
Active hydrogen tuning by copper–cobalt bimetal catalysts for boosting ammonia electrosynthesis from simulated wastewater†
Chunqi
Yang
a,
Chang
Liu
a,
Jingwen
Zhuang
a,
Ziyan
Yang
a,
Aiping
Chen
a,
Yuhang
Li
 *a and
Chunzhong
Li
*abc
*a and
Chunzhong
Li
*abc
aKey Laboratory for Ultrafine Materials of Ministry of Education, Shanghai Engineering Research Center of Hierarchical Nanomaterials, School of Materials Science and Engineering, East China University of Science and Technology, Shanghai 200237, China. E-mail: yuhangli@ecust.edu.cn
bSchool of Chemical Engineering, East China University of Science and Technology, Shanghai 200237, China. E-mail: czli@ecust.edu.cn
cDepartment of Chemical Engineering, School of Chemistry and Chemical Engineering, Shanghai Jiao Tong University, Shanghai, 200240, China. E-mail: czlilab@sjtu.edu.cn
First published on 7th November 2024
Abstract
The electrochemical nitrate reduction reaction (NO3RR) represents a promising approach to balance the nitrogen cycle, converting environmental pollutant NO3− to valuable ammonia (NH3). However, the whole reaction involves complex proton-coupled electron transfer processes, requiring the development of efficient catalysts. Owing to unique d-orbitals, Cu-based catalysts exhibit excellent performance. Here, we design a Cu5–Co5 bimetal nanocomposite that achieves a high FENH3 of 94.1%, a yield rate of 14.8 mg h−1 cm−2 and great stability over twenty hours. The yield rate can be enhanced in a flow cell and reach 30.9 mg h−1 cm−2. We test the performance of the Cu5–Co5 catalyst for simulated wastewater treatment, exhibiting a yield rate of 6.7 mg h−1 cm−2 at −100 mA cm−2. Furthermore, in situ ATR-SEIRAS and Raman spectra reveal the reaction pathway on the Cu5–Co5 catalyst. The Cu can adsorb NO3− and convert to *NO2−, while Co(OH)2 derived from metallic Co can promote water spillover and facilitate the subsequent *NO2−-to-NH3 conversion.
1 Introduction
The natural nitrogen cycle is regarded as one of the most essential biochemical cycles, closely related to human beings and ecological environments.1,2 Versatile nitrogen-containing species within the nitrogen cycle can be mutually interconverted to maintain this balance.3 However, this natural nitrogen balance has been disrupted by excessive human activity, such as excessive use of fertilizer, emission of substandard industrial waste and combustion of fuels. Nitrate (NO3−) is widespread in agricultural and industrial wastewater as the primary nitrogenous pollutant, which poses a significant hazard. The over-accumulation of NO3− in water will affect water quality to damage ecosystems and induce cancer to endanger human health.4,5 The current treatment methods are biological denitrification and ion exchange, which are inefficient for high-concentration pollutants.6 Biological denitrification is an important route for removing NO3−: denitrifying bacteria in nature can convert N in harmful NO3− to harmless nitrogen (N2).7 Ammonia (NH3), which is a fundamental and adaptable nitrogen-containing feedstock for fertilizers, pharmaceuticals and chemicals, is a higher value-added product compared to the worthless product N2.8 Meanwhile, NH3 with high hydrogen (H2) storage capacity and zero emission is also regarded as a next-generation energy carrier.9 The annual demand for ammonia exceeded 150 million tons in 2023 and its annual production is expected to exceed 200 million tons globally in 2030 and 560 million tons in 2050.10 The Haber–Bosch method is the current method for industrial NH3 production, requiring harsh reaction conditions (over 400–600 K and 200–400 atm) and causing serious pollution (releasing more than 1.2% of greenhouse gas).11 Therefore, exploring a green and sustainable approach to highly efficient NH3 production is very desirable. The conversion of NO3− to NH3 driven by renewable electricity is a promising alternative to the conventional NH3 manufacturing process and an effective method for the removal of nitrogen-containing wastewater. Hence, achieving a high faradaic efficiency (FE) of NH3 (>90%) under high current density (>200 mA cm−2) is necessary.12The electrochemical NO3− reduction reaction (NO3RR) is an eight-electron process, involving a series of by-products (NO2−, N2, N2H4, etc.) and competitive hydrogen evolution reaction (HER) with a two-electron transfer.13,14 The catalyst is the key factor in the whole reaction. Thus, designing an efficient catalyst for active NO3RR is meaningful and imperative. Based on theoretical and experimental results, copper (Cu)-based catalysts can inject electrons into the N–O bond of NO3− to adsorb NO3− and activate NO3− to *NO2−.15,16 Meanwhile, the NO3RR is also a complicated nine-proton coupled process. The following *NO2−-to-*NH3 conversion is a multi-step deoxygenation and hydrogenation process, while Cu-based catalysts have a weak ability to generate active hydrogen (*H) from water dissociation.17,18 The moderate ability of water spillover to provide *H plays an important role, where insufficient *H supply may produce by-product NO2− and the excessive accumulation of *NO2− will deactivate Cu sites leading to sluggish reaction kinetics and inferior selectivity.19 Sufficient *H supply can promote the *NO2− conversion to achieve high performance for NH3 generation.20
Enhancement of FE and reaction kinetics is important for catalyst design.21 Various design strategies have been adapted for Cu-based catalysts (alloying, heteroatom doping, amorphous structure, oxygen vacancy defects, etc.).22,23 Li et al. have introduced amorphous CeOx to regulate the electronic structure of Cu (Cu/a-CeOx), achieving a dynamic balance between *H generation and consumption.24 Li et al. have anchored Cu in MnO6 octahedral molecular sieve (Cu-OMS), where dispersed Cu adsorbs NO3− and one-dimensional OMS provides enough *H for sequent protonation.25 However, metal oxide catalysts suffer from poor stability (mostly less than 10 hours) due to reconstruction and active metal site leaching.26 Compounding with high HER active elements is another feasible strategy for tuning of *H.27,28 Zeng et al. have introduced Co3O4 with Cu1–N–C, where Cu1–N–C sites adsorb NO3− to generate *NO2− and adjacent Co3O4 sites can accelerate *NO2− reduction to NH3.29 Sun et al. have alloyed nickel (Ni) with solid-solution Cu to produce a Janus Cu@Ni catalyst. Ni sites can accelerate H2O dissociation to provide *H for the hydrogenation of *NOx, which is generated on the adjacent Cu sites.30
Cobalt-based catalysts are regarded as active catalysts for HER and NO2− reduction reaction (NO2RR). Inspired by the above, we have successfully introduced Co to synthesize Cu–Co bimetal catalysts (denoted as Cux–Coy) through annealing CuCo benzene-1,3,5-tricarboxylate (CuCo-BTC) precursors in a H2/Ar atmosphere. The Cu5–Co5 catalyst exhibits a remarkable NO3RR performance, FENH3 > 80% from −0.2 to −0.8 V versus RHE (reversible hydrogen electrode; the following potentials have been converted into reversible hydrogen electrode values unless otherwise stated), reaching a maximum FENH3 of 94.1% and a yield rate of 14.8 mg h−1 cm−2. Furthermore, the NH3 FE reaches 94.8% and the yield rate can be enhanced to 30.9 mg h−1 cm−2 in a flow cell. Besides, the Cu5–Co5 catalyst exhibits excellent performance in stimulated wastewater where different concentrations of NO3− and NO2− coexist. In 30 mM NOx− electrolyte, the yield rate reaches a maximum of 6.7 mg h−1 cm−2. In situ Raman and XRD results show the metallic Co can transfer Co(OH)2 during the reaction. Besides, in situ ATR-SEIRAS (in situ attenuated total reflection surface-enhanced infrared absorption spectroscopy) combined with in situ Raman results are used to reveal the reaction mechanism and pathway of the NO3RR on the Cu5–Co5 catalyst. The introduction of Co can facilitate the reduction of *NO2− and promote the dissociation of water to provide enough *H for subsequent hydrogenation, improving NH3 generation and exhibiting excellent NO3RR performance. This work provides an efficient catalyst for green NH3 generation through recycling pollutant NO3−.
2 Discussion
2.1 Synthesis and characterization
Various ratios of Cu–Co catalysts are synthesized via a two-step strategy: hydrothermal reaction and calcination post-treatment. Briefly, CuCo-BTC precursors are synthesized through the hydrothermal method and the precursors are subsequently calcined under a H2/Ar atmosphere to obtain Cu–Co nanocomposites (see details in Supporting Information†). A series of characterizations were conducted to investigate the composition and morphology of the obtained catalyst. XRD (X-ray diffraction) patterns of pure Cu and Co are respectively ascribed to metallic Cu and metallic Co (Fig. S1a and S2a†). The peaks of pure Cu at 74.0°, 50.4°, and 43.3° are ascribed to (220), (200) and (111) planes of FCC (face-centered cubic) Cu (JCPDS no. 04-0836). In addition, the diffraction peaks of Co at 75.9°, 51.5° and 44.2° are indexed to (220), (200) and (111) planes of FCC Co (JCPDS no. 15-0806). The XRD pattern of Cu5–Co5 in Fig. 1a demonstrates the purity of the obtained catalyst and there are only normal diffraction peaks belonging to metallic Cu and Co and no obvious peaks for their oxides, implying the successful synthesis of the composite of Cu and Co. The results of ICP-OES (inductively coupled plasma optical emission spectrometry) can prove that the molar ratio of Cu![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Co is close to 5:5 (Table S1†). The morphology and structure of the catalysts were investigated through SEM (scanning electron microscopy) and TEM (transmission electron microscopy). The SEM images show the nanoparticle structure of the pure Cu and pure Co (Fig. S1b and S2b†). In the TEM images, the 0.208 nm lattice spacing of pure Cu corresponds to metallic Cu(110) and the 0.180 nm lattice spacing of pure metallic Co corresponds to Co(110) (Fig. S1c, d and S2c, d†). The SEM results of the Cu5–Co5 catalyst also exhibit nanoparticles and HRTEM images show that Cu5–Co5 has a great crystallinity, the lattice spacings measured to be 0.208 nm and 0.180 nm being respectively ascribed to the (111) facets of metallic Cu and the (200) facets of metallic Co (Fig. 1b and Fig. S3†). The EDX (energy-dispersive X-ray) elemental mapping images demonstrate that Cu and Co are uniformly dispersed (Fig. 1c). The results match well with the XRD observation, proving the successful fabrication of Cu–Co bimetal composition. XPS (X-ray photoelectron spectroscopy) is used to analyze the chemical state of surface elements of the as-prepared Cu5–Co5 catalyst (Fig. S4a†). The measured C 1s high-resolution spectra show two peaks at 284.8 and 285.7 eV, attributed to C–C and C–O respectively (Fig. S4b†).31,32 No obvious peak ascribed to O–C
:Co is close to 5:5 (Table S1†). The morphology and structure of the catalysts were investigated through SEM (scanning electron microscopy) and TEM (transmission electron microscopy). The SEM images show the nanoparticle structure of the pure Cu and pure Co (Fig. S1b and S2b†). In the TEM images, the 0.208 nm lattice spacing of pure Cu corresponds to metallic Cu(110) and the 0.180 nm lattice spacing of pure metallic Co corresponds to Co(110) (Fig. S1c, d and S2c, d†). The SEM results of the Cu5–Co5 catalyst also exhibit nanoparticles and HRTEM images show that Cu5–Co5 has a great crystallinity, the lattice spacings measured to be 0.208 nm and 0.180 nm being respectively ascribed to the (111) facets of metallic Cu and the (200) facets of metallic Co (Fig. 1b and Fig. S3†). The EDX (energy-dispersive X-ray) elemental mapping images demonstrate that Cu and Co are uniformly dispersed (Fig. 1c). The results match well with the XRD observation, proving the successful fabrication of Cu–Co bimetal composition. XPS (X-ray photoelectron spectroscopy) is used to analyze the chemical state of surface elements of the as-prepared Cu5–Co5 catalyst (Fig. S4a†). The measured C 1s high-resolution spectra show two peaks at 284.8 and 285.7 eV, attributed to C–C and C–O respectively (Fig. S4b†).31,32 No obvious peak ascribed to O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O is observed, demonstrating the transformation of carbon after the calcination treatment.33 The Co 2p high-resolution spectrum exhibits the presence of metallic Co and oxidized Co2+ and Co3+ (Fig. 1d). The superficial oxidized Cox+ originates from the oxidation of the surface exposed to air.34 A similar result is observed in the Cu 2p high-resolution spectrum and AES (Auger electron spectroscopy, Fig. 1e and f) of Cu LMN. A set of Cu 2p satellite feature peaks can be observed, which shows that Cu2+ and Cu+ originate from the partial oxidation of the catalyst surface.35,36
O is observed, demonstrating the transformation of carbon after the calcination treatment.33 The Co 2p high-resolution spectrum exhibits the presence of metallic Co and oxidized Co2+ and Co3+ (Fig. 1d). The superficial oxidized Cox+ originates from the oxidation of the surface exposed to air.34 A similar result is observed in the Cu 2p high-resolution spectrum and AES (Auger electron spectroscopy, Fig. 1e and f) of Cu LMN. A set of Cu 2p satellite feature peaks can be observed, which shows that Cu2+ and Cu+ originate from the partial oxidation of the catalyst surface.35,36
 | ||
| Fig. 1 Morphological and structural characterization of the Cu5–Co5 nanocomposite. (a) XRD patterns. (b) HRTEM image. (c) HAADF-STEM image and corresponding EDX mappings. (d) XPS peaks of Co 2p. (e) XPS peaks of Cu 2p. (f) Cu LMN spectrum. | ||
2.2 Electrocatalytic NO3RR performance
The NO3RR performance of as-prepared Cu–Co catalysts has been investigated in an H-type electrolytic reactor. We initially confirm the NO3RR activity through linear scanning voltammetry (LSV) in 1 M KOH with and without NO3− (Fig. 2a). The LSV curves exhibit that the Cu5–Co5 catalyst possesses the highest current density while Cu possesses the lowest current density, suggesting the incorporation of Co can improve the intrinsic activity of Cu (Fig. S5a†). The reduction peak (S1) near 0 V is ascribed to the adsorption of NO3− (NO3− to *NO3−) and the peak (S2) at −0.3 V is accorded to the reduction of *NO3− (*NO3− to *NO2−).37,38 The peaks of Cu located at approximately −0.2 V and −0.6 V indicate that the composition of Co can promote the reaction to occur at lower potentials. Besides, various atom ratios of Cu–Co catalysts display better current densities than pure Cu and Co, showing that the introduction of Co can improve the NO3RR performance (Fig. S5b†). | ||
| Fig. 2 Electrochemical performances. (a) LSV curves of the Cu5–Co5 catalyst at a scan rate of 10 mV s−1 in 1 M KOH with and without 0.1 M NO3− (the shading S1 and S2 representing peaks near 0 V and −0.3 V). (b) NH3 FEs and yield rates under different potentials (−0.2 V to −0.8 V). (c) NH3 FEs and yield rates of Cu, Co and Cu5–Co5 catalysts under −0.6 V. (d) Curves of capacitance Δj as a function of different scan rates. (e) Nyquist comparison plots at −0.3 V. (f) Tafel slopes. (g) Stability of the Cu5–Co5 catalyst during cyclic reduction tests at −0.6 V. | ||
Chronoamperometry measurements are performed to test the NO3RR activity of the Cu5–Co5 catalyst under various applied potentials. The liquid products (NO2−, NH3 and N2H4) are quantified using various colorimetric methods (Fig. S6†). The gaseous products (H2 and N2) generated during the reaction are detected by gas chromatography. Trace amounts of N2 and N2H4 are detected, which demonstrate that no N–N coupling products are generated during the whole reaction. In a wide potential window (from −0.2 V to −0.8 V), FENH3 are all over 80% which exhibits a volcano-like shape and reaches the highest FENH3 of 94.1% under −0.6 V (Fig. 2b and Fig. S7†). Besides, the yield rate gradually increases and reaches a maximum of 14.8 mg h−1 cm−2, outperforming greatly pure Cu and Co (Fig. 2c). Besides, the Cu5–Co5 catalyst shows the highest FE and yield rate compared to Cu–Co catalysts of other atomic ratios as shown in Fig. S8.† As the percentage of Co increases, the yield rate also increases and then decreases, with the best performance at Cu:Co = 5:5 ratio. We have further compared the Cu5–Co5 catalyst with physically mixed Cu and Co (mixed Cu5–Co5), where Cu5–Co5 exhibits better performance (Fig. S9†). Due to intrinsic retarded kinetics for spillover, pure Cu exhibits the lowest yield and yield increases with the introduction of Co. The introduction of Co sites can promote water dissociation to provide sufficient hydrogen while excessive hydrogen will cause the HER to be the dominant reaction.39 The above results suggest that the presence of Co can improve the NO3RR performance of Cu-based catalysts. We have also synthesized and tested the performance of Cu5Co5 alloy (see details in Supporting Information†). The results demonstrate that the Cu5–Co5 composition exhibits better performance (Fig. S10†).
The Cu5–Co5 catalyst is utilized for further intrinsic electrocatalytic activity analysis. We measure electrochemical double-layer capacitance to evaluate the electrochemical active surface area (ECSA).40 Pure Cu exhibits the lowest ECSA value (0.02 mF cm−2) while Co has a slightly higher ECSA value (0.74 mF cm−2), which suggests that metallic Cu possesses inferior kinetics.16 The Cu5–Co5 catalyst has the highest ECSA value (5.89 mF cm−2), much higher than that of metallic Co and Cu (Fig. 2d and Fig. S11†). The results of ECSA measurements indicate that the addition of Co can expose more active sites. The charge-transfer resistance is compared by electrochemical impedance spectroscopy (EIS) measurements conducted from 105 to 10−1 Hz. The charge-transfer resistance (Rct) and the series resistance (Rs) are obtained through an equivalent circuit fitting, showing that the charge-transfer resistance of the Cu5–Co5 catalyst is smaller than that of metallic Cu and Co (Fig. 2e and Table S2†). Furthermore, the reaction kinetics of catalysts are also analyzed through Tafel slopes. The smallest value for Cu5–Co5 reflects the faster intrinsic kinetics compared to pure Cu and Co, which is favorable for the NO3RR and is consistent with chronoamperometry results (Fig. 2f). The above results imply that the rapid reaction kinetics benefits from the synergy effect between metallic Cu and Co. The above results imply that combining Co with Cu can increase the acceleration of the electron transfer, which can greatly enhance the entire NO3RR performance.41
Consecutive electrolysis cycles under constant potential (−0.6 V) are performed to test the stability of the Cu5–Co5 catalyst. The results display great stability of the obtained catalyst, FENH3 exceeding 90% and yield rate reaching 10 mg h−1 cm−2 before the penultimate cycle (Fig. 2g). The gradual increase of yield during the initial cycles originates from the activation under reduction potential. The Cu5–Co5 catalyst can be operated stably for close to 20 cycles. XPS and XRD are also used to analyse the Cu5–Co5 catalyst after consecutive stability testing (Fig. S12a–c†). The results imply that the decay stems from leaching of Cu in alkaline and air environments, which demonstrates that Cu acts as the predominant sites for the high performance of the NO3RR.42,43 The Co(OH)2 appearing after the reaction originated from the oxidation of metallic Co by NO3− during the reaction (Fig. S12d†).44 Besides, the stability of the Cu5–Co5 catalyst outperforms that of most reported Cu-based catalysts.45–47
Based on the excellent performance of the Cu5–Co5 catalyst in the H-cell, a flow electrolytic cell assembled with an NH3 capture chamber is used to meet the requirements of industrialization for higher current densities (Fig. S13†). In the LSV curve (Fig. 3a), approximately quadruple current densities can be achieved in the flow system due to lower ohmic resistances and faster mass transfer. Chronopotentiometry was used to test the NO3RR performances and the results show that FENH3 reaches up to 94.8% (Fig. 3b and Fig. S14†), while still maintaining high NH3 selectivity. Besides, the yield rate increases with the current density, reaching 30.9 mg h−1 cm−2 under 600 mA cm−2. These results show the promising potential of the Cu5–Co5 catalyst for NO3RR application at industrial current density levels. Considering the coexistence of NO3− and NO2− in wastewater, we stimulate and test the performance of NOx−-to-NH3 conversion in wastewater at different concentrations (varying from 10 mM to 100 mM NOx−). The results display a curve with a volcano shape under different concentrations, with a maximum yield of 6.7 mg h−1 cm−2 at 30 mM NOx− concentration (Fig. 3c). Low concentration will cause the HER to become the dominant reaction while high concentration will cause diffusion problems.48 The performance of Cu5–Co5 in the NOxRR implies the potential ability for the conversion of N-containing wastewater to value-added NH3.
 | ||
| Fig. 3 Electrochemical performances. (a) LSV curves of the Cu5–Co5 catalyst in H-cell and flow cell. (b) NH3 yield rates under different current densities in the flow cell. (c) NH3 FEs and yield rates in various concentrations of NOx−. (d) 1H NMR spectra of produced 15NH4+ and 14NH4+. (e) Comparison of the Cu5–Co5 catalyst with other recently reported electrocatalysts. | ||
To investigate whether there is external NH3 interference, 15N isotope labeling experiments are conducted to confirm the true nitrogen origin of NH3. In the 1H nuclear magnetic resonance spectra (1H NMR) of the electrolyte after 1 h reaction, a triplet coupling peak attributed to 14NH4+ can be observed while a double peak appears using K15NO3 as the reagent (Fig. 3d). These results demonstrate the generated NH3 originates from the electroreduction of NO3− ions rather than from environmental contamination. Meanwhile, we have also utilized a control experiment to confirm the results, which is performed in 1 M KOH without NO3− as the catholyte. The UV-visible spectrum shows that there is a trace amount of NH3, further precluding external interference and demonstrating the reliability of our experiments (Fig. S15a and b†). Furthermore, ion chromatography is also utilized to prove the accuracy and reliability of the colorimetric method, showing that the quantitative results of both methods are close and indicating the accuracy and reliability of the indophenol blue method (Fig. S15c†). Fig. 3e and Table S3† compare the NO3RR performance of the Cu5–Co5 catalyst with those of other catalysts, its performance being superior to those of most recently reported catalysts.49–56
2.3 Reaction pathway
The generation of *H during the reaction can be detected through EPR (electron paramagnetic resonance, Fig. S16a and b†). DMPO (5,5-dimethyl-1-pyrroline-N-oxide) as a trapping agent can combine with *H to form DMPO-H. Besides, the increased signal intensity of Cu5–Co5 demonstrates the introduction of Co can facilitate water dissociation to generate *H. The reaction mechanism is investigated through in situ ATR-SEIRAS spectra in the same electrolyte. ATR-SEIRAS spectra are measured ranging from 0.3 V to −0.6 V (Fig. 4a). The peak near 1350 cm−1 is ascribed to the N–O bond of *NO3− asymmetric stretching vibration and the peak near 1220 cm−1 is attributed to the anti-symmetric stretching vibration of *NO2−.57 The negative trends indicate that NO3− is adsorbed on the Cu5–Co5 surface to generate *NO3− and then reduces to *NO2−. The appearance of NO scaling mode of *NO at approximately 1550 cm−1 shows that consumed *NO2− is converted to *NO.58 Then, the distinct peaks at 1280 cm−1 of *NH, 1150 cm−1 of *NH2 and 1440 cm−1 of NH3 can demonstrate the following deoxygenation and hydrogenation to generate *NH4+.59 The obvious *NO2− and *NO peaks in the spectrum of pure Cu imply the weak ability for *NO2− reduction and hydrogenation, which leads to NO2− as the main by-product (Fig. 4b). The peaks at approximately 3600 cm−1 are ascribed to weak hydrogen bonds of surface adsorption of H2O, which is considered the key to providing *H (Fig. 4c). The peaks shift to lower wavenumber as the applied potential gradually increases while no obvious change is observed for pure Cu (Fig. 4d), showing the introduction of Co sites can improve the adsorption and cleavage of surface H2O to generate more *H to promote hydrogenation steps.60 Consequently, the whole reaction can be regarded as a preliminary deoxygenation and subsequent hydrogenation process. Furthermore, time-dependent ATR-SEIRAS spectra of the Cu5–Co5 catalyst show increasingly negative peaks of *NO2− and increasingly positive peaks of *NH2, indicating the continuous *NO2− consumption and reduction to generate *NH2 (Fig. S16c†). These results demonstrate that *NO2− and *NHx are essential intermediates in the whole reaction.
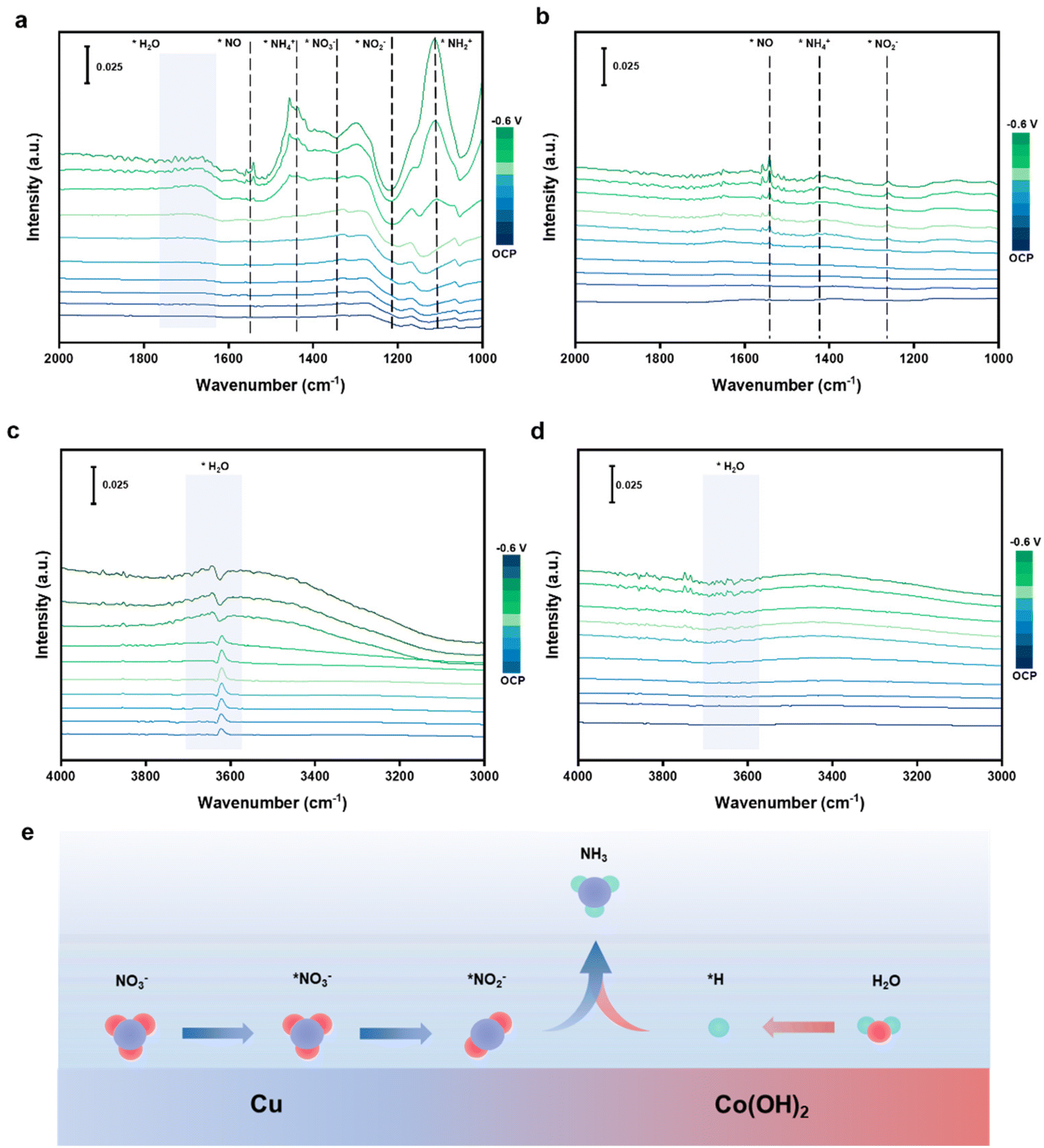 | ||
| Fig. 4 (a–d) In situ ATR-SEIRAS spectra of NO3RR on Cu5–Co5 and Cu catalysts under different potentials. (e) Scheme of the reaction mechanism on the Cu5–Co5 catalyst. | ||
Furthermore, in situ Raman spectra are also used to investigate the surface of the catalyst and reaction mechanism. For the pure Cu catalyst, the characteristic peak of CuO at 290 cm−1 and the characteristic peak of Cu2O at 620 cm−1 due to partial oxidation are observed, which are consistent with the XPS results.24 The peaks gradually shrink while a peak at 690 cm−1 ascribed to Cu-OH is observed, demonstrating the oxide layer on the surface is gradually reduced to metallic Cu (Fig. S17a†).57 For the pure Co catalyst, the characteristic peak of CoOx at 680 cm−1 is observed, which decreases in intensity under the reduction potential and then metallic Co is oxidized to Co(OH)2 by NO3− (Fig. S17b†).44 The oxidized Cu and Co are also observed on the Cu5–Co5 catalyst, proving the negligible effect of the oxide species on the performance (Fig. S17c†). Besides, a similar phenomenon of reconstruction is also observed for the Cu5–Co5 catalyst, which is consistent with the XRD results after reaction (Fig. S12d†). In the Raman spectra measured from OCP to −0.3 V, the characteristic peaks at 1325 cm−1 of NO2−, 1528 cm−1 of *NO and 1610 cm−1 of *NH2 can indicate the N-intermediate reaction pathway, which is consistent with the results of in situ ATR-SEIRAS spectra.24,51
Based on the in situ ATR-SEIRAS and in situ Raman spectra, the NO3RR reaction mechanism on the Cu5–Co5 catalyst can be considered as a series of deoxygenation and proton-coupled electron transfer steps with the following pathway: NO3− → *NO3− → *NO2− → *NO → *NH → *NH2 → *NH3 → NH3. We propose a possible reaction mechanism over the Cu5–Co5 catalyst. NO3− can be adsorbed on Cu sites to generate *NO2−, while the Co(OH)2 sites facilitate the conversion of *NO2− to *NO and the generation of moderate protons to drive subsequent hydrogenation of *NO through water spillover (Fig. 4e).
3 Conclusions
In summary, we have successfully prepared bimetal Cu–Co nanocomposites. The obtained Cu5–Co5 catalyst can exhibit an outstanding performance for the NO3RR to produce valuable NH3, which shows great selectivity for NH3 in a wide potential window ranging from −0.2 V to −0.8 V. Besides, the Cu5–Co5 catalyst can reach a maximum FE of 94.8% and a maximum yield rate of 30.9 mg h−1 cm−2. It also shows great stability of NH3 electrosynthesis for 20 h without obvious decay, outperforming most of the recently reported catalysts. The Cu5–Co5 catalyst can also exhibit a great performance in simulated wastewater electrolytes containing different NOx− concentrations, displaying the potential to convert pollutants to value-added NH3. In situ Raman spectra and XRD reveal that metallic Co can produce Co(OH)2 during the reaction. Based on the in situ ATR-SEIRAS and Raman results, we propose a possible reaction pathway on the Cu5–Co5 catalyst. The Cu sites activate NO3− to form *NO2−. The incorporation of Cu can not only promote the reduction of *NO2− but also facilitate the water dissociation to provide sufficient *H for subsequent hydrogenation, which is beneficial for NH3 generation. Our work can provide a promising and available strategy for the development of efficient NO3RR catalysts, aiming to deal with the treatment of nitrogenous pollutants and maintenance of the nitrogen cycle.Author contributions
C. L. and Y. L. conceived, designed and supervised the project. C. Y. prepared the catalysts, performed the characterizations, conducted electrochemical tests and wrote the paper. C. L., J. Z., Z. Y. and A. C. assisted in experiments and characterization. All authors discussed the results and contributed to the manuscript.Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its ESI.†Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22322805, 22178104, U22B20143), the Shanghai Municipal Science and Technology Major Project, the Shanghai Scientific and Technological Innovation Project (22dz1205900), the “Fundamental Research Funds for the Central Universities”, and the Shanghai Rising-Star Program (23QA1402200).References
- J. Li, H. Al-Mahayni, D. Chartrand, A. Seifitokaldani and N. Kornienko, Nat. Synth., 2023, 2, 757–765 CrossRef.
- X. Fu, J. B. Pedersen, Y. Zhou, M. Saccoccio, S. Li, R. Sažinas, K. Li, S. Z. Andersen, A. Xu, N. H. Deissler, J. B. V. Mygind, C. Wei, J. Kibsgaard, P. C. K. Vesborg, J. K. Nørskov and I. Chorkendorff, Science, 2023, 379, 707–712 CrossRef CAS.
- P. Xu, G. Li, Y. Zheng, J. C. H. Fung, A. Chen, Z. Zeng, H. Shen, M. Hu, J. Mao, Y. Zheng, X. Cui, Z. Guo, Y. Chen, L. Feng, S. He, X. Zhang, A. K. H. Lau, S. Tao and B. Z. Houlton, Nature, 2024, 626, 792–798 CrossRef CAS PubMed.
- B. Gu, X. Zhang, S. K. Lam, Y. Yu, H. J. M. van Grinsven, S. Zhang, X. Wang, B. L. Bodirsky, S. Wang, J. Duan, C. Ren, L. Bouwman, W. de Vries, J. Xu, M. A. Sutton and D. Chen, Nature, 2023, 613, 77–84 CrossRef CAS PubMed.
- H. M. Baulch, E. H. Stanley and E. S. Bernhardt, Nature, 2011, 477, E3–E3 CrossRef CAS.
- J. W. Palko, D. I. Oyarzun, B. Ha, M. Stadermann and J. G. Santiago, Chem. Eng. J., 2018, 334, 1289–1296 CrossRef CAS.
- J. S. Graf, S. Schorn, K. Kitzinger, S. Ahmerkamp, C. Woehle, B. Huettel, C. J. Schubert, M. M. M. Kuypers and J. Milucka, Nature, 2021, 591, 445–450 CrossRef CAS.
- F.-Y. Chen, Z.-Y. Wu, S. Gupta, D. J. Rivera, S. V. Lambeets, S. Pecaut, J. Y. T. Kim, P. Zhu, Y. Z. Finfrock, D. M. Meira, G. King, G. Gao, W. Xu, D. A. Cullen, H. Zhou, Y. Han, D. E. Perea, C. L. Muhich and H. Wang, Nat. Nanotechnol., 2022, 17, 759–767 CrossRef CAS PubMed.
- J. Guo and P. Chen, Chem, 2017, 3, 709–712 CAS.
- M. Shi, D. Bao, J. Yan, H. Zhong and X. Zhang, Acc. Mater. Res., 2024, 5, 160–172 CrossRef CAS.
- S. Z. Andersen, V. Čolić, S. Yang, J. A. Schwalbe, A. C. Nielander, J. M. McEnaney, K. Enemark-Rasmussen, J. G. Baker, A. R. Singh, B. A. Rohr, M. J. Statt, S. J. Blair, S. Mezzavilla, J. Kibsgaard, P. C. K. Vesborg, M. Cargnello, S. F. Bent, T. F. Jaramillo, I. E. L. Stephens, J. K. Nørskov and I. Chorkendorff, Nature, 2019, 570, 504–508 CrossRef CAS PubMed.
- J. Liang, Z. Li, L. Zhang, X. He, Y. Luo, D. Zheng, Y. Wang, T. Li, H. Yan, B. Ying, S. Sun, Q. Liu, M. S. Hamdy, B. Tang and X. Sun, Chem, 2023, 9, 1768–1827 CAS.
- Z. Gong, W. Zhong, Z. He, Q. Liu, H. Chen, D. Zhou, N. Zhang, X. Kang and Y. Chen, Appl. Catal., B, 2022, 305, 121021 CrossRef CAS.
- H. Zhang, C. Wang, H. Luo, J. Chen, M. Kuang and J. Yang, Angew. Chem., Int. Ed., 2023, 62, e202217071 CrossRef CAS.
- Y. Wang, W. Zhou, R. Jia, Y. Yu and B. Zhang, Angew. Chem., Int. Ed., 2020, 59, 5350–5354 CrossRef CAS.
- R. Daiyan, T. Tran-Phu, P. Kumar, K. Iputera, Z. Tong, J. Leverett, M. H. A. Khan, A. Asghar Esmailpour, A. Jalili, M. Lim, A. Tricoli, R.-S. Liu, X. Lu, E. Lovell and R. Amal, Energy Environ. Sci., 2021, 14, 3588–3598 RSC.
- K. Wang, R. Mao, R. Liu, J. Zhang, H. Zhao, W. Ran and X. Zhao, Nat. Water, 2023, 1, 1068–1078 CrossRef.
- K. Fan, W. Xie, J. Li, Y. Sun, P. Xu, Y. Tang, Z. Li and M. Shao, Nat. Commun., 2022, 13, 7958 CrossRef CAS.
- Y. Wang, A. Xu, Z. Wang, L. Huang, J. Li, F. Li, J. Wicks, M. Luo, D.-H. Nam, C.-S. Tan, Y. Ding, J. Wu, Y. Lum, C.-T. Dinh, D. Sinton, G. Zheng and E. H. Sargent, J. Am. Chem. Soc., 2020, 142, 5702–5708 CrossRef CAS PubMed.
- G. Zhang, X. Li, K. Chen, Y. Guo, D. Ma and K. Chu, Angew. Chem., Int. Ed., 2023, 62, e202300054 CrossRef CAS.
- P. Li, R. Li, Y. Liu, M. Xie, Z. Jin and G. Yu, J. Am. Chem. Soc., 2023, 145, 6471–6479 CrossRef CAS PubMed.
- G. Zhang, F. Wang, K. Chen, J. Kang and K. Chu, Adv. Funct. Mater., 2024, 34, 2305372 CrossRef CAS.
- W. Gao, K. Xie, J. Xie, X. Wang, H. Zhang, S. Chen, H. Wang, Z. Li and C. Li, Adv. Mater., 2023, 35, 2202952 CrossRef CAS.
- Y. Li, C. Wang, L. Yang, W. Ge, J. Shen, Y. Zhu and C. Li, Adv. Energy Mater., 2024, 14, 2303863 CrossRef CAS.
- S. Li, C. Xiao, R. Chen, M. Wang, Y. Ma, K. Luo, M. Shen, Y. Zhu, Y. Li and C. Li, Green Chem., 2023, 25, 10549–10555 RSC.
- J. Huo, J.-P. Tessonnier and B. H. Shanks, ACS Catal., 2021, 11, 5248–5270 CrossRef CAS.
- L. Wang, Z. Zeng, W. Gao, T. Maxson, D. Raciti, M. Giroux, X. Pan, C. Wang and J. Greeley, Science, 2019, 363, 870–874 CrossRef CAS PubMed.
- Y. Wang, L. Cao, N. J. Libretto, X. Li, C. Li, Y. Wan, C. He, J. Lee, J. Gregg, H. Zong, D. Su, J. T. Miller, T. Mueller and C. Wang, J. Am. Chem. Soc., 2019, 141, 16635–16642 CrossRef CAS PubMed.
- Y. Liu, J. Wei, Z. Yang, L. Zheng, J. Zhao, Z. Song, Y. Zhou, J. Cheng, J. Meng, Z. Geng and J. Zeng, Nat. Commun., 2024, 15, 3619 CAS.
- Y.-Y. Lou, Q.-Z. Zheng, S.-Y. Zhou, J.-Y. Fang, O. Akdim, X.-Y. Ding, R. Oh, G.-S. Park, X. Huang and S.-G. Sun, ACS Catal., 2024, 14, 5098–5108 CAS.
- R. Chen, L. Cheng, J. Liu, Y. Wang, W. Ge, C. Xiao, H. Jiang, Y. Li and C. Li, Small, 2022, 18, 2200720 CAS.
- J. Cheng, G. Dai, W. Sun, X. Yang, R. Xia, Y. Xu and Y. Mao, Energy Fuels, 2024, 38, 2501–2510 CrossRef CAS.
- Y. Qu, T. Dai, Y. Cui, G. Ding, Y. Zhu, Z. Wang and Q. Jiang, Small, 2024, 20, 2308246 CrossRef CAS PubMed.
- M. Shui, C. Huang, P. Ma, W. Li, Q. He, W. Wu, Y. Tan and J. Bao, Chin. Chem. Lett., 2021, 32, 2203–2206 CrossRef CAS.
- M. Jo and A. Tanaka, Appl. Surf. Sci., 1996, 100–101, 11–14 CrossRef.
- Y. Wang, S. Xia, R. Cai, J. Zhang, J. Wang, C. Yu, J. Cui, Y. Zhang, J. Wu, S. Yang, H. H. Tan and Y. Wu, Small, 2023, 19, 2302295 CrossRef CAS PubMed.
- J.-Y. Fang, Q.-Z. Zheng, Y.-Y. Lou, K.-M. Zhao, S.-N. Hu, G. Li, O. Akdim, X.-Y. Huang and S.-G. Sun, Nat. Commun., 2022, 13, 7899 CrossRef CAS PubMed.
- E. B. Molodkina, M. R. Ehrenburg, Yu. M. Polukarov, A. I. Danilov, J. Souza-Garcia and J. M. Feliu, Electrochim. Acta, 2010, 56, 154–165 CrossRef CAS.
- G. Zhang, X. Li, K. Chen, Y. Guo, D. Ma and K. Chu, Angew. Chem., Int. Ed., 2023, 62, e202300054 CrossRef CAS.
- X. Peng, L. Zeng, D. Wang, Z. Liu, Y. Li, Z. Li, B. Yang, L. Lei, L. Dai and Y. Hou, Chem. Soc. Rev., 2023, 52, 2193–2237 RSC.
- K. Zhang, C. Xiao, Y. Li and C. Li, Nanoscale, 2023, 15, 15649–15655 RSC.
- Y. Liu, J. Wei, Z. Yang, L. Zheng, J. Zhao, Z. Song, Y. Zhou, J. Cheng, J. Meng, Z. Geng and J. Zeng, Nat. Commun., 2024, 15, 3619 CrossRef CAS PubMed.
- Z. Jia, T. Feng, M. Ma, Z. Li and L. Tang, Surf. Interfaces, 2024, 48, 104294 CrossRef CAS.
- S. Han, H. Li, T. Li, F. Chen, R. Yang, Y. Yu and B. Zhang, Nat. Catal., 2023, 6, 402–414 CrossRef CAS.
- Z. Gong, X. Xiang, W. Zhong, C. Jia, P. Chen, N. Zhang, S. Zhao, W. Liu, Y. Chen and Z. Lin, Angew. Chem., Int. Ed., 2023, 62, e202308775 CrossRef CAS PubMed.
- C. Wang, Z. Liu, T. Hu, J. Li, L. Dong, F. Du, C. Li and C. Guo, ChemSusChem, 2021, 14, 1825–1829 CrossRef CAS.
- G.-F. Chen, Y. Yuan, H. Jiang, S.-Y. Ren, L.-X. Ding, L. Ma, T. Wu, J. Lu and H. Wang, Nat. Energy, 2020, 5, 605–613 CrossRef CAS.
- Y. Zhou, R. Duan, H. Li, M. Zhao, C. Ding and C. Li, ACS Catal., 2023, 13, 10846–10854 CrossRef CAS.
- J. Xu, S. Zhang, H. Liu, S. Liu, Y. Yuan, Y. Meng, M. Wang, C. Shen, Q. Peng, J. Chen, X. Wang, L. Song, K. Li and W. Chen, Angew. Chem., Int. Ed., 2023, 62, e202308044 CrossRef CAS PubMed.
- J. Cai, S. Qin, M. A. Akram, X. Hou, P. Jin, F. Wang, B. Zhu, X. Li and L. Feng, J. Mater. Chem. A, 2022, 10, 12669–12678 RSC.
- R. Zhao, Q. Yan, L. Yu, T. Yan, X. Zhu, Z. Zhao, L. Liu and J. Xi, Adv. Mater., 2023, 35, 2306633 CrossRef CAS PubMed.
- Z.-H. Xue, H.-C. Shen, P. Chen, G.-X. Pan, W.-W. Zhang, W.-M. Zhang, S.-N. Zhang, X.-H. Li and C. T. Yavuz, ACS Energy Lett., 2023, 8, 3843–3851 CrossRef CAS.
- R. Zhang, Y. Guo, S. Zhang, D. Chen, Y. Zhao, Z. Huang, L. Ma, P. Li, Q. Yang, G. Liang and C. Zhi, Adv. Energy Mater., 2022, 12, 2103872 CrossRef CAS.
- R. Jia, Y. Wang, C. Wang, Y. Ling, Y. Yu and B. Zhang, ACS Catal., 2020, 10, 3533–3540 CrossRef CAS.
- L. Yang, C. Wang, Y. Li, W. Ge, L. Tang, J. Shen, Y. Zhu and C. Li, Adv. Funct. Mater., 2024, 2401094 CrossRef CAS.
- Y. Wang, M. Sun, J. Zhou, Y. Xiong, Q. Zhang, C. Ye, X. Wang, P. Lu, T. Feng, F. Hao, F. Liu, J. Wang, Y. Ma, J. Yin, S. Chu, L. Gu, B. Huang and Z. Fan, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2306461120 CrossRef CAS.
- J.-Y. Fang, Q.-Z. Zheng, Y.-Y. Lou, K.-M. Zhao, S.-N. Hu, G. Li, O. Akdim, X.-Y. Huang and S.-G. Sun, Nat. Commun., 2022, 13, 7899 CrossRef CAS PubMed.
- G. Zhang, F. Wang, K. Chen, J. Kang and K. Chu, Adv. Funct. Mater., 2024, 34, 2305372 CrossRef CAS.
- L. Tian, R. Hao, Y.-X. Zhou, Y. Zhang, Q.-X. Guan, Y.-P. Liu and W. Li, ACS Sustainable Chem. Eng., 2023, 11, 15865–15875 CrossRef CAS.
- Y. Zhou, R. Duan, H. Li, M. Zhao, C. Ding and C. Li, ACS Catal., 2023, 13, 10846–10854 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc04120h |
| This journal is © The Royal Society of Chemistry 2025 |