
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
One-pot one-step enzymatic synthesis of 5-(aminomethyl)-2-furancarboxylic acid from 5-(hydroxymethyl) furfural†
Eleonora Fornoni a,
Ammar Al-Shameria,
Pablo Domínguez de Maríab and
Volker Sieber*acde
a,
Ammar Al-Shameria,
Pablo Domínguez de Maríab and
Volker Sieber*acde
aChair of Chemistry of Biogenic Resources, Technische Universität München, Campus Straubing, Schulgasse, 16, 94315 Straubing, Germany. E-mail: sieber@tum.de
bSustainable Momentum, SL. Av. Ansite 3, 4-6. – 35011 Las Palmas de Gran Canaria, Canary Is, Spain
cSynBioFoundry@TUM, Technische Universität München, Campus Straubing, Schulgasse, 22, 94315 Straubing, Germany
dTUM Catalysis Research Center, Technische Universität München, Campus Straubing, Ernst-Otto-Fischer-Straße, 1, 85748 Garching, Germany
eSchool of Chemistry and Molecular Biosciences, The University of Queensland, Copper Road, 68, 4072 St. Lucia 4072, Australia
First published on 23rd May 2025
Abstract
5-(Aminomethyl)-2-furancarboxylic acid (AMFCA) represents a biogenic replacement to fossil-based monomers for the production of semi-aromatic polyamides, thermoplastics widely used in the automotive, electrical and packaging industries. AMFCA has been synthesized via chemical, chemo-enzymatic and enzymatic one-pot two-step transformations. Herein, the conversion of 5-(hydroxymethyl)-furfural (HMF) to AMFCA is explored through a one-pot, one-step, cell-free, enzymatic process, reaching titers in the range of 3.35–4.62 g L−1. The four-enzyme cascade includes an engineered HMF oxidase from Methylovorus sp. MP688, an aldehyde dehydrogenase from Sphingobium sp. SYK-6 and an omega-transaminase from Chromobacterium violaceum as main biocatalysts. L-Alanine is used as an amine donor, sustainably regenerated by an alanine dehydrogenase, which also guarantees an intrinsic nicotinamide cofactor balance. As environmental metrics, the E-factor and the “Global Warming Potential” (GWP, kg CO2 per kg AMFCA) are introduced for both the upstream and downstream processes of AMFCA production for the first time. The sustainability assessment provides valuable insights into the hotspots to be improved for minimized environmental burden: operating at high substrate loadings, generation of wastewater effluents that can be mildly treated, optimized volume-to-product ratios in chromatographic steps, and introduction of renewable energy in the chemical plant.
Green foundation1. The polyamide monomer 5-(aminomethyl)-2-furancarboxylic acid (AMFCA) is synthesized from cellulose-derived substrates (HMF and HMFCA) in a one-pot one-step system via enzymatic catalysis in aqueous media, under benign reaction conditions.2. AMFCA syntheses are performed without the use of organic solvents and with purified enzymes as sole catalysts. The biogenic amino acid L-alanine is employed as an amine donor and it is enzymatically regenerated from pyruvate and ammonia as the nitrogen source providing redox neutrality. 3. Increasing the reaction titer would reduce the E-factor and Global Warming Potential of AMFCA syntheses. The potential of the developed system should be assessed for the treatment of HMF-rich aqueous effluents of biorefineries. Alternative formulations for the employed bio-catalysts (e.g. crude extracts of recombinant cells and immobilized enzymes) would also contribute to a greener process. |
Introduction
In today's world, an increasing awareness and sense of responsibility for environmental concerns are driving the development of sustainable production routes. For the chemical industry, a promising platform chemical is 5-(hydroxymethyl)furfural (HMF). Often referred to as the “sleeping giant” because of its high untapped potentiality,1 HMF has been included in the top 14 most important bio-based molecules by the US Department of Energy.2 HMF is obtained via acidic dehydration of C6 sugars (especially fructose). In well-established multi-step processes, it is directly derived from cellulose hydrolysis.3 In a downstream process, HMF can be functionalized to a broad panel of fine and bulk chemicals, which represent sustainable replacements for many petrol-based products.3,4Some HMF-derivatives have been explored as bio-based polymer components. For example, 2,5-furandicarboxylic acid (FDCA), obtained via multiple HMF oxidations, has been employed for poly(ethylene-2,5-furandicarboxylate) (PEF) synthesis on a pilot-plant scale.5 PEF is a bio-based and biodegradable alternative to fossil-derived polyethylene terephthalate (PET). More recently, an oxidized and aminated derivative of HMF, 5-(aminomethyl)-2-furancarboxylic acid (AMFCA), has been included in the structure of aromatic and semi-aromatic polyamides. The obtained poly(5-aminomethyl-2-furoic acid) (PAMF) showed thermal and mechanical characteristics comparable to commercially available polymers.6 These materials are highly thermo- and chemical-resistant thermoplastics used in the automotive, electronics and packaging industries. Fossil-based terephthalic acid and hexamethylene diamine are the monomers usually condensed to yield the most commercialized semi-aromatic polyamides.7 Other than for polymer production, AMFCA was also studied for applications in the pharma industry as an inhibitor of the γ-aminobutyric acid aminotransferase8 or of telomer elongation in cancer cells.9
Over the past few years, few studies have been dedicated to AMFCA production from HMF. Two chemical synthetic routes were developed by Lankenau and Kanan (with furfural as a starting material)10 and by Zhu and co-workers.11 In both cases, high temperatures (90–155 °C), strong acids, high-pressure H2 atmospheres (3 MPa H211) and/or large volumes of organic solvents (e.g. methanol11) were employed. In more recent works, milder chemo-enzymatic routes have been proposed. The Froidevaux and Heuson group showed the possibility of a Pt/SiO2 oxidation of HMF to 2-formyl-5-furancarboxylic acid (FFCA) followed by an enzymatic amination to AMFCA. For the biocatalytic step, an immobilized omega-transaminase from Chromobacterium violaceum (Cv-ωTA) and isopropylamine (IPA) or (S)-α-methylbenzylamine as amine donors was used.12 Similarly, Ning Li's group established a laccase-TEMPO oxidation of HMF to FFCA coupled to a subsequent amination with resting E. coli whole-cells expressing Cv-ωTA, using L-alanine (L-Ala) as an amine donor.13 Recently, Hyungdon Yun and collaborators reported the first fully enzymatic synthesis of AMFCA from HMF in two steps.14 The HMF oxidation was performed via an engineered galactose oxidase15 combined with an aldehyde reductase from Synechocystis sp. PCC 6906. Horseradish peroxidase (HRP) and catalase were added as auxiliary catalysts for galactose oxidase activation. Transamination was granted by a transaminase from Shimia marina. Isopropylamine served as an amine donor and dimethyl sulfoxide (DMSO) was employed as (co)solvent for all substrate solutions.
These works ventured towards a more sustainable AMFCA synthesis, yet with some limitations. In the chemo-enzymatic systems, the unspecificity of chemical oxidation methods led to the accumulation of over-oxidized FFCA (FDCA). This side-product's high concentrations mainly reflected in lowered reaction yields.13 Moreover, all conversions were performed in a one-pot two-step fashion due to the incompatibility of reaction conditions between the oxidation and amination steps.
Inspired by these previous works, and for further advancement in AMFCA sustainable production, we designed a purely enzymatic synthesis route starting from HMF. In the system (Scheme 1), HMF is firstly oxidized by either an HMF oxidase (HMFO) or an aldehyde dehydrogenase (ALDH) to respectively yield 2,5-diformylfuran (DFF) or 5-hydroxymethyl-2-furancarboxylic acid (HMFCA).16 These intermediates are oxidized to FFCA by the same enzymes. Once formed, FFCA is aminated to AMFCA by Cv-ωTA with L-Ala as an amine donor. L-Ala was chosen because of its possible enzymatic regeneration from ammonia with an alanine dehydrogenase (AlaDH) and its biogenic origin. Moreover, the process occurs in aqueous media, which may enable the treatment/valorization of crude aqueous effluents from lignocellulose biorefineries. Despite the low furan concentrations, a pre-treatment with our envisioned cascade would simultaneously create valuable and cleaner effluents (e.g. for fermentations without furan inhibition issues).17 The proposed enzymatic catalysis may bring advantages to AMFCA preparation since HMF can be selectively oxidized to FFCA, the oxidation and amination can simultaneously occur in the same reaction vessel, and mild conditions and coupled co-factors and amine-donors are employed. Moreover, when compared to whole-cell catalysis, a cell-free enzymatic approach corresponds to a tighter carbon flux control, smoother mass-transfer, easier catalyst fine-tuning and milder treatment of wastewater streams.18–20
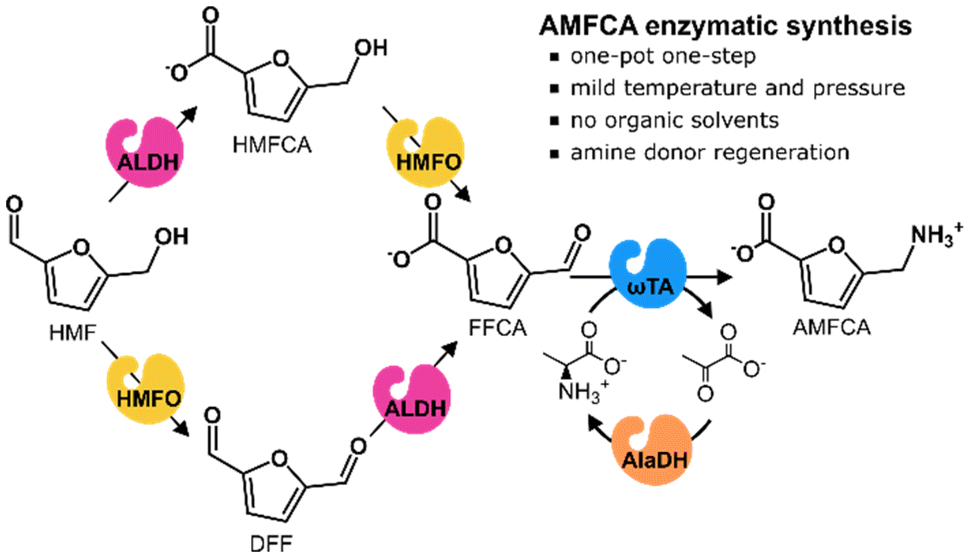 | ||
| Scheme 1 Enzymatic conversion of HMF to AMFCA proposed in this work. HMFO: HMF oxidase. ALDH: aldehyde dehydrogenase. ωTA: omega-transaminase. AlaDH: alanine dehydrogenase. | ||
Results and discussion
HMF oxidase selection
For the cascade development, we firstly compared a pool of seven oxidases. An HMFO from Methylovorus sp. MP688 (MetspHMFO)21 and one from Mycobacterium sp. MS1601 (MycspHMFO, mutant Y444F)22 have previously been described to efficiently catalyze HMF oxidation. Other five putative HMFOs were found through pBLAST research on the NCBI database using the MetspHMFO sequence as a query. All selected HMFOs and their relative sequence similarities to MetspHMFO are reported in Table S5 (ESI).† HMFO genes were all cloned in pET28a vectors with N-terminal hexahistidine tags. MycspHMFO presented an additional maltose-binding domain (MBP) at the N-terminus. The constructs were recombinantly expressed in E. coli BL21(DE3). Only three of the oxidases (MetspHMFO, MycspHMFO and PcHMFO) were expressed in soluble form (Fig. S1, ESI†). These HMFOs were further purified via affinity chromatography and kinetically characterized on all substrates of interest.The kinetic data (Table S6, ESI†) showed that all HMFOs displayed the highest catalytic efficiencies (kcat/KM) for the oxidation of the hydroxy groups of HMF and HMFCA, in line with their addition to the family of glucose-methanol-choline (GMC) oxidoreductases.23,24 Overall, MetspHMFO registered the highest turnover numbers (kcat) and high kcat/KM for the oxidations of interest in Scheme 1. Thus, MetspHMFO was the oxidase of choice for further experiments.
HMFCA conversion to AMFCA
AMFCA production was initially studied in a simplified enzymatic cascade starting from HMFCA as the substrate (Scheme 2). As a main catalyst, MetspHMFO was employed for HMFCA oxidation to FFCA and Cv-ωTA for FFCA amination.12,25,26 The amine donor (L-Ala) regeneration was guaranteed by an alanine dehydrogenase from Bacillus subtilis (BsAlaDH)27 and a formate dehydrogenase from Candida bouidinii (CbFDH)28 allowed NADH regeneration through formate oxidation to CO2.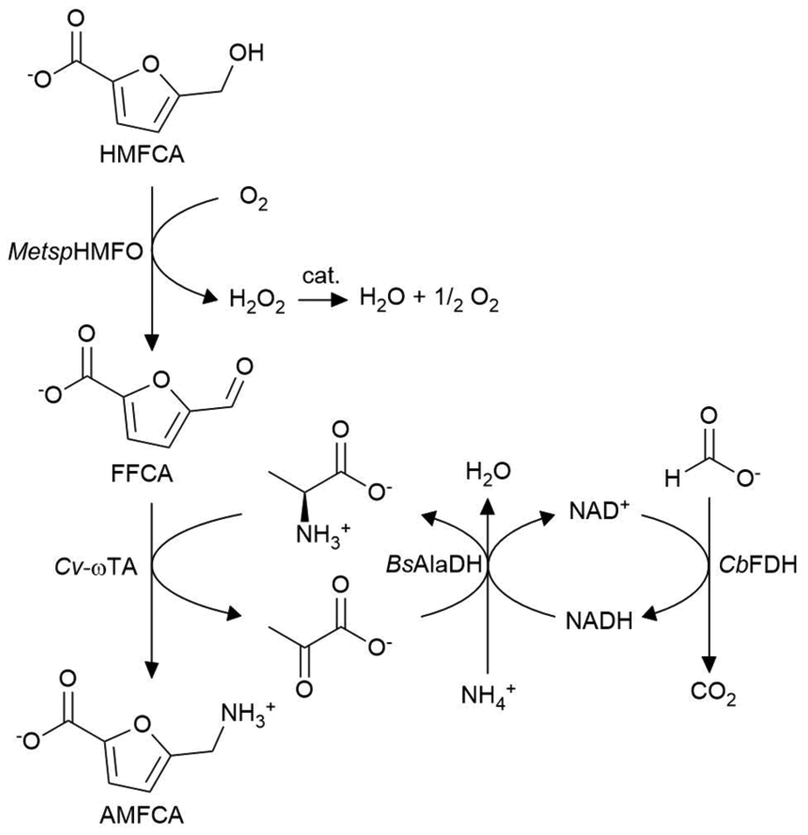 | ||
| Scheme 2 Reaction scheme for the enzymatic conversion of HMFCA to AMFCA. MetspHMFO: Methylovorus sp. MP688 HMF oxidase. Cv-ωTA: Chromobacterium violaceum omega-transaminase. BsAlaDH: Bacillus subtilis alanine dehydrogenase. CbFDH: Candida bouidinii formate dehydrogenase. cat.: catalase. | ||
The enzymatic cascade was firstly implemented in one-pot with 20 mM HMFCA (Fig. 1a). Within the first 6 h, complete conversion to AMFCA was observed. In this reaction time, a titer of 2.78 g L−1 and a space–time yield of 0.44 g L−1 h−1 were reached. To the best of our knowledge, this is the first time that a direct conversion of HMFCA to AMFCA is reported. Other enzymatic processes for AMFCA synthesis started from either FFCA or HMF.14,29
 | ||
| Fig. 1 Time courses for HMFCA conversions to AMFCA with a 20 mM (a) or 60 mM (b) initial substrate concentration. Reactions performed in a 1 mL final volume with 50 mM L-Ala in 100 mM NaPi buffer, pH 8.0, at 30 °C, under constant oscillation. Enzyme concentrations: 4.19(a)–5.77(b) μM MetspHMFO, 0.58(a)–9.53(b) μM Cv-ωTA, and 0.56 μM BsAlaDH. Further details are reported in Table S3, ESI.† | ||
Starting from higher substrate loadings (40–50 mM), yields of maximum 36% could be obtained under the same reaction conditions (Fig. S2, ESI†). The reason behind such low yields could be the inhibition of Cv-ωTA by FFCA with an inhibition constant (Ki) of 8.63 ± 2.39 mM (Table S8, ESI†). Consistently, previous works have also reported very low yields for FFCA amination with Cv-ωTA when the substrate was applied at concentrations much higher than the FFCA inhibition constant.13,29
Accounting for Cv-ωTA FFCA inhibition, the enzyme ratio Cv-ωTA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MetspHMFO in the reaction was adjusted to 62:38. This resulted in the conversion of 60 mM HMFCA to AMFCA with a yield of 87% (Fig. 1b). After 8 h, an AMFCA titer of 7.37 ± 0.38 g L−1 and a space–time yield (STY) of 0.92 g L−1 h−1 were reached. Other chemo-enzymatic or purely enzymatic syntheses of AMFCA resulted in STYs in the range 0.40–0.47 g L−1 h−1 even when 99% yields were reached.11–14,29,30
:MetspHMFO in the reaction was adjusted to 62:38. This resulted in the conversion of 60 mM HMFCA to AMFCA with a yield of 87% (Fig. 1b). After 8 h, an AMFCA titer of 7.37 ± 0.38 g L−1 and a space–time yield (STY) of 0.92 g L−1 h−1 were reached. Other chemo-enzymatic or purely enzymatic syntheses of AMFCA resulted in STYs in the range 0.40–0.47 g L−1 h−1 even when 99% yields were reached.11–14,29,30
HMFCA-cascade transfer to HMF conversion
The substrate promiscuity of MetspHMFO (Table S6, ESI†) theoretically allows the synthesis of AMFCA from HMF with the same pool of enzymes used for HMFCA functionalization. MetspHMFO oxidizes HMF to DFF and further to FFCA, which is then aminated to AMFCA by Cv-ωTA.With these assumptions, a series of reactions were performed for the conversion of 18 mM HMF employing the same enzymes used for HMFCA conversions. However, no AMFCA could ever be formed (example reaction in Fig. 2d). HMF was consumed over time, but only accumulations of FFCA and the side-product 5-(aminomethyl)furan-2-carbaldehyde (AMFA) were detected (Fig. S8, ESI†). AMFA presence suggests the formation of its precursor DFF, which is unfortunately not detectable with the HPLC analysis used here.
 | ||
| Fig. 2 Time courses of 16 mM HMF conversions to AMFCA using different ALDHs: EcALDH (a), BovALDH (b), SphALDH (c), and no ALDH (d). Reactions were performed with 50 mM L-Ala in 100 mM NaPi buffer, pH 8.0, at 30 °C, under constant oscillation. Enzyme concentrations: 0.33(a–c)–1.53(d) μM MetspHMFO, 6.66 μM EcALDH (a), 6.51 μM BovALDH (b), 6.28 μM SphALDH (c), 0.58(d)–1.61(a–c) μM Cv-ωTA, and 0.57–0.68 μM BsAlaDH. Details in Table S4, ESI.† | ||
DFF and AMFA formations certainly were two of the main factors hindering any AMFCA formation. DFF most likely accumulated due to its slow oxidation by MetspHMFO (low kcat, Table S6, ESI†). DFF is widely known to lead to protein cross-linking, especially in the presence of primary amines.31 DFF was also found to strongly inhibit Cv-ωTA already at low concentrations (Ki = 2.20 ± 0.66 mM, Table S8, ESI†). On the other hand, AMFA represents a particularly reactive species: its amino and aldehyde functional groups can spontaneously react to give imines. AMFA can polymerize with other AMFA molecules or interact with any other amino- or aldehyde molecule in the reaction and thus prevent any further functionalization to the desired product.25
ALDH selection
The accumulation of DFF leading to the formation of AMFA seems the bottleneck of the cascade starting from HMF. One possibility to circumvent such accumulation would be a deviation of HMF oxidation to preferably form HMFCA instead of DFF. The addition of an aldehyde dehydrogenase (ALDH) to the cascade can ensure the formation of HMFCA from HMF. Additionally, ALDHs can oxidize DFF to FFCA and thus support DFF removal from the system. Including an ALDH also eliminates the need for cofactor regeneration resulting in a cofactor-balanced cascade with no net production of CO2.Hence, we investigated various ALDHs. An ALDH from E. coli (EcALDH) and one from Bos taurus (BovALDH) were previously reported for the efficient oxidation of HMF.32 A third ALDH from Sphingobium sp. SYK-6 (SphALDH) was studied for its high catalytic efficiency on substrates similar to HMF: m-hydroxybenzaldehyde, vanillin and syringaldehyde among others.33 The genes for the three variants were cloned in pET28a vectors with an N-terminal hexahistidine tag for purification; they were recombinantly expressed in E. coli BL21(DE3) and purified through affinity chromatography. The purified enzymes were kinetically characterized for the oxidation of the aldehyde functions on HMF, DFF and FFCA.
The kinetic parameters measured on the three ALDHs are summarized in Table 1. From a substrate perspective, FFCA appeared to be the least favorable substrate for all ALDHs (highest KM and lowest kcat/KM). On the other side, both HMF and DFF were well accepted by all three enzymes, with SphALDH registering the highest catalytic efficiencies on both HMF and DFF (10938 M−1 s−1 and 19220 M−1 s−1, respectively). Better conversions with SphALDH might be related to the higher similarity between its native substrate (syringaldehyde) and the furan derivatives used here.33 In contrast, EcALDH was originally reported for its activity on aliphatic aldehydes, such as 3-hydroxypropionaldehyde, isovaleraldehyde and propionaldehyde.34 BovALDH is naturally responsible for retinaldehyde oxidation in the bovine retina.35,36
| Enzyme | Substrate | KM [mM] | kcat [s−1] | kcat/KM [M−1 s−1] | Ki [mM] |
|---|---|---|---|---|---|
| n.d. = not detected. | |||||
| EcALDH | HMF | 0.51 ± 0.09 | 1.14 ± 0.04 | 2255 | n.d. |
| DFF | 0.32 ± 0.17 | 2.81 ± 0.62 | 8882 | n.d. | |
| FFCA | 10.53 ± 3.53 | 0.57 ± 0.11 | 54 | n.d. | |
| BovALDH | HMF | 0.004 ± 0.010 | 0.024 ± 0.001 | 5573 | n.d. |
| DFF | 0.02 ± 0.01 | 0.024 ± 0.002 | 1166 | n.d. | |
| FFCA | 17.80 ± 13.05 | 0.053 ± 0.025 | 2.96 | n.d. | |
| SphALDH | HMF | 0.07 ± 0.02 | 0.78 ± 0.08 | 10938 |
3.29 ± 1.17 |
| DFF | 0.03 ± 0.002 | 0.49 ± 0.01 | 19220 |
2.31 ± 0.11 | |
| FFCA | 0.05 ± 0.15 | 0.01 ± 0.01 | 245 | n.d. | |
Despite the highest catalytic efficiencies, SphALDH was also the only ALDH showing substrate inhibition at low concentrations of HMF and DFF (<3.5 mM). However, the inhibition was not complete, but partial37 (Fig. S6, ESI†).
All considered, each tested dehydrogenase actually represented a suitable candidate for our purposes.
AMFCA enzymatic production from HMF
The cascade in Scheme 3 represents the enzymatic synthesis of AMFCA from HMF when an ALDH is included in the biocatalyst pool. ALDHs mainly catalyze the oxidations of HMF to HMFCA, DFF to FFCA and AMFA to AMFCA. Different routes coexist to yield the final product. The direction prevalently followed by the carbon flux is determined by the fine balance between the enzyme activities. The routes result in a NAD+/NADH cofactor balance, except when HMF is oxidized to FFCA solely by MetspHMFO and FFCA is then aminated to AMFCA by Cv-ωTA. In this case, a net production of NAD+ is expected. However, DFF oxidation by MetspHMFO is unlikely to occur if we consider the much lower catalytic efficiency (kcat/KM) measured on DFF with MetspHMFO (Table S6, ESI†) compared to the efficiencies registered with the ALDHs tested here (Table 1).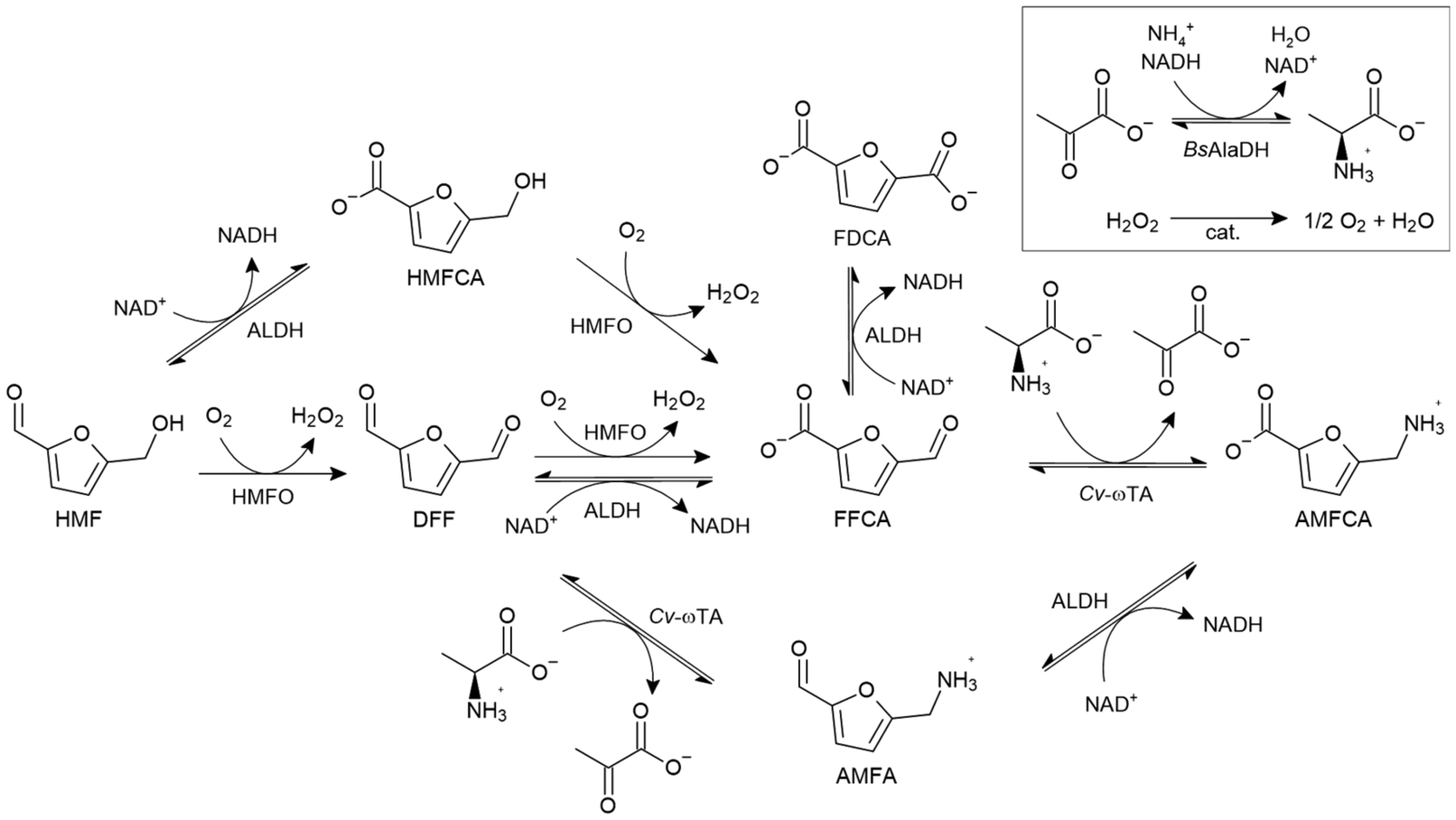 | ||
| Scheme 3 Reaction scheme of the enzymatic cascade for the conversion of HMF to AMFCA. HMFO: HMF oxidase. ALDH: aldehyde dehydrogenase. Cv-ωTA: omega-transaminase from Chromobacterium violaceum. BsAlaDH: alanine dehydrogenase from Bacillus subtilis. cat.: catalase. | ||
In light of these considerations on the cascade (Scheme 3) and the kinetic data registered for each of the involved enzymes, an in-silico model was firstly prepared with COPASI38 to optimize the enzyme ratios to be employed to yield the highest amount of AMFCA (data not shown). With the determined enzyme concentrations, the potential of the three selected ALDHs was separately assessed for the conversion of 16 mM HMF in one-pot one-step reactions (Fig. 2a–c). Each synthesis yielded almost full conversions (92–100% yield) to the final product within 24 h. A high accumulation of the intermediate HMFCA (∼8 mM) was observed after 2 h in the reactions with EcALDH and SphALDH. For both these reactions, very little or no AMFA formation was detected on HPLC after 4 h (Fig. S9, ESI†). These findings hint at an actual role of the ALDHs in pulling the carbon flux from HMF to HMFCA. However, when BovALDH was used, HMFCA accumulated to a maximum of ∼1.3 mM and AMFA formation was detected (Fig. S9, ESI†). The reduced impact of BovALDH on HMF oxidation is explained by its lower kcat for HMF compared to the other ALDHs (Table 1). The final conversion from HMF to AMFCA was anyways achieved in 8 h because of BovALDH contribution to DFF and AMFA oxidation.
Clearly, the carbon flux through the cascade simultaneously followed all the routes presented in Scheme 3. The ALDHs influenced the flux according to their specific activities on the aldehydes involved. Each of the presented conversions reached almost the full extent to form AMFCA with titers up to 2.41 g L−1.
To increase AMFCA titers, reactions with the different ALDHs and fed with 45 mM HMF were set up. The results (Fig. S3†) highlighted a clear difference depending on the included ALDH: EcALDH- and BovALDH-catalyzed reactions yielding 12% and 21% conversion to AMFCA, respectively. Only SphALDH could bring about a 62% yield. Compared to the other ALDHs, SphALDH was the only dehydrogenase that resulted in a total depletion of AMFA in 24 h (Fig. S10, ESI†). In this reaction (Fig. S3c†), a final titer of 4.10 ± 0.26 g L−1 was achieved in 24 h. In an AMFCA synthesis by the Ning Li group, titers up to 10.3 g L−1 were obtained.13 The AMFCA enzymatic synthesis by Hyungdon Yun and colleagues resulted in a final product concentration of ∼14.11 g L−1.14 The higher titers registered in these syntheses result from a higher substrate (HMF) load and higher conversions. To achieve more comparable production parameters, our synthesis needed further optimization
Biocatalyst optimization for AMFCA synthesis
In the reaction catalyzed by SphALDH (Fig. S3c†), around 40% of the initial carbon accumulated in intermediates (HMFCA and FFCA) and side products (5-(aminomethyl)-2-(hydroxymethyl)-furan (AMHMF) and FDCA). Different reaction engineering strategies were considered to funnel the carbon flux to AMFCA. A higher MetspHMFO concentration was applied to accelerate HMFCA oxidation to FFCA. The addition of CbFDH and formate after 8 h of reaction was tested to secure an efficient cofactor regeneration through the entire reaction and minimize FDCA formation. To recreate conditions similar to those of the reactions starting from HMFCA (Fig. 1b), a mixture of 50:50 L-Ala and pyruvate was employed for an efficient cofactor recirculation at the beginning of the reactions and favor HMF oxidation to HMFCA. Combinations of the strategies were tested as well. However, only comparable AMFCA yields could be obtained (Fig. S4, ESI†). The major limitation encountered under these conditions could be traced back to the broad promiscuity of the employed enzymes, especially MetspHMFO. Its higher activity and lower KM on HMF compared to those of HMFCA (Table 1) make it a far better catalyst for DFF rather than FFCA production.
Thus, we opted for a biocatalyst engineering approach. Dijkman et al. previously engineered MetspHMFO to specifically increase its activity on furans bearing a carboxylic acid.39 The mutation of valine 367 to an arginine (V367R) was found to be responsible for stabilizing carboxylic acid functions in the substrate binding pocket. Therefore, a site-directed mutagenesis was performed on the wild-type MetspHMFO to obtain MetspHMFO(V367R). The mutant was expressed in E. coli BL21(DE3), purified with affinity chromatography and kinetically characterized for HMF, DFF, FFCA and HMFCA oxidation. The results (Table S9†) show that the mutation did not particularly affect HMF and DFF oxidation kinetics. On the other side, it highly improved the enzyme catalytic efficiencies (kcat/KM) on FFCA and HMFCA. Considering the ratio between the catalytic efficiencies for HMF and HMFCA, MetspHMFO seems to prefer HMF oxidation over HMFCA with a factor of 6.45 (Table S6, ESI†). For the mutant, the efficiency ratio reduces to 0.48, meaning that HMFCA is the preferred oxidation substrate, in line with our synthetic purposes.
With its improved kinetic properties, MetspHMFO(V367R) was then employed to set up new reactions for AMFCA synthesis form HMF. The results (Fig. 3a) clearly show the advantage in HMFCA oxidation brought about by MetspHMFO(V367R). HMFCA as an intermediate peaked at ∼14 mM and was almost completely consumed within 24 h. Nevertheless, the reaction yield was comparable to the results obtained with the wild-type MetspHMFO (Fig. S3c, ESI†). The main limitations appeared to be the slower HMF oxidation and FFCA amination (Fig. 3a).
 | ||
| Fig. 3 Time courses of HMF conversions to AMFCA with the mutant MetspHMFO(V367R). Reactions were performed with 45(b)–50(a) mM HMF and 50 mM L-Ala in 100 mM NaPi buffer, pH 8.0, at 30 °C, under constant oscillation. Enzyme concentrations in a: 0.32 μM MetspHMFO(V367R), 8.98 μM SphALDH, 1.61 μM Cv-ωTA, and 1.64 μM BsAlaDH. In the reaction presented in (b), the concentrations of SphALDH, Cv-ωTA and BsAlaDH were increased 1.5 times. Further details are presented in Table S4, ESI.† | ||
Our strategy to improve AMFCA production consisted in increasing the biocatalyst concentrations: more SphALDH for a faster HMF oxidation, more Cv-ωTA to accelerate FFCA amination and more BsAlaDH to ensure an efficient nicotinamide cofactor recirculation. New cascade reactions were performed with 1.5-times increased concentrations of these catalysts. The reaction time course (Fig. 3b) shows that HMF was oxidized faster and completely depleted already after 4 h. HMFCA accumulated up to ∼16 mM and was totally consumed within 8 h. FFCA peaked at 8 h (∼15 mM) and was almost fully aminated within 24 h. As previously observed, FDCA formed starting from 8 h. Around 7% of the initial carbon from HMF accumulated in other products like DFF, AMHMF and AMFA. A final AMFCA yield of 72% was achieved, with FDCA representing the major side product (15% yield). The observed FDCA formation may be related to two factors. Firstly, FFCA could be oxidized to FDCA by SphALDH to provide reduced NADH needed by BsAlaDH for pyruvate amination. Secondly, FFCA oxidation could be attributed to MetspHMFO(V367R), which in fact showed a 57-fold improved catalytic efficiency on FFCA compared to the wild-type MetspHMFO (Tables S9 and S6, respectively, ESI†). Further studies would be needed to better understand the FDCA formation to tailor the carbon fluxes through the cascade to avoid unwanted reactivities. As an alternative, considering its importance for the polymer industry,16 FDCA could be considered a secondary product along with AMFCA as a primary product, as suggested by others as well.12
In our study, the cascade reaction with MetspHMFO(V367R) and the 1.5-times higher load of other biocatalysts represents the optimal condition for AMFCA enzymatic production. Starting from 5.67 g L−1 HMF (45 mM), a product titer of 4.62 ± 0.17 g L−1 was obtained. Our system titer remains lower than the values previously reported in the literature (9.31–14.11 g L−1).11,13,14 The closest system to ours, an enzymatic cascade presented by Hyungdon Yun's group, reported the need for separation into two-steps: the oxidation of HMF to FFCA and FFCA amination to AMFCA. The herein presented AMFCA enzymatic synthesis follows a one-pot one-step reaction, which can simplify the process set-up while enabling the valorization of crude biorefinery effluents with low-but-inhibitory furan concentrations. Previously, the main bottleneck for a one-pot one-step synthesis was identified in the promiscuity of the employed transaminase (a variant from Shimia marina).14 In Hyungdon Yun's system, IPA was employed in a 10-fold excess as an amine donor. In our system, an equimolar amount of L-Ala was sufficient to guarantee FFCA amination, particularly thanks to the in situ regeneration of the amine donor. In another chemo-enzymatic AMFCA synthesis by Ning Li's group, a titer of 10.3 g L−1 was also achieved in a two-step reaction.13 The two steps were separated by heat inactivation of the oxidizing laccase at 100 °C, pH adjustment and a 1:2 dilution of the reaction solution to allow transamination. While this approach showed remarkable high efficiency, avoiding intermediate steps would certainly benefit AMFCA production, especially when considering its applicability at industrial scale within synthetic pipelines.
AMFCA enzymatic production at a higher scale
After process optimization, the potential of the developed enzymatic cascade for AMFCA enzymatic synthesis in one-pot one-step was assessed at a higher scale. A reaction cascade was conducted starting from 46 mM HMF in 50 mL reaction volume with a similar composition to that of the synthesis in Fig. 3b and reduced enzyme load. The reaction was performed in a gas washing bottle with a filter plate with a constant moisturized-air flow, at 30 °C, while stirring. The time course of the reaction over 48 h is reported in Fig. 4a. HMF was completely depleted in 6 h. The intermediate HMFCA accumulated up to ∼23 mM at 6 h and was as well totally consumed in 24 h. Overall, the oxidations proceeded slower when compared to the performances in Fig. 3b. This was presumably due to the lower enzyme loadings and the remarkable differences in reaction conditions: in this system, the oxygen transfer rate was lower and enzyme inactivation was more significant. The AMFCA direct precursor, FFCA, accumulated during the first 24 h of reaction. Its slow amination by Cv-ωTA can be attributed to the enzyme's lower stability under the reaction conditions and/or to a slower L-Ala regeneration, in turn due to a slower NADH formation (slower SphALDH oxidation). FDCA generation in the final hours was presumably caused by the system's need for NADH regeneration and/or FFCA over-oxidation by MetspHMFO(V367R), as discussed above. However, the proposed system comprises various (sub)cascades, and further research would be necessary to fully understand all the carbon fluxes observed. Overall, a 51% yield and an AMFCA titer of 3.35 g L−1 were achieved in 48 h. | ||
| Fig. 4 a: time course of 46 mM HMF conversions to AMFCA on a 50 mL scale. The reaction was performed with 50 mM L-Ala in 100 mM NaPi buffer, pH 8.0, at 30 °C, under constant stirring and with a regular moisturized-air flow above the reaction solution. Enzyme concentrations were: 0.19 μM MetspHMFO(V367R), 6.76 μM SphALDH, 2.45 μM Cv-ωTA, and 1.3 μM BsAlaDH. Further details are presented in the ESI.† b: GWP and E-factor calculations for AMFCA syntheses in aqueous media and at different substrate (HMF) loads. Full conversions to the product were considered. Three different wastewater treatment paths were evaluated: mild WWTP, a “recommended” path and water incineration. For the GWP derived from the energy impact, heating of a reaction tank for 24 h at 30 °C was considered. | ||
AMFCA was purified from the reaction solution through two steps of cation exchange chromatography. In the first purification, AMFCA was obtained as a dry powder with 33.5% purity (LC-MS). L-Ala was the main other impurity, observed via thin-layer chromatography (Fig. S11, ESI†). Thus, after resuspension of the obtained material in aqueous buffer, L-Ala was enzymatically deaminated to pyruvate. BsAlaDH was employed in combination with a water-producing NADH oxidase (LpNOX, from Lactobacillus pentosus)40 for cofactor recycling. After incubation for 3 h at 30 °C, the solution containing the AMFCA–pyruvate mixture underwent a second cation-exchanger purification step. The obtained dry powder resulted in an AMFCA purity of 98.6%, measured via LC-MS and NMR (Fig. S13 and S14, ESI†).
Thus, the AMFCA enzymatic production from HMF at a higher scale resulted in an isolated yield of 38.4% (purified product mass/initial substrate mass). This value is lower than other achievements previously reported, e.g. 84% isolated yield for a one-pot two-step chemo-enzymatic AMFCA synthesis30 and 99% isolated yield for an enzymatic production.14 However, compared to both systems, the one-step one-pot synthesis presented here employs a biogenic amine donor (L-Ala) in a fully enzymatic fashion, without using organic solvents in the downstream process (DSP). In related works, despite the enzymatic production of AMFCA, large volumes of methanol and ethyl acetate and a protection/de-protection with Boc-anhydride were employed in DSP.14
Considering our AMFCA production, further optimization both in the upstream process (USP) and downstream process would certainly be necessary to improve the synthesis efficacy. The possibility of applying enzymes in aqueous (secondary) effluents containing traces of furans from biorefineries may contribute to adding more value through carbon-use optimization in future circular economy cycles. Another potential approach would consist in improving the biocatalysts’ robustness by either employing different homologs or engineering the current variants for higher stability. For transamination catalysis, different transaminases have been explored, immobilized and engineered in several works on HMF and DFF amination.25,41–43 Assessing the potential of these biocatalysts for FFCA amination in the cascade presented here certainly represents a promising strategy to improve AMFCA production at a higher scale.
Sustainability analysis of AMFCA syntheses
Along with the above-discussed technical considerations, assessing the environmental impact of (bio)chemical processes with quantitative metrics results is fundamental nowadays. To our knowledge, no studies related to the environmental impact of AMFCA synthesis have been reported hitherto. Given expected potential interest and future marketability of AMFCA, herein some environmental considerations are discussed.For the USP for AMFCA production, a synthesis in aqueous medium was considered, with substrate (HMF) loads spanning from 5 to 20 g L−1, including as well the other works reported for AMFCA synthesis. Total E-factors were first calculated. Yet, the E-factor, being valuable and intuitive, is an absolute metric that does not assess the CO2 produced at the end life of produced waste.44,45 Therefore, the Global Warming Potential (GWP) was measured as well, since it offers a comparable metric to different benchmark (bio)chemical processes through a common waste (CO2). The GWP, expressed as kg of CO2 per kg product, accounts for the CO2 released in the environment (secondary waste) during the treatment of waste produced during the process (primary waste).46 For AMFCA, the principal waste stream will be wastewater as spent reaction media. Herein, three possible waste treatments were considered.47–49 In the first scenario, the effluents are directly sent to a Wastewater Treatment Plant (WWTP) as the constituent chemicals are all biodegradable. In the second case, the effluents are incinerated because of recalcitrant elements that cannot be cost-efficiently removed (worst-case scenario). In an intermediate situation, the waste stream is firstly cost-efficiently treated to remove chemicals that hamper a mild WWTP and the treated aqueous fraction is then sent to WWTP. The last path is assumed to be the case for most biocatalytic processes as impurities, co-solvents, by-products, etc. are still present in produced wastewaters. Therefore, it is named a “recommended” scenario. In addition, the GWP deriving from the energy needed for our AMFCA production (one kilogram product) was also calculated. The equations50 used for the calculations are reported in the ESI.†
The calculated sustainability metrics are summarized in Fig. 4b. Total E-factors ranged from ∼50 to ∼200, mostly wastewater from reaction media, and correlated with the substrate load. Lower GWPs were also measured for higher substrate loads, thus emphasizing the importance of performing AMFCA syntheses at higher HMF concentrations. However, broadly different GWPs were registered for a specific HMF load depending on the wastewater treatment path chosen. A mild WWTP clearly represented the most environmentally friendly option at any HMF load. Assuming the “recommended” path for wastewater treatment of biocatalytic processes, the total GWP spans in the range 73–18 kg CO2 per kg AMFCA. Conversely, when incineration represents the only possibility for wastewater treatment, the GWP range shifts to almost 2-fold higher values. This highlights the heavier environmental footprint of AMFCA production if severe processing conditions are applied, e.g. in chemical syntheses,10,11 which may generate untreatable by-products and humins. From a different perspective, the energy impact component in the GWPs is less prominent, considering the reaction conditions of the synthesis presented in this work (mild temperature). When higher temperatures are required, the GWP related to the heating process would certainly result in a more significant impact. This may presumably be the case for two-step AMFCA syntheses where a heat treatment at 100 °C separates the two production steps,13,30 or the chemical syntheses performed at 90 °C.11 Overall, a trade-off between substrate loadings and temperature should be considered for the reported processes for AMFCA. Looking beyond, an attractive scenario would be valorizing furans from biorefinery effluents, and using the treated effluent as a sugar source for subsequent fermentations (without inhibitory furans). In those cases, the wastewater would not be treated, but directly reused as media again, diminishing the GWP considerably.
Similar calculations were also performed to assess the sustainability of the DSP for AMFCA isolation. As stated above, two cation-exchange chromatography steps were followed, with pure water and ammonia-containing water as main effluents. A mild WWTP was considered for the treatment of such aqueous waste streams. The data (Fig. S15, ESI†) clearly highlight the strong impact of DSP on the GWP of the entire process.45 Assuming a single use of aqueous effluents (worst case), a total GWP for DSP of ∼1000 kg CO2 per kg AMFCA should be assumed. It must be noted, though, that the DSP has not been optimized at this stage. Assuming, for instance, the complete recycling of the water streams from the equilibration and wash steps of the chromatography column, cumulative GWPs of ∼175.21 kg CO2 per kg AMFCA would be obtained (5-fold less than the worst case). Notably, very similar DSPs were followed as well in other works to obtain pure AMFCA.12,30 Thus, comparable GWPs can be assumed as well for other AMFCA productions where a cation-exchanger purification is included. More sustainable DSP designs should be developed to reduce the total GWP of AMFCA syntheses in particular and for other biorefinery-like processes in general. Several options for such improvement may be: (i) optimizing the volume-to-product ratio in the chromatography steps; (ii) recycling effluents during the DSP; and (iii) introducing renewable energy in the chemical plant. Data provided are calculated assuming the European standard GWP (0.25 kg CO2 per kW h).50 The introduction of hydropower energy would reduce 6-fold the energy environmental impact for the overall process (0.04 kg CO2 per kW h).51
Conclusions
Exploring the possibilities to exploit the “sleeping giant” HMF, in this paper, we studied a one-pot one-step, cell-free, fully enzymatic HMF functionalization to AMFCA. Two synthetic pathways were developed: one starting from HMFCA and another from HMF. Both biotransformations were performed in one-pot one-step systems, under mild reaction conditions, in aqueous media and employing the recyclable and biogenic L-Ala as an amine donor. The AMFCA synthesis from HMF was successfully scaled to higher volumes, with titers in the range of 4 g AMFCA per L. The approach may become useful to valorize furan-containing aqueous effluents from biorefineries. A sustainability assessment focusing on CO2 production (GWP) was conducted, considering the impact of the energy and the fate of the spent wastewater created in USP and DSP. The assessment reflected the hotspots for environmental improvement: namely, higher substrate loadings, mild conditions, effluent(s) recycling and renewable energy. Looking beyond, further process optimization and better understanding of all (sub)mechanisms and paths in the cascade, and scale-up to pilot volumes are crucial to foster the actual application of the AMFCA syntheses presented here. Considering the limitations observed for AMFCA production from HMF, further reaction and/or biocatalyst engineering approaches will be key to achieving industrially sound conditions.Author contributions
E. F. contributed to the methodology, validation, visualization and writing (original draft) of this work. P. D. d. M. contributed to conceptualization, methodology, visualization and writing (review and editing) of the sustainability assessment for the presented cascade. A. A. and V. S. contributed to conceptualization, project administration, resources, funding acquisition, supervision and writing (review and editing).Data availability
The data supporting this article are available in the ESI.†Conflicts of interest
The authors have no conflicts of interest to declare.Acknowledgements
The authors acknowledge the German Federal Ministry of Education and Research (Bundesministerium für Bildung und Forschung, BMBF) for funding the present research work (project: SynHydro3, FK2: 031B1123A). A.A. thanks the Bundesministerium für Bildung und Forschung (BMBF), Project REDEFINEH2 (FZ: 01DD21005) for the financial support. The authors want to thank Anna Ganter for the cloning, expression and purification of the oxidase PcHMFO.References
- K. I. Galkin and V. P. Ananikov, ChemSusChem, 2019, 12, 2976–2982 CrossRef CAS PubMed.
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539–554 RSC.
- Q.-S. Kong, X.-L. Li, H.-J. Xu and Y. Fu, Fuel Process. Technol., 2020, 209, 106528 CrossRef CAS.
- A. A. Rosatella, S. P. Simeonov, R. F. M. Frade and C. A. M. Afonso, Green Chem., 2011, 13, 754–793 RSC.
- E. de Jong, H. R. A. Visser, A. S. Dias, C. Harvey and G. M. Gruter, Polymers, 2022, 14, 943 CrossRef CAS PubMed.
- C. P. Woroch, I. W. Cox and M. W. Kanan, J. Am. Chem. Soc., 2023, 145, 697–705 CrossRef CAS PubMed.
- S. Djukic, A. Bocahut, J. Bikard and D. R. Long, Heliyon, 2020, 6, e03857 CrossRef CAS PubMed.
- D. D. Hawker and R. B. Silverman, Bioorg. Med. Chem., 2012, 20, 5763–5773 CrossRef CAS PubMed.
- T. K. Chakraborty, A. Arora, S. Roy, N. Kumar and S. Maiti, J. Med. Chem., 2007, 50, 5539–5542 CrossRef CAS PubMed.
- A. W. Lankenau and M. W. Kanan, Chem. Sci., 2019, 11, 248–252 RSC.
- C. Zhu, K. Wang, F. Gao, Z. Sun, M. Chen, J. Fei, C. Chen, H. He, Y. Liu and Y. Cao, Chem. Commun., 2024, 60, 7483–7486 RSC.
- A. Lancien, R. Wojcieszak, E. Cuvelier, M. Duban, P. Dhulster, S. Paul, F. Dumeignil, R. Froidevaux and E. Heuson, ChemCatChem, 2020, 13, 247–259 CrossRef.
- Z. C. Wu, W. W. Li, M. H. Zong and N. Li, Chem. – Eur. J., 2024, 30, e202400269 CrossRef CAS PubMed.
- P. Giri, S. Lim, T. P. Khobragade, A. D. Pagar, M. D. Patil, S. Sarak, H. Jeon, S. Joo, Y. Goh, S. Jung, Y. J. Jang, S. B. Choi, Y. C. Kim, T. J. Kang, Y. S. Heo and H. Yun, Nat. Commun., 2024, 15, 6371 CrossRef CAS PubMed.
- W. R. Birmingham, A. Toftgaard Pedersen, M. Dias Gomes, M. Bøje Madsen, M. Breuer, J. M. Woodley and N. J. Turner, Nat. Commun., 2021, 12, 4946 CrossRef CAS PubMed.
- D. Troiano, V. Orsat and M.-J. Dumont, ACS Catal., 2020, 10, 9145–9169 CrossRef CAS.
- Y. Jia, S. Maitra, L. Kudli, J. S. Guest and V. Singh, Green Chem., 2024, 26, 11340–11350 RSC.
- B. J. Rasor, B. Vogeli, G. M. Landwehr, J. W. Bogart, A. S. Karim and M. C. Jewett, Curr. Opin. Biotechnol., 2021, 69, 136–144 CrossRef CAS PubMed.
- N. J. Claassens, S. Burgener, B. Vogeli, T. J. Erb and A. Bar-Even, Curr. Opin. Biotechnol., 2019, 60, 221–229 CrossRef CAS PubMed.
- M. Teshima, V. P. Willers and V. Sieber, Curr. Opin. Biotechnol., 2023, 79, 102868 CrossRef CAS PubMed.
- W. P. Dijkman and M. W. Fraaije, Appl. Environ. Microbiol., 2014, 80, 1082–1090 CrossRef PubMed.
- M. Sayed, Y. Gaber, F. Junghus, E. V. Martin, S. H. Pyo and R. Hatti-Kaul, Microb. Biotechnol., 2022, 15, 2176–2190 CrossRef CAS PubMed.
- V. B. Urlacher and K. Koschorreck, Appl. Microbiol. Biotechnol., 2021, 105, 4111–4126 CrossRef CAS PubMed.
- M. Viñambres, M. Espada, A. T. Martínez and A. Serrano, Appl. Environ. Microbiol., 2020, 86, e00842-20 CrossRef PubMed.
- A. Pintor, N. Cascelli, A. Volkov, V. Gotor-Fernandez and I. Lavandera, ChemBioChem, 2023, 24, e202300514 CrossRef CAS PubMed.
- K. E. Cassimjee, C. Branneby, V. Abedi, A. Wells and P. Berglund, Chem. Commun., 2010, 46, 5569–5571 RSC.
- C. E. Grimshaw and W. W. Cleland, Biochemistry, 1981, 20, 5650–5655 CrossRef CAS PubMed.
- Q. Guo, L. Gakhar, K. Wickersham, K. Francis, A. Vardi-Kilshtain, D. T. Major, C. M. Cheatum and A. Kohen, Biochemistry, 2016, 55, 2760–2771 CrossRef CAS PubMed.
- A. Dunbabin, F. Subrizi, J. M. Ward, T. D. Sheppard and H. C. Hailes, Green Chem., 2017, 19, 397–404 RSC.
- Q. Wu, M.-H. Zong and N. Li, ACS Sustainable Chem. Eng., 2024, 12, 17869–17877 CrossRef CAS.
- C. Danielli, L. van Langen, D. Boes, F. Asaro, S. Anselmi, F. Provenza, M. Renzi and L. Gardossi, RSC Adv., 2022, 12, 35676–35684 RSC.
- T. Knaus, V. Tseliou, L. D. Humphreys, N. S. Scrutton and F. G. Mutti, Green Chem., 2018, 20, 3931–3943 RSC.
- N. Kamimura, T. Goto, K. Takahashi, D. Kasai, Y. Otsuka, M. Nakamura, Y. Katayama, M. Fukuda and E. Masai, Sci. Rep., 2017, 7, 44422 CrossRef CAS PubMed.
- J. E. Jo, S. Mohan Raj, C. Rathnasingh, E. Selvakumar, W. C. Jung and S. Park, Appl. Microbiol. Biotechnol., 2008, 81, 51–60 CrossRef CAS PubMed.
- J. C. Saari, R. J. Champer, M. A. Asson-Batres, G. G. Garwin, J. Huang, J. W. Crabb and A. H. Milam, Vis. Neurosci., 1995, 12, 263–272 CrossRef CAS PubMed.
- H.-H. Ting and M. J. C. Crabbe, Biochem. J., 1983, 215, 351–359 CrossRef CAS PubMed.
- M. Yoshino and K. Murakami, SpringerPlus, 2015, 4, 292 CrossRef PubMed.
- S. Hoops, S. Sahle, R. Gauges, C. Lee, J. Pahle, N. Simus, M. Singhal, L. Xu, P. Mendes and U. Kummer, Bioinformatics, 2006, 22, 3067–3074 CrossRef CAS PubMed.
- W. P. Dijkman, C. Binda, M. W. Fraaije and A. Mattevi, ACS Catal., 2015, 5, 1833–1839 CrossRef CAS.
- C. Nowak, B. Beer, A. Pick, T. Roth, P. Lommes and V. Sieber, Front. Microbiol., 2015, 6, 957 CrossRef PubMed.
- H. Yang, J. Jiang, C. Ma, Y.-C. He and Z. Yang, Fuel, 2025, 388, 134429 CrossRef CAS.
- T. Heinks, L. M. Merz, J. Liedtke, M. Höhne, L. M. van Langen, U. T. Bornscheuer, G. Fischer von Mollard and P. Berglund, Catalysts, 2023, 13, 875 CrossRef CAS.
- C. Wu, Q. Li, J. Di, Y.-C. He and C. Ma, Fuel, 2023, 343, 127830 CrossRef CAS.
- P. Domínguez de María, Curr. Opin. Green Sustain. Chem., 2021, 31, 100514 CrossRef.
- P. Domínguez de María, Green Chem., 2022, 24, 9620–9628 RSC.
- P. Domínguez de María, Curr. Opin. Green Sustain. Chem., 2025, 52, 101003 CrossRef.
- U. Onken, A. Koettgen, H. Scheidat, P. Schueepp and F. Gallou, Chimia, 2019, 3, 730 CrossRef PubMed.
- C. Krell, R. Schreiber, L. Hueber, L. Sciascera, X. Zheng, A. Clarke, R. Haenggi, M. Parmentier, H. Baguia, S. Rodde and F. Gallou, Org. Process Res. Dev., 2021, 25, 900–915 CrossRef CAS.
- P. Domínguez de María, S. Kara and F. Gallou, Molecules, 2023, 28, 6452 CrossRef PubMed.
- P. Domínguez de María, RSC Sustainability, 2024, 2, 3817–3825 RSC.
- M. A. F. Delgove, A.-B. Laurent, J. M. Woodley, S. M. A. De Wildeman, K. V. Bernaerts and Y. van der Meer, ChemSusChem, 2019, 12, 1349–1360 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc06452f |
| This journal is © The Royal Society of Chemistry 2025 |