
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
N-Heterocyclic carbene-/photoredox-catalyzed regioselective carbonylation of alkenes†
Mao-Lin
Yang
a and
Xiao-Feng
Wu
 *abc
*abc
aLeibniz-Institut für Katalyse e.V., Albert-Einstein-Str. 29a, 18059 Rostock, Germany. E-mail: xiao-feng.wu@catalysis.de
bDalian National Laboratory for Clean Energy, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, 116023 Liaoning, China
cState Key Laboratory of Elemento-Organic Chemistry, Nankai University, Tianjin, 300071, China
First published on 2nd April 2025
Abstract
Besides its special reactivities, organocatalysis offers the notable advantage of avoiding metal residue compared with metal catalysis, and N-heterocyclic carbenes are important organocatalysts. Recently, the combination of NHC organocatalysis and photoredox catalysis has emerged as a promising strategy for C–C bond formation via radical intermediates. However, very few organocatalysis strategies can be used in carbonylation chemistry, especially using N-heterocyclic carbene as it gets deactivated by carbon monoxide. Herein, for the first time, we developed a catalytic carbonylation strategy combining NHC catalysis with photocatalysis to enable CO-involved regioselective synthetic transformation. Under standard conditions, carbonylative diacylation of alkenes was realized to afford valuable 1,4-dicarbonyl compounds in good yields. This strategy offers novel insights into the design of photoinitiated organocatalytic transformations of carbon monoxide.
Green foundation1. Organocatalysis avoids the use of transition-metal catalysts, and photocatalysis makes use of light.2. A catalytic carbonylation strategy by combining NHC catalysis with photocatalysis to enable CO-involved regioselective synthetic transformation was developed. 3. The combination of NHC catalysis and photocatalysis would inspire further developments in CO chemistry. |
Introduction
Carbon monoxide (CO) has been recognized as an abundant, cost-effective, and versatile C1 feedstock in carbonylation chemistry.1 Since the beginning of 20th century, various novel transition metal-catalyzed carbonylation reactions have been developed, such as the Mond–Langer process,2a Gattermann-Koch reaction,2b Roelen's oxo process,2c the Monsanto/Cativa process,2d and Heck's palladium-catalyzed methodologies (Scheme 1a).2e Since then, transition metal catalysts have been regarded as essential for facilitating carbonylation reactions.2 However, organocatalysts have been demonstrated to be powerful in catalyzing organic transformations owing to their lone pairs of electrons and accessible vacant orbitals (Scheme 1b).3 Accordingly, innovative transformations were achieved, and their importance was recognized with a Nobel prize in chemistry in 2021.3d However, owing to its strong coordinating ability (σ-donating and π-back donating), CO readily interacts with organocatalysts, which contain lone pairs of electrons, forming ketene complexes that lead to catalyst deactivation.3b Thus, the appropriate selection of small-molecule catalysts and the design of an effective catalytic system are significant challenges in carbonylation chemistry. | ||
| Scheme 1 Challenges and our design. | ||
N-Heterocyclic carbenes (NHC) are powerful organic catalysts capable of cleaving enthalpically strong bonds, such as Si–H,4 C–H5 and C–F6 bonds, and even challenging molecules like H2 and NH3.3a,7 Most NHC-catalyzed reactions are ion-based processes involving electron pair transfer, which is a concept that has been extensively studied over the past few decades.8 However, radical reactions driven by single electron transfer (SET) processes remain relatively underexplored. Recent advancements in this area, particularly the achievements of Studer,9a–c Ye,9d Chi,9e,f Rovis,9g Ohmiya9h Glorius,9k Scheidt,9i,j and other research groups, have significantly expanded the scope of NHC catalysis; however, further exploration is still required to tackle more challenging transformations in gas capture reactions, such as carbonylation.
In recent decades, visible light photocatalysis has emerged as a powerful tool for organic synthesis, effectively converting light energy into chemical energy and offering an environmentally benign approach for the construction of complex molecular frameworks.10 This process typically involves the photoredox-driven SET mechanism, wherein an excited photocatalyst transfers an electron to a substrate, generating a radical ion that subsequently undergoes transformation in radical-based reactions.11 The dynamic nature of reactive sites in radical coupling reactions often leads to the occurrence of undesirable side reactions, which become particularly problematic in multi-component radical processes. Consequently, achieving regioselective control in radical coupling to precisely synthesize target compounds remains a formidable challenge in this field. Combined photoredox and carbene organocatalysis is recognized as an advanced strategy enabling radical–radical coupling. Notably, in multicomponent systems, the strategic application of NHC-catalysts plays a key role in mitigating the side reactions that arise from multiple reactive sites during photocatalytic processes, leveraging their regioselective catalytic effects. This approach is particularly advantageous in reactions that require prolonged reaction time, such as those involving gas capture, as it promotes efficient and selective transformations.
Considering the above discussed background, the application of NHC-/photo-catalysis in carbonylation appears attractive. Exploration in this direction offers several distinct advantages: (1) the employment of NHC small-molecule catalysts effectively mitigates the issue of metal contamination in the target product; (2) NHC catalysis facilitates enhanced regioselectivity in gas-involved (requiring prolonged reaction time) multi-radical coupling reactions due to its high reactivity; (3) in CO-capture-based carbonylation systems, the development of appropriate NHC catalysts not only prevents catalyst deactivation due to CO's strong coordination ability but also enables the efficient synthesis of the desired products; (4) the advanced carbene/photon co-catalysis system represents the first reported strategy for CO-based carbonylation, representing a significant milestone to further advance the field of sustainable and green carbonylation processes.
The design of suitable carbene catalysis system, in conjunction with a compatible photocatalytic system for the carbonylation of CO, presents several key challenges, as outlined in Scheme 1c. First, precise control of CO concentration is critical, as excessive CO may deactivate the carbene catalyst, while insufficient CO could reduce the efficiency of its capture as formed acyl radical intend to occur reverse decarbonylation. Second, the polarity matching within the catalytic system, coupled with the presence of multiple reactive sites, may restrict the carbene catalyst to two-component reactions9i,f,j,12 or lead to undesired radical coupling side reactions,13 thereby suppressing the formation of the desired four-component product. Third, incompatibilities between the carbene catalytic system and the photocatalytic system may disrupt the SET relay, thereby impeding the overall catalytic cycle.
Based on the above considerations and our ongoing exploration of green carbonylation,14 as well as integrating the valuable applications of 1,4-dicarbonyl compounds in heterocyclic synthesis and diol preparation,15 we designed a new carbonylation catalytic system which leverages the regioselective catalysis of NHC small molecules and visible light to achieve the efficient and sustainable conversion of CO (Scheme 1d). In this process, NHC serves as a catalyst, driving the reaction with high catalytic activity and regioselectivity. Photocatalysis functions as a radical-initiating method, generating the radicals required for CO capture while facilitating the catalytic cycle of the carbene catalyst. CO, serving as a carbonyl source, is captured by radicals to generate acyl radicals, which then regioselectively activate alkenes. The carbene catalytic process and the photocatalytic process operate in tandem, complementing each other to facilitate the efficient CO-inclusive four-component radical coupling reaction, offering a series of substituted 1,4-dicarbonyl compounds. The success of this transformation provides new insights into the application of organic small-molecule catalysis in carbonylation chemistry, promoting the further development of green CO conversion.
Results and discussion
To realize the above design of an NHC-/photoredox-catalyzed carbon monoxide-inclusive four-component diacylation process (for more details, see the ESI†), we initially employed styrene 1a, acyl imidazole 2a, and Hantzsch ester 3a as model substrates under blue light-emitting diode (LED) irradiation, wherein we explored the carbonylation of styrene under 40 bar of CO, as shown in Table 1. At the beginning, IrdF(CF3)ppy2(dtbpy)PF6 (PC-1, 1.5 mol%) was identified as a more effective photocatalyst than the alternatives (Table 1, entries 2–5), and the optimal reaction temperature was found to be 40 °C (Table 1, entry 1). Then, we focused on the chemical structure of NHC molecules, the leaving group (LG) of acyl radical donors, and the Hantzsch esters (DHP) that capture CO molecules. Through systematic experimental investigations, we identified NHC-9, LG1, and DHP2 as the most suitable components for our carbonylation system (Table 1, entries 6–14). Additional control experiments underscored the essential roles of light, the photocatalyst, the NHC-catalyst, and the base in achieving a successful transformation (Table 1, entry 16). Finally, the optimal reaction conditions are shown in Table 1, entry 15.|
|
||
|---|---|---|
| Entry | Variations as shown | Yield (%) |
| The reaction was conducted using 1a (0.1 mmol), 2a (0.2 mmol), 3a (0.25 mmoln, 0.15 mmolo), MeCN (1.0 mL) irradiation with blue LEDs at room temperature for 24 h. CO (30a, 40b, 50c, 60d bar). PC (1 mol%e, 1.5 mol%f, 2 mol%g, and 3 mol%h). NHC (10 mol%i, 20 mol%j, 25 mol%k, 30 mol%l, and 50 mol%m), determined using GC with hexadecane as internal standard. Isolated yield is shown in parentheses. | ||
| 1b,f,k,o | r.t., 40 °C or 50 °C | 59, 65 or 40 |
| 2b,g,k,o | PC-2 instead of PC-1 | 30 |
| 3b,g,k,o | PC-3 instead of PC-1 | 22 |
| 4b,g,k,o | 4CzIPN instead of PC-1 | 10 |
| 5b,g,k,o | PC-1 | 52g, 56f |
| 6b,i,g,o | NHC-3 instead of NHC-9 | 15 |
| 7b,i,g,o | NHC-6 instead of NHC-9 | 13 |
| 8b,i,o | NHC-9 | 38g, 43f |
| 9b,i,g,o | NHC-10 instead of NHC-9 | 30 |
| 10b,i,f,o | NHC-11, 12 or 13 instead of NHC-9 | — |
| 11b,k,f,o | LG2 instead of LG1 | 22 |
| 12b,k,f,o | LG3 instead of LG1 | 35 |
| 13b,k,f,o | LG4 instead of LG1 | 32 |
| 14b,k,f,o | LG1 | 52 |
| 15b,f,k,n | 40 °C and 36 h, instead of r.t. and 24 h | 90 (82) |
| 16b,f,k,n | 40 °C, 36 h; without PC, NHC, light or base | — |

|
||

With the optimal conditions in hand, we subsequently explored the applicability of the carbon monoxide-inclusive four-component diacylation method for synthesizing 1,4-diones (Schemes 2 and 3). A series of substituted alkenes were transformed successfully and the desired dicarbonyl products 4a–z were formed in moderate to good yields (40–85%), including 5-vinylbenzo[d][1,3]dioxole (1m), which was efficiently converted to 4m in 70% yield. Fluorine-substituted styrenes, such as 2,3,4,5,6-pentafluorostyrene (1i), 4-difluoromethoxy styrene (1o) and 3-trifluoromethylstyrene (1w), also produced the corresponding products 4i, 4o and 4w in yields of 59%, 56% and 51%, respectively.
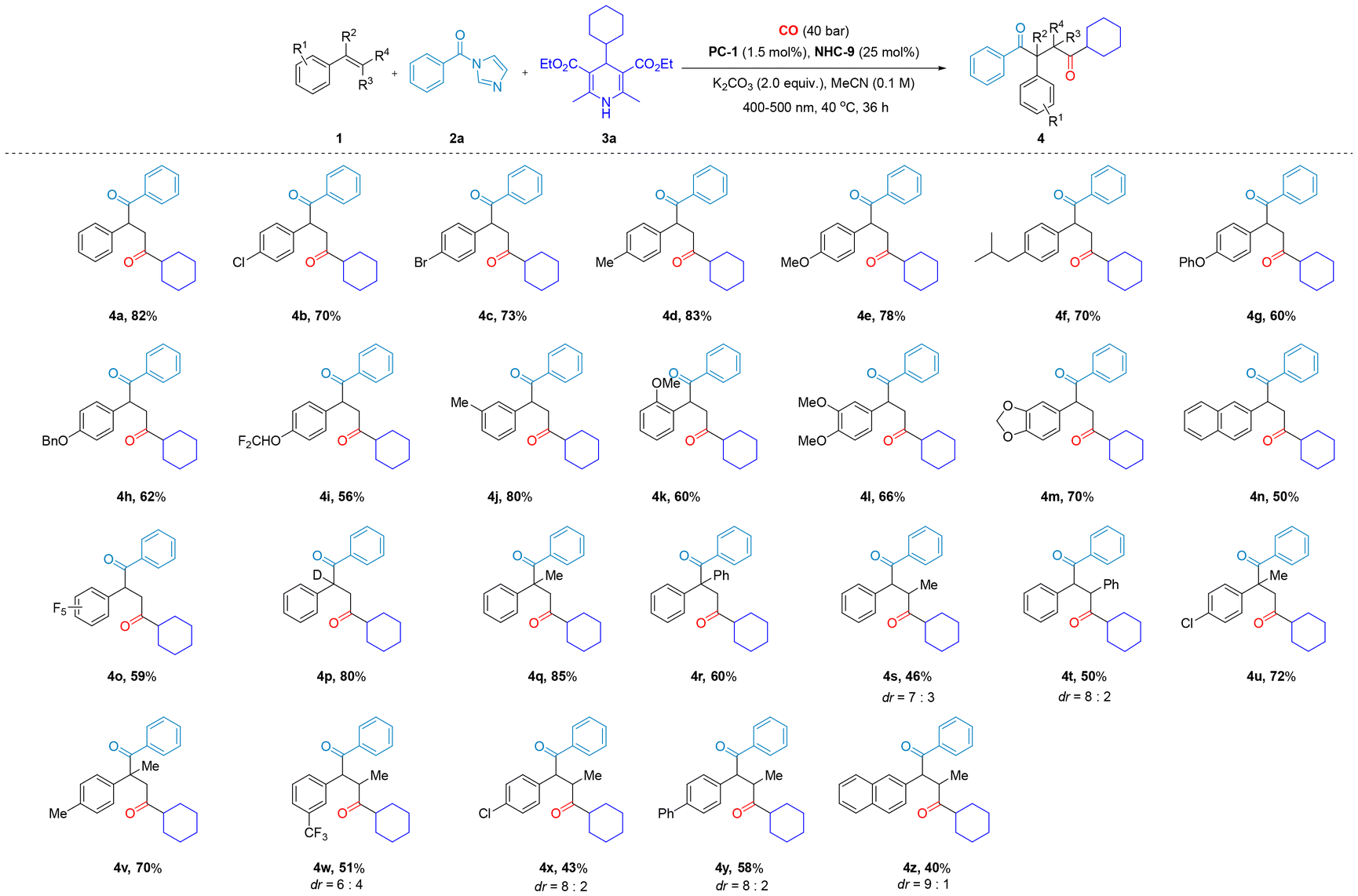 | ||
| Scheme 2 Substrate scope of various alkenes, acyl imidazoles and Hantzsch esters to synthesize 1,4-diketones. Reaction conditions: 1 (0.2 mmol), 2 (0.4 mmol), 3 (0.5 mmol), MeCN (2.0 mL), K2CO3 (0.4 mmol), 400–500 nm, isolated yield. | ||
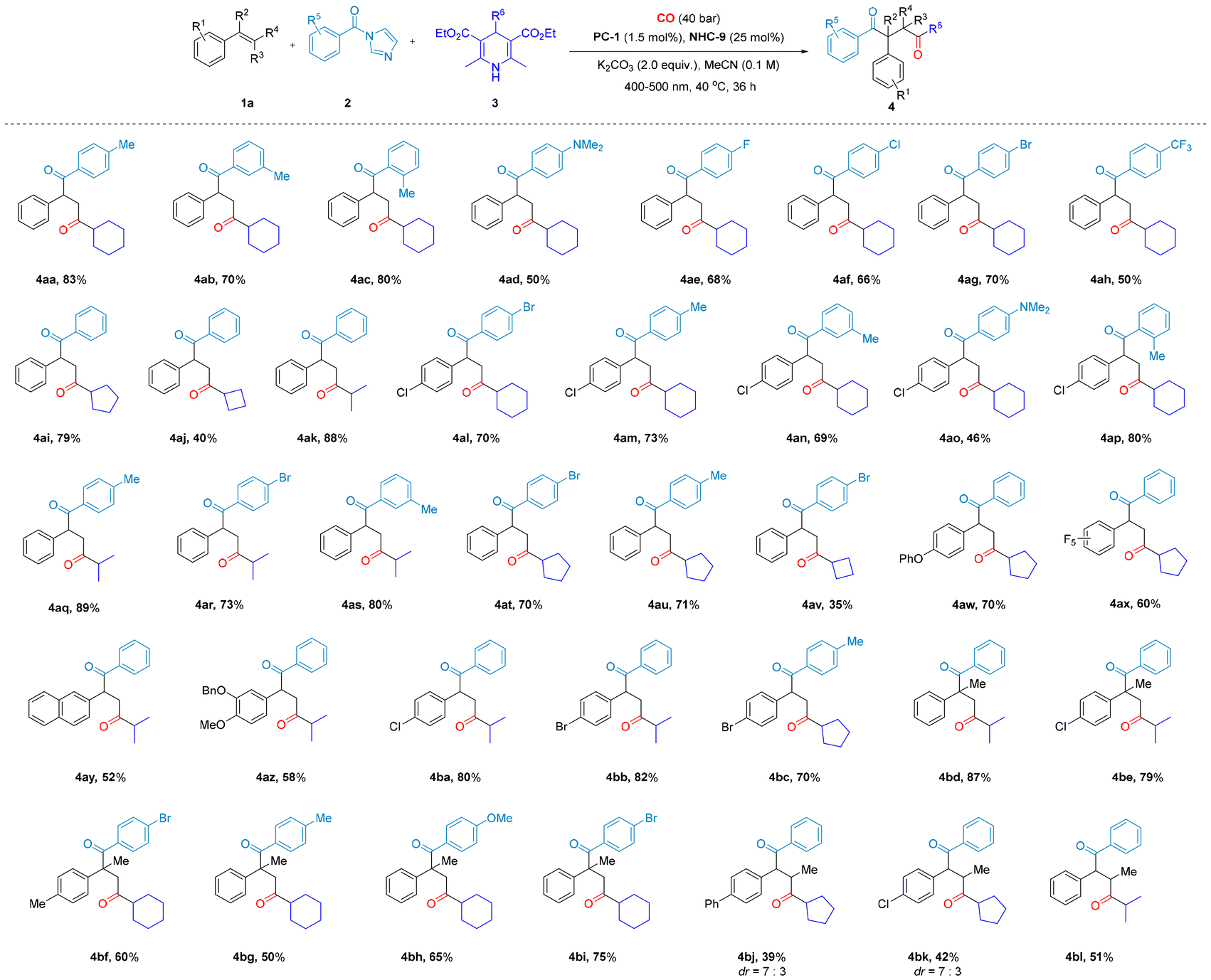 | ||
| Scheme 3 Substrate scope of various styrenes, acyl imidazoles and Hantzsch esters to synthesize 1,4-diketones. Reaction conditions: 1 (0.2 mmol), 2 (0.4 mmol), 3 (0.5 mmol), MeCN (2.0 ml), K2CO3 (0.4 mmol), 400–500 nm, isolated yield. | ||
In addition, α,β-disubstituted styrenes also qualified as substrates that delivered the desired dicarbonyl products 4q–z. β-Substituted styrenes, such as 1-propenylbenzene (1s), 1,2-diphenylethene (1t), 1-chloro-4-(prop-1-en-1-yl)benzene (1x), and 2-(prop-1-en-1-yl)naphthalene (1z), delivered the corresponding dicarbonyl products 4s, 4t, 4x, and 4z in yields of 46%, 50%, 43%, and 40%, respectively, with diastereomeric ratios (dr) ranging from 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 to 9:1. α-Disubstituted styrenes, such as 4-isopropenyltoluene (1q), 1,1-diphenylethene (1r), 1-chloro-4-(prop-1-en-2-yl)benzene (1u), 1-methyl-4-(prop-1-en-2-yl)benzene (1v) and 1-methyl-4-(prop-1-en-2-yl)benzene (1w), delivered the targeted dicarbonyl products 4q, 4r, 4u, 4v and 4w in 85%, 60%, 72%, 70% and 51% yields, respectively.
:4 to 9:1. α-Disubstituted styrenes, such as 4-isopropenyltoluene (1q), 1,1-diphenylethene (1r), 1-chloro-4-(prop-1-en-2-yl)benzene (1u), 1-methyl-4-(prop-1-en-2-yl)benzene (1v) and 1-methyl-4-(prop-1-en-2-yl)benzene (1w), delivered the targeted dicarbonyl products 4q, 4r, 4u, 4v and 4w in 85%, 60%, 72%, 70% and 51% yields, respectively.
Remarkably, α-substituted styrenes exhibit excellent performance in our catalytic system, both in terms of product conversion and complete regioselectivity. This outcome is particularly surprising, as in traditional free radical reactions, α-substituted styrenes—especially phenyl-substituted variants—tend to effectively capture free radicals due to their electron-donating properties, conjugation effects, and stabilization of free radical intermediates.16 As a result, they typically serve as efficient radical scavengers, terminating the propagation of the radical chain in the SET process. However, in our catalytic system, instead of inhibiting the reaction, the α-substituted styrene facilitates the process and enables the successful regioselective formation of the target dicarbonyl compound, which is particularly valuable for applications requiring precise structural control. Furthermore, the substrate scopes of acyl imidazoles and Hantzsch esters were also examined. When acyl imidazoles functionalized with various substituents on the benzene ring were investigated, the influence of the electronic properties of the aromatic ring became evident. Substituents with electron-donating and electron-withdrawing characteristics were found to significantly impact the reaction efficiency. Among these, electron-donating substituents enhance the reaction outcome. For instance, employing methyl-substituted benzene ring series acyl imidazoles as the substrate resulted in the formation of 1,4-diketone products 4aa–4ac, achieving moderate to excellent yields (70–83%). In contrast, the reaction involving acyl imidazoles with electron-withdrawing substituents on the benzene ring yielded lower quantities of 4ae–4ah (50–70%). The reaction efficiency was observed to decrease in the order of 4-position, 2-position, and 3-position methyl substitution, further supporting the notion that stronger electron-donating effects enhance the reactivity of acyl imidazoles. Then, different substituted Hantzsch esters were also successfully employed for this conversion into an array of 1,4-diones (4aa, 4ai–4ak). In light of the overall results, the reactions involving a range of substituted styrenes, acyl imidazoles, and Hantzsch ester substrates afforded product yields spanning from moderate to excellent. These results underscore the broad substrate scope and robust catalytic performance of the novel carbonylation system we have developed.
To gain more insight into the mechanism of this reaction, several mechanistic experiments were conducted (Scheme 4). Firstly, we used the highly reactive free radical scavenger 2,2,6,6-tetramethyl-1-piperidinoxy (TEMPO). Under standard reaction conditions, no product 4a was obtained and product cyclohexyl-TEMPO 5 as the main product was detected by GC-MS, which validated the presence of the cyclohexyl radical group. Control experiments are essential to verify the necessity of reaction parameters, and several control experiments were performed (Scheme 4, eqn (b) and (e)). When the reactions were performed in the absence of light, base, CO, photocatalyst or NHC-catalyst, the desired product was not found. These results suggested that light, base, CO, photocatalyst and NHC-catalyst are necessary to induce the production of the 1,4-diketone compounds. Given the potential for NHC-catalyst deactivation at high CO concentrations, we systematically evaluated the effect of CO pressure on the catalytic performance under the optimized conditions. Notably, a CO pressure of 40 bar proved optimal for our system, while increasing the CO pressure to 60 bar led to a marked decrease in product yield (Scheme 4, eqn (f)). While investigating the influence of the NHC catalyst loading and the optimal substrate stoichiometry on our catalytic system, we conducted a series of control-variable experiments. Based on the experimental feedback, we observed that the NHC catalyst demonstrated moderate catalytic activity at 10 mol%, and the highest yield was achieved with 25 mol% NHC-catalyst. However, increasing the NHC-catalyst loading beyond this point led to a gradual decrease in product yield (Scheme 4, eqn (h)). This trend can be attributed to the excess NHC catalyst, which may reduce the overall catalytic activity of the dual-catalytic system, thereby hindering the reaction progress.17 Furthermore, the optimal substrate ratio of 2:1:2.5 (1a/2a/3a) resulted in the most efficient reaction (Scheme 4, eqn (g)). This composition appears to represent an ideal balance in the concentrations of NHC catalyst and the substrates, thereby promoting the reaction kinetics and driving the system toward the desired transformation.17 These results not only corroborate our previous hypothesis but also highlight the significant influence of NHC design and catalytic system optimization on the efficiency of the carbonylation process.
 | ||
| Scheme 4 Radical capture experiments, exploration of influencing factors of catalytic system and synthetic transformations of 1,4-diones. (e) 1a/2a/3a = 2:1:2:5, MeCN (0.1 M), K2CO3 (2 eq.), PC-1 (1.5 mol%), NHC-9 (25 mol%), 40 °C, 400–500 nm, determined using GC. (f) 1a/2a/3a = 1:1:1.5, MeCN (0.1 M), K2CO3 (2 eq.), PC-1 (1.5 mol%), NHC-9 (25 mol%), r.t., 24 h, 400–500 nm, determined using GC. (g) CO (40 bar), MeCN (0.1 M), K2CO3 (2 eq.), PC-1 (1.5 mol%), NHC-9 (25 mol%), 40 °C, 24 h, 400–500 nm, determined using GC. (h) 1a/2a/3a = 1:1:1.5, MeCN (0.1 M), CO (40 bar), MeCN (1.0 mL), K2CO3 (2 eq.), PC-1 (2 mol%), r.t., 24 h, 400–500 nm, determined using GC. | ||
To highlight the simplicity and efficiency of the method, the standard procedure for synthesizing compound 4a was executed beginning with benzoic acid as the starting material (Scheme 4, eqn (c)). In situ generation of benzoyl imidazole via carbonyldiimidazole (CDI) was confirmed by gas chromatography-mass spectrometry (GC-MS). Subsequent reaction under the standard conditions afforded compound 4a in 75% yield (compared to 82% when starting from benzoyl imidazole). This result further highlights the practicality of our NHC/photocatalytic carbonylation system in facilitating the transformation of inexpensive and readily available carboxylic acid compounds. Furthermore, the application and subsequent transformation of the dicarbonylation product (4a) is also a key focus of our research (Scheme 4, eqn (d)). The condensation of 4a, facilitated by 4-methylbenzenesulfonic acid (PTSA) in ethanol at 60 °C, resulted in the formation of 2,3,5-trisubstituted furan 6a with an 88% yield. Reactions of 4a with aniline in water at 100 °C, as well as with aniline in toluene containing PTSA at 110 °C, both yielded 1,2,3,5-tetrasubstituted pyrrole 6b, in 85% and 87% yields, respectively. Notably, the 1,4-diketone intermediate we synthesized also contributed to the formation of the 1,4-diol skeleton. Specifically, 4a was reduced to the 1,4-diol 6c in 90% yield using sodium borohydride in methanol at room temperature. Treatment of 4a with hydrazine hydrate at room temperature afforded 3,5,6-trisubstituted pyridazine 6d in 83% yield. Finally, the reactions of 4a with ammonium acetate or ammonium formate in acetic acid at 100 °C led to the formation of 2,3,5-trisubstituted pyrrole 6e in yields of 94% and 90%, respectively. These results not only validate the potential applications of 1,4-diketone-based skeleton compounds, but also offer novel insights into the efficient conversion of carbon monoxide under mild conditions, further underscoring the efficacy and value of our NHC/photocatalytic carbonylation system.
Based on our mechanistic experiments for this reaction and previous reports,9,12,13 a possible mechanism for this dual N-heterocyclic carbene-/photoredox-catalyzed carbonylation was proposed (Scheme 5). Initially, under light irradiation, the photosensitizer is excited, leading to the oxidation of Hantzsch esters (3a) by the photoactivated species, generating intermediate A. This process results in the formation of a cyclohexyl radical and the release of diethyl 2,6-dimethylpyridine-3,5-dicarboxylate after further deprotonation. The cyclohexyl radical subsequently captures CO to form the acyl radical B, which undergoes radical addition to styrene (1a), yielding intermediate C. Simultaneously, NHC-9 is activated by a base to generate the active carbene intermediate H. This intermediate H then catalyzes the conversion of acyl imidazole 2a (derived from the transformation of benzoic acid D) to intermediate acyl azolium salts E. Subsequently, with the assistance of photocatalytic single-electron transfer, the reactive intermediate F can be generated. Finally, intermediates F and C couple to form intermediate G, which subsequently undergoes further transformation to afford the target compound 4a while releasing the free carbene catalyst.
 | ||
| Scheme 5 Plausible reaction mechanism for the dual NHC-/photoredox-catalyzed carbonylation reaction. | ||
Conclusions
In summary, we developed a novel dual catalytic strategy combining NHC catalysis with photocatalysis for the carbonylation of alkenes, enabling the synthesis of valuable 1,4-dicarbonyl compounds. In this reaction, three new C–C bonds and two carbonyl groups were constructed in one reaction. Moreover, the synthetic practicality of the protocol was demonstrated through the efficient construction of significant heterocycles (furan, pyrrole, and pyridazine) as well as 1,4-diol derivatives. This strategy provides new insights into the design of NHC-/photoredox-catalysis-driven multi-catalytic systems for CO utilization under economical and sustainable conditions, thereby fostering advancements in the field of small molecule synthesis combined with photocatalytic gas capture.Author contributions
X.-F. W. conceived and directed the project and revised the manuscript. M.-L. Y. performed all the experiments and prepared the manuscript and the ESI.†Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We appreciate the financial support provided by National Key R&D Program of China (2023YFA1507500), the Chinese Scholarship Council. We also thank the analytical department of Leibniz-Institute for Catalysis for their excellent analytical service here. We appreciate the general support from Professor Armin Börner in Leibniz-Institute for Catalysis.References
- (a) W. Cruickshank, J. Nat. Philos., Chem., Arts, 1801, 5, 201–211 Search PubMed; (b) X.-F. Wu, H. Neumann and M. Beller, Chem. Soc. Rev., 2011, 40, 4986–5009 Search PubMed; (c) Y.-H. Li, Y.-Y. Hu and X.-F. Wu, Chem. Soc. Rev., 2018, 47, 172–194 Search PubMed; (d) J.-B. Peng, H.-Q. Geng and X.-F. Wu, Chem, 2019, 5, 526–552 CrossRef CAS; (e) J.-B. Peng, F.-P. Wu and X.-F. Wu, Chem. Rev., 2019, 119, 2090–2127 Search PubMed; (f) C. Zhu, J. Liu, M.-B. Li and J.-E. Bäckvall, Chem. Soc. Rev., 2020, 49, 341–353 Search PubMed; (g) M. Yang, Y. Liu, X. Qi, Y. Zhao and X. Wu, Green Synth. Catal., 2024, 5, 211–269 Search PubMed.
- (a) L. Mond, C. Langer and F. Quincke, J. Chem. Soc., Trans., 1890, 57, 749–753 RSC; (b) L. Gattermann and J. A. Koch, Ber. Dtsch. Chem. Ges., 1897, 30, 1622–1624 CrossRef CAS; (c) O. Roelen, German Pat., DE 849.548, filed 1938 and granted 1952 Search PubMed; (d) D. S. Noyce, J. Tsuji, D. R. Kearns, A. E. Goetz, J. J. Mond, L. S. Hegedus and D. W. Brown, J. Am. Chem. Soc., 1969, 91, 5090–5092 CrossRef; (e) A. Schoenberg, I. Bartoletti and R. F. Heck, J. Org. Chem., 1974, 39, 3318–3326 CrossRef CAS; (f) F.-P. Wu, Y. Yuan, C. Schünemann, P. C. J. Kamer and X.-F. Wu, Angew. Chem., Int. Ed., 2020, 59, 10451–10455 CrossRef CAS PubMed; (g) Y. Yuan, F.-P. Wu, J.-X. Xu and X.-F. Wu, Angew. Chem., Int. Ed., 2020, 59, 17055–17061 CrossRef CAS PubMed.
- (a) G. D. Frey, V. Lavallo, B. Donnadieu, W. W. Schoeller and G. Bertrand, Science, 2007, 316, 439–441 CrossRef CAS PubMed; (b) V. Lavallo, Y. Canac, C. Präsang, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2005, 44, 5705–5709 CrossRef CAS PubMed; (c) D. Martin, M. Soleilhavoup and G. Bertrand, Chem. Sci., 2011, 2, 389–399 RSC; (d) Nobel Prize in Chemistry 2021, https://www.nobelprize.org/prizes/chemistry/2021/summary/.
- (a) G. D. Frey, J. D. Masuda, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 9444–9447 CrossRef CAS PubMed; (b) A. V. Zhukhovitskiy, M. G. Mavros, K. T. Queeney, T. Wu, T. Van Voorhis and J. A. Johnson, J. Am. Chem. Soc., 2016, 138, 8639–8652 CrossRef CAS PubMed.
- Z. R. Turner, Chem. – Eur. J., 2016, 22, 11461–11468 CrossRef CAS PubMed.
- E. Pietrasiak and E. Lee, Synlett, 2020, 1349–1360 CAS.
- J. P. Moerdyk, G. A. Blake, D. T. Chase and C. W. Bielawski, J. Am. Chem. Soc., 2013, 135, 18798–18801 CrossRef CAS PubMed.
- (a) D. Wei, Y. Zhu, C. Zhang, D. Sun, W. Zhang and M. Tang, J. Mol. Catal. A: Chem., 2011, 334, 108–115 CrossRef CAS; (b) L. Yang, F. Wang, R. Lee, Y. Lv, K.-W. Huang and G. Zhong, Org. Lett., 2014, 16, 3872–3875 CrossRef CAS PubMed; (c) A. Lee, A. Younai, C. K. Price, J. Izquierdo, R. K. Mishra and K. A. Scheidt, J. Am. Chem. Soc., 2014, 136, 10589–10592 CrossRef CAS PubMed; (d) K. Zhao and D. Enders, Angew. Chem., Int. Ed., 2017, 56, 3754–3756 CrossRef CAS PubMed; (e) H. Ohmiya, ACS Catal., 2020, 10, 6862–6869 CrossRef CAS.
- (a) J. Guin, S. De Sarkar, S. Grimme and A. Studer, Angew. Chem., Int. Ed., 2008, 47, 8727–8730 CrossRef CAS PubMed; (b) S. De Sarkar, S. Grimme and A. Studer, J. Am. Chem. Soc., 2010, 132, 1190–1191 CrossRef CAS PubMed; (c) Q. Y. Meng, N. Döben and A. Studer, Angew. Chem., Int. Ed., 2020, 59, 19956–19960 CrossRef CAS PubMed; (d) X. Y. Chen, K. Q. Chen, D. Q. Sun and S. Ye, Chem. Sci., 2017, 8, 1936–1941 Search PubMed; (e) Y. Zhang, Y. Du, Z. Huang, J. Xu, X. Wu, Y. Wang, M. Wang, S. Yang, R. D. Webster and Y. R. Chi, J. Am. Chem. Soc., 2015, 137, 2416–2419 CrossRef CAS PubMed; (f) S.-C. Ren, W.-X. Lv, X. Yang, J.-L. Yan, J. Xu, F.-X. Wang, L. Hao, H. Chai, Z. Jin and Y. R. Chi, ACS Catal., 2021, 11, 2925–2934 Search PubMed; (g) N. A. White and T. Rovis, J. Am. Chem. Soc., 2014, 136, 14674–14677 CrossRef CAS PubMed; (h) T. Ishii, K. Nagao and H. Ohmiya, Chem. Sci., 2020, 11, 5630–5636 RSC; (i) A. V. Bay, K. P. Fitzpatrick, R. C. Betori and K. A. Scheidt, Angew. Chem., Int. Ed., 2020, 59, 9143–9148 CrossRef CAS PubMed; (j) S. Byun, M. U. Hwang, H. R. Wise, A. V. Bay, P. H. Y. Cheong and K. A. Scheidt, Angew. Chem., Int. Ed., 2023, 62, e202312829 CrossRef CAS PubMed; (k) M. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485 CrossRef CAS PubMed.
- (a) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC; (b) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed; (c) D. M. Schultz and T. P. Yoon, Science, 2014, 243, 1239176 Search PubMed; (d) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed; (e) K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035–10074 Search PubMed; (f) L. Zhang and E. Meggers, Acc. Chem. Res., 2017, 50, 320–330 CrossRef CAS PubMed; (g) J. Twilton, C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. Macmillan, Nat. Rev. Chem., 2017, 1, 0052 CrossRef CAS.
- (a) D. Ravelli, S. Protti and M. Fagnoni, Chem. Rev., 2016, 116, 9850–9913 CrossRef CAS PubMed; (b) Y. Sumida and H. Ohmiya, Chem. Soc. Rev., 2021, 50, 6320–6332 RSC.
- (a) A. V. Bay, K. P. Fitzpatrick, G. A. González-Montiel, A. O. Farah, P. H. Y. Cheong and K. A. Scheidt, Angew. Chem., Int. Ed., 2021, 60, 17925–17931 CrossRef CAS PubMed; (b) Y. Sato, Y. Goto, K. Nakamura, Y. Miyamoto, Y. Sumida and H. Ohmiya, ACS Catal., 2021, 11, 12886–12892 CrossRef CAS; (c) J. Reimler, X. Y. Yu, N. Spreckelmeyer, C. G. Daniliuc and A. Studer, Angew. Chem., Int. Ed., 2023, 62, e202303222 CrossRef CAS PubMed; (d) K. Liu and A. Studer, J. Am. Chem. Soc., 2021, 143, 4903–4909 CrossRef CAS PubMed.
- (a) L. Marzo, Eur. J. Org. Chem., 2021, 4603–4610 Search PubMed; (b) Q.-Z. Li, R. Zeng, B. Han and J.-L. Li, Chem. – Eur. J., 2021, 27, 3238–3250 CrossRef CAS PubMed; (c) J. Liu, X. Xing, J. Huang, L. Lu and W. Xiao, Chem. Sci., 2020, 11, 10605 RSC; (d) L. Dai and S. Ye, Chin. Chem. Lett., 2021, 32, 660–667 CrossRef CAS.
- (a) Y. Wang, H. Yang, Y. Zheng, M. Hu, J. Zhu, Z.-P. Bao, Y. Zhao and X.-F. Wu, Nat. Catal., 2024, 7, 1065–1075 CrossRef CAS; (b) H. Yang, Y. Wang, L.-C. Wang and X.-F. Wu, Chem. Sci., 2024, 15, 14304–14309 RSC; (c) J. Zhang, L.-C. Wang, Y. Wang and X.-F. Wu, Green Chem., 2024, 26, 11686–11694 RSC.
- (a) Modern Heterocyclic Chemistry, ed. J. Alvarez-Builla, J. J. Vaquero and J. Barluenga, Wiley-VCH, 2011 Search PubMed; (b) H. Renata, Q. Zhou, G. Dünstl, J. Felding, R. R. Merchant, C.-H. Yeh and P. S. Baran, J. Am. Chem. Soc., 2015, 137, 1330–1340 CrossRef CAS PubMed; (c) A. Deepthi, B. P. Babu and A. L. Balachandran, Org. Prep. Proced. Int., 2019, 51, 409–442 CrossRef CAS; (d) M. K. Hunjan, S. Panday, A. Gupta, J. Bhaumik, P. Das and J. K. Laha, Chem. Rec., 2021, 21, 715–780 CrossRef CAS PubMed.
- (a) F. Zhao, H.-J. Ai and X.-F. Wu, Angew. Chem., Int. Ed., 2022, 61, e202200062 CrossRef CAS PubMed; (b) M. Yang, Y. Liu, P. Yang, Y. Zhao and X.-F. Wu, Sci. China: Chem., 2025, 68 DOI:10.1007/s11426-024-2382-y.
- (a) M. Fèvre, J. Pinaud, Y. Gnanou, J. Vignolle and D. Taton, Chem. Soc. Rev., 2013, 42, 2142–2172 RSC; (b) J. M. Jeffrey and J. W. Bode, Acc. Chem. Res., 2014, 47, 696–707 CrossRef PubMed; (c) I. C. Gerber and P. Serp, Chem. Rev., 2020, 120, 1250–1349 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: General comments, general procedure, analytic data, and NMR spectra. See DOI: https://doi.org/10.1039/d5gc00609k |
| This journal is © The Royal Society of Chemistry 2025 |