
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Biomass-derived sustainable hypergolic rocket propellants with hydrogen peroxide†
Ramlithin Mavila
Chathoth
a,
Charlie
Oommen
a,
Michael
Gozin
 bcd,
Srinivas
Dharavath
e,
Manojkumar
Jujam
e,
Deepan
Chowdhury
b and
Jagadish
Das
*f
bcd,
Srinivas
Dharavath
e,
Manojkumar
Jujam
e,
Deepan
Chowdhury
b and
Jagadish
Das
*f
aDepartment of Aerospace Engineering, Indian Institute of Science, Karnataka 560012, India
bSchool of Chemistry, Faculty of Exact Sciences, Tel Aviv University, Tel Aviv 69978, Israel
cCenter for Nanoscience and Nanotechnology, Tel Aviv University, Tel Aviv 69978, Israel
dCenter for Advanced Combustion Science, Tel Aviv University, Tel Aviv 69978, Israel
eDepartment of Chemistry, Indian Institute of Technology Kanpur, Kanpur 208016, Uttar Pradesh, India
fDepartment of Chemistry, Indian Institute of Technology Patna, Bihar 801106, India. E-mail: jdas@iitp.ac.in
First published on 22nd April 2025
Abstract
Sustainable fuels, derived from various renewable biological sources, are having a significant impact on land and marine transportation, as well as on aviation. However, in the case of the space sector, this advancement is limited. In an effort to prepare a sustainable rocket fuel (SRF) from readily available bioresources, herein we report for the first time the valorization of a widely available biomass – coconut husk into hypergolic composite fuel. We showed that the hypergolic reactions of various formulations of coconut husk-derived SRFs with a green oxidizer – H2O2 (95%) could be promoted with the addition of catalytic amounts of guanine-containing polymeric complexes of manganese or copper (Mn–GU or Cu–GU). It was found that the top-performing fuel formulation, with a total manganese content of just 2 wt%, showed an impressive ignition delay time below 50 ms. Mechanistic studies exploring the structure–ignition capability relationships of coconut husk-derived SRFs and Mn–GU and Cu–GU materials revealed that the nitrate content of coconut husk-derived SRFs and the metal–ligand cooperation in the Mn–GU and Cu–GU complexes played important roles in the ignition process of our novel SRF formulations.
Green foundation1. The traditional ammonium perchlorate-based rocket propellants, highly toxic and carcinogenic hydrazine, and corrosive fuming nitric acid/nitrogen tetroxide-based propellants produce environmentally hazardous greenhouse gases and chlorinated combustion products. In contrast, hydrogen peroxide (H2O2) produces only water and oxygen as green decomposition products. Therefore, developing H2O2-based propulsion systems is highly desired for future green space missions.2. Herein, we have developed novel chlorine-free composite materials utilizing renewable biomass coconut-husk-derived fuel and guanine-derived metal complexes that exhibit excellent hypergolic ignition with a green oxidizer – H2O2. The study of the materials’ ignition, energetic, and physical properties showed that they have the potential to be used as green and non-hazardous hypergolic propellants in hybrid rocket motors. 3. The present work will pave the way for the development of other renewable biomass-derived advanced green rocket propellants that will have higher performance than traditional rocket propellants. |
Introduction
In recent years, the global space industry has seen exponential growth due to the rapid rise in the deployment of novel satellite technologies for both civil and defense applications, including surveillance, weather monitoring, global positioning, internet, and even cellular connectivity.1,2 In 2023, the space economy was valued at $0.63 trillion, and it is anticipated to increase at a compound annual growth rate (CAGR) of 9%, reaching $1.80 trillion by 2035.3 Due to the rapid increase in the pace and number of satellites launched, it is estimated that by the year 2050, the level of ozone layer depletion as a result of these launches could be much higher than that ever done by restricted-for-use chlorofluorocarbons (CFCs).4 Therefore, there is a high demand for greener and sustainable propulsion systems that can reduce harmful air pollutants and greenhouse gases arising from the burning of tons of traditional propellants.5,6One of the propulsion systems used in spacecraft is based on a hypergolic reaction between a fuel and an oxidizer. The hypergolic reaction is a spontaneous, exothermic redox reaction that occurs when a suitable oxidizer is mixed with an appropriate liquid or solid fuel, leading to the ignition of the fuel. Such hypergolic propulsion systems could be advantageous as they eliminate the need for a separate igniter and can even offer higher performance with better control depending on the choice of fuel and oxidizer.7–9 For hypergolic propulsion systems, hydrazine and its derivatives [monomethyl-hydrazine (MMH) and unsymmetrical dimethylhydrazine (UDMH)] are commonly used as fuels, and nitrogen tetroxide (N2O4) or fuming nitric acid (HNO3) is used as an oxidizer.10,11 These fuels are highly toxic and harmful to humans and the environment, and the oxidizers are highly corrosive, which creates severe problems in use, handling, storage, and disposal.12 In an effort to develop more advanced semi-green hypergolic propulsion, energetic ionic liquids (EILs), metal organic frameworks (MOFs), and metal carborane clusters were studied as hypergolic fuels for their ignition with fuming HNO3.13–21 To further achieve a “fully green” propulsion system, several green oxidizers were recently investigated including hydroxylammonium nitrate (HAN), ammonium dinitramide (ADN), and hydrogen peroxide (H2O2).22–27 Several groups reported that the hypergolic reaction of H2O2 (>90%) was achieved with some fuels including borohydride, cyano-borohydride, amino-borane, or bis-borane compounds.28–51 Since H2O2 produces only water and oxygen during its decomposition, H2O2 is considered as the next-generation green oxidizer. The use of H2O2 as an oxidizer has many other advantages in terms of its relatively low cost, handling, storage, and disposal, in comparison with N2O4 and HNO3.52,53
Recent studies showed that air- and water-stable transition metal complexes of energetic nitrogen-rich ligands exhibited impressive capability to promote hypergolic reactions with H2O2 (98%).54,55 However, these complexes’ relatively high cost and higher metal content (>15 wt%) may hamper their application in the next-generation sustainable hypergolic propulsion systems. One of the innovative solutions to this problem could be the utilization of agricultural waste and other biomasses to prepare cost-effective, green, and sustainable rocket fuels.
Bioenergy that could be obtained from biomass is an important pillar of a circular economy, which presently accounts for 55% of all global renewable energy supply and is about 6% of total energy produced worldwide.56 The emission of greenhouse gases from fossil fuels is much higher than that from biofuels, which has triggered prolific and intense research related to the development of cost-effective processes for biomass conversion.57,58 The advantages of using renewable biomass are that these feedstocks are available in huge quantities, including agricultural waste, forestry and wood processing residues, food waste, algae, and many others.59–61 Due to microalgal and lignocellulose abundance, low cost, and non-edibility, these biomaterials are considered among the most promising feedstocks for the production of biofuels for land and marine transportation, as well as for sustainable aviation fuels (SAFs).62,63
The currently utilized processes for the latter feedstocks’ conversion into biofuels are based on thermochemical processes that include hydrothermal liquefaction (HTL) and gasification, and on biochemical processes, such as fermentation (ethanol production) and anaerobic digestion (biogas production).64,65 Although biomass has the potential to decentralize energy production from fossil fuels, the poor yields of bioconversion processes impede these processes’ large-scale industrial production. The poor yields of biofuels are mainly due to complex structures of cell walls present in the biomass, which require additional pre-treatments to efficiently break down these cell walls.66
To the best of our knowledge, the utilization of biomass emanating from agricultural waste for the preparation of rocket propellants has not been systematically explored yet.67 One of the widely available agricultural products is coconut, which is produced at about 62 million tons per year.68 Coconut husk (CH), which is the main agricultural waste of coconut-derived food production, contains about 42% of cellulose and 45% of lignin, making it a suitable and abundant source of biofuel.69
With the aim to prepare green and low-cost sustainable rocket fuels (SRFs), here we report an unprecedented approach for the conversion of CH into valuable “fully green” rocket propellant formulations F1–F11, which were shown to be hypergolic with a H2O2 oxidizer. A comprehensive structural characterization and thermal and energetic property evaluation of CH-derived fuels NCH1–NCH4 and guanine (GU)-based combustion catalysts – Mn–GU and Cu–GU complexes were performed. Ignition delay studies, isothermal stability studies, and mechanistic studies to explore the nature of the ignition process of novel propellant formulations containing Mn–GU and Cu–GU complexes were also performed.
Results and discussion
Materials and methods
All commercially available reagents were used without further purification. FT-IR spectra were recorded using an ATR unit on a PerkinElmer Frontier spectrometer. Densities were measured at 25 °C by employing an Ultrapyc 1200E Quantachrome helium gas pycnometer. Elemental Analysis was performed using a Thermo Scientific Flash 2000 Organic Elemental Analyzer. DSC analyses were carried out using a PerkinElmer DSC 8000, at a heating rate of 10 °C min−1. TG analyses were carried out by using a high temperature TA (SDT 650) instrument at a heating rate of 10 °C min−1 and under a N2 atmosphere (flow rate: 100 mL min−1). EPR measurements were performed by using a JEOL Model JES FA200 instrument with an operating frequency of 9.452 GHz. SEM-EDS measurements were performed using a ZEISS Ultra-55 scanning electron microscope. The samples were subjected to gold sputtering prior to SEM-EDS measurements to prevent charging and improve image quality. The peak at 0 keV in the EDS spectra is the noise peak or strobe peak and stems from the acquisition electronics of the EDS spectrometer. XPS measurements were performed using a Thermo Scientific K-Alpha Surface Analysis using an Al Kα monochromatic excitation source of energy (1486.6 eV). The heat of combustion was measured by bomb calorimetry (Parr 6200), under O2 pressure of 32 MPa. Solid-state UV-Vis (DRS) measurements were performed using a Shimadzu MPC3600, and UV-vis absorbance of liquid samples were measured using an Analytik Jena SPECORD S-600. Ignition tests were filmed using a Chronos (1.4) high-speed video camera operating at 2000 fps. Contact angle measurements were performed using a Rame-Hart goniometer (250-F1).Synthesis
To convert the agricultural waste, coconut husk, into rocket fuel, we took a sample of raw coconut husk (25.0 g) from matured coconut, and washed it with water at room temperature and then with hot water to remove most of the unwanted impurities. The washed coconut husk was dried under air and ground to powder, and dry particles were sieved through a metal sieve to obtain a uniform coconut husk (CH) brown powder (23.0 g; Fig. 1). Subsequently, this CH powder (3.0 g) was slowly added to a mixture of HNO3 and concentrated sulfuric acid (H2SO4) at 0 °C. At the end of the addition, the reaction mixture was stirred for 15 min, and then allowed to warm up to room temperature and kept for various periods of time (from 1 day to 4 days). Upon nitration process termination and acids dilution, the insoluble materials were collected by filtration and washed with cold water and aqueous sodium bicarbonate (NaHCO3). The resulting nitrated coconut husk (NCH1–NCH4; Fig. 1, ESI†) yellow powders were dried under air to obtain materials that were used in all further studies. The reduction in the weights of the obtained NCH1–NCH4 nitrated products versus the starting CH material could be explained by the degradation of most of the lignin biopolymers present in CH during the nitration process, where the resulting lignin-derived compounds became soluble in the nitrating mixture. The chemical compositions of NCH1–NCH4 were comprehensively characterized by infrared spectroscopy (Fig. 3 and S3–S6; ESI†), elemental analysis (Table 1), scanning electron microscopy (SEM), and energy-dispersive X-ray spectroscopy (EDS) (Fig. 5 and 6 and S20–S23; ESI†), and UV-vis spectroscopy (Fig. 4). | ||
| Fig. 1 Schematic of the preparation of NCH4 fuel. | ||
 | ||
| Fig. 2 Synthesis of the Mn–Gu and Cu–Gu complexes, their proposed molecular structures, and physical appearance. | ||
Guanine (GU; 2-amino-1H-purin-6(9H)-one) is practically insoluble in most common organic solvents. However, it could be solubilized in acidic or basic aqueous solutions.70 Although Fe, Ru, Pd, Pt, and Cu transition metal complexes of guanine (as a functional group of the nucleotide base of DNA or RNA) and its derivatives were described, these reports are mostly limited to the structure of complexes in which guanine is present as a neutral ligand.71–75 The preparation of deprotonated GU transition metal complexes is highly challenging, due to the related solubility issues. In our initial attempts for the preparation of the Mn–GU complex, we first deprotonated this ligand by dissolving GU in aqueous KOH (2.0 M), which was followed by slow addition of aqueous MnCl2·4H2O. This approach was unsuccessful due to the formation of a black Mn3O4 precipitate under these basic conditions, as was observed in the infrared spectroscopy analysis of this precipitate (Fig. S7; ESI†).76 A similar problem was encountered during our attempts to prepare a Fe–GU complex, using FeCl3·6H2O as a starting material. We observed that under the used basic reaction conditions, generation of iron hydroxides was preferred over the formation of Fe–GU complexes.
In our further efforts to counter the formation of metal oxides, we discovered that the use of a specific ratio between KOH (2.5 equivalents), GU (4.0 equivalents), and metal precursor salts (1.0 equivalent) prevented the preferential formation of metal oxides and directed the reaction towards the metal–GU complex formation. Thus, an aqueous solution of a metal precursor (MnCl2·4H2O or CuCl2·2H2O) was slowly added to the basic GU suspension and the reaction mixture was stirred to yield the desired Mn–Gu and Cu–Gu complexes (Fig. 2 and ESI†). Notably, in contrast to the Mn–GU compound that was formed (in 49% yield) as an insoluble material (most probably, a coordination polymer), a Cu–GU complex was initially formed (in 55% yield) as a soluble compound. Upon solvent removal, during the final purification step, the Cu–GU complex became insoluble (forming a film), which strongly suggests its conversion into a coordination polymeric structure. The chemical structures of Mn–Gu and Cu–Gu complexes were comprehensively characterized by infrared spectroscopy (Fig. S8–S10; ESI†), elemental analysis (Table 1), SEM (Fig. 5; Fig. S20; ESI†), electron paramagnetic resonance (EPR) spectroscopy (Fig. 7), X-ray photoelectron spectroscopy (XPS; Fig. 8; Fig. S13; ESI†), solid-state UV-vis (DRS) spectroscopy (Fig. 4), and thermogravimetric (TG) analysis (Fig. 9).
FT-IR analysis
To determine the presence of different functional groups in CH nitration products NCH1–NCH4, a comparative analysis between the infrared spectra of CH and NCH1–NCH4 was performed (Fig. 3 (left), Fig. S2–S6; ESI†). The spectrum of CH was found to fully match the previously reported spectra of coconut husk.77 Peaks observed in the range of 1110–950 cm−1 in the spectrum of CH could be assigned to the C–O bond stretching of the glycosidic C–O–C bonds of cellulose, and hemicellulose, as well as to the phenolic C–OH and etheric C–O–C bonds present in the lignin component of CH.77–79 Since the infrared spectra of compounds NCH1–NCH4 were found to be practically indistinguishable from one another, the NCH4 compound was used for further analysis and comparison. In the spectra of this compound, a significant reduction (versus the CH spectrum) in the intensity of the cellulose and hemicellulose peaks was observed at 3302 cm−1 (OH groups), and 2919 cm−1 (CH and CH2 groups).77–79 A reduction in the intensity of another peak at 1726 cm−1, characteristic of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds in hemicellulose, pectin, lignin, and wax (all found in CH), showed that the nitration process of CH led to a significant removal of these components from the surface of the coir fibers.76–78 An appearance of three new intense peaks in the spectrum of NCH4 (versus the CH spectrum) at 1640 cm−1 (asymmetric N–O stretching), 1276 cm−1 (symmetric N–O stretching), and 825 cm−1 (O–NO2 stretching) was a clear indication of the presence of NO3 groups in the nitrated compounds, as a result of the conversion of OH groups in CH to the corresponding NO3 groups.80 Furthermore, in the spectrum of NCH4, peaks at 1606 cm−1 (CC bonds of aromatic rings) and 1256 cm−1 (C–O stretching of acetyl groups), both belonging to lignin, present in the spectrum of parent CH, practically disappeared. The latter observation strongly suggests degradation of the lignin component of CH through the nitration process, with a subsequent dissolution and removal of resulted lignin derivatives during the nitration reaction work-up. To further confirm the degradation of lignin during the CH nitration, we selectively extracted lignin from CH (Fig. S11, ESI†),81 and allowed to react the extracted lignin with a mixture of HNO3 and H2SO4 for 4 days. After completion of the reaction, about 97.0 wt% of the lignin was found to be soluble in the nitration reaction mixture, indicating this bio-polymer fragmentation into smaller molecules and/or acid-soluble products. The infrared spectral analysis clearly indicates that the prepared NCH1–NCH4 materials are mostly composed of nitrocellulose,80 along with some small amounts of nitro-lignin derivatives trapped in it.
O bonds in hemicellulose, pectin, lignin, and wax (all found in CH), showed that the nitration process of CH led to a significant removal of these components from the surface of the coir fibers.76–78 An appearance of three new intense peaks in the spectrum of NCH4 (versus the CH spectrum) at 1640 cm−1 (asymmetric N–O stretching), 1276 cm−1 (symmetric N–O stretching), and 825 cm−1 (O–NO2 stretching) was a clear indication of the presence of NO3 groups in the nitrated compounds, as a result of the conversion of OH groups in CH to the corresponding NO3 groups.80 Furthermore, in the spectrum of NCH4, peaks at 1606 cm−1 (CC bonds of aromatic rings) and 1256 cm−1 (C–O stretching of acetyl groups), both belonging to lignin, present in the spectrum of parent CH, practically disappeared. The latter observation strongly suggests degradation of the lignin component of CH through the nitration process, with a subsequent dissolution and removal of resulted lignin derivatives during the nitration reaction work-up. To further confirm the degradation of lignin during the CH nitration, we selectively extracted lignin from CH (Fig. S11, ESI†),81 and allowed to react the extracted lignin with a mixture of HNO3 and H2SO4 for 4 days. After completion of the reaction, about 97.0 wt% of the lignin was found to be soluble in the nitration reaction mixture, indicating this bio-polymer fragmentation into smaller molecules and/or acid-soluble products. The infrared spectral analysis clearly indicates that the prepared NCH1–NCH4 materials are mostly composed of nitrocellulose,80 along with some small amounts of nitro-lignin derivatives trapped in it.
The infrared spectra of Mn–GU and Cu–GU complexes showed similar patterns to the spectrum of the parent GU ligand, in the range of 2500–3500 cm−1, corresponding to the NH2 and NH groups of GU (Fig. 3 (right), Fig. S8–S10; ESI†). The peaks at 1691 cm−1 (CO) and 1668 cm−1 (NH2 scissoring) did not show any significant shift versus the parent GU,82 indicating that both NH2 and CO groups of GU did not have any significant interaction with the metal centers in Mn–GU and Cu–GU complexes. In the case of the Cu–GU complex, appearance of new peaks at 1591 cm−1 (CC bond) and 1555 cm−1 (CN bond) was observed, while peaks at 1635 cm−1 and 1563 cm−1, belonging to the vibrations of the same bonds in the parent GU, disappeared.82 This significant change (shift) could be explained by the binding of the GU ligand in its deprotonated form, via its N7 and/or N9 nitrogens, to the Cu metal center in the Cu–GU complex. In contrast, in the infrared spectrum of the Mn–GU complex, the intensity of 1635 cm−1 and 1563 cm−1 peaks was only reduced, and no shifts due to GU complexation with the Mn metal center via its N7 and/or N3 nitrogens were observed. These results clearly indicate the difference in the GU ligand binding modes in the Mn–GU and Cu–GU complexes and different molecular structures of these complexes. In the fingerprint region (1000–500 cm−1), in the spectra of both Mn–GU and Cu–GU complexes, significant changes in the peak pattern and intensity (versus the parent GU) were observed, indicating that heterocyclic rings of the GU ligand had participated in the metal center coordination.
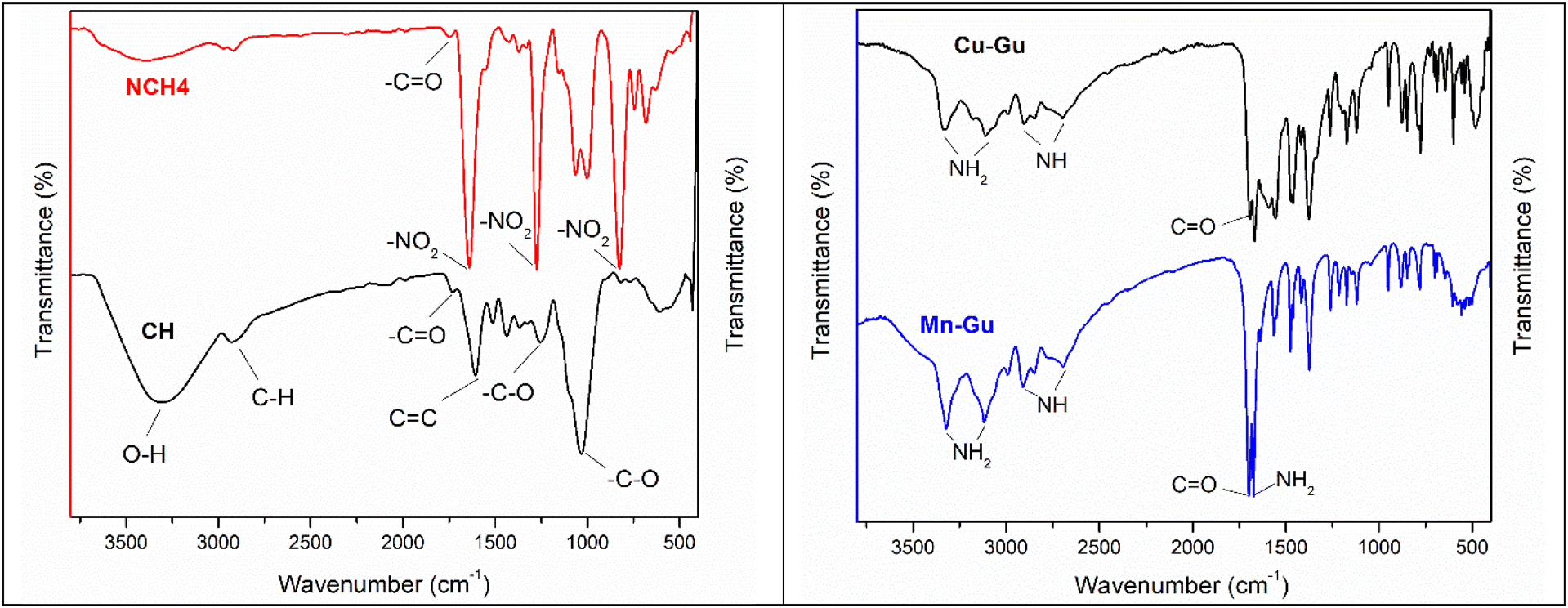 | ||
| Fig. 3 FT-IR spectra. (Left): CH and NCH4 and (right): Cu–GU and Mn–GU complexes. | ||
Elemental analysis
The elemental composition of NCH1–NCH4, Cu–GU and Mn–GU complexes was obtained from elemental analysis (CHN) measurements (Table 1). Since the main components of the dried coconut husk were cellulose, lignin, and hemicellulose, no nitrogen-containing molecules should be present in CH.69 Indeed, our analysis of the CH samples showed only the presence of carbon (32.44 wt%) and hydrogen (4.49 wt%); the rest were oxygen atoms. In comparison with CH, samples of NCH1–NCH4 showed significantly lower carbon and hydrogen content. This observation could be the result of lignin and hemicellulose removal from CH upon its nitration. Another important observation was found in NCH1–NCH4 materials, where their nitrogen content increased from 11.75 wt% in NCH1 (product of nitration for 24 h) to 13.91 wt% in NCH4 (product of nitration for 96 h), indicating that the longer nitration time led to a higher number of NO3 groups. These results were further supported by carbon-to-nitrogen (C/N) ratios calculated for the NCH1–NCH4 material, which were showing a gradual decrease as the nitrogen content in the material was increased due to the prolonged nitration time (Table 1). Empirical formulas derived from the elemental analyses of the NCH1–NCH4 material are presented in Table 1. The empirical formula calculated for the Mn–GU and Cu–GU complexes indicates the presence of two GU ligands per each Mn and Cu metal centers, respectively (Table 1).| Comp. | Formula | Carbon (C) calc./found (%) | Hydrogen (H) calc./found (%) | Nitrogen (N) calc./found (%) | C/N ratio |
|---|---|---|---|---|---|
| CH | NA | 32.01/32.44 | 4.03/4.49 | 0/0 | 0 |
| NCH1 | (C13H17N5O22)n | 26.23/26.31 | 2.88/2.84 | 11.76/11.75 | 2.24 |
| NCH2 | (C10H13N4O18)n | 25.17/25.47 | 2.75/2.73 | 11.74/11.69 | 2.18 |
| NCH3 | (C10H13N4O17)n | 26.04/25.78 | 2.84/2.84 | 12.15/12.22 | 2.11 |
| NCH4 | (C9H11N4O14)n | 27.08/27.38 | 2.78/2.80 | 14.04/13.91 | 1.97 |
| Mn–GU | C10H14N10MnO5 | 29.35/29.75 | 3.45/3.05 | 34.23/34.13 | 0.87 |
| Cu–GU | C10H14N10CuO5 | 28.75/29.29 | 3.38/3.02 | 33.52/33.03 | 0.89 |
UV-vis spectroscopy studies
The UV-vis spectrum of NCH4 (1.0 mg mL−1 in DMSO) showed a peak at 252 nm, adjacent to a broad peak/plateau (between 314 and 453 nm) with a small maximum at 373 nm (Fig. 4 (left)), where the spectrum of coconut husk-extracted lignin (1.0 mg mL−1 in DMSO) also showed a peak at 252 nm, adjacent to even broader plateau (between 314 and 537 nm) (Fig. 8 (left)). A comparison between the spectra of NCH4 and lignin in the 200–300 nm region indicated the presence of phenolic moieties in both materials.83 The appearance of the new peak at 373 nm and disappearance of peak at 537 nm in NCH4 indicated that the lignin present in NCH4 probably undergoes nitration to form nitro-lignin. | ||
| Fig. 4 (Left): UV-vis spectra (baseline subtracted) of NCH4 and lignin in DMSO; (right): solid state UV-vis (DRS) spectra of GU, Mn–GU, and Cu–GU. | ||
To assess the concentration of nitro-lignin in NCH4, we selectively extracted the nitro-lignin from NCH4 using a similar procedure previously reported for lignin extraction from cellulose–lignin mixtures (ESI†).81 We were able to extract 0.3 wt% of nitro-lignin from NCH4. The FT-IR analysis of the extracted nitro-lignin showed peaks at 1631 cm−1 that correspond to nitro groups, confirming the formation of nitro-lignin (Fig. S12; ESI†). Therefore, a small and broad peak at 373 nm in the UV-vis spectrum of NCH4 could be attributed to the formed nitro-lignin.84 Such a low content of nitro-lignin in the NCH4 material could be attributed to degradation of lignin during the nitration process.
Due to a lack of solubility of polymeric Mn–GU and Cu–GU complexes in common solvents, solid-state UV-vis (diffuse reflectance spectroscopy, DRS) analysis of these complexes was performed (Fig. 4 (right)). Using the Kubelka–Munk function, F(Rα) (eqn (1)), the measured reflectance spectra were converted into absorption spectra as shown in Fig. 4 (right).85
 | (1) |
The UV-vis DRS spectrum of solid GU showed two broad peaks with absorption maxima (λmax) of 244 and 276 nm. These peaks could be assigned to the π → π* transition of GU.82 Upon the formation of Mn–GU and Cu–GU complexes, only a slight change in the λmax of these peaks was observed. While three new peaks appeared for both Mn–GU and Cu–GU complexes with a λmax of 378, 480 and 512 nm (for Mn–GU), and 378, 482, and 522 nm (for Cu–GU). These peaks in the range of 375–540 nm could be assigned to metal-to-ligand charge transfer (MLCT) transitions, further proving the successful formation of metal–GU complexes.82
Scanning electron microscopy–energy dispersive X-ray spectroscopy studies
The sizes and shapes of solid fuel particles, as well as the presence of cracks, pores and voids in composite propellants may affect these propellants’ performance.86 We used SEM equipped with EDS to study the morphology of CH and NCH1–NCH4 materials (Fig. 5 and Fig. S20; ESI†). The SEM images of the CH surface revealed its superimposed lamellar-shaped structure, with a number of cracks and defects and the presence of some small particulate impurities. The irregular distribution of fats, wax and other materials (including impurities) represented a non-uniform structure of CH. Upon nitration of CH, the surface roughness of the produced NCH1–NCH4 materials became smoother and flatter, with a much lower number of defects on it. The smoothness of the NCH1–NCH4 material surface could be the result of efficient removal of surface-adhered impurities, as well as of wax and fatty acids. It was found that with the prolongation of the time of nitration, the smoothness of the surface changed. The longer nitration time and the higher degree of nitration led to smoother surfaces in the NCH1–NCH4 materials (Fig. S20; ESI†), which is probably the result of better removal of hemicellulose and lignin, along with wax and fats from CH. | ||
| Fig. 5 SEM images. (Top): CH; (middle): NCH4; (bottom): Mn–GU complex. | ||
EDS analyses were also performed to identify the elements present in both CH and NCH1–NCH4 materials. Only carbon and oxygen were found for CH, while samples of NCH1–NCH4 showed the presence of carbon, nitrogen, and oxygen (Fig. 6 and Fig. S21–S23; Tables S1–S4; ESI†). The obtained C/N ratios (wt%) for NCH1–NCH4 were found to be in good agreement with the C/N ratios that were obtained from the elemental analysis of these materials (Table 1). SEM analysis of Mn–GU and Cu–GU complexes revealed that their surfaces had amorphous structures.
 | ||
| Fig. 6 Representative EDS spectra. (Left): CH; (right): NCH4. | ||
Electron paramagnetic resonance studies
X-band electron paramagnetic resonance (EPR) spectrometry measurements (at room temperature) of both Mn–GU and Cu–GU complexes (in the solid state) were performed to study the oxidation states of the metal centers and their molecular geometry. The EPR spectrum of Mn–GU showed a broad signal with a g value of 1.98 (Fig. 7, left). This observation indicates the presence of the Mn(II) metal center in the Mn–GU complex.87 The absence of fine or hyperfine splitting also suggests that upon coordination of the Mn(II) metal center with the GU ligand, the symmetry around the Mn(II) center became distorted. Due to this distorted symmetry, the resonances become anisotropic, leading to a broad line. The presence of the broad lines in the EPR spectrum of the Mn–GU complex may indicate a polymeric nature of this complex.88 The analysis of the EPR spectrum of the Cu–GU complex showed a typical pattern of the Cu(II) metal center, with g∥ and g⊥ values of 2.28 and 2.06, respectively, and a gav of 2.13 (Fig. 7, right).89 The deviation of gav of the Cu–GU complex from the free electron (2.0023) was due to the coordination of the Cu(II) metal center with the GU ligand. In the case of the latter complex, the following trend for g∥ and g⊥ values was observed – g∥ > g⊥ > 2.0023 – which is characteristic of the axial symmetry of Cu(II) complexes.89 | ||
| Fig. 7 EPR spectra of powdered Mn–GU and Cu–GU complexes. | ||
X-ray photoelectron spectroscopy studies
A single crystal X-ray crystallography analysis of both Mn–GU and Cu–GU complexes could not be obtained due to the lack of solubility of these complexes in water and all evaluated organic solvents. Both the X-ray powder diffraction and SEM analyses showed that these complexes are amorphous in their nature. Therefore, X-ray photoelectron spectroscopy (XPS) analyses were used to obtain information regarding the Mn–GU and Cu–GU structures and the metal–GU bonding arrangements. The XPS spectra (survey) of Mn–GU showed the presence of Mn, C, N, and O elements in this complex structure (Fig. 8, top left). The deconvoluted high-resolution spectra of Mn 2p showed two main peaks at 641.88 and 653.68 eV, corresponding to Mn 2p3/2 and Mn 2p1/2, respectively (Fig. 8, top middle).90 The energy difference between the Mn 2p3/2 and Mn 2p1/2 states was found to be 11.8 eV. This energy difference strongly suggested the presence of Mn(II) metal center.90 A satellite peak was also observed at 643.48 eV, which was characteristic of the Mn(II)–O bond.91 In the case of the C 1s spectrum of Mn–GU, three peaks at 284.99, 286.40, and 288.05 eV were observed (Fig. S13a, ESI†). The peak at 284.99 eV could be assigned to the C–C and C–H bonds of GU. The other peaks at 286.40 eV corresponded to the C–N–C and N–C–N bonds, while the peak at 288.05 eV corresponded to the N–CO bonds of GU. The N 1s spectrum showed two peaks at 398.84 and 400.24 eV, which could be assigned to the NC and NH2–C bonds of GU, respectively (Fig. S13b, ESI†).92 In terms of its spectral pattern and binding energy, both the C 1s and N 1s spectra of Mn–GU were found to be different from the pure GU spectrum. This observation further confirmed the coordination of the Mn center with the N-ring atoms of GU.93 The deconvoluted spectrum of O 1s showed three peaks, where the peak at 529.68 eV could be assigned to the oxo-bridged Mn–O–Mn bonds (Fig. 8, top right).94 The other two peaks at 531.26 and 531.8 eV corresponded to the CO bonds of GU and water molecules, present in the structure of the Mn–GU complex, respectively.
 | ||
| Fig. 8 XPS spectra of Mn–GU (top) and Cu–GU (bottom) complexes. | ||
The XPS spectra (survey) of Cu–GU showed the presence of Cu, C, N, and O in its structure (Fig. 8, bottom left). The deconvoluted spectra of Cu 2p showed two major peaks at 932.85 and 952.63 eV, corresponding to the Cu 2p3/2 and Cu 2p1/2 states of Cu(II), respectively (Fig. 8, bottom middle).95,96 Similar to Mn–GU, the C 1s spectrum of Cu–GU also showed three peaks at 287.86, 286.28 and 285.2; the N 1s spectrum showed two peaks at 400.15 and 398.8 eV and the O 1s spectrum showed two peaks at 531.48 and 531.15 eV (Fig. 8, bottom right, and Fig. S13c and S13d, ESI†). The shape of the N 1s and C 1s spectra of Cu–GU were found to be different from the spectra of Mn–GU and GU, and the binding energies were also found to be different. This could be the effect of the different binding modes of GU to Cu, where the NH ring proton of GU was probably deprotonated by KOH. While, in the case of Mn–GU, no deprotonation occurred, and GU bound to the Mn center as a neutral ligand.
Study of thermal and energetic properties
To study the thermal properties of NCH1–NCH4 fuels and Mn–GU and Cu–GU complexes, differential scanning calorimetry (DSC) and thermogravimetric (TG) analyses were performed. The TG thermogram of the Mn–GU complex showed that this material was highly thermostable, with its peak decomposition temperature at about 500 °C (Fig. 9, left). Decomposition occurred in two continuous steps, leading to 82% mass loss of the sample. Upon heating, the sample of Mn–GU lost its coordinatively bound water molecules, well above 100 °C. This dehydration process resulted in 16.8% (observed experimentally) mass loss of the samples, which corresponded to four molecules of water, loss of which should lead to a calculated weight reduction of 17.12%. Our predicted structure of Mn–GU, where two water molecules were coordinatively bound to each Mn metal center, was in line with these TGA results (Fig. 2). It was reported that high temperature (about 800 °C) decomposition under an inert atmosphere of oxygen/water-containing Mn complexes typically produces MnO as the final product.97 Considering the formation of MnO residue in the case of Mn–GU thermal degradation, 17.34% (calculated) of mass should remain after the TG analysis of this complex. This calculated value of the MnO residue was found to be in accordance with our experimental TG results (observed 18% residue), further confirming the proposed chemical composition of the Mn–GU complex.In contrast to Mn–GU, the TG thermogram of Cu–GU showed a two-stage decomposition process (Fig. 9, right). The two-step decomposition process could be due to the presence of the deprotonated GU ligand in the Cu–GU complex. The first-stage decomposition showed a mass loss of 27.51%, with a peak temperature of 458 °C, while most of the mass was lost in the second stage (45.99%), with a peak temperature of 660 °C. The residual mass could be attributed to the formation of copper oxides (CuO or Cu2O).
 | ||
| Fig. 9 (Left): TGA thermograms of the Mn–GU complex; (right): TGA thermograms of the Cu–GU complex. | ||
The DSC thermograms of NCH1–NCH4 showed a similar decomposition pattern, with the peak decomposition temperature of about 207 °C (Fig. 10). Although these decomposition patterns were similar, the total amount of energy released was found to vary from 1574 J g−1 for NCH1 to 1544 J g−1 for NCH4 (Fig. S14–S17; ESI†). This small change in energy release could be the effect of variation in the degree of nitration and percentage of nitro-lignin present in each sample. To further evaluate the energetic properties of NCH1–NCH4 materials, bomb calorimetry analyses under an O2 atmosphere were performed. These analyses showed that upon removal of lignin, hemicellulose and other impurities, during the nitration process, the heat release of NCH4 was reduced to almost half of the untreated CH (Table 2). Due to the differences in the structure, the total heat release of the Mn–GU complex was found to be slightly higher than that of Cu–GU.
 | ||
| Fig. 10 (Left): DSC thermograms of CH and NCH1–NCH4 materials; (right): TGA thermograms of CH and NCH1–NCH4. | ||
| Materials | Density (g cm−3) | T p (°C) | Energy density (MJ kg−1) | ΔHc (kJ mol−1) | ΔHf (kJ mol−1) |
|---|---|---|---|---|---|
| CH | 1.23 | 367 | 17.82 | NA | NA |
| NCH4 | 1.38 | 207 | 8.64 | −3433.6 | −1680.06 |
| Mn–GU | 1.80 | 500 | 10.01 | −4088.96 | −2106.95 |
| Cu–GU | 1.74 | 458 | 9.34 | −3893.83 | −2066.65 |
From the heat release data obtained by bomb calorimetry analyses, the enthalpies of formation of NCH4, Mn–GU, and Cu–GU were calculated (Table 2 and ESI†). The heat release of the formulations F1 and F8 was found to be 1442 and 1444 J g−1, respectively (Fig. S18 and S19; ESI†). The reduction in heat release (by 100 J g−1) than that of NCH4 was due to the presence of non-energetic Mn–GU and Cu–GU complexes.
Ignition studies with hydrogen peroxide
After comprehensive structural characterization and thermal and energetic property evaluation of the prepared materials, their ignition tests were performed using high grade H2O2 (95%) to show their potential application as hypergolic fuels in hybrid rocket engines. All the ignition events were recorded using a high-speed video camera, operating at 2000 frames per second (fps). For these tests, powdered samples (about 30 mg) were placed in a white transparent glass vial and one drop of H2O2 (15 μL) was added. The time difference between the initial contact of oxidizer droplet with the surface of the material, until the appearance of a clearly visible flame was determined as an ignition delay (ID) time. An ID time below 50 ms is crucial for proper functioning of hypergolic propulsion systems.14Although both the Mn–GU and Cu–GU complexes were found to be highly reactive with H2O2, no clearly visible flame was observed during the addition of H2O2 (95%) to the compounds (Fig. S24†). The high decomposition temperature of both Mn–GU and Cu–GU and the absence of energetic functional groups in their structure could be the main reason for lack of any visible flames’ appearance.
Thereafter, we prepared formulations of Mn–GU (promoter) and NCH1–NCH4 (fuels) at a promoter-to-fuel ratio of 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :75 wt%, by grinding both the materials together using a mortar and pestle. The SEM analysis of F1 formulation showed a homogeneous distribution of Mn–GU particles over the surface of the NCH4 fuel (Table 3; Fig. S20; ESI†). The prepared Mn–GU-based (25 wt%) formulations of NCH1–NCH4 (F2, F5, F6 and F7; ESI†) were found to exhibit fast hypergolic ignitions with H2O2 (95%), showing ID times below 50 ms, where F2 showed the lowest ID time of 42 ms (Table 3, Fig. 11, and S24; ESI†). Among these formulations, the shortest ID time for F2 could be attributed to the higher degree of nitration of NCH4 in comparison with NCH1–NCH3.
:75 wt%, by grinding both the materials together using a mortar and pestle. The SEM analysis of F1 formulation showed a homogeneous distribution of Mn–GU particles over the surface of the NCH4 fuel (Table 3; Fig. S20; ESI†). The prepared Mn–GU-based (25 wt%) formulations of NCH1–NCH4 (F2, F5, F6 and F7; ESI†) were found to exhibit fast hypergolic ignitions with H2O2 (95%), showing ID times below 50 ms, where F2 showed the lowest ID time of 42 ms (Table 3, Fig. 11, and S24; ESI†). Among these formulations, the shortest ID time for F2 could be attributed to the higher degree of nitration of NCH4 in comparison with NCH1–NCH3.
 | ||
| Fig. 11 Hypergolic ignition images of F1–F4 formulations with H2O2 (95%). The ID values reported are an average of three experiments. | ||
| Fuel name | Composition | ID time (ms) | Specific impulse (Isp; s) | Oxidizer-to-fuel ratio (O/F) | Density specific impulse (ρIsp; s g cm−3) | |
|---|---|---|---|---|---|---|
| 1 | Mn–GU | — | NI | 228 | 2.00 | 347 |
| 2 | Cu–GU | — | NI | 223 | 2.00 | 341 |
| 3 | F1 | NCH4 (70 wt%) + Mn–GU (30 wt%) | 36 | 237 | 1.25 | 341 |
| 4 | F2 | NCH4 (75 wt%) + Mn–GU (25 wt%) | 42 | 238 | 1.15 | 343 |
| 5 | F3 | NCH4 (80 wt%) + Mn–GU (20 wt%) | 47 | 239 | 1.10 | 342 |
| 6 | F4 | NCH4 (85 wt%) + Mn–GU (15 wt%) | 49 | 240 | 1.00 | 341 |
| 7 | F8 | NCH4 (70 wt%) + Cu–GU (30 wt%) | 159 | 236 | 1.15 | 340 |
| 8 | F9 | NCH4 (75 wt%) + Cu–GU (25 wt%) | 158 | 237 | 1.10 | 339 |
| 9 | F10 | NCH4 (75 wt%) + JN15 (25 wt%) | 19 | 251 | 1.50 | 354 |
| 10 | F11 | NCH4 (85 wt%) + JN15 (15 wt%) | 38 | 248 | 1.20 | 350 |
To study the influence of Mn–GU content on ID time, three additional formulations, namely, F1, F3, and F4, were prepared (Table 3). The formulation F1, with the highest Mn–Gu content, showed the lowest ID time of 36 ms, while the formulations F3 and F4 showed ID time of 47 and 49 ms, respectively. The ID time of 49 ms for F4 formulation was highly impressive, as it contained only 2 wt% of Mn metal in its composition, making it very promising for application as a hypergolic fuel in hybrid rocket motors.
In the case of Cu–GU formulations F8 and F9, comparatively higher ID times of 159 and 158 ms were observed. The longer ID times observed for Cu–GU formulations (F8 and F9) versus similarly composed Mn–GU formulations (F1 and F2) could be explained by a lower reactivity of the Cu–GU complex with H2O2, in comparison with Mn–GU. To further demonstrate the potential application of NCH4 as a hypergolic fuel, it was mixed with Janus-type H2O2 ignitophore (JN14 and JN15) and [FcCH2N(Me)3][I], previously reported by us.54 The formulations F10 and F11 exhibited impressive ID times of 19 and 38 ms with H2O2 (95%), respectively (Fig. S24; ESI†). However, in the case of formulations F20–F22, ID times above 100 ms were observed (Fig. S24; ESI†). This showed that as per operational requirements, the NCH4 fuel could be mixed with any suitable ignitophores, to tune and obtain the desired ignition delay time. To further demonstrate the ignition capability of our prepared materials under an inert atmosphere, we performed an ignition test of formulation F11 under an N2 atmosphere (Fig. S25; ESI†). Under the N2 atmospheric conditions, F11 showed an ID time of 40 ms, which was similar to the ID time under open-air conditions.
We observed an orange-coloured flame in most of the formulations during their hypergolic ignition with H2O2. Only CuCl2·2H2O containing formulation F18 showed a greenish coloured flame. Thereafter, we studied the flame propagation of formulation F4 with time. Soon after the appearance of a clearly visible flame at 49 ms, the flame continued to grow at a very fast rate, reaching a maximum height of about 9.3 cm within 6 ms (Fig. S26, ESI†). After 75 ms, the flame started decreasing gradually, and the process continued to 202 ms, until the flame disappeared. For F1–F4, an ignition duration of about 200 ms was observed, while for F10–F11, a relatively longer duration of about 300 ms was observed.
Specific impulse calculations
Specific impulse is one of the most important properties of a propellant, which determines the efficiency of a rocket motor in generating thrust. For solid propellants, a specific impulse close to 250 s is highly desired.98 The specific impulses of Mn–GU, Cu–GU, and their formulations F1–F4, and F8–F9, as well as for JN15-based formulations F10 and F11 were calculated using EXPLO 5 (V 6.04) software (Table 3 and Fig. S27–S36; ESI†). Both Mn–GU and Cu–GU showed lower specific impulses of 228 and 223 s, while their formulations F1–F4, F8 and F9 showed higher specific impulses in the range of 236–240 s. In the case of formulation F10, a maximum specific impulse of 251 s was observed. The specific impulse curves showed a parabolic pattern, and the maximum specific impulse was observed when the O/F ratio was either 1 or slightly above 1 (Fig. S27–S36; ESI†). The density specific impulse of formulations F1–F4 was calculated to be in the range of 341–343 s g cm−3. Similar to the specific impulse (Isp) of the F10 formulation, its density specific impulse (ρIsp) was also found to be higher (ρIsp = 354 s g cm−3) than other prepared formulations. The specific impulse of F4 and F10 was further calculated with liquid oxygen (LOx), and the results were found to be slightly higher than that obtained for H2O2 (95%) (Fig. S37 and S38; ESI†). ID time and Isp and ρIsp values showed that formulations F4, F10, and F11 have the potential to be used as high-performing hypergolic fuels.Stability studies
An important criterion of an efficient propellant is to have good chemical and thermal stability under prolonged storage conditions.99 To evaluate the potential use of various materials prepared in this study, as prospective propellants, isothermal TG measurements were performed. In these analyses, the mass loss of NCH4, NC–CH (Fig. 12), NC–COT (nitrocellulose obtained from cotton), and F1 and F8 formulations were monitored for 90 min, at a constant temperature of 145 °C, which was 5 °C below the auto-ignition temperature of NC–COT (150 °C) (Fig. 12).100 In the case of NCH4, only 5.1% mass loss was observed, in comparison with NC–COT (mass loss of 13.5%). The higher stability of NCH4 could be attributed to the presence of nitro-lignin in this material. NC–CH also showed a mass loss of 6.5%, which was also significantly lower than that of NC–COT. This result could be explained by incomplete removal of lignin during the extraction of cellulose from CH.100 Similarly, the best performing hypergolic formulation F1 exhibited excellent thermostability in our TG analyses, with mass loss of just 4.3%. Apart from nitro-lignin, the presence of amino groups (from GU) in highly thermostable metal complexes Mn–GU and Cu–GU can trap the formed nitroxyl or NOx radicals, during the decomposition of nitrocellulose, thus improving chemical and thermal stabilities of F1 and F8 formulations under elevated temperature conditions. | ||
| Fig. 12 TGA thermograms of NCH4, F1, F8, NC–CH, and NC–COT materials. | ||
Mechanistic studies
The characterization of the NCH4 composition showed that this material mainly comprises nitrocellulose and a small amount of nitro-lignin. Since Mn–GU, Cu–GU, and their formulations with CH did not show any hypergolic ignitions with H2O2 (95%), the nitrocellulose presence in NCH4 could be the main reason for the successful hypergolic ignitions of formulations F1–F9 with H2O2 (Table 3, Fig. 11 and S24; ESI†). To validate this hypothesis, we extracted cellulose from CH and nitrated it to prepare nitrocellulose (NC–CH; ESI†). Extraction of cellulose from CH was performed in two steps, which included delignification, followed by a bleaching process (ESI†). The infrared spectrum of the CH-extracted cellulose matched the reported spectra of the cellulose obtained from other resources, such as cotton, wood, and bacteria (Fig. 13, top left).101 The infrared spectrum, DSC and TGA thermograms, and the elemental analysis of the NC–CH material [N (12.07 wt%), C (25.92 wt%) and H (2.73 wt%)] revealed its chemical composition (Fig. 13). | ||
| Fig. 13 FT-IR spectra and the image of the materials. (Top left): CH-extracted cellulose; (top right): NC–CH. (Bottom left): DSC thermogram of NC–CH; (bottom right): TGA thermogram of NC–CH. | ||
Thereafter, we prepared formulations F12–F19 and evaluated their capability to show hypergolic ignition with H2O2 (95%). Among these formulations, F12 exhibited the ID time (40 ms), which was even slightly shorter than that of formulation F2 (Table 4 and Fig. S24; ESI†). This ID time of F12 could be explained by an absence or very low content of nitro-lignin in NC–CH. Similarly, F19 also exhibited an ID time of 26 ms, which was 12 ms shorter than formulation F11. This observation and the lack of Mn–GU and Cu–GU ignitability with H2O2 proved that nitrocellulose present in NCH1–NCH4 (and the corresponding F1–F9 formulations) was the critical component responsible for the hypergolic ignition of these fuels. However, for the nitro-lignin (NL) formulation F23, no ignition was observed.
| Sl. no. | Fuel name | Composition | ID time (ms) |
|---|---|---|---|
| 1 | F12 | NC–CH (75 wt%) + Mn–GU (25 wt%) | 40 |
| 2 | F13 | NCH4 (75 wt%) + GU (25 wt%) | NR |
| 3 | F14 | CH (75 wt%) + Mn–GU (25 wt%) | NI |
| 4 | F15 | CH (75 wt%) + Cu–GU (25 wt%) | NI |
| 5 | F16 | NCH4 (75 wt%) + MnCl2·2H2O (25 wt%) | NI |
| 6 | F17 | NCH4 (75 wt%) + MnO2 (25 wt%) | 733 |
| 7 | F18 | NCH4 (75 wt%) + CuCl2·2H2O (25 wt%) | 510 |
| 8 | F19 | NC–CH (85 wt%) + JN15 (15 wt%) | 26 |
| 9 | F20 | NCH4 (85 wt%) + JN14 (15 wt%) | 115 |
| 10 | F21 | NCH4 (85 wt%) + [FcCH2N(Me3)][I] (15 wt%) | 278 |
| 11 | F22 | NCH4 (90 wt%) + JN15 (10 wt%) | 162 |
| 12 | F23 | NL (85 wt%) + Mn–GU (15 wt%) | NI |
To further study the importance of metal–ligand cooperativity (MLC) on ID time, ignition tests with the precursors of Mn–GU and Cu–GU were performed (Table 4). The MnCl2·2H2O-containing formulation F16 exhibited only a slow decomposition of H2O2 without any flame appearance. In contrast, MnO2-containing formulation F17 and CuCl2·2H2O-containing formulation F18 showed slow hypergolic ignitions, with ID times of 733 ms and 510 ms, respectively, showing a critical role of the MLC on the catalysed ignition processes. The metal (Mn and Cu) and GU ligand MLC is involved in the decomposition of H2O2 to generate active radical or anionic species (˙OH or HOO−), which reacts with nitrocellulose. These exothermic reactions generate sufficient amounts of heat to initiate the ignition process.55,102 The DSC analyses of formulations F1 and F8 showed a single stage decomposition process with a peak temperature at about 205 °C, which is close to the decomposition temperature of NCH4 (Fig. 14 and S18 and S19; ESI†). It means that the synergistic effect of the exothermic decomposition of NCH4 also leads to the decomposition of Mn–GU and Cu–GU complexes at a much lower temperature than their individual decomposition temperatures.
 | ||
| Fig. 14 (Left): DSC and TGA thermograms of F1 formulation; (right): SEM image of F1 formulation. | ||
The ID time of a hypergolic solid fuel not only depends on the chemical composition of this fuel, but also on the structure and chemistry of this fuel surface, and how fast the liquid oxidizer can interact with the surface of the fuel. A fuel having hydrophilic surface can interact at a faster rate with highly polar oxidizers, such as H2O2, leading to rapid decomposition of H2O2 and thus to a shorter ID time.54 The hydrophilicity or hydrophobicity (wettability) of a flat surface could be studied by contact-angle measurements, where a low contact angle (for example, with water) represents a more hydrophilic surface.103 We studied the surface wettability of our complexes and formulations by the sessile-drop contact angle technique, where deionized water was used instead of H2O2 (Fig. 15). The water contact angles of Mn–GU and Cu–GU complexes were found to be 70.1°, and 82.1°, respectively. The lower contact angle of Mn–GU than that of Cu–GU could be attributed to the differences of their chemical structures and therefore to the differences of their surface structures. Due to the higher hydrophilicity of Mn–GU, its formulation F1 also showed a relatively lower contact angle of 83.4° than that of the corresponding Cu–GU formulation F8 (87.5°). The lower contact angle or better interaction of F1 with H2O2 oxidizer was manifested in a shorter ID time than of F8 formulation (Table 3). Moreover, the SEM images of F1 and F8 formulations showed that particles of both Mn–GU and Cu–GU complexes were evenly distributed over the surface of NCH4, making the surfaces of these formulations active for their hypergolic reactions with H2O2 (Fig. 14 and S20; ESI†).
 | ||
| Fig. 15 Contact angles of Mn–GU and Cu–GU complexes and F1 and F8 formulations, all measured with deionized water. | ||
Conclusions
We report a cost-effective methodology for the utilization of a biomass, available in huge quantities – coconut husk, and its conversion into inexpensive and sustainable rocket fuels (SRFs) – NCH1–NCH4. In addition to these SRFs, we also prepared Mn–GU or Cu–GU combustion catalysts, capable of promoting the hypergolic ignition of NCH1–NCH4 fuels with an H2O2 (95%) oxidizer. Mn–GU or Cu–GU polymeric complexes were prepared in a single step from a guanine biomaterial and a simple salt of manganese or copper. The structures of NCH1–NCH4 materials and Mn–GU and Cu–GU polymeric complexes were comprehensively characterized by FT-IR, UV-vis, XPS, and EPR spectroscopic analyses, elemental analysis, and SEM-EDS, DSC and TG analyses. Ignition studies of F1–F11 formulations, containing NCH1–NCH4 with Mn–GU, Cu–GU, or JN15 complexes, with H2O2 (95%), showed impressive ID times down to 19 ms (for formulation F10), while EXPLO5 calculations of these formulations exhibited specific impulses up to 251 s and density specific impulses up to 354 s g cm−3. High temperature stability studies of F1 and F8 formulations (without any added stabilizers) by isothermal TGA showed only 4.3% mass loss in both these formulations, which was 3.1 times lower than that of the military grade non-stabilized nitrocellulose. We believe that the presence of nitro-lignin and catalytic amounts of Mn–GU and Cu–GU polymeric complexes led to the improved thermostability of our formulations. Mechanistic studies showed that the degree of nitration of nitrocellulose in NCH1–NCH4 SRFs, as well as MLC in Mn–GU and Cu–GU complexes played important roles in the hypergolic ignition processes of F1–F8 formulations. Surface wettability (contact angle) studies of Mn–GU and Cu–GU complexes and F1 and F8 formulations showed that not only the chemical structures of the materials, but also their surface characteristics made contributions to the ignition properties of these materials, where the lower contact angle led to a shorter ID time. In conclusion, our valorization methodology of conversion of coconut husk into green hypergolic propellants, with an H2O2 oxidizer, is a conceptually new approach that can pave a way for the preparation of highly useful and economical sustainable rocket fuels (SRFs) from bio-waste and other bioresources.Author contributions
Jagadish Das contributed to the supervision, analysis, resources, writing of the manuscript and funding acquisition. Ramlithin Mavila Chathoth synthesized and characterized the materials and wrote the manuscript. Charlie Oommen and Michael Gozin contributed to the resources and writing of the manuscript. Srinivas Dharavath, Manojkumar Jujam, and Deepan Chowdhury contributed to experimental characterization.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. D. acknowledges financial support from the Department of Science and Technology, INSPIRE Division (Reference No. DST/INSPIRE/04/2022/000772), S. D. acknowledges SERB (ANRF) (No. CRG/2023/000007) and M. J. thanks PMRF for the fellowship. We acknowledge analytical support from I-STEM, CENSE IISc Bangalore, SATF IISc Bangalore, SSCU IISc Bangalore, SAIF IIT Madras, Department of Chemistry – IIT Patna, and SAIF IIT Patna. We also acknowledge Prof. Jagadeesh Gopalan for his help in carrying out the ignition tests.References
- O. Kodheli, N. Maturo, B. Shankar, J. C. M. Duncan, S. Chatzinotas, J. Querol and G. Goussetis, IEEE Commun. Surv. Tutorials, 2021, 23(1), 70–109 Search PubMed.
- C. Schmierer, M. Kobald, K. Tomilin, U. Fischer and S. Schlechtriem, Acta Astronaut., 2019, 159, 578–583 CrossRef CAS.
- A. Acket-Goemaere, R. Brukardt, J. Klempner, A. Sierra and B. Stokes, In McKinsey & Company, 2024, https://www.mckinsey.com/industries/aerospace-and-defense/our-insights/space-the-1-point-8-trillion-dollar-opportunity-for-global-economic-growth.
- M. Ross, D. Toohey, M. Peinemann and P. Ross, Astropolitics, 2009, 7(1), 50–82 CrossRef.
- A. Sarritzu and A. Pasini, Acta Astronaut., 2024, 217, 100–115 CrossRef CAS.
- R. G. Ryan, E. A. Marais, C. J. Balhatchet and S. D. Eastham, Earth’s Future, 2022, 10(6), e2021EF002612 CrossRef CAS PubMed.
- G. Silva and K. Iha, J. Aerosp. Technol. Manage., 2012, 4, 407 CrossRef.
- I. I. Sam, S. Gayathri, G. Santhosh, J. Cyriac and S. Reshmi, J. Mol. Liq., 2022, 350, 118217 CrossRef.
- S. M. Davis and N. Yilmaz, Adv. Aerosp. Eng., 2014, 29313 Search PubMed.
- C. Hampton, K. Ramesh and J. Smith, 41stAerospace Sci. Meeting and Exhibit, 2003, 1359, DOI:10.2514/6.2003-1359.
- S. A. Stern and J. T. Mullhaupt, Chem. Rev., 1960, 60(2), 185 CrossRef CAS.
- D. M. Mason, J. Jet Propul., 1956, 26(9), 741 CrossRef CAS.
- S. Li, H. Gau and J. M. Shreeve, Angew. Chem., 2014, 126, 3013 CrossRef.
- H. M. Titi, J. M. Marrett, G. Dayaker, M. Arhangelskis, C. Mottillo, A. J. Morris, G. P. Rachiero, T. Friščić and R. D. Rogers, Sci. Adv., 2019, 5, 9044 CrossRef PubMed.
- Q. Wang, J. Wang, S. Wang, Z. Wang, M. Cao, C. He, J. Yang, S. Zang and T. C. W. Mak, J. Am. Chem. Soc., 2020, 142, 12010 CrossRef CAS PubMed.
- H. M. Titi, M. Arhangelskis, G. P. Rachiero, T. Friscic and R. D. Rogers, Angew. Chem., Int. Ed., 2019, 58, 18399 CrossRef CAS PubMed.
- O. Jobin, C. Mottillo, H. M. Titi, J. M. Marrett, M. Arhangelskis, R. D. Rogers, B. Elzein, T. Friščić and E. Robert, Chem. Sci., 2022, 13, 3424 RSC.
- S. Huang, W. Zhang, T. Liu, K. Wang, X. Qi, J. Zhang and Q. Zhang, Chem. – Asian J., 2016, 11, 3528–3533 CrossRef CAS PubMed.
- Q. Lai, Y. Long, P. Yin, J. M. Shreeve and S. Pang, Acc. Chem. Res., 2024, 14031–14035 Search PubMed.
- T. L. Pu, X. Y. Wang, Z. B. Sun, X. Y. Dong, Q. Y. Wang and S. Q. Zang, Angew. Chem., Int. Ed., 2024, 63, e202402 Search PubMed.
- C. Wang, C. Li, Z. Duan, Z. F. Wang, Q. Y. Wang and S. Q. Zang, Small, 2024, 20, 231097 Search PubMed.
- D. Trache, T. M. Klapötke, L. Maiz, M. A. Elghany and L. T. DeLuca, Green Chem., 2017, 19, 4711 RSC.
- D. Freudenmann and H. K. Ciezki, Propellants, Explos., Pyrotech., 2019, 44, 1084–1089 CrossRef CAS.
- A. A. Esparza, R. E. Ferguson, A. Choudhuri, N. D. Love and E. Shafirovich, Combust. Flame, 2018, 193, 417–423 CrossRef CAS.
- R. Amrousse, T. Katsumi, N. Itouyama, N. Azuma, H. Kagawa, K. Hatai, H. Ikeda and K. Hori, Combust. Flame, 2015, 162(6), 2686–2692 CrossRef CAS.
- A. J. Bennett, L. M. Foroughi and A. J. Matzger, J. Am. Chem. Soc., 2024, 146, 1771–1775 CrossRef CAS PubMed.
- D. M. Le, A. Renault, A. Delage, P. Ducos, C. M. Sabate, N. Pelletier, E. Lacôte and G. Jacob, Chem. – Eur. J., 2024, 30, e202303965 CrossRef CAS PubMed.
- W. Florczuk and G. Rarata, 53rdAIAA/SAE/ASEE Joint Propul. Conf. AIAA, 2017, 2017–4849.
- D. A. Castaneda and B. Natan, Acta Astronaut., 2019, 158, 286–295 CrossRef CAS.
- V. K. Bhosale, J. Jeong and S. Kwon, Fuel, 2019, 255, 115729 CrossRef CAS.
- M. A. Pfeil, A. S. Kulkarni, P. V. Ramachandran, S. F. Son and S. D. Heister, J. Propul. Power, 2016, 32, 23–31 CrossRef CAS.
- M. A. Pfeil, J. D. Dennis, S. F. Son, S. D. Heister, T. L. Pourpoint and P. V. Ramachandran, J. Propul. Power, 2015, 31, 365–372 CrossRef.
- J. John, P. Nandagopalan, S. W. Baek and S. J. Cho, Combust. Flame, 2020, 212, 205–215 CrossRef CAS.
- V. K. Bhosale, K. Lee, H. Yoon and S. Kwon, Fuel, 2024, 376, 132688 CrossRef CAS.
- Y. J. Wang, X. Y. Wang, H. Xu, W. W. Ren, R. Pang, L. Yang, W. C. Tong, Q. Y. Wang and S. Q. Zang, Chem. Eng. J., 2023, 455, 140587 CrossRef CAS.
- J. H. Huang, A. Q. Ji, Z. Y. Wang, Q. Y. Wang and S. Q. Zang, Adv. Sci., 2024, 11, 2401861 CrossRef CAS PubMed.
- F. A. S. Mota, G. S. Dias, D. A. Machado, J. C. Andrade, F. S. Costa, L. Fei and C. Tang, Combust. Flame, 2024, 267, 113567 CrossRef CAS.
- G. S. Dias, F. A. S. Mota, L. Fei, M. Liu, C. Tang and F. S. Costa, Proc. Combust. Inst., 2024, 40(1–4), 105269 CrossRef CAS.
- S. Nath, L. Mallick and J. K. Lefkowitz, Combust. Flame, 2023, 258(2), 113061 CrossRef CAS.
- A. V. Shaferov, S. T. Arakelov, F. E. Teslenko, A. N. Pivkina, N. V. Muravyev and L. L. Fershtat, Chem. – Eur. J., 2023, 29, e202300948 CrossRef CAS PubMed.
- D. Zhang, D. Yu, Y. Yuan, P. Zhang and X. Fan, Fuel, 2023, 342, 127788 CrossRef CAS.
- F. A. S. Mota, L. Fei, C. Tang, Z. Huang and F. S. Costa, Fuel, 2023, 336, 127086 CrossRef CAS.
- S. C. Ricker, D. Brüggemann, D. Freudenmann, R. Ricker and S. Schlechtriem, Fuel, 2022, 328, 125290 CrossRef CAS.
- K. Wang, Z. Wang, X. Zhao, X. Qi, S. Song, Y. Jin and Q. Zhang, Combust. Flame, 2022, 244, 112235 CrossRef CAS.
- D. A. Castaneda and B. Natan, Fuel, 2022, 316, 123432 CrossRef CAS.
- X. Zhao, Z. Wang, X. Qi, S. Song, S. Huang, K. Wang and Q. Zhang, Inorg. Chem., 2021, 60(22), 17033–17039 CrossRef CAS PubMed.
- F. Lauck, J. Balkenhohl, M. Negri, D. Freudenmann and S. Schlechtriem, Combust. Flame, 2021, 226, 87–97 CrossRef CAS.
- M. B. Padwal, D. A. Castaneda and B. Natan, Proc. Combust. Inst., 2021, 38(4), 6703–6711 CrossRef CAS.
- K. Wang, T. Liu, Y. Jin, S. Huang, N. Petrutik, D. Shem-Tov, Q. Yan, M. Gozin and Q. Zhang, J. Mater. Chem. A, 2020, 8, 14661–14670 RSC.
- K. Wang, A. K. Chinnam, N. Petrutik, E. P. Komarala, Q. Zhang, Q. Yan, R. Dobrovetsky and M. Gozin, J. Mater. Chem. A, 2018, 6, 22819–22829 RSC.
- A. K. Chinnam, N. Petrutik, K. Wang, A. Shlomovich, O. Shamis, D. Shem-Tov, M. Suceska, Q. Yan, R. Dobrovetsky and M. Gozin, J. Mater. Chem. A, 2018, 6, 19989–19997 RSC.
- J. H. Corpening, R. K. Palmer, S. D. Heister and J. J. Rusek, Int. J. Altern. Propul., 2007, 1, 154–173 CrossRef CAS.
- A. A. Chandler, B. J. Cantwell, G. S. Hubbard and A. Karabeyoglu, Acta Astronaut., 2011, 69, 1066–1072 CrossRef CAS.
- N. Petrutik, I. Kaminker, E. Flaxer, D. Shem-Tov, T. Giladi, Y. Bar-Bechor, J. Das and M. Gozin, Chem. Eng. J., 2023, 454(2), 140170 CrossRef CAS.
- J. Das, D. Shem-Tov, S. Wang, L. Zhang, E. Flaxer, S. Zhang, J. Stierstorfer, K. Wang, Q. L. Yan, R. Dobrovetsky and M. Gozin, Chem. Eng. J., 2021, 426, 131806 CrossRef CAS.
- https://www.iea.org/energy-system/renewables/bioenergy .
- M. G. Contreras, H. H. Escoto and E. A. Garnica, Bioresour. Technol., 2023, 385, 129470 CrossRef PubMed.
- F. Cheng and C. E. Brewer, Renewable Sustainable Energy Rev., 2017, 72, 673–722 CrossRef CAS.
- N. Gaurav, S. Sivasankari, G. S. Kiran, A. Ninawe and J. Selvin, Renewable Sustainable Energy Rev., 2017, 73, 205–214 CrossRef CAS.
- M. A. A. El-Khair, S. A. Hanafi, M. G. A. El-Moghny, M. El-Saied, M. S. Elmelawy and M. S. El-Deab, Chem. Eng. J., 2024, 490, 151716 CrossRef.
- C. Liu, P. Luan, Q. Li, Z. Cheng, P. Xiang, D. Liu, Y. Hou, Y. Yang and H. Zhu, Adv. Mater., 2021, 33, 2001654 CrossRef CAS PubMed.
- A. J. Ragauskas, G. T. Beckham, M. J. Biddy, R. Chandra, F. Chen, M. F. Davis, B. H. Davison, R. A. Dixon, P. Gilna, M. Keller, P. Langan, A. K. Naskar, J. N. Saddler, T. J. Tschaplinski, G. A. Tuskan and C. E. Wyman, Science, 2014, 344, 1246843 CrossRef PubMed.
- G. De Bhowmick, A. K. Sarmah and R. Sen, Bioresour. Technol., 2018, 247, 1144–1154 CrossRef CAS PubMed.
- D. Lee, H. Nam, M. W. Seo, S. H. Lee, D. Tokmurzin, S. Wang and Y. K. Park, Chem. Eng. J., 2022, 447, 137501 CrossRef CAS.
- C. Sawatdeenarunat, K. C. Surendra, D. Takara, H. Oechsner and S. K. Khanal, Bioresour. Technol., 2015, 178, 178–186 CrossRef CAS PubMed.
- A. R. Mankar, A. Pandey, A. Modak and K. K. Pant, Bioresour. Technol., 2021, 334, 125235 CrossRef CAS PubMed.
- N. Chalmpes, M. Baikousi, T. Giousis, P. Rudolf, C. E. Salmas, D. Moschovas, A. Avgeropoulos, A. B. Bourlinos, I. Tantis, A. Bakandritsos, D. Gournis and M. A. Karakassides, Micro, 2022, 2, 137–153 CrossRef.
- https://www.fao.org/faostat/en/#data/QCL .
- S. O. Anuchi, K. L. S. Campbell and J. P. Hallett, Sci. Rep., 2022, 12, 6108 CrossRef CAS PubMed.
- E. Colacio, O. Crespo, R. Cuesta, R. Kivek and A. Lagun, J. Inorg. Biochem., 2004, 98, 595–600 CrossRef CAS PubMed.
- C. M. Mikulski, L. Mattucci, Y. Smith, T. B. Tran and N. M. Karyannis, Inorg. Chim. Acta, 1982, 66, L71–L73 CrossRef CAS.
- T. F. Mastropietro, D. Armentano, E. Grisolia, C. Zanchini, F. Lloret, M. Julve and G. De Munno, Dalton Trans., 2008, 4, 514–520 RSC.
- A. P. Novikov, M. A. Volkov, A. V. Safonov, M. S. Grigoriev and E. V. Abkhalimov, Crystals, 2021, 11, 1417 CrossRef CAS.
- M. Rohner, A. Medina-Molner and B. Spingler, Inorg. Chem., 2016, 55, 6130–6140 CrossRef CAS PubMed.
- M. Orts-Arroyo, N. Moliner, F. Lloret and J. Martínez-Lillo, Magnetochemistry, 2022, 8, 93 CrossRef CAS.
- W. Z. Wang, C. K. Xu, G. H. Wang, Y. K. Liu and C. L. Zheng, Adv. Mater., 2002, 14(11), 837–840 CrossRef CAS.
- M. P. R. Guambo, L. Spencer, N. S. Vispo, K. Vizuete, A. Debut, D. C. Whitehead, R. Santos-Oliveira and F. Alexis, Polymers, 2020, 12, 3042 CrossRef CAS PubMed.
- M. Ichim, L. Stelea, I. Filip, G. Lisa and E. Ioan Muresan, Crystals, 2022, 12, 1249 CrossRef CAS.
- B. R. Freitas, J. O. Braga, M. P. Orlandi, B. P. da Silva, I. V. Aoki, V. F. C. Lins and F. Cotting, J. Mater. Res. Technol., 2022, 19, 1332–1342 CrossRef CAS.
- V. I. Kovalenko, R. M. Mukhamadeeva, L. N. Maklakova and N. G. Gustova, J. Struct. Chem., 1994, 34, 540–547 CrossRef.
- H. Váquez-Torres, G. Canché-Escamilla and C. A. Cruz-Ramos, J. Appl. Polym. Sci., 1992, 45, 633–644 CrossRef.
- C. M. Mikulski, L. Mattucci, L. Weiss and N. M. Karayannis, Inorg. Chim. Acta, 1985, 107(1), 81–85 CrossRef CAS.
- J. Ruwoldt, M. Tanase-Opedal and K. Syverud, ACS Omega, 2022, 7(50), 46371–46383 CrossRef CAS PubMed.
- Y. G. Khabarov, N. Y. Kuzyakov, V. A. Veshnyakov, G. V. Komarova and A. Y. Garkotin, Russ. Chem. Bull., 2016, 65, 2925–2931 CrossRef CAS.
- P. Makuła, M. Pacia and W. Macyk, J. Phys. Chem. Lett., 2018, 9(23), 6814–6817 CrossRef PubMed.
- T. Li, J. Xu, J. Han and Y. He, Micromachines, 2021, 12, 1378 CrossRef PubMed.
- D. M. Gagnon, R. C. Hadley, A. Ozarowski, E. M. Nolan and R. D. Britt, J. Phys. Chem. B, 2019, 123(23), 4929–4934 CrossRef CAS PubMed.
- M. S. Reddya, B. Prathimaa, M. Saraswathib, S. Babuc, Y. Saralad and A. V. Reddy, J. Appl. Pharm. Sci., 2016, 6(05), 090–096 CrossRef CAS.
- Ş. Ç. Özkan, A. Yilmaz, E. Arslan, L. Açık, Ü. Sayın and E. G. Mutlu, Supramol. Chem., 2014, 27(4), 255–267 CrossRef.
- M. Li, W. Lei, Y. Yu, W. Yang, J. Li, D. Chen, S. Xu, M. Fenga and H. Li, Nanoscale, 2018, 10, 15926–15931 RSC.
- A. Ghods, C. Zhou and I. T. Ferguson, J. Vac. Sci. Technol., A, 2020, 38, 042408 CrossRef CAS.
- S. Ptasińska, A. Stypczyńska, T. Nixon, N. J. Mason, D. V. Klyachko and L. Sanche, J. Chem. Phys., 2008, 129, 065102 CrossRef PubMed.
- H. Zhou, H. Xu and Y. Liu, Appl. Catal., B, 2019, 244, 965–973 CrossRef CAS.
- Z. Yang, J. Gong, C. Tang, W. Zhu, Z. Cheng, J. Jiang, A. Ma and Q. Ding, J. Mater. Sci.: Mater. Electron., 2017, 28, 17533–11754 CrossRef CAS.
- M. Ganiga and J. Cyriac, ChemPhysChem, 2016, 17, 2315–2321 CrossRef CAS PubMed.
- M. Fujiwara, T. Matsushita and S. Ikeda, Anal. Sci., 1993, 9, 289–291 CrossRef CAS.
- R. Mrozek, Z. Rzączyńska and M. Sikorska-Iwan, J. Therm. Anal. Calorim., 2001, 63, 839–846 CrossRef CAS.
- D. Frem, J. Aerosp. Technol. Manage., 2018, 10, e3318 CrossRef.
- S. N. A. Rusly, S. H. Jamal, A. Samsuri, S. A. M. Noor and K. S. A. Rahim, Energ. Mater. Front., 2024, 5(1), 52–69 CrossRef CAS.
- M. F. Cherif, D. Trache, F. Benaliouche, A. F. Tarchoun, S. Chelouche and A. Mezroua, Int. J. Biol. Macromol., 2020, 164, 794–807 CrossRef PubMed.
- M. F. Rosa, E. S. Medeiros, J. A. Malmonge, K. S. Gregorski, D. F. Wood, L. H. C. Mattoso, G. Glenn, W. J. Orts and S. H. Imam, Carbohydr. Polym., 2010, 81(1), 83–92 CrossRef CAS.
- R. Serra-Maia, M. Bellier, S. Chastka, K. Tranhuu, A. Subowo, J. D. Rimstidt, P. M. Usov, A. J. Morris and F. M. Michel, ACS Appl. Mater. Interfaces, 2018, 10, 21224–21234 CrossRef CAS PubMed.
- T. Huhtamäki, X. Tian, J. T. Korhonen and R. H. A. Ras, Nat. Protoc., 2018, 13(7), 1521–1538 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5gc00255a |
| This journal is © The Royal Society of Chemistry 2025 |