
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceWaste-minimized access to diarylamines and triarylamines via Csp2–N coupling under batch and flow conditions†
Giulia
Brufani‡
ab,
Shaomin
Chen‡
bc,
Maria Teresa
Tiberi
b,
Filippo
Campana
b,
Emilia
Paone
 a,
Yanlong
Gu
c,
Francesco
Mauriello
a and
Luigi
Vaccaro
*b
a,
Yanlong
Gu
c,
Francesco
Mauriello
a and
Luigi
Vaccaro
*b
aDipartimento di Ingegneria Civile, dell'Energia, dell'Ambiente e dei Materiali, Università degli Studi Mediterranea di Reggio Calabria, 89122 Reggio Calabria, Italy
bLaboratory of Green S.O.C. – Dipartimento di Chimica, Biologia e Biotecnologie, Università degli Studi di Perugia, Via Elce di Sotto 8, 06123-Perugia, Italy. E-mail: luigi.vaccaro@unipg.it; Web: https://greensoc.chm.unipg.it
cKey Laboratory of Material Chemistry for Energy Conversion and Storage, Ministry of Education, Hubei Key Laboratory of Material Chemistry and Service Failure, School of Chemistry and Chemical Engineering, Huazhong University of Science and Technology, Wuhan 430074, China
First published on 28th January 2025
Abstract
In this study, we present a waste-minimized strategy for synthesising diarylamines and triarylamines via Buchwald–Hartwig coupling. These structural motifs rank among the top 20 most prevalent functional groups in biologically active molecules, and they are widely used in fields ranging from medicinal chemistry to materials science. The heterogeneous catalyst Pd/C was effectively employed under batch conditions and packed into the reactor for the flow setup. Our waste-reduction strategy utilises an azeotropic mixture of cyclopentyl methyl ether (CPME) derived from petrochemical waste and water. To enhance process circularity, the heterogeneous catalyst, phosphine-based ligand, and CPME were recovered and reused. The use of a biphasic CPME azeotrope as the reaction medium facilitated the process under flow conditions by enabling the solubilisation of all reaction components. Final product isolation was achieved thanks to an in-line liquid–liquid separator, which allowed for a significant reduction in waste generation.
Green foundation1. This study outlines a waste-minimization strategy for synthesising diarylamines and triarylamines via Buchwald–Hartwig coupling, a widely useful and utilised synthetic tool. By employing the commercially and widely available Pd/C catalyst alongside the CPME![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O azeotrope, our protocol proposes a greener entry to this process. :H2O azeotrope, our protocol proposes a greener entry to this process.
2. The protocol has been studied and developed to adapt and be readily usable under flow conditions, allowing the reduction of waste and ensuring high efficiency. This approach significantly lowers the environmental impact while ensuring scalable and reliable production. 3. Green metrics (E-factors, RME, MRP, BI, and SHI) highlight the environmental and efficiency improvements of our method. The E-factor, reduced to 2.2–2.8, reflects the waste minimisation enabled by recycling CPME, the ligand, and Pd/C. It should also be considered that with our approach, column chromatography is no longer necessary. |
Introduction
Diarylamines and triarylamines are of great interest due to their versatile properties and extensive applications across various branches of chemistry, from medicinal chemistry to materials science. Notably, diarylamines rank among the top 20 most prevalent functional groups in biologically active molecules (Fig. 1A).1 The molecular structure and electronic characteristics of triarylamines significantly influence the aggregation state and charge transport properties, making them an adaptable platform for use in organic, dye-sensitized, and perovskite solar cells (Fig. 1B).2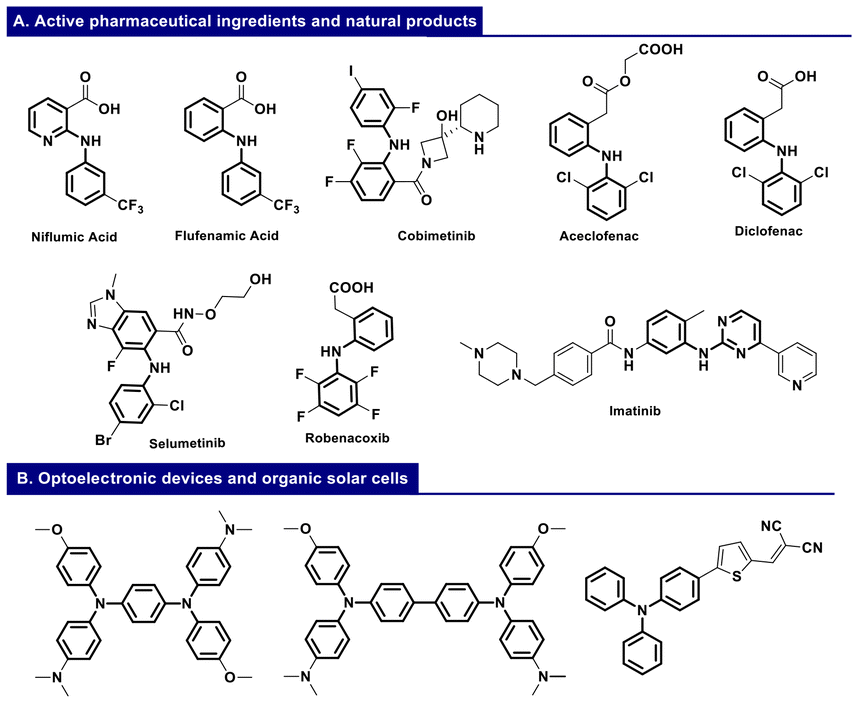 | ||
| Fig. 1 (A) Diarylamines renowned for their versatile properties as active pharmaceutical ingredients (APIs) and natural products. (B) Triarylamines renowned for their applications in optoelectronic and organic cell devices. | ||
The Pd-catalysed Buchwald–Hartwig cross-coupling of amines and aryl halides to form Csp2–N bonds has become a preferred method in both basic and applied chemistry, establishing itself as the favoured technique for synthesising arylamines.3,4 Since its discovery,5–8 the scientific community has invested considerable effort in developing novel homogeneous Pd-based catalysts and phosphine-based ligands to enhance process efficiency.9,10 Despite the wide applicability of such robust Csp2–N coupling at the industrial scale,11–13 there is room for improvement in terms of its “greenness”.
With the aim of recovering valuable Pd metal catalysts and meeting the demand for isolating products with minimal Pd-contamination, the exploration of Pd-heterogeneous supported catalysts for the Buchwald–Hartwig reaction is of significant interest. This involves employing various forms of heterogeneous Pd-based catalysts such as nanocrystals and nanoparticles,14,15 as well as nanoparticles immobilised on carbon nanotubes, graphene,16–18 carbon nitrides,19 alumina,20,21 polymers,22 silica23–25 and inorganic oxide.26–29 Surprisingly, the use of readily available and commercially accessible palladium on charcoal (Pd/C) has been limited to one example that employs ferrocene-1,1′-bis-(diphenylphosphine) as a ligand.30
It is well established that flow chemistry may provide several advantages over batch synthesis, including improved safety, better scalability, process automation, and possibly reduced waste production.31–33 The major challenge in implementing the Buchwald–Hartwig coupling under flow conditions is the use of inorganic bases, which results in the formation of a stoichiometric insoluble halide salt as a by-product.34 To address this issue, sonication of the reactor channels has been proposed to prevent aggregation and reactor clogging using a homogeneous Pd-based catalyst.35,36
Highly active N-heterocyclic carbene (NHC) ligand-based homogeneous Pd-catalysts have been employed in continuous flow amination performed in a microreactor using dimethoxyethane (DME). However, the use of this solvent, with its undesired benign and safety properties and its miscibility with water, complicates the product stream and makes catalyst recovery challenging.37 Alternatively, a change of nature of the base and the use of an organic base, such as 1,5-diazabicyclo(4.3.0)non-5-ene (DBN) or 1,8-diazabicyclo(5.4.0)undec-7-ene (DBU), eliminates the formation of precipitates.38 However, such an approach has been scarcely investigated due to its limitations in applicability and economic non-viability for large-scale operations. Therefore, the identification of effective biphasic conditions could deliver an effective solution and circumvent these limitations.
Common solvents such as DME, 1,4-dioxane, and toluene, often used in Pd-catalysed Buchwald–Hartwig cross-coupling, are both hazardous and environmentally harmful. Increasing interest is therefore directed towards the use of waste-derived reaction media that offer a lower ecological footprint and effective performances.39,40 CPME has recently emerged as a viable alternative, showing favourable toxicity and safety profiles and intriguing chemical properties. It is derived from petrochemical waste via a 100% atom economical process. Moreover, possible CPME production can also be envisaged, starting from biomass and further enhancing its appeal. This solvent forms a heterogeneous aqueous azeotrope with a boiling point of 83 °C, facilitating its utilisation as a biphasic system with water.41–43
After considering all the information described above, we decided to investigate the conditions for the definition of a waste-minimised strategy to access di- and triarylamines via Buchwald–Hartwig coupling. We planned our study on the use of Pd/C as a commercial and widely accessible heterogeneous catalyst. We planned to study and compare batch and flow conditions. Our proposed strategy involved using an azeotropic mixture of CPME and water. The adoption of such a biphasic reaction medium facilitates the application under flow conditions, enabling the solubilisation of all the reaction components.
We also studied the incorporation of an in-line workup methodology, resulting in a general protocol that allows the selective extraction of the product.44 Herein, we report the final isolation of the product realised by setting up an in-line workup constituted by a liquid–liquid separator, which also allowed CPME recovery with a significant reduction of the waste generated and the recycling of the phosphine ligand used (Fig. 2).
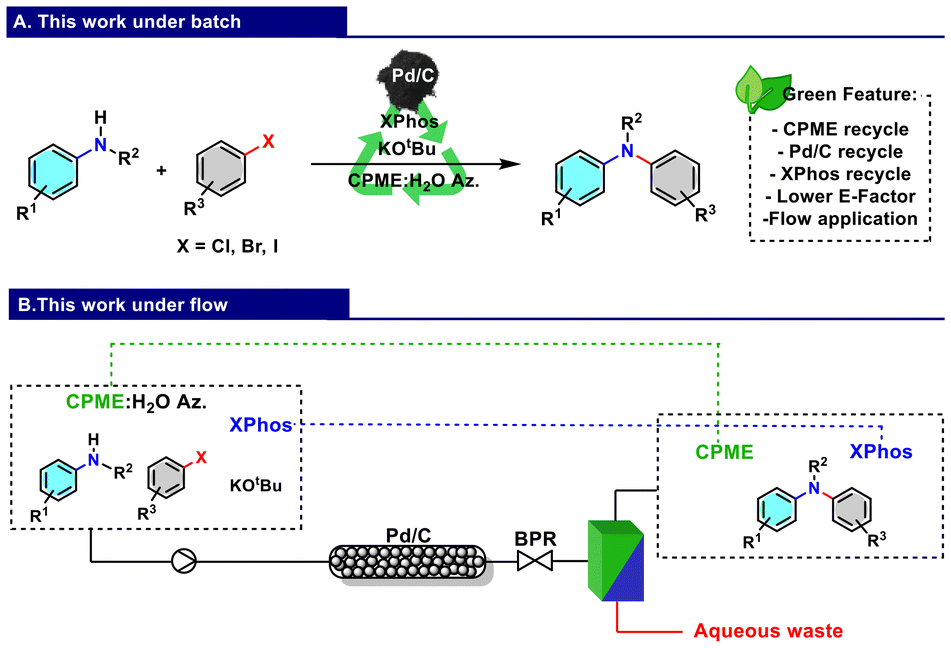 | ||
| Fig. 2 (A) Waste-minimised process for the synthesis of di- and triarylamines under batch conditions. (B) Application of the developed protocol under flow conditions using a recoverable CPME:H2O azeotrope and employing Pd/C as a heterogeneous catalyst. | ||
Results and discussion
We started the optimisation of the Pd/C 10 wt% (1 mol%) catalysed Buchwald–Hartwig coupling by selecting p-toluidine (1a) and p-bromotoluene (2a) as preferred substrates. Aiming to utilise a commercially available and readily accessible phosphine ligand, XPhos (3 mol%) was initially selected due to its effectiveness with aryl chlorides and aryl bromides. The solubility of the base sodium tert-butoxide (NaOtBu) and the associated inorganic halide (NaBr) is crucial for reactivity, requiring a careful choice of reaction medium. We began the solvent screening by testing polar protic solvents, which resulted in moderate conversions (Table 1, entries 1 and 2). The polar solvent acetonitrile gave no conversion (Table 1, entry 3). Petroleum-based apolar solvents gave satisfactory conversion, especially xylene (Table 1, entries 4–6). CPME, which is an eco-friendly alternative to petroleum-based apolar solvents, is widely used as a green alternative to more problematic ethereal solvents such as diethyl ether, tetrahydrofuran (THF), DME, 1,4-dioxane, and methyl tert-butyl ether (MTBE). CPME afforded complete conversion to the desired product 3a (Table 1, entry 7). The amount of 2a was reduced from 1.4 equiv. to 1.1 equiv. without any loss of efficiency (Table 1, entries 8 and 9). Among the inorganic bases screened, KOtBu gave comparable results to NaOtBu (Table 1, entry 10).|
|
|||||
|---|---|---|---|---|---|
| Entry | 2a (equiv.) | Solvent | Ligand | Base | GC_Conv.b (%) |
| a Reaction conditions: 1a (0.5 mmol), 2a (1.4 equiv.), Pd/C 10 wt% (1 mol%), XPhos (3 mol%), and solvent (1 mol L−1) under Ar at 110 °C for 24 h. b The conversion was determined via gas–liquid chromatography using samples of pure compounds as reference standards. c 0.5 mol L−1. d 0.7 mol L−1. EtOH = ethanol; iPrOH = isopropanol; and CH3CN = acetonitrile. | |||||
| 1 | 1.4 | EtOH | XPhos | NaOtBu | 60 |
| 2 | 1.4 | iPrOH | XPhos | NaOtBu | 50 |
| 3 | 1.4 | CH3CN | XPhos | NaOtBu | — |
| 4 | 1.4 | Toluene | XPhos | NaOtBu | 81 |
| 5 | 1.4 | Mesitylene | XPhos | NaOtBu | 79 |
| 6 | 1.4 | Xylene | XPhos | NaOtBu | 95 |
| 7 | 1.4 | CPME | XPhos | NaOtBu | >99 |
| 8 | 1.2 | CPME | XPhos | NaOtBu | >99 |
| 9 | 1.1 | CPME | XPhos | NaOtBu | >99 |
| 10 | 1.1 | CPME | XPhos | KOtBu | 94 |
| 11 | 1.1 | CPME:H2O (50:50)c |
XPhos | NaOtBu | 83 |
| 12 | 1.1 | CPME:H2O Az.c |
XPhos | NaOtBu | 80 |
| 13 | 1.1 | CPME:H2O Az.d |
XPhos | NaOtBu | 80 |
| 14 | 1.1 | CPME:H2O Az.d |
XPhos | KOtBu | >99 |
| 15 | 1.1 | Toluene:H2O Az.d |
XPhos | KOtBu | 78 |

Given our interest in performing the reaction under flow conditions, the solubility of all reaction components is crucial. Therefore, we opted for a biphasic immiscible reaction mixture, which effectively solubilises both the inorganic and organic components, as well as the inorganic salt by-product, thereby preventing catalyst inhibition. Encouraged by the optimal results obtained with CPME, we tested a mixture of CPME:H2O (50:50 wt%). However, a decrease in conversion was observed (Table 1, entry 11). The amount of water was decreased to the azeotropic ratio (84:16 wt%), but no complete conversion was obtained, even after varying the concentration (Table 1, entries 12 and 13). Using KOtBu as a base resulted in complete conversion, confirming the crucial role of the inorganic salt (Table 1, entry 14). When the reaction was performed with a toluene:H2O azeotrope (80:20 wt%), a decrease in conversion was observed (Table 1, entry 15). XPhos played a crucial role, and performing the reaction without it did not result in conversion. Moreover, we tested SPhos as a representative third-generation ligand along with various phosphine ligands from the second and first generations. The results indicate that XPhos provides the best performance, while SPhos is also active but leads to lower conversion. Ligands from the first and second generations showed significantly lower activity (see the ESI for further optimisation†). The blank experiment (without Pd/C) showed no conversion to 3a.
We also studied the reusability of Pd/C under the optimised reaction conditions (Table 1, entry 14) and observed good conversion in the 4th run and a slight decrease in conversion in the 5th run (Fig. 3). The amount of Pd-loss in each run is about 1% of the total amount used (see the ESI for further information†).
 | ||
| Fig. 3 Recycling of Pd/C under the optimised reaction conditions. | ||
The morphology of Pd/C 10 wt% was analysed using high-resolution transmission electron microscopy (HR-TEM), energy dispersive X-ray spectroscopy (EDS), and X-ray diffraction (XRD). HR-TEM images of the fresh Pd/C catalyst reveal well-dispersed nanoparticles across the carbon support, with an average diameter of approximately 2.7 ± 0.6 nm. After the first run, an increase in nanoparticle size is observed, reaching an average of 4.6 ± 1.5 nm. Following five consecutive runs, the analysis shows that the nanoparticles begin to aggregate significantly, accompanied by a further increase in size, resulting in an approximate diameter of 7.2 ± 2 nm. This growth correlates with the decrease in conversion observed during the 5th run. The aggregation complicates the precise measurement of the average particle size. EDS correlates with the Pd distribution (Fig. 4A). The XRD patterns of fresh Pd/C and Pd/C after the 1st run show two characteristic peaks for Pd nanoparticles at 40° and 46°, reflecting the crystalline structure of Pd on the carbon support. These results, along with HR-TEM analysis, confirm that the nanometric particle size is maintained throughout the process (Fig. 4B).
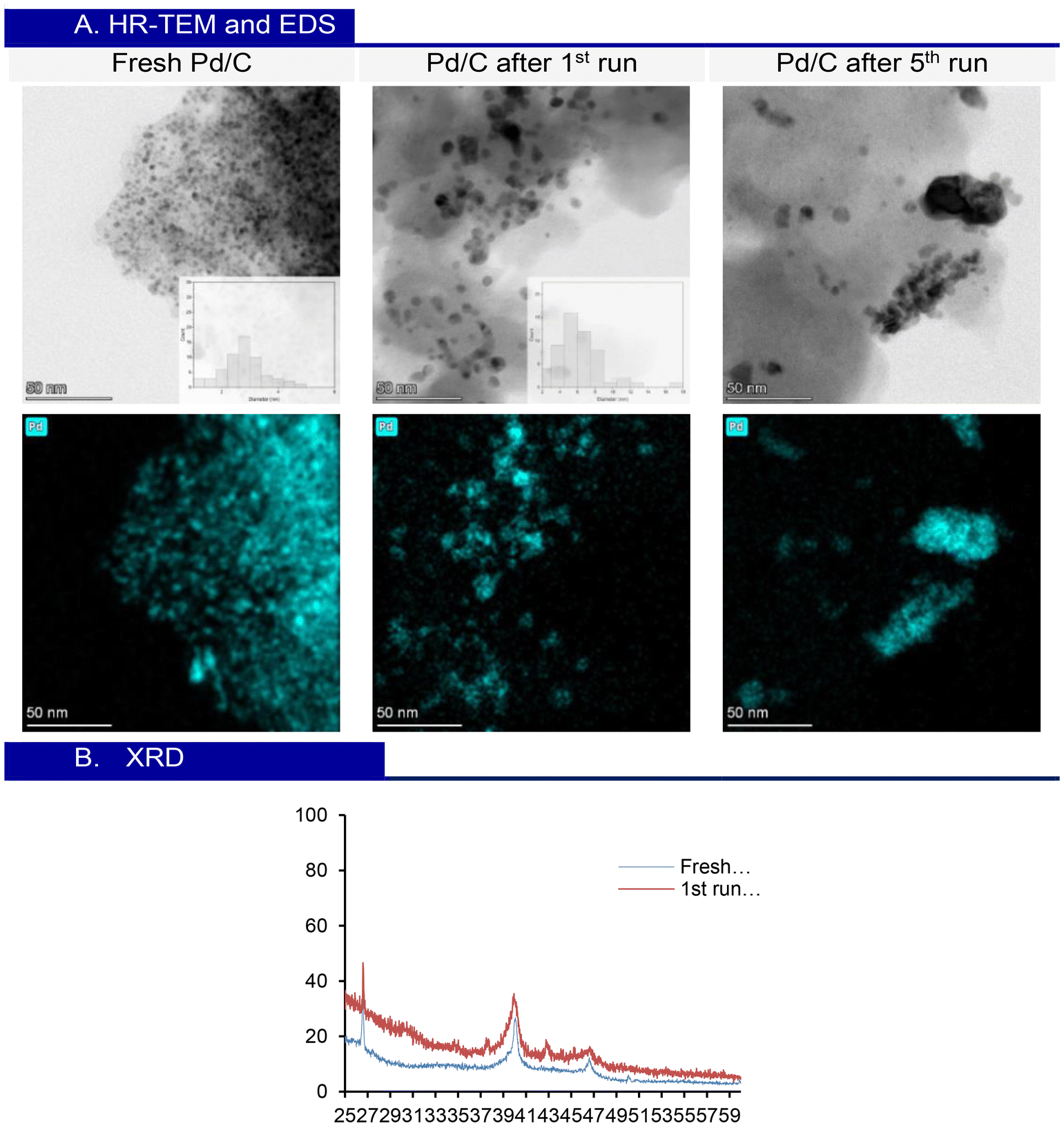 | ||
| Fig. 4 (A) HR-TEM analysis of fresh Pd/C and Pd/C after the 1st run and 5th run, and the respective EDS analysis. (B) XRD patterns of fresh Pd/C and Pd/C after the 1st run. | ||
To clarify the behaviour of the catalytic system in the process, a hot filtration test at 3 h and a Hg test were conducted, both suggesting an operative heterogeneous mechanism. Microwave plasma atomic emission spectroscopy (MP-AES) analysis of the crude reaction mixture after 3 h revealed a Pd loss of 0.3% (1.5 × 10−5 mmol of Pd) in solution, coherent with the observed interruption of reactivity following both tests (see the ESI for further information†).
Aiming to minimise the waste-formation of the overall process, a careful optimisation of the workup procedure was performed. The catalyst was filtered and washed with CPME. The organic layer was extracted from the water to eliminate KBr and the excess base. CPME was distilled from the crude reaction mixture (95%), followed by the addition of cold ethanol (EtOH) to recover XPhos (91%). After distilling EtOH (95%), a pure product was obtained. The recovered solvents, CPME and EtOH, were characterised by nuclear magnetic resonance (NMR) and reused. The XPhos ligand was recovered in pure crystalline form, characterised by NMR, and reused over 10 times without any loss in efficiency. This approach allowed us to avoid purification through column chromatography.
Various aryl halides were tested under the optimised reaction conditions using our workup, and higher isolated yields were observed with bromides and iodides (Fig. 5). In this context, it should be noted that the choice of aryl halides has already been quantified in terms of its impact by life cycle assessment (LCA), showing that for variously substituted substrates, bromide and iodide can also be the overall best-balanced choices.45
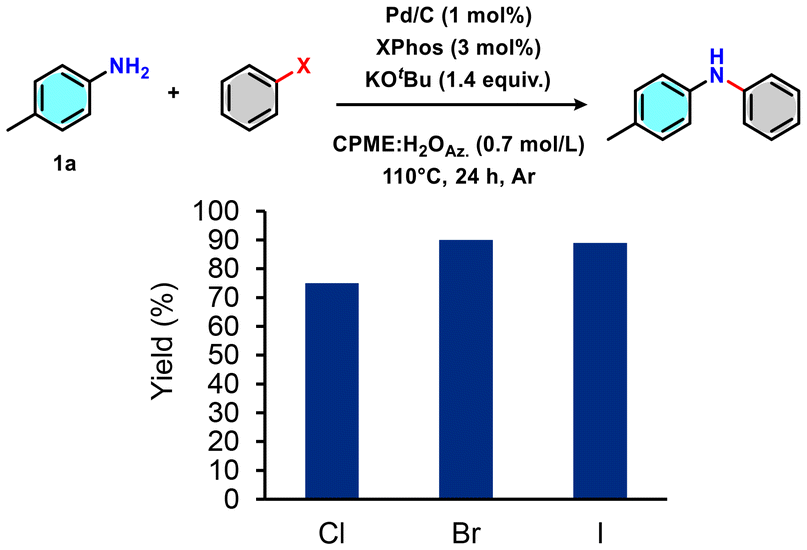 | ||
| Fig. 5 Optimised reaction conditions and workup applied with different aryl halides. | ||
The optimised reaction conditions and workup were extended to the synthesis of 19 diarylamines. Anilines with both alkyl and methoxy substituents proved to be effective starting materials and led to excellent yields (3a–3k). Anilines with electron-donating groups in conjugative positions are activated, consistent with the increased availability of the electron pair on the nitrogen, which facilitates their coordination to the metal centre. As expected, lower reactivity was observed for anilines substituted with electron-withdrawing groups, such as trifluoromethyl, or with species that can contribute to greater delocalisation of the lone pair on nitrogen (3l–3o). In these cases, it is necessary to increase the amount of catalyst, often requiring purification of the reaction product via a chromatographic column (3m, 3o, 3r and 3s). Variously functionalised aryl bromides were tested (3p–3r). The reactivity was found to be strictly related to the electrophilicity of the aryl halide. When 3-bromopyridine was used, the desired product (3r) was obtained with a moderate yield. The optimised reaction conditions were applied with N-methylaniline, obtaining product 3s with a reasonable yield.
The process was further applied to the synthesis of triarylamines from diarylamines, and N-aryl substituted carbazoles, obtaining satisfactory results in terms of isolated yield.
The only representative scale-up to 5 mmol for product 3j was achieved without any loss of efficacy, thanks to the reduced amount of Pd/C, which ensured efficient mass transfer.
To provide a more comprehensive evaluation of the applicability of the optimised protocol to various classes of amines, the process was implemented with cyclohexylamine (7a) and morpholine (7b), providing good yields of the corresponding arylamines (8a and 8b) (Scheme 1).
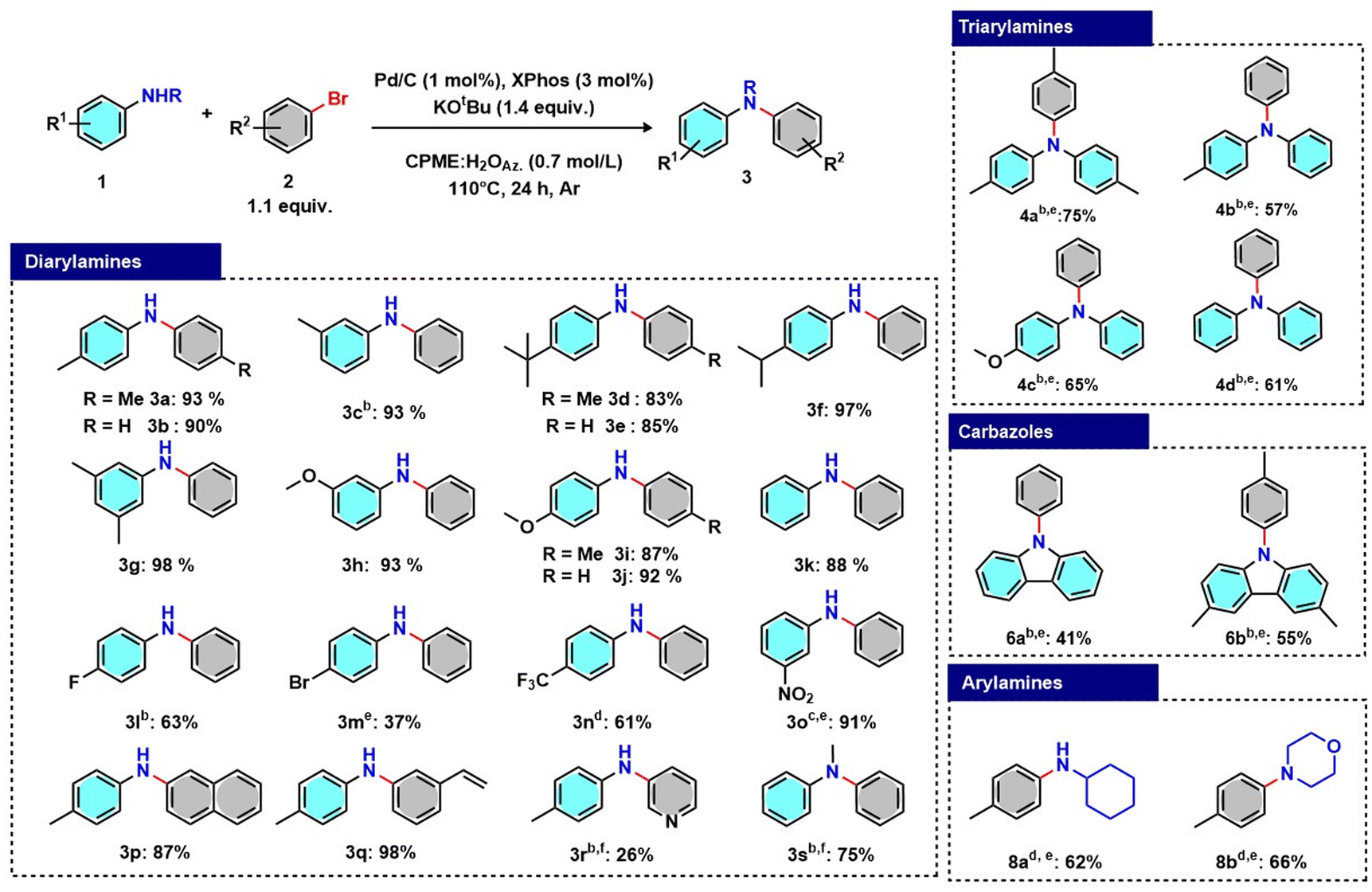 | ||
| Scheme 1 Substrate scope for the synthesis of diarylamines, triarylamines, N-aryl carbazoles and arylamines under the optimised reaction conditions. Reaction conditions: Pd/C 10 wt% (1 mol%), XPhos (3 mol%,), 1a (1 mmol), 2a (1.1 equiv.), KOtBu (1.4 equiv.), and CPME:H2O azeotrope (1.4 ml, 0.7 mol L−1) were consecutively added under an Ar atmosphere. The reaction was performed at 110 °C for 24 h, obtaining complete conversion. aPd/C (2 mol%) and XPhos (6 mol%). bPd/C (2.5 mol%) and XPhos (7.5 mol%). cPd/C (5 mol%) and XPhos (15 mol%). dThe chromatographic column was used due to incomplete conversion. | ||
Aiming to increase the efficiency of the process and find conditions that allow easier scale-up and limit exposure to the reagent, a continuous flow protocol was designed using a packed reactor with a blend of Pd/C and quartz powder equipped with an appropriate back pressure regulator (BPR). An in-line continuous liquid–liquid membrane separator (Zaiput Inc.) was connected to the line to allow the recovery of CPME, which was further reused. The water in which salt KBr and the excess of the base are solubilised, therefore, represents the inevitable major waste associated with the reaction.
In continuous flow experiments, a minimal amount of sodium dodecyl sulphate (SDS) was added as an emulsifier (1 × 10−4 mol L−1) to maintain the stability of the CPME:H2O azeotropic emulsion and to avoid any potential separation and blockage during the process.46
We investigated different conditions to optimise the flow protocol and started by using a 25 cm reactor with an internal diameter of 6 mm packed using 0.6 g of Pd/C 10 wt%. Due to the low residence time, a conversion of 11% to 3a was obtained (Table 2, entry 1). Increasing the residence time resulted in a slight increase in conversion (Table 2, entries 2 and 3). By duplicating the amount of catalyst in the blend, a substantial increase in conversion was observed (Table 2, entry 4). An increase in conversion was observed by increasing the reactor length (Table 2, entry 5) and by increasing the concentration from 0.06 mol L−1 to 0.12 mol L−1 (Table 2, entry 6). Complete conversion was observed using the conditions reported in Table 2, entry 10. These conditions enabled the quantitative conversion of 5 mmol 1a into the desired product 3a, achieving 95% yield with a productivity of 0.6 mmol h−1, while in the batch process was 0.2 mmol h−1. The complete elution required a total time of 520 minutes instead of 24 h. The reactors remained active throughout the optimisation process and substrate scope studies. The Pd content in the desired product was monitored relative to the mmol eluted, revealing a reduced value ranging from 3.6 ppm to 5.2 ppm (see the ESI for further details†).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Coil length (cm) | Pd/C (g) | Concentration (mol L−1) | Residence time (min) | Flow rate (mL min−1) | GC_conv.b (%) |
|
a Reaction conditions: in a 0.06 mol L−1 solution of 1a (5 mmol) in CPME:H2O Az. + SDS (1 × 10−4 mol L−1), 2a (1.1 equiv.), KOtBu (1.4 equiv.) and XPhos (0.5 equiv.) were dissolved at 45 °C. The reaction mixture was pumped through a packed reactor thermoset at 110 °C.
b The conversion was determined via gas chromatography using samples of pure compounds as reference standards.
c BPR 100 psi.
d XPhos 3 mol%.
|
||||||
| 1 | 25 | 0.6 | 0.06 | 30 | 1 | 11 |
| 2 | 25 | 0.6 | 0.06 | 40 | 0.5 | 19 |
| 3c | 25 | 0.6 | 0.06 | 75 | 0.5 | 23 |
| 4c | 25 | 1.2 | 0.06 | 100 | 0.5 | 52 |
| 5 | 50 | 0.6 | 0.06 | 165 | 1 | 41 |
| 6 | 50 | 0.6 | 0.12 | 140 | 1 | 52 |
| 7 | 50 | 1.2 | 0.12 | 180 | 1 | 58 |
| 8 | 75 | 1.2 | 0.12 | 200 | 1 | 75 |
| 9d | 75 | 1.2 | 0.12 | 200 | 1 | 12 |
| 10 | 100 | 1.2 | 0.12 | 290 | 1 | 99 (95) |
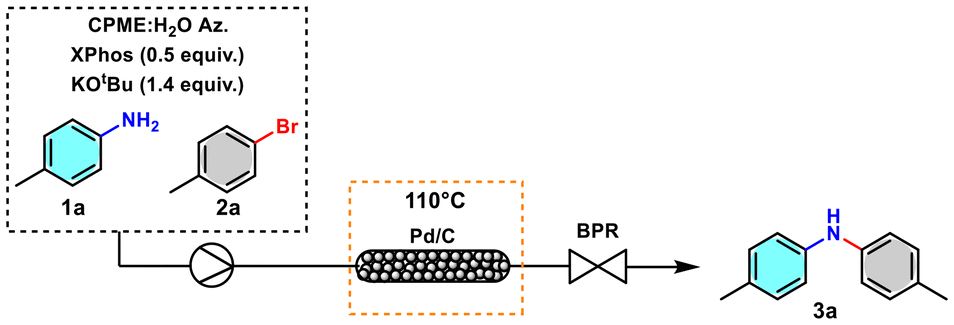
The in-line continuous liquid–liquid membrane separator connected to the flow reactor enables the recovery of product 3a and XPhos in CPME, which was then distilled and consequently reused. XPhos was precipitated from cold EtOH in a pure crystalline form and reused with no change in efficiency (96%). Then, the pure product (3a) was obtained by distilling EtOH with an isolated yield of 95% (Fig. 6).
 | ||
| Fig. 6 Developed flow setup applied in the synthesis of representative diarylamines. | ||
The developed setup was applied representatively for the synthesis of products 3a, 3b, and 3j, obtaining yields higher than those of the batch process and improving the efficacy over the reaction time (Fig. 6).
At this stage, we compared various protocols reported in the literature with the optimised new batch and flow protocols for the selected product 3j. The careful optimisation of the workup procedures for both our methods significantly allowed us to reduce waste by eliminating the need for column chromatography purification and enabling the recovery of all reaction components. This allowed us to achieve E-factor values around 2–3, a remarkable improvement from the typical values in the hundreds of literature processes.21,27,47,48 The E-factors under batch (Fig. 7A, e) and flow (Fig. 7A, f) conditions were comparable, with a slight increase observed in the flow protocols due to the diluted conditions necessary to solubilise all reaction components, as reported by the E-factor profile (Fig. 7B and C) (see the ESI† for E-factor calculation details).
 | ||
| Fig. 7 (A) Comparison of E-factors between literature protocols and our developed batch and flow processes. (B) E-factor profiles of this work under batch conditions. (C) E-factor profiles of this work under flow conditions. | ||
The well-established protocols a and b were selected to compare atom economy (AE), reaction mass efficiency (RME), material recovery parameter (MRP), stoichiometric factor (SF), benign index (BI), and safety hazard index (SHI) with our batch and flow processes.
The seven-pointed radial polygon analysis, supported by the vector magnitude ratio (VMR), demonstrated a larger area for our processes. This improvement is primarily attributed to the successful recovery of solvents and ligands, along with the avoidance of chromatographic columns, which significantly reduced the E-factor and enhanced both RME and MRP. Additionally, the SHI and BI metrics highlighted the overall environmental benefits of the optimised processes. In the flow process, a higher BI was observed due to the diluted reaction conditions, while the SHI was lower, reflecting the increased solvent usage. However, the SHI value only considers the intrinsic properties of the chemicals used. It does not account for the safety advantages of the confined reaction environment in tubes and reactors, which minimise operator exposure and enhance overall process safety (Fig. 8) (see the ESI for green metrics evaluation†).
 | ||
| Fig. 8 Seven-pointed radial polygon analysis evaluation. | ||
Conclusions
In conclusion, we developed a waste-minimized Buchwald–Hartwig coupling process for synthesising diarylamines and triarylamines using a heterogeneous Pd/C catalyst in both batch and flow setups. Our waste-reduction strategy employs an azeotropic mixture of CPME derived from petrochemical waste and water. This biphasic reaction mixture effectively solubilises the base and inorganic halides, overcoming the primary challenge of adapting this reaction for flow applications.To enhance process circularity, we optimised the workup to recover and reuse the CPME, XPhos ligand, and Pd/C catalyst. The latter was successfully recycled for 4 consecutive runs.
Additionally, the optimised workup eliminated the need for chromatographic purification, significantly reducing waste generation.
This protocol was successfully extended to the synthesis of 19 diarylamines, 6 triarylamines, and N-aryl-substituted carbazoles. Aliphatic amines were also tested.
The flow setup was designed with an in-line workup methodology, incorporating a liquid–liquid separator that enabled selective product extraction and CPME recovery. The green metrics, including E-factor, RME, MRP, BI, and SHI, demonstrated significant improvements in terms of greenness compared to well-established protocols.
Overall, the presented approach minimises waste and also provides a method for producing diarylamines and triarylamines with high efficiency and reduced environmental impact.
Author contributions
G. B. and S. C. contributed equally. The manuscript was written with contributions from all authors. All authors have approved the final version of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This publication was prepared with support and funding from the European Union – NextGenerationEU under the Italian Ministry of University and Research (MUR) National Innovation Ecosystem grant ECS00000041 – VITALITY and also “Ecosistema TECH4YOU – (Spoke 3 – Goal 3.5). MUR is thanked for the PRIN-2022 project “20223ARWAY – REWIND”. The University of Perugia is acknowledged for financial support to the university project “Fondo Ricerca di Ateneo, edizione 2022”. Zeon Corporation, Japan, is acknowledged for kindly supporting with free samples of CPME. This research was also supported by MAECI with the project HYDROFA.References
- S. D. Roughley and A. M. Jordan, J. Med. Chem., 2011, 54, 3451–3479 CrossRef CAS PubMed.
- J. Wang, K. Liu, L. Ma and X. Zhan, Chem. Rev., 2016, 116, 14675–14725 CrossRef CAS.
- P. Ruiz-Castillo and S. L. Buchwald, Chem. Rev., 2016, 116, 12564–12649 CrossRef CAS.
- A. Fnaiche, L. Mélin, N. G. Suárez, A. Paquin, V. Vu, F. Li, A. Allali-Hassani, A. Bolotokova, F. Allemand, M. Gelin, P. Cotelle, S. Woo, S. R. LaPlante, D. Barsyte-Lovejoy, V. Santhakumar, M. Vedadi, J.-F. Guichou, B. Annabi and A. Gagnon, Bioorg. Med. Chem. Lett., 2023, 95, 129488 CrossRef CAS PubMed.
- A. S. Guram and S. L. Buchwald, J. Am. Chem. Soc., 1994, 116, 7901–7902 CrossRef CAS.
- A. S. Guram, R. A. Rennels and S. L. Buchwald, Angew. Chem., Int. Ed. Engl., 1995, 34, 1348–1350 CrossRef CAS.
- F. Paul, J. Patt and J. F. Hartwig, J. Am. Chem. Soc., 1994, 116, 5969–5970 CrossRef CAS.
- J. Louie and J. F. Hartwig, Tetrahedron Lett., 1995, 36, 3609–3612 CrossRef CAS.
- R. Dorel, C. P. Grugel and A. M. Haydl, Angew. Chem., Int. Ed., 2019, 58, 17118–17129 CrossRef CAS PubMed.
- J. Tian, G. Wang, Z.-H. Qi and J. Ma, ACS Omega, 2020, 5, 21385–21391 CrossRef CAS PubMed.
- G. E. Robinson, O. R. Cunningham, M. Dekhane, J. C. McManus, A. O'Kearney-McMullan, A. M. Mirajkar, V. Mishra, A. K. Norton, B. Venugopalan and E. G. Williams, Org. Process Res. Dev., 2004, 8, 925–930 CrossRef CAS.
- D. Mitchell, K. P. Cole, P. M. Pollock, D. M. Coppert, T. P. Burkholder and J. R. Clayton, Org. Process Res. Dev., 2012, 16, 70–81 CrossRef CAS.
- J. B. Sperry, K. E. Price Wiglesworth, I. Edmonds, P. Fiore, D. C. Boyles, D. B. Damon, R. L. Dorow, E. L. Piatnitski Chekler, J. Langille and J. W. Coe, Org. Process Res. Dev., 2014, 18, 1752–1758 CrossRef CAS.
- M. Kim, Y. Kim, J. W. Hong, S. Ahn, W. Y. Kim and S. W. Han, Chem. Commun., 2014, 50, 9454 RSC.
- I. C. Watson, A. Schumann, H. Yu, E. C. Davy, R. McDonald, M. J. Ferguson, C. Hering-Junghans and E. Rivard, Chem. – Eur. J., 2019, 25, 9678–9690 CrossRef CAS PubMed.
- H. Veisi, P. Safarimehr and S. Hemmati, Mater. Sci. Eng., C, 2019, 96, 310–318 CrossRef CAS PubMed.
- M. Sarvestani and R. Azadi, Appl. Organomet. Chem., 2018, 32, e3906 CrossRef.
- R. Fareghi-Alamdari, M. G. Haqiqi and N. Zekri, New J. Chem., 2016, 40, 1287–1296 RSC.
- D. Nandi, R. U. Islam, N. Devi, S. Siwal and K. Mallick, New J. Chem., 2018, 42, 812–816 RSC.
- S. G. Babu, B. Emayavaramban, P. Jerome and R. Karvembu, Catal. Lett., 2017, 147, 2619–2629 CrossRef.
- J. B. Ernst, C. Schwermann, G. Yokota, M. Tada, S. Muratsugu, N. L. Doltsinis and F. Glorius, J. Am. Chem. Soc., 2017, 139, 9144–9147 CrossRef CAS.
- R. Nishio, S. Wessely, M. Sugiura and S. Kobayashi, J. Comb. Chem., 2006, 8, 459–461 CrossRef CAS PubMed.
- F. Panahi, F. Daneshgar, F. Haghighi and A. Khalafi-Nezhad, J. Organomet. Chem., 2017, 851, 210–217 CrossRef CAS.
- H. Veisi, T. Tamoradi, B. Karmakar and S. Hemmati, J. Phys. Chem. Solids, 2020, 138, 109256 CrossRef CAS.
- T. Mizusaki, K. Matsumoto, K. Takeuchi, N. Fukaya, Y. Takagi and J.-C. Choi, Organometallics, 2019, 38, 1872–1876 CrossRef CAS.
- S. Fekri, Y. Mansoori, D. Esquivel and M. A. Navarro, ChemistrySelect, 2023, 8, e202204378 CrossRef CAS.
- S. Sá, M. B. Gawande, A. Velhinho, J. P. Veiga, N. Bundaleski, J. Trigueiro, A. Tolstogouzov, O. M. N. D. Teodoro, R. Zboril, R. S. Varma and P. S. Branco, Green Chem., 2014, 16, 3494–3500 RSC.
- H. Veisi, P. Sarachegol and S. Hemmati, Polyhedron, 2018, 156, 64–71 CrossRef CAS.
- R. S. Shelkar, S. H. Gund and J. M. Nagarkar, RSC Adv., 2014, 4, 53387–53396 RSC.
- Y. Monguchi, K. Kitamoto, T. Ikawa, T. Maegawa and H. Sajiki, Adv. Synth. Catal., 2008, 350, 2767–2777 CrossRef CAS.
- M. Baumann, T. S. Moody, M. Smyth and S. Wharry, Org. Process Res. Dev., 2020, 24, 1802–1813 CrossRef CAS.
- F. Ferlin, D. Lanari and L. Vaccaro, Green Chem., 2020, 22, 5937–5955 RSC.
- Sustainable Flow Chemistry: Methods and Applications, ed. L. Vaccaro, Wiley, 1st edn, 2017 Search PubMed.
- Y. Sunesson, E. Limé, S. O. Nilsson Lill, R. E. Meadows and P.-O. Norrby, J. Org. Chem., 2014, 79, 11961–11969 CrossRef CAS.
- P. Yaseneva, P. Hodgson, J. Zakrzewski, S. Falß, R. E. Meadows and A. A. Lapkin, React. Chem. Eng., 2016, 1, 229–238 RSC.
- T. Noël, J. R. Naber, R. L. Hartman, J. P. McMullen, K. F. Jensen and S. L. Buchwald, Chem. Sci., 2011, 2, 287–290 RSC.
- A. Pommella, G. Tomaiuolo, A. Chartoire, S. Caserta, G. Toscano, S. P. Nolan and S. Guido, Chem. Eng. J., 2013, 223, 578–583 CrossRef CAS.
- S. K. Kashani, J. E. Jessiman and S. G. Newman, Org. Process Res. Dev., 2020, 24, 1948–1954 CrossRef CAS.
- F. P. Byrne, S. Jin, G. Paggiola, T. H. M. Petchey, J. H. Clark, T. J. Farmer, A. J. Hunt, C. R. McElroy and J. Sherwood, Sustainable Chem. Processes, 2016, 4, 7 CrossRef.
- R. K. Henderson, C. Jiménez-González, D. J. C. Constable, S. R. Alston, G. G. A. Inglis, G. Fisher, J. Sherwood, S. P. Binks and A. D. Curzons, Green Chem., 2011, 13, 854 RSC.
- U. Azzena, M. Carraro, L. Pisano, S. Monticelli, R. Bartolotta and V. Pace, ChemSusChem, 2019, 12, 40–70 CrossRef CAS.
- K. Watanabe, N. Yamagiwa and Y. Torisawa, Org. Process Res. Dev., 2007, 11, 251–258 CrossRef CAS.
- G. de Gonzalo, A. R. Alcántara and P. DomínguezdeMaría, ChemSusChem, 2019, 12, 2083–2097 CrossRef CAS PubMed.
- A. Chartoire, C. Claver, M. Corpet, J. Krinsky, J. Mayen, D. Nelson, S. P. Nolan, I. Peñafiel, R. Woodward and R. E. Meadows, Org. Process Res. Dev., 2016, 20, 551–557 CrossRef CAS.
- J. L. Osorio-Tejada, F. Ferlin, L. Vaccaro and V. Hessel, Green Chem., 2023, 25, 9760–9778 RSC.
- F. Ferlin, F. Valentini, D. Sciosci, M. Calamante, E. Petricci and L. Vaccaro, ACS Sustainable Chem. Eng., 2021, 9, 12196–12204 CrossRef CAS.
- B. P. Fors, N. R. Davis and S. L. Buchwald, J. Am. Chem. Soc., 2009, 131, 5766–5768 CrossRef CAS PubMed.
- S. Pradhan, A. Bhattacharyya and R. P. John, Tetrahedron Lett., 2016, 57, 1532–1536 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: General procedures, full characterization of the synthesized compounds and copies of 1H, 13C and 19F NMR spectra. See DOI: https://doi.org/10.1039/d4gc06065b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |