
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEngineered enzymatic cascade converts diols to amino alcohols†
Hannah R.
Valentino
 a,
Liangyu
Qian
a,
Jerry M.
Parks
a,
Erin E.
Drufva
a,
Ada
Sedova
a,
Pankti S.
Mehta
b,
Mary P.
Watson
b,
Richard J.
Giannone
a,
Stephanie S.
Galanie‡
a and
Joshua K.
Michener
*a
a,
Liangyu
Qian
a,
Jerry M.
Parks
a,
Erin E.
Drufva
a,
Ada
Sedova
a,
Pankti S.
Mehta
b,
Mary P.
Watson
b,
Richard J.
Giannone
a,
Stephanie S.
Galanie‡
a and
Joshua K.
Michener
*a
aBiosciences Division, Oak Ridge National Laboratory, 1 Bethel Valley RD, Oak Ridge, TN 37831, USA. E-mail: michenerjk@ornl.gov
bDepartment of Chemistry and Biochemistry, University of Delaware, Newark, Delaware 19716, USA
First published on 6th May 2025
Abstract
Aliphatic amino alcohols such as 6-amino-1-hexanol are potential platform chemicals for a variety of advanced materials, but applications are currently limited by reagent costs. Aliphatic amino alcohols can currently be synthesized from biomass-derived diols at elevated temperatures and pressures using Ru-based catalysts that produce a mixture of amino-alcohol, diamine, and cyclic amine products. Replacing chemical amination with an enzymatic cascade would reduce resource needs and enable reactions under milder conditions. In this work, we characterized a two-enzyme cascade that selectively converts C4–C7 diols to the corresponding amino alcohols under aqueous conditions at room temperature and pressure. By engineering the rate-limiting enzyme and optimizing reaction conditions, we increased amino alcohol production nearly 30-fold, achieving a selectivity of 99%. The same enzyme cascade could also be used to convert amino alcohols into cyclic amines through reduction of the corresponding cyclic imine. This engineered cascade provides a green opportunity to sustainably synthesize asymmetric bifunctional platform chemicals.
Green foundation1. An enzyme cascade was engineered for selective conversion of renewable biomass-derived diols to aliphatic amino alcohols2. This enzymatic cascade can replace transition metal catalysts that require flammable reagents with renewable biocatalysts operating under mild conditions 3. Further efforts to increase reaction yield would improve industrial viability and simplify purification |
Introduction
Amino alcohols are important industrial chemicals due to their mixed functional groups. Short-chain amino alcohols such as ethanolamine are readily available, produced at the million ton per year scale, and used in the production of surfactants, as corrosion inhibitors, and for gas purification including CO2 capture.1–3 Longer chain aliphatic amino alcohols such as 6-amino-1-hexanol have a broad range of potential applications, including the synthesis of advanced polymers such as poly(ester amides),4 poly(β-amino ester)s,5 and poly(urea vinyl ether ester)s;6 liquid crystalline elastomers;7 zeolite catalysts;8 and improved solar cells.9 However, applications of aliphatic amino alcohols are limited by cost and availability, since synthetic routes to these compounds are challenging.Short-chain amino alcohols such as ethanolamine are synthesized by direct amination of cyclic ethers (e.g. ethylene oxide or propylene oxide),1 but aliphatic amino alcohols are typically synthesized from diols.10 These diols can be produced from renewable sources, for example through chemical, enzymatic, or chemoenzymatic conversion of biomass-derived sugars into 1,5-pentanediol or 1,6-hexanediol.10–15 Conversion of diols to amino alcohols has generally been studied as a byproduct of diamine production.16–18 While conversions are high, yields are typically low, producing a mixture of amino alcohol, diamine, and cyclic amine products.10 These reactions also require elevated temperatures (∼200 °C) and pressure (10–25 bar H2), with associated flammability risks.15–17 These limitations highlight the need to identify greener and more selective strategies for amino alcohol production.
Biocatalysis offers a potential alternative route for selective monoamination of diols to amino alcohols under mild conditions, since enzymes can be identified or engineered to minimize further amination of the amino alcohol products. A biocatalytic approach would be greener by virtue of replacing transition metal catalysts such as ruthenium with renewable biocatalysts while also reducing the safety risks by eliminating the use of high pressure hydrogen and flammable solvents. However, while the selectivity of biocatalytic approaches can minimize byproduct formation, these methods frequently use relatively low substrate concentrations that also complicate product isolation and wastewater treatment. Therefore, enzyme and process engineering would be needed to minimize separations costs and process waste. Overall, enabling the replacement of petroleum-derived polymer precursors with biomass-derived alternatives will also increase overall materials sustainability.
Biocatalytic conversion of diols to amino alcohols has been demonstrated using a three-enzyme cascade combining an alcohol dehydrogenase (ADH) with a transaminase (TA) and alanine dehydrogenase (AlaDH).19 The ADH first oxidizes the alcohol to an aldehyde, which is then converted to an amine by the TA using L-alanine as the amine donor and generating pyruvate as a byproduct. L-Alanine is regenerated from pyruvate and ammonia by the action of AlaDH. Though effective, this process requires three biocatalysts plus the addition of L-alanine. Alternately, an enzyme cascade has been demonstrated for amino alcohol production from amino acids using a carboxylic acid reductase and aldehyde dehydrogenase, but the ATP requirement for this pathway limits in vitro applications.20 Another two-enzyme cascade has been demonstrated for the conversion of mono-alcohols to mono-amines.21 The first oxidation step is again catalyzed by an ADH, but the amination step is performed by an engineered chimeric amine dehydrogenase (AmDH) that directly converts an aldehyde to a primary amine using ammonia.22 This cascade was successful at conversion of many mono-alcohols with high yield and has recently been demonstrated to regioselectively convert vicinal diols to amino alcohols.23 However, activity with 1,n-diols has not been demonstrated, and high conversion rates required a large amount of enzyme, 3.7 g L−1 of ADH and an unspecified concentration of AmDH.
Our goal for this project was to optimize the minimal ADH/AmDH enzyme cascade for the conversion of 1,n-diols to amino alcohols. This cascade converted a variety of diols into their corresponding amino alcohols, though initially at very low conversion. Combining process optimization with enzyme engineering of the rate-limiting AmDH increased amino alcohol production nearly 30-fold. Intriguingly, our cascade also produced cyclic amines under certain conditions, and cyclic amine production could be increased further through enzyme optimization. Thus, we have engineered an improved enzymatic cascade that converts a variety of 1,n-diols to amino alcohols under moderate conditions.
Experimental
Synthesis and expression of enzymes
Genes encoding ADH24 and AmDH22 were synthesized by GenScript (Piscataway, NJ). ADH was subcloned into pET24b(+) between the restriction sites NheI and DpnII with a C-terminal His6 tag for purification. AmDH was subcloned into pET28a(+) between the restriction sites NcoI and XhoI with a N-terminal His6 tag. Plasmids containing ADH and AmDH were transformed into electrocompetent BL21 (DE3) Escherichia coli cells (Sigma-Aldrich, St Louis, MO) according to the manufacturer's specifications.Expression and purification of ADH, AmDH, and any variants was conducted following previously published methods.21 A colony or glycerol stock of BL21(DE3) E. coli containing plasmid DNA was used to inoculate 10 mL of LB medium supplemented with 100 mg L−1 kanamycin. The culture was incubated at 37 °C and 250 rpm overnight. The overnight culture was then diluted 100-fold into 300 mL of TB medium with 100 mM potassium phosphate pH 7.5 and 100 mg L−1 kanamycin. The culture was incubated at 37 °C at 350 rpm until an OD of 0.6–0.8 was reached. Protein expression was induced by adding IPTG to a final concentration of 0.5 mM. The temperature was decreased to 30 °C and the culture was grown with shaking overnight. Cells were harvested by centrifugation for 30 minutes at 3750 rpm and 4 °C, the supernatant was discarded, and the pellets were stored at −80 °C until further use.
Frozen cell pellets were resuspended in 100 mL of 20 mM Tris-Cl (pH 8), 10 mM imidazole, and 300 mM NaCl supplemented with 25 μL of 1 U μL−1 DNase (Thermo Scientific), 50 ng mL−1 lysozyme, and one SIGMAFAST™ protease inhibitor cocktail tablet (Sigma). Cell suspensions were sonicated using a MISONIX 3000 sonicator for a total of 10 minutes in bursts of 5 seconds on/10 seconds off at 5–7 amplitude. Lysates were centrifuged at 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 rpm and 4 °C for 45 min and the supernatant was removed for purification.
500 rpm and 4 °C for 45 min and the supernatant was removed for purification.
Purification of ADH and AmDH
ADH and AmDH were purified under similar conditions.21 Supernatants were loaded at 5 mL min−1 onto an ÄKTA Start FPLC (Cytiva Marlburough, MA) equipped with a 5 mL HisTrap™ column (Cytiva). The column was washed with 20 mM Tris-Cl (pH 8), 70 mM imidazole, and 300 mM NaCl before eluting with 20 mM Tris-Cl (pH 8), 300 mM imidazole, and 300 mM NaCl at 2 mL min−1. Collected fractions were pooled and exchanged into a storage buffer containing 20 mM Tris-Cl (pH 8) and 10% glycerol using a HiPrep™ 26/10 desalting column (Cytiva). Enzymes were concentrated using an Amicon® Pro centrifugal filter with a 10 kDA cutoff (Millipore). The final purified product was analyzed by SDS-PAGE (ESI Fig. 1 and 2†) and enzyme concentration was measured using the Bradford reagent (Sigma) with a BSA standard curve (OZ Biosciences). Pure enzymes were stored at −80 °C for subsequent activity assays.Enzymatic assays
All tested substrates and their amine products were purchased at analytical grade from standard scientific suppliers. We purchased 6-hydroxyhexanal from Enamine (Kyiv, Ukraine). To test the published ADH/AmDH cascade's activity toward diol substrates, purified enzymes were reacted for 24 h with 5-aminopentanol, 6-aminohexanol, hexanediol, heptanediol, and octanediol individually as substrates. Assay conditions were 2 M ammonium chloride (pH 8), 50 mM substrate, 1 mM NAD+, 10 μM ADH, and 10 μM AmDH. Individual 500 μL enzyme reactions were incubated at 30 °C with shaking for 24 h. Reactions were quenched with 150 μL of 40 mM amylamine in 50% methanol. Quenched reactions were centrifuged for 10 min at 4000 rpm, diluted 20-fold in ultrapure water, and analyzed by LC-MS/MS.Enzyme mutagenesis
Single site-saturation mutagenesis, multi-site-saturation mutagenesis, and random mutagenesis were performed using the Q5 ® Site-Directed Mutagenesis Kit (NEB, Ipswich, MA), QuikChange Multi Site-Directed Mutagenesis Kit (Agilent, Santa Clara, CA), and GeneMorph II EZClone Domain Mutagenesis Kit (Agilent), respectively, according to the manufacturer's directions. The primers used for each mutagenesis library are detailed in the ESI (ESI Tables 1–4†). For random domain mutagenesis conducted with AmDH, 700 ng of template DNA was used to construct a library with low mutation frequency. The library was purified using the QIAquick PCR Purification Kit (Qiagen Germantown, MD) to remove any salts before transformation.Libraries were transformed into BL21(DE3) E. coli by electroporation according to the manufacturer's specifications. Briefly, 2 μL of DNA was added to 50 μL of competent cells, the mixture was transferred to a 1 mm gap cuvette, and electroporated by a Gene Pulser Xcell Microbial System (Biorad Hercules, CA) at 1.8 kV. Cells were immediately diluted with 960 μL of recovery media (Sigma St Louis, MO) and transferred to a sterile 10 mL culture tube. Cells were incubated with shaking at 37 °C for 1 hour, plated onto LB agar plates supplemented with 100 μg mL−1 kanamycin, and incubated at 37 °C overnight.
Enzyme mutant screens
All 96-well plate liquid transfers were conducted with a MicroPro300 (Rainin, Emeryville, CA). Mutant colonies were transferred from agar plates using a PIXL colony picker (Singer Instruments, UK) into deep well 96-well plates (Thermo Scientific) with 350 μL LB media containing 100 μg mL−1 kanamycin and 1% dextrose in each well. Each plate contained 10 control wells with 4 negative and 6 parent or best variant controls. Plates were sealed with AeraSeal film (Excel Scientific, Victorville, CA) and incubated overnight at 37 °C and 80% humidity at 300 rpm in an LT-X shaking incubator (Kuhner, Basel, Switzerland). The next day, 20 μL of each overnight culture was added to 380 μL of TB media containing 100 μg mL−1 kanamycin in a 2.2 mL 96-well deep well plate (VWR) for enzyme expression. Expression plates were sealed and incubated at 37 °C and 300 rpm with 80% humidity until an OD of 0.6–1 was reached. IPTG was then added to each well to a final concentration of 0.5 mM IPTG. Plates were incubated at 30 °C and 300 rpm with 80% humidity overnight. The next day, cell pellets were harvested by centrifugation at 3750 rpm and 4 °C for 20 minutes. The supernatant was discarded and the plates were frozen at −80 °C for at least 30 minutes before proceeding. Cell pellets were lysed by transferring 250 μL of 2 M NH4Cl pH 8, 0.1 mM Ca2Cl, 2.5 mM MgCl, 1 mg mL−1 lysozyme, and 5 μL of 1 U μL−1 DNase into each well. During lysis, plates were agitated using a titer plate shaker for approximately 2 h at room temperature. Lysates were clarified by centrifugation at 3750 rpm and 4 °C for 20 min. The resulting supernatants were used in all screening assays to test variant libraries.AmDH variant activity was determined by measuring 6-aminohexanol production. A stock of 0.25 mM 6-hydroxyhexanal was prepared in DMSO and used immediately. Assay conditions were 2 M NH4Cl (pH 8), 2.5 mM 6-hydroxyhexanal, 0.3 mM NADH, 10% DMSO, and 20% cell lysate at a final reaction volume of 50 μL. Reactions were incubated at 30 °C for 30–60 minutes before quenching in 150 μL of 50% MeOH containing 400 μM amylamine as an internal standard. Quenched reactions were centrifuged at 4000 rpm for 10 min and 10 μL of the reaction was diluted in 190 μL of ultrapure water. Diluted samples were sealed with silicone mats and the abundance of 6-aminohexanol was determined by LC-MS/MS (see analytics). The signal was normalized to the abundance of amylamine for analysis.
The comparison of AmDH+ and AmDH+/L146V activities was determined by measuring 6-aminohexanol production. A stock of 0.25 M 6-hydroxyhexanal was prepared in DMSO and used immediately. Assay conditions were 0.7 M NH4Cl (pH 8), 2.5 mM 6-hydroxyhexanal, 0.7 mM NADH, 10% DMSO, and 2 μM enzyme at a final reaction volume of 50 μL. Reactions were incubated at 30 °C for 15 minutes before quenching in 150 μL of 50% MeOH containing 40 μM amylamine as an internal standard. The sample preparation procedure for LC-MS/MS was the same as the above description for AmDH variants.
ADH kinetics
ADH activity was measured using NAD+ reduction with UV-visible spectrophotometry. All kinetic assays were performed in 2 M NH4Cl (pH 8) with 0.5 μM enzyme at a final volume of 50 μL. Alcohol saturation was measured with a fixed concentration of 10 mM NAD+, and NAD+ saturation was determined in the presence of 100 mM 1,6-hexanediol. Measurements of C4–C7 diols were performed at concentrations of 1–200 mM, amino alcohols at concentrations of 0.5–500 mM, and NAD+ at concentrations of 0.125–19 mM. Data were normalized by subtracting the absorbance at 800 nm from the absorbance at 340 nm and converted to [NADH] using the Beers–Lambert equation with b = 1 cm and εNADH = 6.22 mM−1 cm−1. The initial rate (μM min−1) of NADH production was calculated as the slope of the linear portion of the curve. The initial activity was plotted against varied substrate concentrations and fit (GraphPad Prism) using eqn (1) for alcohol saturation and eqn (2) for NAD+ with the substrate-inhibition model, where v0 is the initial velocity of the enzyme, [E] is the concentration of enzyme, [S] is the concentration of substrate, kcat is the maximal rate of enzyme at saturating substrate concentration, KM is Michaelis–Menton constant, and Ki is the substrate inhibition constant. | (1) |
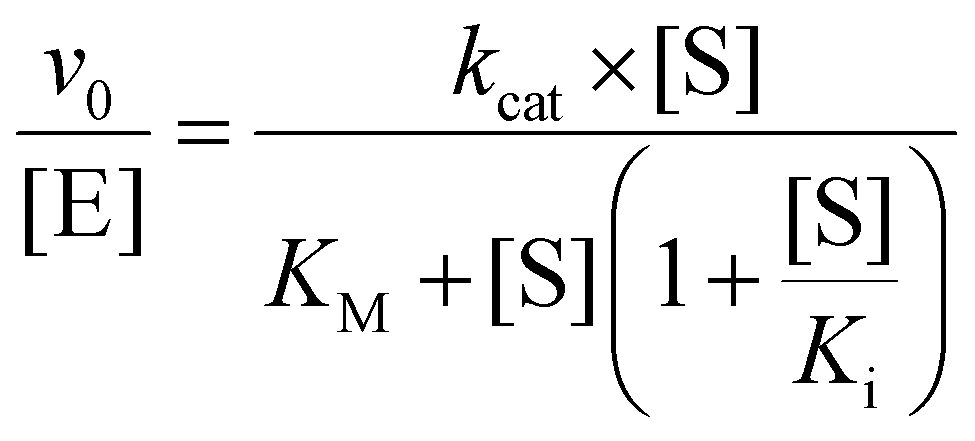 | (2) |
AmDH kinetics
The activity of AmDH and AmDH+ with 6-hydroxyhexanal was measured by 6-aminohexanol detection. A stock of 0.25 mM 6-hydroxyhexanal in DMSO was prepared and used immediately. Assay conditions were 2 M NH4Cl (pH 8), 10% DMSO, 0.7 mM NADH, 10 μM AmDH or 2 μM AmDH+, and 0.05–5 mM of 6-hydroxyhexanal at a final volume of 50 μL. A standard curve was prepared, consisting of 0.01–0.5 mM 6-aminohexanol in 2 M NH4Cl (pH 8) and incubated in 50 μL volumes with the reactions. The reactions and standard curves were incubated at 30 °C and 120 rpm and quenched after 15, 30, 45, and 60 min for analysis. The initial rate as a function of substrate concentration was fit with the Michaelis–Menton model (eqn (1)) to determine steady-state kinetic parameters of AmDH and AmDH+ with 6-hydroxyhexanal. To determine optimal pH, assays were performed with 2 M NH4Cl at pH of 8, 8.5, 9, 9.5, and 10; 5 μM AmDH; 0.3 mM NADH; 2.5 mM 6-hydroxyhexanal; and 10% DMSO at 45 °C and 150 rpm for 1 h.Dual enzyme assays
Initial assay conditions were 2 M NH4Cl (pH 8), 1 mM NAD+, 50 mM substrate, 10 μM ADH, and 10 μM AmDH at a reaction volume of 50 or 75 μL. Reactions were incubated at 30 °C for up to 24 hours before quenching and analysis. Reaction conditions were optimized by varying temperature in a thermal cycler with a gradient from 35–60 °C. Reactions were performed with 2 M NH4Cl (pH 8), 5 mM NAD+, 10 μM ADH, 10 μM AmDH, and 50 mM 1,6-hexanediol for 2.5 hours. For varying NAD+, reactions containing 2 M NH4Cl (pH 8), 0.1–10 mM NAD+, 10 μM ADH, 10 μM AmDH, and 50 mM 1,6-hexanediol were incubated at 40 °C and 150 rpm for 2.5 h. Final reactions with ADH and AmDH/AmDH+ used 2 M NH4Cl (pH 8.5), 1 mM NAD+, 50 mM alcohol or amino alcohol, 10 μM ADH, and 10 μM AmDH and were incubated at 45 °C for 5, 24, and 48 h and then quenched and analyzed.For large scale of 6-aminohexanol production, 2.5 mL reactions were performed with 0.7 M NH4Cl (pH 8.5), 10 mM NAD+, 50 mM 1,6-hexanediol, 10 μM ADH, and 20 μM AmDH+ at 45 °C for 24 h, then quenched and analyzed.
Analytics
Products were detected with an Agilent Infinity II HPLC paired with a 6470 Triple Quad mass spectrometer. Multiple reaction monitoring (MRM) was used to measure the amine products 4-aminobutanol, 5-aminopentanol, 6-aminohexanol, 7-aminoheptanol, pyrrolidine, piperidine, azepane, and azocane (ESI Tables S1 and S2†) without column separation (direct injection through union). Each sample was analyzed for 1 min with an isocratic gradient of 30:70 water:acetonitrile with 0.1% formic acid at a flow rate of 0.4 mL min−1 and a 2 μL sample injection. Data were analyzed using Agilent MassHunter Quantitative Analysis to determine the integrated areas of the MRM ion response. All data were normalized to the response of amylamine and quantified using standard curves.
Azepane production was verified by high-resolution LC-MS/MS using a Vanquish UHPLC directly coupled to a Q Exactive Plus mass spectrometer (Thermo Scientific). For each sample, 5 μL was injected onto a nanospray emitter (75 μM inner diameter packed with 15 cm of Kinetex C18 resin [1.7 μM; Phenomenex]) and separated over a 15 min gradient from 100% solvent A (98:2 water:acetonitrile, 0.1% formic acid) to 75% solvent B (30:70 water:acetonitrile, 0.1% formic acid) utilizing split-flow to adjust flow rates to 300 nL min−1. Parallel Reaction Monitoring (PRM) was used to both determine the elution time and fragmentation profile of the azepane standard ([M + H]+ at 100.1121 m/z) and compare it to samples to verify its production (MS parameters: resolution 17500; AGC target 1e6 with 100 ms max fill time; isolation window of 2.0 m/z with a 0.5 m/z offset; HCD with stepped normalized collision energy of 30/35/40).
6-Aminohexanol production was verified by Agilent Infinity II HPLC paired with a 6470 Triple Quad mass spectrometer. For compound separation, a mixed mode ion-exchange reversed-phase Primesep 100 column (3 μm, 4.6 × 50 mm, SIELC Technologies, Wheeling, IL) and a linear gradient of 2–10% (vol/vol) acetonitrile (MeCN) in water with 20 mM ammonium formate (AmFm) over 18 min at a flow rate of 0.4 mL min−1 were used. The LC condition was as follows: 0 to 3 min, 2% MeCN/20 mM AmFm; 3 to 9 min, 2–10% MeCN/20 mM AmFm; 9 to 12 min, 10% MeCN/20 mM AmFm; 12.01 min to 18 min, 2% MeCN/20 mM AmFm. An MS2 scan was used to determine the elution time and fragmentation profile of a 6-aminohexanol standard ([M + H]+ = 118.1 m/z) and, in future runs, to identify the 6-aminohexanol reaction product. A collision energy of 20 V was used for LC-MS/MS analysis.
Measurement of thermal melting
To determine the melting temperature (Tm) of AmDH and AmDH+, the purified enzymes were diluted to 1 mg mL−1 in 2 M NH4Cl (pH 8). Protein thermal shift assays (PTSAs) were conducted using a QuantStudio 3 Real-Time PCR System instrument (Thermo Fisher Scientific, Waltham, MA) with a final reaction volume of 20 μL per well. Each well contained 5 μL of Thermo Fisher Protein Thermal Shift Buffer (as received), 2.5 μL of Thermo Fisher Protein Thermal Shift Dye (diluted from stock solution, as received, to 8× with distilled water), and 12.5 μL of protein solution. The sample fluorescence was measured while being heated from 25–99 °C at a constant rate of 0.05 °C s−1 after a 2-minute hold at 25 °C, using the Melt Curve experiment settings in the QuantStudio Design and Analysis software, and settings as described in the Thermo Fisher Protein Thermal Shift Studies user guide, after incubation with all well components for 30 minutes on ice. Melting curves were processed and analyzed with Thermo Fisher Protein Thermal Shift software and the Tm was determined using the derivative method in the software.Protein modeling
ColabFold version 1.5.525,26 was used to generate models of AmDH and AmDH+ with default settings. For both proteins the model confidence was high (pLDDT = 93.1 and pTM = 0.89 in each case). Based on a BLAST27 search of the Protein Data Bank (https://www.rcsb.org), the closest homolog with a known structure is a leucine dehydrogenase from Geobacillus stearothermophilus (PDB entry 6ACH).28 We superimposed the cofactor binding domain of each model onto the leucine dehydrogenase template structure to place the NADH cofactor into the models. We then generated force field parameters for NADH with antechamber and sqm from AmberTools2329 using the AM1-BCC charge model30 and the General Amber Force Field.31 The Amber ff19SB force field32 was used to describe the protein. Next, we performed an energy minimization consisting of 500 steps of steepest descent followed by 1500 steps of conjugate gradient with sander.Docking
PDBQT files for the 6-hydroxyhexanal substrate and the receptor were prepared with Meeko (https://github.com/forlilab/Meeko) and prepare_receptor from the ADFR suite,33 respectively. The docking box was centered at C4N of NADH and had dimensions of 16 × 16 × 16 Å. Docking of 6-hydroxyhexanal to both AmDH and AmDH+ was performed with AutoDock Vina 1.2.534 with an exhaustiveness of 32. A total of 100 poses were generated in each case.Results and discussion
To test the utility of the ADH/AmDH cascade for generating aliphatic amino alcohols, we first assessed its activity with diol substrates (Fig. 1A). We purified the two enzymes and incubated them for 24 hours with C5–C7 diols. We detected conversion of all three diol substrates, with the highest production of 250 ± 60 μM 6-amino-1-hexanol from 1,6-hexanediol (Fig. 1B and ESI Table 3†). To identify pathway bottlenecks, we next determined steady state kinetic parameters using ADH and AmDH with the corresponding diol and hydroxyaldehyde substrates (ESI Tables 4 and 5†). We found that the ADH diol binding affinity was poor, with KM values exceeding 30 mM for all tested substrates (Fig. 1C and ESI Table 4†). Similarly, AmDH had much lower activity with 6-hydroxyhexanal than with the previously-tested substrate hexanal (Fig. 1D and ESI Table 5†). | ||
| Fig. 1 Initial pathway characterization. (A) A two-enzyme cascade converts a representative diol, 1,6-hexanediol, to the corresponding amino alcohol. (B) Conversion of diol substrates by the parental enzyme cascade. Purified enzymes (10 μM each) were incubated with 50 mM of the indicated diol substrate. Amino alcohol formation was monitored by HPLC-MS/MS. Error bars show one standard deviation, calculated from three biological replicates. (C) Initial reaction rate for ADH with the indicated diol substrate. Reaction kinetics were determined spectrophotometrically by monitoring NAD+ reduction. (D) Initial reaction rate for AmDH with hexanal or 6-hydroxyhexanal. Reaction kinetics were determined by HPLC-MS detection of the reaction product, hexylamine or 6-amino-1-hexanol. | ||
These enzymes have previously been used to convert hexanol to hexylamine at high yield,21 so we hypothesized that amino alcohol production is limited by the low catalytic efficiency of AmDH with 6-hydroxyhexanal compared to hexanal. Therefore, we chose to target AmDH for mutagenesis to increase activity with hydroxy aldehyde substrates. To this end, we constructed a series of site-saturation mutagenesis libraries targeting residues near the AmDH substrate binding pocket (Fig. 2A). We screened each mutant in cell lysate by HPLC-MS/MS for conversion of 6-hydroxyhexanal to 6-amino-1-hexanol. We then rescreened the most active enzymes and purified those with consistently improved activity for further characterization. We identified two different mutants with approximately 2-fold improved conversion of 6-hydroxyhexanal, containing either the T124C or N303D amino acid replacements (Fig. 2B).
 | ||
| Fig. 2 Initial amine dehydrogenase mutagenesis. (A) Structural model of AmDH with NADH (stick, cyan) bound in the active site. The residues targeted for site-saturation mutagenesis (green) and random domain mutagenesis (blue) are highlighted. (B) Fold improvement of AmDH variants compared to parent. Purified enzymes (10 μM) were incubated with 2.5 mM 6-hydroxyhexanal and production of 6-amino-1-hexanol was monitored by HPLC-MS/MS. Error bars show the standard error, calculated from the indicated biological replicates. Horizontal dashed line indicates parental activity. p values were calculated from a two-tailed t-test. *p < 0.05; **p < 0.01, ***p < 0.001. | ||
We then constructed a random domain mutagenesis library of AmDH in the substrate binding domain, specifically targeting residues 37–168 (Fig. 2A). We assayed 1400 variants for improved amino alcohol production following the same conditions as previously described. From this assay we identified four mutants that resulted in greater than 2-fold improvement over parent, containing the N155S, P158L, G177A, and M130V/C142Y/Y162H amino acid replacements (Fig. 2B).
To identify combinations of amino acid replacements that further improve AmDH activity, we randomly recombined the mutations identified in the previous screens and assayed 700 variants for conversion of 6-hydroxyhexanal to 6-amino-1-hexanol (ESI Table 6†). The top hits all contained a trio of amino acid replacements, T124C/C142Y/N155S, and two isolates with just these three changes had 6.0- and 7.8-fold improvement in activity. Four isolates containing the three core mutations as well as additional P158L or P158R replacements had similar increases in activity. The best mutant, which we termed AmDH+, had five amino acid replacements, T124C/M130 V/C142Y/V146L/N155S, and an overall improvement over parent for 6-hydroxyhexanal conversion of approximately 12-fold (Fig. 3A).
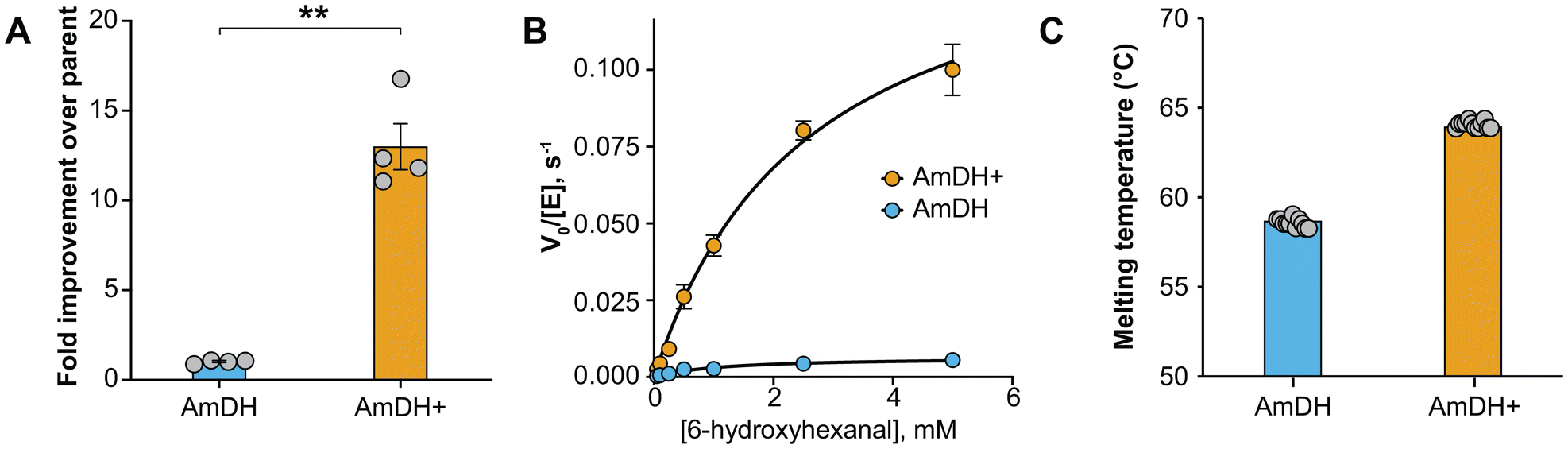 | ||
| Fig. 3 Characterization of improved amine dehydrogenase mutant. (A) Fold improvement of recombined AmDH variants compared to parent. Error bars show the standard error, calculated from the four replicates shown. **p < 0.01, based on a two-tailed t-test. (B) Initial reaction rate for parental and engineered AmDH with 6-hydroxyhexanal. Formation of the 6-amino-1-hexanol product was monitored by HPLC-MS. Error bars show the standard error, calculated from three biological replicates. (C) Thermostability of parental and engineered AmDH. Unfolding was monitored fluorimetrically and the melting temperature was calculated as the temperature that maximized the derivative of fluorescence with respect to temperature. Error bars show one standard deviation, calculated from twelve replicates. | ||
The V146L replacement seen in AmDH+ was the result of a spontaneous mutation during library construction. We reverted this mutation and observed a small but statistically significant decrease in activity, so we proceeded with AmDH+ (ESI Fig. 3†). Characterization of AmDH+ showed that the increased activity was due to a 22 ± 3-fold increase in kcat, partially compensated by a small increase in KM (Fig. 3B). The turnover number for AmDH+ with 6-hydroxyhexanal is comparable to that of wild-type AmDH with hexanal (ESI Table 5†). We also determined that the mutations in AmDH+ produced a significant change in thermal stability, with the Tm increasing by 5 °C compared to the parent, suggesting that AmDH+ is a good candidate for further enzyme engineering (Fig. 3C and ESI Fig. 4†). We note that the parental AmDH is an enzyme chimera, so its folding is likely sub-optimal and readily improved through mutation. For example, the N155S replacement occurs immediately adjacent to the splice site between protein domains and is hypothesized to increase stability by improving domain alignment. Consistent with this hypothesis, the AlphaFold2 model of AmDH+ is slightly more compact than the parent AmDH (ESI Fig. 5†) and has a more enclosed active site (ESI Fig. 6†).
Other replacements, such as T124C and M130V, occur near the active site of the protein and likely improve activity with 6-hydroxyhexanal by restructuring the binding pocket (ESI Fig. 7†). To test this hypothesis, we constructed models of NADH-bound WT AmDH and AmDH+ and docked 6-hydroxyhexanal in the active site (Fig. 4). The aldehyde moiety was located closer to C4 of the nicotinamide ring in the AmDH+ model (3.7 Å) than in the AmDH model (4.2 Å). In addition, the docking score was slightly more favorable for AmDH+ (−4.6 kcal mol−1) than in AmDH (−4.1 kcal mol−1).
 | ||
| Fig. 4 Docked poses of 6-hydroxyhexanal (cyan sticks) to (A) AmDH and (B) AmDH+. Colors: Domain 1, light blue; Domain 2, wheat. NADH is shown in green sticks. | ||
We next optimized reaction temperature, pH, and NAD+ concentration to improve product yield using AmDH+ (Fig. 5). The assays showed a mild effect from varying the NAD+ concentration and clear optima for temperature and pH. The final optimized reaction conditions were 1 mM NAD+, 45 °C, and pH 8.5.
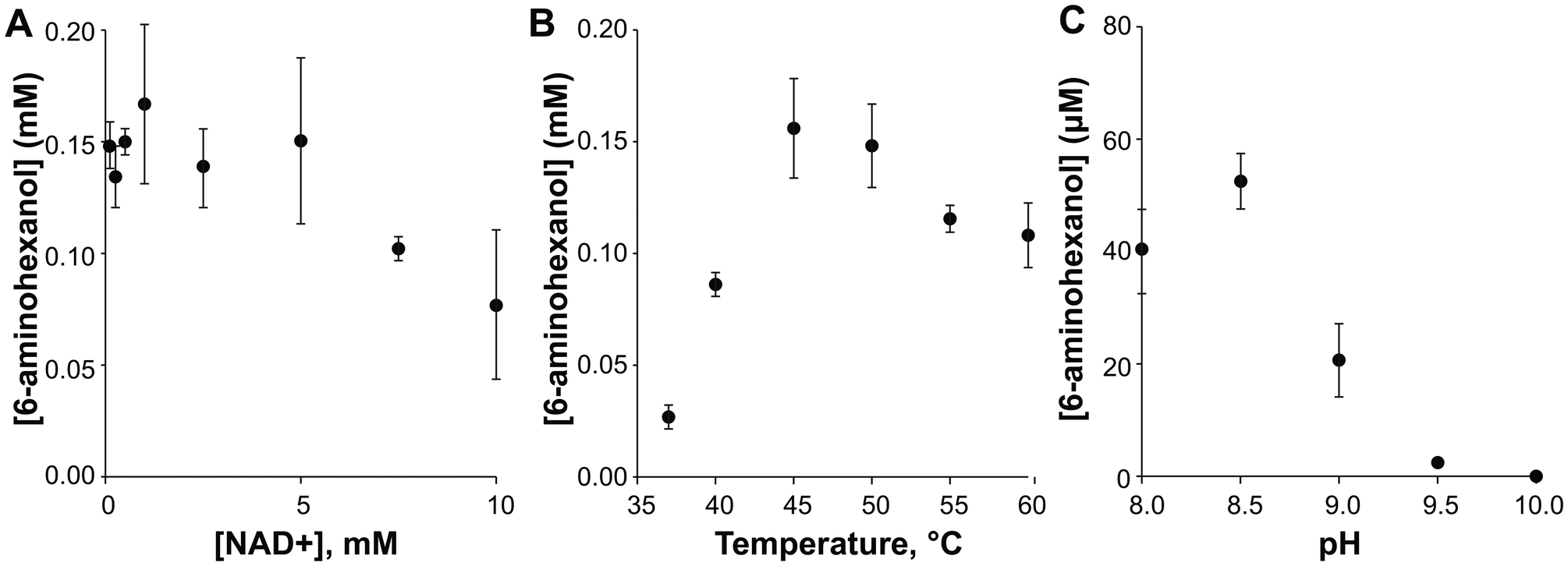 | ||
| Fig. 5 Optimization of reaction conditions for 6-aminohexanol production. In all cases, production of 6-aminohexanol was monitored by LC-MS. Error bars show one standard deviation, calculated from three biological replicates. (A) Optimization of NAD+. Reaction conditions were 10 μM ADH, 10 μM AmDH+, 50 mM 1,6-hexanediol, 2 M ammonium chloride at pH 8.0, and 0.5–10 mM NAD+. Assays were incubated for 2.5 h at 40 °C. (B) Temperature optimization. Reaction conditions were 10 μM ADH, 10 μM AmDH+, 50 mM 1,6-hexanediol, 2 M ammonium chloride at pH 8.0, and 1 mM NAD+ at 35–60 °C. Assays were incubated for 2.5 h. (C) pH optimization for AmDH+. Reaction conditions were 5 μM AmDH+, 2.5 mM 6-hydroxyhexanal, 1 mM NADH, and 2 M ammonium chloride at pH 8–10. Assays were incubated for 1 h at 45 °C. | ||
We tested the parent and engineered AmDH with ADH and a range of C4–C7 diols under these improved conditions and monitored amino alcohol production by HPLC-MS/MS (Fig. 6, ESI Fig. 8, and Table 7†). Introduction of the engineered AmDH+ significantly improved productivity, for example increasing conversion of 1,6-hexanediol to 6-aminohexanol by more than 8-fold. Using ADH and AmDH+, 6-aminohexanol production was then scaled from 50 μL to 2.5 mL with no change in yield (ESI Fig. 9†). The volumetric productivity of 6-amino-1-hexanol production was 0.3 mM h−1.
 | ||
| Fig. 6 Diol conversion using the optimized enzyme cascade. Conversion of indicated diol substrates to (A) amino alcohol products and (B) cyclic amine byproducts by the parental and engineered cascades. Reactions were conducted for 24 h with 10 μM of each enzyme and 50 mM substrate. (C) Proposed reaction scheme for cyclic amine production from amino alcohols, using 6-amino-1-hexanol as a representative substrate. Error bars show one standard deviation, calculated from the indicated biological replicates. N.D.: Not detected. *p < 0.05; **p < 0.01; ***p < 0.001, based on a two-tailed t-test. | ||
Considering potential scale-up, we demonstrated that the diol and amino alcohol can be separated by liquid chromatography. However, at a larger scale, distillation would be the most efficient way to purify the amino alcohol product. Additional enzyme engineering to further improve conversion at higher substrate concentrations would minimize the need to remove residual diol and treat process wastewater. Enzyme immobilization could also improve process economics by enabling enzyme recycling and potentially improving substrate and cofactor channeling through co-localization.35
Under the optimized reaction conditions, we also observed production of the C5 and C6 cyclic amines, piperidine and azepane, from the corresponding diols (Fig. 6B). No diamine production was observed, giving a final selectivity for 6-aminohexanol production of 99%.
To confirm that the cyclic products were generated enzymatically from amino alcohol intermediates, we repeated the conversion assays using C4–C7 amino alcohols as substrates (ESI Fig. 10 and Table 8†). We saw production of all four expected cyclic amines, with highest production of piperidine and azepane. We hypothesize that cyclic amines are generated through spontaneous cyclization of the amino aldehyde followed by imine reduction by AmDH (Fig. 6C). Cyclic amine production increased by as much as 20-fold using AmDH+ as compared to parental AmDH, suggesting that cyclic amine production could also be further increased by enzyme engineering.
Cyclic amines are important precursors in pharmaceutical synthesis, since saturated cyclic amines such as pyrrolidine and piperidine are some of the most frequently used heterocycles in small molecule drugs.36 Industrial synthesis of these compounds uses toxic metal catalysts and harsh reaction conditions.37,38 Recent efforts have sought to identify sustainable routes for N-heterocycles using chemical, enzymatic, and chemoenzymatic approaches.38–40 Typically, these cascades are limited due to narrow substrate scope, expensive cofactors, low substrate tolerance, and low protein expression.39 While preliminary, the further improvements to the redox-neutral cascade reported in this study could produce industrially relevant saturated cyclic amines of various ring sizes with applications in green synthesis.
Conclusions
Combining process optimization with enzyme engineering, we increased 6-aminohexanol production by nearly 30-fold, from 250 μM to 7.3 mM, for a final yield of 15%. Selectivity remained high, with amino alcohol comprising more than 99% of products. Further engineering, particularly using the combined cascade, could increase production of either amino alcohols or cyclic amines.Author contributions
HRV: investigation, methodology, writing – original draft; LQ: investigation, writing – review and editing; JMP: investigation, writing – review and editing; EED: investigation, writing – original draft; AS: investigation, writing – review and editing; PSM: investigation; MPW: supervision, writing – review and editing; RJG: investigation, writing – review and editing; SSG: conceptualization, funding acquisition; JKM: conceptualization, funding acquisition, project administration, supervision, writing – original draft. HRV performed the majority of the enzyme characterization and engineering. LQ compared AmDH variant activity, performed reaction scale-up, and analytical method development. JMP performed protein modeling and docking analyses. EED constructed and purified select AmDH mutants. AS performed thermostability analyses. PSM tested approaches for product isolation. RJG performed mass spectrometry to confirm cyclic amine production.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported as part of the Center for Plastics Innovation, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Basic Energy Sciences at the University of Delaware under award #DE-SC0021166 and at Oak Ridge National Laboratory under contract #ERKCK55.References
-
M. Ernst, J.-P. Melder, F. I. Berger and C. Koch, Ullmann's Encyclopedia of Industrial Chemistry, 2022, pp. 1–30 Search PubMed
.
- B. Mandald and S. S. Bandyopadhyay, Environ. Sci. Technol., 2006, 40, 6076–6084 CrossRef PubMed
- E. B. Rinker, S. S. Ashour and O. C. Sandall, Ind. Eng. Chem. Res., 2000, 39, 4346–4356 CrossRef CAS
- M. Vera, A. Almontassir, A. Rodríguez-Galán and J. Puiggalí, Macromolecules, 2003, 36, 9784–9796 CrossRef CAS
- Y. Liu, Y. Li, D. Keskin and L. Shi, Adv. Healthcare Mater., 2019, 8, e1801359 CrossRef PubMed
- A. Maity, J. Chen, N. Wilson-Faubert, A. Laventure and A. Nazemi, Macromolecules, 2024, 57, 305–316 CrossRef
- H.-H. Yoon, D.-Y. Kim, K.-U. Jeong and S.-K. Ahn, Macromolecules, 2018, 51, 1141–1149 CrossRef CAS
- Y. Wang, J. Yu, F. Yang, Y. Zhao, J. Zhuang, X. Lu, X. Zhu and K. Zhu, Ind. Eng. Chem. Res., 2023, 62, 10012–10023 CrossRef CAS
- W. Pan, P. Chen, S. Zhu, R. He, Q. Zheng, F. Cao, Z. Lan, J. Wu, W. Sun and Y. Li, Inorg. Chem. Front., 2023, 10, 5734–5744 RSC
- N. K. Gupta, P. Reif, P. Palenicek and M. Rose, ACS Catal., 2022, 12, 10400–10440 CrossRef CAS
- S. Wang, A. Cheng, F. Liu, J. Zhang, T. Xia, X. Zeng, W. Fan and Y. Zhang, Ind. Chem. Mater., 2023, 1, 188–206 RSC
- X. Cen, Y. Liu, B. Chen, D. Liu and Z. Chen, ACS Synth. Biol., 2021, 10, 192–203 CrossRef CAS PubMed
- Z. Zhang, Q. Li, F. Wang, R. Li, X. Yu, L. Kang, J. Zhao and A. Li, Green Chem., 2020, 22, 7476–7483 RSC
- Q. Li, Z. Zhang, J. Zhao and A. Li, Green Chem., 2022, 24, 4270–4303 RSC
- J. Yang, J. Zhang, E. Benassi, X. Li, H. Liu, W. Fang, J. Tian, C. Xia and Z. Huang, Green Chem. Eng., 2024, 5, 119–131 CrossRef
- N. K. Gupta, P. Palenicek, L. Nortmeyer, G. M. Meyer, T. Schäfer, T. Hellmann, J. P. Hofmann and M. Rose, ACS Sustainable Chem. Eng., 2022, 10, 14560–14567 CrossRef CAS
- L. Zhang, Y. Yang, L. Zhou, F. Zhao and H. Cheng, Appl. Catal., A, 2024, 669, 119509 CrossRef CAS
- Y. Li, H. Cheng, C. Zhang, B. Zhang, T. Liu, Q. Wu, X. Su, W. Lin and F. Zhao, Sci. China: Chem., 2017, 60, 920–926 CrossRef CAS
- J. H. Sattler, M. Fuchs, K. Tauber, F. G. Mutti, K. Faber, J. Pfeffer, T. Haas and W. Kroutil, Angew. Chem., Int. Ed., 2012, 51, 9156–9159 CrossRef CAS PubMed
- S. Sarak, H. Jeon, M. D. Patil, T. P. Khobragade, A. D. Pagar, S. Sung, H.-W. Yoo, B.-G. Kim, S. H. Yoon and H. Yun, Catal. Lett., 2020, 150, 3079–3085 CrossRef CAS
- F. G. Mutti, T. Knaus, N. S. Scrutton, M. Breuer and N. J. Turner, Science, 2015, 349, 1525–1529 CrossRef CAS PubMed
- B. R. Bommarius, M. Schürmann and A. S. Bommarius, Chem. Commun., 2014, 50, 14953–14955 RSC
- M. L. Corrado, T. Knaus and F. G. Mutti, Green Chem., 2019, 21, 6246–6251 RSC
- X. Guo, Y. Feng, X. Wang, Y. Liu, W. Liu, Q. Li, J. Wang, S. Xue and Z. K. Zhao, Bioorg. Med. Chem. Lett., 2019, 29, 1446–1449 CrossRef CAS PubMed
- M. Mirdita, K. Schütze, Y. Moriwaki, L. Heo, S. Ovchinnikov and M. Steinegger, Nat. Methods, 2022, 19, 679–682 CrossRef CAS PubMed
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Žídek, A. Potapenko, A. Bridgland, C. Meyer, S. A. A. Kohl, A. J. Ballard, A. Cowie, B. Romera-Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. Silver, O. Vinyals, A. W. Senior, K. Kavukcuoglu, P. Kohli and D. Hassabis, Nature, 2021, 596, 583–589 CrossRef CAS PubMed
- S. F. Altschul, W. Gish, W. Miller, E. W. Myers and D. J. Lipman, J. Mol. Biol., 1990, 215, 403–410 CrossRef CAS PubMed
- H. Yamaguchi, A. Kamegawa, K. Nakata, T. Kashiwagi, T. Mizukoshi, Y. Fujiyoshi and K. Tani, J. Struct. Biol., 2019, 205, 11–21 CrossRef CAS PubMed
- D. A. Case, H. M. Aktulga, K. Belfon, D. S. Cerutti, G. A. Cisneros, V. W. D. Cruzeiro, N. Forouzesh, T. J. Giese, A. W. Götz, H. Gohlke, S. Izadi, K. Kasavajhala, M. C. Kaymak, E. King, T. Kurtzman, T.-S. Lee, P. Li, J. Liu, T. Luchko, R. Luo, M. Manathunga, M. R. Machado, H. M. Nguyen, K. A. O'Hearn, A. V. Onufriev, F. Pan, S. Pantano, R. Qi, A. Rahnamoun, A. Risheh, S. Schott-Verdugo, A. Shajan, J. Swails, J. Wang, H. Wei, X. Wu, Y. Wu, S. Zhang, S. Zhao, Q. Zhu, T. E. Cheatham 3rd, D. R. Roe, A. Roitberg, C. Simmerling, D. M. York, M. C. Nagan and K. M. Merz Jr, J. Chem. Inf. Model., 2023, 63, 6183–6191 CrossRef CAS PubMed
- A. Jakalian, D. B. Jack and C. I. Bayly, J. Comput. Chem., 2002, 23, 1623–1641 CrossRef CAS PubMed
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed
- C. Tian, K. Kasavajhala, K. A. A. Belfon, L. Raguette, H. Huang, A. N. Migues, J. Bickel, Y. Wang, J. Pincay, Q. Wu and C. Simmerling, J. Chem. Theory Comput., 2020, 16, 528–552 CrossRef PubMed
- P. A. Ravindranath, S. Forli, D. S. Goodsell, A. J. Olson and M. F. Sanner, PLoS Comput. Biol., 2015, 11, e1004586 CrossRef PubMed
- J. Eberhardt, D. Santos-Martins, A. F. Tillack and S. Forli, J. Chem. Inf. Model., 2021, 61, 3891–3898 CrossRef CAS PubMed
- S. Velasco-Lozano, J. Santiago-Arcos, J. A. Mayoral and F. López-Gallego, ChemCatChem, 2020, 12, 3030–3041 CrossRef CAS
- R. D. Taylor, M. MacCoss and A. D. G. Lawson, J. Med. Chem., 2014, 57, 5845–5859 CrossRef CAS PubMed
-
P. Roose, K. Eller, E. Henkes, R. Rossbacher and H. Höke, Ullmann's Encyclopedia of Industrial Chemistry, pp. 2015, 1–55 Search PubMed
- C.-B. Hong, G. Li and H. Liu, Green Chem., 2023, 25, 3515–3523 RSC
- J. Feng, W.-C. Geng, H. Jiang and B. Wu, Biotechnol. Adv., 2022, 54, 107813 CrossRef CAS PubMed
- H. Li, H. Guo, Z. Fang, T. M. Aida and R. L. Smith, Green Chem., 2020, 22, 582–611 RSC
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc02141j |
| ‡ Present affiliation: Merck & Co., Inc. |
| This journal is © The Royal Society of Chemistry 2025 |