
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recovering copper from e-waste: recyclable precipitation versus solvent extraction with carbon emission assessment†
Susanna S. M.
Vance
a,
Efthalia
Chatzisymeon
b,
Carole A.
Morrison
 *a and
Jason B.
Love
*a
*a and
Jason B.
Love
*a
aEaStCHEM School of Chemistry, University of Edinburgh, Edinburgh EH9 3FJ, UK. E-mail: Carole.Morrison@ed.ac.uk; jason.Love@ed.ac.uk
bSchool of Engineering, University of Edinburgh, Edinburgh EH9 3FJ, UK
First published on 12th March 2025
Abstract
As the demand for copper continues to rise, so too does the need for sustainable methods for its recovery from waste streams. Taking inspiration from phenolic oxime reagents used in solvent extraction, the development of two recyclable ligands that act as selective precipitants for the recovery of copper from aqueous mixed-metal acidic solutions is reported. Switching the mode of action from traditional solvent extraction to precipitation eliminates the need for an organic solvent, fulfilling an important principle of green chemistry. The ligand designs feature ditopic phenolic oxime or pyrazole units that result in metal coordination at two sites, thereby promoting the formation of infinite coordination polymers that precipitate from solution. Complete copper recovery from single-metal solutions of CuSO4 and from mixed-metal solutions that contain nickel, zinc, cobalt and iron is demonstrated, under mildly acidic conditions. The copper was recovered from the loaded precipitates by washing with dilute sulfuric acid, and the ligands reused directly for multiple cycles without loss of performance. Furthermore, 96% of the copper present in a leachate solution derived from waste printed circuit boards was recovered using the phenolic pyrazole ligand. The carbon emissions of this process were also estimated by life cycle assessment and compared with those generated from the recovery of copper by ACORGA M5910, a typical phenolic oxime solvent extractant, with the precipitation process found to be more environmentally benign.
Green foundation1. This research evaluates a new precipitation approach for the recovery and recycling of copper, a technologically important metal. Current methods are either energy intense or use significant quantities of chemicals and reagents derived from fossil resources.2. The precipitation approach negates the need for organic solvents used in traditional solvent extraction processes and reduces the quantity of chemical extractant needed, while maintaining selectivity for copper over other metals. A life-cycle assessment comparing precipitation with solvent extraction methods highlights the reduced carbon emissions using the precipitation method. 3. The life-cycle analysis shows a significant contribution is made to carbon emissions by the laboratory glassware and so an engineered solution to develop a viable, scalable process is needed. This method has the potential to link into new approaches to metal dissolution that avoid toxic chemicals and harsh acids, so further enhancing its sustainability. These aspects lie beyond the scope of the current research. |
Introduction
Copper is a vital technology metal in modern society, as its high electrical conductivity and ability to form robust alloys with other metals results in its widespread application in many key industries, including infrastructure, renewables, transport, medicine, and electronics.1–3 In 2023, the European Commission added copper to the critical raw materials list as a strategic raw material, highlighting how fundamental this element is to all aspects of human life.4 However, continued industrial growth has created extensive solid and aqueous copper-containing waste streams,5 with the latter posing serious environmental and health risks if remediation is not successful.6–8 Copper mines encompass a large portion of these wastewaters, with the production of copper tailings in China reaching 1.6 billion tons in 2016.9 Storing mine tailings consumes valuable resources, while current protocols to extract further metal from tailings typically involve smelting, which results in secondary pollution.9 In addition to wastewaters, other secondary sources, such as copper scrap10 and waste electronic and electrical equipment (WEEE or e-waste)11,12 have garnered attention in the field of metal recycling. E-waste is of particular interest, given that the concentration of copper can be 30–40 times higher than in the primary ore.13 Clearly, the design of sustainable and environmentally benign processes for the recovery of copper from secondary sources is essential to combat increasing demand and growing environmental concerns.14To-date, several processes originally developed for the treatment of low-grade ores15 have demonstrated success in the recovery of copper from secondary sources. This includes cementation, which while shown to be very effective in the recovery of high purity copper from e-waste,16 consumes other metals such as zinc in the process, leading to further challenges from a sustainability perspective. Another facile recovery route is chemical precipitation, in which pH adjustment can afford precipitation of the target metal. However, its application is highly dependent on the composition of the leach solution and poor selectivity was observed when mixtures of metals with similar chemical properties are present.17 Similar selectivity issues have been encountered with the emerging field of electrochemical recovery methods, which are gaining popularity due to their mild operating conditions and minimal chemical usage.18–20 Adsorption technologies can be highly effective for the removal of pollutants from wastewater, including copper.21,22 However, many adsorbents used for this purpose are metal composites, which again raise sustainability concerns, and these reagents often operate more effectively at high pH, rendering them unsuitable for metal recovery from acidic leach streams.23–25 Success in the removal of copper from wastewater and e-waste has also been achieved with ion exchange technologies, but these processes involve complex set-ups and sometimes require activation of the exchange resin.26,27 Finally, extensive research in the field of hydrometallurgy has led to the development of sophisticated systems capable of complex separations,28–31 which are achieved through ligand design, using supramolecular concepts and coordination chemistry to afford metal selectivity.32–39 Solvent extraction processes are widely employed in the metal recovery industry and can afford high selectivity for the target metal and facilitate reuse of the ligand, thereby offering strong sustainability credentials.40 Phenolic oximes are used extensively as reagents for the recovery of copper by this route, accounting for ca. 25% of global production.41–43 While recent research employing phenolic oximes to recover copper from e-waste streams has shown promising results,44–46 these processes use large quantities of organic solvents, which are detrimental to both human health and the environment.47 Although efforts are being made to develop greener solvents for use in solvent extraction,48 the process would be simpler and more sustainable if the organic solvent could be dispensed with altogether.
Thus, a route towards achieving a more sustainable process for copper recovery is to capitalise on the selectivity achieved by phenolic oximes in solvent extraction processes, but to switch to precipitation, thereby removing the need for an organic solvent.49,50 Here, we present two simple, recyclable precipitants which achieve high selectivity for copper over other base metals commonly present in copper leach streams under conditions comparable with those employed in solvent extraction. Furthermore, we show quantitative precipitation of copper from an e-waste leachate with high selectivity for copper. In addition, a life cycle assessment (LCA) was conducted on this system which confirms that the precipitation process is more environmentally benign than a typical solvent extraction system, based on carbon emissions. We also demonstrate that the precipitants may be recycled for use in subsequent cycles by a simple stripping process using dilute sulfuric acid. Finally, characterisation of the copper-containing precipitates by Fourier-transform infra-red (FTIR) spectroscopy, powder X-ray diffraction (PXRD), scanning electron microscopy with energy dispersive spectroscopy (SEM-EDS), X-ray fluorescence (XRF) spectroscopy, and matrix assisted laser desorption/ionisation (MALDI) mass spectrometry confirms complex formation and informs on their likely chemical structures. Thus, the simple ditopic oxime and pyrazole ligands employed in this work present sustainable routes to copper recovery from both primary and secondary sources through facile precipitation using recyclable ligands.
Results and discussion
Ligand design
In this work, two simple ligands based on phenolic oximes were developed to target selective copper precipitation from aqueous waste streams without the need for organic solvents. This was achieved by modifying the basic monotopic ligand framework to ditopic structures that would promote the formation of long extended chains on Cu(II) coordination (see Fig. 1). Our previous work has indicated that this strategy is favourable for precipitation,51,52 while reports show that Cu(II) complexes of rigid phenolic oxime ligands can result in water-insoluble products.53–57 Two ligands were developed, the symmetric phenolic oxime LO and its pyrazole analogue LP (Fig. 1, top), the latter was identified as being more resistant to hydrolysis.58 Both were tested as copper precipitants under conditions comparable with those used in solvent extraction processes for copper recovery. The single crystal X-ray structure was obtained for LO (Fig. 1, bottom left) and shows that the ligand does indeed form long extended chains due to intermolecular hydrogen bonding between the phenolic oxygen atom and the oximic functionality of the adjacent chain (Fig. 1, bottom right). | ||
| Fig. 1 (Top left) Chemical structure of the oxime (LO) and (top right) pyrazole (LP) ligands developed in this work as selective precipitants for copper. (Bottom left) Single crystal X-ray structure of LO showing the intramolecular hydrogen bonds formed in the monomer between N1–H3 and N2–H4. Thermal displacement ellipsoids are drawn at 50% probability. (Bottom right) Presentation of the packing in the crystal lattice, highlighting neighbouring chains that interact through intramolecular hydrogen bonding (O–H⋯O 2.762(2)), and the cavity created within the chains that can accommodate the copper metal. | ||
Precipitation experiments
In the first instance, the precipitation of Cu(II) from aqueous acidic solution was investigated over the pH range 1–5. This pH regime is similar to the typical pH range of copper feed streams from primary ores (ca. pH 2),59 and mine tailings (pH 3–8).60 Addition of the ditopic phenolic oxime LO (0.1 mmol, 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 LO:Cu) to a solution of CuSO4 (0.02 mmol, variable H2SO4 concentration, see ESI Table S1†) results in near quantitative precipitation of copper at pH ≥ 3 (Fig. 2, blue line) after 1 h, as evidenced by the reduction in copper concentration in the supernatant by ICP-OES. Under similar conditions, the maximum precipitation of copper by the phenolic pyrazole LP occurs at pH ≥ 1.75 (Fig. 2, pink line) after 24 h; note the time for this experiment was extended for LP to achieve quantitative precipitation, as only 40% uptake is observed after 1 h (see ESI Fig. S5†).
:1 LO:Cu) to a solution of CuSO4 (0.02 mmol, variable H2SO4 concentration, see ESI Table S1†) results in near quantitative precipitation of copper at pH ≥ 3 (Fig. 2, blue line) after 1 h, as evidenced by the reduction in copper concentration in the supernatant by ICP-OES. Under similar conditions, the maximum precipitation of copper by the phenolic pyrazole LP occurs at pH ≥ 1.75 (Fig. 2, pink line) after 24 h; note the time for this experiment was extended for LP to achieve quantitative precipitation, as only 40% uptake is observed after 1 h (see ESI Fig. S5†).
 | ||
| Fig. 2 Precipitation plot (%) of Cu from acidified 0.01 M CuSO4 solution (0.02 mmol Cu) for LO (blue) and LP (pink) across the pH range 1–5 using an excess of ligand (0.1 mmol) and stirring for 1 h (oxime) or 24 h (pyrazole). The solid lines are drawn for ease of interpretation. | ||
With the pH range over which the ligands are effective thus established, the ligand to metal ratio was explored to find the optimum conditions for precipitation. For both ligands, an excess of ligand is required to achieve quantitative precipitation, with an optimum ratio of 5:1 (see ESI Fig. S6 and S7†). This ratio is significantly higher than suggested by the empirical formula of the expected complex [Cu(L)]n and is attributed to the very low solubility of the ligands in aqueous media (see ESI section 2†).
Selectivity of precipitation
With the ability of LO and LP for Cu(II) precipitation verified, the selectivity for Cu(II) in a competitive environment was evaluated by conducting precipitation experiments from mixed-metal solutions. To this end, mixed-metal solutions of equimolar Cu(II), Ni(II), Zn(II), and Co(II) were prepared, given they present the greatest challenge to achieving selective Cu(II) separation, and are commonly found in copper leach streams. Addition of 0.1 mmol of solid LO to a solution of Cu(II), Ni(II), Zn(II), and Co(II) (0.02 mmol each) results in near quantitative precipitation of copper (90%) over the pH range 3–5 (Fig. 3a, blue line) with no co-precipitation observed for any other metal. When the excess of ligand is increased (10:1 LO:Cu, 0.2 mmol), no change in selectivity occurs but an improvement in performance is observed as a 10% increase in precipitation at pH 2 (Fig. 3b). LO therefore affords similar selectivity to commercial phenolic oximes used in solvent extraction regimes, with selectivity for copper observed at low pH. Furthermore, LO affords selectivity for copper over a broader pH range, with near quantitative and selective precipitation of copper observed from pH 3–5. Conversely, commercial phenolic oximes such as ACORGA P50 achieve quantitative copper extraction at a lower pH (pH1/2 = 0) but are selective over a much narrower range (pH 1–2).41 Experiments were performed under the same conditions for LP, resulting in quantitative precipitation of copper over the pH range 2–5 after 24 h, with precipitation possible at a lower pH than the oxime (Fig. 3c). However, when the ratio of this ligand is increased (10:1 LP:Cu, 0.2 mmol) some co-precipitation of Ni(II) occurs (Fig. 4d, green line), confirming that the optimum ligand to metal ratio for selective copper precipitation is 5:1.
 | ||
| Fig. 3 Precipitation plots (%) for LO (a, b) and LP (c, d) at a 5:1 ligand to copper ratio (a, c) and a 10:1 ligand to copper ratio (b, d) from equimolar mixed-metal solutions over the pH range 1–5, performed in duplicate. The solid lines are drawn for ease of interpretation. | ||
 | ||
| Fig. 4 Schematic of the overall precipitation process from a mixed-metal solution (pH ≤ 5), highlighting ligand reuse in multiple cycles. | ||
Selectivity for copper in a mixed metal solution containing Fe(III) was also investigated, given that iron can be present in high concentrations in many copper-containing leach streams.41 However, as Fe(III) precipitates from aqueous solutions at pH > 2 as insoluble oxyhydroxides,41 only the low pH range (0.2–2) was investigated. In this case, addition of 0.1 mmol of solid LO to an equimolar solution of Cu(II), Ni(II), Zn(II), Co(II) and Fe(III) (0.02 mmol each) affords a precipitation plot identical to that in Fig. 3(b), with uptake for copper only observed from pH > 1.5 (see ESI Fig. S8†). Analogous experiments for LP show (after 24 h) that while selectivity for Cu(II) is maintained at pH 1 (90% precipitation), when the pH rises to 1.5–2 this selectivity falls, with a small amount of co-precipitation of Fe(III) (30%) and Ni(II) (10%) observed (ESI Fig. S8†). This loss in selectivity for LP likely occurs due to the lower overall pH1/2 of LP compared with LO.
Finally, the optimal conditions required to strip the metals from the precipitates were established. For LO, quantitative stripping of copper from a copper loaded precipitate was achieved after two, 1 h contacts (ESI Fig. S10†) with 2 M H2SO4. Decreasing the acid concentration to 1 M did not result in quantitative stripping after two 1 h contacts (Fig. S10†). Near quantitative stripping was achieved with either 1 M or 2 M H2SO4 for LP (Fig. S11†), with 2 M H2SO4 chosen as the optimum strip solution to allow for direct comparison between LO and LP.
Recycling experiments
Having established that LO and LP are selective precipitants for Cu(II) from a mixed-metal waste stream and that the copper can be stripped by 2 M H2SO4, it is important to establish whether the recovered ligands can be recycled for subsequent precipitation cycles. To this end, a process was developed (Fig. 4) in which the solid ligand (either LO or LP, 0.1 mmol) was stirred with an aqueous solution of copper (0.02 mmol CuSO4, pH 5) for either 1 h (LO) or 24 h (LP). The raffinate was decanted and the solids washed with water to remove any entrained copper (see ESI Tables S2 and S3†). The copper-loaded precipitate was then washed with sulfuric acid (2 M, ×3) to strip the copper and release the ligand for use in the next cycle. Before the start of the next cycle, the ligand was washed with water (×3) to remove any entrained H2SO4. If this step is not carried out prior to the start of the next cycle, the complete precipitation of copper does not occur as the pH remains too low due to the entrained acid (see ESI Fig. S12 and S13†).High precipitation of copper was achieved using both LO and LP (>80%) across all three cycles, with complete stripping achieved by three H2SO4 strip steps, and the majority of the metal recovered in the first strip step (see Fig. 5 and Tables S2 and S3†). The high percentage precipitation and near complete recovery of precipitated copper show that both ligands are robust. This process is also comparable in performance to commercial phenolic oxime solvent extractants over similar pH ranges that are typical of sulfuric acid leach streams,61 and displays similar selectivity and recyclability but without the need for organic solvents. The process therefore fulfils a green chemistry principle, by eliminating the need for organic solvents in this recovery process, thereby overcoming safety and environmental issues typically associated with using organic solvents on a large scale.
 | ||
| Fig. 5 Recovery plots showing the performance of (a) LO and (b) LP over three cycles. Dark blue bars show the percentage precipitation of Cu(II) using an excess of ligand (5:1 L:Cu, 0.1 mmol) and stirring for 1 h (oxime) or 24 h (pyrazole). Light blue bars show the copper percentage recovered from the first strip with 2 M H2SO4, purple from the second strip, and pink from the third strip. | ||
Selective precipitation of copper from e-waste
Having established that LO and LP act as selective precipitants for copper from model mixed-metal solutions, their ability to recover copper from e-waste was tested. Given the variable nature of e-waste, additional parameters need to be factored into the recovery process, such as the usually unknown concentration of the variety of metals present in the leachate and an unknown pH. Samples of waste printed circuit boards (PCBs, 1 cm2) were leached with aqua regia for 24 h and the resulting solution was analysed by ICP-OES to obtain the initial metal concentrations (see ESI Table S4†). The leachate solution was then diluted in ultra-pure water to afford a solution of appropriate 0.01 M copper concentration for recovery using LO and LP, both of which need to be in excess. The pH of the diluted leachate was determined as ca. 1.5, which falls in the range appropriate for precipitation by LP only (see Fig. 2). Thus, an excess of LP (0.1 mmol) was added to the diluted leach solution for 24 h which results in the complete precipitation of copper (Fig. 6a), with minimal co-precipitation of Fe and Sn (<2% of total metal). Furthermore, near quantitative stripping (>90%) of the copper-loaded precipitate is achieved using the conditions defined above, confirming the suitability of LP as a selective precipitant for the recovery of copper from e-waste. | ||
| Fig. 6 (a) Metal recovery plot showing the selective uptake of copper (96% Cu precipitation) from an e-waste leachate (wt% of each metal shown), with minimal co-precipitation of Fe (0.6% total metal) and Sn (0.9% total metal). (b) Metal recovery plot showing the near quantitative stripping of copper by 2 M H2SO4 from the loaded precipitate, with the majority of the copper (>70%) recovered in the first strip. | ||
Characterisation
The copper complexes formed with LO and LP, with an expected empirical formula of [Cu(L)]n, are highly insoluble in aqueous and most organic solvents (e.g., methanol, acetonitrile, chloroform) and consequently no single crystals suitable for crystallographic analysis were obtained. The complexes were therefore characterised by a variety of analytical techniques, including FT-IR spectroscopy to confirm complex formation, PXRD and SEM to inform on the crystallinity and morphology of the solid precipitates, respectively, and XRF spectroscopy to confirm that the copper lost from solution was contained in the precipitate. Finally, mass spectrometry measurements supported the formation of extended ligand–copper chains, consolidating the hypothesis that extended structures are formed upon precipitation.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching band at 1630 cm−1 (black asterisk, Fig. 7a), which shifts to a lower stretching frequency (1583 cm−1) upon complex formation, confirming coordination of the ligand to copper through the oxime nitrogen. A small peak at 1630 cm−1 remains, arising either from residual ligand or the end groups of the coordination polymer chain. The phenolic O–H stretching band is observed as a weak peak at 2650 cm−1 (purple asterisk, Fig. 7a). This peak is very small in the complex spectrum indicating that deprotonation of the OH group has occurred, again indicating complex formation. The oxime ν(O–H) stretch is found at 3412 cm−1 (green asterisk Fig. 7a) in the IR spectrum of the ligand and at 3362 cm−1 in the spectrum of the complex, with peak broadening also observed in the latter spectrum. This shift to a lower wavenumber, combined with the peak broadening, is associated with intramolecular hydrogen bonding, as is seen in the structures of square-planar Cu(II) complexes of phenolic oxime ligands and postulated above.62,63
N stretching band at 1630 cm−1 (black asterisk, Fig. 7a), which shifts to a lower stretching frequency (1583 cm−1) upon complex formation, confirming coordination of the ligand to copper through the oxime nitrogen. A small peak at 1630 cm−1 remains, arising either from residual ligand or the end groups of the coordination polymer chain. The phenolic O–H stretching band is observed as a weak peak at 2650 cm−1 (purple asterisk, Fig. 7a). This peak is very small in the complex spectrum indicating that deprotonation of the OH group has occurred, again indicating complex formation. The oxime ν(O–H) stretch is found at 3412 cm−1 (green asterisk Fig. 7a) in the IR spectrum of the ligand and at 3362 cm−1 in the spectrum of the complex, with peak broadening also observed in the latter spectrum. This shift to a lower wavenumber, combined with the peak broadening, is associated with intramolecular hydrogen bonding, as is seen in the structures of square-planar Cu(II) complexes of phenolic oxime ligands and postulated above.62,63
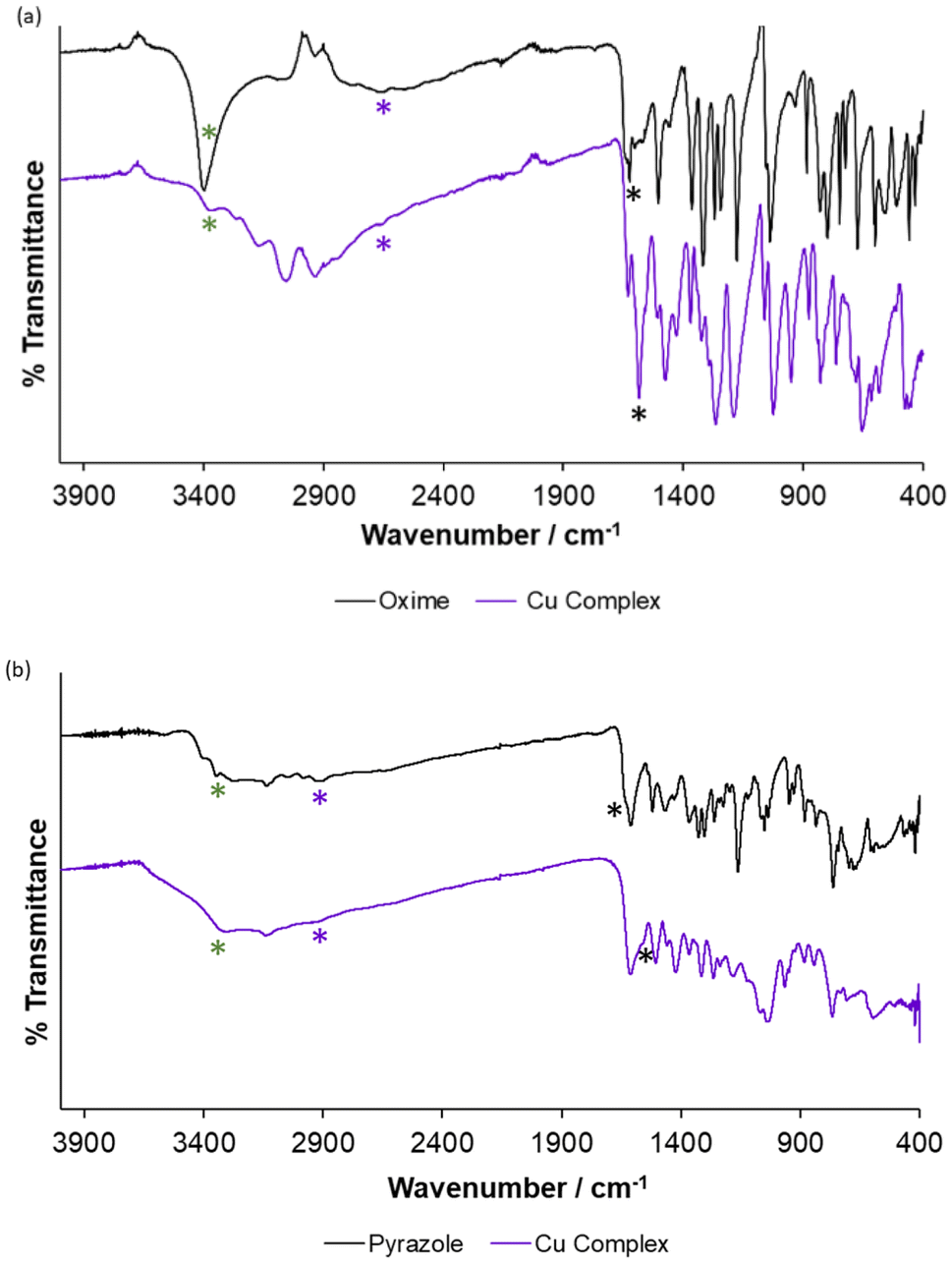 | ||
| Fig. 7 ATR FT-IR spectra recorded for LO and Cu–LO (a) and LP and Cu–LP (b) over the range 4000–400 cm−1. | ||
| Compound | ν(OH)/cm−1 | ν(NH)/cm−1 |
ν(CN)/cm−1 |
|---|---|---|---|
| LO | 2650 (phenol) | n/a | 1630 |
| 3412 (oxime) | |||
| LP | 2908 | 3373 | 1630 |
| Cu–LO | 2650 (phenol) | n/a | 1583 |
| 3362 (oxime) | |||
| Cu–LP | — | 3330 | 1556 |
The FT-IR spectrum of LP shows the CN stretch as a shoulder (1630 cm−1, black asterisk Fig. 7b) on the CC stretch of the pyrazole carbons (1620 cm−1). This shoulder moves to a lower frequency (1556 cm−1) upon formation of the copper complex, which supports coordination of the pyrazole nitrogen to the metal centre. The phenolic O–H stretch is observed as a broad band centred at 2908 cm−1 (purple asterisk Fig. 7b). This band is virtually absent in the spectrum of the complex, which indicates deprotonation upon complex formation. Finally, the ν(N–H) stretch is observed at 3373 cm−1 (green asterisk Fig. 8b) in the IR spectrum recorded for LP. This shifts to a lower, broader, adsorption (3330 cm−1) upon complexation with Cu(II), which can be attributed to intramolecular hydrogen bonding.
 | ||
| Fig. 8 The PXRD patterns of (a) LO, (b) Cu–LO complex, (c) LP and (d) Cu–LP complex recorded over a 2θ range of 5°–30°. | ||
 | ||
| Fig. 9 SEM images of (a) LO, (b) Cu–LO complex, (c) LP and (d) Cu–LP complex recorded at a magnification level of between 600–800×. Reference scales are given at the bottom of each image. | ||
The SEM images of LP show small blocks varying in size from approximately 20 μm in diameter to 60 μm, whereas [Cu(LP)]n presents as a fine powder clumped into larger aggregates, approx. 200 μm in size. Again, the copper-containing material appears semi-crystalline, in accordance with the PXRD pattern. EDS confirms the presence of C, N and O in the sample of LP and the presence of Cu, C, N and O in [Cu(LP)]n, with the presence of copper in the complex further confirmed by XRF (see ESI Table S8†).
 | ||
| Fig. 10 (Top) Full mass spectrum of the Cu–LP precipitate using 4-cyano-2-nitrophenol matrix prepared in acetonitrile, alongside isotope pattern matching of ions with two LP ligands coordinating to (a) one, (b) two, or (c) three copper ions. | ||
Carbon accounting
A life cycle assessment (LCA) in terms of carbon emissions was conducted to provide a benchmark for comparing precipitation and conventional solvent extraction processes. In order to obtain comparable data, the same waste PCB leach solution discussed above was processed by the standard solvent extractant ACORGA M5910, as outlined in the Experimental section. ISO 40040 and 40044 protocols were followed for the LCA. This benchmark study is not intended as an in-depth analysis of the two methods but rather serves as an indication of the carbon emissions associated with each process and provides a baseline for comparing the two processes on an industrial scale. The functional unit for this study was defined as the production of 1 g of pure copper metal, given this would be the desirable end product from the complete recovery cycle. The post-processing (electroplating) of the pure acidic copper solution and the preparation of the leachate solution were defined as outside of the system boundary (Fig. 11), given these steps would be the same for both processes. Thus, this study focuses on providing a comparison of the separation and purification steps only. The life-cycle inventory is outlined in the Experimental section and ESI (section 8†). | ||
| Fig. 11 System boundaries for the precipitation and solvent extraction separation and purification cycles for copper, with elements that only appear in one of the cycles highlighted (solvent and centrifuge). | ||
 | ||
| Fig. 12 (a) Chart showing the contributions to the total CO2e emissions of all the inputs/outputs for the selective recovery of copper by LP, with those contributing less than 0.5% shown together as the ‘remaining’ section. (b) Chart showing the breakdown of the contributions of each input/output grouped in the ‘remaining’ section of (a). (c) Chart showing the contributions to the total CO2e emissions of all the inputs/outputs for the selective recovery of copper by ACORGA M5910 in kerosene, with those contributing less than 1% shown together as the ‘remaining’ section. (d) Chart showing the breakdown of the contributions of each input/output grouped in the ‘remaining’ section of (c). | ||
 | ||
| Fig. 13 (a) Plot showing the total kg CO2e emissions associated with both the precipitation and solvent extraction recovery processes, including all listed inventory items. (b) Comparison of the kg CO2e emissions for the precipitation and solvent extraction processes, taking only the separation reagents into account. (c) Comparison of the precipitation and solvent extraction processes including all reagents, where 3 strip steps are employed in the precipitation process. (d) Comparison of the precipitation and solvent extraction processes, again including all reagents, with only one strip step employed in the precipitation process. | ||
This lab-scale study provides only a preliminary comparison of the recovery of copper from e-waste by recyclable precipitation or solvent extraction. In particular, the equipment used in this study varies significantly from industrial-scale processes, with the vessel used accounting for the largest portion of emissions due to its disposal after use (analytical glassware is not reused to avoid contamination); in contrast, on an industrial scale, a large, long-lifespan reactor vessel would be used. Furthermore, more specialised equipment is employed in industry such as mixer-settlers, making the use of a centrifuge to separate precipitates from solution unnecessary. The study can however inform on lab practices and highlight areas that result in significant environmental pollution.
Conclusions
Taking inspiration from phenolic oxime ligands that are used extensively in copper extraction, two recyclable ligands have been developed for the selective recovery of copper by precipitation from aqueous base metal acidic solutions. This is relevant to the processing of leach streams created as wastewater tailings in primary copper mining and in the processing of secondary sources, such as copper scrap and electronic waste. Switching the mode of action from traditional solvent extraction to precipitation eliminates the need for an organic solvent, fulfilling an important principle of green chemistry.The ligand design features symmetric oxime or pyrazole functional groups which allows for metal coordination at two sites, thus promoting the formation of coordination polymers that precipitate from aqueous solutions. While this design feature was successful, the rapid precipitation generates largely amorphous solids that are insoluble in common organic solvents and hinders their structural characterisation. However, the combination of FT-IR spectroscopy and MALDI mass spectrometry concurs with the formation of [Cu(L)]n chains, while SEM-EDS and XRF spectroscopy show that the precipitates contain the copper lost from solution.
When used in five-fold excess, both ligands achieve complete copper recovery from single and mixed-metal solutions under mild conditions. The precipitants are straightforwardly stripped to recover the copper by washing with dilute H2SO4 and the ligands may be reused for multiple cycles. Furthermore, the pyrazole-based ligand is highly effective in the precipitation of Cu(II) from waste printed circuit boards. These ligands therefore present an environmentally benign alternative to traditional solvent extractants, offering the potential for a more sustainable route to extracting and recycling high purity copper from waste.
The LCA study supports the claim that switching to a precipitation process from solvent extraction on a lab scale results in less environmental pollution, as quantified by lower CO2e emissions. Furthermore, the sensitivity analysis shows that the contributions to emissions are minimal for the precipitation reagent (LP), whereas they are more significant in the solvent extraction process (ACORGA M5910 and kerosene) due to the use of solvent and the need for a larger amount of ligand to achieve high copper recovery. Finally, when all reagents used in the two processes are considered and the same number of strip steps are employed, the precipitation system results in lower emissions, which, given the processes would use similar assemblies on an industrial scale, suggests that the precipitation process is a viable recovery method for copper and offers a more environmentally benign recovery process, regarding carbon emissions.
Experimental methods
All solvents and reagents were used as received from Sigma-Aldrich, Fisher Scientific UK, Alfa Aesar, Acros Organics or VWR International. Ultra-pure water was obtained from a Milli-Q purification system.Preparation of 1,1′-(4,6-dihydroxy-1,3-phenylene)bis[ethenone] 1,1′-dioxime (LO)
LO was prepared according to an adapted method from the literature.57 4,6-Diacetylresorcinol (5.0 g, 25.75 mmol) was added to a round-bottomed flask with NaOH (1.25 M, 100 mL). A solution of hydroxylamine hydrochloride (5.7 g, 82 mmol) in ultra-pure H2O was then added and the solution heated to 80 °C for 1 h. After cooling, the solution was neutralized with HCl (5 M, <10 mL) and the product precipitated. The solids were washed with warm H2O to afford LO as a pale pink solid (5.6 g, 96%). 1H NMR (500.12 MHz, acetone-d): δH = 11.83 (s, 2H, OH (oxime)), 10.43 (s, 2H, OH (phenolic)), 7.63 (s, 1H, aromatic CH), 6.30 (s, 1H, aromatic CH), 2.36 (s, 6H, CH3); 13C{1H}: NMR (125.76 MHz, acetone-d): δC = 161.75, 159.53, 129.04, 112.77, 105.27, 11.17 ppm; FTIR: ν = 3412, 3051, 2931, 2650, 1630, 1504, 1365, 1348, 1268, 1242, 1177 cm−1; MALDI-MS (m/z): C10H12N2O4 [M + H]+, calcd 225.08698, found 225.08735 (mass error ±1.64 ppm).Preparation of 4,6-di-1H-pyrazol-3-yl-1,3-benzenediol (LP)
LP was prepared according to adapted procedures from the literature.58,64,65 4,6-Diacetylresorcinol (2.0 g, 10.3 mmol) was added to a round-bottomed flask with toluene (20 mL) and dimethylformamide-dimethylacetal (5.3 mL, 40 mmol) at room temperature. The reaction was then heated to 80 °C for 6 h. The resulting precipitate was filtered and used in the next step without further purification. The product from step 1 (1.3 g, 4.27 mmol) was added to a round-bottomed flask with ethanol (25 mL) and hydrazine hydrate (0.83 mL, 17 mmol). The solution was stirred under reflux for 20 h. The solvent was then removed under vacuum. The yellow residue was recrystallized in EtOH/H2O to afford LP as a yellow solid (1.0 g, 40%). 1H NMR (500.12 MHz, acetone-d): δH = 12.32 (s, 2H, NH), 11.19 (s, 2H, OH), 8.08 (s, 1H, aromatic CH), 7.88 (s, 2H, pyrazole CH), 6.94 (s, 2H, pyrazole CH), 6.48 (s, 1H, aromatic CH); 13C{1H} NMR (125.76 MHz, acetone-d): δC = 157.97, 130.05, 127.89, 125.41, 110.33, 104.31, 101.44 ppm; FTIR: ν = 3373, 3130, 3055, 2975, 2908, 1630, 1607, 1518, 1461, 1368, 1323, 1300, 1258, 1160 cm−1; MALDI-MS (m/z): C12H10N4O2 [M + H]+, calcd 243.08765, found 243.08795 (mass error ±1.23 ppm), [M + Na]+, calcd 265.06960, found 265.06996 (mass error ±1.36 ppm).Preparation of [Cu(L)]n complexes
Samples of [Cu(LP)]n and [Cu(LO)]n for characterization were prepared by contacting solid ligand (0.5 mmol) with a solution of CuSO4 in H2O (1 M, 5 mL). Samples were stirred for 1 h and then the solids collected and washed with water and acetone to remove any excess ligand.pH measurements
The pH of each stock solution was measured using a Mettler Toledo T5 titrator with a pH electrode attachment. Prior to measurements, the pH probe was calibrated using pH 4.01 and pH 7.00 buffer solutions. Sample measurements were taken until a constant pH value was recorded (no more than 2 minutes). The pH electrode was rinsed with ultra-pure water between measurements and stored in a KCl buffer solution.Precipitation of Cu (single metal) pH 1–5
Stock solutions of CuSO4 (0.01 M) were prepared in ultra-pure H2O (0.01 M CuSO4/H2O = pH 5) and H2SO4 (0.01 M CuSO4/H2SO4; 0.1 M = pH 1, 0.01 M = pH 2, pH 3 and pH 4 stock solutions prepared from pH 1 and 5 stocks, see ESI†) to afford five stock solutions of pH 1–5. Solid ligand (either LO (0.0224 g) or LP (0.0242 g), 0.1 mmol) was added to a vial with a magnetic stir bar followed by a solution of CuSO4 (0.01 M, 2 mL, pH 1–5). Samples were stirred for 1 h (oxime) or 24 h (pyrazole) at 500 rpm at room temperature (20 °C). The stir bar was removed and samples centrifuged for 5 min at 4000 rpm to separate the solids from the supernatant. The supernatants and feed samples were diluted ×100 in 2% nitric acid for analysis by ICP-OES to determine metal content. All experiments were carried out in duplicate.Precipitation of Cu varying L![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :M ratio
:M ratio
Solid LP (0.2 mmol, 0.0484 g, 1:1; 0.4 mmol, 0.0969 g, 2:1; 0.06 mmol, 0.0145 g, 3:1; 0.08 mmol, 0.0194 g, 4:1; 0.1 mmol, 0.0242 g, 5:1; 0.14 mmol, 0.0339 g, 7:1; 0.2 mmol, 0.0484 g, 10:1) was added to a vial with a magnetic stir bar followed by a solution of CuSO4 (2 mL, pH 5, 0.1 M 1:1/2:1, 0.01 M 3:1–10:1). Samples were stirred for 24 h at 500 rpm at room temperature (20 °C). The stir bar was removed and samples centrifuged for 5 min at 4000 rpm to separate the solids from the supernatant. The supernatants and feed samples were diluted ×100 in 2% nitric acid for analysis by ICP-OES to determine metal content. All experiments were carried out in duplicate.
Precipitation of Cu (mixed metal, no Fe) pH 1–5
Mixed-metal stock solutions of CuSO4/NiSO4/CoSO4/ZnSO4 (0.01 M in each metal) were prepared in ultra-pure H2O (pH 5) and H2SO4 (0.1 M = pH 1, 0.01 M = pH 2, pH 3 and pH 4 stock solutions prepared from pH 1 and 5 stocks) to afford five stock solutions of pH 1–5. Solid ligand (either LO (0.0224 g) or LP (0.0242 g), 0.1 mmol, 5:1 L:Cu) was added to a vial with a magnetic stir bar followed by the mixed-metal solution (0.01 M, 2 mL, pH 1–5). Samples were stirred for 1 h (oxime) or 24 h (pyrazole) at 500 rpm at room temperature (20 °C). The stir bar was removed and samples centrifuged for 5 min at 4000 rpm to separate the solids from the supernatant. This experiment was repeated with a larger excess of ligand (LO (0.0448 g) or LP (0.0484 g) 0.2 mmol, 10:1 L:Cu) under similar conditions. The supernatants and feed samples were diluted ×100 in 2% nitric acid for analysis by ICP-OES to determine metal content. All experiments were carried out in duplicate.
Precipitation of Cu (mixed metal, with Fe) pH 0.2–2
Mixed-metal stock solutions of CuSO4/NiSO4/CoSO4/ZnSO4/FeCl3 (0.01 M in each metal) were prepared in ultra-pure H2O and H2SO4 to afford five stock solutions between pH 0.2–2 (0.2, 0.9, 1, 1.5 and 2). Solid ligand (either LO (0.0224 g) or LP (0.0242 g), 0.1 mmol, 5:1 L:Cu) was added to a vial with a magnetic stir bar followed by the mixed-metal solution (0.01 M, 2 mL, pH 0.2–2). Samples were stirred for 1 h (oxime) or 24 h (pyrazole) at 500 rpm at room temperature (20 °C). The stir bar was removed and samples centrifuged for 5 min at 4000 rpm to separate the solids from the supernatant. The supernatants and feed samples were diluted ×100 in 2% nitric acid for analysis by ICP-OES to determine metal content. All experiments were carried out in duplicate.
Stripping of copper loaded precipitates
Solid ligand, LO (0.0224 g, 0.1 mmol) or LP (0.0242 g, 0.1 mmol) was added to a vial with a magnetic stir bar followed by a solution of CuSO4 in ultra-pure H2O (2 mL, 0.01 M) and stirred for either 1 h (LO) or 24 h (LP) at 500 rpm at room temperature (20 °C). The stir bar was removed and samples centrifuged for 5 min at 4000 rpm to separate the solids from the supernatant. The loaded precipitates were then stripped by contacting with a solution of H2SO4 (2 mL, either 1 M or 2 M) and stirring for 1 h at 500 rpm at room temperature (20 °C). The stir bar was removed and samples centrifuged for 5 min at 4000 rpm to separate the solids from the supernatant. The supernatants and feed samples were diluted ×100 in 2% nitric acid for analysis by ICP-OES to determine metal content. All experiments were carried out in duplicate.Precipitation of Cu from e-waste
An e-waste leach solution derived from waste PCBs dissolved in aqua regia was diluted in ultra-pure H2O to afford a leachate of suitable pH for precipitation by LP (pH ∼ 1.5). Solid LP (0.0242 g, 0.1 mmol) was added to a vial with a magnetic stir bar followed by the e-waste solution (approx. 0.01 M Cu, 2 mL, pH ∼ 1.5). Samples were stirred for 24 h at 500 rpm at room temperature (20 °C). The stir bar was removed and samples centrifuged for 5 min at 4000 rpm to separate the solids from the supernatant. The solids were then washed with ultra-pure H2O (4 mL) to remove any entrained metal solution. The copper-loaded precipitates were contacted with H2SO4 (2 M, 2 mL) for 1 h at room temperature. The samples were centrifuged, solids separated, and the strip procedure repeated twice. The supernatants and feed samples were diluted ×1000 in 2% nitric acid for analysis by ICP-MS to determine metal content. All experiments were carried out in duplicate.Solvent extraction of Cu from e-waste (for LCA study)
A solution of ACORGA M5910 (30% in kerosene) was prepared and added to a vial (2 mL) with the e-waste leach solution (as before, 2 mL) and stirred for 1 h at 500 rpm at room temperature (20 °C). The stir bar was removed and samples left to stand for approx. 2 min to allow phases to settle. The organic and aqueous phases were then separated. The loaded organic phase (1 mL) was then contacted with H2SO4 (2 M, 1 mL) for 1 h at room temperature. The samples were then allowed to settle and then the phases were separated. The aqueous phases and feed samples were diluted ×100 in 2% nitric acid for analysis by ICP-OES to determine metal content. All experiments were carried out in duplicate.Ligand recycling experiments
Solid ligand (either LO (0.0224 g) or LP (0.0242 g), 0.1 mmol) was added to a vial with a magnetic stir bar followed by a solution of CuSO4 (0.01 M/H2O, 2 mL, pH 5). Samples were stirred for 1 h (oxime) or 24 h (pyrazole) at 500 rpm at room temperature (20 °C). The stir bar was removed and samples centrifuged for 5 min at 4000 rpm to separate the solids from the supernatant. Samples were then washed with ultra-pure water (2 mL, 15 min) to remove any residual metal. The solids were then contacted with dilute H2SO4 (2 M, 2 mL) for 30 min at room temperature (20 °C). The stir bar was removed and the samples centrifuged for 5 min at 4000 rpm to separate the solids from the supernatant. This strip procedure was repeated twice to ensure complete stripping of the ligand. The solids were then washed with ultra-pure H2O (2 mL, ×3) for 1 h to remove any entrained H2SO4. The complete cycle was then repeated ×2. The supernatants and feed samples were diluted ×100 in 2% nitric acid for analysis by ICP-OES to determine metal content. All experiments were carried out in duplicate.ICP-OES/ICP-MS analysis
Analysis of metal content in all samples except those from the precipitation of copper from e-waste was carried out using ICP-OES on a PerkinElmer Optima 8300 Inductively Coupled Plasma Optical Emission Spectrometer. All samples were diluted in 2% nitric acid and taken up at a rate of 1.3 mL min s−1, with the following argon gas flow parameters: 12 L min−1 plasma, 0.2 min−1 auxiliary and 0.6 L min−1 nebulizer. See Table S5† for summary of wavelengths used for ICP-OES analysis. All samples were run with Y internal standard. Analysis of metal content in the samples from the precipitation of copper from e-waste was carried out using ICP-MS on an Agilent 7900 Inductively Coupled Plasma Mass Spectrometer. All samples were diluted in 2% nitric acid and taken up by a peristaltic pump at a rate of 0.3 rps with the following argon plasma conditions: 1550 W RF power, 15 L min−1 plasma flow, 0.9 L min−1 auxillary flow and 1.09 L min−1 nebulizer flow. The instrument was operated in ‘spectrum multi-tune acquisition mode’ and three replicate runs per sample were employed to ensure good counting statistics. Each isotope was integrated for 0.1 s per point giving a total integrations time of 0.3 s per unit mass. A pulse counting to analogue counting factor was determined across the entire mass range at the start of the analysis day. See Table S6† for summary of ICP-MS isotopes used for analysis. ICP calibration standards were obtained from VWR International, SCP science or Sigma-Aldrich.FTIR spectroscopy
Fourier transform-infrared measurements (ATR FT-IR) were collected on a Thermo Scientific Nicolet Summit X FT-IR instrument over the range 4000–400 cm−1 at a resolution of 4 cm−1 or on a Shimadzu IRSpirit over the range 4000–400 cm−1 at a resolution of 4 cm−1.Powder X-ray diffraction (PXRD)
Powder X-Ray Diffraction (PXRD) data of LO, [Cu(LO)]n and [Cu(LP)]n were recorded on a Bruker D8 Advance diffractometer in transmission geometry with Cu Kα radiation (λ = 1.5406 Å). Data were collected over the 2θ range 5°–30° for 1 h. PXRD data of LP were collected on a Rigaku Miniflex diffractometer in reflection geometry with Cu Kα1 and Kα2 radiation (Kα1 = 1.540593 Å, Kα2 = 1.544414 Å) over the 2θ range 5°–30° for 40 min. Data were analysed using the Topas Academic (version 6) software suite and plotted using DAWN (version 2.35) science software.66Single crystal X-ray diffraction
Colourless plates of LO were grown by slow evaporation from a saturated solution of LO in ethanol. Crystals suitable for X-ray diffraction formed over the course of one week. X-ray crystallographic data of LO were collected at 120 K on a Bruker APEX-II CCD diffractometer using graphite monochromated Mo-Kα radiation (λ = 0.71073 Å). The structure was solved using SHelXT direct methods and refined using a full matrix least-square refinement using ShelXL.67–69 All programs were used within the Olex Suites.70Scanning electron microscopy-energy dispersive X-ray spectroscopy (SEM-EDS)
SEM images of LO, [Cu(LO)]n, LP and [Cu(LP)]n were collected on a ThermoScientific Phenom G6 Pure Microscope equipped with a backscattered electron detector and energy dispersive X-ray spectroscopy (EDS) detector. Samples were loaded onto carbon tabs attached to SEM pin stubs. Images were taken with a resolution of ≤15 nm at an energy of 15 kV and magnification level of between 660× and 800×.X-ray fluorescence (XRF)
XRF data of [Cu(LO)]n and [Cu(LP)]n were collected on a Bruker S2 Puma fitted with a HighSense Peltier cooled silicon drift detector with an element range of Na–Am. Samples were loaded into the sample holders and sealed using Mylar foil with a thickness of 3.6 μm and run with air mode.Mass spectrometry (matrix assisted laser desorption/ionisation; MALDI)
MALDI-TOF MS measurements of LO, [Cu(LO)]n, LP and [Cu(LP)]n were collected on a SolariXR FT-ICR mass spectrometer with a Bruker Daltonics 7 T superconducting magnet. Samples were added to a solution of 4-cyano-2-nitrophenol (matrix) in acetonitrile (solvent) and loaded onto the analysis plate dropwise. Samples were also prepared in 2,5-dihydroxybenzoic acid or sinapinic acid in TFA.Life cycle inventory (LCI)
The raw materials and process inputs for both the precipitation and solvent extraction processes were defined (see ESI Table S9†). The data used to compile the inventory were taken from lab-scale experimental results, reported above. In both processes, the same volume and concentration of leach solution were used to allow a direct comparison to be made. The quantities for all materials and assemblies were then adjusted according to the functional unit (see ESI Table S9†). The ligands used (LP for precipitation and ACORGA M5910 for solvent extraction) were assigned the same lifespan in addition to the solvent (kerosene) used in the solvent extraction process. The lifespan was set as 30 days, assuming 1 use per day. Approximate emission factors for the ligands were used71 due to the scarce availability of emission factors in accessible databases for complex organic molecules. For example, an emission factor for toluene was used71 to estimate the CO2e emissions for kerosene, given that a factor could not be found for kerosene. The same dataset was also used to provide a CO2e emission factor for the sulfuric acid used in both processes.71 Emission factors for water, electricity and lab equipment used in both processes were obtained from the UK government reports of greenhouse gas conversion factors for 2024.72 Finally, literature values were used to provide estimates of CO2e emission factors for the waste raffinate solution obtained after separating the copper,73 and e-waste, which was included in the calculations as an avoided impact.74 The stir plate and centrifuge used were given the same lifespan of 20 years, assuming 8 h per day usage for 220 days a year, accounting for weekends and shutdown time. The capacities of the centrifuge (8 mL) and stir plate (20 mL) were also taken into account. The magnetic stir bar used in conjunction with the stir plate was omitted from the LCI given the equipment was the same for both processes.Data availability
Raw data associated with ICP, FTIR, PXRD, XRF, and life-cycle analysis are deposited in the Edinburgh DataShare repository at https://doi.org/10.7488/ds/7889.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. S. M. V. thanks the School of Chemistry for an Edinburgh Doctoral College Scholarship. We thank Dr Lorna Eades for assistance with ICP-MS analysis, Dr Rebecca Rae for assistance with SEM and XRF measurements, Rhona Cowan for assistance with MALDI measurements, Dr Andrew Hall for assistance with NMR spectroscopy measurements and Dr Gary Nichol for assistance with single crystal X-ray diffraction measurements.References
- W. Zhou, X. Liu, X. Lyu, W. Gao, H. Su and C. Li, J. Cleaner Prod., 2022, 368, 133095 CrossRef CAS.
- K. J. J. Kuipers, L. F. C. M. van Oers, M. Verboon and E. van der Voet, Glob. Environ. Change., 2018, 49, 106–115 CrossRef.
- G. M. Mudd, Z. Weng and S. M. Jowitt, Econ. Geol., 2013, 108(5), 1163–1183 CrossRef CAS.
- E. Commission, https://single-market-economy.ec.europa.eu/sectors/raw-materials/areas-specific-interest/critical-raw-materials_en, 2024.
- M. Jun, R. R. Srivastava, J. Jeong, J.-c. Lee and M.-s. Kim, Green Chem., 2016, 18, 3823–3834 RSC.
- Y. Liu, H. Wang, Y. Cui and N. Chen, Int. J. Environ. Res. Public Health, 2023, 20, 3885 CrossRef CAS PubMed.
- Q. Li, Y. Wang, Z. Chang, W. El Kolaly, F. Fan and M. Li, J. Water Proc. Eng., 2024, 58, 104746 CrossRef.
- N. A. A. Qasem, R. H. Mohammed and D. U. Lawal, npj Clean Water, 2021, 4, 36 CrossRef CAS.
- J. Chen, Z. Wang, Y. Wu, L. Li, B. Li, D. a. Pan and T. Zuo, Resour., Conserv. Recycl., 2019, 146, 35–44 CrossRef.
- X. Li, B. Ma, C. Wang and Y. Chen, Sustainable Mater. Technol., 2024, 41, e01026 CrossRef CAS.
- J. A. Barragan, C. Ponce de Leon, J. R. Aleman Castro, A. Peregrina-Lucano, F. Gomez-Zamudio and E. R. Larios-Duran, ACS Omega, 2020, 5, 12355–12363 CrossRef CAS PubMed.
- A. M. Elgarahy, M. G. Eloffy, A. K. Priya, A. Hammad, M. Zahran, A. Maged and K. Z. Elwakeel, Sustainable Chem. Environ., 2024, 7, 100124 Search PubMed.
- E. Hsu, K. Barmak, A. C. West and A.-H. A. Park, Green Chem., 2019, 21, 919–936 RSC.
- A. Elshkaki, T. E. Graedel, L. Ciacci and B. K. Reck, Glob. Environ. Change., 2016, 39, 305–315 Search PubMed.
- Q. Xie, Y. Liu, Q. Chen, X. Geng, C. Liang, P. Zhang and L. Wang, Hydrometallurgy, 2023, 221, 106128 Search PubMed.
- I. Birloaga and F. Vegliò, J. Environ. Chem. Eng., 2016, 4, 20–29 CrossRef CAS.
- C. Yang, J. Li, Q. Tan, L. Liu and Q. Dong, ACS Sustainable Chem. Eng., 2017, 5, 3524–3534 Search PubMed.
- Y. Xiao, Y. Yang, J. van den Berg, J. Sietsma, H. Agterhuis, G. Visser and D. Bol, Hydrometallurgy, 2013, 140, 128–134 CrossRef CAS.
- Y. Xue and Y. Wang, Green Chem., 2020, 22, 6288–6309 Search PubMed.
- A. Fathima, J. Y. B. Tang, A. Giannis, I. Ilankoon and M. N. Chong, Chemosphere, 2022, 298, 134340 CrossRef CAS PubMed.
- M. I. A. Abdel Maksoud, A. M. Elgarahy, C. Farrell, A. a. H. Al-Muhtaseb, D. W. Rooney and A. I. Osman, Coord. Chem. Rev., 2020, 403, 213096 Search PubMed.
- Y. Gu, Y. Sun, Y. Zhang, H. Chi, W. Zhang, Q. Liang and R. Jing, Nano–Micro Lett., 2014, 6, 80–87 Search PubMed.
- A. E. Chávez-Guajardo, J. C. Medina-Llamas, L. Maqueira, C. A. S. Andrade, K. G. B. Alves and C. P. de Melo, Chem. Eng. J., 2015, 281, 826–836 CrossRef.
- Y. Zhang, Z. Xie, Z. Wang, X. Feng, Y. Wang and A. Wu, Dalton Trans., 2016, 45, 12653–12660 Search PubMed.
- A. Iqbal, M. R. Jan, J. Shah and M. N. Sarwar, J. Chem. Technol. Biotechnol., 2023, 98, 967–977 CrossRef CAS.
- S. Mariyam, S. Zuhara, T. Al-Ansari, H. Mackey and G. McKay, Adsorption, 2022, 28, 185–196 CrossRef CAS.
- E. A. Ajiboye, V. Aishvarya and J. Petersen, Minerals, 2023, 13, 1285 CrossRef CAS.
- A. I. Carrick, E. D. Doidge, A. Bouch, G. S. Nichol, J. Patrick, E. R. Schofield, C. A. Morrison and J. B. Love, Chemistry, 2021, 27, 8714–8722 Search PubMed.
- Y. Hong, D. Thirion, S. Subramanian, M. Yoo, H. Choi, H. Y. Kim, J. F. Stoddart and C. T. Yavuz, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 16174–16180 CrossRef CAS PubMed.
- A. I. Carrick, J. Patrick, E. R. Schofield, P. O’Shaughnessy, B. Breeze, J. B. Love and C. A. Morrison, Sep. Purif. Technol., 2024, 333, 125893 CrossRef CAS.
- J. G. O'Connell-Danes, B. Ozen Ilik, E. E. Hull, B. T. Ngwenya, C. A. Morrison and J. B. Love, ACS Sustainable Chem. Eng., 2024, 12, 9301–9305 CrossRef.
- I. Carson, K. J. MacRuary, E. D. Doidge, R. J. Ellis, R. A. Grant, R. J. Gordon, J. B. Love, C. A. Morrison, G. S. Nichol, P. A. Tasker and A. M. Wilson, Inorg. Chem., 2015, 54, 8685–8692 CrossRef CAS PubMed.
- E. A. Mowafy and D. Mohamed, Sep. Purif. Technol., 2016, 167, 146–153 CrossRef CAS.
- E. D. Doidge, L. M. M. Kinsman, Y. Ji, I. Carson, A. J. Duffy, I. A. Kordas, E. Shao, P. A. Tasker, B. T. Ngwenya, C. A. Morrison and J. B. Love, ACS Sustainable Chem. Eng., 2019, 7, 15019–15029 CrossRef CAS.
- J. J. M. Nelson and E. J. Schelter, Inorg. Chem., 2019, 58, 979–990 CrossRef CAS PubMed.
- J. G. O'Connell-Danes, B. T. Ngwenya, C. A. Morrison and J. B. Love, Nat. Commun., 2022, 13, 4497 CrossRef PubMed.
- H. Wu, Y. Wang, C. Tang, L. O. Jones, B. Song, X. Y. Chen, L. Zhang, Y. Wu, C. L. Stern, G. C. Schatz, W. Liu and J. F. Stoddart, Nat. Commun., 2023, 14, 1284 CrossRef CAS PubMed.
- J. O'Connell-Danes, B. T. Ngwenya, C. A. Morrison, G. S. Nichol, L. H. Delmau and J. B. Love, JACS Au, 2024, 4, 798–806 CrossRef PubMed.
- S. S. M. Vance, M. Mojsak, L. M. M. Kinsman, R. Rae, C. Kirk, J. B. Love and C. A. Morrison, Inorg. Chem., 2024, 63, 9332–9345 CrossRef CAS PubMed.
- J. R. Turkington, P. J. Bailey, J. B. Love, A. M. Wilson and P. A. Tasker, Chem. Commun., 2013, 49, 1891–1899 RSC.
- A. M. Wilson, P. J. Bailey, P. A. Tasker, J. R. Turkington, R. A. Grant and J. B. Love, Chem. Soc. Rev., 2014, 43, 123–134 RSC.
- R. S. Forgan, D. K. Henderson, F. E. McAllister, P. A. Tasker, F. J. White, J. Campbell and R. M. Swart, Can. Metall. Q., 2013, 47, 293–300 CrossRef.
- R. S. Forgan, B. D. Roach, P. A. Wood, F. J. White, J. Campbell, D. K. Henderson, E. Kamenetzky, F. E. McAllister, S. Parsons, E. Pidcock, P. Richardson, R. M. Swart and P. A. Tasker, Inorg. Chem., 2011, 50, 4515–4522 CrossRef CAS PubMed.
- H. Long Le, J. Jeong, J.-C. Lee, B. D. Pandey, J.-M. Yoo and T. H. Huyunh, Miner. Process. Extr. Metall. Rev., 2011, 32, 90–104 CrossRef CAS.
- A. Kumari, M. K. Jha, J.-c. Lee and R. P. Singh, J. Cleaner Prod., 2016, 112, 4826–4834 CrossRef CAS.
- M. D. Rao, K. K. Singh, C. A. Morrison and J. B. Love, Sep. Purif. Technol., 2021, 263, 118400 CrossRef CAS.
- N. S. Babu and S. M. Reddy, presented in part at the National Seminar on Impact of Toxic Metals, Minerals and Solvents Leading to Environmental Pollution, 2014.
- M. R. S. Foreman, R. K. Johansson, G. Mariotti, I. Persson, B. E. Tebikachew and M. S. Tyumentsev, RSC Sustainability, 2024, 2, 655–675 RSC.
- Q. Shi, S. Yan, C. Wang, C. Zeng, H. Hu, M. Chen, M. Chen and Q. Zhang, Environ. Technol., 2023, 44, 1798–1807 CrossRef CAS PubMed.
- H. Zhang, J. Chen, S. Ni, C. Bie, H. Zhi and X. Sun, J. Environ. Manage., 2022, 304, 114164 CrossRef CAS PubMed.
- L. M. M. Kinsman, B. T. Ngwenya, C. A. Morrison and J. B. Love, Nat. Commun., 2021, 12, 6258 CrossRef CAS PubMed.
- A. Nag, M. K. Singh, C. A. Morrison and J. B. Love, Angew. Chem., Int. Ed., 2023, 62, e202308356 CrossRef CAS PubMed.
- Y. Yamini, J. Hassan and M.-H. Karbasi, Microchim. Acta, 2004, 148, 305–309 CrossRef CAS.
- T. C. Stamatatos, G. Vlahopoulou, C. P. Raptopoulou, V. Psycharis, A. Escuer, G. Christou and S. P. Perlepes, Eur. J. Inorg. Chem., 2012, 2012, 3121–3131 CrossRef CAS.
- L. Stoica and I. Lacatusu, Int. J. Environ. Waste Manag., 2012, 9, 293–312 CrossRef CAS.
- E. Yilmaz and M. Soylak, Environ. Monit. Assess., 2014, 186, 5461–5468 CrossRef CAS PubMed.
- A. N. Abu-Baker and M. A. Al-Qudah, Appl. Phys. A, 2016, 122, 765 CrossRef.
- M. R. Healy, J. W. Roebuck, E. D. Doidge, L. C. Emeleus, P. J. Bailey, J. Campbell, A. J. Fischmann, J. B. Love, C. A. Morrison, T. Sassi, D. J. White and P. A. Tasker, Dalton Trans., 2016, 45, 3055–3062 RSC.
- K. C. Sole and O. Tinkler, presented in part at the Copper Cobalt Africa, incorporating the 8th Southern African Base Metals Conference, Livingstone, Zambia, 2015.
- W. Zhang, J. Long, Z. Wei and L. Alakangas, Environ. Earth Sci., 2016, 75, 1462 CrossRef.
- A. Smith, Coord. Chem. Rev., 2003, 241, 61–85 CrossRef CAS.
- N. B. Colthup, L. H. Daly and S. E. Wiberley, Introduction to Infrared and Raman Spectroscopy, Academic Press, 3rd edn, 1990 Search PubMed.
- B. S. Shyamala and V. Jayatyagaraju, Synth. React. Inorg. Met.-Org. Chem., 2006, 33, 63–75 CrossRef.
- Z. H. You, Y. H. Chen, Y. Tang and Y. K. Liu, Org. Lett., 2019, 21, 8358–8363 Search PubMed.
- T. E. Ali, M. A. Assiri, M. A. Ibrahim, E. M. El-Amin and I. S. Yahia, Heterocycles, 2019, 98, 114–125 CrossRef.
- M. Basham, J. Filik, M. T. Wharmby, P. C. Chang, B. El Kassaby, M. Gerring, J. Aishima, K. Levik, B. C. Pulford, I. Sikharulidze, D. Sneddon, M. Webber, S. S. Dhesi, F. Maccherozzi, O. Svensson, S. Brockhauser, G. Naray and A. W. Ashton, J. Synchrotron Radiat., 2015, 22, 853–858 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- Winnipeg sewage treatment program, SEWPCC Process Selection Report, 2011.
- UK Government greenhouse gas reporting: conversion factors 2024, https://www.gov.uk/government/publications/greenhouse-gas-reporting-conversion-factors-2024, 2024.
- S. Çapa, A. Özdemir, Z. Günkaya, A. Özkan and M. Banar, J. Water Process Eng., 2022, 49, 103002 CrossRef.
- J. Hong, W. Shi, Y. Wang, W. Chen and X. Li, Waste Manag., 2015, 38, 357–365 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Extended precipitation experiments, analytical ICP data, PXRD refinement data, extended XRF data, extended MALDI mass spectrometry data, life cycle inventory and crystallographic tables. CCDC 2404167. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4gc06466f |
| This journal is © The Royal Society of Chemistry 2025 |