
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
In situ amino–lignin production via biomass fractionation for high-efficacy CO2 capture†
Ruijie
Wu
 a,
Caiyun
Liu
b,
Yongchao
Zhang
*b,
Jiayun
Xu
a,
Andrey
Pranovich
a,
Jarl
Hemming
a,
Teija
Tirri
a,
Xiaoju
Wang
ac and
Chunlin
Xu
*a
a,
Caiyun
Liu
b,
Yongchao
Zhang
*b,
Jiayun
Xu
a,
Andrey
Pranovich
a,
Jarl
Hemming
a,
Teija
Tirri
a,
Xiaoju
Wang
ac and
Chunlin
Xu
*a
aLaboratory of Natural Materials Technology, Åbo Akademi University, Henrikinkatu 2, Turku FI-20500, Finland. E-mail: Chunlin.Xu@abo.fi
bState Key Laboratory of Green Papermaking and Resource Recycling, Qilu University of Technology, Shandong Academy of Sciences, Jinan 250353, Shandong, China. E-mail: yczhang@qlu.edu.cn
cPharmaceutical Sciences Laboratory, Faculty of Science and Engineering, Åbo Akademi University, Tykistökatu 6A, Turku FI-20520, Finland
First published on 8th April 2025
Abstract
Amine-functionalized solid adsorbents (NFSAs) from green and renewable biomasses have been proven to be a significant breakthrough for utilizing renewable resources in the carbon capture and storage field. Lignin, a major biomass component, is an auspicious biomass-based adsorbent material owing to its rich chemical structure, abundant functional groups, and high stability. Inevitably, the carbon–carbon condensation during technical lignin isolation leads to heterogeneity, which restricts the lignin amination modification in NFSAs. Herein, we present a technology breakthrough in the direct production of aminated lignins with high N-density via the reactive fractionation of biomasses using an aniline–formic acid solvent system. This innovative strategy enabled in situ amination of lignins in high weight percent yields during the acid delignification of different biomasses and holistically produced high-purity cellulose (glucose yield above 95%). Here, aniline acted as a nucleophile to capture Cα carbocations in the lignin structure and selectively directed the condensation towards dominant α-amination, effectively inhibiting the uncontrolled lignin condensation to retain the chemical reactivity of lignin macromolecules. The nanomaterials further derived from these aminated lignin macromolecules offered promising perspectives as bio-based adsorbents for efficient CO2 capture. Nanoparticles of amino–lignin in the size range of 339 ± 72 nm derived from the bamboo biomass demonstrated an exceptional adsorption capacity of 324 mg CO2 per gram of adsorbent in a wet capture method. Comparison with existing industrial processes has demonstrated that our approach is green and sustainable, with potential commercial value for subsequent industrial applications and large-scale utilization. This proof-of-concept method highlights a significant advancement in carbon capture technologies, utilizing sustainable and biodegradable nanomaterials to address the pressing environmental challenges.
Green foundation1. Our novel strategy facilitated the simultaneous production of high-N-density aminated lignins and high-purity cellulose (glucose yield above 95%) via in situ reactive fractionation of biomasses using a recyclable organosolv system. Compared with the traditional multi-step methods (chemical treatments and delignification), our method reduced the separation and purification steps of lignins, significantly reducing energy consumption and production cost.2. This method directly produced high-N-density aminated lignin (i.e., by introducing 1 formanilide per 1.2 native lignin subunits). The further processed nanomaterials from aminated lignins demonstrated an exceptional adsorption capacity of 324 mg CO2 per gram of lignin in a wet capture method. 3. For future studies, physical methods such as ultrasonication-aided processing can be considered to reduce the use of solvents in the fabrication of nanoparticles, thereby improving the ‘greenness’ and promoting the application of these renewable adsorbers for large-scale utilization. |
Introduction
Carbon capture and storage (CCS) is key to achieving net zero emissions and ceasing human-driven climate change.1,2 New approaches for CCS are prominently innovated with a focus on developing efficient scrubber systems that directly bind CO2 from the air or flue gases and offer cost-effective operability of the absorption process.3,4 The current use of aqueous amine solutions (e.g. for capturing post-combustion CO2) is a benchmark for CCS in industries.5,6 The main drawbacks of these liquid solvents include toxicity, secondary pollution, and equipment corrosion.2 In contrast, the implementation of solid adsorbents (e.g., activated carbons, zeolites, and metal–organic frameworks) minimizes the production of secondary wastes; however, the cost and efficiency of carbon capture remain primary concerns when using them.2,7 Thus, bio-based adsorbents have gained more momentum owing to the merits of renewable and sustainable resources, reducing the dependence on petrochemicals for adsorbent development.8,9 In this field, new developments largely focus on biochars and hydrochars that are synthesized via the pyrolysis of various biomass waste materials, which need to be further activated physically or chemically for high CO2 capture efficiency. Fewer efforts have focused on chemically modifying the biomass for the production of biopolymeric adsorbents that can effectively capture CO2 while reducing the overall cost of adsorbent synthesis and making the operation of CO2 capture competitive for industrial scalability. More recently, amine-functionalized solid adsorbents (NFSAs) have become a promising avenue for CO2 capture by functionalizing a mass of amine groups onto solid supports as binding sites.7 NFSAs could achieve fast adsorption, high capacity, low desorption temperature, and enhanced moisture resistance in terms of CO2 adsorption. Lignocelluloses preserved in natural biomasses serve as valuable polymeric feedstocks and are geographically abundant. When considered as the solid support in an NFSA, lignin presents intrinsic advantages owing to the hydrophobicity of polyphenolic macromolecules and their insensitivity to moisture compared with polysaccharides such as cellulose and hemicellulose.9,10 Developing a holistic and green method to directly produce lignin-based NFSAs from biomasses would mark a significant breakthrough in the utilization of renewable resources as the method could produce amine-functionalized lignins for effective use in CO2 capture while reducing the overall cost of adsorbent synthesis and making CO2 capture operation competitive for industrial scale applications.8Native lignins are biosynthesized as amorphous and crosslinked polyphenols within the plant cell wall of biomasses formed by three phenylpropanoid derivatives (p-coumaryl, coniferyl, and sinapyl alcohols) via enzymatic dehydrogenation polymerization. Compared with native lignins, technical lignins are largely obtained as a byproduct from pulping and biorefinery industries, such as kraft, sulfite, and alkaline pulping as well as pilot-scale organosolv fractionation processes.11 Depending on the severity of the fractionation condition, benzylic functionalization (e.g., thiolation, sulfonylation, and etherification) readily occurs, and uncontrolled lignin condensation is inevitable in these processes, which significantly alters the lignin structure to yield highly recalcitrant materials.12,13 Thus, recalcitrant carbon–carbon linkage is formed in the macromolecular structure, and it suppresses the chemical reactivity (specifically the number of reactive phenolic hydroxyl groups). This increases the structure heterogeneity (high dispersity of Mw in the resulting lignin) and consequently compromises the downstream utilization of technical lignins.14,15 Complex chemical modification techniques are often employed to obtain lignin products with specific functionalities, thereby enhancing the application value of technical lignins. For example, one effective amination route for technical lignins deploys the Mannich addition between amines (including diethylamine, ethylenediamine, long-chain alkylamine, and ethanolamine) and formaldehyde to produce amides, which then react further with carbon with high electron density in lignins to yield aminated lignin polymers (Fig. 1a).16–18 However, the severe condensation of technical lignins leads to low modification efficiency.19,20
 | ||
| Fig. 1 Representative amination modification pathways. (a) Mannich amination of technical lignin (already existing method) and (b) in situ amination of lignin during acid delignification of biomass (this work). | ||
The emergence of ‘lignin-first’ biorefinery concepts in recent years reflects a growing consensus that how to prevent the condensation reactions needs to be prioritized within the lignin valorization paradigm in the holistic valorization of lignocellulosic biomasses.14,15,21,22 The formation of Cα carbocations by the dehydration of Cα–OH in the lignin structure is endorsed as a critical reaction intermediate when forming recalcitrant carbon–carbon linkage.12,14 Consequently, effective strategies have emphasized inhibiting the condensation either by (1) chemical functionalization to the benzylic alcohols through etherification, oxidation, and acetalization of the hydroxyl group at the Cα position to prevent the formation of Cα carbocation, or by (2) adding nucleophiles to capture carbocation for preventing the condensation.14,15 For instance, in a mild organosolv system of H2O/HCl/alcohols (secondary ethanol and tertiary n-pentanol), alcohol incorporation at the benzylic α-position of β-aryl ether (β-O-4) enabled extracting high-Mw lignin with increased solubility while retaining the β-O-4 linkage.23 Similarly, in the acid-based organosolv-fraction system (HCl/1,4-dioxane), the addition of aldehydes (formaldehyde, acetaldehyde, and propionaldehyde) has been demonstrated effective in protecting Cα- and Cγ–OH by adding aldehydes to prevent carbocation formation, thereby stabilizing the lignin with high retention of β-O-4 linkage.15 Meanwhile, Chen et al. found that selectively carboxylating the Cγ–OH position with citric acid and etherifying the Cα–OH position with butanol during acid fractionation can reduce the spatial constraints of lignin and stabilize its active intermediates, thereby facilitating lignin extraction and achieving a high yield of light-colored lignin.24 A seminal work recently reported by N. Li et al. demonstrated the effectiveness of deploying phenolic monomers such as syringol and phenol in a formic acid system to selectively inhibit uncontrolled condensation.14 This state-of-the-art research shows that the phenolic monomers with high nucleophilicity could trap the reactive benzylic carbocation, selectively directing lignin condensation towards α-arylation rather than aryl–ether cleavage and g-arylation. As nesting independently in our laboratory research in parallel, the formic acid-based fractionation system has been progressively explored as a ‘lignin-first’ biorefinery approach aimed at holistic and green valorization of lignocellulosic biomasses.25 To create bio-based adsorbents for CO2 capture, we also identified reactive fractionation with the formic-acid system as a viable pathway for directly producing amine-functionalized lignin from biomasses. Our innovation lies in leveraging aniline as the explicit amination reagent within the formic acid system, which captures carbonation during fractionation to minimize lignin condensation (Fig. 1b). Importantly, this reactive fractionation introduces amino groups into the lignin structure in situ, enabling the direct production of highly reactive aminated lignin from various botanic sources. To integrate amino–lignin materials into an effective and operational CO2 adsorption process, our endeavors have further developed nanoscale or micron-scale solid particles (LMNPs) of the aminated lignin, achieving a stable colloidal suspension. We successfully illustrated a correlation between the physical attributes of the LMNPs, such as particle size, and their CO2 adsorption efficacy. This proof-of-concept data represent a significant advancement for CCS technologies enabled by sustainable and biodegradable nanomaterials.
Experimental
Materials and methods
Three different biomass resources (poplar, pine, and bamboo) were selected as starting materials. Formic acid (≥98%), aniline (≥99.5%), and tetrahydrofuran (THF, HPLC grade, ≥99.8%) were purchased from Sigma-Aldrich and used directly without any purification.In situ reactive fractionation of biomass
The fractionation process was performed in a 250 mL hydrothermal reactor with a tetrafluoroethylene lining using 85% formic acid/aniline as a reaction reagent. The chips were fully mixed with the reaction reagent at a solid-to-liquid ratio of 1/10 and reacted in an oil bath (140 °C) for 2 h. The volume ratio of formic acid and aniline is 90/10. After the reaction, the slurry was filtered and fully washed with hot formic acid (70 °C, 4 times) until the filtrate was colorless. The filtrate was collected and evaporated at 40 °C to recycle the formic acid. The consented liquid was dropped into ether (10 times ether per 1 volume of liquid). The residue was collected by filtration, dried, and named aminated lignin. The ether was evaporated, and the residual formanilide was collected. The characterization methods of aminated lignin are shown in the ESI.†Formation of the amino-LMNPs
LMNPs were formed by solvent–antisolvent methods. THF was used as a solvent to dissolve aminated lignin at different initial concentrations. Then the antisolvent was added by different methods. The different methods were named accordingly, and more details are shown in Fig. 4a. More details can be found in the ESI.† To provide additional CO2 binding sites, acid hydrolysis (HCl solution, 1 M) was employed to convert the amide groups to amino groups. More details can be found in the ESI.†CO2 adsorption and regeneration performance
The amino-LMNP colloidal suspension was used to adsorb CO2 at room temperature, and the schematic diagram of the experimental setup is shown in Fig. S16.† The CO2 gas was purged into LMNP dispersion at 1 bar and 100 mL min−1 for 30 min. After the adsorption, the CO2 adsorption capacity was calculated by a gravimetric method. Before testing, the amino-LMNP colloidal suspension was degassed by an ultrasonic bath. The effect of the resources, formation methods, and the LMNP concentrations were detected. The adsorbed LMNPs were desorbed by using an HCl solution (1 M) at a concentration of 10 mg mL−1. The regenerated LMNP colloidal suspension was performed by testing the cycling capacity for 5 cycles.Characterization
The chemical components of lignocellulosic biomass and lignin mainly include extractives, cellulose, hemicelluloses, and lignin (Klason lignin and acid-soluble lignin). More analytical details are given in the ESI.† The structural characteristics of aminated lignin were determined by nuclear magnetic resonance (NMR) spectroscopy, e.g.1H, 13C, 1H–13C HSQC and 31P NMR, according to the previous literature.26 The molecular weight of aminated lignin was analyzed using an HPLC system equipped with a differential refractive index (RI) concentration detector and a multi-angle light scattering detector (MALS) in a DMSO/LiBr (0.05 M) eluent. The element content of the aminated lignin was analyzed using an organic elemental analyzer (Thermo Scientific FLASH 2000). The identification of the model compounds after amination was realized by gas chromatography-mass spectrometry (GC-MS) and liquid chromatography-electrospray ionization-mass spectrometry (LC-ESI-MS) combined with NMR. The morphology of amino-LMNPs was analyzed by transmission electron microscopy (TEM, Jeol JEM-1400 plus). For the sample preparation, 100 μL of LMNP suspension was diluted to 1 mL with MilliQ water. Then 5 μL of diluted amino-LMNP suspension (about 0.1 mg mL−1) was dropped onto the carbon film-supporting grid. After deposition for 3 min, the excess water was carefully absorbed by blotting a sharp end of filter paper on the edge of the copper grid. In addition, differential light scattering (DLS, Zetasizer nano series, Malvern) was used to detect the zeta potential of amino-LMNPs.Results and discussion
In situ lignin amination during the formic acid fractionation of different biomasses
The recalcitrant cell wall structure of carbohydrates (cellulose and hemicellulose) and lignin intertwined requires a fractionation process to separate the compounds for full utilization.27 The formic acid delignification process has been proven effective in crumbling lignocellulosic biomass's cell wall resistance.28 During the acid-based delignification process, the protonating agent (H+) can break the ester bond (lignin-carbohydrate complex and β-O-4 linkage in lignin), so that hemicelluloses and lignin are dissolved in the liquid stream. However, under severe conditions, the repolymerization of lignin is inevitable, resulting in a decrease in the reactivity (lower functional groups and higher Mw) and separation efficiency of lignin. Aniline, possessing strong nucleophilicity and active amino group, was used to stabilize benzyl carbocations during formic acid delignification of lignocellulosic biomass, simultaneously achieving the in situ lignin amination, which imparted high reactivity and adsorption properties to lignin (Fig. 2). Under severe conditions (140 °C for 2 h), the aniline–formic acid solvent system can efficiently fractionate all types of wood-based biomass (softwood, hardwood, and bamboo), where lignin and hemicellulosic sugars were dissolved into solvents, and cellulose was kept as residual. Based on the starting materials (biomasses), the weight percent (wt%) contents of lignin in poplar, pine and bamboo were 24.57, 27.60 and 27.22 wt%, respectively. The weight percent yields of the aminated lignin were higher than the origin lignin content, and the nitrogen content in the aminated lignins were higher than 5% (Fig. 2b and c), which indicated the successful in situ amination during the formic acid delignification of biomass. The % yields of the aminated lignin from poplar, pine, and bamboo were 46.77, 39.82, and 46.41 wt%, respectively, while the % yields of FA lignin were 25.09, 16.91, and 26.13 wt%, respectively, representing increases of 86.41, 135.48, and 77.61%, respectively. Calculated against the wt% content of elemental N content in the aminated lignin fractions, formanilide has reacted as about 44–46 wt% in mass content to the aminated lignin, indicative of a high degree of amination to the final lignin product through this approach. The yields and purities of cellulose fractionated by the aniline–formic acid system from different biomass resources were as well higher than that of formic acid, especially the softwood (pine) (Fig. 2d and Table S1†), which indicated that the addition of aniline could promote lignin removal and avoid the degradation of cellulose. During the acid-catalyzed degradation of lignocellulosic biomass, the destruction of the hydrogen-bonding network in microfibrils would expose the OH groups, thereby accepting hydrogen bonds with amino groups to form a stable cellulose-formanilide complex, thus reducing the degradation of cellulose.29 The high purities of the cellulose fractions enabled high enzymatic hydrolysis efficiency (Fig. S2†), consistent with previous research results.30,31 During the formic acid fractionation, the hemicelluloses will degrade into oligosaccharides, monosaccharides, and degradation products (furfural and hydroxymethylfurfural (HMF)), and dissolve into the liquid phase of hydrolysate (Table S2†). After fractionation with the aniline–formic acid system, the hydrolyzed hemicellulosic sugar products were found in much lower concentrations, compared with the only formic acid fractionation system, especially for poplar and bamboo. Moreover, neither furfural nor HMF was detected as the degradation product after formic acid-aniline fractionation. The disappearance of hemicellulose sugars and their degradation products can be attributed to the reaction between formanilide and hemicellulose degradation products during the aniline–formic acid fractionation process.29,32 This reaction led to the formation of a lignin-like product, referred to as pseudo-lignin. This phenomenon explained the higher acid-soluble lignin content observed in the obtained aminated lignin. Furthermore, the Py-GC/MS results of the aminated lignin provide additional confirmation of these results (Fig. S3†). As the dose of aniline is excessive in the reaction system, the reaction between furfural and formanilide reduced the concentration of furfural as a hemicellulose degradation product, further shifting the reaction equilibrium in the degradation of hemicellulose as furfural products, instead of monosaccharides. A Sankey diagram is presented in Fig. S1† to illustrate the mass flow and solvent recovery scenarios involved in this reaction fractionation process for different types of biomasses. | ||
| Fig. 2 In situ reactive fractionation of biomass produces high-reactivity aminated lignin and high-purity cellulose fraction. (a) In situ reactive fractionation pathway of different biomasses; (b) weight percent yields of lignin and Klason lignin (numerical data presented as navy blue dots) (“N&FA process” denotes aniline addition in the formic acid fractionation; “FA process” denotes the fractionation, the yield was calculated based on the freeze-dry weight of starting materials; the red stars represent the lignin content in the starting materials); (c) nitrogen content in lignin (the content was determined using an elemental analyzer); (d) weight percent yield of cellulose fraction (the yield was calculated based on the freeze-dry weight of starting materials). | ||
In situ lignin amination mechanism
It has been proven that the aniline–formic acid solvent system can efficiently degrade and dissolve the lignin from different biomasses. The lignin dimer model compound (veratrylglycerol β-O-4-guaiacyl ether, VG) was used to reveal the amination mechanism using the formic acid-aniline solvent system (Fig. 3a). In contrast to the traditional lignin amination method by the Mannich reaction, the in situ lignin amination using a formic acid–aniline system arose in the course of acid-catalyzed depolymerization of lignin. The cleavage of α-aryl ether bonds and β-aryl ether linkages in the lignin structure is the primary approach for acid-catalyzed depolymerization of lignin.12,28 The cleavage of α-aryl ether bonds and β-aryl ether linkages not only leads to the lignin depolymerization, but also the lignin condensation under severe acidic conditions.14,22 The dehydration of Cα–OH in the lignin structure will form an important reaction intermediate, Cα carbocation, which has strong electrophilicity and can react with the aromatic rings of other small lignin fragments to form C–C bonds.15,21ortho-Alkylation or condensation reactions may also occur in this process, thus forming cross-linked lignin polymers. Compared with other small lignin fragments, the aromatic ring of formanilide (the reaction product of formic acid and aniline) has stronger activity and stronger competitiveness in capturing carbocations. Mechanistic characterization using VG showed that the three nucleophilic sites of formanilide (ortho and para positions of the amino group) can couple with the active intermediate carbocation, resulting in the aminated lignin trimer model as the main product (products 2 and 3). The products were structurally identified and quantified by GCMS, LCMS, and NMR (Fig. S4–7†). The difference between products 2 and 3 is whether the γ-hydroxyhydroxyl group is formylated in a formic acid solution. At the same time, no obvious release of guaiacol monomer was observed in the GCMS and LCMS chromatography, indicating that formanilide can prevent the cleavage of the β-aryl ether linkages and the resulting aminated lignin trimer model remained stable under severe acidic conditions. New signals were observed from the 2D HSQC NMR spectrum, which confirms this phenomenon. Compared with the 2D HSQC NMR spectrum of VG, new signals appeared in the aromatic region of the aminated lignin trimer model, which belonged to formanilide, indicating that formanilide was successfully introduced into VG and stabilized the aryl ether linkages of VG. Moreover, similar signals to the model compound (α-arylated structure, similar to the structure of the aminated lignin trimer model compound) were observed in the aminated lignin, indicating that the in situ amination using the aniline–formic acid organic solvent system is fully compatible with the formic acid delignification from lignocellulosic biomass. Quantitative analysis by 2D HSQC combined with 13C NMR indicated that the contents of the aminated β-ether structures in the aminated lignin obtained from poplar, pine, and bamboo were 10.25, 9.04, and 7.11/100 Ar, respectively, partly remaining the native lignin structure. In contrast, the signal in the aliphatic region of the linkage in formic acid lignin disappeared completely (Fig. S9†), indicating that all the linkages were destroyed.28 Meanwhile, complex signals were observed in the aromatic region of aminated lignin, while few signals were found for formic acid lignin, which not only indicated the presence of obvious aminated β-ether structures in aminated lignin, but also demonstrated that amination could effectively avoid the condensation of lignin, especially the carbon–carbon condensation between the aromatic ring of lignin and the carbocation. The molecular weight of lignin also proved that the aniline–formic acid solvent system can effectively inhibit the condensation of lignin (Fig. S10†). Quantitative analysis of the hydroxyl groups of lignin by 31P NMR revealed that the content of C5-substituted OH in aminated lignin was lower than that in formic acid lignin, indicating that aminated lignin contained fewer C–C condensed structures (Fig. S11†). Structural identification based on the NMR results confirmed: (1) selective amination substitution occurring at the Cα position, (2) the β-ether linkage preservation, and (3) less lignin condensation.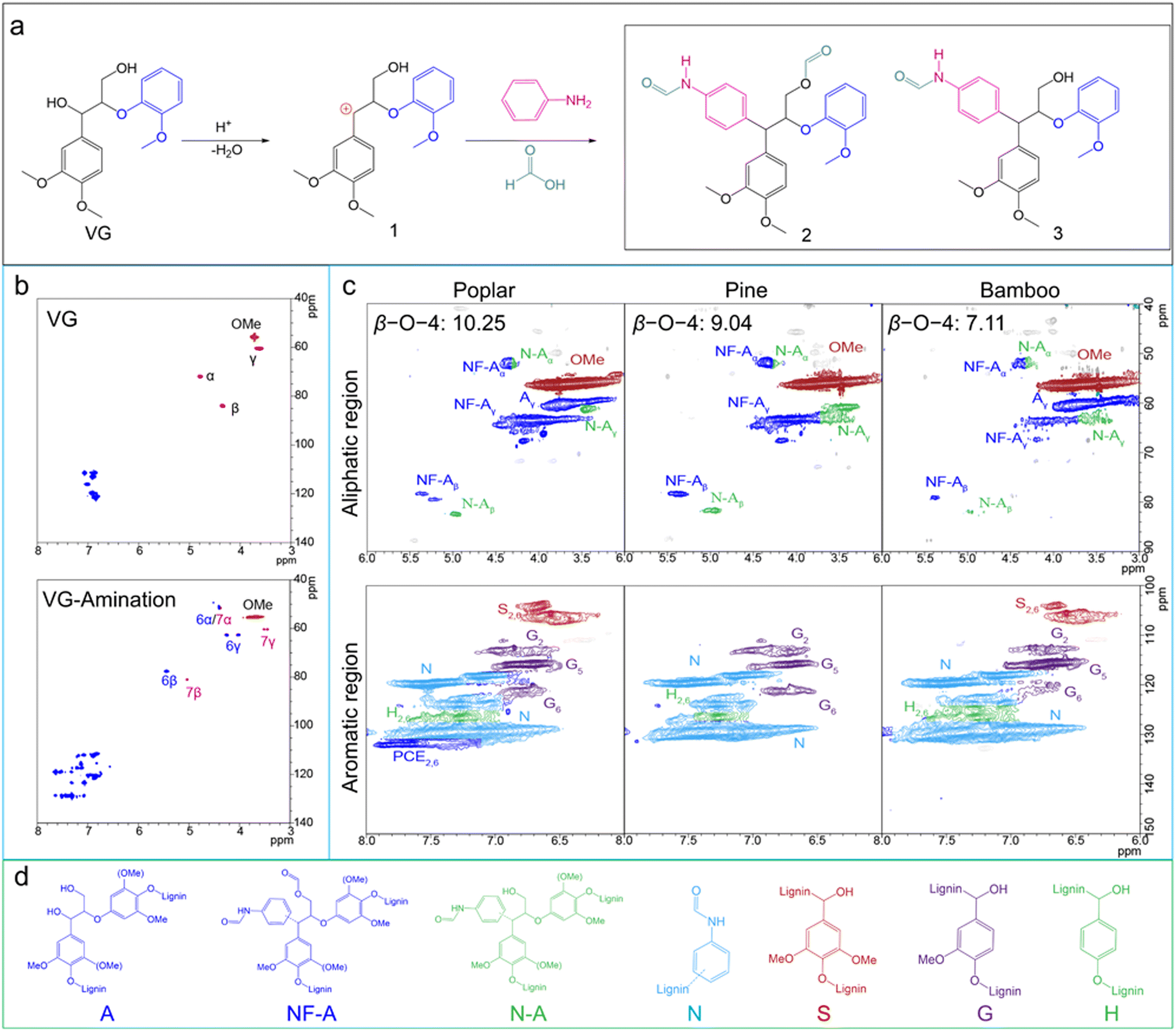 | ||
| Fig. 3 In situ amination and mechanistic and structural characterization of aminated lignin. (a) Amination pathway of veratrylglycerol β-O-4-guaiacyl ether (VG) using an aniline-formic acid solvent system. The intermediate 1 (Cα-Carbocation) was the key for lignin condensation. Two products (product 2 and 3) were detected using GC-MS, LC-MS and NMR. More details can be seen in the ESI;† (b) comparison of whole regions in the 2D HSQC NMR spectrum of VG and mixed products after amination; (c) 2D HSQC NMR spectra of different aminated lignins. All aminated lignins were fully dissolved in dimethylsulfoxide (DMSO)-d6. The spectrum showed an obvious arylation-stabilized β-ether bond signal (aliphatic regions) and formanilide signals (aromatic regions); and (d) main structures in the aminated lignin. | ||
 | ||
| Fig. 4 Formation strategies of amino–lignin micro-nanoparticles (LMNPs) with different particle sizes. (a) Formation of the amino-LMNPs via antisolvent shifting methods. The amino-LMNP sizes were controlled by adjusting the addition methods and types of antisolvents. The HCl solution (1 M) was used to remove the formyl groups; (b) morphologies of amino-LMNPs. TEM images were used to observe the morphology of amino-LMNPs, and the particle sizes were calculated using ImageJ. The zeta potentials of amino-LMNPs were determined using dynamic light scattering. | ||
Application of aminated lignin in CO2 adsorption
To efficiently utilize the aminated lignin as a NFSA, we produced its micro- or nanometer-scale particles. The high surface-area-to-volume ratio of these nanomaterials has beneficial implications in adsorption applications.33,34 Due to the amphiphilic properties of lignin, the lignin micro/nanoparticles (LMNPs) can be readily manufactured with protocols extensively reported elsewhere.10,35 During the self-assembly formation of LMNPs, the hydrophobic part (phenylpropane subunits) in the lignin structure becomes the nucleation of LMNPs, while the hydrophilic part (amino, hydroxyl, carboxyl groups, etc.) turns to its surface.36,37 Importantly, the LMNPs prepared by the aminated lignin are hypothesized to expose more amide groups on the surface for CO2 adsorption.7 The THF solvent was used to dissolve aminated lignin, and the particles with sizes ranging from micrometers to nanometers were successfully prepared by controlling formation methods.34 Meanwhile, as mentioned in the characterization of aminated lignins, the amino group of aniline reacted with formic acid to convert it into amide. To provide more CO2 adsorption sites, acid hydrolysis (HCl solution, 1 M) was used to break –NH–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O linkage on the surface of LMNPs under mild conditions (room temperature), thereby converting amide (Ar–NH–R) into amine (Ar–NH2), which was verified by 1H NMR (Fig. S13†). The particle sizes of amino-LMNPs processed with higher initial concentrations (20 mg mL−1 THF) were larger than those of the amino-LMNPs with lower initial concentrations (10 mg mL−1 THF), which was attributed to the presence of more lignin molecules in the solvent, which can increase the interactions, allowing the particles to adsorb more lignin units and grow.37 This phenomenon existed in all amino-LMNPs, especially the amino-LMNPs from hardwood (poplar) with a particle size of 1129 nm. The sequence order in which the antisolvent (H2O) is mixed with lignin solution (lignin-THF) has a considerable influence on the particle size distribution of amino-LMNPs. As discussed earlier by Almeida et al.,38 the size of lignin particles can be regulated from micrometers (antisolvent to lignin solution) to nanometers (lignin solution to antisolvent) by controlling the order of adding lignin solution and antisolvent. Our results are consistent with their findings and the tuning on mixing order is applicable to size control over different aminated lignins. The formation of the smaller particle size distribution of amino-LMNPs is mainly because when the lignin solution is slowly dropped into excess water and water molecules compete with THF for intermolecular bonds, thereby prompting the lignin macromolecules to quickly nucleate into LNPs.39 However, when water is slowly dropped into the lignin solution, the low anti-solvent content cannot cause the lignin macromolecules to immediately nucleate, precipitate and dissolve in THF. The strong intermolecular bond competition between THF, lignin molecules, and water causes the lignin macromolecules to continuously rearrange in the solution before precipitating to form critical nuclei, which then collide with small molecular lignin and agglomerate, thereby prompting them to grow into LMPs. Meanwhile, compared with neutral water, acidic water (HCl solution, pH 2.5) can yield LNPs with a smaller particle size and their colloidal suspension allows higher light transmittance. The pH-dependent size variation of LNPs is more profound with the ones prepared from aminated lignins from softwood (pine) and bamboo. It is speculated that HCl protonates the hydrophilic groups (secondary amine groups, hydroxyl groups, and carboxyl groups) of lignin, thereby increasing the repulsion between lignin macromolecules, so that lignin macromolecules cannot form a dense core structure.33 In addition to the formation methods, the botanic origin of bioresources also affects the particle size of amino-LMNPs. The particle size of amino-LMNPs from different bioresources decreased following the order of hardwood (poplar) > softwood (pine) > bamboo using the same formation method, which might be attributed to the different lignin properties. It is known that non-covalent forces (such as hydrogen bonds, hydrophobic and π–π interactions) drive the self-assembly process of LMNP formation, and the non-covalent π–π interactions between lignin subunits decrease in the order of syringyl (S) > guaiacyl (G) > p-hydroxyphenyl (H) units.40 Compared with softwood and bamboo, hardwood contained more S units, which led to the larger particle size of LMNPs prepared from hardwood aminated lignin, while bamboo contained a large number of H units, which led to its smaller particle sizes. In summary, amino-LMNPs can be manufactured from micrometer to nanometer scale by regulating the formation conditions (order for the addition of lignin solution and antisolvent, initial lignin concentration, and antisolvent pH).
O linkage on the surface of LMNPs under mild conditions (room temperature), thereby converting amide (Ar–NH–R) into amine (Ar–NH2), which was verified by 1H NMR (Fig. S13†). The particle sizes of amino-LMNPs processed with higher initial concentrations (20 mg mL−1 THF) were larger than those of the amino-LMNPs with lower initial concentrations (10 mg mL−1 THF), which was attributed to the presence of more lignin molecules in the solvent, which can increase the interactions, allowing the particles to adsorb more lignin units and grow.37 This phenomenon existed in all amino-LMNPs, especially the amino-LMNPs from hardwood (poplar) with a particle size of 1129 nm. The sequence order in which the antisolvent (H2O) is mixed with lignin solution (lignin-THF) has a considerable influence on the particle size distribution of amino-LMNPs. As discussed earlier by Almeida et al.,38 the size of lignin particles can be regulated from micrometers (antisolvent to lignin solution) to nanometers (lignin solution to antisolvent) by controlling the order of adding lignin solution and antisolvent. Our results are consistent with their findings and the tuning on mixing order is applicable to size control over different aminated lignins. The formation of the smaller particle size distribution of amino-LMNPs is mainly because when the lignin solution is slowly dropped into excess water and water molecules compete with THF for intermolecular bonds, thereby prompting the lignin macromolecules to quickly nucleate into LNPs.39 However, when water is slowly dropped into the lignin solution, the low anti-solvent content cannot cause the lignin macromolecules to immediately nucleate, precipitate and dissolve in THF. The strong intermolecular bond competition between THF, lignin molecules, and water causes the lignin macromolecules to continuously rearrange in the solution before precipitating to form critical nuclei, which then collide with small molecular lignin and agglomerate, thereby prompting them to grow into LMPs. Meanwhile, compared with neutral water, acidic water (HCl solution, pH 2.5) can yield LNPs with a smaller particle size and their colloidal suspension allows higher light transmittance. The pH-dependent size variation of LNPs is more profound with the ones prepared from aminated lignins from softwood (pine) and bamboo. It is speculated that HCl protonates the hydrophilic groups (secondary amine groups, hydroxyl groups, and carboxyl groups) of lignin, thereby increasing the repulsion between lignin macromolecules, so that lignin macromolecules cannot form a dense core structure.33 In addition to the formation methods, the botanic origin of bioresources also affects the particle size of amino-LMNPs. The particle size of amino-LMNPs from different bioresources decreased following the order of hardwood (poplar) > softwood (pine) > bamboo using the same formation method, which might be attributed to the different lignin properties. It is known that non-covalent forces (such as hydrogen bonds, hydrophobic and π–π interactions) drive the self-assembly process of LMNP formation, and the non-covalent π–π interactions between lignin subunits decrease in the order of syringyl (S) > guaiacyl (G) > p-hydroxyphenyl (H) units.40 Compared with softwood and bamboo, hardwood contained more S units, which led to the larger particle size of LMNPs prepared from hardwood aminated lignin, while bamboo contained a large number of H units, which led to its smaller particle sizes. In summary, amino-LMNPs can be manufactured from micrometer to nanometer scale by regulating the formation conditions (order for the addition of lignin solution and antisolvent, initial lignin concentration, and antisolvent pH).
The colloidal suspension of this series of LMNPs was applied to adsorb CO2 and their adsorption capacities were investigated under the same condition (concentration of 2 mg LMNPs per mL, CO2 flow rate of 100 mL min−1, 25 °C) (Fig. 5). The CO2 adsorption capacities in the series of LMNPs increased following the order of hardwood (poplar) < softwood (pine) < bamboo and the adsorption capacity of the LMNPs dramatically increased with the decrease in particle sizes. This is ascribed to LMNPs with smaller particle sizes, which can release more amino groups on the surface of LMNPs (higher zeta potential), thereby providing more CO2 adsorption sites. Compared with hardwood (poplar) and softwood (pine), the CO2 adsorption capacity of LMNPs prepared from bamboo was higher, and the maximum CO2 adsorption capacity can reach 386.51 mg g−1 LMNPs. However, the particle size of this specific LMNP (produced by method 4) is too small to be recovered by centrifugation or filtration. Considering the large-scale application, the bamboo-LMNPs prepared by method 3 are more suitable with a high CO2 adsorption capacity (324.12 mg g−1 LMNPs) and are easy to recover by centrifugation at 5040 g. Therefore, this specific type of amino-LMNPs were used for subsequent study. The CO2 capture capacity of amino-LMNPs initially increased and then decreased with the rise in the initial concentration of LMNPs, reaching the highest CO2 adsorption at 2 mg mL−1 (Table S7†). This indicated that an excessive LMNP content led to aggregation among the LMNPs, which, in turn, reduced the interaction between amino groups and CO2. Meanwhile, bamboo-LMNPs with varied surface chemistry exhibited varying CO2 adsorption capacities, with the CO2 adsorption performance following the order: amino-LMNPs > aminated LMNPs > FA LMNPs. To further test the stability of LMNPs, the adsorbed LMNPs were desorbed using a 1 M hydrochloric acid solution and then re-adsorbed. LMNPs demonstrated excellent regenerative performance, maintaining a high CO2 capture capacity even after five cycles. The CO2 adsorption capacity of freeze-dried LMNPs was examined by TGA (Table S1†0). No obvious difference was observed among different LMNPs, which may be due to the aggregation or collapse of lignin nanoparticles during the drying process. Based on the above discussion, LMNPs showed a good CO2 adsorption capacity in the colloidal suspension, and the adsorption capacity reached 324.12 mg g−1 LMNPs, which was consistent with the high density of the amino groups (about 1 formanilide can be introduced into every 1.2 native subunits, as calculated by N elemental analysis). Drying will significantly affect the physical status of LMNPs, which is a common challenge for nanomaterials built with hydroxyl-rich biomacromolecules. This will prompt our future perspectives to engineer LMNPs onto a porous solid support to gain accessibility to their nano-dimensions in a dry product, which is considered critical to bind CO2 directly from the air or flue gases.
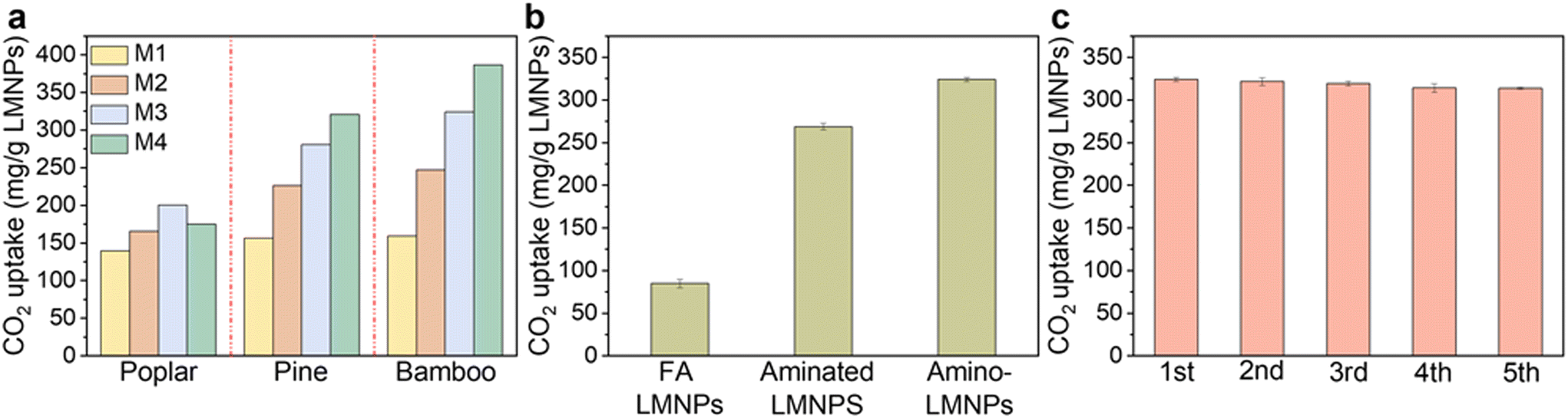 | ||
| Fig. 5 CO2 adsorption capacity of amino-LMNPs using a dispersion system (CO2 flow rate was 100 mL min−1 at 1 bar, and the adsorption testing was performed at 25 °C). (a) Comparison of the CO2 adsorption capacity of amino-LMNPs from different biomass types and preparation methods at a concentration of 2 mg mL−1; (b) comparison of CO2 adsorption capacity of different LMNPs prepared from bamboo using method 3 at a concentration of 2 mg mL−1 (“FA LMNPs” denotes LMNPs prepared from the lignin fractionated by only formic acid; “Aminated LMNPs” denotes the LMNPs prepared from aminated lignin without deformylation during the formation; “Amino-LMNPs” denotes the LMNPs prepared from aminated lignin with deformylation during the formation); (c) cyclic capacity of 5 adsorption–desorption cycles. The adsorbed LMNPs were desorbed using HCl solution (1 M) at a concentration of 10 mg mL−1. | ||
Sustainability evaluations to the as-reported reactive fractionation method for biomass to produce lignin nanomaterials as NFSAs
The above discussion has demonstrated the overall process of in situ lignin amination during the acid delignification of biomass and preparation of amino-LMNPs. In the regard of resource efficiency, the one-pot reactive fractionation of biomass using the organic solvent system of aniline–formic acid can simultaneously produce aminated lignin in high reactivity and cellulose fractions in high purities. Moreover, the organic solvent can be readily recycled by evaporation and reused. We then deployed several green metrics (E-factor, mass intensity, and reaction mass efficiency) as simple and easy-to-use indicators to compare our approach with existing industrial methods to probe the process's green sustainability and the subsequent industrial relevance and potential commercial value for large-scale utilization.41,42 The process details are shown in Fig. 6a. Compared with the traditional methods that utilize lignin as a feedstock for amination modification, this work yields aminated lignin directly from biomass in a one-step process. For the assessment of sustainability, all processes are initiated from the biomass, with all products exclusively focused on LMNPs. The E-factor or environmental factor covers all wastes generated by a particular method. It can be used as a simple and easy-to-use indicator as a convenient tool for comparing different procedures. Compared with other processes using industrial lignin as raw material for amination modification, the Esimple and Ecomplex values of this process were lower. This is because, compared with the pulping process that required large amounts of water as a solvent, this process used recyclable organic solvents, thereby reducing waste generation, which was conducive to promoting the sustainability of the process and reducing production costs. Mass intensity (MI) and reaction mass efficiency (RME) are two other important green chemistry indicators that can reflect the evaluation of resource utilization efficiency and waste generation. The MI of our approach was comparable to that of other processes. The RME of our approach is much higher than that of other processes, as the aniline–formic acid solvent can be readily recycled by evaporation and reused, while the existing industrial methods required significant water consumption. | ||
| Fig. 6 Mass flow and metrics of the reactive fractionation method for biomass to produce lignin nanomaterials as NFSAs. (a) Mass flow of the reactive fractionation method for biomass to produce lignin nanomaterials as NFSAs (recycle notes were created for the recycle solvent during the whole process and the safety note was addressed for the deployment of THF as the solvent for the LNP production); (b) mass-based green metrics for evaluating “sustainability” for the whole process (Esimple, E-factor calculated without any solvent other than the reactants; Ecomplex, E-factor calculated with solvents and reactants; MI, mass intensity; RME, reaction mass efficiency; more details can be found in the ESI†). | ||
This work developed an efficient and sustainable one-pot method for direct utilization of lignocellulosic biomass feedstock to produce aminated lignin. Compared to the traditional multi-step method (lignin separation followed by chemical modification) for preparing aminated lignin from lignocellulosic biomass, this method is operated at lower temperatures and utilizes recyclable organic solvents. Moreover, the one-step process reduces lignin separation and purification steps, significantly reducing energy consumption and production costs. Aminated lignin-based LMNPs have been proven with an outstanding CO2 capture performance under wet conditions. Compared to LMNPs produced via the aminated modification of technical lignin, the LMNPs with amino groups obtained through this method possess a higher density of CO2-binding sites, enabling enhanced CO2 capture capacity. In the current study, THF was used in the LNP production to achieve a better control over the LMNP morphology, as we aimed to elucidate the explicit impact of botanic origin and particle size on the performance of CO2 adsorption for amino-LMNPs. In addition, our subsequent studies showed that the CO2 adsorption capacity was not strictly associated with the integrity of the LMNP spheres. Therefore, physical methods such as ultrasonication-aided processing to obtain nanoparticles will be further considered to reduce the use of solvents in the fabrication of nanoparticles of aminated lignin, aiming for improved ‘greenness’ and promoting the relevance of these renewable NFSAs for large-scale utilization in the future.
Conclusions
We proposed a technological breakthrough to directly produce high N-density aminated lignins by in situ reactive fractionation of biomass using an aniline–formic acid organic system. Notably, our approach achieved selective α-arylation using aniline to capture Cα carbocations during the formic acid delignification of biomasses, thereby enabling in situ amination of lignins with high weight percent yields and simultaneously producing high-purity cellulose (i.e., maximum glucose yield of 95.15%). Our approach is applicable to different biomass types (hardwood, softwood, and bamboo). We prepared stable colloidal suspensions of micro- or nano-sized solid particles (LMNPs) using aminated lignins and applied LMNPs to CO2 adsorption. We successfully demonstrated the correlation between the physicochemical attributes of LMNPs (e.g., particle size and aminated lignin) and their CO2 adsorption efficiencies. LMNPs prepared from aminated lignin of bamboo exhibited an excellent CO2 adsorption capacity of 324 mg g−1 LMNPs and regeneration performance. This in situ reactive fractionation to produce aminated lignins is holistically simple in the operation and eco-friendly considering both the chemicals and end products involved in the preparatory process, with scalability ensured using biomass as the raw materials to produce highly efficient NFSAs. In the current study, THF was used in LMNP production to better control the LMNP morphology, as we aimed to elucidate the explicit impact of botanic origin and particle size on the CO2 adsorption of amino-LMNPs. For future outlook, such a physical method as ultrasonication-aided processing will be considered to reduce the solvent use in the fabrication of nanoparticles, aiming for improved ‘greenness’ and promoting the relevance of these renewable NFSAs for large-scale utilization in the future. We anticipate that these aminated lignin-derived solid adsorbents will broaden carbon capture applications by integrating them more seamlessly into various sectors, underscoring the importance of sustainable bio-based materials in reducing greenhouse gas emissions.Author contributions
Y.Z., X.W., and C.X. conceived and designed the experiments. R.W. performed most of the experiments collected and analyzed the data, and wrote the first draft. C.L. contributed to the biomass fractionation experiment. J.X. contributed to the TEM analytical. A.P., J.H., and T.T. contributed the analysis tools.Data availability
The data that support the findings of this study are available from the corresponding author upon request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We (C. X. and R. W.) would like to acknowledge funding from Business Finland (5855/31/2023). R. W. would also like to acknowledge funding support from the China Scholarship Council (202209370020). C. L. and Y. Z. would like to acknowledge funding from the National Natural Science Foundation of China (No. 32001269 and No. 32271821), China Postdoctoral Science Foundation (No. 2021M701287), and Shandong Province High Education Youth Innovation Team Project (2023KJ133). We particularly thank Prof. Ron Zevenhoven and Dr. Jens Back for their guidance on CO2 adsorption. We specially thank Dr. Hao Zhang for coaching in XPS and BET measurements. TEM imaging was processed and analyzed in the Electron Microscopy Laboratory, Institute of Biomedicine, University of Turku, which receives financial support from Biocenter Finland. Parts of the research used Research Council of Finland Research Infrastructure “Printed Intelligence Infrastructure” (PII-FIRI).References
- H. Li, M. E. Zick, T. Trisukhon, M. Signorile, X. Liu, H. Eastmond, S. Sharma, T. L. Spreng, J. Taylor and J. W. Gittins, Nature, 2024, 630, 654–659 CrossRef CAS PubMed.
- B. Dziejarski, J. Serafin, K. Andersson and R. Krzyżyńska, Mater. Today Sustain., 2023, 100483 Search PubMed.
- J. Gao, Y. Hoshino and G. Inoue, Chem. Eng. J., 2020, 383, 123123 CrossRef CAS.
- E. García-Bordejé and R. González-Olmos, Prog. Energy Combust., 2024, 100, 101132 CrossRef.
- M. Chen and J. Xu, Molecules, 2023, 28, 5461 CrossRef CAS PubMed.
- N. S. Mohamed Hatta, M. K. Aroua, F. Hussin and L. T. Gew, Energies, 2022, 15, 3753 CrossRef CAS.
- J. Feng, L. Fan, M. Zhang and M. Guo, Colloids Surf., A, 2023, 656, 130510 CrossRef CAS.
- R. M. Cigala, G. De Luca, I. Ielo and F. Crea, Polymers, 2024, 16, 1063 CrossRef CAS PubMed.
- J. Carrier, C. Y. Lai and D. Radu, ACS Environ. Au, 2024, 4, 196–203 CrossRef CAS PubMed.
- B. Zhao, M. Borghei, T. Zou, L. Wang, L. S. Johansson, J. Majoinen, M. H. Sipponen, M. Österberg, B. D. Mattos and O. J. Rojas, ACS Nano, 2021, 15, 6774–6786 CrossRef CAS PubMed.
- Z. Luo, C. Liu, A. Radu, D. F. de Waard, Y. Wang, J. T. Behaghel de Bueren, P. D. Kouris, M. D. Boot, J. Xiao and H. Zhang, Nat. Chem. Eng., 2024, 1, 61–72 CrossRef.
- Z. Gong and L. Shuai, Trends Chem., 2023, 5, 163–166 CrossRef CAS.
- S. D. Shinde, X. Meng, R. Kumar and A. J. Ragauskas, Green Chem., 2018, 20, 2192–2205 RSC.
- N. Li, K. Yan, T. Rukkijakan, J. Liang, Y. Liu, Z. Wang, H. Nie, S. Muangmeesri, G. Castiella-Ona and X. Pan, Nature, 2024, 630, 381–386 CrossRef CAS PubMed.
- L. Shuai, M. T. Amiri, Y. M. Questell-Santiago, F. Héroguel, Y. Li, H. Kim, R. Meilan, C. Chapple, J. Ralph and J. S. Luterbacher, Science, 2016, 354, 329–333 CrossRef CAS PubMed.
- M. Kollman, X. Jiang, S. J. Thompson, O. Mante, D. C. Dayton, H. Chang and H. Jameel, Green Chem., 2021, 23, 7122–7136 RSC.
- X. Du, J. Li and M. E. Lindström, Ind. Crops Prod., 2014, 52, 729–735 CrossRef CAS.
- L. Liu, X. Wan, S. Chen, P. Boonthamrongkit, M. Sipponen and S. Renneckar, ChemSusChem, 2023, 16, e202300276 CrossRef CAS PubMed.
- G. J. Jiao, P. Peng, S. L. Sun, Z. C. Geng and D. She, Int. J. Biol. Macromol., 2019, 127, 544–554 CrossRef CAS PubMed.
- J. Chen, L. An, J. H. Bae, J. W. Heo, S. Y. Han and Y. S. Kim, Ind. Crops Prod., 2021, 173, 114102 CrossRef CAS.
- Y. Liu, N. Deak, Z. Wang, H. Yu, L. Hameleers, E. Jurak, P. J. Deuss and K. Barta, Nat. Commun., 2021, 12, 5424 CrossRef CAS PubMed.
- W. Lan, M. T. Amiri, C. M. Hunston and J. S. Luterbacher, Angew. Chem., Int. Ed., 2018, 57, 1356–1360 CrossRef CAS PubMed.
- K. J. Yong and T. Y. Wu, Bioresour. Technol., 2023, 384, 129238 CrossRef CAS PubMed.
- J. Zheng, L. Chen, X. Qiu, S. Sun and X. Lin, Green Chem., 2023, 25, 10415–10423 RSC.
- Y. Zhang, R. Wu, S. Ni, X. Chen, Z. Wang, Z. Li, Y. Fu, M. Qin and C. Xu, Under review.
- L. Wang, L. Tan, L. Hu, X. Wang, R. Koppolu, T. Tirri, B. Van Bochove, P. Ihalainen, L. S. Seleenmary Sobhanadhas, J. V. Seppälä, S. Willför, M. Toivakka and C. Xu, ACS Sustainable Chem. Eng., 2021, 9, 8770–8782 CrossRef CAS.
- Z. H. Liu, N. Hao, Y. Y. Wang, C. Dou, F. Lin, R. Shen, R. Bura, D. B. Hodge, B. E. Dale and A. J. Ragauskas, Nat. Commun., 2021, 12, 3912 CrossRef CAS PubMed.
- A. Rahimi, A. Ulbrich, J. J. Coon and S. S. Stahl, Nature, 2014, 515, 249–252 CrossRef CAS PubMed.
- S. Ntakirutimana, T. Xu, H. Liu, J. Q. Cui, Q. J. Zong, Z. H. Liu, B. Z. Li and Y. J. Yuan, Green Chem., 2022, 24, 5460–5478 RSC.
- C. Cai, C. Zhang, N. Li, H. Liu, J. Xie, H. Lou, X. Pan, J. Y. Zhu and F. Wang, Renewable Sustainable Energy Rev., 2023, 183, 113445 CrossRef CAS.
- R. Wu, W. Liu, Z. Li and Q. Hou, Ind. Crops Prod., 2023, 194, 116353 CrossRef CAS.
- M. Kevin, G. Y. Lim and G. W. Ho, Green Chem., 2015, 17, 1120–1126 RSC.
- P. Jiang, B. Pang, G. Li, Y. Han and F. Chu, Int. J. Biol. Macromol., 2024, 254, 127948 CrossRef CAS PubMed.
- J. H. Lee, K. Kim, X. Jin, T. M. Kim, I. G. Choi and J. W. Choi, Int. J. Biol. Macromol., 2021, 183, 660–667 CrossRef CAS PubMed.
- F. Xiong, Y. Han, S. Wang, G. Li, T. Qin, Y. Chen and F. Chu, ACS Sustainable Chem. Eng., 2017, 5, 2273–2281 CrossRef CAS.
- P. Figueiredo, M. H. Lahtinen, M. B. Agustin, D. M. De Carvalho, S. Hirvonen, P. A. Penttilä and K. S. Mikkonen, ChemSusChem, 2021, 14, 4718–4730 CrossRef CAS PubMed.
- W. D. H. Schneider, A. J. P. Dillon and M. Camassola, Biotechnol. Adv., 2021, 47, 107685 CrossRef CAS PubMed.
- F. Almeida, A. Margarida, N. Seixas, R. J. B. Pinto, A. J. D. Silvestre and A. M. Da Costa Lopes, ChemNanoMat, 2024, 10, e202400010 CrossRef CAS.
- M. H. Sipponen, H. Lange, M. Ago and C. Crestini, ACS Sustainable Chem. Eng., 2018, 6, 9342–9351 CrossRef CAS PubMed.
- L. Wang, R. Wu, Q. Wang, O. Backman, P. Eklund, X. Wang and C. Xu, Adv. Funct. Mater., 2024, 34, 2315679 CrossRef CAS.
- C. Libretti, L. S. Correa and M. A. Meier, Green Chem., 2024, 26, 4358–4386 RSC.
- C. R. McElroy, A. Constantinou, L. C. Jones, L. Summerton and J. H. Clark, Green Chem., 2015, 17, 3111–3121 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5gc00429b |
| This journal is © The Royal Society of Chemistry 2025 |