
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDe novo biosynthesis of 4,6-dihydroxycoumarin in Escherichia coli†
Qi
Gan‡
,
Tian
Jiang‡
,
Chenyi
Li
,
Xinyu
Gong
,
Jianli
Zhang
,
Bhaven K.
Desai
and
Yajun
Yan
 *
*
School of Chemical, Materials and Biomedical Engineering, College of Engineering, University of Georgia, Athens, GA 30602, USA. E-mail: yajunyan@uga.edu.cn
First published on 13th February 2025
Abstract
Coumarins and their derivatives possess crucial biochemical and pharmaceutical properties. However, the exploration of the coumarin biosynthesis pathways remains limited, restricting their microbial biosynthesis, especially for hydroxycoumarins. In this work, we designed and verified novel artificial pathways to produce a valuable compound 4,6-dihydroxycoumarin (4,6-DHC) in Escherichia coli. Based on the retrosynthesis analysis, multiple routes were designed and verified by extending the shikimate pathway, screening the potential enzymes, and characterizing the enzymes involved. Rare codon optimization and protein engineering strategies were applied to optimize the rate-limiting steps. De novo biosynthesis of 4,6-DHC was achieved using the cheap carbon source glycerol, and the titer can reach 18.3 ± 0.7 mg L−1. Ultimately, inducible regulation of critical pathway genes with a tetracycline-inducible controller yielded a significant boost in 4,6-DHC production, achieving a titer of 56.7 ± 2.1 mg L−1. This research successfully created a microbial platform for 4,6-dihydroxycoumarin production and demonstrated a generalizable strategy for synthesizing valuable compounds.
Green foundation(1) This work developed novel biosynthetic pathways to de novo produce valuable 4,6-dihydroxycouamrin in E. coli from glycerol, offering an environmentally friendly and sustainable approach.(2) Through protein engineering and pathway regulation, we reduced by-product formation and improved carbon efficiency, achieving a 4,6-DHC production of 56.7 ± 2.1 mg L−1 with a yield of 3.84 mg g−1 glycerol. (3) Further optimization through metabolic engineering could enhance production even more. Our research offers valuable insights into the sustainable biosynthesis of other valuable hydroxycoumarin compounds. |
Introduction
Coumarins and their derivatives are primarily plant secondary metabolites with a wide range of pharmacological activities, such as anti-inflammatory, antiviral, anti-cancer, and anti-oxidative activities.1–6 For instance, 6,7-dehydroxycoumarin (esculetin) holds promise as a chemotherapeutic agent.7 7,8-Dihydroxycoumarin (daphnetin) exhibits various neurotrophic effects.8 7-Hydroxycoumarin (umbelliferone) is utilized as the active ingredient in sunscreens, fluorescence indicators, and dye layers.9 Beyond the pharmacological applications, coumarins with hydroxyl groups at various positions are crucial for the synthesis of a diverse array of valuable compounds, serving as versatile building blocks.6,10 For example, 4-hydroxycoumarin (4-HC) is known as the precursor to synthesize warfarin, one of the most widely used oral anticoagulants.11 4,6-Dihydroxycoumarin (4,6-DHC) is indeed a significant compound in organic chemistry, particularly in the synthesis of various coumarin-related compounds. It serves as a key starting material or intermediate in the synthesis of these compounds, including isocoumarins and furochromenes, which are known for their bioactive properties.12,13 Also, 4,6-DHC plays a pivotal role as an ingredient in the synthesis of a class of Furo[3,2-c] coumarin derivatives. These derivatives are valued for their photoactive properties, like 3-diazo-4-oxo-3,4-dihydrocoumarins, making them valuable across a diverse range of applications.13,14Direct acquisition of coumarins from nature is constrained by low yields, long life cycle periods, seasonal and regional limitations, and complex extraction and purification processes.15 Over the past few decades, various methods were developed for the chemical synthesis of hydroxycoumarins, utilizing petrochemicals like phenol and acetosalicylate as starting materials, under harsh reaction conditions.16–19 This process can generate waste products and pose adverse effects on the environment. Additionally, the chemical synthesis of coumarin-based compounds entails an intramolecular cyclization reaction catalyzed by chemical oxidants, which is especially preferred for forming the C–O bond.20 However, achieving selective hydroxylation of the aromatic ring poses a significant challenge within this process.
Biosynthesis offers a sustainable and eco-friendly approach to produce valuable compounds and has proven to be effective in synthesizing a growing array of significant metabolites, such as caffeic acid,21 naringin,22 terpenoids,23 resveratrol,24etc. However, a significant challenge in the heterologous production of natural products lies in the insufficient characterization of their inherent biosynthetic pathways and the limited identification of enzymes involved. Hydroxylated coumarins are primarily produced through the metabolism of coumarins catalyzed by certain cytochromes P450 (CYPs), such as CYP2A6. The hydroxy group was predominantly introduced at positions 4, 5, 6, and 8, although to a lesser extent than at position 7.25–27 However, membrane associated P450 monooxygenases are difficult to be functionally expressed in prokaryotic systems. An alternative approach is substituting plant cytochrome P450 enzymes with a promiscuous bacterial hydroxylase, introducing hydroxyl groups onto the aromatic rings before lactonization. Lin et al. successfully engineered novel biosynthetic pathways in Escherichia coli to produce umbelliferone and scopoletin from p-coumaric acid and caffeic acid, respectively.28 Additionally, a de novo 4-hydroxycoumarin biosynthetic pathway was designed and constructed in E. coli.11
To expand the availability of hydroxycoumarins, in this study, we developed innovative artificial pathways for synthesizing 4,6-dihydroxycoumarin (4,6-DHC) in E. coli. Initially, we explored enzyme promiscuity to synthesize novel hydroxycoumarins by analyzing retrosynthetic pathways and catalytic mechanisms. We then designed and confirmed multiple routes to produce the precursor 2,5-dihydroxybenzoic acid (2,5-DHBA) for 4,6-DHC synthesis. This involved extending the shikimate pathway, enzyme screening, and enzyme optimization through rare codon utilization and protein engineering. We successfully assembled complete artificial pathways in E. coli, achieving the de novo biosynthesis of 4,6-DHC using glycerol as a cost-effective carbon source, yielding 18.3 ± 0.7 mg L−1. Furthermore, by controlling the expression of key pathway genes using a tetracycline-inducible controller, we significantly increased 4,6-DHC production to 56.7 ± 2.1 mg L−1, indicating a yield of 3.84 mg per g-glycerol. Our work establishes a microbial platform for 4,6-dihydroxycoumarin production and demonstrates a versatile pathway design approach for the de novo biosynthesis of valuable compounds.
Results
Leveraging enzyme promiscuity for the biosynthesis of novel dihydroxycoumarins
Dihydroxycoumarins (DHCs) hold promise as potential pharmaceutical and agrochemical agents due to their structural versatility and pharmacological significance (Fig. 1a), yet their biosynthesis remains relatively unexplored. Previous research has shown that SdgA, a CoA ligase, converts salicylic acid (2-HBA) to salicylic acid-CoA (2-HBA-CoA). Then the anthraniloyl-CoA anthraniloyltransferase PqsD converts 2-HBA-CoA and malonyl-CoA to produce 4-hydroxycoumarin.11 Based on the catalytic mechanism (Fig. 1b) and the similarity of the substrate structures, we hypothesized that SdgA and PqsD can catalyze the conversion of other dihydroxybenzoic acids (DHBAs) to related DHCs. To achieve this, a range of DHBAs including 2,3-DHBA, 2,4-DHBA, 2,5-DHBA, and 2,6-DHBA were evaluated in the feeding experiments using the strain DX1 expressing PqsD and SdgA. Referring to the feeding-test results (Fig. 1c), the addition of 200 mg L−1 2,5-DHBA resulted in the detection of the corresponding product, 4,6-DHC, with a titer of 26.3 ± 1.9 mg L−1. This observation suggested that the enzymes PqsD and SdgA possess the promiscuity to convert 2,5-DHBA to the corresponding dihydroxycoumarin 4,6-DHC. No consumption of 2,4-DHBA or 2,6-DHBA and the formation of related dihydroxycoumarins were observed. When feeding 2,3-DHBA, although there was a decrease in the concentration of the substrate, no corresponding product, 4,8-DHC, was detected. This phenomenon may be attributed to the potential involvement of 2,3-DHBA in the biosynthesis of the siderophore enterobactin in E. coli.29 Taken together, the results above indicated that a new biosynthetic step was achieved to produce a novel dihydroxycoumarin, 4,6-DHC, using the enzyme promiscuities of SdgA and PqsD toward 2,5-DHBA.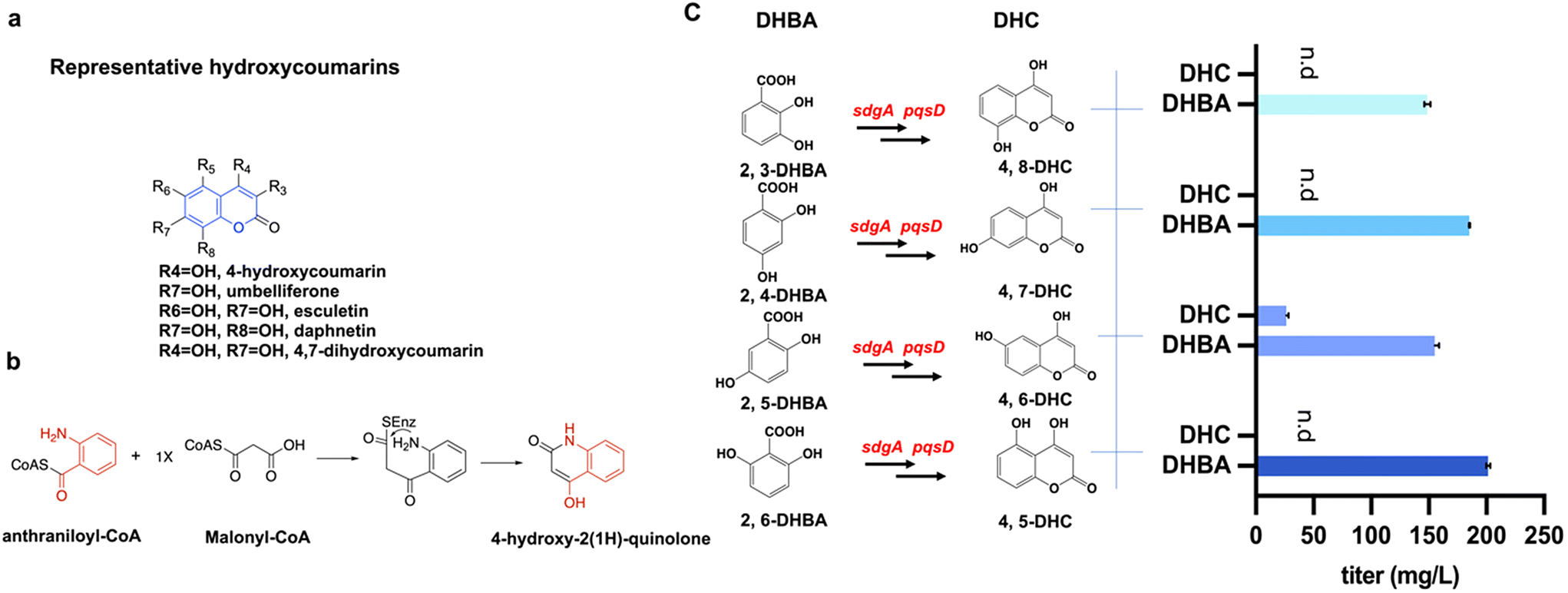 | ||
| Fig. 1 Conversion of dihydroxybenzoic acids (DHBAs) to value-added dihydroxycoumarins (DHCs). a. Representative biologically active hydroxycoumarins. The coumarin moiety is highlighted in blue. b. PqsD-catalyzed 4-hydroxy-2(1H)-quinolone generation pathway and mechanism. c. Conversion from different DHBAs to the corresponding DHCs by feeding 200 mg L−1 DHBA. The gene sdgA encodes salicylate-CoA ligase; the gene pqsD encodes anthraniloyl-CoA anthraniloyltransferase. n.d: not detected. The data presented represent the mean ± standard deviation (SD) from three independent experiments, with error bars indicating the SD. | ||
Design and verification of various pathways for the biosynthesis of 4,6-DHC
With the success in the biosynthesis of 4,6-DHC from 2,5-DHBA, our next emphasis is to develop the pathways for the precursor 2,5-DHBA biosynthesis, to enable de novo production of 4,6-DHC. Based on the previous research, 2,5-DHBA can be generated from chorismate using the corresponding chorismate lyases in various hydroxylation manners with different intermediates, like 2-hydroxybenzoic acid (2-HBA), 3-hydroxybenzoic acid (3-HBA), and 4-hydroxybenzoic acid (4-HBA).30–32 Thus, we designed three parallel routes to generate 2,5-DHBA from chorismate through 2-HBA catalyzed by EntC (from E. coli), PchB (from Pseudomonas aeruginosa) and SalABCD (from Ralstonia eutropha) (route I), 3-HBA catalyzed by Hyg5 (from Streptomyces rapamycinicus) and 3HB6H (from Rhodococcus jostii) (route II) and 4-HBA catalyzed by Ubic (from E. coli) and PhgABC (from Brevibacillus laterosporus) (route III) (Fig. 2). Except for 3HB6H, we explored the enzyme Re3HB6H from Ralstonia eutropha to convert 3-HBA to 2,5-DHBA (Table 1 and ESI Fig. 1†). Feeding experiments were conducted to comprehensively evaluate the efficiency of these routes to produce 2,5-DHBA, utilizing 2-HBA, 3-HBA, and 4-HBA as substrates respectively (Fig. 3b). The plasmids pZE-SalABCD, pHA-3HB6H, and pHA-PhgC-PhgB-PhgA were transferred into E. coli BW25113 (F′), yielding the strains DX2, DX3, and DX4, respectively. The empty plasmid pHA-MCS was transferred into E. coli BW25113 (F′) as a control. When feeding 1 g L−1 of different HBAs, the strains DX2, DX3 and DX4 produced 611.8 ± 16.4 mg L−1, 987.4 ± 4.2 mg L−1 and 512.7 ± 16.9 mg L−1 2,5-DHBA, respectively at 24 h (Fig. 3b). These results demonstrated the superior efficiency of biosynthesizing 2,5-DHBA via the 3-HBA pathway, compared with the other two pathways. | ||
| Fig. 2 Novel artificial pathways for the de novo biosynthesis of 4,6-DHC. 2-HBA, 2-hydroxybenzoic acid; 3-HBA, 3-hydroxybenzoic acid; 4-HBA, 4-hydroxybenzoic acid; 2,5-DHBA-CoA, 2,5-dihydroxybenzoate-CoA; 2,5-DHBA, 2,5-dihydroxybenzoic acid; 4-HC, 4-hydroxycoumarin; 4,6-DHC, 4,6-dihydroxycoumarin. entC encodes isochorismate synthase; pchB encodes isochorismate lyase; hyg5 encodes phosphoenolpyruvate synthetase; ubiC encodes chorismate lyase; Pteto1 encodes salicylate 5-hydroxylase; 3hb6h encodes 3-hydroxybenzoate 6-monooxygenase; phgC encodes p-hydroxybenzoyl-CoA ligase; phgA encodes p-hydroxybenzoyl-CoA 1-hydroxylase; phgB encodes gentisyl-CoA thioesterase; sdgA encodes 2-HBA-CoA synthase; and pqsD encodes anthraniloyl-CoA anthraniloyltransferase. In route I, chorismate is converted to 2-HBA and further converted to 2,5-DHBA. In route II, chorismate is converted to 3-HBA and further converted to 2,5-DHBA. In route III, chorismate is converted to 4-HBA and further converted to 2,5-DHBA. | ||
 | ||
| Fig. 3 Schematic of engineering strategies to optimize 4,6-DHC production in E. coli. a. The conversion from 2,5-DHBA to 4,6-DHC through feeding experiments. b. Comparison of the production of 2,5-DHBA from different HBAs through feeding experiments. c. Comparison of the production of 4,6-DHC from different HBAs through feeding experiments. d. De novo biosynthesis of different HBAs by three different routes. e. De novo biosynthesis of 4,6-DHC by three different artificial pathways. f. Improving 4,6-DHC production by PqsD optimization. g. Improving 4,6-DHC production by regulating the pathway genes and PqsD optimization. Solid arrows represent single-step reactions, while dashed arrows represent multi-step reactions. The data presented represent the mean ± standard deviation (SD) from three independent experiments, with error bars indicating the SD. Note: hyg5: the codon-optimized version for E. coli; hyg5*: incorporates rare codons at special sites based on hyg5; and pqsD: wild-type pqsD; pqsD (P259A): a variant of pqsD at site P259. | ||
| Enzyme | Substrate | K m (μM) | k cat (min−1) | k cat/Km (μM−1 min−1) |
|---|---|---|---|---|
| SdgA | 2-HBA | 26.6 ± 2.0 | 886.67 ± 66.33 | 33.33 |
| 2,5-DHBA | 52.6 ± 9.1 | 828.00 ± 32.33 | 15.74 | |
| PqsD | 2-HBA-CoA | 27.2 ± 3.4 | 103.56 ± 2.72 | 3.81 |
| 2,5-DHBA-CoA | 97.3 ± 16.7 | 63.28 ± 4.17 | 0.65 | |
| PqsD (P259A) | 2-HBA-CoA | 32.9 ± 4.9 | 154.25 ± 3.25 | 4.69 |
| 2,5-DHBA-CoA | 58.8 ± 8.1 | 140.92 ± 10.16 | 2.40 | |
| 3HB6H | 3-HBA | 79.5 ± 7.9 | 194.24 ± 5.76 | 2.44 |
| Re3HB6H | 3-HBA | 33.5 ± 4.3 | 34.23 ± 0.58 | 1.02 |
To further test the transformation from different HBAs to the final product 4,6-DHC, the strains DX5, DX6, and DX7 were constructed by co-transformation of the plasmids pZE-SalABCD, pHA-3HB6H, and pHA-PhgC-PhgB-PhgA combined with pCS-PqsD-SdgA into E. coli BW25113 (F′), respectively. When we provided the strain DX6 with 1 g L−1 3-HBA as a substrate, we observed the production of 4,6-DHC at a concentration of 158.2 ± 1.9 mg L−1. When we supplied the strain DX5 with 1 g L−1 2-HBA as a substrate, we observed the production of 4,6-DHC at 0.7 ± 0.1 mg L−1 and the byproduct 4-HC at 61.8 ± 5.1 mg L−1 (Fig. 3c). These findings implied the presence of a competitive reaction favoring the generation of the byproduct 4-HC over the desired product 4,6-DHC. The in vitro enzyme assays (Table 1 and ESI Fig. 2, 3†) revealed that the enzyme PqsD exhibited a substrate preference toward 2-HBA-CoA, with a higher Km value for 2,5-DHBA-CoA compared to 2-HBA-CoA (97.3 ± 16.7 μM vs. 27.2 ± 3.4 μM) (Table 1 and ESI Fig. 3†). Protein engineering aimed at enhancing the selectivity and catalytic activity of PqsD for 2,5-DHBA-CoA, along with improving the flux toward this substrate, presents a promising strategy to boost the production of 4,6-DHC. Upon feeding 4-HBA into the cultures, the strain DX7 produced only 0.5 ± 0.1 mg L−1 4,6-DHC (Fig. 3c and ESI Fig. 4a†). This could be attributed to the activity of PhgB, gentisyl-CoA thioesterase, which may reduce the formation of the direct precursor 2,5-DHBA-CoA. To mitigate the impact of PhgB and streamline the 4-HBA pathway, we opted to eliminate the enzymes PhgB and SdgA in the optimized route III. Subsequently, the strain DX7S was constructed by transferring the plasmid pHA-PhgC-PhgA-PqsD into E. coli BW25113 (F′). However, despite these efforts, the feeding experiment still resulted in minimal 4,6-DHC production (ESI Fig. 4b†). The potential reason is that there might be a competitive inhibition effect on the enzyme PqsD caused by 4-HBA-CoA. To validate this hypothesis, we conducted a feeding experiment by simultaneously adding 200 mg L−1 of both 2,5-DHBA and 4-HBA. The results of this feeding experiment clearly indicated a significant reduction in 4,6-DHC production when 4-HBA was present, especially when the p-hydroxybenzoyl-CoA ligase PhgC was expressed (ESI Fig. 4c†). This finding supported our hypothesis that 4-HBA-CoA may act as a competitive inhibitor of PqsD towards 2,5-DHBA-CoA. It's possible that the 2,5-DHBA-CoA analog could occupy the 2,5-DHBA-CoA channel in PqsD and act as a channel blocker, preventing 2,5-DHBA-CoA from entering the catalytic site.33
After exploring the feasibility of 4,6-DHC biosynthesis through different routes and to enable de novo 4,6-DHC production in E. coli, we next evaluated HBA de novo production using glycerol as the carbon source. As shown in Fig. 3d, at 48 h, the strain DX8 (E. coli BW25113 (F′) carrying pZE-EP) accumulated 134.3 ± 3.2 mg L−1 2-HBA, the strain DX9 (E. coli BW25113 (F′) carrying pHA-Hyg5) only generated 5.4 ± 0.5 mg L−1 3-HBA and the strain DX11 (E. coli BW25113 (F′) carrying pHA-Ubic) produced 49.4 ± 4.4 mg L−1 4-HBA. Afterward, we evaluated the entire production process, carefully considering the advantages and disadvantages of each pathway, with particular emphasis on route I (2-HBA pathway) and route II (3-HBA pathway) for further investigation.
Enzyme engineering to increase the pathway efficiency
In route II, as evidenced by the stepwise transformation test results (Fig. 3a–d), the low activity of Hyg5 and PqsD appears to limit the overall conversion efficiency. To enhance the efficiency of the desired reaction, a commonly employed strategy involves codon optimization to boost the expression of heterologous genes.34 Multiple pathway genes underwent codon optimization for synthesis, including hyg5 and 3hb6h. However, this approach can potentially result in protein misfolding and insolubility due to accelerated translation rates and the elimination of essential translation pauses.35,36 The SDS-PAGE results revealed that the Hyg5 protein predominantly formed inclusion bodies, suggesting its poor solubility (Fig. 4b). Thus, we hypothesized that codon optimization may accelerate protein translation, potentially adversely affecting enzyme folding and solubility. Recent studies suggested the deliberate incorporation of specific codons known as “synonymous rare codons” (SRCs) can facilitate the co-translational folding of polypeptide chains, ultimately enhancing the expression of functional proteins.37 Therefore, we proposed introducing rare codons at strategic positions within hyg5 to help balance the translation rates and potentially enhance protein solubility and activity. When introducing SRCs, it is recommended to focus on regions encoding the start of α-helices or β-strands, particularly in the initial portion of the protein and the 5′ coding sequence.37,38 So, after analyzing the secondary structure of Hyg5 and comparing the hyg5 sequence with the original sequence without codon optimization, we selected positions 4, 13, 24, 28, 52, 63, 125, 173, 192 and 260 for the replacement of SRCs (Fig. 4a). By incorporating rare codons into the hyg5 gene, we generated hyg5*. According to Fig. 4b, the gene with synonymous substitutions (hyg5*) led to a roughly four-fold increase in protein solubility compared to the original gene, hyg5. We subsequently established the strain DX10 by transferring the plasmid pHA-Hyg5*. In the strain DX10, the titer of 3-HBA reached 37.4 ± 3.8 mg L−1 at 24 h, which is approximately 6.9-fold higher than that of the strain DX9 (Fig. 4c). The SDS-PAGE analysis revealed that the rare codon-optimized gene hyg5* led to improved solubility of the Hyg5 protein compared to the initially synthesized gene, hyg5. Taken together, the rare codon optimization strategies we used here improved the expression level of Hyg5 in E. coli as well as increased the accumulation of 3-HBA.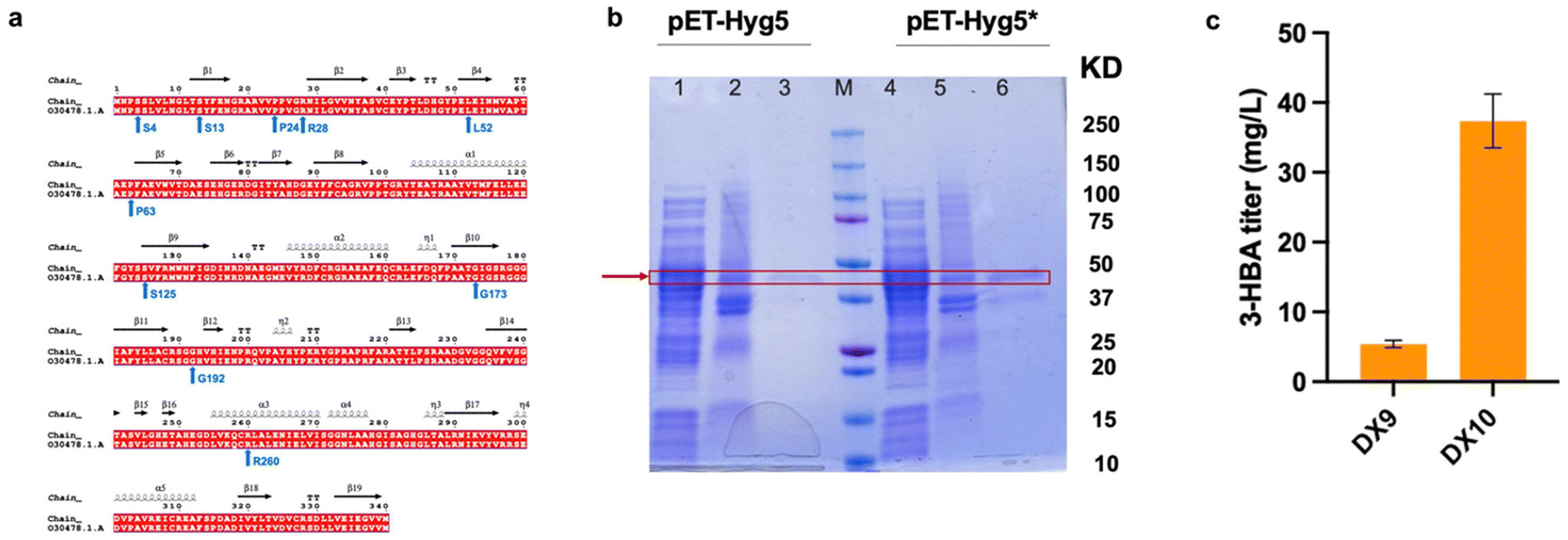 | ||
| Fig. 4 Optimization of Hyg5 and production of 3-HBA through feeding experiments. a. Hyg5 sequence and its secondary structure. b. SDS-PAGE analysis was used to determine the solubility levels of Hyg5 and Hyg5*. The bands corresponding to Hyg5 and Hyg5* are indicated by red arrows. 1/4: supernatants, 2/5: pellets, 3/6: purified protein, M: marker. c. The titer of 3-HBA in different strains. Strain DX9: E. coli BW25113 (F′) carrying pHA-Hyg5; strain DX10: E. coli BW25113 (F′) carrying pHA-Hyg5*. The data presented represent the mean ± standard deviation (SD) from three independent experiments, with error bars indicating the SD. | ||
In addition to employing rare codon optimization on hyg5 to enhance 3-HBA production, we attempted to reduce the IPTG induction concentration to lower the expression of Hyg5, but this did not improve the 3-HBA titer (ESI Fig. 5a†). In E. coli, pheA and tyrA, downstream genes in the shikimate pathway are essential for phenylalanine and tyrosine biosynthesis. So, we tried to boost 3-HBA titers by disrupting competitive pathways, including pheA and tyrA. Introducing pHA-Hyg5 into the strain E. coli ATCC 31884 ΔpheA Δtyr did result in improved 3-HBA titer (ESI Fig. 5b†). However, when we introduced the entire 3-HBA pathways (pHA-Hyg5-3HB6H and pCS-PS) into E. coli ATCC 31884 ΔpheA ΔtyrA, we failed to detect 4,6-DHC production (ESI Fig. 5c†). This failure may be attributed to the pheA and tyrA knockouts, which likely affected enzyme expression and impaired cell growth.
To address by-product formation in route I, the PqsD enzyme was rationally engineered to improve its substrate selectivity and activity for 2,5-DHBA-CoA. Kinetic studies revealed that PqsD displayed a greater substrate preference for 2-HBA-CoA (Kcat = 103.56 ± 2.72 min−1, Km = 27.2 ± 3.4 μM) over 2,5-DHBA-CoA (Kcat = 63.28 ± 4.17 min−1, Km = 97.3 ± 16.7 μM) (Table 1). By performing rational protein engineering on key residues in the PqsD binding pocket (ESI Fig. 6a†) and introducing random mutations at P259 of PqsD (ESI Fig. 7†), the P259A mutant was identified as the sole variant that enhanced the 4,6-DHC titer compared to the wild-type enzyme (1.45-fold, 38.4 ± 1.8 mg L−1vs. 26.3 ± 1.9 mg L−1) (ESI Fig. 6b†). Kinetic studies with PqsD-P259A (Kcat = 140.92 ± 10.16 min−1, Km = 58.8 ± 8.1 μM) (Table 1 and ESI Fig. 8†) showed an increase in Kcat and a decrease in Km toward 2,5-DHBA-CoA compared to wild-type PqsD (Kcat = 63.28 ± 4.17 min−1, Km = 97.3 ± 16.7 μM). The combination of increased Kcat and Km signifies that PqsD-P259A mutation improves both the binding affinity and the catalytic activity of the enzyme toward 2,5-DHBA-CoA. Further details on pqsD protein engineering are provided in the ESI.†
De novo biosynthesis of 4,6-DHC in E. coli
With the verified enzyme activity in each step and engineered enzymes, we designed the de novo biosynthesis of 4,6-DHC from glycerol in E. coli. To accomplish this, we successfully created the producing strain DX12 containing the plasmids pZE-EntC-PchB-SdgA-PqsD and pCS-SalABCD, the strain DX13 containing the plasmids pHA-Hyg5*-3HB6H and pCS-SdgA-PqsD and the strain DX15 containing the plasmids pHA-Hyg5*-3HB6H and pCS-SdgA-PqsD (P259A). The strain DX12 produced only 1.0 ± 0.3 mg L−1 of 4,6-DHC, but 18.5 ± 1.9 mg L−1 of 4-HC (Fig. 3e), suggesting the need for additional strategies to optimize the biosynthetic pathway through 2-HBA and decrease the production of byproduct 4-HC. For the 3-HBA pathway, the strain DX15 yielded 4,6-DHC at a titer of 22.7± 2.1 mg L−1, surpassing the strain DX13, which produced it at 15.5 ± 2.6 mg L−1, indicating a 1.46-fold increase (Fig. 3e and f).Pathway regulation to further improve 4,6-DHC production
In comparison with the 3-HBA pathway, the 2-HBA pathway exhibits advantages in high yield hydroxybenzoic acid production (Fig. 3d). However, as previously mentioned, the issue of by-product formation caused by the promiscuity of PqsD towards 2-HBA-CoA poses a bottleneck in 4,6-DHC production. Since 2-HBA serves as the precursor for 2,5-DHBA, we hypothesized that the by-product formation could be controlled by delaying the expression of SdgA and PqsD, which can rewire more 2-HBA for 2,5-DHBA biosynthesis (Fig. 5a). To achieve this, inducible expression was employed to control the expression of SdgA and PqsD. The tetR-tetO1 regulatory system, comprising PtetO1 promoters and the tetR repressor, is a well-established method for regulating gene expression by introducing the inducer tetracycline.39,40 To establish the inducible regulation system, we positioned the sdgA and pqsD genes under the control of the PtetO1 promoter, while tetR was regulated by the Plpp0.2 promoter. This resulted in the creation of the plasmids pZE-PtetO1-PqsD-SdgA, pZE-PLacO1-EP-PtetO1-PqsD-SdgA and pCS-Plpp0.2-tetR-SalABCD. In the absence of the inducer tetracycline, the tetR protein binds to the PtetO1 promoter, effectively suppressing the expression of sdgA and pqsD. Upon introducing the inducer into the culture, the tetR repression can be released, enabling the initiation of sdgA and pqsD gene expression. | ||
| Fig. 5 Inducible regulation of the de novo biosynthesis of 4,6-DHC in the strain DX16 and the strain DX17. a. Pathway regulation in pqsD and sdgA expression to reduce byproduct 4-HC production. b. The titer of 4,6-DHC by using different induction times. c. The titer of 4-HC by using different induction times. d. The titer of 2,5-DHBA by using different induction times. e. The titer of 4,6-DHC using the strain DX16 and the strain DX17. The data presented represent the mean ± standard deviation (SD) from three independent experiments, with error bars indicating the SD. | ||
To apply this inducible regulation system, we co-transferred the plasmids pZE-PLacO1-EP-PtetO1-PqsD-SdgA and pCS-Plpp0.2-tetR-SalABCD into E. coli BW25113 (F′), generating the strain DX16. We explored various tetracycline induction concentrations (ESI Fig. 9†) and induction times (0 h, 3 h, 6 h, 9 h, 12 h) to identify the optimal production conditions. The addition of tetracycline at different time points resulted in varying levels of 4,6-DHC, 4-HC and 2,5-DHBA accumulation (Fig. 5b–d). Remarkably, when tetracycline was introduced at 3 h, the strain DX16 achieved the highest 4,6-DHC titer of 18.3 ± 0.7 mg L−1 at 48 h, representing an 18.3-fold increase compared to the static control strain DX12 lacking the delay in sdgA and pqsD expression (Fig. 5b and 3e). Furthermore, compared with the strain DX12, we observed a reduction in the by-product 4-HC, with its titer decreasing from 18.5 ± 1.9 mg L−1 to 6.6 ± 0.4 mg L−1 at 48 h (Fig. 5d). These findings clearly demonstrate that delaying the expression of sdgA and pqsD is advantageous for by-product reduction and enhanced 4,6-DHC production. For further enhancement of 4,6-DHC production, the PqsD (P259A) mutant was incorporated into the plasmid pZE-PLacO1-EP-PtetO1-PqsD-SdgA to replace PqsD. We co-transferred the plasmids pZE-PLacO1-EP-PtetO1-PqsD (P259A)-SdgA and pCS-Plpp0.2-tetR-SalABCD into E. coli BW25113 (F′), resulting in the strain DX17. After 48 hours of cultivation, the strain DX17 successfully produced more 4,6-DHC with a titer of 56.7 ± 2.1 mg L−1, marking a 3.1-fold increase compared to the strain DX16 and yielding 3.84 mg per g-glycerol (Fig. 3g, 5e and ESI Fig. 10, 11†). Overall, the novel 4,6-DHC synthetic pathway has been validated and the engineered PqsD enabled a higher titer for de novo 4,6-DHC biosynthesis in E. coli.
To further investigate pathway regulation in 4,6-DHC biosynthesis at the translation level, we conducted SDS-PAGE analysis using the strain DX17 to examine variations in the expression of the enzymes SdgA and PqsD under tetR-PtetO1 regulation. The accumulation of 2,5-DHBA, 4-HC, and 4,6-DHC and the OD600 values were tested (ESI Fig. 12a–12d†). As shown in ESI Fig. 12e–12g,† at 6 h, protein expression levels were relatively low, and the SdgA and PqsD band intensity did not show a clear trend, likely due to the rapid cell growth. At 12 and 18 h, the expression levels of SdgA and PqsD were higher when induced at 0 and 3 h compared to induction at 6 and 9 h, with band intensities following the trend: 0 h ≈ 3 h > 6 h > 9 h. As shown in ESI Fig. 12g,† after 24 h, the expression levels of SdgA and PqsD began to decline. These protein expression results correspond to the observed 4,6-DHC production patterns. Specifically, 4,6-DHC accumulated rapidly before 24 h, followed by a slower increase to reach a relatively stable level. Induction at 3 h resulted in the highest 4,6-DHC production performance. These findings are consistent with our proposed mechanism and are supported by the experimental data presented. Directing carbon flux toward 2,5-DHBA while maintaining optimal expression levels of SdgA and PqsD is crucial for achieving higher target product titers.
Discussion
Many valuable natural products are difficult to obtain by chemical synthesis due to the requirements of specific catalysts, reagents, and harsh reaction conditions.41 Biosynthesis provides an alternative method to produce valuable molecules in an efficient and eco-friendly manner using engineered microorganisms. To broaden the spectrum of potential metabolic pathways, efforts have been made with the help of bioinformatics and computer science analysis, including predicting new enzymes, exploring new reactions using natural enzymes, optimizing existing enzymes for novel reactions, and even de novo designing enzymes with specific target functions.42–45 The primary principles to be considered in circuit design are the recruitment of highly efficient and host-suitable enzymes.Nevertheless, integrating novel synthetic pathways into metabolic networks presents challenges, as they often disrupt endogenous metabolism, raise growth burden and generate unwanted side reactions. Also, while certain enzymes within these constructed pathways may exhibit high catalytic efficiency, they might not properly align with other intermediates or could be susceptible to inhibition by other intermediates within the pathway, thus acting as bottlenecks within biosynthetic pathways. Within the designed pathways, various analogs of 2,5-DHBA-CoA are generated. Specifically, 2-HBA-CoA can also be catalyzed by PqsD, resulting in the undesired by-product 4-HC. Additionally, 4-HBA-CoA may exhibit an inhibitory effect on PqsD. Sometimes, integrating all precursor pathways within a single strain would circumvent issues related to protein expression burden, cofactor deficiency, and side reactions in a single cell. As a result, the titer of the target products may not be improved under these conditions. These findings highlight the importance of carefully considering the enzyme's substrate promiscuity and the potential formation of undesired by-products when designing and optimizing biosynthetic pathways.
Strategies to address these challenges may include enzyme engineering to improve substrate specificity and reduce side reactions, incorporating an inducible regulation system to control the timing of critical gene expression, and optimizing processes to alleviate inhibitory effects on enzyme activity. In a heterologous expression system, enzymes frequently face low solubility issues. To address this, we modulated the translation rate by introducing SRCs. More strategies can be employed to enhance protein solubility, such as fusion tags, co-expression of chaperones, directed evolution, buffer optimization, expression conditions, and host strain selection.46–48 Additionally, targeted enzyme modification of PqsD was performed to improve substrate specificity and activity, resulting in a mutant with increased enzyme activity towards 2,5-DHBA-CoA. Using the two different circuits through 2-HBA and 3-HBA respectively, we were able to carry out the designed route for the biosynthesis of 4,6-DHC in E. coli through a whole-cell transformation process.
Conclusion
In this study, we devised novel biosynthetic pathways to synthesize 4,6-DHC from the simple carbon source glycerol by discovering novel biological reactions rooted in known enzymatic mechanisms. A rare codon introduction strategy was applied to significantly improve the solubility of Hyg5 which led to a 6.9-fold increase in 3-HBA production compared to the original codon-optimized enzyme Hyg5. Additionally, targeted enzyme modifications were performed on PqsD to enhance substrate specificity and activity, resulting in a mutant with increased enzyme activity towards 2,5-DHBA-CoA. Combined with pathway regulation, the titer of 4,6-DHC was greatly improved to 56.7 ± 2.1 mg L−1, with a yield of 3.84 mg g−1 glycerol under shake flask culture conditions. Importantly, this is the first reported biosynthesis of 4,6-dihydroxycoumarin. Achieving selective hydroxylation of the aromatic ring for the chemical synthesis of hydroxycoumarins presents a significant challenge. Compared to chemical synthesis methods, biosynthesis in E. coli offers a more environmentally friendly and sustainable approach. Further optimization through metabolic engineering could enhance its yield even more. To further explore the potential applications of our approach, additional precursors such as amino, nitro, or chloro salicylic acid could be tested to produce a wider range of hydroxycoumarins. Our research offers valuable insights into the sustainable synthesis of valuable compounds.Materials and methods
Medium, strains, plasmids, and chemicals
To perform various tasks such as inoculating cells, propagating plasmids, and expressing proteins, a lysogeny broth (LB) medium (10 g L−1 tryptone, 5 g L−1 yeast extract and 10 g L−1 NaCl) was used. An optimized M9Y medium (20 g L−1 glycerol, 5 g L−1 yeast extract, 1 g L−1 NH4Cl, 6 g L−1 Na2HPO4, 3 g L−1 KH2PO4, 0.5 g L−1 NaCl, 246.5 mg L−1 MgSO4·7H2O and 14.7 mg L−1 CaCl2·2H2O) was employed for the feeding tests and for the de novo biosynthesis. Antibiotics such as ampicillin (100 μg L−1) and kanamycin (50 μg L−1) were supplied to the culture if necessary. To induce protein overexpression, IPTG (isopropyl-β-D-thiogalactose) was added. Plasmid construction and replication were performed using the E. coli strain XL1-blue. E. coli BW25113 (F′) was utilized for feeding tests and de novo fermentation, while E. coli BL21 (DE3) was employed for protein expression. The plasmids pZE12-luc/pHA-egfp-MCS and pCS27/pMK-egfp-MCS served as the backbones for high/medium-copy number plasmids, respectively.49 The plasmid pETDuet-1 served as the vector for protein expression and purification. ESI Table 1† provides details of the strains and plasmids used in this study. All primers employed in this investigation were synthesized by Integrated DNA Technologies (IDT) and Eurofins Genomics.2-HBA (2-hydroxybenzoic acid), 3-HBA (3-hydroxybenzoic acid), 4-HBA (4-hydroxybenzoic acid), 2,3-DHBA (2,3-dihydroxybenzoic acid), 2,4-DHBA (2,4-dihydroxybenzoic acid), 2,5-DHBA (2,5-dihydroxybenzoic acid), 2,6-DHBA (2,6-dihydroxybenzoic acid), and 4,6-DHC (4,6-dihydroxycoumarin) were purchased from Sigma-Aldrich, USA, while Fisher Chemicals, USA, provided methanol. New England Biolabs, USA, provided High Fidelity Phusion DNA polymerase, the restriction enzyme and the fast ligation kit. In addition, the Plasmid Miniprep Kit and Gel DNA Recovery Kit were purchased from Zymo Research, USA.
DNA manipulation
Genes were synthesized for 3hb6h originating from Rhodococcus jostii RHA1 and hyg5 from Streptomyces rapamycinicus. These synthetic genes were optimized for the codon usage of Escherichia coli and were acquired from a commercial source (Eurofins Genomics, USA). Re3hb6h was cloned from Ralstonia eutropha genomic DNA. phgA, phgB, and phgC were cloned from Brevibacillus laterosporus PHB-7a (ATCC29653) genomic DNA. The DNA sequences corresponding to these genes are provided in ESI Table 2.† In the previous study, the high-copy plasmid pHA-eGFP-MCS was created, featuring a ColE1 origin, an ampicillin resistance gene, and the PLlacO1 promoter. Additionally, the plasmid includes a synthetic multicloning site (MCS) with sequential recognition sites for KpnI, MscI, NdeI, BsrGI, SalI, ClaI, HindIII, NheI, BamHI, and MluI. Five plasmids pZE-EP, pZE-EPPS, pCS-PS, pZE-SalABCD, and pCS-Plpp0.2-RBS1.0-K127Y were constructed in our previous research.11,30,40 To examine the conversion of various HBAs to DHBAs, we constructed the plasmids pHA-3HB6H and pHA-PhgC-PhgB-PhgA. The plasmid pHA-3HB6H was constructed by inserting the gene 3hb6h into pHA-MCS using KpnI/HindIII. To obtain the plasmid pHA-PhgC-PhgB-PhgA, phgC was inserted into pHA-eGFP-MCS using KpnI/BsrGI, phgA was inserted into pHA-eGFP-MCS using KpnI/SalI, and phgB was inserted into pHA-eGFP-MCS using KpnI/XbaI, resulting in the plasmids pHA-PhgA, pHA-PhgB, and pHA-PhgC, respectively. Next, the operon PLlacO1-phgB-terminator, containing the PLlacO1 promoter, phgB gene, and terminator, was inserted into pHA-PhgC using BsrGI/XhoI to create pHA-PhgC-PhgB. Subsequently, the operon PLlacO1-phgA-terminator was cloned from pHA-PhgA, digested using XbaI/SacI, and the plasmid pHA-PhgC-PhgB was digested using SpeI/SacI. The resulting digested fragment was then ligated with the vector. To construct the plasmids for the de novo synthesis of HBAs, the genes hyg5, hyg5*, and ubic were inserted into the plasmid pHA-eGFP-MCS using KpnI/SalI, resulting in the formation of the plasmids pHA-Hyg5, pHA-Hyg5*, and pHA-Ubic, respectively. To construct the plasmids pHA-Hyg5-3HB6H and pHA-Hyg5*-3HB6H, the 3hb6h gene was inserted into the plasmids pHA-Hyg5 and pHA-Hyg5* using SpeI/SacI, respectively. The plasmid pCS-PS-Ubic was constructed by inserting ubic into the plasmid pCS-PS using SpeI/SacI. To obtain the plasmid pHA-PhgC-PhgA-PqsD, the operon PLlacO1-phgA-terminator was inserted into the plasmid pHA-PhgC using BsrGI/XhoI, resulting in the plasmid pHA-PhgC-PhgA. Next, the operon PLlacO1-pqsD-terminator was introduced into pHA-PhgC-PhgA using XbalI/SacI. To obtain the plasmid pZE-EP-Pteto1-pqsD-sdgA, the operon Pteto1-pqsD-sdgA-terminator was inserted into pZE-EP using SpeI/SacI. The plasmid pCS-Plpp0.2-tetR was constructed by inserting tetR into the plasmid pCS-Plpp0.2-RBS1.0-K127Y using KpnI/BamHI. Next, the operon PLlacO1-SalABCD-terminator was cloned from pCS-SalABCD and was inserted into pCS-Plpp0.2-tetR using SpeI/SacI, resulting in the plasmid pCS-Plpp0.2-tetR-SalABCD. To obtain the proteins, the genes hyg5, hyg5*, sdgA, 3hb6h, Re3hb6h, pqsD and pqsD (P259A) were individually sub-cloned into pETDuet-1. Each gene was incorporated in-frame with the his-tag DNA sequence using BamHI/HindIII.Feeding tests
The plasmid pCS-PqsD-SdgA was transferred into E. coli BW25113 (F′) to examine the conversion from 2,5-DHBA to DHC, resulting in the strain DX1. 200 mg L−1 2,5-DHBA was added as the substrate. The plasmid pZE-SalABCD, pHA-3HB6H, or pHA-PhgC-PhgB-PhgA was individually transferred into E. coli BW25113 (F′) to assess the conversion from HBA to DHBA, yielding the strains DX2, DX3, and DX4, respectively. 1 g L−1 of 2-HBA, 3-HBA, or 4-HBA was added as the respective substrate. To evaluate the conversion from HBA to 4,6-DHC, the plasmid pZE-SalABCD, pHA-3HB6H, or pHA-PhgC-PhgB-PhgA along with pCS-PqsD-SdgA was introduced into E. coli BW25113 (F′), resulting in the strains DX5, DX6, and DX7, respectively. 1 g L−1 of 2-HBA, 3-HBA, or 4-HBA was added as the respective substrate. The strains were cultured overnight at 37 °C in the LB medium (3.5 mL per tube). A volume of 1 mL of bacterial seed solution was inoculated into 20 mL of the optimized M9Y medium containing ampicillin and kanamycin, followed by incubation at 37 °C and 270 rpm for 3 hours. Subsequently, protein overexpression was induced by adding 0.5 mM IPTG along with substrates at a suitable concentration. The cultures were then transferred to 30 °C for further growth. An HPLC analysis was performed on samples taken at 24 h. Feeding experiments were carried out in triplicate.De novo fermentation
To achieve the de novo biosynthesis of HBA, the strains DX8, DX9, DX10, and DX11 were cultured in the optimized M9Y medium. For the de novo biosynthesis of 4,6-dihydroxycoumarin, the strains DX12, DX13, DX14, DX15, DX16, and DX17 were employed (Table S1†). Initially, a single colony from the producing strain was selected and cultured in 3.5 mL of the LB medium at 37 °C and 270 rpm overnight. Subsequently, 1 mL of the strain seed liquid was shifted into the optimized M9Y medium (20 mL per flask) and kept at 37 °C and 270 rpm for 3 h. Next, IPTG was supplied to the medium with a suitable concentration. The strains were kept growing at 30 °C for 72 h. Samples were collected and subjected to HPLC analysis. Each experiment was carried out in triplicate.Protein expression and purification
To obtain these proteins, we initiated the process by transferring the plasmids pET-Hyg5, pET-Hyg5*, pET-SdgA, pET-3HB6H, pET-Re3HB6H, pET-PqsD and pET-PqsD (P259A) into E. coli BL21 Star (DE3). Subsequently, single transformants were cultivated overnight in 3 mL of LB medium containing 100 μg mL−1 of ampicillin at a temperature of 37 °C. 1% seed culture was then transferred to a 250 mL shake flask containing 50 mL of LB medium. This culture was allowed to grow at 37 °C until the OD600 values reached 0.6–0.8 and was induced with 0.5 mM isopropyl-β-D-thiogalactoside (IPTG). Then the flask was maintained at 30 °C for an additional 5 hours with agitation at 270 rpm.The cells were harvested and disrupted using a Mini Bead Beater from Biospec. Subsequently, the proteins were purified using a His-Spin™ Protein Miniprep Kit (Zymo Research) in accordance with the manufacturer's provided protocol. To ensure the quality and quantity of the obtained proteins, we validated them through Tricine-SDS-polyacrylamide gel electrophoresis (Tricine-SDS-PAGE) utilizing a 12% protein gel (ESI Fig. 13†). Additionally, we quantified the protein concentrations using a Pierce BCA Protein Assay Kit from Thermo Scientific, following the manufacturer's recommended procedures. The protein expression level was assessed by expressing the protein, taking equal sample volumes, and loading them onto an SDS-PAGE gel. The relative levels of soluble hyg5 and hyg5* proteins were quantified using grayscale scanning, followed by analysis of the protein band intensity using ImageJ software.
Enzyme assays
The kinetic parameters of the enzyme SdgA were tested using the optimized method from Ishiyama.50 The total 1 mL reaction system included 780 μL of Tris-HCl (100 mM, pH = 7.5), 10 μL of MgCl2 (0.5 M), 50 μL of coenzyme A (5 mM), 50 μL of ATP (100 mM), 10 μL of the purified enzyme SdgA, and 100 μL of salicylate or 2,5-dihydroxybenzoic acid (100 μM, 200 μM, 500 μM, 1 mM, 2 mM, 4 mM, and 5 mM). The reactions lasted for 0.5 min at 30 °C and then were terminated by adding 20 μL of 20% HCl. The reaction rates were calculated according to the rates of salicylate consumption, which were measured by HPLC.Coupled assays were utilized to ascertain the kinetic parameters of the enzymes PqsD and PqsD (P259A) based on the method described by Lin.11 For this purpose, SdgA was employed to convert salicylate and 2,5-dihydroxybenzoic acid to salicylate-CoA and 2,5-dihydroxybenzoic acid-CoA, respectively. The reaction system, consisting of 200 μL volume, comprised the following components: 145 μL of Tris-HCl (pH = 7.5, 100 mM), 10 μL of MgCl2 (100 mM), 5 μL of CoA (20 mM), 10 μL of ATP (100 mM), 10 μL of purified SdgA, and 20 μL of salicylate or 2,5-dihydroxybenzoic acid (20–400 μM). The reaction was first incubated at 30 °C for 30 minutes. Subsequently, the purified PqsD or PqsD (P259A) was introduced into the reaction system and further incubated for 1 minute. The reactions were halted by acidification using 20 μL of 20% HCl. The enzyme parameters were determined based on the production of 4-hydroxycoumarin and 4,6-dihydroxycoumarin, which were quantified using HPLC.
The activities of 3HB6H and Re3HB6H were assayed based on the method described by Chen.51 A 200 μL reaction mixture was prepared, consisting of 400 μM NADH, varying concentrations of 3-HBA (20 μM, 50 μM, 100 μM, 200 μM, 300 μM, 400 μM, 600 μM), and the purified enzyme extract in 50 mM phosphate buffer (pH 8.0) at 30 °C. The rate of 3-HBA consumption was used to determine the reaction rate of 3-HBA. The reaction proceeded for 30 s and was halted by adding 20% HCl. The analysis of enzyme assay data was conducted using OriginPro 2020 software. The experiments were conducted in triplicate.
Molecular docking
Molecular docking analyses were conducted using AutoDock Vina in this study.52 The structure of PqsD utilized in the docking analysis was sourced from the Protein Data Bank (PDB ID: 3H76). All water molecules and ligands present in the crystal structure were eliminated, and hydrogen atoms were added to the protein. Following this, 2,5-DHBA-CoA was prepared for docking using ChemDraw and Avogadro. The docking site was defined based on the substrate anthranilate coordination from 3H76, with a docking radius set to 12.0 Å to fully cover the substrate pocket's cavity volume. After performing the docking simulation to generate 3H76/2,5-DHBA-CoA complexes, the results were analyzed to identify the most favorable binding poses of the ligand, and appropriate complexes were selected for further analysis based on energy scores and geometric conformations.Site-specific mutagenesis
Site-directed mutagenesis for the generation of PqsD variants with specific changes was achieved through the SLIM method.53 The pCS-PqsD-SdgA plasmid served as the template. The pCS-PqsD(P259A)-SdgA plasmid served as the template for the combination variant construction. The validity of all mutants was through DNA sequencing.Random mutagenesis of pqsD
Random mutagenesis was performed with E. coli XL1-Blue competent cells. Mutation procedures followed the error-prone PCR manual. After mutation, plasmids were extracted and further transformed with E. coli BW25113 (F′) competent cells. After cultivation at 37 °C for 1 h, cells were plated on the LB agar plate with 50 μg mL−1 kanamycin. The plates were cultivated at 37 °C overnight. Twenty-five single colonies were selected and inoculated in 10 mL of TB broth with 50 μg mL−1 kanamycin at 37 °C overnight for the feeding test.Protein expression analysis
1 mL samples were collected during different fermentation times from the shaking flask cultures during the de novo fermentation using the strain DX17. The cells were harvested and disrupted using a Mini Bead Beater from Biospec. The protein expression level was assessed by loading equal volumes of the supernatant, obtained after lysing the harvested cells, onto an SDS-PAGE gel.Metabolite analysis
Samples were collected, centrifuged to filter the membranes, and then subjected to HPLC analysis. HPLC analysis (Agilent, 1260 Infinity II, ZORBAX SB-C18) was used for the quantification of 2-HBA, 3-HBA, 4-HBA, 2,5-DHBA, 4-HC and 4,6-DHC. The samples were analyzed using methanol (solution A) and water (solution B) with 0.1% TFA. The HPLC program consisted of a gradient methanol: 0–2 min, 5% MeOH; 2–30 min, 5%–60% MeOH; 30–32 min, 60%–90% MeOH; and 32–35 min, 90%–5% MeOH. The column oven was set at 45 °C. UV absorbance at 280 nm was utilized for the quantification of 4-HBA, 4-HC and 4,6-DHC and at 300 nm for the quantification of 2-HBA, 3-HBA and 2,5-DHBA.Author contributions
Qi Gan: conceptualization, data curation, software, formal analysis, investigation, methodology, validation, visualization, writing – original draft, and writing – review & editing. Tian Jiang: conceptualization, formal analysis, investigation, methodology, validation, and writing – review & editing. Chenyi Li: investigation and methodology. Xinyu Gong: formal analysis and writing – review & editing. Jianli Zhang: formal analysis and writing – review & editing. Bhaven K. Desai: investigation and writing – review & editing. Yajun Yan: conceptualization, funding acquisition, investigation, methodology, project administration, supervision, and writing – review & editing.Data availability
The data supporting the findings of this work are available within the paper and the ESI files.† Source data will be provided upon request.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award number R35GM128620. We also acknowledge the support from the College of Engineering, The University of Georgia, Athens.References
- E. Kupeli Akkol, Y. Genc, B. Karpuz, E. Sobarzo-Sanchez and R. Capasso, Coumarins and Coumarin-Related Compounds in Pharmacotherapy of Cancer, Cancers, 2020, 12(7), 1959 CrossRef PubMed.
- M. E. Riveiro, N. De Kimpe, A. Moglioni, R. Vazquez, F. Monczor, C. Shayo and C. Davio, Coumarins: old compounds with novel promising therapeutic perspectives, Curr. Med. Chem., 2010, 17(13), 1325–1338 CrossRef CAS PubMed.
- A. Tosun, E. K. Akkol and E. Yesilada, Anti-inflammatory and antinociceptive activity of coumarins from Seseli gummiferum subsp. corymbosum (Apiaceae), Z. Naturforsch., C:J. Biosci., 2009, 64(1–2), 56–62 CrossRef CAS PubMed.
- K. N. Venugopala, V. Rashmi and B. Odhav, Review on natural coumarin lead compounds for their pharmacological activity, BioMed Res. Int., 2013, 2013, 963248 CAS.
- D. R. Vianna, L. Hamerski, F. Figueiro, A. Bernardi, L. C. Visentin, E. N. Pires, H. F. Teixeira, C. G. Salbego, V. L. Eifler-Lima, A. M. Battastini, G. L. von Poser and A. C. Pinto, Selective cytotoxicity and apoptosis induction in glioma cell lines by 5-oxygenated-6,7-methylenedioxycoumarins from Pterocaulon species, Eur. J. Med. Chem., 2012, 57, 268–274 CrossRef CAS PubMed.
- F. Annunziata, C. Pinna, S. Dallavalle, L. Tamborini and A. Pinto, An Overview of Coumarin as a Versatile and Readily Accessible Scaffold with Broad-Ranging Biological Activities, Int. J. Mol. Sci., 2020, 21(13), 4618 CrossRef CAS PubMed.
- J. H. Cho, J. C. Shin, J. J. Cho, Y. H. Choi, J. H. Shim and J. I. Chae, Esculetin (6,7-dihydroxycoumarin): a potential cancer chemopreventive agent through suppression of Sp1 in oral squamous cancer cells, Int. J. Oncol., 2015, 46(1), 265–271 CrossRef CAS PubMed.
- L. Yan, X. Zhou, X. Zhou, Z. Zhang and H. M. Luo, Neurotrophic effects of 7,8-dihydroxycoumarin in primary cultured rat cortical neurons, Neurosci. Bull., 2012, 28(5), 493–498 CrossRef CAS PubMed.
- A. Kornicka, L. Balewski, M. Lahutta and J. Kokoszka, Umbelliferone and Its Synthetic Derivatives as Suitable Molecules for the Development of Agents with Biological Activities: A Review of Their Pharmacological and Therapeutic Potential, Pharmaceuticals, 2023, 16(12), 1732 CrossRef CAS PubMed.
- P. Y. Lin, K. S. Yeh, C. L. Su, S. Y. Sheu, T. Chen, K. L. Ou, M. H. Lin and L. W. Lee, Synthesis and antibacterial activities of novel 4-hydroxy-7-hydroxy- and 3-carboxycoumarin derivatives, Molecules, 2012, 17(9), 10846–10863 CrossRef CAS PubMed.
- Y. Lin, X. Shen, Q. Yuan and Y. Yan, Microbial biosynthesis of the anticoagulant precursor 4-hydroxycoumarin, Nat. Commun., 2013, 4, 2603 CrossRef PubMed.
- V. Flores-Morales, A. P. Villasana-Ruiz, I. Garza-Veloz, S. Gonzalez-Delgado and M. L. Martinez-Fierro, Therapeutic Effects of Coumarins with Different Substitution Patterns, Molecules, 2023, 28(5), 2413 CrossRef CAS PubMed.
- K. Neog, D. Dutta, B. Das and P. Gogoi, Coumarin to Isocoumarin: One-Pot Synthesis of 3-Substituted Isocoumarins from 4-Hydroxycoumarins and Benzyne Precursors, Org. Lett., 2017, 19(3), 730–733 CrossRef CAS PubMed.
- X. Feng, Z. Qin, X. Cheng, D. Liu, Y. Peng, H. Huang, B. Song, J. Bian and Z. Li, Copper(II)-Catalyzed Tandem Reaction: Synthesis of Furo[3,2-c]coumarin Derivatives and Evaluation for Photophysical Properties, J. Org. Chem., 2021, 86(18), 12537–12548 CrossRef CAS PubMed.
- M. Ervina, V. K. C. Wea, A. Oktaviani, L. Hartanti, R. Sinansari and Y. R. Wilianto, Steven; Caroline, Solvent preextraction influenced to coumarin and glucose binding capacity of cinnamomi's extracts, J. Adv. Pharm. Technol. Res., 2023, 14(2), 69–74 CrossRef CAS PubMed.
- E. H. Avdovic, I. P. Petrovic, M. J. Stevanovic, L. Saso, J. M. Dimitric Markovic, N. D. Filipovic, M. Z. Zivic, T. N. Cvetic Antic, M. V. Zizic, N. V. Todorovic, M. Vukic, S. R. Trifunovic and Z. S. Markovic, Synthesis and Biological Screening of New 4-Hydroxycoumarin Derivatives and Their Palladium(II) Complexes, Oxid. Med. Cell. Longevity, 2021, 2021, 8849568 CrossRef PubMed.
- S. M. Hashemi, Z. Hosseini-Khah, F. Mahmoudi and S. Emami, Synthesis of 4-Hydroxycoumarin-Based Triazoles/Oxadiazoles as Novel Anticancer Agents, Chem. Biodivers., 2022, 19(10), e202200043 CrossRef CAS PubMed.
- Q. Wang, Y. Chen, G. Li, Z. Liu, J. Ma, M. Liu, D. Li, J. Han and B. Wang, Synthesis and evaluation of bi-functional 7-hydroxycoumarin platinum(IV) complexes as antitumor agents, Bioorg. Med. Chem., 2019, 27(10), 2112–2121 CrossRef CAS PubMed.
- L. Fernandez-Pena, M. J. Matos and E. Lopez, Recent Advances in Biologically Active Coumarins from Marine Sources: Synthesis and Evaluation, Mar. Drugs, 2022, 21(1), 37 CrossRef PubMed.
- S. K. Gadakh, S. Dey and A. Sudalai, Rh-Catalyzed Synthesis of Coumarin Derivatives from Phenolic Acetates and Acrylates via C-H Bond Activation, J. Org. Chem., 2015, 80(22), 11544–11550 CrossRef CAS PubMed.
- H. Li, C. B. Li, X. L. Xu and G. H. Zhou, Effects of illumination and packaging on non-heme iron and color attributes of sliced ham, Meat Sci., 2012, 91(4), 521–526 CrossRef CAS PubMed.
- T. Jiang, C. Li, Y. Zou, J. Zhang, Q. Gan and Y. Yan, Establishing an Autonomous Cascaded Artificial Dynamic (AutoCAD) regulation system for improved pathway performance, Metab. Eng., 2022, 74, 1–10 CrossRef CAS PubMed.
- S. Moser and H. Pichler, Identifying and engineering the ideal microbial terpenoid production host, Appl. Microbiol. Biotechnol., 2019, 103(14), 5501–5516 CrossRef CAS PubMed.
- M. Li, K. R. Kildegaard, Y. Chen, A. Rodriguez, I. Borodina and J. Nielsen, De novo production of resveratrol from glucose or ethanol by engineered Saccharomyces cerevisiae, Metab. Eng., 2015, 32, 1–11 CrossRef PubMed.
- B. G. Lake, Coumarin metabolism, toxicity and carcinogenicity: relevance for human risk assessment, Food Chem. Toxicol., 1999, 37(4), 423–453 CrossRef CAS PubMed.
- C. J. Hsieh, M. Sun, G. Osborne, K. Ricker, F. C. Tsai, K. Li, R. Tomar, J. Phuong, R. Schmitz and M. S. Sandy, Cancer Hazard Identification Integrating Human Variability: The Case of Coumarin, Int. J. Toxicol., 2019, 38(6), 501–552 CrossRef CAS PubMed.
- S. L. Born, D. Caudill, K. L. Fliter and M. P. Purdon, Identification of the cytochromes P450 that catalyze coumarin 3,4-epoxidation and 3-hydroxylation, Drug Metab. Dispos., 2002, 30(5), 483–487 CrossRef CAS PubMed.
- Y. Lin, X. Sun, Q. Yuan and Y. Yan, Combinatorial biosynthesis of plant-specific coumarins in bacteria, Metab. Eng., 2013, 18, 69–77 CrossRef CAS PubMed.
- A. M. Gehring, K. A. Bradley and C. T. Walsh, Enterobactin biosynthesis in Escherichia coli: isochorismate lyase (EntB) is a bifunctional enzyme that is phosphopantetheinylated by EntD and then acylated by EntE using ATP and 2,3-dihydroxybenzoate, Biochemistry, 1997, 36(28), 8495–8503 CrossRef CAS PubMed.
- X. Shen, J. Wang, B. K. Gall, E. M. Ferreira, Q. Yuan and Y. Yan, Establishment of Novel Biosynthetic Pathways for the Production of Salicyl Alcohol and Gentisyl Alcohol in Engineered Escherichia coli, ACS Synth. Biol., 2018, 7(4), 1012–1017 CrossRef CAS PubMed.
- H. Zhao, Y. Xu, S. Lin, J. C. Spain and N. Y. Zhou, The molecular basis for the intramolecular migration (NIH shift) of the carboxyl group during para-hydroxybenzoate catabolism, Mol. Microbiol., 2018, 110(3), 411–424 CrossRef CAS PubMed.
- S. Noda, T. Shirai, Y. Mori, S. Oyama and A. Kondo, Engineering a synthetic pathway for maleate in Escherichia coli, Nat. Commun., 2017, 8(1), 1153 CrossRef PubMed.
- E. Weidel, J. C. de Jong, C. Brengel, M. P. Storz, A. Braunshausen, M. Negri, A. Plaza, A. Steinbach, R. Muller and R. W. Hartmann, Structure optimization of 2-benzamidobenzoic acids as PqsD inhibitors for Pseudomonas aeruginosa infections and elucidation of binding mode by SPR, STD NMR, and molecular docking, J. Med. Chem., 2013, 56(15), 6146–6155 CrossRef CAS PubMed.
- C. Elena, P. Ravasi, M. E. Castelli, S. Peiru and H. G. Menzella, Expression of codon optimized genes in microbial systems: current industrial applications and perspectives, Front. Microbiol., 2014, 5, 21 Search PubMed.
- G. Zhang, M. Hubalewska and Z. Ignatova, Transient ribosomal attenuation coordinates protein synthesis and co-translational folding, Nat. Struct. Mol. Biol., 2009, 16(3), 274–280 CrossRef CAS PubMed.
- A. K. Hess, P. Saffert, K. Liebeton and Z. Ignatova, Optimization of translation profiles enhances protein expression and solubility, PLoS One, 2015, 10(5), e0127039 CrossRef PubMed.
- C. Zhong, P. Wei and Y. P. Zhang, Enhancing functional expression of codon-optimized heterologous enzymes in Escherichia coli BL21(DE3) by selective introduction of synonymous rare codons, Biotechnol. Bioeng., 2017, 114(5), 1054–1064 CrossRef CAS PubMed.
- D. E. Cheong, K. C. Ko, Y. Han, H. G. Jeon, B. H. Sung, G. J. Kim, J. H. Choi and J. J. Song, Enhancing functional expression of heterologous proteins through random substitution of genetic codes in the 5′ coding region, Biotechnol. Bioeng., 2015, 112(4), 822–826 CrossRef CAS PubMed.
- C. Berens and W. Hillen, Gene regulation by tetracyclines. Constraints of resistance regulation in bacteria shape TetR for application in eukaryotes, Eur. J. Biochem., 2003, 270(15), 3109–3121 CrossRef CAS PubMed.
- T. Jiang, Y. Teng, C. Li, Q. Gan, J. Zhang, Y. Zou, B. K. Desai and Y. Yan, Establishing Tunable Genetic Logic Gates with Versatile Dynamic Performance by Varying Regulatory Parameters, ACS Synth. Biol., 2023, 12(12), 3730–3742 CrossRef CAS PubMed.
- C. J. Li and B. M. Trost, Green chemistry for chemical synthesis, Proc. Natl. Acad. Sci. U. S. A., 2008, 105(36), 13197–13202 CrossRef CAS PubMed.
- G. A. Prosser, G. Larrouy-Maumus and L. P. de Carvalho, Metabolomic strategies for the identification of new enzyme functions and metabolic pathways, EMBO Rep., 2014, 15(6), 657–669 CrossRef CAS PubMed.
- M. Cuperlovic-Culf, Machine Learning Methods for Analysis of Metabolic Data and Metabolic Pathway Modeling, Metabolites, 2018, 8(1), 4 CrossRef PubMed.
- G. B. Kim, W. J. Kim, H. U. Kim and S. Y. Lee, Machine learning applications in systems metabolic engineering, Curr. Opin. Biotechnol, 2020, 64, 1–9 CrossRef CAS PubMed.
- D. Lee, O. Redfern and C. Orengo, Predicting protein function from sequence and structure, Nat. Rev. Mol. Cell Biol., 2007, 8(12), 995–1005 CrossRef CAS PubMed.
- S. R. Trevino, J. M. Scholtz and C. N. Pace, Measuring and increasing protein solubility, J. Pharm. Sci., 2008, 97(10), 4155–4166 CrossRef CAS PubMed.
- V. Paraskevopoulou and F. H. Falcone, Polyionic Tags as Enhancers of Protein Solubility in Recombinant Protein Expression, Microorganisms, 2018, 6(2), 47 CrossRef PubMed.
- R. Arya, J. S. Sabir, R. S. Bora and K. S. Saini, Optimization of culture parameters and novel strategies to improve protein solubility, Methods Mol. Biol., 2015, 1258, 45–63 CrossRef CAS PubMed.
- Y. Lin and Y. Yan, Biosynthesis of caffeic acid in Escherichia coli using its endogenous hydroxylase complex, Microb. Cell Fact., 2012, 11, 42 CrossRef CAS PubMed.
- D. Ishiyama, D. Vujaklija and J. Davies, Novel pathway of salicylate degradation by Streptomyces sp. strain WA46, Appl. Environ. Microbiol., 2004, 70(3), 1297–1306 CrossRef CAS PubMed.
- X. Chen, H. Tang, Y. Liu, P. Xu, Y. Xue, K. Lin and C. Cui, Purification and Initial Characterization of 3-Hydroxybenzoate 6-Hydroxylase From a Halophilic Martelella Strain AD-3, Front. Microbiol., 2018, 9, 1335 CrossRef PubMed.
- O. Trott and A. J. Olson, AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading, J. Comput. Chem., 2010, 31(2), 455–461 CrossRef CAS PubMed.
- J. Chiu, P. E. March, R. Lee and D. Tillett, Site-directed, Ligase-Independent Mutagenesis (SLIM): a single-tube methodology approaching 100% efficiency in 4 h, Nucleic Acids Res., 2004, 32(21), e174 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc05694a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |