass of catalyst (0.01 g), H2O2 (50 wt% 0.68 g) H2O (2.22 g), MeOH (5.6 g), 5%H2/CO2 (420 psi), 0.5 h, 20 °C, 1200 rpm. B.D.L: below the detection limit. | |||||
| 1%Au/Al2O3 | 2 | 0.001 | B.D.L | — | 22 |
| 0.75%Au–0.25%Pd/Al2O3 | 70 | 0.14 | 21 | 59 | 287 |
| 0.5%Au–0.5%Pd/Al2O3 | 62 | 0.12 | 33 | 34 | 366 |
| 0.25%Au–0.75%Pd/Al2O3 | 59 | 0.12 | 32 | 34 | 289 |
| 1%Pd/Al2O3 | 43 | 0.09 | 26 | 30 | 200 |
| 0.5%Pd/Al2O3 | 32 | 0.07 | 17 | 34 | 119 |
:Pd ratio on mean nanoparticle size of 1%AuPd/Al2O3 catalysts
| Catalyst | Mean particle size/nm (S.D) | Reaction rate/mmolaldehyde mmolmetal−1 h−1 |
|---|---|---|
| Catalysts exposed to a reductive heat treatment (5%H2/Ar, 500 °C, 4 h, 10 °C min−1). Reaction rate is based on theoretical metal loading, at a reaction time of 0.5 h. | ||
| 1%Au/Al2O3 | 28.9 (26.1) | 0 |
| 0.75%Au–0.25%Pd/Al2O3 | 2.4 (1.4) | 2.06 × 103 |
| 0.5%Au–0.5%Pd/Al2O3 | 3.5 (2.2) | 5.92 × 103 |
| 0.25%Au–0.75%Pd/Al2O3 | 3.1 (1.8) | 5.82 × 103 |
| 1%Pd/Al2O3 | 5.1 (4.8) | 3.4 × 103 |
Again, we highlight the discrepancy in catalytic activity towards H2O2 direct synthesis and the oxidative valorisation of benzyl alcohol over this series of catalyst formulations, which we consider further implicates the contribution of alternative oxidative agents in the observed catalysis. Indeed, the 0.25%Au–0.75%Pd/Al2O3 formulation can be seen to offer a significantly improved performance towards benzyl alcohol oxidation compared to the 0.5%Au–0.5%Pd/Al2O3 analogue, while also offering high H2 efficiency (81%), despite the similar rates of H2O2 synthesis observed over both formulations, which may indicate the potential secondary role of H2O2 in benzyl alcohol oxidation.
With the clear enhancement in catalytic activity identified upon the introduction of Au into supported Pd catalysts, we were motivated to further investigate a subset of these formulations (i.e. the 1%Pd/Al2O3 and 0.5%Pd–0.5%Au/Al2O3 catalysts), in order to broaden our understanding of the underlying cause for the observed trends in catalytic performance.
Further evaluation focussing on these key catalysts established the negligible contribution of purely oxidative (using a 25%O2/CO2 atmosphere) (0.2–0.8% product yield) or reductive (using a 5%H2/CO2 atmosphere) (0.4–1.0% product yield), pathways towards benzyl alcohol conversion (Figure S5†). These observations, particularly the low product yields detected under an aerobic atmosphere, may be unsurprising given the reaction temperatures within this study (50 °C), with temperatures exceeding 80 °C typically required for aerobic benzyl alcohol oxidation over AuPd surfaces.55,56 When utilising a H2-only gaseous atmosphere, relatively low concentrations of benzaldehyde were detected (approx. 0.1% yield), which can be attributed to the incomplete purging of dissolved oxygen from the reaction medium and the resulting production of low concentrations of H2O2 and related radical species. Notably, in the absence of exogenous O2, toluene was also observed and indeed was found to be the major product (0.3–0.8% yield), which can be related to the known reactivity of Pd-based catalysts towards disproportionation pathways.1,57 Subsequent studies also indicated that a significant improvement in benzyl alcohol conversion may be obtained via in situ H2O2 production, compared to that observed when using the preformed oxidant at a concentration identical to that which would be obtained if all the H2 present in the in situ system was selectively converted to H2O2. This is likely a result of the complete addition of the ex situ generated H2O2 at the start of the reaction, although the effect of the proprietary stabilising agents present in commercially available H2O2 on catalytic performance should also be considered. Additionally, such observations again further highlight the potential for reactive oxygen species, rather than H2O2 itself, to be primarily responsible for the observed catalysis. Regardless, given the relatively high costs associated with commercial H2O2, the comparative economic and environmental benefits of the in situ approach are clear and we consider these will only strengthen with the application of non-fossil derived H2 sources.
Extended reaction time studies comparing the catalytic efficacy of key formulations are reported in Fig. 3 (additional data are presented in Table S5A and B,† with determination of catalyst stability, through ICP-MS analysis of post-reaction solutions, at key time points reported in Table S6†). As over our standard reaction time (0.5 h), the higher catalytic activity of the 0.5%Au–0.5%Pd/Al2O3 catalyst was clear (27.1% product yield, 97% benzaldehyde selectivity), significantly outperforming the 1%Pd/Al2O3 analogue (19.0% product yield, 100% benzaldehyde selectivity), over 1.5 h of reaction. The high benzaldehyde selectivity offered by both formulations indicates the suppression of competitive reaction pathways, and can primarily be related to the presence of unconverted substrate, with earlier works focussed on aerobic oxidation of benzyl alcohol, revealing that the presence of benzyl alcohol (at concentrations as low as 2%), in addition to a range of other alcohols (although notably not methanol, the solvent used in this study), can inhibit the overoxidation of benzaldehyde to the corresponding acid.58
 | ||
| Fig. 3 Comparison of catalytic performance towards the selective oxidation of benzyl alcohol via in situ H2O2 synthesis over the (A) 1%Pd/Al2O3 and (B) 0.5%Au–0.5%Pd/Al2O3 catalysts, as a function of reaction time. Key: benzaldehyde (black bars), benzoic acid (red bars), H2 selectivity (purple circles). Reaction conditions: mass of catalyst (0.01 g), benzyl alcohol (1.04 g, 9.62 mmol), MeOH (7.1 g), 5%H2/CO2 (420 psi), 25%O2/CO2 (160 psi), 50 °C, 1200 rpm. Note: H2 selectivity is determined based on the mol of H2 utilised in the formation of benzaldehyde. | ||
Notably, catalytic performance was found to plateau after relatively short reaction times (45 minutes), with minimal additional conversion of benzyl alcohol observed beyond this time point. While this may be indicative of catalyst deactivation, it is important to highlight the relatively high rates of H2 conversion (approx. 80%) and the potential for the reaction to become limited by H2 availability, particularly given the excess of benzyl alcohol (in comparison to H2) present in the system. Interestingly, the 1%Pd/Al2O3 and 0.5%Au–0.5%Pd/Al2O3 catalysts displayed similar H2 conversion rates over the course of the reaction (Table S5A and B†), with this metric reaching approximately 90% over 1.5 h. However, as with our standard reaction time, the bimetallic formulation offered significantly improved selectivity based on H2 compared to the Pd-only analogue (69 and 50% H2 selectivity for the 0.5%Au–0.5%Pd/Al2O3 and 1%Pd/Al2O3 catalysts respectively). Again we consider the improved efficiency of the AuPd formulation noteworthy given the concerns associated with selective H2 utilisation during the valorisation of chemical feedstocks via in situ H2O2 production.13,23
Given the plateau in catalytic performance and the high rates of H2 conversion observed for the AuPd and Pd-only formulations during our time-on-line study and the potential for the reaction to become limited by H2 availability, we next investigated catalytic performance over sequential in situ benzyl alcohol experiments, where gaseous reagents were replaced at 0.5 h intervals (Fig. 4, with additional information reported in Table S7†). For both catalysts, the yield of benzaldehyde increased considerably over four successive reactions, although it should be noted that this increase was not linear, which may suggest some loss in catalyst performance or restructuring of catalytic sites with time. However, the influence of reagent availability, particularly in the case of the AuPd catalyst should also be considered given the relatively high rates of benzyl alcohol conversion observed upon sequential reactions (>40% over two successive reactions). Regardless, the reactivity of the 0.5%Au–0.5%Pd/Al2O3 catalyst is noteworthy achieving a product yield of 62% (98% benzaldehyde selectivity) over four successive reactions, significantly outperforming the 1%Pd/Al2O3 analogue (33% product yield, 100% benzaldehyde selectivity) as well as previous reports in the literature (Table S8†), highlighting the potential improvements in the in situ approach to alcohol valorisation that may be achieved through rational catalyst design. Further comparison to aerobic approaches benzyl alcohol oxidation over AuPd surfaces are reported in Table S9,† and indicate the competitiveness of the in situ approach, although it is clear that further technical evaluation is required (life cycle analysis, process safety techno-economic analysis) to fully understand the technical feasibility of the in situ approach.
 | ||
| Fig. 4 Comparison of catalytic performance towards the selective oxidation of benzyl alcohol via in situ H2O2 synthesis, over the (A) 1%Pd/Al2O3 and (B) 0.5%Au–0.5%Pd/Al2O3 catalysts, over successive reactions. Key: benzaldehyde (black bars), benzoic acid (red bars), H2 selectivity (purple circles). Reaction conditions: mass of catalyst (0.01 g), benzyl alcohol (1.04 g, 9.62 mmol), MeOH (7.1 g), 5%H2/CO2 (420 psi), 25%O2/CO2 (160 psi), 50 °C, 0.5 h, 1200 rpm. | ||
Catalyst stability was subsequently evaluated through re-use experiments (Figure S6†). While a loss in catalytic activity was observed over both formulations, the high selectivity towards benzaldehyde and in the case of the AuPd catalyst, selectivity towards H2 observed upon initial use was largely retained. Interestingly, the AuPd formulation was also found to retain a greater proportion of its initial activity over three uses (approx. 80%) compared to the Pd-only analogue (50%). In part, this can be attributed to the increased stability of the bimetallic catalyst, as evidenced by ICP-MS analysis of post-reaction solutions, where the presence of Au was found to considerably inhibit Pd leaching (Table S10†), which aligns well with previous studies into AuPd-based formulations,47 although it is likely that other factors beyond metal loss are responsible for the variation in performance upon reuse.
Evaluation of the as-prepared catalytic materials via STEM-HAADF (Fig. 5) imaging revealed a relatively broad particle size distribution, which is typical of the wet impregnation route to catalyst synthesis, particularly for bimetallic AuPd-based formulations. However, here we highlight that for both formulations mean particle size was found to be between approximately 3 and 5 nm (Table 3 and Figure S4†). Notably, unlike in our earlier studies which have focussed on Pd-based catalysts prepared by a similar wet impregnation technique, the monometallic catalyst also displays a bimodal particle size distribution, which may evidence the role of nanoparticle carrier in dictating particle size, with these previous works primarily focussed on the use of TiO2 as the catalyst support.27 Subsequent STEM-XEDS mapping of individual nanoparticles within the bimetallic formulation (Fig. 6, with additional data reported in Fig. S7 and S8†), confirmed the presence of AuPd random alloys, regardless of particle size, although the larger particles (>10 nm) were found to be Au-rich, again in keeping with earlier works into analogous formulations.33
 | ||
| Fig. 5 Representative HAADF-STEM of the as-prepared (A and B) 1%Pd/Al2O3 and (C and D) 0.5%Au–0.5%Pd/Al2O3 catalysts, demonstrating the bimodal distribution in particle size. | ||
 | ||
| Fig. 6 HAADF-STEM and corresponding X-EDS analysis of the as-prepared (A) 1%Pd/Al2O3 and (B) 0.5%Au–0.5%Pd/Al2O3 catalysts. | ||
The alloying of Au with Pd has been well-reported to improve catalytic selectivity towards H2O2 through the inhibition of competitive degradation pathways that lead to the formation of H2O (i.e. O–O bond dissociation), as well as promoting desorption of the synthesised H2O2.37 Recently, the ability of AuPd surfaces to also promote the desorption of reaction intermediates (i.e. ˙O2−, ˙OH and ˙OOH) formed during H2O2 synthesis has also been described,38 with further works demonstrating the efficacy of these highly reactive oxygen-based species to catalyze both selective oxidation59 and total degradation of chemical feedstocks.60 With these earlier works in mind, we subsequently conducted a series of spin-trapping electron paramagnetic resonance (EPR) spectroscopy measurements using 5,5-dimethyl-1-pyrroline N-oxide (DMPO) as the radical trapping agent.
Fig. 7A reports the EPR spectra obtained under in situ benzyl alcohol oxidation reaction conditions, with control experiments conducted in the presence of a H2O-only solvent reported in Fig. 7B (additional blank reactions in Fig. S9†). Over both catalyst formulations, a clear signal attributed to an O-centred methoxy radical (CH3O˙) trapped by DMPO, forming a DMPO-OCH3 aminoxyl radical adduct (with giso = 2.006, aiso(14N) = 1.559 mT and aiso(1Hβ) = 2.260 mT), when a methanol solvent was utilised. The presence of such methoxy species is considered to result from the ability of methanol to act as a scavenger for oxygen-based radicals generated during H2O2 synthesis or via catalytic degradation of the synthesised H2O2.61,62
 | ||
| Fig. 7 Experimental (black) and simulated (red) EPR spectra of DMPO-radical adducts formed during the in situ oxidation of benzyl alcohol. (A) Reactions conducted in methanol at 50 °C, with benzyl alcohol (9.62 mmol), 5% H2/CO2 (420 psi), and 25% O2/CO2 (160 psi) in the presence of DMPO and the (i) 1%Pd/Al2O3 or (ii) 0.5%Au–0.5%Pd/Al2O3 catalysts. (B) Control reactions conducted in water at 50 °C, in the presence of DMPO and (i) 1%Pd/Al2O3 or (ii) 0.5%Au–0.5%Pd/Al2O3 under in situ conditions and the absence of benzyl alcohol, with analogous experiments over the (iii) 1%Pd/Al2O3 and (iv) 0.5%Au–0.5%Pd/Al2O3 catalysts under in situ conditions and in the presence of benzyl alcohol. | ||
To confirm the presence of a reactive oxygen progenitor species further studies were conducted, utilising a H2O-only solvent (Fig. 7B). For both catalysts, experiments conducted in the absence of benzyl alcohol (spectra i and ii), the spectra were dominated by a signal symptomatic of trapped oxygen-based radicals (giso = 2.006, aiso(14N) = 1.493 mT, and aiso(1Hβ) = 1.493 mT). However, we were unable to distinguish between trapped hydroxyl (˙OH) and hydroperoxyl (˙OOH) species due to the short half-life of the DMPO-OOH adduct (1–4 min), which rapidly decays to DMPO-OH in the presence of unreacted DMPO. Unsurprisingly, in the absence of the gaseous reagents (Fig. 7B spectra iii and iv) the signal associated with the DMPO-OH adduct decreased significantly, and the spectra was dominated by a new signal consistent with a DMPO-trapped C-centred species (PhCH˙(OH))63 (characterised by giso = 2.006, aiso(14N) = 1.559 mT, and aiso(1Hβ) = 2.260 mT), suggesting that PhCH˙(OH) is the first intermediate in the oxidation of benzyl alcohol. Control experiments (Figure S6†) in the absence of the catalyst revealed no signal from either trapped O- or C-centred radical species, indicating that the generation of such reactive species is a catalytic process, with additional experiments further identifying the presence of the DMPO-OCH3 nitroxide radical.
To support our EPR studies, we subsequently investigated the effect of radical quenching agents (Na2SO3 and NaNO2 separately and at a concentration of 0.05 M), on catalytic performance towards in situ benzyl alcohol oxidation (Table S11†). A substantial decrease in benzyl alcohol conversion was observed, further indicating the role of radical species in the reaction mechanism. Notably, when taken together with our EPR investigations, these experiments indicate that the oxygen-centred radicals are the primary species responsible for the observed catalysis. The non-innocent nature of the methanol solvent is also indicated. However, the role of the solvent-based radicals is unclear, that is we were unable to determine if the methanol simply acts as a radical propagating agent or if there is also involvement in the reaction mechanism, possibly through the promotion of H-abstraction from the alcohol moiety, which is considered key in the formation of the aldehyde.64,65 Furthermore, we recognise the direct role of the solvent in the synthesis of H2O2 and associated ROS, as outlined by Flaherty and co-workers;66 as such, there is clearly a need to develop a detailed understanding of solvent involvement in the reaction mechanism.
Conclusions and future perspectives
The in situ synthesis and efficient utilisation of H2O2 (and associated reactive oxygen species), over AuPd nanoalloys has been demonstrated to offer high efficacy in the selective oxidation of benzyl alcohol to the corresponding aldehyde, offering improved performance compared to the use of preformed H2O2 or aerobic oxidative pathways. In particular, the role of the support was found to be crucial in dictating catalytic performance towards benzyl alcohol valorisation, with the γ-Al2O3 supported formulation outperforming analogous catalysts prepared on a range of other common oxides by a factor of 10, despite the increased activity of alternative formulations towards H2O2 direct synthesis. Such observations suggest that H2O2 itself is not the primary species responsible for the observed catalysis, with further detailed analysis by electron paramagnetic spectroscopy indicating the role of both reactive oxygen species (e.g. ˙OOH, ˙OH, and ˙O2− generated as intermediates during H2O2 synthesis, via H2O2 decomposition or a combination of both processes) and methoxy-based (˙CH3O) radicals (resulting from the radical scavenging ability of the methanol solvent) within the reaction mechanism. Although the extent to which these methoxy-based species contribute directly to the observed catalysis is not fully understood, one can hypothesise that they contribute to the initial proton-abstraction from the alcohol moiety (PhCH2OH) or may act as a propagating agent for subsequent ROS formation. However, with the known ability of the solvent to play a crucial role in the formation of H2O2 (and associated ROS), as well as in the inhibition of benzaldehyde overoxidation, there is clear potential for the solvent to be involved in numerous mechanistic steps, which must be fully understood if this approach to chemical synthesis is to be fully developed.The alloying of Pd with Au was found to considerably enhance the selective utilisation of H2, overcoming a key hurdle which has limited the adoption of the in situ approach for the oxidative valorisation of many chemical feedstocks and the resulting AuPd/Al2O3 catalyst was found to significantly outperform previous examples reported in the literature, achieving yields of benzaldehyde in excess of 60%, with near total selectivity. However, there is still scope for further improvements in catalytic design in order to improve H2 utilisation rates and catalyst stability.
Our observation of relatively large concentrations of residual H2O2 within benzyl alcohol oxidation product streams and the demonstration that H2O2 itself it not the primary oxidative species in benzyl alcohol valorisation also indicates that there is scope for further improvement in catalyst design, to shift selectivity away from H2O2 and towards ROS formation. However, there is a need to ensure effective utilisation of the radical flux and minimise competitive termination reactions, which lead to the unselective utilisation of H2.
There are now a growing number of reports which outline the potential benefits of the in situ approach to chemical synthesis, particularly for oxime manufacture and the valorisation of methane. Indeed, such chemistry may find particular application in the production of low-value/high-volume commodity chemicals, such as adipic acid, cyclohexanone, cyclohexanol and phenol, as well as other benzyl alcohol derivatives, where the high cost of pre-formed H2O2, relative to that of the desired product, has prevented progression to industrial production, despite promising results at the laboratory scale. However, it is clear that in many cases, concerns around catalytic selectivity and deactivation, often resulting from the presence of H2, required to generate the oxidant in situ, must be addressed.
While the potential of the in situ technology is particularly exciting, it is important to note that several hurdles must first be overcome if it is to rival industrially operated processes. From a safety perspective, it is imperative that explosive mixtures of H2/O2 are avoided. Additionally, as with industrial processes that currently utilise preformed H2O2 (e.g. cyclohexanone ammoximation and propylene oxide manufacture), there is a need to ensure hot-spots of H2O2 are avoided, both from a safety aspect and process efficiency standpoint. There is clearly also a need to further enhance catalyst stability to ensure lifetimes required for industrial application are met.
Author contributions
G.S and R.J.L conducted catalyst synthesis, testing, and corresponding data analysis. G.S., R.J.L, J.L, G.M., D.J.M., T.E.D., A.L, and X.L. conducted catalyst characterisation and corresponding data processing. R.J.L., D.M.M, A.F., L.C. and X.L provided technical advice. R.J.L and G.J.H contributed to the design of the study and results interpretation. R.J.L wrote the manuscript and ESI,† with all authors commenting on and amending both documents. All authors discussed and contributed to this work.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
G. S., R. J. L and G. J. H gratefully acknowledge Cardiff University and the Max Planck Centre for Fundamental Heterogeneous Catalysis (FUNCAT) for financial support. A. F. thanks the Net Zero Innovation Institute of Cardiff University for a University Research Fellowship X. L. acknowledges financial support from the National Key R&D Program of China (2021YFA1500300 and 2022YFA1500146) and the National Natural Science Foundation of China (22272106). The authors would like to thank the CCI-Electron Microscopy Facility which has been part-funded by the European Regional Development Fund through the Welsh Government and The Wolfson Foundation. XPS data collection was performed at the EPSRC National Facility for XPS (‘HarwellXPS’).References
- C. E. Chan-Thaw, A. Savara and A. Villa, Catalysts, 2018, 8, 431 CrossRef.
- B. L. Ryland and S. S. Stahl, Angew. Chem., Int. Ed., 2014, 53, 8824–8838 CrossRef CAS PubMed.
- P. Wu, Y. Cao, L. Zhao, Y. Wang, Z. He, W. Xing, P. Bai, S. Mintova and Z. Yan, J. Catal., 2019, 375, 32–43 CrossRef CAS.
- W. Zhou, G. Chen, B. Yu, J. Zhou, J. Qian, M. He and Q. Chen, Appl. Catal., A, 2020, 592, 117417 CrossRef CAS.
- H. Göksu, H. Burhan, S. D. Mustafov and F. Şen, Sci. Rep., 2020, 10, 5439 CrossRef PubMed.
- A. Savara, I. Rossetti, C. E. Chan-Thaw, L. Prati and A. Villa, ChemCatChem, 2016, 8, 2482–2491 CrossRef CAS.
- W. Hou, N. A. Dehm and R. W. J. Scott, J. Catal., 2008, 253, 22–27 CrossRef CAS.
- K. Kawamura, T. Yasuda, T. Hatanaka, K. Hamahiga, N. Matsuda, M. Ueshima and K. Nakai, Chem. Eng. J., 2016, 285, 49–56 CrossRef CAS.
- C. Xu, L. Zhang, Y. An, X. Wang, G. Xu, Y. Chen and L. Dai, Appl. Catal., A, 2018, 558, 26–33 CrossRef CAS.
- A. L. Cánepa, V. R. Elías, V. M. Vaschetti, E. V. Sabre, G. A. Eimer and S. G. Casuscelli, Appl. Catal., A, 2017, 545, 72–78 CrossRef.
- N. O'Callaghan and J. A. Sullivan, Appl. Catal., B, 2014, 146, 258–266 CrossRef.
- B. Puértolas, A. K. Hill, T. García, B. Solsona and L. Torrente-Murciano, Catal. Today, 2015, 248, 115–127 CrossRef.
- R. J. Lewis and G. J. Hutchings, Acc. Chem. Res., 2024, 57, 106–119 CrossRef CAS PubMed.
- T. Hayashi, K. Tanaka and M. Haruta, J. Catal., 1998, 178, 566–575 CrossRef CAS.
- J. Huang, E. Lima, T. Akita, A. Guzmán, C. Qi, T. Takei and M. Haruta, J. Catal., 2011, 278, 8–15 CrossRef CAS.
- S. Kanungo, D. M. Perez Ferrandez, F. Neira D’Angelo, J. C. Schouten and T. A. Nijhuis, J. Catal., 2016, 338, 284–294 CrossRef CAS.
- S. Quon, D. Y. Jo, G.-H. Han, S. S. Han, M.-G. Seo and K. Lee, J. Catal., 2018, 368, 237–247 CrossRef CAS.
- Q. Liu, J. C. Bauer, R. E. Schaak and J. H. Lunsford, Appl. Catal., A, 2008, 339, 130–136 CrossRef CAS.
- S. Yu, X. Cheng, Y. Wang, B. Xiao, Y. Xing, J. Ren, Y. Lu, H. Li, C. Zhuang and G. Chen, Nat. Commun., 2022, 13, 4737 CrossRef CAS PubMed.
- L. F. D. L. e Freitas, B. Puértolas, J. Zhang, B. Wang, A. S. Hoffman, S. R. Bare, J. Pérez-Ramírez, J. W. Medlin and E. Nikolla, ACS Catal., 2020, 10, 5202–5207 CrossRef.
- F. Wang, C. Xia, S. P. de Visser and Y. Wang, J. Am. Chem. Soc., 2019, 141, 901–910 CrossRef CAS PubMed.
- D. W. Flaherty, ACS Catal., 2018, 8, 1520–1527 CrossRef CAS.
- G. Wang, W. Du, X. Duan, Y. Cao, Z. Zhang, J. Xu, W. Chen, G. Qian, W. Yuan, X. Zhou and D. Chen, Chem. Catal., 2021, 1, 885–895 CrossRef CAS.
- Q. Chen and E. J. Beckman, Green Chem., 2008, 10, 934–938 RSC.
- S.-Y. Ye, S. Hamakawa, S. Tanaka, K. Sato, M. Esashi and F. Mizukami, Chem. Eng. J., 2009, 155, 829–837 CrossRef CAS.
- J. Lyu, L. Niu, F. Shen, J. Wei, Y. Xiang, Z. Yu, G. Zhang, C. Ding, Y. Huang and X. Li, ACS Omega, 2020, 5, 16865–16874 CrossRef CAS PubMed.
- C. M. Crombie, R. J. Lewis, R. L. Taylor, D. J. Morgan, T. E. Davies, A. Folli, D. M. Murphy, J. K. Edwards, J. Qi, H. Jiang, C. J. Kiely, X. Liu, M. S. Skjøth-Rasmussen and G. J. Hutchings, ACS Catal., 2021, 11, 2701–2714 CrossRef CAS.
- M. Santonastaso, S. J. Freakley, P. J. Miedziak, G. L. Brett, J. K. Edwards and G. J. Hutchings, Org. Process Res. Dev., 2014, 18, 1455–1460 CrossRef CAS.
- Benzaldehyde Market – Growth, Industry Applications & Market Trends, viahttps://www.futuremarketinsights.com/reports/benzaldehyde-market, accessed 07.02.2025.
- L. Kesavan, R. Tiruvalam, M. H. A. Rahim, M. I. bin Saiman, D. I. Enache, R. L. Jenkins, N. Dimitratos, J. A. Lopez-Sanchez, S. H. Taylor, D. W. Knight, C. J. Kiely and G. J. Hutchings, Science, 2011, 331, 195–199 CrossRef CAS PubMed.
- C. M. Crombie, R. J. Lewis, D. Kovačič, D. J. Morgan, T. E. Davies, J. K. Edwards, M. S. Skjøth-Rasmussen and G. J. Hutchings, Catal. Lett., 2021, 151, 164–171 CrossRef CAS.
- J. K. Edwards, B. Solsona, E. N. Ntainjua, A. F. Carley, A. A. Herzing, C. J. Kiely and G. J. Hutchings, Science, 2009, 323, 1037 CrossRef CAS PubMed.
- D. Kovačič, R. J. Lewis, C. M. Crombie, D. J. Morgan, T. E. Davies, Á. López-Martín, T. Qin, C. S. Allen, J. K. Edwards, L. Chen, M. S. Skjøth-Rasmussen, X. Liu and G. J. Hutchings, Green Chem., 2023, 25, 10436–10446 RSC.
- T. Ricciardulli, S. Gorthy, J. S. Adams, C. Thompson, A. M. Karim, M. Neurock and D. W. Flaherty, J. Am. Chem. Soc., 2021, 143, 5445–5464 CrossRef CAS PubMed.
- N. M. Wilson, P. Priyadarshini, S. Kunz and D. W. Flaherty, J. Catal., 2018, 357, 163–175 CrossRef.
- S. Kanungo, L. van Haandel, E. J. M. Hensen, J. C. Schouten and M. F. Neira d'Angelo, J. Catal., 2019, 370, 200–209 CrossRef CAS.
- J. Li, T. Ishihara and K. Yoshizawa, J. Phys. Chem. C, 2011, 115, 25359–25367 CrossRef CAS.
- T. Richards, J. H. Harrhy, R. J. Lewis, A. G. R. Howe, G. M. Suldecki, A. Folli, D. J. Morgan, T. E. Davies, E. J. Loveridge, D. A. Crole, J. K. Edwards, P. Gaskin, C. J. Kiely, Q. He, D. M. Murphy, J.-Y. Maillard, S. J. Freakley and G. J. Hutchings, Nat. Catal., 2021, 4, 575–585 CrossRef CAS.
- R. J. Lewis, K. Ueura, X. Liu, Y. Fukuta, T. Qin, T. E. Davies, D. J. Morgan, A. Stenner, J. Singleton, J. K. Edwards, S. J. Freakley, C. J. Kiely, L. Chen, Y. Yamamoto and G. J. Hutchings, ACS Catal., 2023, 13, 1934–1945 CrossRef CAS.
- A. Santos, R. J. Lewis, G. Malta, A. G. R. Howe, D. J. Morgan, E. Hampton, P. Gaskin and G. J. Hutchings, Ind. Eng. Chem. Res., 2019, 58, 12623–12631 CrossRef CAS.
- J. K. Edwards, A. Thomas, A. F. Carley, A. A. Herzing, C. J. Kiely and G. J. Hutchings, Green Chem., 2008, 10, 388–394 RSC.
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42–55 CrossRef CAS PubMed.
- E. N. Ntainjua, J. K. Edwards, A. F. Carley, J. A. Lopez-Sanchez, J. A. Moulijn, A. A. Herzing, C. J. Kiley and G. J. Hutchings, Green Chem., 2008, 10, 1162–1169 RSC.
- T. Richards, R. J. Lewis, D. J. Morgan and G. J. Hutchings, Catal. Lett., 2022, 153, 32–40 CrossRef.
- G. Sharp, R. J. Lewis, J. Liu, G. Magri, D. J. Morgan, T. E. Davies, Á. López-Martín, R. Li, C. R. Morris, D. M. Murphy, A. Folli, A. I. Dugulan, L. Chen, X. Liu and G. J. Hutchings, ACS Catal., 2024, 14, 15279–15293 CrossRef CAS.
- F. Ni, R. J. Lewis, Á. López-Martín, L. R. Smith, D. J. Morgan, T. E. Davies, S. H. Taylor and G. J. Hutchings, Catal. Today, 2024, 442, 114910 CrossRef CAS.
- R. J. Lewis, K. Ueura, X. Liu, Y. Fukuta, T. E. Davies, D. J. Morgan, L. Chen, J. Qi, J. Singleton, J. K. Edwards, S. J. Freakley, C. J. Kiely, Y. Yamamoto and G. J. Hutchings, Science, 2022, 376, 615–620 CrossRef CAS PubMed.
- H. Xu, D. Cheng and Y. Gao, ACS Catal., 2017, 7, 2164–2170 CrossRef CAS.
- F. Menegazzo, M. Manzoli, M. Signoretto, F. Pinna and G. Strukul, Catal. Today, 2015, 248, 18–27 CrossRef CAS.
- J. Pritchard, L. Kesavan, M. Piccinini, Q. He, R. Tiruvalam, N. Dimitratos, J. A. Lopez-Sanchez, A. F. Carley, J. K. Edwards, C. J. Kiely and G. J. Hutchings, Langmuir, 2010, 26, 16568–16577 CrossRef CAS PubMed.
- A. Savara, C. E. Chan-Thaw, J. E. Sutton, D. Wang, L. Prati and V. Alberto, ChemCatChem, 2017, 9, 253–257 CrossRef CAS.
- Y. Chen, H. Lim, Q. Tang, Y. Gao, T. Sun, Q. Yan and Y. Yang, Appl. Catal., A, 2010, 380, 55–65 CrossRef CAS.
- J. Long, H. Liu, S. Wu, S. Liao and Y. Li, ACS Catal., 2013, 3, 647–654 CrossRef CAS.
- P. Tian, L. Ouyang, X. Xu, C. Ao, X. Xu, R. Si, X. Shen, M. Lin, J. Xu and Y. F. Han, J. Catal., 2017, 349, 30–40 CrossRef CAS.
- J. Luo, H. Yu, H. Wang, H. Wang and F. Peng, Chem. Eng. J., 2014, 240, 434–442 CrossRef CAS.
- A. Constantinou, G. Wu, B. Venezia, P. Ellis, S. Kuhn and A. Gavriilidis, Top. Catal., 2019, 62, 1126–1131 CrossRef CAS.
- C. Keresszegi, D. Ferri, T. Mallat and A. Baiker, J. Phys. Chem. B, 2005, 109, 958–967 CrossRef CAS PubMed.
- W. Partenheimer, Adv. Synth. Catal., 2006, 348, 559–568 CrossRef CAS.
- M. H. Ab Rahim, M. M. Forde, R. L. Jenkins, C. Hammond, Q. He, N. Dimitratos, J. A. Lopez-Sanchez, A. F. Carley, S. H. Taylor, D. J. Willock, D. M. Murphy, C. J. Kiely and G. J. Hutchings, Angew. Chem., Int. Ed., 2013, 52, 1280–1284 CrossRef CAS PubMed.
- A. Santos, R. J. Lewis, D. J. Morgan, T. E. Davies, E. Hampton, P. Gaskin and G. J. Hutchings, Catal. Sci. Technol., 2021, 11, 7866–7874 RSC.
- J. Marugán, D. Hufschmidt, M.-J. López-Muñoz, V. Selzer and D. Bahnemann, Appl. Catal., B, 2006, 62, 201–207 CrossRef.
- M. R. Billany, K. Khatib, M. Gordon and J. K. Sugden, Int. J. Pharm., 1996, 137, 143–147 CrossRef CAS.
- G. R. Buettner, Free Radical Biol. Med., 1987, 3, 259–303 CrossRef CAS PubMed.
- J. Zhang, S. Xiao, R. Chen and F. Chen, J. Chem. Technol. Biotechnol., 2019, 94, 1613–1621 CrossRef CAS.
- Y. Zhao, C. Yu, S. Wu, W. Zhang, W. Xue and Z. Zeng, Catal. Lett., 2018, 148, 3082–3092 CrossRef CAS.
- N. M. Wilson and D. W. Flaherty, J. Am. Chem. Soc., 2016, 138, 574–586 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5gc00680e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |