
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Methyl viologen as a catalytic acceptor for electron donor–acceptor photoinduced cyclization reactions†
Rubaishan
Jeyaseelan‡
,
Wenheng
Liu‡
 ,
Jens
Zumbusch
and
Line
Næsborg
*
,
Jens
Zumbusch
and
Line
Næsborg
*
Westfälische Wilhelms-Universität Münster, Organisch-Chemisches Institut, Corrensstraße 36, 48149 Münster, Germany. E-mail: lnaesbor@uni-muenster.de; Web: https://www.unimuenster.de/Chemie.oc/naesborg/index.html
First published on 3rd December 2024
Abstract
We present a new method for [2 + 2] and [4 + 2] cyclizations via a radical cation formation driven by methyl viologen mediated electron donor–acceptor complexes. This approach enables the synthesis of cyclobutane and cyclohexene derivatives using a catalytic amount of methyl viologen enabled by oxygen as the terminal oxidant.
The concept of electron donor–acceptor (EDA) complexation, also known as charge-transfer complexation, has been explored by scientists since the 1970s.1–6 Initially investigated from a quantum-mechanical perspective by Mulliken and co-workers in the 1950s,7,8 an EDA complex is formed when an electron-rich molecule (the donor) interacts non-covalently with an electron-deficient molecule (the acceptor). Unlike the individual donor or acceptor, the resulting complex typically absorbs visible light through the newly formed charge transfer band. Upon irradiation a single electron transfer (SET) process between the donor and acceptor occurs. The formed radical ion pair can undergo one of two competing pathways: it can either continue to generate the desired intermediate, or it can undergo back electron transfer (BET), which suppresses the formation of the intended product. Early research attempted to overcome BET by incorporating leaving groups to facilitate the generation of new radicals, thus promoting the desired reaction pathway.
Since 2013, the concept of EDA complexation witnessed its renaissance and many combinations of various electron-rich donors and electron-poor acceptors were investigated.9,10 These EDA pairs required the use of both donor and acceptor in stoichiometric amounts in order to achieve the desired product in sufficient yields.11–15 In 2019, Bosque, Bach and co-workers reported an EDA complex using 3-acetoxyquiniculidine as a catalytic electron donor with a tetrachlorophthalamide ester as the electron-poor acceptor (Scheme 1A).16 In 2022, Melchiorre and co-workers applied tetrachlorophthalimides as a catalytic acceptor (Scheme 1B).17 A Giese-type reaction was performed after irradiation of the EDA complex. The phthalimide radical anion was oxidized by the secondary radical, formed the Giese-type product and regenerated the catalytic donor.
 | ||
| Scheme 1 Catalytic donor (A) and acceptor (B) for EDA-complexations. This work on methyl viologen as a catalytic acceptor, oxygen as the terminal oxidant and no use of a leaving group. A = acceptor, D = donor, LG = leaving group, RA = redox auxiliary. | ||
In previous studies, leaving groups are usually essential when applying EDA complexes to synthesis practice,18 and the reactions often required stoichiometric amounts of both electron donors and acceptors. Methyl viologen, which is known as an antimicrobial agent and herbicide, offers a promising alternative to generate EDA complexes and improve atom economy by avoiding the use of leaving groups. It was found to have three thermodynamically stable oxidation states (Scheme 2).19 These three oxidation states are the di-cation MV2+, radical cation MV˙+, and neutral methyl viologen MV. The MV2+ can be reduced to the radical cation state MV˙+, and the reduction potential of MV2+ to MV˙+ is more feasible, which reveals the capability of being a single electron-acceptor.20 Furthermore, previous studies have also demonstrated that methyl viologen can act as an electron acceptor for many substrate classes to form EDA complexes,21–44 but, to the best of our knowledge, synthetic protocols have not been demonstrated. Although, pyridinium salts have been used for radical formations in EDA-complex based strategies.45,46
 | ||
| Scheme 2 Three oxidation states of methyl viologen. | ||
We set out to investigate methyl viologen as a suitable electron-poor acceptor for EDA complexation with different electron donors for application in synthesis.47 Herein, we explore methyl viologen in catalytic amounts for the photoinduction of formal [2 + 2] and [4 + 2] photocycloadditions via EDA complexes.
As our target reaction we selected the radical cationic cyclization reaction between anethole and isoprene to form the desired cyclohexenes.48 In order to enable the desired reactivity, we propose the formation of a charge transfer complex between the electron-rich anethole and the electron-poor methyl viologen, followed by radical addition of the anethole radical cation to isoprene. Therefore, we conducted UV/Vis-studies of this pair in different solvents. In these studies (Fig. 1), we observed the formation of charge transfer bands in trifluoroethanol (TFE), aqueous sodium dodecyl sulfate (SDS) solution and methanol. The formation of the EDA complex was more pronounced in the aqueous micellar solution compared to the organic solvents. This is likely due to the increased local concentration in the aqueous micellar solution leading to a stronger signal for the EDA complexation. Preliminary studies show that no product formation was observed in the aqueous SDS solution (details available in ESI†). However, in TFE the desired product was observed, allowing us to investigate the formal [4 + 2] cycloaddition.
 | ||
| Fig. 1 UV/Vis spectroscopy of trans-anethole and methyl viologen in different solvents. | ||
All UV/Vis spectra exhibited absorption around λmax = 450 nm which could relate to the predicted absorption of the methyl viologen radical cation MV˙+.20 To confirm this hypothesis, a spectroelectrochemical measurement of methyl viologen in TFE was conducted (Fig. 2) and as the reduction potential decreased, an absorption band emerged around λmax = 450 nm. The single reduced methyl viologen correlates to the signal observed in the UV/vis spectra of the mixture of anethole and methyl viologen. Consequently, the presence of this absorption band correlates with the formation of the MV˙+.
 | ||
| Fig. 2 Spectroelectrometric measurements of MVCl2 illustrating the appearance of MV˙+ in TFE. | ||
Our optimization starts with an equimolar amount of MVCl2, 3 equivalents of isoprene in 1 mL TFE (dried over molecular sieves overnight), irradiated with 520 nm green light for 24 h, under these conditions, the expected [4 + 2] cycloadduct was formed in 60% NMR yield (Table 1, entry 1). After successful product formation, we gradually reduced the amount of MVCl2 to 5 mol% (entries 2 and 3), the NMR yield remained constant (entry 3, 60% NMR yield). After decreasing the amount of solvent (entry 4) and reaction time (entry 5), the desired product was isolated in a good yield (entry 6, 72%). We tested various solvents that also exhibit potential ability to stabilize formed radical cation intermediates such as MeNO2 and MeOH (entries 7 and 8), but the desired product was not observed.49 Control reactions (entries 9–11) showed that methyl viologen and light are crucial to form the desired product and oxygen was found to play a vital role in the reaction cycle, likely to regenerate the methyl viologen acceptor. Oxygen has been previously reported to react fast with the methyl viologen radical cation (MV˙+).50–52
|
|
||||
|---|---|---|---|---|
| Entry | MVCl2 | Solvent | Time | Yieldb |
| Reactions were performed with trans-anethole (0.2 mmol), methyl viologen and isoprene (3.0 eq.) in solvent irradiated with a 520 nm LED (10 W) at 20 °C.a Full optimization table is available in the ESI.†b NMR yield (isolated yield).c Dark.d Without MVCl2.e Degassed solvent. | ||||
| 1 | 100 mol% | TFE (2 mL) | 24 h | 60% |
| 2 | 50 mol% | TFE (2 mL) | 24 h | 56% |
| 3 | 5 mol% | TFE (2 mL) | 24 h | 60% |
| 4 | 5 mol% | TFE (1 mL) | 24 h | 64% |
| 5 | 5 mol% | TFE (1 mL) | 16 h | 66% (72%) |
| 6 | 5 mol% | MeOH (1 mL) | 16 h | 0% |
| 7 | 5 mol% | MeNO2 (1 mL) | 16 h | 0% |
| 8 | 5 mol% | DCE![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :TFE (4:1) (1 mL) :TFE (4:1) (1 mL) |
16 h | 3% |
| 9c | 5 mol% | TFE (1 mL) | 16 h | 0% |
| 10d | — | TFE (1 mL) | 16 h | 1% |
| 11e | 5 mol% | TFE (1 mL) | 16 h | 14% |
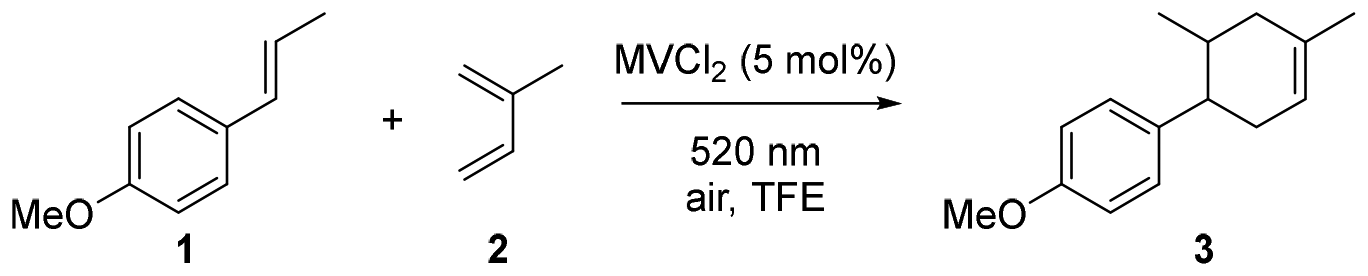
The radical cation cyclization reaction Initiated by the methyl viologen mediated EDA complex formation demonstrates a relatively broad generality for this type of radical chain reaction (Scheme 3). Anethole and compounds with similar structures efficiently formed the [4 + 2] cycloadduct as anticipated (6a–6e). Previous studies have exhibited the crucial role of the alkoxy group on the benzene ring in visible light induced radical cation-based cyclization reactions.53–59 The methyl viologen mediated EDA complexes demonstrate an improved tolerance to other para substituents compared to most strategies and substrates substituted with para-phenyl and para-methyl groups could be converted to the desired products in good yields (6f–6g). Electron-deficient olefins typically favor thermal or acid-mediated [4 + 2] cycloadditions and their exploration in photochemical reactions has been limited.60 In this context, electron-deficient olefins generally do not function well as electron donors in EDA complex formations. However, in this study, we found that these electron withdrawing group (EWG)-conjugated olefins exhibited yields of up to 90% (6h–6j), with aldehyde groups being well-tolerated. N-Vinylcarbazole could also be successfully employed in the [4 + 2] cyclization as an electron-rich olefin (6k), broadening the scope of potential electron donors to be paired with methyl viologen as the acceptor.
 | ||
| Scheme 3 Scope of the radical cation formal Diels–Alder reaction. Reactions were performed with methyl viologen (5 mol%) and diene (3.0 eq.) in dry TFE irradiated with LED light (10 W) at 20 °C. a520 nm LED light (10 W) for 16 h. b520 nm LED light (10 W) for 72 h. c445 nm LED light (10 W) for 24 h. | ||
We propose that the mechanism of the electron rich olefin–MVCl2 EDA complex induced oxidative cycloaddition begins with a charge transfer complex formation with the olefin and methyl viologen, followed by visible light irradiation, which initiates single electron transfer generating a radical ion pair. In the presence of oxygen, the radical cation state of methyl viologen is rapidly oxidized back to its di-cation state, effectively suppressing back electron transfer. The formed radical cation 10 then undergoes radical cyclization to afford the product radical cation 11. This product radical cation can be further oxidized by O2˙− or another equivalent of neutral 7, initiating another round of the radical chain reaction and generating the product 9 (Scheme 4).
 | ||
| Scheme 4 Proposed mechanism. | ||
After successfully exploring the radical cation-based [4 + 2] cyclization, we propose that the radical cation 10 can also be applied in the radical cation-based [2 + 2] cyclization (Scheme 5). We started with anethole and styrene, resulting in a moderate yield with regioselectivity being strictly controlled (13a). Substituting styrene with para-methoxy group led to a decline in yield (13b). When using an electron-deficient ketone, the yield was modest after prolonged irradiation (13c). We found that dimerization is also feasible, 13d was isolated after 48 h irradiation. To inhibit oxidative cleavage of the substrates, we tested degassed solvent, but this only resulted in trace amounts of the product, supporting the proposed role of oxygen in the reaction mechanism. Another dimer 13e was also isolated in a modest yield, which was suppressed by competitive polymerization.
 | ||
| Scheme 5 Scope of the radical cation [2 + 2] cycloaddition. Reactions were performed with methyl viologen (5 mol%) and diene (3.0 eq.) in dry TFE irradiated with LED light (10 W) at 20 °C. a520 nm LED light (10 W) for 48 h. b445 nm LED light (10 W) for 72 h. | ||
Conclusions
In summary, methyl viologen mediated EDA complexes provide an easily available and atom economical method for synthesizing cyclobutane and cyclohexene molecules under very mild conditions. This approach requires a catalytic amount of methyl viologen, which can be regenerated using air as the terminal oxidant. The process effectively inhibits back electron transfer without the need for leaving groups, further improving atom economy. Additionally, the reaction is tolerant to atmospheric conditions and utilizes environmentally friendly visible light for irradiation, making it both efficient and sustainable.Author contributions
R. J., W. L. and L. N. contributed to writing the manuscript. Experiments were performed by R. J., W. L. and J. Z. The project was guided by L. N.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge Elena Horst and Lena Lezius for the spectroelectrochemical experiment. We are grateful for financial support from FCI (Liebig fellowship for L. N. and R. J.) and from DFG (GRK 2678-437785492 for W. L.).References
- D. Cantacuzène, C. Wakselman and R. Dorme, J. Chem. Soc., Perkin Trans. 1, 1977, 1365–1371 RSC.
- J. F. Bunnett, Acc. Chem. Res., 1978, 11, 413–420 CrossRef CAS.
- S. Fukuzumi, K. Mochida and J. K. Kochi, J. Am. Chem. Soc., 1979, 101, 5961–5972 CrossRef CAS.
- S. Sankararaman, W. A. Haney and J. K. Kochi, J. Am. Chem. Soc., 1987, 109, 7824–7838 CrossRef CAS.
- P. A. Wade, H. A. Morrison and N. Kornblum, J. Org. Chem., 1987, 52, 3102–3107 CrossRef CAS.
- G. A. Russell and K. Wang, J. Org. Chem., 1991, 56, 3475–3479 CrossRef CAS.
- R. S. Mulliken, J. Am. Chem. Soc., 1952, 74, 811–824 CrossRef CAS.
- R. S. Mulliken, J. Am. Chem. Soc., 1950, 72, 600–608 CrossRef CAS.
- E. Arceo, I. D. Jurberg, A. Álvarez-Fernández and P. Melchiorre, Nat. Chem., 2013, 5, 750–756 CrossRef CAS.
- M. Tobisu, T. Furukawa and N. Chatani, Chem. Lett., 2013, 42, 1203–1205 CrossRef CAS.
- A. Postigo, Eur. J. Org. Chem., 2018, 6391–6404 CrossRef CAS.
- G. E. M. Crisenza, D. Mazzarella and P. Melchiorre, J. Am. Chem. Soc., 2020, 142, 5461–5476 CrossRef CAS PubMed.
- Y. Yuan, S. Majumder, M. Yang and S. Guo, Tetrahedron Lett., 2020, 61, 151506 CrossRef CAS.
- Z. Yang, Y. Liu, K. Cao, X. Zhang, H. Jiang and J. Li, Beilstein J. Org. Chem., 2021, 17, 771–799 CrossRef CAS.
- L. Zheng, L. Cai, K. Tao, Z. Xie, Y.-L. Lai and W. Guo, Asian J. Org. Chem., 2021, 10, 711–748 CrossRef CAS.
- I. Bosque and T. Bach, ACS Catal., 2019, 9, 9103–9109 CrossRef CAS.
- W. Zhou, S. Wu and P. Melchiorre, J. Am. Chem. Soc., 2022, 144, 8914–8919 CrossRef CAS.
- For laving group-free reactions in electron donor–acceptor complex photochemistry, see: (a) W. Yuan, J. Huang, X. Xu, L. Wang and X.-Y. Tang, Org. Lett., 2021, 23, 7139–7143 CrossRef CAS PubMed; (b) R. Sun, X. Yang, Y. Ge, J. Song, X. Zheng, M. Yuan, R. Li, H. Chen and H. Fu, ACS Catal., 2021, 11, 11762–11773 CrossRef CAS; (c) E. Yoshioka, S. Kohtani, T. Hashimoto, T. Takebe and H. Miyabe, Chem. Pharm. Bull., 2017, 65, 33–35 CrossRef CAS.
- R. Rajaram and L. Neelakantan, Results Chem., 2023, 5, 100703 CrossRef CAS.
- J. Ding, C. Zheng, L. Wang, C. Lu, B. Zhang, Y. Chen, M. Li, G. Zhai and X. Zhuang, J. Mater. Chem. A, 2019, 7, 23337–23360 RSC.
- A. Nakahara and J. H. Wang, J. Phys. Chem., 1963, 67, 496–498 CrossRef CAS.
- F. M. Martens and J. W. Verhoeven, J. Phys. Chem., 1981, 85, 1773–1777 CrossRef CAS.
- F. M. Martens and J. W. Verhoeven, Recl. Trav. Chim. Pays-Bas, 1981, 100, 228–236 CrossRef CAS.
- A. S. N. Murthy and A. P. Bhardwaj, Spectrochim. Acta, Part A, 1982, 38, 207–212 CrossRef.
- M. Z. Hoffman, D. R. Prasad, G. Jones and V. Malba, J. Am. Chem. Soc., 1983, 105, 6360–6362 CrossRef CAS.
- L. Y. C. Lee, J. K. Hurst, M. Politi, K. Kurihara and J. H. Fendler, J. Am. Chem. Soc., 1983, 105, 370–373 CrossRef CAS.
- G. Jones, S. F. Griffin, C. Y. Choi and W. R. Bergmark, J. Org. Chem., 1984, 49, 2705–2708 CrossRef CAS.
- D. R. Prasad and M. Z. Hoffman, J. Phys. Chem., 1984, 88, 5660–5665 CrossRef CAS.
- G. Jones and V. Malba, Chem. Phys. Lett., 1985, 119, 105–110 CrossRef CAS.
- S. G. Bertolotti, J. J. Cosa, H. E. Gsponer, M. Hamity and C. M. Previtali, Can. J. Chem., 1986, 64, 845–848 CrossRef CAS.
- M. Hamity and R. H. Lema, Can. J. Chem., 1991, 69, 146–150 CrossRef CAS.
- M. Hamity and R. H. Lema, J. Photochem. Photobiol., A, 1993, 76, 83–90 CrossRef CAS.
- T. M. Bockman, S. M. Hubig and J. K. Kochi, J. Org. Chem., 1997, 62, 2210–2221 CrossRef CAS.
- M. A. Biasutti, S. G. Bertolotti and C. M. Previtali, J. Braz. Chem. Soc., 1998, 9, 63–68 CrossRef CAS.
- P. M. S. Monk and N. M. Hodgkinson, Electrochim. Acta, 1998, 43, 245–255 CrossRef CAS.
- P. M. S. Monk, N. M. Hodgkinson and R. D. Partridge, Dyes Pigm., 1999, 43, 241–251 CrossRef CAS.
- E. B. de Borba, C. L. C. Amaral, M. J. Politi, R. Villalobos and M. S. Baptista, Langmuir, 2000, 16, 5900–5907 CrossRef CAS.
- C. Prayer, T.-H. Tran-Thi, S. Pommeret, P. d'Oliveira and P. Meynadier, Chem. Phys. Lett., 2000, 323, 467–473 CrossRef CAS.
- H. Kunkely and A. Vogler, Chem. Phys. Lett., 2001, 345, 309–311 CrossRef CAS.
- N. Kakegawa, T. Kondo and M. Ogawa, Langmuir, 2003, 19, 3578–3582 CrossRef CAS.
- Y. Imai, S. Kido, K. Kamon, T. Kinuta, T. Sato, N. Tajima, R. Kuroda and Y. Matsubara, Org. Lett., 2007, 9, 5047–5050 CrossRef CAS.
- Y. Zhu, T. Yu, P. Hao, J. Shen and Y. Fu, J. Cluster Sci., 2016, 27, 1283–1291 CrossRef CAS.
- A. K. Jeevan and K. R. Gopidas, J. Phys. Chem. B, 2021, 125, 4428–4437 CrossRef CAS.
- A. S. Jalilov, S. Patwardhan, A. Singh, T. Simeon, A. A. Sarjeant, G. C. Schatz and F. D. Lewis, J. Phys. Chem. B, 2014, 118, 125–133 CrossRef CAS PubMed.
- S. Cuadros, G. Goti, G. Barison, A. Raulli, T. Bortolato, G. Pelosi, P. Costa and L. Dell'Amico, Angew. Chem., Int. Ed., 2023, 62, e202303585 CrossRef.
- J. Paut, S. Baldon, E. Anselmi, L. Dell'Amico, G. Dagousset and E. Magnier, Adv. Synth. Catal., 2024, 366, 3500–3504 CrossRef CAS.
- S. V. Rosokha and J. K. Kochi, Acc. Chem. Res., 2008, 41, 641–653 CrossRef CAS PubMed.
- F. Mueller and J. Mattay, Chem. Rev., 1993, 93, 99–117 CrossRef CAS.
- N. Shida, Y. Imada, S. Nagahara, Y. Okada and K. Chiba, Chem. Commun., 2019, 2, 1–8 CrossRef.
- C. L. Bird and A. T. Kuhn, Chem. Soc. Rev., 1981, 10, 49 RSC.
- W. H. Koppenol, D. M. Stanbury and P. L. Bounds, Free Radicals Biol. Med., 2010, 49, 317–322 CrossRef CAS.
- P. Wardman, Free Radical Res. Commun., 1991, 14, 57–67 CrossRef CAS.
- J. H. Shin, E. Y. Seong, H. J. Mun, Y. J. Jang and E. J. Kang, Org. Lett., 2018, 20, 5872–5876 CrossRef CAS.
- S. Lin, M. A. Ischay, C. G. Fry and T. P. Yoon, J. Am. Chem. Soc., 2011, 133, 19350–19353 CrossRef CAS.
- M. A. Cismesia and T. P. Yoon, Chem. Sci., 2015, 6, 5426–5434 RSC.
- S. M. Stevenson, M. P. Shores and E. M. Ferreira, Angew. Chem., Int. Ed., 2015, 54, 6506–6510 CrossRef CAS.
- S. S. Rozenel, C. R. Azpilcueta, M. M. Flores-Leonar, J. P. F. Rebolledo-Chávez, L. Ortiz-Frade, C. Amador-Bedolla and E. Martin, Catal. Today, 2018, 310, 2–10 CrossRef CAS.
- K. Tanaka, M. Kishimoto, M. Sukekawa, Y. Hoshino and K. Honda, Tetrahedron Lett., 2018, 59, 3361–3364 CrossRef CAS.
- Y. J. Jang, H. An, S. Choi, J. Hong, S. H. Lee, K.-H. Ahn, Y. You and E. J. Kang, Org. Lett., 2022, 24, 4479–4484 CrossRef CAS.
- S. M. Stevenson, R. F. Higgins, M. P. Shores and E. M. Ferreira, Chem. Sci., 2017, 8, 654–660 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc05481d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |