
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA H2-free heterogeneous route to glycerol-based acrylics via Re-based deoxydehydration†
Maja
Gabrič‡
 ab,
Florian M.
Harth‡
a,
Brigita
Hočevar
a,
Sašo
Gyergyek
c,
Blaž
Likozar
a and
Miha
Grilc
*ab
ab,
Florian M.
Harth‡
a,
Brigita
Hočevar
a,
Sašo
Gyergyek
c,
Blaž
Likozar
a and
Miha
Grilc
*ab
aDepartment of Catalysis and Chemical Reaction Engineering, National Institute of Chemistry, Hajdrihova 19, 1000 Ljubljana, Slovenia. E-mail: miha.grilc@ki.si
bUniversity of Nova Gorica, Vipavska Cesta 13, 5000 Nova Gorica, Slovenia
cDepartment for Materials Synthesis, Jožef Stefan Institute, Jamova 39, 1000 Ljubljana, Slovenia
First published on 20th March 2025
Abstract
A novel catalytic route from bio-based glyceric acid to acrylic acid and its esters, key polymer precursors, has been developed. This sustainable process involves rhenium-catalyzed deoxydehydration (DODH) in an alcoholic medium, avoiding gaseous hydrogen or other hazardous reagents. Using a supported Re/C catalyst and methanol as solvent, a combined yield of acrylic acid and methyl acrylate of up to 65% was obtained at 150 °C after 72 h under an inert N2 atmosphere. Moreover, conducting the same reaction under 5 bar of H2 at an increased temperature of 180 °C boosts the DODH rate constant from 0.21 ± 0.03 to 4.08 ± 0.22 h−1 gcat−1, thus yielding more than 90% of DODH products in just 10 h. This sustainable process is easily transferable to other alcohols that yield alkyl acrylates, further expanding the applicability of the presented invention to other monomers and products.
Green foundation1. We report on an environmentally friendly, sustainable and novel heterogeneous catalytic process that converts glyceric acid to acrylic acid, a precursor for many polymers. Not only is this the first reported route to produce bio-based acrylic acid from glyceric acid, a glycerol derivative, but this deoxydehydration reaction does not require a hazardous hydrogen source or other external reducing agents.2. We achieved over 90% of deoxydehydrated products of sustainable bio-based acrylic acid and its ester under H2 pressure. Notably, the alcohol is used as both the solvent and reducing agent for deoxydehydration, resulting in a yield of over 60% of acrylates and their derivatives at relatively low temperatures and without the need for gaseous hydrogen. 3. Future research should focus on the development of catalysts with enhanced deoxydehydration activity. Furthermore, the reusability of catalysts must be improved for sustainable processes. |
Acrylic acid (AA) and acrylate esters are highly sought-after chemical building blocks in the polymer industry. To reduce the dependency on fossil resources, bio-based production methods are being explored to replace the traditional propylene oxidation pathway.1 Alternatively, much cheaper glycerol can be used as a (C3) feedstock for catalytic conversion into AA via various routes as reported in previous reviews2–6 and detailed studies.7–13Fig. 1 summarizes that these processes generally require two steps: (i) selective oxidation of one terminal hydroxyl group to a carboxylic acid and (ii) dehydration of adjacent hydroxyl group(s) to a vinyl moiety. Subsequent esterification yields the corresponding acrylates. The first pathway involves acid-catalyzed5,14–16 or Fe-catalyzed gas phase dehydration,17–19 while the second pathway features deoxydehydration (DODH) to allyl alcohol.20–23 Both intermediates are further oxidized to AA.22,24 Catalytic conversion of glycerol into lactic acid was also reported,25 along with further acid-catalyzed dehydration using zeolites or solid acids to AA.26,27 Another well-explored oxidation pathway of bio-based glycerol leads to glyceric acid (GA),28–30 which is the starting point for our process. This communication is the first demonstration of a successful heterogeneously catalyzed conversion of GA into valuable acrylic acid or its esters, as no such studies have been reported in the literature so far. To the best of our knowledge, the only remotely similar approach has been described by Boucher-Jacobs and Nicholas.31 Although olefins were targeted from GA, DODH products were also obtained but with low selectivity due to dominant side reactions. However, the DODH reaction was only possible with the use of toxic indoline as a reducing agent and by relying on a homogeneous catalyst.31 While the use of a solid catalyst facilitates product separation, catalyst recycling and continuous operation (fixed bed), the elimination of reducing agents such as external H2 or indoline offers additional safety and environmental and techno-economic advantages. A patent application for DODH of GA, including the use of a solid catalyst and the elimination of reducing agents, is pending.32 These aspects are also a considerable advantage compared to the approaches reported by Yang et al., relying on (over-)stoichiometric use of HI and H2 to initially form 3-iodopropionic acid and subsequent hydrolysis to AA.33
 | ||
| Fig. 1 Overview of catalytic routes from glycerol to acrylic acid and acrylate esters. | ||
The DODH reaction has previously been studied on various vicinal groups of polycarboxylic acids, with either a homogeneous catalyst or a heterogeneous rhenium catalyst.34–38 This preliminary study explores DODH on glyceric acid, screening a range of molecular and supported Re catalysts in a total of 26 different experiments. Re/C was identified as the most promising catalyst, consistent with our previous DODH studies on C6 systems.39,40 The reaction kinetics were studied experimentally and in silico at various temperatures under a hydrogen or an inert atmosphere. Finally, the role of different alcohols was systematically investigated, to gain insight into their intricate role as solvents, reducing agents and carboxyl moiety protecting agents. All the experimental procedures are described in the ESI.†
Rhenium catalyst screening
The catalyst screening test for glyceric acid DODH in MeOH was performed at 150 °C for 72 h under an inert atmosphere with eight different homogeneous and supported Re-based materials, as presented in Fig. 2 (detailed data in Table S1†). The heterogeneous catalyst underwent reductive pre-treatment in a tubular furnace at 400 °C (3 h, 200 mL min−1 flow of pure H2). Among the three homogeneous catalysts, Re2O7 and (NH)4ReO4 were able to convert GA into MA at 150 °C, but with low yields of 8.5 and 13.4% after 72 h, respectively. The general suitability of high-oxidation-state Re compounds has been demonstrated for several DODH reactions of similar compounds.41–44 Differences between the Re salts are suggested to be a result of solubility differences.45,46 In general, experiments also show that methanol is suitable as a reducing agent for the DODH reaction of GA, while esterification of GA, AA and PA to the respective methyl esters occurs in parallel. Compared to homogeneous catalysts as well as other supported Re catalysts, Re/C showed superior activity, yielding 60.6% DODH products after 72 h, primarily MA (46.6%), but also free AA (10.0%) and saturated MP (4.4%). The repeatability of these results was confirmed, and the discrepancy margin was estimated to be within 10% (Table S2†). Over Re/SiO2, Re/Al2O3, and Re/H-ZSM-5, only methyl glycerate (MG) and hardly any DODH products were obtained, while Re/TiO2 showed marginal DODH activity. In contrast, Re/C again exhibited well balanced DODH, hydrogenation, esterification and MeOH dehydrogenation activity, which is crucial for this rather complex cascade catalytic system.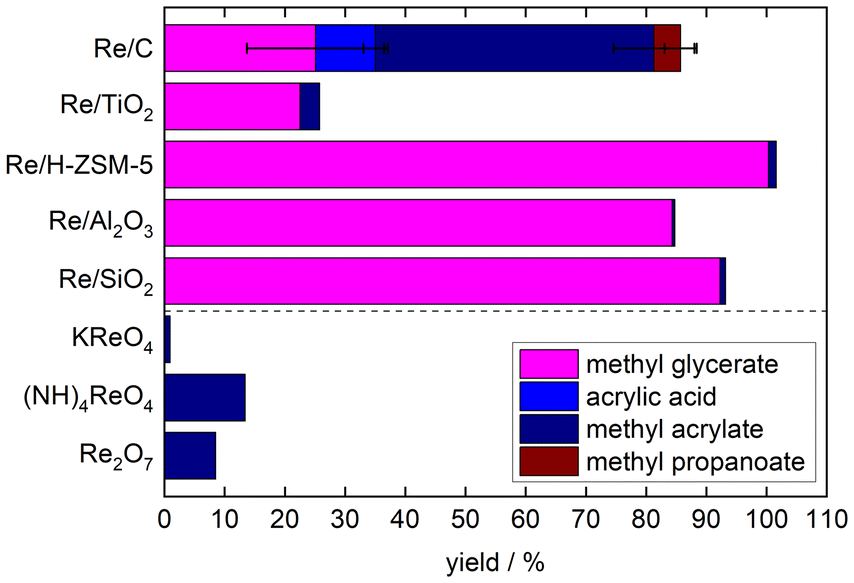 | ||
Fig. 2 Yields of DODH products after 72 h of glyceric acid over supported and unsupported Re catalysts in methanol. Reaction conditions: V(MeOH) = 45 mL, n(glyceric acid) = 0.001 mol, n(Re) = 0.04 mmol, molar ratio (glyceric acid![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Re) = 25:1, T = 150 °C, and 5 barg N2. :Re) = 25:1, T = 150 °C, and 5 barg N2. | ||
To investigate the reason for this exceptional performance, particularly the outstanding DODH activity, the solid catalysts were thoroughly characterized by H2-TPR, TEM, N2-physisorption, and XPS.40 The characterization data of the different supported Re catalysts are summarized in Tables S8 and S9.† While most catalyst properties do not seem to directly affect catalytic activity, high Re dispersion and suitable Re oxidation states (Re4+ and Re6+) and metallic Re appear to be essential for DODH activity. Thus, the observed effect of the support material in the catalytic reaction most importantly manifests itself in the form of Re–support interactions that can stabilize the required ensemble of Re oxidation states. Carbonaceous materials have proven to be suitable supports for aldaric acid DODH,39,40 as both oxidized and metallic Re species are simultaneously stabilized by carbon and residual oxygen containing functionalities in the matrix (XPS results in Table S9†). TEM observations show that Re nanoparticles are uniformly dispersed on the carbon support (Fig. 3), and this correlates with our previous findings that additional Re or Re-oxide species are likely atomically dispersed on the carbon surface.40 TEM was used to observe the Re nanoparticles deposited on support materials (Fig. 3). For the Re/C catalyst, the agglomerated Re nanoparticles are clearly visible as a much darker area on a lightly grey background (Fig. 3). The specimen support is made of a C film and therefore the C support (marked with a black arrow) appears only slightly darker than the background. The SiO2 particles are shapeless and appear darker than the background, while Re nanoparticles are well visible as dark particles. TiO2 nanoparticles are crystalline, and the contrast varies depending on their relative orientation to the electron beam and local thickness. Small, a few nm in diameter, Re nanoparticles are still relatively well seen (marked with white arrows). Al2O3 is in the form of platelet nanoparticles of approx. a few 10 nm in size. Depending on the orientation, they appear as platelets or needles. Much smaller and isotropic dark particles (Re) are well visible (marked with white arrows). H-ZSM-5 appears rather like SiO2 with clearly visible Re nanoparticles and their agglomerates. The results of the characterization have shown that the oxidation states of the active phase, rhenium, and its dispersion on the support are decisive for the occurrence of the DODH reaction.40 However, a more comprehensive investigation is required including different carbon supports to gain further insight into the role of, e.g., the surface functionality as well as the metal–support interactions with Re. Such studies exceed the scope of this short communication.
 | ||
| Fig. 3 TEM images of the supported Re catalysts used in the DODH of GA. | ||
Screening of reaction conditions
Based on the promising results with Re/C, the catalyst was further tested at various temperatures under a H2 or N2 atmosphere (Fig. 5 and Table S3†). These results were also subjected to regression analysis in accordance with the kinetic model formulated based on the reaction pathway network (Fig. 4). The kinetic rate constants of the model are shown in Tables S10a and b.† Under an inert N2 atmosphere, DODH of GA already takes place at temperatures as low as 120 °C. The DODH reaction rate (constant) to acrylic acid (k2 = 0.012 h−1 gcat−1) is approximately 3 times lower compared to esterification to MA, while the subsequent DODH reaction rate of MA is even an order of magnitude lower. The yields of DODH products are only around 10% after 72 h, while the selectivity is high, due to a negligible hydrogenation reaction rate (<0.001 h−1 g cat−1) at 120 °C. A temperature increase results in the following yield of DODH products within 10 h: 22.4% (150 °C), 46.9% (165 °C) and 65.1% (180 °C). At temperatures above 150 °C, the saturation of the vinyl group (especially r6) becomes quite noticeable after long reaction times, so reaction time or selectivity should be compromised if acrylic functionality needs to be retained. Furthermore, long reaction times at elevated temperatures >150 °C are detrimental, since they result in lower product yields due to undesired side reactions such as the addition of methanol to the double bond of MA to form 3-methoxy methyl propanoate (Fig. S5†). However, considering the superior market value of the unsaturated products AA and MA, the optimal temperature for the N2 atmosphere can be considered around 150 °C, where the yield of the unsaturated products was near 50% after 72 h. Nevertheless, further research into the catalyst material (including synthesis and pre-treatment protocols), solvent type and process conditions and a holistic kinetic study have the potential to further optimize the outcome of the process. Under a H2 atmosphere, a two-fold effect can be seen from Fig. 5 and the kinetic parameters in Table S10b.† On the one hand, the presence of the reducing agent H2 considerably enhances the DODH reaction, which can be clearly observed after 10 hours of reaction at each tested temperature (Table S3†). This is likely due to the high availability of hydrogen species on the catalyst surface compared to the inert conditions, where solely in situ formed H2 can be used from the hydrogen-donor solvent. On the other hand, and in agreement with these studies,36,37,47–51 H2 also boosts the (undesired) hydrogenation of the double bond of AA and MA. Thus, providing H2 as an additional reducing agent generally boosts DODH, especially for MG (k2 is 3 to 4 times higher at all temperatures under H2 compared to N2), but it also tends to over-hydrogenate products to MP and PA. For that reason, the temperature, H2 pressure and reaction time need to be precisely optimized if AA or MA is the targeted product or should be considered for yielding PA or MP. Nevertheless, it is worth highlighting that at 180 °C in the presence of H2, a total yield of >90% DODH products was obtained in just 10 h, with around 20% of these products having hydrogenated double bonds. | ||
| Fig. 4 Proposed reaction pathway network of GA by esterification, DODH and hydrogenation reactions. | ||
 | ||
| Fig. 5 Yields of DODH products after 10 and 72 h of glyceric acid DODH over Re/C in methanol at different reaction temperatures and under either an inert N2 gas (A) or a reducing H2 gas atmosphere at 5 barg (B). Reaction conditions: V(MeOH) = 45 mL, n(glyceric acid) = 0.001 mol, n(Re) = 0.04 mmol, molar ratio (glyceric acid:Re) = 25:1, and T = 150 °C. The striped bars on the right represent modelled values. | ||
Solvent screening
The alcoholic solvent, which until now has always been MeOH, evidently plays a crucial role in the process overall and in the catalytic cycle in particular. Besides being a solvent, it also acts as the reducing agent for the DODH reaction, and thus, the reaction is possible without the addition of molecular H2 (or other organic reducing agents). The role of the hydrogen-donor solvent is evident from the detection of its dehydrogenated product, methylal, as well as the formed H2 during the reaction under an inert atmosphere. The precise amounts of H2 formed and methylal formed during a typical experiment are detailed in the ESI (Fig. S5 and Tables S5 and S7†). Moreover, protection of the free carboxylic group (GA, PA and MP) with an alcohol solvent is desired to prevent intramolecular esterification or even spontaneous polymerization. Esters are less reactive than the free carboxylic acid group, resulting in a lower tendency for the molecule to undergo uncontrolled (exothermic) polymerization, not only under reaction conditions, but also under typical storage and handling conditions, further contributing to the stability and extended shelf life of the product.52 The results in Fig. 6 and kinetic parameters in Tables S10a and b† indicate that esterification reactions proceed sufficiently fast in relation to the DODH reaction under the tested operation window; however, the addition of an acidic esterification catalyst such as the commercial acidic resin Amberlyst can be beneficial as seen in Table S1.† Acrylic acid can be esterified with alcohols such as methanol, ethanol, isopropanol, n-propanol, n-butanol and n-pentanol to produce acrylate esters with special properties that can be used in UV-resistant paints, flexible elastomers, fast-curing coatings and specialty adhesives.53–55 However, the multifunctional role of alcohol in the reaction can influence the process in other ways. Therefore, the suitability of other short-chain alcohols was investigated (Fig. 6 and Table S4†). Among the linear primary alcohols, the total yield of DODH products decreases from ca. 60% (methanol and ethanol) to ca. 33% (n-pentanol and n-butanol), indicating that with increasing chain length, they become less suitable as reducing agents. Interestingly, despite the similar total yield, the yield of esterified products is higher over methanol than over ethanol, indicating that esterification is also more facile with a shorter chain length. It should be noted, however, that for n-butanol and n-pentanol, acrylic acid and propanoic acid could not be quantified by GCMS due to overlap with the alcohol or the corresponding oxidation product. The secondary alcohol isopropanol behaves significantly differently from primary alcohols. It is a far stronger reducing agent42 and thus significantly more DODH products are formed (77.7%) as well as more hydrogenated products (60.1% combined yield). At the same time, less than 1/8 of the products were esterified, showing that esterification with the secondary alcohol is expectedly less pronounced than with any of the primary alcohols. Despite the high DODH activity, the use of isopropanol is detrimental to the selectivity for the primary target product MA, as it promotes strong hydrogenation reactions similar to those observed in experiments using H2 as an additional, more potent reducing agent. From that viewpoint, MeOH is the best-suited alcohol for this reaction. | ||
| Fig. 6 Yields of DODH products after 72 h of glyceric acid over Re/C in different solvents. Reaction conditions: V(solvent) = 45 mL, n(glyceric acid) = 0.001 mol, n(Re) = 0.04 mmol, molar ratio (glyceric acid:Re) = 25:1, T = 150 °C, and 5 barg N2. aEsters with the respective alcohol that was used as solvent, e.g., ethyl esters in the case of ethanol. | ||
Conclusions
Overall, the results presented in this innovative article show that the Re-catalyzed deoxydehydration (DODH) reaction is a promising pathway for converting (bio)glycerol-derived glyceric acid into acrylates, presenting a viable and sustainable alternative to previously reported methods. First and foremost, the DODH of glyceric acid was successfully implemented as a heterogeneously catalyzed process using different supported Re catalysts. Re/C showed the highest DODH activity, even exceeding typical homogeneous catalysts. The DODH reaction was conducted in alcohols, which also serve as reducing agents, making the process feasible without the addition of hazardous reducing agents like H2. Moreover, glyceric acid and the DODH reaction product acrylic acid readily form esters with the alcohol, which leads to alkyl acrylates being the primary product of the reaction. Varying the alcohol also allows for the formation of a range of different alkyl acrylates. The highest yields of the target product methyl acrylate (>45%) were obtained over Re/C at 150 °C in methanol after 72 h.In addition to enhancing the efficiency and sustainability of acrylate production, our approach opens new avenues for exploration. Future efforts should concentrate on developing catalysts with enhanced DODH activity and diversified product selectivity, possibly through metal doping, to optimize and broaden the applications of this transformative technology. Furthermore, optimizing the catalyst for stability and reusability could address concerns such as catalyst leaching, further advancing our bio-based process. To deepen our understanding of acrylate formation mechanisms and refine product control, employing a microkinetic model will provide comprehensive insights and enhance the practical application of our findings.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the Slovenian Research and Innovation Agency (research projects N2-0242 and J1-3020 and research core funding P2-0152). Nives Kokol, Urška Kavčič, Simon Starašinič and Vili Resnik are acknowledged for their excellent support in the lab. Janvit Teržan is acknowledged for conducting the XPS analysis.References
- R. Beerthuis, G. Rothenberg and N. R. Shiju, Green Chem., 2015, 17, 1341–1361 RSC.
- R. K. Grasselli and F. Trifirò, Rend. Lincei, 2017, 28, 59–67 CrossRef.
- K. Avasthi, A. Bohre, M. Grilc, B. Likozar and B. Saha, Catal. Sci. Technol., 2020, 10, 5411–5437 RSC.
- D. Sun, Y. Yamada, S. Sato and W. Ueda, Green Chem., 2017, 19, 3186–3213 RSC.
- B. Katryniok, S. Paul, V. Bellière-Baca, P. Rey and F. Dumeignil, Green Chem., 2010, 12, 2079–2098 RSC.
- S. T. Wu, Q. M. She, R. Tesser, M. Di Serio and C. H. Zhou, Catal. Sci. Eng., 2020, 62, 481–523 CrossRef CAS.
- N. Razali and A. Z. Abdullah, Appl. Catal., A, 2017, 543, 234–246 CrossRef CAS.
- K. Wang, Z. Yang, Y. Ma, W. Zhao, J. Sun, T. Lu and H. He, Biofuels, Bioprod. Biorefin., 2022, 16, 1428–1454 CrossRef CAS.
- P. Mäki-Arvela, I. L. Simakova, T. Salmi and D. Yu. Murzin, Chem. Rev., 2014, 114, 1909–1971 Search PubMed.
- A. Villa, N. Dimitratos, C. E. Chan-Thaw, C. Hammond, L. Prati and G. J. Hutchings, Acc. Chem. Res., 2015, 48, 1403–1412 CrossRef CAS PubMed.
- K. S. Vargas, J. Zaffran, M. Araque, M. Sadakane and B. Katryniok, Mol. Catal., 2023, 535, 112856 Search PubMed.
- Y. Kon, M. Araque, T. Nakashima, S. Paul, F. Dumeignil and B. Katryniok, ChemistrySelect, 2017, 2, 9864–9868 Search PubMed.
- Y. Kon, B. Katryniok, F. Dumeignil, M. Araque-Marin and S. Paul, United States Patent, US10500567, 2019 Search PubMed.
- A. Abdullah, A. Z. Abdullah, M. Ahmed, J. Khan, M. Shahadat, K. Umar and M. A. Alim, J. Cleaner Prod., 2022, 341, 130876 Search PubMed.
- L. Liu, X. P. Ye and J. J. Bozell, ChemSusChem, 2012, 5, 1162–1180 Search PubMed.
- A. Galadima and O. Muraza, J. Taiwan Inst. Chem. Eng., 2016, 67, 29–44 Search PubMed.
- A. Kostyniuk, D. Bajec, P. Djinović and B. Likozar, Chem. Eng. J., 2020, 397, 125430 CrossRef CAS.
- G. Sánchez, B. Z. Dlugogorski, E. M. Kennedy and M. Stockenhuber, Appl. Catal., A, 2016, 509, 130–142 CrossRef.
- G. Sánchez, J. Friggieri, C. Keast, M. Drewery, B. Z. Dlugogorski, E. Kennedy and M. Stockenhuber, Appl. Catal., B, 2014, 152–153, 117–128 Search PubMed.
- F. C. Jentoft, Catal. Sci. Technol., 2022, 12, 6308–6358 RSC.
- J. R. Dethlefsen and P. Fristrup, ChemSusChem, 2015, 8, 767–775 Search PubMed.
- S. Yang, M. Kim, S. Yang, D. S. Kim, W. J. Lee and H. Lee, Catal. Sci. Technol., 2016, 6, 3616–3622 RSC.
- M. Kim and H. Lee, ACS Sustainable Chem. Eng., 2017, 5, 11371–11376 CrossRef CAS.
- X. Li and Y. Zhang, ACS Catal., 2016, 6, 143–150 CrossRef CAS.
- N. Razali and A. Z. Abdullah, Appl. Catal., A, 2017, 543, 234–246 CrossRef CAS.
- E. Blanco, S. Loridant and C. Pinel, in Reaction Pathways and Mechanisms in Thermocatalytic Biomass Conversion II, ed. M. Schlaf and Z. C. Zhang, Springer, Singapore, 2016, pp. 39–62 Search PubMed.
- L. Huang, M. H. Wai and S. Kawi, React. Chem. Eng., 2023, 8, 502–537 RSC.
- L. Wen, X. Zhang and F. F. Abdi, Mater. Today Energy, 2024, 44, 101648 CrossRef CAS.
- K. Wang, Z. Yang, Y. Ma, W. Zhao, J. Sun, T. Lu and H. He, Biofuels, Bioprod. Biorefin., 2022, 16, 1428–1454 CrossRef CAS.
- L. Fan, B. Liu, X. Liu, N. Senthilkumar, G. Wang and Z. Wen, Energy Technol., 2021, 9, 2000804 CrossRef CAS.
- C. Boucher-Jacobs and K. M. Nicholas, Organometallics, 2015, 34, 1985–1990 CrossRef CAS.
- F. M. Harth, B. Likozar and M. Grilc, European Patent Office, PCT/EP2023/070258, 2023 Search PubMed.
- W. Yang, L. Teng and L. Shengqin, China National Intellectual Property Administration, CN108997108A, 2018 Search PubMed.
- B. E. Sharkey and F. C. Jentoft, ACS Catal., 2019, 9, 11317–11328 CAS.
- B. E. Sharkey, A. L. Denning, F. C. Jentoft, R. Gangadhara, T. V. Gopaladasu and K. M. Nicholas, Catal. Today, 2018, 310, 86–93 CAS.
- S. Tazawa, N. Ota, M. Tamura, Y. Nakagawa, K. Okumura and K. Tomishige, ACS Catal., 2016, 6, 6393–6397 CAS.
- K. Yamaguchi, J. Cao, M. Betchaku, Y. Nakagawa, M. Tamura, A. Nakayama, M. Yabushita and K. Tomishige, ChemSusChem, 2022, 15, e202102663 CAS.
- J. Gan, Y. Nakagawa, M. Yabushita and K. Tomishige, Green Chem., 2024, 26, 8267–8281 RSC.
- B. Hočevar, A. Prašnikar, M. Huš, M. Grilc and B. Likozar, Angew. Chem., Int. Ed., 2021, 60, 1244–1253 Search PubMed.
- F. M. Harth, M. Gabrič, J. Teržan, B. Hočevar, S. Gyergyek, B. Likozar and M. Grilc, Catal. Today, 2024, 441, 114879 CAS.
- J. R. Dethlefsen and P. Fristrup, ChemSusChem, 2015, 8, 767–775 CrossRef CAS PubMed.
- S. Raju, M.-E. Moret and R. J. M. Klein Gebbink, ACS Catal., 2015, 5, 281–300 CrossRef CAS.
- L. J. Donnelly, S. P. Thomas and J. B. Love, Chem.– Asian J., 2019, 14, 3782–3790 CrossRef CAS PubMed.
- M. Shiramizu and F. D. Toste, Angew. Chem., Int. Ed., 2012, 51, 8082–8086 CrossRef CAS PubMed.
- S. Orda, M. Drzazga, K. Leszczyńska-Sejda, M. Ciszewski, A. Kocur, P. Branecka, K. Gall, M. Słaboń and M. Lemanowicz, Materials, 2023, 16(15), 5481 CrossRef CAS PubMed.
- J. M. Casas, E. Sepúlveda, L. Bravo and L. Cifuentes, Hydrometallurgy, 2012, 113–114, 192–194 Search PubMed.
- K. Tomishige, Y. Nakagawa and M. Tamura, Chin. Chem. Lett., 2020, 31, 1071–1077 Search PubMed.
- N. Ota, M. Tamura, Y. Nakagawa, K. Okumura and K. Tomishige, ACS Catal., 2016, 6, 3213–3226 CrossRef CAS.
- L. Denning, H. Dang, Z. Liu, K. M. Nicholas and F. C. Jentoft, ChemCatChem, 2013, 5, 3567–3570 CrossRef.
- Y. Nakagawa, S. Tazawa, T. Wang, M. Tamura, N. Hiyoshi, K. Okumura and K. Tomishige, ACS Catal., 2018, 8, 584–595 CrossRef CAS.
- K. Yamaguchi, Y. Nakagawa, C. Li, M. Yabushita and K. Tomishige, ACS Catal., 2022, 12, 12582–12595 CrossRef CAS.
- W. Bauer, in Encyclopedia of Chemical Technology, John Wiley & Sons, 2003 Search PubMed.
- S. Karakuş, E. Sert, A. D. Buluklu and F. S. Atalay, Ind. Eng. Chem. Res., 2014, 53, 4192–4198 CrossRef.
- G. Jyoti, A. Keshav, J. Anandkumar and S. Bhoi, Int. J. Chem. Kinet., 2018, 50, 370–380 Search PubMed.
- E. Sert, A. D. Buluklu, S. Karakuş and F. S. Atalay, Chem. Eng. Process., 2013, 73, 23–28 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5gc00445d |
| ‡ Both authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |