
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA green and sustainable catalytic protocol for methoxymethylation of primary amides using methanol with dihydrogen release†
Reshma
Babu
a,
Ganesan
Sivakumar
a,
Smruti
Rekha Padhy
a and
Ekambaram
Balaraman
 *ab
*ab
aDepartment of Chemistry, Indian Institute of Science Education and Research (IISER) Tirupati, Tirupati-517619, India. E-mail: eb.raman@iisertirupati.ac.in
bDST-Nodal Center for APIs and KSM Production Under the Therapeutic Chemicals Program, IISER-Tirupati, Tirupati-517619, India. E-mail: eb.raman@iisertirupati.ac.in
First published on 15th January 2025
Abstract
A highly efficient and selective catalytic method for the methoxymethylation of primary amides with the liberation of dihydrogen under Mn(I) catalysis is reported. This marks the first report on the synthesis of N-(methoxymethyl)benzamide derivatives, including the synthesis of pharmaceutically important amides through the interrupted borrowing hydrogen (IBH) strategy. The current method showcases a broad substrate scope and utilizes methanol as a methoxy methylating agent and solvent medium. The present unprecedented strategy obviates the need for toxic reagents and multi-step synthesis protocols. Mechanistic, kinetic studies and isotopic labeling experiments were also performed to gain mechanistic insights.
Green foundation1. A highly efficient and selective catalytic method for the methoxymethylation of primary amides, with the concurrent liberation of dihydrogen under Mn(I) catalysis, is reported. This represents the first synthesis of N-(methoxymethyl)benzamide derivatives, including pharmaceutically significant amides, through the interrupted borrowing hydrogen (IBH) strategy.2. The method demonstrates a broad substrate scope, utilizing methanol both as a methoxymethylating agent and solvent. 3. This novel approach eliminates the need for toxic reagents and multi-step synthesis protocols, offering a greener and more sustainable alternative. Additionally, the reusability of the homogeneous catalyst is successfully demonstrated. |
Nitrogen-containing motifs are pivotal in medicinal chemistry and drug discovery, with over 80% of FDA-approved small molecule drugs incorporating at least one nitrogen atom. This underscores their critical role in pharmaceutical research and development.1 The development of atom- and step-efficient catalytic methods for functionalizing nitrogen-containing compounds has significantly advanced organic synthesis. Recently, attention has been directed toward simple modifications of nitrogen-containing molecules, such as amines, amides, sulfonamides, and N-heterocycles, which can dramatically influence the biological and physiological properties of active pharmaceutical compounds.2 In this context, the synthesis of N-(methoxymethyl) amides, using methanol as a greener C1 building block, is particularly noteworthy. Methanol is widely available, cost-effective, and recognized as a greener liquid organic hydrogen carrier, with an annual global production of around 70 million tons.3 However, significant challenges exist in synthesizing N-(methoxymethyl) amides due to the low nucleophilicity of amides and the high activation energy required for methanol dehydrogenation. N-(Methoxyalkyl)benzamides, important for their presence in natural toxins like pederin and their use in synthetic herbicides, illustrate the broader significance of this motif.4 Despite its potential, and to the best of our knowledge, a green and step-economical synthesis method for N-(methoxymethyl) amides using methanol has not yet been realized.
In recent years, significant attention has been given to transition metal-catalyzed C–X (X = carbon or a heteroatom) bond-forming reactions through the borrowing hydrogen (BH) or hydrogen auto-transfer (HA) strategy, which is regarded as a highly promising and environmentally friendly method with exceptional atom economy.5 Numerous studies have highlighted the use of 3d-transition metals in borrowing hydrogen catalysis, particularly in facilitating C–C and C–N bond formation.6 In contrast, the interrupted borrowing hydrogen (IBH) strategy remains relatively underexplored and is rarely reported in the field of transition metal catalysis.7 The HA/BH strategy begins by dehydrogenating easily accessible alcohols, which then combine with various nucleophilic partners (C or N) to form an unsaturated intermediate. Following this, the intermediate undergoes selective hydrogenation to yield the desired end product. This approach involves borrowing hydrogen from one of the starting materials, primarily alcohols, acting as the hydrogen donor. Conversely, in the IBH strategy, the reduction of unsaturated intermediates is interrupted, enabling the in situ conjugate addition of various nucleophiles, ultimately leading to the formation of diversified products.8 A limited number of eco-benign synthesis methods for N-methylated amides under transition-metal catalysis via the HA/BH approach have been reported.9 Herein, we report a highly efficient and selective catalytic method for the methoxymethylation of primary amides using methanol with the liberation of dihydrogen. This marks the first report on the synthesis of N-(methoxymethyl)benzamide derivatives through the IBH strategy. The current method showcases a broad substrate scope and operates under molecular Mn-catalysis. Interestingly, we illustrated the extensive utility of this approach by broadening its application to the functionalization of biologically active compounds (Scheme 1).
 | ||
| Scheme 1 (a) General scheme of the IBH strategy, (b) Mn-catalyzed N-methoxymethylation of primary amides using methanol. | ||
Recently, molecularly defined PNP-Mn(I) complexes have emerged as significant catalysts in (de)hydrogenation and related transformations.10 Building on this, we have developed various synthetic organic transformations, including C–C and C–N bond-forming reactions, using a dehydrogenation strategy facilitated by a robust PNP-Mn(I) catalytic system under mild conditions.11 Inspired by these advancements, we established a novel synthetic approach for the functionalization of nitrogen-containing motifs, such as amides. Specifically, we systematically explored several key parameters for the Mn-catalyzed synthesis of N-(methoxymethyl)amide derivatives using methanol (Table 1).
|
|
||
|---|---|---|
| Entry | Reaction conditions | Yield of 1a (%) |
| a Reaction conditions: substrate 1 (0.5 mmol), 2 (3 mL), cat. Mn(I) (5 mol%), ligand L7 (5 mol%) and K2CO3 (1.5 eq.) were heated at 130 °C (silicone oil bath temperature) for 12 h under an argon atmosphere. Yields were determined with a GC instrument using m-xylene as an internal standard. b Isolated yield. n.d = not detected. | ||
| 1 | Standard conditions | 83b |
| 2 | MnCl2 instead of MnBr(CO)5 | n.d |
| 3 | Cp*Mn(CO)3 instead of MnBr(CO)5 | n.d |
| 4 | NiCl2 instead of MnBr(CO)5 | n.d |
| 5 | CoCl2 instead of MnBr(CO)5 | n.d |
| 6 | Cs2CO3 instead of K2CO3 | 78 |
| 7 | KOH instead of K2CO3 | 26 |
| 8 | KOtBu instead of K2CO3 | 42 |
| 9 | L4 instead of L7 | n.d |
| 10 | L5 instead of L7 | 56b |
| 11 | L6 instead of L7 | 32b |
| 12 | At 150 °C | 84 |
| 13 | Absence of catalyst | n.d |
| 14 | No K2CO3 | n.d |
| 15 | 1 eq. of K2CO3 | 65b |

|
||

To investigate the [Mn]-catalyzed synthesis of N-(methoxymethyl)benzamide, benzamide 1 was chosen as the model substrate. We thoroughly screened various [Mn] catalysts, ligands, bases, solvents, and reaction temperatures (Table 1). After systematic studies, the optimal conditions for this transformation were found to be a catalytic amount of [Mn]/L7 (5 mol% Mn(CO)5Br and 5 mol% L7) along with K2CO3 (1 mmol) in methanol (3 mL) at 130 °C (oil-bath temperature). Under these conditions, the desired product 1a was obtained in an 83% isolated yield (Table 1, entry 1). The product was completely analyzed using GC-MS, HRMS, and NMR spectroscopy techniques. The necessity of each compound was systematically studied under standard conditions. It was observed that, under the current Mn-catalyzed conditions, ligands L5 and L6 (a two methylene-bridged PNP ligand) produced the desired product 1a, in decent yield, demonstrating similar reactivity compared to the current phosphine-free NNN ligand system (L1–L4). Notably, under the optimized reaction conditions, no product formation (1a) was observed in the absence of the [Mn] catalyst and base, and both unreacted starting materials were recovered from the reaction mixture (Table 1, entries 13 and 14). Performing the reaction at 150 °C resulted in a very slight increase in the yield of 1a (Table 1, entry 12). Among the various [Mn] catalysts screened for the present interrupted borrowing hydrogenation (IBH) reaction, freshly prepared Mn(CO)5Br exhibited superior catalytic activity, providing the highest yield of 1a (Table 1, entries 1–3). Subsequently, we investigated the influence of various bases, and K2CO3 was found to be an optimal base for facilitating this reaction (Table 1, entries 1 and 6–8). Thus, K2CO3 was identified as the optimal base for this transformation, yielding the maximum amount of 1a under the optimal reaction conditions (Table 1, entry 1). Notably, a maximum yield of 1a was achieved by using 1 equivalent of K2CO3 (Table 1, entry 15).
In order to show the generality of the present catalytic protocol, diverse benzamides were reacted with methanol under optimal reaction conditions (Scheme 2). The present catalytic system exhibited excellent tolerance towards a wide array of amides, including aromatic, aliphatic, cyclic, acyclic, conjugated, di- and tri-substituted amides, yielding up to 89% of the corresponding N-(methoxymethyl)benzamide derivatives. The present catalytic conditions show excellent reactivity towards amide derivatives containing electron-releasing groups at different positions (especially para-, meta-, and ortho-positions) on the aromatic ring. These substrates yielded the corresponding moderate to good yields (products 1a–3a, 10a–12a, and 14a–15a), with up to 89% isolated yield. Observations revealed that benzyl alcohols substituted with halides, such as p-chloro (4a), p-bromo (7a), p-iodo (9a), o-bromo (13a) and m-iodo (18a) groups, yielded the products in moderate yields. Interestingly, p-fluoro, p-nitro, and o-fluoro substituted amides (5 and 6) yielded the unsubstituted N-(methoxymethyl)benzamide. Indeed, under our standard catalytic conditions, the fluoro and nitro groups underwent an SNAr-type reaction with methanol. Gratifyingly, benzamides containing amino groups underwent tandem N-methylation (via the BH strategy) and N-methoxymethylation reactions (via the IBH strategy) and yielded products 8a (71%) and 25a (76%) in very good yields. Furthermore, the cyclic amide, cyclohexyl carboxamide, smoothly underwent N-methoxymethylation and yielded the product 16a in 51% yield. Notably, aliphatic amides, including hexanamide and pivalamide, yielded the corresponding products 19a and 20a in yields of 54% and 56%, respectively. Multi-substituted amides (di/tri-substituted; 22a–26a) with electron-donating groups at various positions yielded the corresponding N-(methoxymethyl) substituted products in moderate to good yields. It is noteworthy that the present catalytic system exhibited good tolerance towards cinnamamide, leading to the desired product 21a (in 63% yield) with a retained olefinic bond. Notably, under the present catalytic conditions, we did not observe any formation of N-methylated amide. In most cases, unreacted primary amide was recovered. Thus, the established catalytic protocol is general, efficient, and selective in producing N-methoxymethylated amides in moderate to very good yields under eco-friendly, step-economical conditions.
 | ||
| Scheme 2 Manganese catalyzed synthesis of N-(methoxymethyl)amide derivatives. Reaction conditions: substrate 1 (0.5 mmol), methanol (3 mL), [Mn-1] (5 mol%), and K2CO3 (1 mmol) were heated at 130 °C (silicone oil-bath temperature) for 12 h under an argon atmosphere. Isolated yield. | ||
A simple alteration to the structure can significantly alter the physiological and biological properties of nitrogen-containing pharmaceutically active molecules. As a result, subjecting 5H-dibenzo[b,f]azepine-5-carboxamide, 27, to methanol under standard reaction conditions yielded the corresponding N-methoxymethylated product 27a, isolated in a yield of 48%. The derivatives of 5H-dibenzo[b,f]azepine-5-carboxamide exhibit anticonvulsant effects and possess properties for blocking brain sodium channels (Scheme 3).12
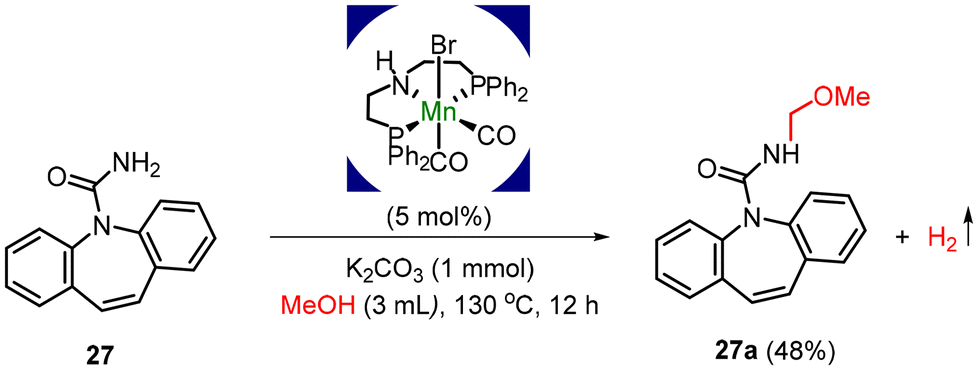 | ||
| Scheme 3 Application of the present protocol: synthesis of pharmaceuticals. | ||
To gain mechanistic insights into the present manganese-catalyzed N-methoxymethylation of primary amides using methanol, several control experiments were performed. Initially, the liberation of molecular H2 from the standard reaction was qualitatively analyzed using an evolved gas analyzer (Scheme 4a). Indeed, when an amide (1) was reacted with formaldehyde (2′) under the current catalytic conditions in the absence of methanol, the expected product (1a) was not obtained. However, in the presence of methanol as the solvent, the expected product was obtained in 31% yield. This result suggests that methanol is indispensable for the reaction and that a slow and steady release of formaldehyde is required for the reaction to proceed (Scheme 4b). Performing the reaction with N-methyl benzamide in the presence of methanol did not yield the corresponding N-(methoxymethyl) substituted product, suggesting that N-methyl benzamide does not act as a reaction intermediate (Scheme 4b). Following this, we conducted deuterium labelling experiments to demonstrate that the reaction proceeds via the IBHS mechanism. Subsequently, two separate deuterium labelling experiments were conducted using methanol-d1 (CH3OD) and methanol-d4 (CD3OD) with benzamide under the current manganese-catalyzed conditions, resulting in the formation of the corresponding deuterium-incorporated products 1a-D and 1a′-D. Confirmation of the formation of two distinct products from these experiments highlighted the role of methanol as both a C1 source and a nucleophile in the reaction, indicating that it follows the IBHS pathway (Scheme 4c). Interestingly, the presence of radical scavengers like TEMPO and BHT resulted in the product being obtained in satisfactory yields. This outcome demonstrates that the current reaction mechanism deviates from the single electron transfer mechanism (Scheme 4d).
 | ||
| Scheme 4 Control experiments. Standard conditions: primary amide (0.5 mmol), methanol (3 mL), [Mn-1] (5 mol%) and K2CO3 (1.5 eq.) heated in an oil bath at 130 °C for 12 h. | ||
Finally, a kinetic analysis was conducted for the Mn-catalysed synthesis of N-(methoxymethyl) benzamide using methanol, as depicted in Fig. 1. The progress of the reaction was monitored using 1H NMR spectroscopy, and the reaction mixture was analyzed at regular intervals. The time-dependent analysis of product formation revealed complete consumption of benzamide within a span of 8 hours. Additionally, the formation of the methoxymethylated product increased gradually over the course of the reaction. The data presented reflect the average of two separate sets of experiments conducted independently.
 | ||
| Fig. 1 Kinetic profile for the Mn-catalysed methoxymethylation of benzamide. | ||
Kinetic studies employing the initial rate approximation were conducted to ascertain the order of each substrate for the synthesis of N-(methoxymethyl)benzamide using methanol (Fig. 2(A–F)). According to graph [A], as the concentration of 1 increases, the reaction rate exhibits dependence, yielding a positive slope of 1.09 when plotting log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) (rate) against log(conc. of 1). Hence, it indicates that the reaction is first order with respect to the concentration of 1. Likewise, as the concentration of the base increases, there is no observable alteration in the reaction rate over time. The plot of log(rate) vs. log(conc. of base) exhibits a negative slope of 6.4. This indicates that the reaction is not influenced by the concentration of the base. Moreover, the relationship between the reaction rate and various loadings of the manganese catalyst revealed a positive slope of 0.67 when plotting log(rate) against log(conc. of cat.[Mn-1]). This implies a fractional order with respect to the concentration of the Mn catalyst. The rate order of the synthesis of N-(methoxymethyl)benzamide using methanol was determined using the initial rate method with various reaction components. The slope of the linear fitting represents the reaction rate. The order of the reaction was then determined by plotting log(rate) vs. log(conc.) for a particular component. Hence, the rate of the reaction depends on the concentration of 1 and the concentration of the manganese catalyst and is independent of the concentration of methanol and the base. From the above kinetic results, the rate equation for the synthesis of N-(methoxymethyl)benzamide using methanol is as follows:
(rate) against log(conc. of 1). Hence, it indicates that the reaction is first order with respect to the concentration of 1. Likewise, as the concentration of the base increases, there is no observable alteration in the reaction rate over time. The plot of log(rate) vs. log(conc. of base) exhibits a negative slope of 6.4. This indicates that the reaction is not influenced by the concentration of the base. Moreover, the relationship between the reaction rate and various loadings of the manganese catalyst revealed a positive slope of 0.67 when plotting log(rate) against log(conc. of cat.[Mn-1]). This implies a fractional order with respect to the concentration of the Mn catalyst. The rate order of the synthesis of N-(methoxymethyl)benzamide using methanol was determined using the initial rate method with various reaction components. The slope of the linear fitting represents the reaction rate. The order of the reaction was then determined by plotting log(rate) vs. log(conc.) for a particular component. Hence, the rate of the reaction depends on the concentration of 1 and the concentration of the manganese catalyst and is independent of the concentration of methanol and the base. From the above kinetic results, the rate equation for the synthesis of N-(methoxymethyl)benzamide using methanol is as follows:
| Rate = k [conc. of 1]1.09 * [conc. of Mn]0.67 |
 | ||
| Fig. 2 Kinetic studies. The graph of [A] conc. of product vs. time with increasing conc. of 1. [B] Log(rate) vs. log(conc. of 1). [C] Conc. of product vs. time with increasing conc. of base. [D] Log(rate) vs. log(conc. of base). [E] Conc. of product vs. time with increasing conc. of Mn catalyst. [F] Log(rate) vs. log(conc. of [Mn-1]). | ||
The reusability of the homogeneous Mn(I)PNP-catalyzed N-methoxymethylation of amides using methanol was effectively demonstrated, as illustrated in Fig. 3. To assess the catalyst's durability, a reaction between benzamide (1) and methanol was conducted under standard conditions. Fresh starting materials were periodically added to the reaction mixture every 12 hours, without introducing an additional Mn(I)PNP complex. Remarkably, the desired N-methoxymethylated product (1a) was successfully obtained in moderate yield after three reaction cycles. However, a noticeable decline in catalytic activity was observed beyond the third cycle. This reduction in efficiency is likely due to the gradual decomposition of the metal complex. It is worth noting that reports on the reusability of soluble homogeneous catalytic systems, particularly for dehydrogenation and related transformations, are exceedingly rare in the literature.13 This study thus highlights a significant step forward in demonstrating the potential for recycling homogeneous catalysts in these types of reactions.
 | ||
| Fig. 3 Recyclability test of the Mn-catalysed N-methoxymethylation reaction (of benzamide with methanol). | ||
Based on control experiments and the literature,10 a plausible mechanism is proposed in Scheme 5. Initially, in the presence of a base, precatalyst Iin situ forms the coordinatively unsaturated reactive amido intermediate II. Subsequently, methanol activation via metal–ligand cooperation (MLC)14 leads to the formation of a methoxy intermediate III. Complex III undergoes β-hydride elimination, resulting in the formation of the corresponding carbonyl compound and intermediate IV. Alternatively, the initial dehydrogenation process may proceed via an outer-sphere mechanism. Upon treatment of the carbonyl compound with benzamide, an N-methylene amide intermediate is generated. This intermediate then follows the IBHS pathway, where methanol acts as a nucleophile. The nucleophilic action of methanol interrupts the reduction of the N-methylene amide intermediate, facilitating an in situ conjugate addition. This process ultimately leads to the formation of an N-methoxymethyl amide.
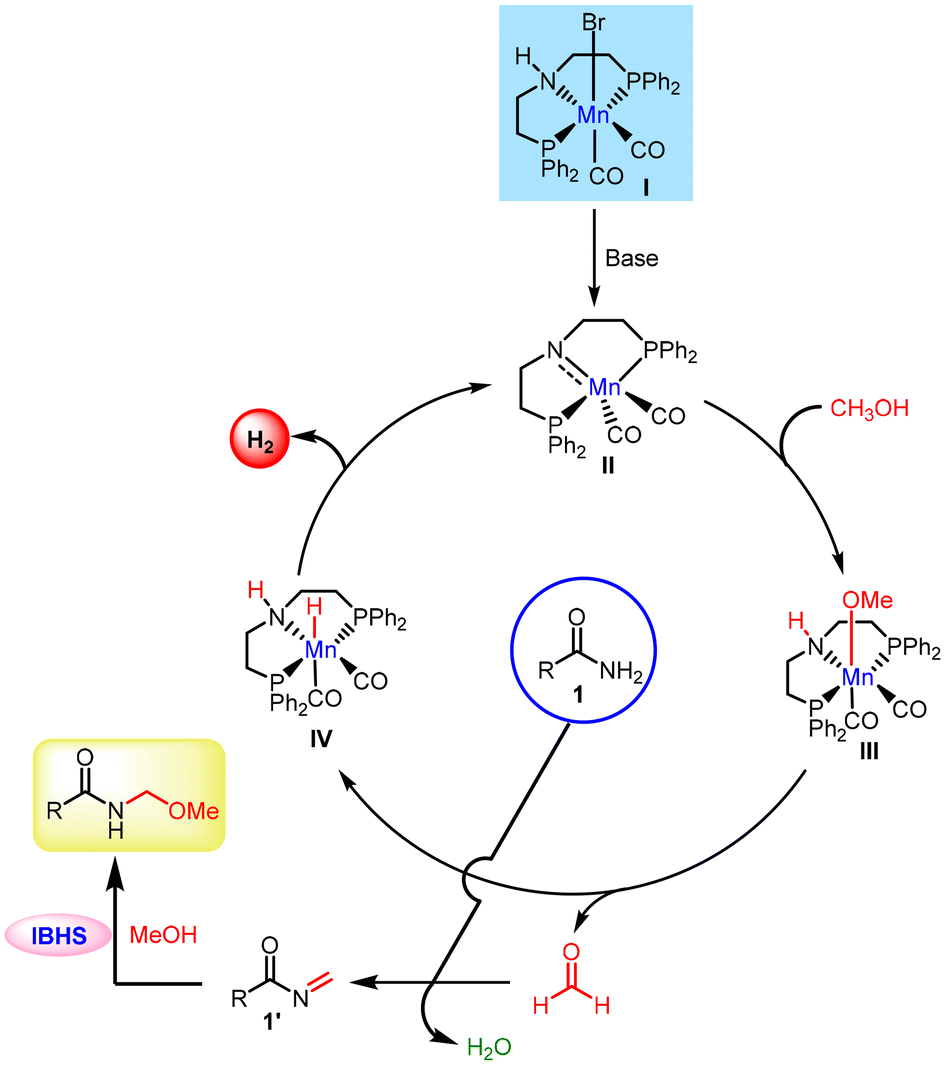 | ||
| Scheme 5 A plausible mechanism for the manganese-catalyzed methoxymethylation of primary amides using methanol with the liberation of dihydrogen. | ||
In conclusion, we report the Mn(I)-catalyzed unprecedented methoxymethylation of primary amides using methanol. This reaction proceeds under benign conditions, with hydrogen gas as the only byproduct. Notably, we demonstrate the broad applicability of this innovative approach by extending its use to the functionalization of biologically active compounds. Mechanistic studies and isotopic labeling experiments suggest that the reaction proceeds via the IBH strategy.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by MoE-STARS (File No.: MoE-STARS/STARS-2/2023-0232). E. B. is a DST-Swarnajayanti Fellow (SERB/F/5892/2020-2021). E. B. also acknowledges the DST-Nodal Center Program (Project no: DST/TDT/TC/KSM/2022/03). R. B. acknowledges the SERB-PMRF, India, for the fellowship. S. R. P. and G. S. thank IISER-Tirupati.References
- E. Vitaku, D. T. Smith and J. T. Njardarson, Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among U.S. FDA approved pharmaceuticals, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed.
- (a) J. Chatterjee, C. Gilon, A. Hoffman and H. Kessler, N-Methylation, of peptides: a new perspective in medicinal chemistry, Acc. Chem. Res., 2008, 41, 1331–1342 CrossRef CAS PubMed; (b) E. J. Barreiro, A. E. Kümmerle and C. A. M. Fraga, The methylation effect in medicinal chemistry, Chem. Rev., 2011, 111, 5215–5246 CrossRef CAS PubMed.
- (a) D. R. Palo, R. A. Dagle and J. D. Holladay, Methanol steam reforming for hydrogen production, Chem. Rev., 2007, 107, 3992–4021 CrossRef CAS PubMed; (b) G. A. Olah, Beyond oil, Angew. Chem., Int. Ed., 2005, 44, 2636 CrossRef CAS PubMed.
- (a) T. Takemura, Y. Nishii, S. Takahashi, J. Kobayashi and T. Nakata, Total synthesis of pederin, a potent insect toxin: the efficient synthesis of the right half, (+)-benzoylpedamide, Tetrahedron, 2002, 58, 6359–6365 CrossRef CAS; (b) B. P. Branchaud and P. Tsai, The relative ease of transient acyl imine formation via selenoxide, sulfoxide, and sulfone. beta. N-H elimination. A feasibility study on the preparation of novel peptide analog, J. Org. Chem., 1987, 52, 5475–5478 CrossRef CAS.
- (a) E. Balaraman, A. Nandakumar, G. Jaiswal and M. K. Sahoo, Iron-catalyzed dehydrogenation reactions and their applications in sustainable energy and catalysis, Catal. Sci. Technol., 2017, 7, 3177–3195 RSC; (b) B. G. Reed-Berendt, D. E. Latham, M. B. Dambatta and L. C. Morrill, Borrowing hydrogen in organic synthesis, ACS Cent. Sci., 2021, 7, 570–585 CrossRef CAS PubMed.
- (a) A. Quintard and J. Rodriguez, A step into an eco-compatible future; iron and cobalt catalyzed borrowing hydrogen transformation, ChemSusChem, 2016, 9, 28–30 CrossRef CAS PubMed; (b) Q. Yang, Q. Wang and Z. Yu, Substitution of alcohols by N-nuclophiles via transition metal-catalyzed dehydrogenation, Chem. Soc. Rev., 2015, 44, 2305–2329 RSC; (c) L. Piccirilli, D. Lobo, J. Pinheiro and M. Nielsen, Recent progress with pincer transition metal catalysts for sustainability, Catalysts, 2020, 10, 773 CrossRef CAS; (d) S. Werkmeister, J. Neumann, K. Junge and M. Beller, Pincer-type complexes for catalytic (De) hydrogenation and transfer (De) hydrogenation reactions: Recent progress, Chem. – Eur. J., 2015, 21, 12226–12250 CrossRef CAS PubMed; (e) F. Kallmeier and R. Kempe, Manganese complexes for (de) hydrogenation catalysis: a comparison to cobalt and iron catalysis, Angew. Chem., Int. Ed., 2018, 57, 46–60 CrossRef CAS PubMed; (f) G. A. Filonenko, R. Van Putten, E. J. M. Hensen and E. A. C. Pidko, Catalytic (de) hydrogenation promoted by non-precious metals-Co, Fe and Mn: recent advances in an emerging field, Chem. Soc. Rev., 2018, 47, 1459–1483 RSC; (g) M. R. Elsby and R. T. Baker, Strategies and mechanisms of metal-ligand cooperativity in first row transition metal complex catalysts, Chem. Soc. Rev., 2020, 49, 8933–8987 RSC; (h) B. G. Reed-Berendt, K. Polidano and L. C. Morrill, Recent advances in homogeneous borrowing hydrogen catalysis using earth-abundant first row transition metals, Org. Biomol. Chem., 2019, 17, 1595–1607 RSC; (i) M. Subaramanian, G. Sivakumar and E. Balaraman, Recent advances in nickel catalyzed C-C and C-N bond formation via ADC reactions, Org. Biomol. Chem., 2021, 19, 4213–4227 RSC; (j) M. Subaramanian, G. Sivakumar and E. Balaraman, First-row transition-metal catalyzed acceptorless dehydrogenation and related reactions: A personal account, Chem. Rec., 2021, 21, 3839–3871 CrossRef CAS PubMed.
- (a) M. Nallagangula, M. Subaramanian, R. Kumar and E. Balaraman, Transition metal-catalysis in interrupted borrowing hydrogen strategy, Chem. Commun., 2023, 59, 7847 RSC; (b) V. Yadav, E. Balaraman and S. B. Mhaske, Phosphine free manganese(II)-catalyst enables acceptorless dehydrogenative coupling of alcohols with indoles, Adv. Synth. Catal., 2021, 363, 4430–4439 CrossRef CAS; (c) G. Wang, Z. Li, C. Li and S. Zhang, Cobalt catalyzed synthesis of α, β-unsaturated esters from esters and alcohols via mild O2-interrupted hydrogen borrowing, J. Catal., 2018, 368, 228–236 CrossRef CAS; (d) C. Sun, X. Zou and F. Li, Direct use of methanol as an alternative to formaldehyde for the synthesis of 3,3′-bisindolylmethanes (3,3′-BIMs), Chem. – Eur. J., 2013, 19, 14030–14033 CrossRef CAS PubMed.
- (a) J. R. Frost, C. B. Cheong, W. M. Akhtar, D. F. J. Caputo, N. G. Stevenson and T. J. Donohoe, Strategic application and transformation of ortho disubstituted phenyl and cyclopropyl ketones to expand the scope of hydrogen borrowing catalysis, J. Am. Chem. Soc., 2015, 137, 15664–15667 CrossRef CAS PubMed; (b) D. Shen, D. L. Poole, C. C. Shotton, A. F. Kornahrens, M. P. Healy and T. J. Donohoe, Hydrogen borrowing and interrupted hydrogen borrowing reactions of ketones and methanol catalyzed by iridium, Angew. Chem., Int. Ed., 2015, 54, 1642–1645 CrossRef CAS PubMed.
- B. Paul, D. Panja and S. Kundu, Ruthenium-Catalyzed Synthesis of N-Methylated Amides using Methanol, Org. Lett., 2019, 21, 5843–5847 CrossRef CAS PubMed.
- (a) M. Peña-López, P. Piehl, S. Elangovan, H. Neumann and M. Beller, Manganese-catalyzed hydrogen-autotransfer C−C bond formation: α-alkylation of ketones with primary alcohols, Angew. Chem., Int. Ed., 2016, 55, 14967–14971 CrossRef PubMed; (b) G. Sivakumar, M. Subaramanian and E. Balaraman, Single-molecular Mn(I)-complex-catalyzed tandem double dehydrogenation cross-coupling of (amino)alcohols under solventless conditions with the liberation of H2 and H2O, ACS Sustainable Chem. Eng., 2022, 10, 7362–7373 CrossRef CAS; (c) A. E. Owen, A. Preiss, A. McLuskie, C. Gao, G. Peters, M. Bühl and A. Kumar, Manganese-catalyzed dehydrogenative synthesis of urea derivatives and polyureas, ACS Catal., 2022, 12, 6923–6933 CrossRef CAS.
- (a) S. Elangovan, J. Neumann, J. B. Sortais, K. Junge, C. Darcel and M. Beller, Efficient and, selective N-alkylation of amines with alcohols catalysed by manganese pincer complexes, Nat. Commun., 2016, 7, 12641–12649 CrossRef PubMed; (b) B. G. Reed-Berendt and L. C. Morrill, Manganese-catalyzed N-alkylation of Sulfonamides using alcohols, J. Org. Chem., 2019, 84, 3715–3724 CrossRef CAS PubMed; (c) Y. Wang, M. Wang, Y. Li and Q. Liu, Homogeneous manganese-catalyzed hydrogenation and dehydrogenation reactions, Chem., 2021, 7, 1180–1223 CrossRef CAS; (d) M. Andérez-Fernández, L. K. Vogt, S. Fischer, W. Zhou, H. Jiao, M. Garbe, S. Elangovan, K. Junge, H. Junge, R. Ludwig and M. Beller, A stable manganese pincer catalyst for the selective dehydrogenation of methanol, Angew. Chem., Int. Ed., 2017, 129, 574–577 CrossRef; (e) A. Mukherjee and D. Milstein, Homogeneous catalysis by cobalt and manganese pincer complexes, ACS Catal., 2018, 8, 11435–11469 CrossRef CAS.
- J. Benes, A. Parada, A. A. Figueiredo, P. C. Alves, A. P. Freitas, D. A. Learmonth, R. A. Cunha, J. Garrett and P. Soares-da-Silva, Anticonvulsant and sodium channel blocking properties of novel 10,11-Dihydro-5H-dibenz[b,f]azepine-5-carboxamidederivatives, J. Med. Chem., 1999, 42, 2582–2587 CrossRef CAS PubMed.
- (a) S. P. Midya, J. Rana, J. Pitchaimani, A. Nandakumar, V. Madhu and E. Balaraman, ChemSusChem, 2018, 11, 3911–3916 CrossRef CAS PubMed; (b) Y. Han, Z. Wu, Z. Wei, Y. Zhai, S. Ru, Q. Zhao, J. Wang, S. Han and Y. Wei, Commun. Chem., 2019, 2, 1–7 CrossRef.
- M. R. Elsby and R. T. Baker, Strategies and mechanisms of metal–ligand cooperativity in first-row transition metal complex catalysts, Chem. Soc. Rev., 2020, 49, 8933–8987 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and spectroscopic data, copies of 1H, and 13C NMR spectra for all compounds. See DOI: https://doi.org/10.1039/d4gc05864j |
| This journal is © The Royal Society of Chemistry 2025 |